Антагонист рецептора il-8
Номер патента: 15520
Опубликовано: 31.08.2011
Авторы: Буш-Петерсен Якоб, Уэбб Эдвард К., Гудмэн Ричард М., Брук Кристофер С.







Формула / Реферат
1. Соединение, которое является солью п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной.
2. Фармацевтическая композиция, содержащая соль п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной и фармацевтически приемлемый носитель или разбавитель.
3. Применение соли п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной в производстве лекарственного средства для применения в лечении хемокинопосредованного заболевания.
4. Применение соли п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной в производстве лекарственного средства для применения в лечении астмы, хронического обструктивного заболевания легких или респираторного дистресс-синдрома у взрослых.
5. Применение соли п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной в производстве лекарственного средства для применения в лечении хронического обструктивного заболевания легких.
6. Фармацевтическая композиция, содержащая соль п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной и один или более дополнительных терапевтических ингредиентов.
7. Композиция по п.6, где дополнительным терапевтическим ингредиентом является антагонист CXCR3 рецептора или антагонист CCR5 рецептора.
8. Способ получения соли п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной, включающий стадии:
a) смешения ацетонитрила и моногидрата п-толуолсульфокислоты;
b) добавления продукта стадии а) к

растворенному в тетрагидрофуране; и
с) кристаллизации продукта, полученного на стадии b), с ацетонитрилом с использованием затравки конечного продукта.
Текст
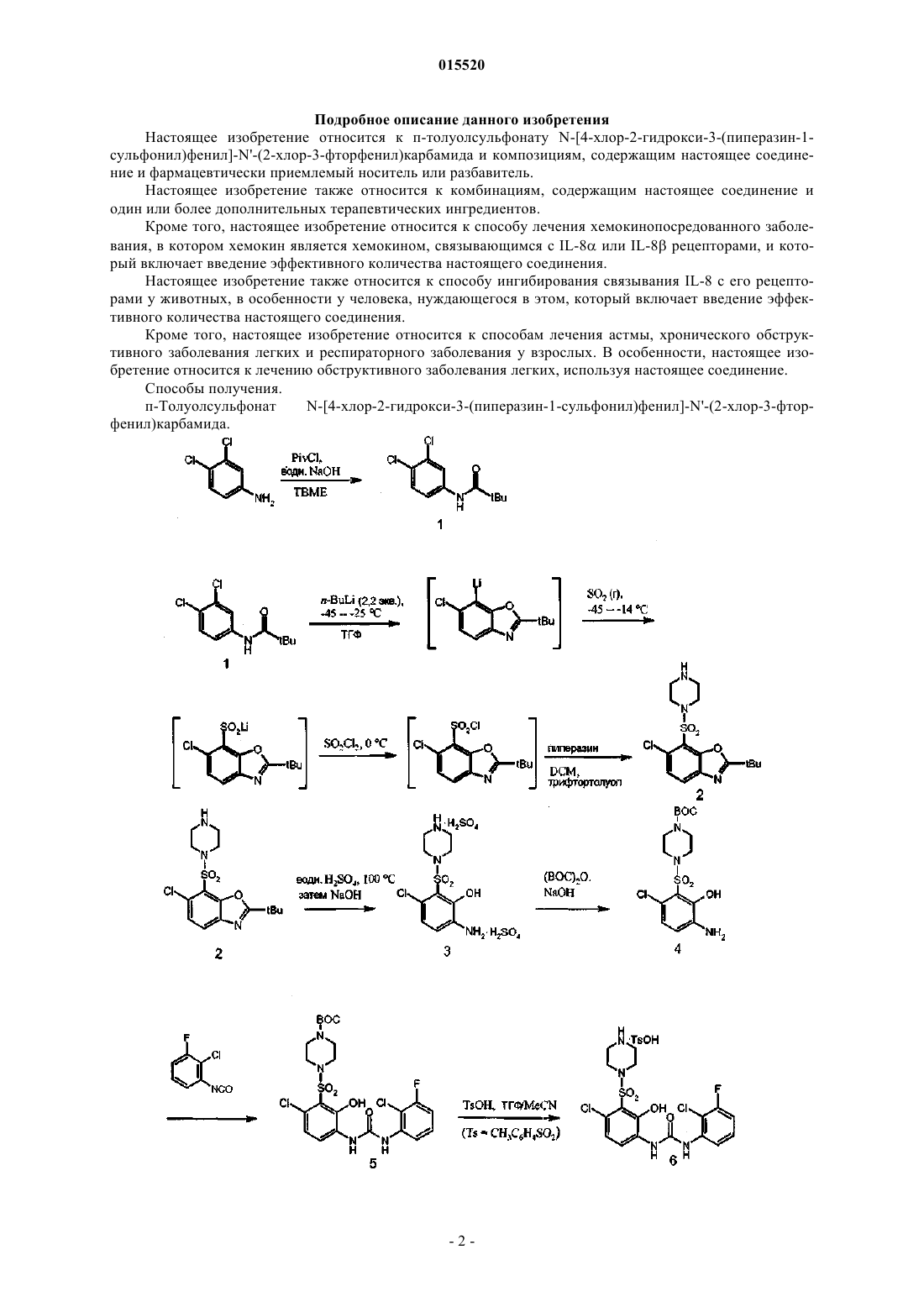
ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ЕВРАЗИЙСКОМУ ПАТЕНТУ Дата публикации и выдачи патента Данное изобретение относится к новому соединению и его композициям, пригодному для лечения болезненного состояния, опосредованного хемокином, интерлейкином-8 (IL-8).(71)(73) Заявитель и патентовладелец: СМИТКЛАЙН БИЧАМ КОРПОРЕЙШН (US) 015520 Область техники, к которой относится изобретение Данное изобретение относится к п-толуолсульфонату N-[4-хлор-2-гидрокси-3-(пиперазин-1 сульфонил)фенил]-N'-(2-хлор-3-фторфенил)карбамида, фармацевтическим композициям, содержащим данное соединение, и его применению для лечения IL-8, GRO, GRO, GRO, NAP-2 и ENA-78 опосредованных заболеваний. Уровень техники Для интерлейкина-8 (IL-8) использовали много различных названий, таких как белок-1 - аттрактант/активатор нейтрофилов (NAP-1), моноцитарный нейтрофильный хемотаксический фактор(MDNCF), нейтрофилактивирующий фактор (NAF) и хемотаксический фактор Т-клеток лимфоцитов. Интерлейкин-8 является хемоаттрактантом для нейтрофилов, базофилов и подкласса Т-клеток. Он вырабатывается большинством ядросодержащих клеток, включая макрофаги, фибробласты, эндотелиальные и эпителиальные клетки, подвергающихся воздействию TNF, IL-1, IL-1 или LPS, и самими нейтрофилами при воздействии на них LPS или хемотаксическими факторами, такими как FMLP.GRO, GRO, GRO и NAP-2 также относятся к хемокиновому -семейству. Подобно IL-8 эти хемокины также имеют различные названия. Например, GRO, ,называют MGSA,исоответственно (активность, стимулирующая рост меланомы). Все хемокины -семейства, которые обладают ELR мотивом, непосредственно предшествующим СХС мотиву, связываются с IL-8 рецептором (CXCR2).IL-8, GRO, GRO, GRO, NAP-2 и ENA-78 стимулируют ряд функций in vitro. Также показано,что все они обладают хемоаттрактантными свойствами для нейтрофилов, тогда как IL-8 и GRO показали Т-лимфоцитную и базофильную хемотаксическую активность. К тому же IL-8 может вызывать высвобождение гистамина из базофилов как у нормальных, так и у атопических индивидов. Кроме того,GRO и IL-8 могут вызывать высвобождение лизосомального фермента и окислительный выброс из нейтрофилов. Также показано, что IL-8 увеличивает поверхностную экспрессию Мас-1 (CD11b/CD18) на нейтрофилах без синтеза белка заново. Это может делать вклад в увеличенную адгезию нейтрофилов к сосудистым эндотелиальным клеткам. Многие известные заболевания характеризуются масштабной нейтрофильной инфильтрацией. Поскольку IL-8, GRO, GRO, GRO и NAP-2 способствуют накоплению и активации нейтрофилов, эти хемокины участвуют в широком диапазоне острых и хронических воспалительных заболеваний, включая псориаз и ревматоидный артрит. Кроме того, ELR хемокины (те, что содержат аминокислотный ELR мотив прямо перед СХС мотивом) также участвуют в ангиостазисе.In vitro IL-8, GRO, GRO, GRO и NAP-2 вызывают изменение формы нейтрофилов, хемотаксис,гранулосекрецию и окислительный выброс, связыванием и активацией рецепторов семитрансмембранного семейства, связанного с G-белком, в особенности связыванием с IL-8 рецепторами, более конкретно сIL-8 рецептором (CXCR2). Имеет место разработка непептидных маленьких молекул - антагонистов для членов этого семейства рецепторов. Следовательно, IL-8 рецептор представляет многообещающую мишень для разработки новых противовоспалительных агентов. В данной области сохраняется необходимость для лечения в соединениях, которые способны связываться с CXCR1 и/или CXCR2 рецепторами. Следовательно, состояния, связанные с увеличением производства IL-8 (которое ответственно за хемотаксис нейтрофильного и Т-клеточного подклассов в воспаленную область) будут улучшаться соединениями, которые являются ингибиторами IL-8 рецепторного связывания. Такие соединения описывают в WO 2004/039775 и в патентах США 6180675 и 6500863. Сущность изобретения Настоящее изобретение относится к п-толуолсульфонату N-[4-хлор-2-гидрокси-3-(пиперазин-1 сульфонил)фенил]-N'-(2-хлор-3-фторфенил)карбамида (настоящее соединение) и композициям, содержащим настоящее соединение и фармацевтически приемлемый носитель или разбавитель. Настоящее изобретение также относится к комбинациям, содержащим настоящее соединение и один или более дополнительных терапевтических ингредиентов. Кроме того, настоящее изобретение относится к способу лечения хемокинопосредованного заболевания, в котором хемокин является хемокином, который связывается с IL-8 или IL-8 рецепторами,который включает введение эффективного количества настоящего соединения. Настоящее изобретение также относится к способу ингибирования связывания IL-8 с его рецепторами у животных, в особенности у человека, нуждающегося в этом, который включает введение эффективного количества настоящего соединения.-1 015520 Подробное описание данного изобретения Настоящее изобретение относится к п-толуолсульфонату N-[4-хлор-2-гидрокси-3-(пиперазин-1 сульфонил)фенил]-N'-(2-хлор-3-фторфенил)карбамида и композициям, содержащим настоящее соединение и фармацевтически приемлемый носитель или разбавитель. Настоящее изобретение также относится к комбинациям, содержащим настоящее соединение и один или более дополнительных терапевтических ингредиентов. Кроме того, настоящее изобретение относится к способу лечения хемокинопосредованного заболевания, в котором хемокин является хемокином, связывающимся с IL-8 или IL-8 рецепторами, и который включает введение эффективного количества настоящего соединения. Настоящее изобретение также относится к способу ингибирования связывания IL-8 с его рецепторами у животных, в особенности у человека, нуждающегося в этом, который включает введение эффективного количества настоящего соединения. Кроме того, настоящее изобретение относится к способам лечения астмы, хронического обструктивного заболевания легких и респираторного заболевания у взрослых. В особенности, настоящее изобретение относится к лечению обструктивного заболевания легких, используя настоящее соединение. Способы получения. п-Толуолсульфонат-2 015520 Получение соединения 1. 3,4-Дихлоранилин (100 г) растворяли в простом трет-бутилметиловом эфире (ТВМЕ) (660 мл) и охлаждали до 10-15 С. Добавляли гидроксид натрия (94 г 30% водного раствора) и раствор энергично перемешивали с помощью механической мешалки. Добавляли триметилацетилхлорид (84 мл) с такой скорость, чтобы поддерживать внутреннюю температуру ниже 35 С. После завершения добавления (10-15 мин) смесь выдерживали при 30-35 С в течение приблизительно 30 мин и затем охлаждали до 0-5C в течение 30-40 мин. Реакционную смесь выдерживали при 0-5 С в течение 1 ч и затем фильтровали, промывали вначале смесью 90:10 вода/метанол(400 мл) и затем водой (600 мл). Сушка при 50-55 С под вакуумом давала продукт в виде грязно-белых кристаллов. Получали выход 127 г. Получение соединения 2. Раствор соединения 1 (300 мл) охлаждали до -(50-40)С в инертной атмосфере азота. Добавляли Nбутиллитий (2,5 M в гексане, 179 мл) с такой скоростью, чтобы поддерживать внутреннюю температуру-(45-30)С (приблизительно 15-30-минутное добавление). Раствор выдерживали при приблизительно-(35-25)С до того момента, пока ВЭЖХ не покажет, что первоначальная реакция завершилась. Затем раствор повторно охлаждали до -(45-40)С и барботировали через раствор диоксид серы (16,9 г), поддерживая внутреннюю температуру ниже приблизительно -14 С, до тех пор, пока раствор не станет кислым. После завершения реакции смесь нагревали до -10-0 С. Затем начиная с -2-3 С, добавляли по каплям сульфурилхлорид (25,2 мл) к тетрагидрофурановому раствору в течение 5-15 мин, поддерживая температуру ниже приблизительно 22 С. Через 5 мин ВЭЖХ подтвердило завершение реакции при выдерживании раствора около 10-15 С. В смеси заменяли растворитель на -трифтортолуол при пониженном давлении, фильтровали, частично концентрировали под вакуумом (до 100 мл) с последующим добавлением дихлорметана (350 мл). К этой смеси добавляли по каплям раствор пиперазина (61,2 г) в дихлорметане (625 мл) при температуре окружающей среды, поддерживая внутреннюю температуру раствора при 15-27 С (2-часовое добавление). Реакционную смесь выдерживали при 20-24 С до завершения реакции. Смесь промывали деионизированной водой (200 мл), органический слой концентрировали с последующим добавлением гептана (450 мл). Продукт (70,5 г) выделяли фильтрацией, промывали гептаном (50-100 мл) и сушили под вакуумом при 50-55 С. Получение соединения 3. Соединение 2 (30 г) добавляли к 16% (по весу в воде) серной кислоте (300 мл). Полученную в результате смесь кипятили с обратным холодильником при 99-103 С в течение 6 ч. После завершения реакции раствор охлаждали до 40-50 С, затем концентрировали до 60 мл при пониженном давлении. Добавляли ацетонитрил (225 мл) и полученную в результате суспензию перемешивали при 20-25 С в течение 1 ч. Продукт выделяли фильтрацией, промывали ацетонитрилом (135 мл) и сушили при 4550 С под вакуумом. Получали выход 33,34 г. Получение соединения 4. Соединение 3 (20 г) добавляли к деионизированной воде (200 мл). рН полученного в результате раствора доводили до 6,5-7,0 добавлением 50% водного гидроксида натрия (6,35 мл) при поддержании внутренней температуры при 20-30 С. Затем добавляли раствор ди-трет-бутилдикарбоната (8,9 г) в этилацетате (80 мл+20 мл промывка). рН полученной в результате смеси доводили до 6,8-7,0 добавлением 50% водного гидроксида натрия (2,45 мл) при поддержании внутренней температуры, равной 20-30 С. После завершения реакции реакционный раствор фильтровали, чтобы удалить небольшое количество осадка. Два слоя фильтрата разделяли и водный слой экстрагировали этилацетатом (140 мл). Объединенные этилацетатные слои промывали водой (40 мл) и концентрировали до 100 мл. Добавляли гептан(100 мл) и полученную в результате суспензию концентрировали до 60 мл. Эту процедуру повторяли еще раз. Затем добавляли гептан (140 мл) и полученную в результате суспензию перемешивали при 20-25 С в течение 1 ч. Продукт выделяли фильтрацией, промывали гептаном (80 мл) и сушили при 40-45 С под вакуумом. Получали выход 15,3 г. Получение соединения 5. Соединение 4 (10 г) добавляли к диметилформамиду (20 мл) и ацетонитрилу (80 мл). Добавляли 2-хлор-3-фторфенилизоционат (4,77 г) при поддержании внутренней температуры между 20-30 С с последующей промывкой 10 мл ацетонитрила. Полученную в результате смесь перемешивали при 20-25 С в течение 2 ч. После завершения реакции добавляли метанол (50 мл). Полученную в результате суспензию перемешивали при 20-25 С в течение 10 мин. Добавляли деионизированную воду (150 мл) и полученную в результате суспензию перемешивали при 20-25 С в течение 1 ч. Продукт выделяли фильтрацией, промывали деионизированной водой (100 мл) и метанолом (15-20 мл) и затем сушили при 40-45 С под вакуумом. Получали выход 14,15 г. Получение соединения 6. Способ 1. Соединение 5 (50 г) растворяли в тетрагидрофуране (ТГФ, 200 мл) и нагревали до 33-37 С и выдерживали при 33-37 С; в другом реакторе готовили раствор ацетонитрила (250 мл), ТГФ (50 мл) и моногидрата п-толуолсульфокислоты (43,9 г). Полученный в результате раствор нагревали до 33-37 С и-3 015520 выдерживали при 33-37 С. Раствор п-толуолсульфокислоты фильтровали и переносили в реактор, содержащий соединение 5 и ТГФ, при поддержании внутренней температуры при 33-37 С. После израсходования исходного материала загружали микронизированную затравку продукта (0,5 г) в минимальном объеме ацетонитрила (5 мл). Затем реакционную смесь нагревали до 53-57 С в течение 40 мин и выдерживали при этой температуре в течение по крайне мере 4 ч. Реакционную смесь охлаждали до 0-5 С,продукт выделяли фильтрацией, промывали ацетонитрилом (250 мл) и сушили под вакуумом при 5560 С. Получали выход 52,24 г. Способ 2. Соединение 5 (500 г) загружали в реактор 1 с последующей загрузкой ацетонитрила (CAN, 3750 мл) и тетрагидрофурана (ТГФ, 1250 мл). Затем раствор нагревали до 60-65 С и сразу наблюдался прозрачный раствор, фильтрацию для осветления проводили в реакторе 2. В реактор 1 добавляли моногидрат п-толуолсульфокислоты (TsOHН 2 О, 439 г) с последующим добавлением ACN (750 мл) и ТГФ (250 мл). Смесь нагревали до 40-45 С и сразу наблюдался прозрачный раствор, проводили фильтрацию для осветления, добавляя раствор в реактор 2 (содержащий раствор исходного материала) и поддерживая температуру в реакторе 2 при 50-60 С. Смесь кипятили с обратным холодильником и выдерживали при 70-80 С до завершения реакции. 3500 мл растворителя удаляли перегонкой при атмосферном давлении. Затем в реактор загружали 2,5 л воды с последующей загрузкой 4 л ACN и температуру выдерживали при 7080 С. После растворения полученный в результате раствор охлаждали до 64-68 С. Через 5-10 мин добавляли в небольшом количестве ацетонитрила измельченную затравку продукта (5 г) и выдерживали при 64-68 С в течение 1 ч. Смесь охлаждали до 0-5 С в течение 2 ч и выдерживали при 0-5C в течение 30 мин перед выделением продукта фильтрацией. Твердый продукт промывали 2,5 л ацетонитрила и сушили под вакуумом при 50-60 С. Получали выход 480 г. Способы лечения. Настоящее соединение является полезным в производстве лекарственного средства для профилактического или терапевтического лечения любого болезненного состояния у человека или другого млекопитающего, которое усиливается или вызывается избыточным или нерегулируемым производством цитокина IL-8 такими клетками млекопитающего, как, но не ограничиваясь ими, моноциты, и/или макрофаги, или другие хемокины, которые связываются с IL-8 или IL-8 рецептором, также называемые как рецептор I или II типа. Соответственно настоящее изобретение относится к способу лечения хемокинопосредованного заболевания, в котором хемокином является хемокин, который связывается с IL-8 или IL-8 рецептором,и способ включает введение эффективного количества настоящего соединения. В частности, хемокинами являются IL-8, GRO, GRO, GRO, NAP-2 или ENA-78. Настоящее соединение вводят в количестве, достаточном для того, чтобы ингибировать цитокиновую функцию, в особенности IL-8, GRO, GRO, GRO, NAP-2 или ENA-78, так что они биологически подавляются до нормальных уровней физиологических функций, или в некоторых случаях до субнормальных уровней так, чтобы облегчать болезненное состояние. Аномальные уровни IL-8, GRO, GRO,GRO, NAP-2 или ENA-78, например, в контексте настоящего изобретения составляют: (i) уровни свободного IL-8, большие чем или равные 1 пг/мл; (ii) любые клетки, связанные с IL-8, GRO, GRO, GRO,NAP-2 или ENA-78, выше нормальных физиологических концентраций или (iii) присутствие IL-8, GRO,GRO, GRO, NAP-2 или ENA-78 выше базальных концентраций в клетках или тканях, в которых IL-8,GRO, GRO, GRO, NAP-2 или ENA-78 соответственно производятся. Существует много болезненных состояний, в которых избыточное или нерегулируемое производство IL-8 влечет за собой ухудшение и/или возникновение заболевания. Хемокинопосредованные заболевания включают псориаз, атопический дерматит, остеоартрит, ревматоидный артрит, астму, хроническое обструктивное заболевание легких, респираторный дистресссиндром у взрослых, воспалительное заболевание кишечника, болезнь Крона, язвенный колит, удар, септический шок, эндотоксический шок, сепсис, вызванный грамотрицательными микроорганизмами, синдром токсического шока, сердечное и почечное реперфузионное повреждение, гломерулонефрит, тромбоз, реакция трансплантат против хозяина, болезнь Альцгеймера, отторжение аллотрансплантанта, малярия, рестеноз, ангиогенезис, атеросклероз, остеопороз, гингивит, вирусные заболевания, такие как риновирус, или нежелательное высвобождение кроветворных стволовых клеток. В частности, соединение настоящего изобретения является полезным для лечения астмы, хронического обструктивного заболевания легких и респираторного дистресс-синдрома у взрослых. Предпочтительно, настоящее соединение является полезным для лечения хронического обструктивного заболевания легких. Заболевания настоящего изобретения в первую очередь характеризуются массивной нейтрофильной инфильтрацией, Т-клеточной инфильтрацией или неоваскулярным ростом и связаны с повышенным производством IL-8, GRO, GRO, GRO, NAP-2 или ENA-78, которое отвечает за хемотаксис нейтрофилов в воспаленную область или направленный рост эндотелиальных клеток. В отличие от других воспалительных цитоксинов (IL-1, TNF и IL-6) IL-8, GRO, GRO, GRO, NAP-2 или ENA-78 обладают-4 015520 уникальным свойством способствовать нейтрофильному хемотаксису, высвобождению фермента, включая, но не ограничиваясь этим, высвобождение эластазы, также как производству и активации супероксида. -Хемокины, особенно GRO, GRO, GRO, NAP-2 или ENA-78, работающие через IL-8 рецепторы I или II типа, могут способствовать неоваскуляризации опухолей, способствуя направленному росту эндотелиальных клеток. Следовательно, ингибирование IL-8-вызванного хемотаксиса или активация приведет к прямому уменьшению нейтрофильной инфильтрации. Недавние данные также подразумевают роль хемокинов в лечение ВИЧ инфекций, Littleman et al.,Nature 381, p. 661 (1996) и Koup et al., Nature 381, p. 667 (1996). Недавние данные также подразумевают применение IL-8 ингибиторов для лечения атеросклероза. Первая ссылка, Boisvert et al., J. Clin. Invest, 1998, 101:353-353, показывает с помощью трансплантации костного мозга, что отсутствие IL-8 рецепторов на стволовых клетках (и, следовательно, на моноцитах/макрофагах) ведет к снижению развития атеросклеротических бляшек у мышей с дефицитом LDL рецепторов. Настоящее изобретение также относится к способам лечения травм ЦНС. Такое лечение осуществляется в острых случаях, также как для предотвращения травмы у тех индивидуумов, которые, как полагают, склонны к травме. Определенные в данном описании травмы ЦНС включают как открытую, так и проникающую травму головы, такую как в результате операции, или закрытую травму головы, такую как травма в области головы. Также включенными в данное определение являются ишемический удар, особенно в области мозга. Ишемический удар можно определить как фокальное неврологическое расстройство, которое является результатом недостаточного снабжения кровью конкретных областей мозга, обычно как следствие эмбол, тромбов или местного атероматозного сужения кровеносных сосудов. Выяснена роль воспалительных цитокинов в этой области и настоящее изобретение относится к способам потенциального лечения этих травм. Доступно относительно непродолжительное лечение для острой травмы, такой как эти.TNF- является цитокином с провоспалительными действиями, включая экспрессию адгезивных молекул эндотелиальных лейкоцитов. Лейкоциты проникают в ишемические повреждения головного мозга и, следовательно, соединения, которые ингибируют или снижают уровни TNF, могли бы быть полезны для лечения ишемического повреждения мозга; см. Liu et al., Stroke, vol. 25., No. 7, p. 1481-88(1994), чье описание вводится в данное описание с помощью ссылки. Модели закрытых повреждений головы и лечение смешанными 5-LO/CO агентами описывают вShohami et al., J. of VaiscClinical Physiology and Pharmacology, vol. 3, No. 2, p. 99-107 (1992). Найдено,что лечение, которое снижает образование отека, улучшает функциональное последствие у животных,подвергнутых лечению. Настоящее соединение вводят в количестве, достаточном для того, чтобы ингибировать IL-8, связанный с IL-8 или IL-8 рецепторами, от связывания с этими рецепторами, такими как доказано снижением нейтрофильного хемотаксиса и активации. Открытие, что настоящее соединение является ингибитором связывания IL-8, основывается на действиях настоящего соединения в испытаниях. Как используется в данном описании, термин "IL-8-опосредованное заболевание или болезненное состояние" относится к любому и всем болезненным состояниям, в которых IL-8, GRO, GRO, GRO, NAP2 или ENA-78 играют роль, или посредством производства самих IL-8, GRO, GRO, GRO, NAP-2 илиENA-78, или IL-8, GRO, GRO, GRO, NAP-2 или ENA-78 по причине высвобождения другого монокина,такого как, но не ограничиваясь ими, IL-1, IL-6 или TNF. Болезненное состояние, в котором, например, IL-1 является основным компонентом и чье производство или действие усиливается или он секретируется в ответ на IL-8, будет, следовательно, считаться болезненным состоянием, опосредованным IL-8. Как используется в данном описании, термин "хемокинопосредованное заболевание или болезненное состояние" относится к любому и всем болезненным состояниям, в которых хемокин, который связывается с IL-8 или IL-8 рецепторами, играет роль, такой как, но не ограничиваясь ими, IL-8, GRO,GRO, GRO, NAP-2 или ENA-78. Оно будет включать болезненное состояние, в котором IL-8 играет роль, или посредством производства самого IL-8, или IL-8 по причине высвобождения другого монокина, такого как, но не ограничиваясь ими, IL-1, IL-6 или TNF. Болезненное состояние, в котором, например, IL-1 является основным компонентом и чье производство или действие усиливается или он секретируется в ответ на IL-8, будет, следовательно, считаться болезненным состоянием, опосредованным IL-8. Как используется в данном описании, термин "цитокин" относится к любому секретируемому полипептиду, который воздействует на функции клеток и является молекулой, которая модулирует взаимодействие между клетками при иммунном, воспалительном или гематопоэтическом ответе. Цитокин включает, но не ограничивается ими, монокины и лимфокины, в зависимости от продуцирующих их клеток. Например, монокином обычно называют цитокин, производимый и секретируемый одноядерной клеткой, такой как макрофаг и/или моноцит. Однако многие другие клетки также производят монокины,такие как природные клетки-киллеры, фибробласты, базофилы, нейтрофилы, эндотелиальные клетки,астроциты мозга, стромальные клетки костного мозга, эпидеральные кератиноциты и B-лимфоциты.-5 015520 Лимфокинами обычно называют цитокин, производимый и секретируемый лимфоцитными клетками. Примеры цитокинов включают, но не ограничиваются ими, интерлейкин-1 (IL-1), интерлейкин-6 (IL-6),интерлейкин-8 (IL-8), фактор альфа некроза опухолей (TNF-) и фактор бета некроза опухолей (TNF-). Как используется в данном описании, термин "хемокин" относится к любому секретируемому полипептиду, который воздействует на функции клеток и является молекулой, которая модулирует взаимодействие между клетками при иммунном, воспалительном или гематопоэтическом ответе, аналогичный термину "цитокин" выше. Хемокин секретируется в основном через клеточные трансмембраны и вызывает хемотаксис и активацию специфических белых клеток крови и лейкоцитов, нейтрофилов, моноцитов, макрофагов, Т-клеток, В-клеток, эндотелиальных клеток и клеток гладких мышц. Примеры хемокинов включают, но не ограничиваются ими, IL-8, GRO, GRO, GRO, NAP-2, ENA-78, IP-10, MIP-1,MIP-, PF-4 и MCP-1, -2 и -3. Для того чтобы применять настоящее соединение в терапии, его будут обычно составлять в фармацевтическую композицию согласно стандартной фармацевтической практике. Данное изобретение, следовательно, также относится к фармацевтической композиции, содержащей эффективное, нетоксичное количество настоящего соединения и фармацевтически приемлемый носитель или разбавитель. Настоящее соединение и фармацевтические композиции, содержащие его, можно удобно вводить любым способом, обычно используемым для введения лекарственного средства, например, перорально,местно, парентерально или ингаляцией. Настоящее соединение можно вводить в обычных дозированных формах, полученных объединением настоящего соединения со стандартными фармацевтическими носителями согласно обычным методикам. Настоящее соединение можно также вводить в обычных дозах в комбинации с известным, вторым терапевтически активным соединением. Эти методики могут включать смешение, гранулирование и прессование или растворение ингредиентов, как подходит для требуемого препарата. Специалистам ясно, что форма и характер фармацевтически приемлемого носителя или разбавителя определяется количеством активного ингредиента, с которым его нужно объединять, способом введения и другими хорошо известными переменными. Носитель (носители) должен быть "приемлемым" в том смысле, что он должен быть совместим с другими ингредиентами композиции и не должен быть вредным для его реципиента. Используемый фармацевтический носитель может быть, например, как твердым, так и жидким. Примерами твердых носителей являются лактоза, гипс, сахароза, тальк, желатин, агар, пектин, акация,стеарат магния, стеариновая кислота и подобные. Примерами жидких носителей являются сироп, арахисовое масло, оливковое масло, вода и подобные. Аналогично, носитель или разбавитель могут включать замедляющий высвобождение материал, хорошо известный в данной области техники, такой как глицерилмоностеарат или глицерилдистеарат отдельно или с воском. Можно использовать большое разнообразие фармацевтических форм. Таким образом, если используют твердый носитель, препарат можно таблетировать, помещать в твердые желатиновые капсулы в виде порошка или в форме гранул или в форме пластинок или пастилок. Количество твердого носителя будет изменяться в широких пределах, но предпочтительно будет от 25 мг до, приблизительно, 1 г. При использовании жидкого носителя, препарат будет в форме сиропа, эмульсии, мягких желатиновых капсул, стерильной жидкости для введения, такой как ампула или неводная жидкая суспензия. Настоящее соединение можно вводить местно, т.е. не системным введением. Оно включает применение настоящего соединения внешне на эпидермис или в полость рта и помещение этого соединения в ухо, глаз и нос так, чтобы соединение не попадало значительно в кровяной поток. В отличие от этого,системное введение относится к пероральному, внутривенному, внутрибрюшинному и внутримышечному введению. Композиции, подходящие для местного введения, включают жидкие или полужидкие препараты,подходящие для проникновения через кожу к месту воспаления, такие как жидкие мази, лосьоны, крема,мази или пасты и капли, подходящие для введения в глаза, уши или нос. Активный ингредиент может содержаться в количестве при местном введении 0,001-10 вес.%, например 1-2 вес.% композиции. Однако он может содержаться вплоть до 10 вес.%, но предпочтительно будет содержаться менее чем 5 вес.%,более предпочтительно 0,1-1 вес.% композиции. Лосьоны согласно настоящему изобретению включают лосьоны, подходящие для нанесения на кожу или в глаза. Лосьон для глаз может содержать стерильный водный раствор, необязательно, содержащий бактерицид, и его можно получить способами, аналогичными способам для получения капель. Лосьоны или жидкие мази для применения на коже могут также содержать агент, чтобы ускорять высыхание и охлаждать кожу, такой как спирт или ацетон, и/или увлажнитель, такой как глицерин или масло, такое как касторовое масло или арахисовое масло. Крема, мази или пасты согласно настоящему изобретению являются полутвердыми композициями активного ингредиента для внешнего применения. Их можно изготовлять смешением активного ингредиента в мелкоизмельченной или порошкообразной форме, отдельно или в растворе или суспензии в водной или неводной жидкости, с помощью подходящего устройства, с жирной или нежирной основой. Основа может содержать углеводороды, такие как твердые, мягкие или жидкие парафины, глицерин,-6 015520 пчелиный воск, металлическое мыло; клейкое вещество; масло природного происхождения, такое как миндальное, кукурузное, арахисовое, касторовое или оливковое масло; шерстяной жир или его производные или жирные кислоты, такие как стеариновая или олеиновая кислота вместе со спиртом, таким как пропиленгликоль или макрогель. В композицию можно вводить любой подходящий поверхностноактивный агент, такой как анионное, катионное или неионное поверхностно-активное вещество, такое как сорбитановый эфир или его полиоксиэтиленовое производное. Можно также включать суспендирующие агенты, такие как природные смолы, производные целлюлозы или неорганические материалы,такие как камнеподобный диоксид кремния и другие ингредиенты, такие как ланолин. Капли согласно настоящему изобретению могут содержать стерильные водные или масляные растворы или суспензии и их можно приготовить растворением активного ингредиента в подходящем водном растворе бактерицидного и/или фунгицидного агента и/или любого другого подходящего консерванта, предпочтительно включает поверхностно-активный агент. Затем полученный в результате раствор можно осветлять фильтрацией, переносить в подходящий контейнер, который затем плотно закрывают и стерилизуют автоклавированием или выдерживанием при 98-100 С в течение получаса. Альтернативно,раствор можно стерилизовать фильтрацией и переносить в контейнер с помощью антисептической методики. Примерами бактерицидных и фунгицидных агентов, подходящих для включения в капли, являются фенилртутные нитраты или ацетаты (0,002%), бензалконийхлорид (0,01%) и хлоргексидинацетат(0,01%). Подходящие растворители для приготовления масляного раствора включают глицерин, разбавленный спирт и пропиленгликоль. Настоящее соединение можно вводить парентерально, т.е. внутривенно, внутримышечно, подкожно, интраназально, интраректально, интравагинально или внутрибрюшинно. Подходящие дозированные формы для этого введения можно приготовлять обычными методиками. Настоящее соединение можно также вводить с помощью ингаляции, т.е. интраназально и пероральной ингаляцией. Подходящие дозированные формы для этого введения, такие как аэрозольная композиция или ингалятор с отмеренными дозами, можно получать обычными методиками. Для всех способов применения, описанных в данном описании для настоящего соединения, режим дневного дозирования при пероральном введении будет предпочтительно составлять от 0,01 до приблизительно 80 мг/кг общего веса тела. Режим дневного дозирования при парентеральном введении составляет от приблизительно 0,001 до приблизительно 80 мг/кг общего веса тела. Режим дневного дозирования при местном введении будет предпочтительно составлять от 0,1 до 150 мг, вводимые 1-4, предпочтительно 2-3 раза в день. Режим дневного дозирования при ингаляции будет предпочтительно составлять от приблизительно 0,01 до приблизительно 1 мг/кг в день. Специалистам в данной области техники ясно,что оптимальное количество и перерывы между конкретными дозами настоящего соединения будут определяться характером и продолжительностью состояния, которое нужно лечить, формой, способом и местом введения, и конкретным пациентом, которого нужно лечить, и что такие оптимумы можно определить обычными методиками. Специалистам в данной области техники также ясно, что оптимальный курс лечения, т.е., количество доз настоящего соединения, даваемое в день в течение определенного количества дней, может быть установлен специалистами в данной области техники, используя обычные испытания на определение курса лечения. Комбинации. Настоящее соединение и фармацевтические композиции согласно данному изобретению можно применять в комбинации с или включать один или более других терапевтических агентов, например,выбранные из противовоспалительных агентов, антихолинергических агентов (в особенности антагонист М 1/М 2/М 3 рецепторов), агонистов 2-адренорецептора, противоинфекционных агентов, таких как антибиотики, противовирусные агенты или антигистаминные препараты. Данное изобретение, таким образом, относится, в дополнительном аспекте к комбинации, включающей настоящее соединение или физиологически функциональное производное настоящего соединения вместе с одним или более другими терапевтически активными агентами, например, выбранными из противовоспалительного агента, таких как кортикостероид или NSAID, антихолинергического агента, агониста 2-адренорецептора, противоинфекционного агента, такого как антибиотик, противовирусный агент или антигистаминный препарат. Один вариант осуществления данного изобретения относится к комбинации, содержащей настоящее соединение или его физиологически функциональное производное вместе с агонистом 2-адренорецептора и/или антихолинергическим и/или PDE-4 ингибитором и/или антигистаминным препаратом. Специалистам в данной области техники ясно, что по необходимости можно использовать другой терапевтический ингредиент (ингредиенты) в форме солей, например в виде солей щелочных металлов или аминов, или солей присоединения кислоты, или пролекарств, или в виде сложных эфиров, например эфиров низших алкилов, или в виде сольватов, например гидратов, чтобы оптимизировать активность,и/или стабильность, и/или физические характеристики, такие как растворимость терапевтического ингредиента. Ясно, что по необходимости можно использовать терапевтический ингредиент в оптически чистой форме. В одном варианте осуществления данное изобретение относится к комбинации, содержащей на-7 015520 стоящее соединение вместе с агонистом 2-адренорецептора. Примеры агониста 2-адренорецептора включают салметерол (который может быть рацематом или отдельным энантиомером, таким какR-энантиомер), салбутамол (который может быть рацематом или отдельным энантиомером, таким какR-энантиомер), формотерол (который может быть рацематом или отдельным диастереомером, таким какR,R-диастереомер), салмефамолом, фенотеролом, кармотеролом, этантеролом, наминтеролом, кленбутеролом, пирбутеролом, флербутеролом, репротеролом, бамбутеролом, индакатеролом, тербуталином и их солями, например ксинафоатом (1-гидрокси-2-нафталинкарбоксилатной солью) салметерола, сульфатной солью или свободным основанием салбутамола или фумаратной солью формотерола. В одном варианте осуществления агонистом 2-адренорецептора является агонист 2-адренорецептора длительного действия, например соединения, которые обеспечивают эффективную бронходилатацию в течение приблизительно 12 ч или более. Другие агонисты 2-адренорецептора включают агонисты, описанные вWO 2002/066422, WO 2002/070490, WO 2002/076933, WO 2003/024439, WO 2003/072539,WO 2003/091204, WO 2004/016578, WO 2004/022547, WO 2004/037807, WO 2004/037773,WO 2004/037768, WO 2004/039762, WO 2004/039766, WO 2001/42193 и WO 2003/042160. Дополнительные примеры агонистов 2-адренорецептора включают 3-(4-[6-2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этиламино)гексил]оксибутил)бензолсульфонамид; 3-(3-[7-2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этиламино)гептил]оксипропил)бензолсульфонамид; 4-(1R)-2-[(6-2-[(2,6-дихлорбензил)окси]этоксигексил)амино]-1-гидроксиэтил-2-(гидроксиметил)фенол; 4-(1R)-2-[(6-4-[3-(циклопентилсульфонил)фенил]бутоксигексил)амино]-1-гидроксиэтил-2(гидроксиметил)фенол;N-22-[4-(3-фенил-4-метоксифенил)аминофенил]этил-2-гидрокси-2-(8-гидрокси-2(1 Н)-хинолинон 5-ил)этиламин и 5-[(R)-2-(2-4-[4-(2-амино-2-метилпропокси)фениламино]фенилэтиламино)-1-гидроксиэтил]-8 гидрокси-1 Н-хинолин-2-он. Агонист 2-адренорецептора может быть в форме соли, образованной с фармацевтически приемлемой кислотой, выбранной из серной, хлористо-водородной, фумаровой, гидроксинафтойной (например,1- или 3-гидрокси-2-нафтойной), коричной, замещенной коричной, трифенилуксусной, сульфаминовой,сульфаниловой, нафталинакриловой, бензойной, 4-метоксибензойной, 2- или 4-гидроксибензойной,4-хлорбензойной и 4-фенилбензойной кислоты. Подходящие противовоспалительные агенты включают кортикостероиды. Примерами кортикостероидов, которые можно использовать в комбинации с соединениями данного изобретения, являются те пероральные и ингаляционные кортикостероиды и их пролекарства, которые обладают противовоспалительной активностью. Примеры включают метилпреднизолон, преднизолон, дексаметазон, флутиказонпропионат, Sфторметиловый эфир 6,9-дифтор-11-гидрокси-16-метил-17-[(4-метил-1,3-тиазол-5-карбонил)окси]-3-оксоандроста-1,4-диен-17-карботиокислоты, S-фторметиловый эфир 6,9-дифтор-17 а-[(2 фуранилкарбонил)окси]-11-гидрокси-16 а-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (флутиказон фуроат), S-(2-оксотетрагидрофуран-3S-иловый) эфир 6,9-дифтор-11-гидрокси-16-метил-3 оксо-17-пропионилоксиандроста-1,4-диен-17-карботиокислоты, S-цианометиловый эфир 6,9 дифтор-11-гидрокси-16-метил-3-оксо-17-(2,2,3,3-тетраметилциклопропилкарбонил)оксиандроста 1,4-диен-17-карботиокислоты и S-фторметиловый эфир 6,9-дифтор-11-гидрокси-16-метил-17-(1 метилциклопропилкарбонил)окси-3-оксоандроста-1,4-диен-17-карботиокислоты, эфиры беклометазона(например, 17-пропионатный эфир или 17,21-дипропионатный эфир), будесонид, флунисолид, эфиры мометазона (например, мометазон фуроат), триамцинолон ацетонид, рофлепонид, циклесонид (16,17(R)-циклогексилметилен]-бис-(окси)]-11,21-дигидроксипрегна-1,4-диен-3,20-дион), бутиксокорт пропионат, RPR-106541 и ST-126. В одном варианте осуществления кортикостероиды включают флутиказонпропионат, S-фторметиловый эфир 6,9-дифтор-11-гидрокси-16-метил-17-[(4-метил-1,3-тиазол 5-карбонил)окси]-3-оксоандроста-1,4-диен-17-карботиокислоты, S-фторметиловый эфир 6,9-дифтор 17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты,S-цианометиловый эфир 6,9-дифтор-11-гидрокси-16-метил-3-оксо-17-(2,2,3,3-тетраметилциклопропилкарбонил)оксиандроста-1,4-диен-17-карботиокислоты и S-фторметиловый эфир 6,9-дифтор 11-гидрокси-16-метил-17-(1-метилциклопропилкарбонил)окси-3-оксоандроста-1,4-диен-17-карботиокислоты. В одном варианте осуществления кортикостероидом является S-фторметиловый эфир 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17 карботиокислоты.-8 015520 Примеры кортикостероидов также включают кортикостероиды, описанные в WO 2002/088167,WO 2002/100879, WO 2002/12265, WO 2002/12266, WO 2005/005451, WO 2005/005452, WO 2006/072599 и WO 2006/072600. Нестероидные соединения, обладающие глюкокортикоидным агонизмом, которые могут обладать селективностью относительно трансрепрессии по сравнению с трансактивацией и которые могут быть полезными в комбинированной терапии, включают соединения, описанные в следующих опубликованных патентных заявках и патентах: WO 2003/082827, WO 1998/54159, WO 2004/005229, WO 2004/009017,WO 2004/018429, WO 2003/104195, WO 2003/082787, WO 2003/082280, WO 2003/059899, WO 2003/101932,WO 2002/02565, WO 2001/16128, WO 2000/66590, WO 2003/086294, WO 2004/026248, WO 2003/061651,WO 2003/08277, WO 2006/000401, WO 2006/000398 и WO 2006/015870. Нестероидные соединения, обладающие глюкокортикоидным агонизмом, которые могут обладать селективностью относительно трансрепрессии по сравнению с трансактивацией и которые могут быть полезными в комбинированной терапии, включают соединения, описанные в следующих патентных заявках: WO 2003/082827, WO 1998/54159, WO 2004/005229, WO 2004/009017, WO 2004/018429,WO 2003/104195, WO 2003/082787, WO 2003/082280, WO 2003/059899, WO 2003/101932,WO 2002/02565, WO 2001/16128, WO 2000/66590, WO 2003/086294, WO 2004/026248, WO 2003/061651 иWO 2003/08277. Примеры противовоспалительных агентов включают нестероидные противовоспалительные лекарственные средства (NSAID). Примеры NSAID включают кромогликат натрия, недокромил натрия, ингибиторы фосфодиэстеразы(PDE) (например, теофиллин, PDE4 ингибиторы или смешанные PDE3/PDE4 ингибиторы), антагонисты лейкотриена, ингибиторы лейкотриенового синтеза (например, монтелукаст), iNOS ингибиторы, ингибиторы триптазы и эластазы, антагонисты бета-2 интегрина и агонисты или антагонисты аденозинового рецептора (например, аденозин 2 а агонисты), цитокиновые антагонисты (например, хемокиновые антагонисты, таких как CCR3 антагонист) или ингибиторы цитокинового синтез, или ингибиторы 5 липоксигеназы. В одном варианте осуществления данное изобретение относится к iNOS (индуцибельная нитрооксидсинтаза) ингибиторам для перорального введения. Примеры iNOS ингибиторов включают ингибиторы, описанные в WO 1993/13055, WO 1998/30537, WO 2002/50021, WO 1995/34534 иWO 1999/62875. Примеры CCR3 ингибиторов включают ингибиторы, описанные в WO 2002/26722. В одном варианте осуществления данное изобретение относится к применению настоящего соединения в комбинации с ингибитором фосфодиэстеразы 4 (PDE4), например в случае композиции, приспособленной для ингаляций. Ингибитором PDE4, полезным в данном аспекте данного изобретения, может быть любое соединение, о котором известно или открыто, что оно действует как ингибитор PDE4, например, как ингибитор PDE4B и/или PDE4D.PDE4 ингибиторные соединения включают цис-4-циано-4-(3-циклопентилокси-4-метоксифенил)циклогексан-1-карбоновую кислоту, 2-карбометокси-4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-он и цис-[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан 1-ол], а также цис-4-циано-4-[3-(циклопентилокси)-4-метоксифенил]циклогексан-1-карбоновую кислоту(также известную как циломиласт) и ее соли, эфиры, пролекарства или физические формы, которые описываются в патенте США 5552438. Другие PDE4 ингибиторные соединения включают AWD-12-281 (N-(3,5-дихлор-4-пиридинил)-1[(4-фторфенил)метил]-5-гидрокси-D-оксо-1 Н-индол-3-ацетамид) от Elbion (Hofgen, N. et al. 15th EFMCV-11294A от Napp (Landells, L.J. et al. Eur. Resp. J. [Annu Cong. Eur. Resp. Soc. (Sept. 19-23, Geneva) 1998],1998, 12 (Suppl. 28): Abst P2393); рофлумиласт (3-(циклопропилметокси)-N-(3,5-дихлор-4-пиридинил)-4(дифторметокси)бензамид) (см. ЕР 0706513 В 1 to Byk Gulden Lomberg, например, см. пример 5 в данном описании); фталазинон (WO 1999/47505) от Byk-Gulden; пумафентрин, (-)-п-[(4aR,10bS)-9-этокси 1,2,3,4,4 а,10b-гексагидро-8-метокси-2-метилбензо[с][1,6]нафтиридин-6-ил]-N,N-диизопропилбензамид,который является смешанным PDE3/PDE4 ингибитором, который получен и опубликован Byk-Gulden, в настоящее время Altana; арофиллин, разрабатываемый Almirall-Prodesfarma; VM554/UM565 от Vernalis; или Т-440 (Tanabe Seiyaku; Fuji, K. et al. J. Pharmacol. Exp. Ther., 1998, 284(1): 162) и Т 2585. Кроме того, PDE4 ингибиторные соединения описываются в опубликованных международных патентных заявках WO 2004/024728, WO 2004/056823, WO 2004/103998 (например, примеры 399 или 544,описанные в них), WO 2005/058892, WO 2005/090348, WO 2005/090353 и WO 2005/090354, все под именем Glaxo Group Limited. Примерами антихолинергических агентов являются такие соединения, которые действуют в качестве антагонистов мускариновых рецепторов, в особенности такие соединения, которые являются антагонистами M1 или M3 рецепторов, двойными антагонистами М 1/М 3 или М 2/М 3, рецепторами или панантагонистами M1/M2/M3 рецепторов. Примеры соединений для введения с помощью ингаляции включают-9 015520 ипратропий (например, в виде бромида, CAS 22254-24-6, продаваемый под названием Atrovent), окситропий (например, в виде бромида, CAS 30286-75-0) и тиотропий (например, в виде бромида,CAS 136310-93-5, продаваемый под названием Spiriva). Также интересующими являются реватропат (например, в виде гидробромида, CAS 262586-79-8) и LAS-34273, который описывается в WO 2001/04118. Примерные соединения для перорального введения включают пирензепин (CAS 28797-61-7), дарифенацин (CAS 133099-04-4, или CAS 133099-07-7 для гидробромида, продаваемый под названием Enablex),оксибутинин (CAS 5633-20-5, продаваемый под названием Ditropan), теродилин (CAS 15793-40-5), толтеродин (CAS 124937-51-5 или CAS 124937-52-6 для тартрата, продаваемый под названием Detrol),отилоний (например, в виде бромида, CAS 26095-59-0, продаваемый под названием Spasmomen), троспийхлорид (CAS 10405-02-4) и солифенацин (CAS 242478-37-1 или CAS 242478-38-2 для сукцината,также известный как YM-905 и продаваемый под названием Vesicare). Дополнительные соединения описывают в WO 2005/037280, WO 2005/046586 и WO 2005/104745. Настоящие комбинации включают, но не ограничиваются:(1R,5S)-3-(2-циано-2,2-дифенилэтил)-8-метил-8-2-[(фенилметил)окси]этил-8 азониабицикло[3.2.1]октанбромид. Другие антихолинергические агенты включают соединения, которые описаны в патентной заявке США 60/487981, введенной в данное описании с помощью ссылки до степени, требуемой для того, чтобы реализовать на практике настоящее изобретение. Они включают, например:(ендо)-3-2,2-дифенил-3-[(1-фенилметаноил)амино]пропил-8,8-диметил-8-азониабицикло[3.2.1]октанбромид. В одном варианте осуществления данное изобретение относится к комбинации, содержащей настоящее соединение вместе с H1 антагонистом. Примеры H1 антагонистов включают, без ограничений,амелексанокс, астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, левоцетиризин,эфлетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дескарбоэтоксилоратадин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокабастин, мизоластин, мехитазин, миансерин, ноберастин, меклизин, норастемизол, олопатадин, пикумаст, пуриламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин, особенно цетиризин, левоцетиризин, эфлетиризин и фексофенадин. В дополнительном варианте осуществления данное изобретение относится к комбинации, содержащей настоящее соединение вместе с Н 3 антагонистом (и/или обратным агонистом). Примеры Н 3 антагонистов включают, например, соединения, описанные в WO 2004/035556 и в WO 2006/045416. Другие антагонисты гистаминовых рецепторов, которые можно использовать в комбинации с соединениями настоя- 10015520 щего изобретения, включают антагонисты (и/или обратные агонисты) Н 4 рецептора, например, соединения, описанные в Jablonowski et al., J. Med. Chem. 46:3957-3960 (2003). В одном варианте осуществления данное изобретение относится к комбинации, содержащей настоящее соединение вместе с антагонистом CCR5 рецептора, таким как 4,4-дифтор-N-1S)-3-3-[3 метил-5-(1-метилэтил)-4 Н-1,2,4-триазол-4-ил]-8-азабицикло[3.2.1]окт-8-ил-1 фенилпропил)циклогексанкарбоксамид: В одном варианте осуществления данное изобретение относится к комбинации, содержащей настоящее соединение вместе с антагонистом CXCR3 рецептора, таким как N-1R)-1-3-[4(этилокси)фенил]-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-илэтил)-N-(3-пиридинилметил)-2-4[(трифторметил)окси]фенилацетамид: Таким образом, данное изобретение относится в дополнительном аспекте к комбинации, содержащей настоящее соединение вместе с PDE4 ингибитором. Таким образом, данное изобретение относится в дополнительном аспекте к комбинации, содержащей настоящее соединение вместе с агонистом 2-адренорецептора. Таким образом, данное изобретение относится в дополнительном аспекте к комбинации, содержащей настоящее соединение вместе с кортикостероидом. Таким образом, данное изобретение относится в дополнительном аспекте к комбинации, содержащей настоящее соединение вместе с нестероидным GR агонистом. Таким образом, данное изобретение относится в дополнительном аспекте к комбинации, содержащей настоящее соединение вместе с антихолинергическим агентом. Таким образом, данное изобретение относится в дополнительном аспекте к комбинации, содержащей настоящее соединение вместе с антигистаминовым агентом. Таким образом, данное изобретение относится в дополнительном аспекте к комбинации, содержащей настоящее соединение вместе с PDE4 ингибитором и агонистом 2-адренорецептора. Таким образом, данное изобретение относится в дополнительном аспекте к комбинации, содержащей настоящее соединение вместе с антихолинергическим агентом и PDE-4 ингибитором. Комбинации, на которые ссылались выше, могут быть удобно представлены для применения в форме фармацевтической композиции и, таким образом, фармацевтическая композиция, содержащая комбинацию, как описано выше, вместе с фармацевтически приемлемым разбавителем или носителем,представляет дополнительный аспект данного изобретения. Отдельные соединения этих комбинаций можно вводить как последовательно, так и одновременно в отдельной или объединенной фармацевтической композиции. В одном варианте осуществления отдельные соединения будут вводить одновременно в комбинированной фармацевтической композиции. Подходящие дозы терапевтических агентов будут легко определены специалистами в данной области техники. Таким образом, данное изобретение относится в дополнительном аспекте к фармацевтической композиции, содержащей комбинацию настоящего соединения вместе с другим терапевтически активным агентом. Таким образом, данное изобретение относится в дополнительном аспекте к фармацевтической композиции, содержащей комбинацию настоящего соединения вместе с PDE4 ингибитором. Таким образом, данное изобретение относится в дополнительном аспекте к фармацевтической композиции, содержащей комбинацию настоящего соединения вместе с агонистом 2-адренорецептора. Таким образом, данное изобретение относится в дополнительном аспекте к фармацевтической ком- 11015520 позиции, содержащей комбинацию настоящего соединения вместе с кортикостероидом. Таким образом, данное изобретение относится в дополнительном аспекте к фармацевтической композиции, содержащей комбинацию настоящего соединения вместе с нестероидным GR агонистом. Таким образом, данное изобретение относится в дополнительном аспекте к фармацевтической композиции, содержащей комбинацию настоящего соединения вместе с антихолинергическим агентом. Таким образом, данное изобретение относится в дополнительном аспекте к фармацевтической композиции, содержащей комбинацию настоящего соединения вместе с антигистаминовым агентом. Таким образом, данное изобретение относится в дополнительном аспекте к фармацевтической композиции, содержащей комбинацию настоящего соединения вместе с антагонистом CXCR3 рецептора. Таким образом, данное изобретение относится в дополнительном аспекте к фармацевтической композиции, содержащей комбинацию настоящего соединения вместе с антагонистом CCR5 рецептора. Данное изобретение в настоящее время будет описываться с помощью ссылки на следующие биологические примеры, которые приводятся просто для иллюстрации и не подразумеваются в качестве ограничения объема настоящего изобретения. Биологические примеры.IL-8 и GRO хемокинингибирующее действие соединений настоящего изобретения определяли с помощью следующих испытаний in vitro. Испытание на связывание с рецепторами.[125I] IL-8 (человеческий рекомбинант) получали от GE Healthcare, с удельной активностью 2000 кюри/ммоль. Все другие химические реагенты являлись химически чистыми. Высокие уровни рекомбинантных человеческих CXCR1 (IL-8 тип ) и CXCR2 (IL-8 тип ) рецепторов индивидуально экспрессировали в свободных клетках яичника китайского хомячка (СНО), как описано ранее (Holmes, et at.,Science, 1991, 253, 1278). Мембраны получали согласно ранее описанному протоколу, Haour, et al., J.Biol. Chem., 249, p. 2195-2205 (1974), вводимому в данное описание с помощью ссылки до степени, требуемой, чтобы получить настоящие мембраны, за исключением того, что буфер для гомогенизации заменяли на 40 мМ Tris-HCl (pH 7,5), 1 мМ MgSO4, 0,5 мМ EGTA (этиленгликоль-бис-(2-аминоэтиловый эфир)-N,N,N',N'-тетрауксусная кислота), 1 мМ PMSF (-толуолсульфонилфторид), 2,5 мг/л леупептин и 0,1 мг/мл апротинин. Клетки гомогенизировали и центрифугировали при 2000 об/мин в течение 10 мин. Надосадочную жидкость центрифугировали при 100000g в течение 1 ч. Надосадочную жидкость удаляли, и мембраны хранили при -80 С. Концентрацию мембранного белка определяли, используя BioRad реагент согласно протоколу производителя, используя альбумин бычьей сыворотки (BSA) в качестве стандарта. Все IL-8 связывания проводили, используя сцинтилляционный анализ близости (SPA), используя шарики агглютинина из проростков пшеницы в 96-луночном планшетном формате. Мембраны CHOCXCR1 или CHO-CXCR2 предварительно выдерживали с шариками в буфере для связывания в течение 30 мин для 4 С. Буфер содержит 20 мМ бис-трис-пропанового буфера, рН 8,0, содержащий 1 мМ MgSO4,0,1 мМ EDTA и 25 мМ NaCl. Соединения разбавляли в ДМСО до 20 конечного разбавления (конечная концентрация соединений между 1 нМ и 30 мкМ, и конечная концентрация ДМСО 5%). Испытания проводили в 96-луночных планшетах (optiplate 96, Packard) при комнатной температуре, в 0,1 мл буфера для связывания с мембранами и 0,04% CHAPS (3-[(3-хлорамидопропил)диметиламмонио]-1 пропансульфонат), 0,0025% BSA и 0,23 нМ [125I] IL-8. Планшеты встряхивали на платформе в течение 1 ч, в конце инкубирования планшеты вращали при 2000 об/мин в течение 5 мин и измеряли в счетчикеTop Count. Рекомбинантный IL-8R, CXCR1 или тип I рецептор также называют в данном описании непермиссивный рецептор и рекомбинантный IL-8R, CXCR2 или тип II рецептор также называют пермиссивный рецептор. Соединение считается активным в данном испытании, если оно показывает значение IC5030 мкМ. Ожидается, что настоящее соединение обладает испытуемой активностью с величиной IC50 приблизительно 13 нМ в настоящем испытании. Испытание на хемотаксис. Проводили испытание на нейтрофильный хемотаксис. Первичные человеческие нейтрофилы выделяли из периферической цельной крови, используя центрифугирование со ступенчатым градиентом перколла, декстрановую седиментацию и гипотонический лизис. Хемоаттрактанты IL-8 (CXCL8) или GRO(CXCL1) помещали в нижнюю камеру 96-луночной камеры (ChemoTx System, Neuro Probe, Gaithersburg,MD). Используемой концентрацией агониста является концентрация ЕС 80. Две камеры разделяли 5 мкм поликарбонатной мембраной. Испытуемое соединение предварительно выдерживали с клетками перед перенесением его наверх фильтра. Хемотаксис продолжался в течение 45 мин во влажном инкубаторе при 37 С с 5% СО 2. В конце периода инкубирования, мембраны удаляли, и мигрировавшие клетки в нижней камере переносили в 96-луночный планшет. Эти клетки измеряли, используя люминесцентный анализ жизнеспособности клеток (Celltiter-Glo, Promega, Madison, WI). Каждый образец испытывали два раза и каждое соединение испытывали по крайне мере три раза. Клетками положительного контроля являются клетки без добавления соединения и обладающие максимальным хемотаксическим ответом. От- 12015520 рицательным контролем (нестимулированным) является испытание без добавления хемокина в нижнюю камеру. Разница между положительным контролем и отрицательным контролем представляет хемотаксическую активность клеток. Соединение считается активным, если оно показывает величину IC505 мкМ. Испытание на CD11b в цельной крови человека. Соединение испытывали на его способность ингибировать GRO-индуцированную экспрессию интегрина CD11b на нейтрофилах цельной крови человека. Кровь отбирали (9 мл), используя линию в виде бабочки и 10 мл шприц, содержащий 0,2 мл рабочего гепарина натрия. Кровь хранили при 37 С до помещения на лед на стадии 5 ниже. Затем базовые растворы соединения разбавляли в 12 раз до максимальной конечной концентрации 120 мкМ. Затем проводили серийное полулогарифмическое разбавление в среде. Затем 10 мкл разбавленного раствора соединения или среду добавляли в подходящие 1275 полипропиленовые пробирки. 100 мкл цельной крови добавляли в каждую пробирку и выдерживали в течение 10 мин на бане с водой при 37 С с первоначальным (мягким) перемешиванием и снова в течение 5 мин. Биомассу GRO разбавляли 1:166,66 в 0,1%BSA-DPBS до "12" концентрации 120 нМ и 10 мкл GRO разбавленного раствора или 0,1% BSA-DPBS добавляли в подходящие пробирки так, чтобы конечная концентрация GRO равнялась 10 нМ. Пробирки инкубировали в течение 10 мин при 37 С с мягким ручным перемешиванием и снова в течение 5 мин. Затем образцы помещали на лед и добавляли 250 мкл охлажденного льдом рабочего разбавленного раствора CellFix, с последующей инкубацией в течение 1 мин на льду. 1,5 мл пробирки Эппендорфа приготовляли в течение GRO инкубации добавлением подходящих антител. В каждую пробирку помещали 10 мкл CD11b-FITC и 5 мкл CD16-PE, за исключением изотипического контроля, при котором добавляли 10 мкл IgG2a-FITC, вместо CD11b. 50 мкл плохо растворяющейся крови из каждой пробирки добавляли в соответствующие пробирки Эппендорфа. Затем образцы инкубировали в течение 20 мин при 4 С в темноте. Добавки смесей крови/антител к 500 мкл охлажденного DPBS добавляли в подходящим образом меченную 1275 полипропиленовую пробирку. Полученную в результате смесь хранили на льду. Добавляли LDS биомассу (10 мкл) и смесь выдерживали в течение 10 мин при 4 С перед анализом потока. Образцы хранили в темном окружении. Добавку LDS трясли, в то время как собирали образцы на проточном цитометре, так что образцы пропускали 10-20 мин после добавления LDS. Использовали скорость потока среды для накопления потока и улучшенную пороговую величинуFL3, чтобы удалить красные кровяные клетки из анализа, используя LDS сигнал. Цветокорректирование правильно устанавливали, используя немеченые образцы и одноцветные образцы, чтобы вычесть потокLDS в РЕ и поток РЕ в FITC и FITC в РЕ. Для BD LSR цитометра, LDS=FL3, PE=FL2, FITC=FL1. Собирали минимум 2000-3000 результатов, которые удовлетворяют гранулоцитному интервалу SSC от FSC, и являлся CD16 положительным с помощью FL2 сигнала. В этом испытании соединение считали активным, если оно показывало величину IC505 мкМ. Мобилизация кальция в клетках CHO-K1, стабильно экспрессирующих CXCR2 и G16.G418, при поддержании температуры 37 С в 5% СО 2 инкубаторе. 3 а двадцать четыре часа перед испытанием клетки собирали и помещали 40000 клеток на одну лунку в 96-луночный темностенный планшет с прозрачным дном (Packard View) и помещали обратно в СО 2 инкубатор. В день испытания соединения последовательно разбавляли 100% ДМСО до 300 требуемой для испытания концентрации. Ростовую среду отсасывали от клеток и заменяли 100 мкл загрузочной среды (ЕМЕМ с солями Эрла w/L-глутамин,0,1% BSA, (Bovuminar Cohen Fraction V от Seriologicals Corp.), 4 мкМ флуо-4-ацетоксиметилэфирного флуоресцентного индикаторного красителя (Fluo-4 AM, от Molecular Probes) и 2,5 мМ пробенецида) и выдерживали в течение 1 ч при 37 С в CO2 инкубаторе. Загрузочную среду отсасывали и заменяли 100 мкл ЕМЕМ с солями Эрла w/L-глутамин, 0,1% желатина и 2,5 мМ пробенецида и выдерживали в течение дополнительных 10 мин. Последовательно разбавленное соединение (3 мкл) в ДМСО при 300 переносили в 96-луночный планшет, содержащий 297 мкл KRH (120 мМ NaCl, 4,6 мМ KCl, 1,03 мМKH2PO4, 25 мМ NaHCO3, 1,0 мМ CaCl2, 1,1 мМ MgCl2, 11 мМ глюкозы, 20 мМ HEPES (рН 7,4 w/2,5 мМ пробенецида и 0,1% желатина (соединение в настоящий момент при 3). Среду отсасывали от клеток, и клетки промывали 3 раза KRH w/2,5 мМ пробенецида, w/0,1% желатина. Добавляли в лунки KRHw/2,5 мМ пробенецида и 0,1% желатина (соединение в настоящий момент при 1) и инкубировали при 37C в СО 2 инкубаторе в течение 10 мин. Планшеты помещали в FLIPR (Fluorometric Imaging PlateReader, Molecular Devices, Sunnyvale CA) для анализа, как описано ранее (Sarau et al., 1999). Процент максимальной Са 2+ мобилизации, вызванной человеческим IL-8, вызванной 1,0 нМ IL-8, EC80 концентрацией для CXCR2, определяли для каждой концентрации соединения и вычисляли IC50, как концентрацию испытуемого соединения, которая ингибирует 50% максимального ответа, вызванного 1,0 нМ IL-8. В этом испытании соединение считали активным, если оно показывало величину IC50 10 мкМ.- 13015520 Нейтрофильная CD11b стимуляция после введение крысам пероральных доз. Крысам Lewis (250-300 г) вводили перорально настоящее соединение или носитель и через один час их умерщвляли с помощью удушения СО 2. Цельную кровь крыс, 3 мл, отбирали пункцией сердца в шприц, содержащий 100 мкл 0,25 М EDTA (GIBCO, Grand Island, NY). Крысиный CXCL2 (PeproTech,Rocky Hill, NJ) исходный раствор получали восстановлением в Кребс/0,1% BSA (KBSA) при 10 мкМ. Исходный раствор разбавляли до "11" используемой максимальной концентрации в DPBS (GIBCO) и последовательно разбавляли в среде KBSA/DPBS. Десять мкл подходящей концентрации настоящего соединения (1,2-100 нМ) или среды добавляли в 1275 мм полипропиленовые пробирки, с последующим добавлением 100 мкл цельной крови. Пробирки выдерживали в течение 30 мин на бане при 37 С, с мягким ручным перемешиванием каждые 10 мин. Затем образцы помещали на лед на 10 мин с последующим добавлением 10 мкл антикрысиный-CD11b-FITC или FITC-меченный мышиный IgG2a изотипический контроль (оба Antigenix America, Huntington Station, NY) и инкубировали в течение 30 мин на льду.FACS лизирующий раствор (Becton Dickinson, San Jose, CA), 1 мл 1, добавляли с немедленным интенсивным перемешиванием, с последующим дополнительным перемешиванием после добавления раствора к последнему образцу. Образцы инкубировали в течение 10 мин при комнатной температуре, и лейкоциты осаждали при 300g и промывали DPBS. Клетки повторно суспендировали в 650 мкл 1% параформальдегида. Лизирующий раствор FACS не полностью лизировал красные клетки крови крыс. По этой причине, для анализов на основе проточной цитометрии 3,5 мкл 1,67 мг/мл этанольного раствора (перенасыщенный; осветленный центрифугированием) LDS-751 (Exciton, Dayton, ОН) добавляли к каждому образцу в течение 1-2 мин проточного анализа, чтобы выделить оставшиеся красные кровяные клетки. Данные по образцам собирали, используя программное обеспечение CellQuest и LSR проточный цитометр (Becton-Dickinson), при настройке на маленькую скорость потока, увеличением FL3 пороговой величины, чтобы удалить LDS-751 отрицательные красные клетки крови, и затем устанавливая популяцию нейтрофилов в боковом рассеивании против графика прямого рассеивания. Затем, измеряли FL1 (зеленаяFITC флуоресценция, непосредственно связанная с содержанием CD11b) этой популяции как среднюю канальную флуоресценцию с помощью анализа с использованием программного обеспечения CellQuest. В данном испытании настоящее соединение испытывали на активность ингибирования нейтрофильной CD11b экспрессии в цельной крови, с последующим пероральным введением доз крысам Lewis при 10 мг/кг. Настоящее соединение значительно сдвигает ЕС 50 кривой чувствительности к концентрации CXCL2 от 4,7 нМ (1,9-7,4; 95% C.I.) у крыс, которым давали носитель, до 12,3 нМ (10,0-14,7),(р 0,001, n=6 на группу). Вышеуказанное описание полностью раскрывает данное изобретение, включая его предпочтительные варианты осуществления. Модификации и улучшения вариантов осуществления, специально раскрытые в данном описании, входят в объем следующей формулы изобретения. Без дополнительных уточнений считается, что специалисты в данной области техники могут, используя предшествующее описание, использовать настоящее изобретение в максимальной степени. Следовательно, примеры данного описания истолковываются как иллюстративные, а не ограничивающие объем настоящего изобретения каким-либо образом. Варианты осуществления данного изобретения, в которых заявляются исключительные свойства и привилегии, определены ниже. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, которое является солью п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной. 2. Фармацевтическая композиция, содержащая соль п-толуолсульфокислоты с N-[4-хлор-2 гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной и фармацевтически приемлемый носитель или разбавитель. 3. Применение соли п-толуолсульфокислоты сN-[4-хлор-2-гидрокси-3-(пиперазин-1 сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной в производстве лекарственного средства для применения в лечении хемокинопосредованного заболевания. 4. Применение соли п-толуолсульфокислоты сN-[4-хлор-2-гидрокси-3-(пиперазин-1 сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной в производстве лекарственного средства для применения в лечении астмы, хронического обструктивного заболевания легких или респираторного дистресс-синдрома у взрослых. 5. Применение соли п-толуолсульфокислоты сN-[4-хлор-2-гидрокси-3-(пиперазин-1 сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной в производстве лекарственного средства для применения в лечении хронического обструктивного заболевания легких. 6. Фармацевтическая композиция, содержащая соль п-толуолсульфокислоты с N-[4-хлор-2 гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной и один или более дополнительных терапевтических ингредиентов. 7. Композиция по п.6, где дополнительным терапевтическим ингредиентом является антагонистCXCR3 рецептора или антагонист CCR5 рецептора.- 14015520 8. Способ получения соли п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1 сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной, включающий стадии:b) добавления продукта стадии а) к растворенному в тетрагидрофуране; и с) кристаллизации продукта, полученного на стадии b), с ацетонитрилом с использованием затравки конечного продукта.
МПК / Метки
МПК: A61K 31/33, A01N 43/00
Метки: рецептора, антагонист
Код ссылки
<a href="https://eas.patents.su/16-15520-antagonist-receptora-il-8.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонист рецептора il-8</a>
Антагонист витронектинового рецептора

Номер патента: 3254
Опубликовано: 27.02.2003
Авторы: Мэнли Питер Дж., Миллер Вилльям Х.
МПК: A61P 9/10, A61K 31/4375
Метки: антагонист, рецептора, витронектинового
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль. 2. Фармацевтическая композиция, включающая соединение по п.1 и фармацевтически приемлемый носитель. 3. Фармацевтическая композиция, включающая соединение по п.1, противоопухолевый агент и фармацевтически приемлемый носитель. 4. Фармацевтическая композиция по п.3, где противоопухолевый агент представляет собой топотекан. 5. Фармацевтическая композиция по п.3, где...
Фармацевтическая композиция, содержащая ингибитор pde4 или рde3/4 и антагонист гистаминового рецептора

Номер патента: 7903
Опубликовано: 27.02.2007
Авторы: Воллин Штефан-Лутц, Бойме Рольф, Ваймар Кристиан, Бундшух Даниела
МПК: A61K 31/4375, A61K 31/4425, A61K 31/4523...
Метки: фармацевтическая, содержащая, ингибитор, гистаминового, композиция, рецептора, антагонист
Формула / Реферат:
1. Фармацевтическая композиция, содержащая первое действующее вещество, выбранное из группы, состоящей из 3-циклопропилметокси-4-дифторметокси-N-(3,5-дихлорпирид-4-ил)бензамида [МНН рофлумиласт], (-)-цис-9-этокси-8-метокси-2-метил-1,2,3,4,4а,10b-гексагидро-6-(4-диизопропиламинокарбонилфенил)бензо-[с][1,6]нафтиридина [МНН пумафентрин] и их фармацевтически приемлемых производных, в смеси со вторым действующим веществом, выбранным из группы,...
Антагонист хемокинового рецептора и циклоспорин в комбинированной терапии

Номер патента: 3181
Опубликовано: 27.02.2003
Авторы: Праудфут Аманда, Веллс Тимоти Н.С., Гроне Херманн-Йозеф, Нельсон Питер Дж.
МПК: A61K 38/19, A61K 38/13
Метки: антагонист, циклоспорин, терапии, хемокинового, комбинированной, рецептора
Формула / Реферат:
1. Применение антагониста хемокинового рецептора в комбинации с циклоспорином с целью получения фармацевтической композиции для лечения или профилактики отторжения трансплантированных органов, тканей или клеток. 2. Применение по п.1, отличающееся тем, что антагонист хемокинового рецептора и циклоспорин используются одновременно, раздельно или последовательно. 3. Применение по любому из предшествующих пунктов, отличающееся тем, что антагонистом...
Композиция, содержащая антагонист рецептора прогестерона и чистый антиэстроген, предназначенная для профилактики и лечения гормонзависимых заболеваний

Номер патента: 11506
Опубликовано: 28.04.2009
Авторы: Шнайдер Мартин, Хоффманн Йенс, Фурманн Ульрике, Зимайстер Герхард
МПК: A61P 5/32, A61P 5/36, A61K 31/567...
Метки: композиция, антагонист, содержащая, антиэстроген, чистый, профилактики, предназначенная, прогестерона, лечения, рецептора, гормонзависимых, заболеваний
Формула / Реферат:
1. Фармацевтическая композиция, содержащая антагонист рецептора прогестерона 11b-(4-ацетилфенил)-17b-гидрокси-17a-(1,1,2,2,2-пентафторэтил)эстра-4,9-диен-3-он и чистый антиэстроген 11b-фтор-17a-метил-7a-{5-[метил(8,8,9,9,9-пентафторнонил)амино]пентил}эстра-1,3,5(10)-триен-3,17b-диол. 2. Фармацевтическая композиция по п.1, в которой массовое соотношение антагониста рецептора прогестерона и чистого антиэстрогена составляет от 1:100 до 100:1. 3....
Антагонист cd 80

Номер патента: 14101
Опубликовано: 30.08.2010
Автор: Мэттьюс Иан Ричард
МПК: A61K 31/5025, A61P 19/00, A61P 17/00...
Метки: антагонист
Формула / Реферат:
1. Холиновая соль 4-(6-фтор-3-оксо-1,3-дигидропиразоло[4,3-с]циннолин-2-ил)-N-(2,2-дифторэтил)бензамида формулы (А)2. Фармацевтическая композиция для приема внутрь, содержащая холиновую соль по п.1 и по меньшей мере один фармацевтически приемлемый носитель.3. Водный раствор указанной холиновой соли по п.1, предназначенный для инъекций.4. Жидкость или мазь на водной основе для местного применения, содержащая холиновую соль по...
Предыдущий патент: Фармацевтическая композиция, содержащая активный ингредиент аторвастатин
Следующий патент: Погружной стакан
Случайный патент: Производные гуанина