Ингибитор регулирующей апоптотические сигналы киназы



















Формула / Реферат
1. Соединение формулы (I)

представляющее собой 5-(4-циклопропил-1Н-имидазол-1-ил)-N-(6-(4-изопропил-4Н-1,2,4-триазол-3-ил)пиридин-2-ил)-2-фтор-4-метилбензамид, или его фармацевтически приемлемая соль.
2. Соединение формулы (I)

представляющее собой 5-(4-циклопропил-1Н-имидазол-1-ил)-2-фтор-N-(6-(4-изопропил-4Н-1,2,4-триазол-3-ил)пиридин-2-ил)-4-метилбензамид.
3. Фармацевтическая композиция для лечения хронической почечной недостаточности, диабетической нефропатии, фиброза почек, фиброза печени, фиброза легких и диабетической почечной недостаточности, содержащая терапевтически эффективное количество соединения или соли по п.1 и фармацевтически приемлемый носитель.
4. Фармацевтическая композиция для лечения хронической почечной недостаточности, диабетической нефропатии, фиброза почек, фиброза печени, фиброза легких и диабетической почечной недостаточности, содержащая терапевтически эффективное количество соединения по п.2 и фармацевтически приемлемый носитель.
5. Способ лечения хронической почечной недостаточности, включающий введение терапевтически эффективного количества соединения по п.2 или его фармацевтически приемлемой соли нуждающемуся в этом пациенту.
6. Способ лечения диабетической нефропатии, включающий введение терапевтически эффективного количества соединения по п.2 или его фармацевтически приемлемой соли нуждающемуся в этом пациенту.
7. Способ лечения фиброза почек, фиброза печени или фиброза легких, включающий введение терапевтически эффективного количества соединения или соли по п.1 нуждающемуся в этом пациенту.
8. Способ лечения диабетического заболевания почек, включающий введение терапевтически эффективного количества соединения или соли по п.1 нуждающемуся в этом пациенту.
9. Применение соединения или соли по п.1 для получения лекарственного средства для лечения хронической почечной недостаточности.
10. Применение соединения или соли по п.1 для лечения диабетической нефропатии, фиброза почек, фиброза печени, фиброза легких и диабетической почечной недостаточности.
11. Промежуточное соединение формулы

представляющее собой 2-амино-5-(4-изопропил-4Н-1,2,4-триазол-3-ил)пиридин или его соль или защищенную форму.
12. Промежуточное соединение формулы

представляющее собой 5-(4-циклопропил-1Н-имидазол-1-ил)-2-фтор-4-метилбензойную кислоту, его соль или защищенную форму.
Текст
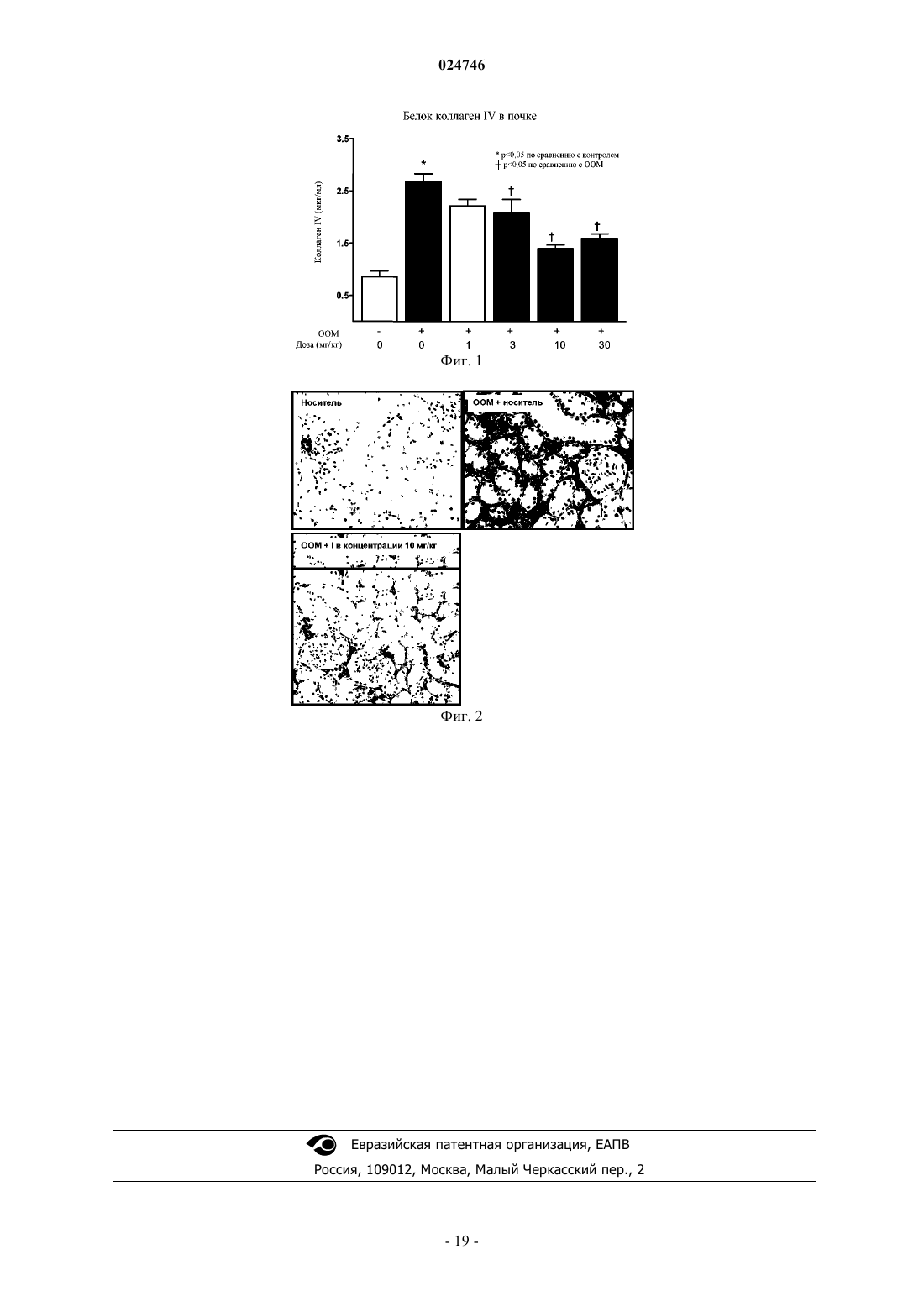
ИНГИБИТОР РЕГУЛИРУЮЩЕЙ АПОПТОТИЧЕСКИЕ СИГНАЛЫ КИНАЗЫ Соединение обладает ингибирующей активностью в отношении регулирующей апоптотические сигналы киназы ("ASK1") и, таким образом, подходит для применения для лечения заболеваний,таких как заболевание почек, диабетическая нефропатия и фиброз почек. Область техники Настоящее изобретение относится к новому соединению для применения для лечения ASK1 опосредованных заболеваний. Изобретение также относится к промежуточным соединениям для получения указанного нового соединения и содержащим его фармацевтическим композициям. Уровень техники Регулирующая апоптотические сигналы киназа 1 (ASK1) является членом семейства киназ киназ митоген-активируемых протеинкиназ ("МАР 3K"), которые активируют c-Jun N-терминальную протеинкиназу ("JNK") и р 38 МАР-киназу (Ichijo H., Nishida E., Irie K., Dijke P.Т., Saitoh M., Moriguchi Т., Matsumoto K., Miyazono K., Gotoh Y. (1997) Science, 275, 90-94). ASK1 активируются различными раздражителями, включая окислительный стресс, активные формы кислорода (АФК), ЛПС, ФНО-, FasL, ЭР-стресс и повышенные концентрации внутриклеточного кальция (Hattori K., Naguro I., Runchel С, Ichijo H. (2009)Cell Comm. Signal. 7:1-10; Takeda K., Noguchi Т., Naguro I., Ichijo H. (2007) Annu. Rev. Pharmacol. Toxicol. 48: 1-8,27; Nagai H., Noguchi Т., Takeda K., Ichijo I. (2007) J. Biochem. Mol. Biol. 40:1-6). Фосфорилирование белка ASK1 может приводить к апоптозу или другим клеточным ответам в зависимости от типа клеток. Было показано, что активация и сигнальный путь ASK1 играют важную роль в широком спектре заболеваний, включая нейродегенеративные, сердечно-сосудистые, воспалительные, аутоиммунные и метаболические расстройства. Кроме того, было показано, что ASK1 принимает участие в опосредовании повреждения органов после ишемии и реперфузии сердца, мозга и почки (Watanabe et al. (2005) BBRC 333, 562-567; Zhang et al., (2003) Life Sci 74-37-43; Terada et al. (2007) BBRC 364: 1043-49). Показано, что АФК связаны с повышенной выработкой воспалительных цитокинов, фиброзом,апоптозом и некрозом в почке. (Singh D.K., Winocour P., Farrington K. Oxidative stress in early diabeticThe suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol 2010 Nov; 6(11):667-678). Более того, окислительный стресс способствует формированию конечных продуктов усиленного гликозилирования (AGE), которые вызывают дальнейшее повреждение почки и выработку АФК. (Hungsynthesis in rat mesangial cells. Am J Nephrol 2009; 29(3): 192-202). Тубулоинтерстициальный фиброз в почке является сильным прогностическим фактором прогрессирования заболевания до почечной недостаточности у пациентов, страдающих хроническими заболеваниями почек (Schainuck L.I., et al. Structural-functional correlations in renal disease. Part II: The correlations.Hum Pathol 1970; 1: 631-641.). Односторонняя обструкция мочеточника (ООМ) у крыс является широко используемой моделью тубулоинтерстициального фиброза. ООМ вызывает тубулоинтерстициальное воспаление, повышенную экспрессию трансформирующего фактора роста бета (ТФР-) и накопление миофибробластов, которые секретируют матриксные белки, такие как коллаген и фибронектин. Модель ООМ может использоваться для исследования потенциальной способности лекарственного средства лечить хроническую почечную недостаточность путем подавления фиброза почек (Chevalier et al., Ureteralobstruction as a model of renal interstitial fibrosis and obstructive nephropathy, Kidney International (2009) 75,1145-1152). Таким образом, терапевтические агенты, действующие в качестве ингибиторов сигналинга ASK1,имеют потенциал в отношении лечения или улучшения качества жизни пациентов, нуждающихся в лечении заболеваний или состояний, таких как нейродегенеративные, сердечно-сосудистые, воспалительные,аутоиммунные и метаболические расстройства. В частности, ингибиторы ASK1 имеют потенциал для лечения кардиоренальных заболеваний, включая заболевание почек, диабетическое заболевание почек,хроническую почечную недостаточность, фиброзные заболевания (включая фиброз легких и почек), респираторные заболевания (включая хроническое обструктивное заболевание легких (ХОЗЛ) и острое повреждение легких), острые и хронические почечной недостаточности. В публикации заявки на патент США 2007/0276050 описаны способы идентификации ингибиторов ASK1, подходящих для предотвращения и/или лечения сердечно-сосудистого заболевания, и способы предотвращения и/или лечения указанного сердечно-сосудистого заболевания у животного. В WO 2009027283 описаны тиазолопиридиновые соединения, способы их получения и способы лечения аутоиммунных расстройств, воспалительных заболеваний, сердечно-сосудистых заболеваний и нейродегенеративных заболеваний. В публикации заявки на патент США 2001/00095410 А 1, опубликованной 13 января 2011 г., описаны соединения, подходящие для применения в качестве ингибиторов ASK-1. Публикация заявки на патент США 2001/00095410 А 1 относится к соединениям формулы (I) где R1 представляет собой алкил, алкенил, алкинил, циклоалкил, арил, гетероарил или гетероциклил, каждый из которых необязательно содержит 1, 2, или 3 заместителя, выбранных из галогена, оксо,алкила, циклоалкила, гетероциклила, арила, арилокси, -NO2, R6, -C(O)-R6, -OC(O)-R6 -C(O)-O-R6, -C(O)N(R6)(R7), -OC(O)-N(R6)(R7), -S-R6, -S(=O)-R6, -S(=O)2R6, -S(=O)2-N(R6)(R7), -S(=O)2-O-R6, -N(R6)(R7), N(R6)-C(O)-R7, -N(R6)-C(O)-O-R7, -N(R6)-C(O)-N(R6)(R7), -N(R6)-S(=O)2-R6, -CN и -O-R6,где указанные алкил, циклоалкил, гетероциклил, фенил и фенокси необязательно содержат 1, 2, или 3 заместителя, выбранных из алкила, циклоалкила, алкокси, гидроксила и галогена; где R6 и R7 независимо выбраны из группы, состоящей из водорода, С 1-С 15 алкила, циклоалкила, гетероциклила, арила и гетероарила, каждый из которых необязательно содержит 1-3 заместителя, выбранных из галогена, алкила, моно- или диалкиламино, амида алкила или арила или гетероарила, -CN, низшего алкокси, -CF3, арила и гетероарила; илиR3 представляет собой арил, гетероарил или гетероциклил, каждый из которых необязательно содержит один или более заместителей, выбранных из алкила, алкокси, циклоалкила, циклоалкилалкила,арила, арилалкила, гетероарила, гетероарилалкила, гетероциклила, гетероциклилалкила, галогена, оксо, NO2, галогеналкила, галогеналкокси, -CN, -O-R6, -O-C(O)-R6, -O-C(O)-N(R6)(R7), -S-R6, -N(R6)(R7), S(=O)-R6, -S(=O)2R6, -S(=O)2-N(R6)(R7), -S(=O)2-O-R6, -N(R6)-C(O)-R7, -N(R6)-C(O)-O-R7, -N(R6)-C(O)N(R6)(R7), -C(O)-R6, -C(O)-O-R6, -C(O)-N(R6)(R7) и -N(R6)-S(=O)2-R7, где указанный алкил, алкокси, циклоалкил, арил, гетероарил или гетероциклил также необязательно содержит один или более заместителей, выбранных из галогена, оксо, -NO2, алкила, галогеналкила, галогеналкокси,-N(R6)(R7), -C(O)-R6, C(O)-O-R6, -C(O)-N(R6)(R7), -CN, -O-R6, циклоалкила, арила, гетероарила и гетероциклила; при условии, что гетероарильный или гетероциклильный фрагмент содержит по меньшей мере один атом азота в кольце;X1, X2, X3, X4, X5, X6, X7 и X8 независимо представляют собой C(R4) или N, где каждый из R4 независимо представляет собой водород, алкил, алкокси, циклоалкил, арил, гетероарил, гетероциклил, галоген, -NO2, галогеналкил, галогеналкокси, -CN, -O-R6, -S-R6, -N(R6)(R7), -S(=O)-R6, -S(=O)2R6, -S(=O)2N(R6)(R7), -S(=O)2-O-R6,-N(R6)-C(O)-R7, -N(R6)-C(O)-O-R7, -N(R6)-C(O)-N(R6)(R7), -C(O)-R6, -C(O)-O-R6,C(O)-N(R6)(R7) или -N(R6)-S(=O)2-R7, где указанный алкил, циклоалкил, арил, гетероарил и гетероциклил также необязательно содержит один или более заместителей, выбранных из галогена, оксо, -NO2, -CF3, O-CF3, -N(R6)(R7), -С(О)-R6, -C(O)-O-R7, -C(O)-N(R6)(R7), -CN, -O-R6; илиX5 и X6 или X6 и X7 соединены с получением необязательно замещенного конденсированного арила или необязательно замещенного конденсированного гетероарила; и при условии, что по меньшей мере один из X2,X3, X4 представляет собой C(R4); по меньшей мере два из X5, X6, X7 и X8 представляют собой C(R4); и по меньшей мере один из X2, X3, X4, X5, X6, X7 и X8 представляет собой N. Несмотря на описанные выше изобретения существует необходимость в соединениях, которые являются высокоактивными и проявляют улучшенные фармакокинетические и/или фармакодинамические свойства для лечения заболеваний, связанных с активацией ASK1. Заявители неожиданно обнаружили в рамках патентной публикации США US 2011/0009410 А новое соединение, проявляющее хорошую активность и в целом улучшенные фармакокинетические и/или фармакодинамические свойства по сравнению с соединениями, предложенными в указанной публикации. Краткое описание изобретения Настоящее изобретение относится к соединению формулы или его фармацевтически приемлемой соли. Согласно одному варианту реализации настоящее изобретение относится к применению соединения формулы (I) для лечения заболевания у пациента, нуждающегося в указанном лечении, с помощью ингибитора ASK1. Согласно другому варианту реализации настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых носителей. Согласно другому варианту реализации настоящее изобретение представляет собой способ лечения диабетической нефропатии или осложнений диабета, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в этом пациенту. Согласно другому варианту реализации настоящее изобретение относится к способу лечения заболевания почек или диабетического заболевания почек, включающему введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в этом пациенту. Согласно другому варианту реализации настоящее изобретение относится к способу лечения фиброза почек, фиброза легких или идиопатического легочного фиброза (ИЛФ), включающему введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в этом пациенту. Согласно другому варианту реализации настоящее изобретение относится к способу лечения диабетической почечной недостаточности, диабетической нефропатии, фиброза почек, фиброза печени или фиброза легких, включающему введение терапевтически эффективного количества соединения или соли формулы (I) нуждающемуся в этом пациенту. Согласно другому варианту реализации настоящее изобретение относится к промежуточным соединениям, подходящим для применения для синтеза соединения формулы (I). Согласно другому варианту реализации настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения хронической почечной недостаточности. Согласно другому варианту реализации настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения диабетического почечной недостаточности. Согласно другому варианту реализации настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления медикамента для лечения хронической почечной недостаточности. Согласно другому варианту реализации настоящее изобретение относится к соединению формулы(I) для применения в терапии. Подробное описание изобретения Фигуры. Фиг. 1 представляет собой гистограмму, показывающую уровни коллагена IV в корковом веществе почки крыс, подверженных семидневной односторонней обструкции мочеточника и получавших лечение носителем или соединением формулы (I) в концентрации 1, 3, 10 или 30 мг/кг два раза в день. На фиг. 2 показаны типичные изображения срезов коркового вещества почки крыс, подверженных семидневной односторонней обструкции мочеточника и получавших лечение носителем или соединением формулы (I) в дозе 1, 3, 10 или 30 мг/кг два раза в день, окрашенных на альфа-гладкомышечный актин(маркер активированных миофибробластов). Определения и общие характеристики Предполагается, что при использовании в настоящей заявке приведенные далее слова и фразы имеют значения, указанные ниже, если иное не следует из контекста, в котором они используются. Если не приводится обозначение или определение слова или фразы, то подразумевается общепринятое значение указанного слова или фразы, которое можно обнаружить в соответствующем словаре или повсеместном использовании, известном специалисту в данной области техники. Термин "хроническая почечная недостаточность" при использовании в настоящей заявке относится к прогрессирующей потере почечной функции в течение продолжительного периода, как правило, нескольких месяцев или даже лет. Хроническую почечную недостаточность (ХПН) диагностирует компетентный субъект, осуществляющий уход, с использованием соответствующей информации, тестов или маркеров, известных специалисту в данной области техники. Хроническая почечная недостаточность подразумевает почечную недостаточность. Термин "диабетическая почечная недостаточность" при использовании в настоящей заявке относится к почечной недостаточности, вызванной диабетом, усугубляемой диабетом или присутствующей совместно с диабетом. Диабетическая почечная недостаточность представляет собой форму хронической почечной недостаточности, которая возникает приблизительно у 30% пациентов, страдающих диабетом. Она определяется как диабет с сопутствующей альбуминурией и/или нарушенной почечной функцией(т.е. сниженной скоростью клубочковой фильтрации (см. de В, I, et al. Temporal trends in the prevalence ofdiabetic kidney disease in the United States. JAMA 2011 Jun 22; 305(24):2532-2539). Термин "фармацевтически приемлемая соль" относится к солям фармацевтических соединений, например соединения формулы (I), которые сохраняют биологическую эффективность и свойства исходного соединения и которые не являются нежелательными с биологической или другой точки зрения. Ука-3 024746 занные соли представляют собой соли присоединения кислот и соли присоединения оснований. Фармацевтически приемлемые соли присоединения кислот могут быть получены из неорганических и органических кислот. Кислоты и основания, применимые для реакции с исходным соединением с образованием фармацевтически приемлемых солей (солей присоединения кислоты или основания соответственно), известны специалисту в данной области техники. Подобным образом, способы получения фармацевтически приемлемых солей из исходного соединения (согласно изобретению) известны специалисту в данной области техники и описаны, например, в источнике Berge, et al. Journal of Pharmaceutical Science, Jan. 1977 vol. 66, No. l и других источниках. Соли, полученные из неорганических кислот, включают, но не ограничиваются указанными, соли соляной кислоты, бромоводородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты и т. д. Соли, полученные из органических кислот, включают, но не ограничиваются указанными, малеиновую кислоту, фумаровую кислоту, винную кислоту, п-толуолсульфоновую кислоту и т. д. Основания, применимые для получения солей присоединения оснований, известны специалисту в данной области техники. Пример фармацевтически приемлемой соли соединения формулы (I) представляет собой гидрохлорид соединения формулы (I). При использовании в настоящей заявке "фармацевтически приемлемый носитель" включает вспомогательные вещества или агенты, такие как растворители, разбавители, диспергирующие агенты, покрывающие агенты, антибактериальные и противогрибковые агенты, изотонические и замедляющие абсорбцию агенты и т. п., которые не оказывают неблагоприятного воздействия на соединение согласно изобретению или его применение. Применение указанных носителей и агентов для получения композиций фармацевтически активных веществ хорошо известно в данной области техники (см., например, Remington's Pharmaceutical Sciences, Mace Publishing Co., Philadelphia, PA 17th Ed. (1985); and Modern Pharmaceutics, Marcel Dekker, Inc. 3rd Ed. (G.S. BankerC.T. Rhodes, Eds.) Термин "кардиоренальные заболевания" при использовании в настоящей заявке относится к заболеваниям, связанным с почечной функцией, вызванным или усугубляемым сердечно-сосудистыми нарушениями, такими как, например, повышенное кровеносное давление или гипертензия. Считается, что гипертензия вносит важный вклад в почечную недостаточность. Термин "респираторные заболевания" при использовании в настоящей заявке относится к заболеваниям, включающим хроническое обструктивное заболевание легких (ХОЗЛ) и идиопатический легочный фиброз (ИЛФ). Термин "терапевтически эффективное количество" относится к количеству соединения формулы(I), которое является достаточным для осуществления лечения согласно приведенному ниже определению при введении пациенту (в частности, человеку), нуждающемуся в указанном лечении, в виде одной или более доз. Терапевтически эффективное количество варьирует в зависимости от пациента, заболевания, которое предполагается лечить, массы тела и/или возраста пациента, тяжести заболевания или способа введения и определяется квалифицированным медицинским работником или лицом, осуществляющим уход. Термин "лечение" или "проведение лечения" означает введение соединения или фармацевтически приемлемой соли формулы (I) для(i) задержки возникновения заболевания, т.е. препятствия развитию клинических симптомов заболевания или задержки их развития;(ii) подавления заболевания, т.е. сдерживания развития клинических симптомов; и/или(iii) облегчения заболевания, т.е. обеспечения регрессии клинических симптомов или их тяжести. Согласно предпочтительному варианту реализации изобретение относится к применению соединения формулы (I) для лечения хронической почечной недостаточности, включающему введение терапевтически эффективного количества нуждающемуся в этом пациенту. Согласно другому предпочтительному варианту реализации изобретение относится к применению соединения формулы (I) для лечения диабетической почечной недостаточности, включающему введение терапевтически эффективного количества нуждающемуся в этом пациенту. Согласно другому предпочтительному варианту реализации изобретение относится к применению соединения формулы (I) для лечения фиброза легких или фиброза почек, включающему введение терапевтически эффективного количества нуждающемуся в этом пациенту. Концентрация полумаксимального ингибирования (IC50) терапевтического агента представляет собой концентрацию указанного терапевтического агента, необходимую для обеспечения 50% максимального ингибирования в отношении фермента-мишени. Поставленной задачей является обеспечение терапевтического агента, например соединения, которое ингибирует регулирующую апоптотические сигналы киназу (ASK1) при низкой IC50. Таким образом, нежелательные побочные действия минимизируются благодаря возможности применения более низкой дозы терапевтического агента для подавления фермента ASK1. Подобным образом, поставленной задачей является обеспечение терапевтического агента, имеющего низкую константу диссоциации (Kd). Kd используется для описания сродства между лигандом (таким как терапевтический агент) и соответствующей киназой или рецептором; т. е. меры того, насколько сильно терапевтический агент связывается с конкретной киназой, например регулирующей апоптотические сигналы киназой ASK1. Таким образом, более низкие значения Kd в целом являются предпочтительным для разработки лекарственного средства. Подобным образом, поставленной задачей является обеспечение соединения, имеющего низкуюEC50. EC50 представляет собой концентрацию лекарственного средства, которая приводит к достижению 50%-ной максимальной эффективности в клетке. Значение ЕС 50 выражается как концентрация соединения в аналитической среде, необходимая для достижения 50%-ной максимальной эффективности. Таким образом, более низкая EC50 в целом является предпочтительной для разработки лекарственного средства. Применимая единица измерения, связанная с EC50, представляет собой скорректированную с учетом связывания с белком ЕС 50 (PBadj ЕС 50 при использовании в настоящей заявке). Указанное значение описывает количество лекарственного средства, например соединения формулы (I), соотнесенное с фракцией лекарственного средства, не связанной с белком, которое обеспечивает 50% максимальной эффективности. Указанное значение показывает эффективность лекарственного средства, коррелированную или скорректированную с учетом количества лекарственного средства, доступного в месте действия-мишени. Другим желаемым свойством соединения является низкое отношение оттока через клеточную мембрану, определяемое с помощью исследований проницаемости на клетках линии САСО. Отношение оттока В/А)/(А/В, составляющее менее 3,0, является предпочтительным. Ожидается, что соединение,отношение оттока которого составляет более 3, активно и быстро выходит из клетки и может не оказывать достаточно продолжительного воздействия в клетке для достижения максимальной эффективности. Другой поставленной задачей является обеспечение лекарственного средства, которое проявляет минимальное нецелевое ингибирование, то есть лекарственное средство, которое минимально ингибирует ферменты Сур 450 (цитохром р 450). Более конкретно, желаемым является лекарственное средство,которое является слабым ингибитором сур 3 А 4 - наиболее важного из ферментов Р 450. Слабый ингибитор представляет собой соединение, которое вызывает по меньшей мере 1,25-кратное, но менее чем 2 кратное повышение значений ППК в плазме крови или 20-50% снижение клиренса (сайт wikipedia.org/wiki/cyp3A4, посещенный 11/12/11). В целом, соединение, IC50 которого в отношении Сур 3 А 4 составляет более 10 мкМ, считается слабым ингибитором. Измерение, применимое для сравнения ингибирования Сур 3 А 4 между кандидатами лекарственных средств представляет собой отношение ингибирования Сур 3 А 4 и скорректированной с учетом связывания с белком EC50. Указанное значение обеспечивает показатель относительной потенциальной способности ингибировать Сур с поправкой на ЕС 50, скорректированную с учетом связывания с белком, специфичную для каждого лекарственного средства. Более высокое отношение для указанного измерения является предпочтительным как показатель более низкой потенциальной способности ингибировать сур 3 А 4. Неожиданно и удачно заявители обнаружили соединение (в настоящей заявке соединение формулы(I в пределах общего объема патентной публикации США 2001/00095410 А 1, которое обеспечивает преимущества по сравнению со структурно близкими соединениями (которые в настоящей заявке обозначаются как соединения А и В), описанными в патентной публикации США 2001/00095410 А 1 Соединение В Таким образом, задача настоящего изобретения включает, но не ограничивается указанными, обеспечение соединения формулы (I) или его фармацевтически приемлемой соли и способы применения указанного соединения формулы (I) для лечения почечной недостаточности, хронической почечной недостаточности, диабетической почечной недостаточности, диабетической нефропатии, фиброза почек или фиброза легких. Комбинированная терапия. Пациенты, проходящие лечение кардиоренальных заболеваний, таких как хроническая почечная недостаточность, могут иметь положительный результат от комбинированного лекарственного лечения. Например, соединение согласно настоящему изобретения можно комбинировать с одним или более из ингибиторов ангиотензинпревращающих ферментов (АСЕ), такими как эналаприл, каптоприл, рамиприл,лизиноприл и хинаприл; или блокаторов рецептора II (ARB), таких как лазартан, олмесартан и ирбесартан; или гипотензивными агентами, такими как амлодипин, нифедипин и фелодипин. Положительным результатом комбинирования может являться повышенная эффективность и/или уменьшение побочных эффектов компонента, так как доза указанного компонента может быть снижена для уменьшения его побочных эффектов, тогда как положительный результат от его эффективности повышает эффективность соединения формулы (I) и/или другого активного компонента (компонентов). Пациенты, страдающие хронической почечной недостаточностью, поддающейся лечению с помощью ингибиторов ASKI, таких как соединение формулы (I), также могут страдать состояниями, на лечение которых благоприятно влияет совместное введение терапевтического агента или агентов (по указанию квалифицированного лица, осуществляющего уход), которые представляют собой антибиотики,анальгетики, антидепрессанты и/или успокоительные средства в комбинации с соединением формулы (I). Комбинированные лекарственные средства можно вводить одновременно или последовательно с временными интервалами по указанию квалифицированного лица, осуществляющего уход, или с помощью лекарственной формы с фиксированными дозами двух или более активных агентов (все активные ингредиенты объединены в одну форму дозирования, например, таблетку). Фармацевтические композиции и введение. Соединение согласно настоящему изобретению можно вводить в форме фармацевтической композиции. В настоящем изобретении, таким образом, предложены фармацевтические композиции, которые содержат в качестве активного ингредиента соединение формулы (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых вспомогательных веществ и/или носителей,включая инертные твердые разбавители и наполнители, разбавители, включая стерильный водный раствор и различные органические растворители, усилители проникновения, солюбилизаторы и адъюванты. Фармацевтические композиции можно вводить отдельно или в комбинации с другими терапевтическими агентами. Композиции для доставки могут быть получены в виде твердых таблеток, капсул, каплетов,мазей, трасдермальных пластырей, форм с замедленным высвобождением, таблеток для рассасывания,лекарственных форм для ингаляции и т. д. Типичные фармацевтические композиции получают и/или вводят с использованием способов и/или процессов, хорошо известных в области фармацевтики (см.,например, Remington's Pharmaceutical Sciences, Mace Publishing Co., Philadelphia, PA 17th Ed. (1985); andModern Pharmaceutics, Marcel Dekker, Inc. 3rd Ed. (G.S. BankerC.T. Rhodes, Eds). Лекарственные формы для комбинированной терапии, содержащие соединение формулы (I), могут быть представлены в виде лекарственных форм с фиксированной дозой, например таблеток, эликсиров,жидкостей, мазей, лекарственных форм для ингаляции, гелей и т. д., с помощью методов, известных специалисту в данной области техники. Фармацевтические композиции соединения формулы (I) можно вводить либо в одной, либо в нескольких дозах с помощью способов, включая, например, ректальный, буккальный, интраназальный и трансдермальный способы введения; путем внутриартериальной инъекции, внутривенно, внутрибрюшинно, парентерально, внутримышечно, подкожно, перорально, местно, с помощью ингалятора или через пропитанное или покрытое соединением устройство, такое как, например, стент или вживляемый в артерию цилиндрический полимерный материал. Наиболее предпочтительные способы введения включают пероральное, парентеральное и внутривенное введение. Соединение формулы (I) можно вводить в фармацевтически эффективном количестве. Каждая единица дозирования для перорального введения предпочтительно содержит от 1 до 500 мг соединения формулы (I). Более предпочтительно доза соединения формулы (I) составляет от 1 до 250 мг. Особо предпочтительно доза соединения формулы (I) варьирует от примерно 20 мг два раза в день до примерно 50 мг два раза в день. Однако необходимо понимать, что реальное вводимое количество соединения, как правило, определяется врачом с учетом соответствующих факторов, включая состояние, которое предполагается лечить, выбранный способ введения, совместное введение соединения, если применимо, возраста, массы тела и ответа на лечение конкретного пациента, тяжести симптомов пациента и т. д. Номенклатура. Соединение согласно настоящему изобретению имеет название, полученное с помощью программы 5-(4-циклопропил-1H-имидазол-1-ил)-N-(6-(4-изопропил-4 Н-1,2,4-триазол-3-ил)пиридин-2-ил)-2 фтор-4-метилбензамид, также известное как 5-4-циклопропил-1 Н-имидазол-1-ил)-2-фтор-N-(6-(4 изопропил-4 Н-1,2,4-триазол-3-ил)пиридин-2-ил)-4-метилбензамид. Синтез соединения формулы (I). Соединение согласно изобретению может быть получено с использованием способов, описанных в настоящей заявке, или их модификаций, которые будут очевидны с учетом описанного в настоящей заявке изобретения. Синтез соединения согласно изобретению можно осуществлять, как описано в следующем примере. Реагенты могут быть получены коммерческим путем, если это доступно, например, из компании Sigma Aldrich или от других производителей химических веществ. Альтернативно, реагенты могут быть получены с использованием схем реакций и способов, известных специалисту в данной об-6 024746(или дихлорметан), диэтиловый эфир, петролейный эфир (ПЭ), метанол, пиридин, этилацетат (ЭА) и т. п. Если иное не указано, растворители, используемые в реакциях согласно настоящему изобретению, представляют собой инертные органические растворители, и реакции осуществляют в условиях инертного газа, предпочтительно азота. Один из способов получения соединений формулы (I) показан на схемах реакций 1 и 2 ниже. Схема 1 К раствору метил 6-аминопиколината (432 г, 2,84 моль) в МеОН (5 л) добавляли NH2NH2.H2O (284 г, 5,68 моль, 2,0 экв.). Реакционную смесь нагревали при температуре обратной конденсации в течение 3 ч и затем охлаждали до комнатной температуры. Образовавшийся в смеси осадок собирали посредством фильтрования, промывали ЭА (2 л 2) и затем сушили в вакууме с получением соединения А (405 г, выход 94%) в виде твердого вещества белого цвета. Получение соединения В. Смесь соединения А (405 г, 2,66 моль) в диметилформамиде-диметилацетале (ДМФА-ДМА) (3,54 л) нагревали при температуре обратной конденсации в течение 18 ч, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Осадок отбирали в ЭА (700 мл) и нагревали при 50 С в течение 20 мин. После охлаждения до комнатной температуры твердое вещество собирали посредством фильтрования и сушили в вакууме с получением соединения В (572 г, выход 82%) в виде твердого вещества белого цвета. Получение соединения С. К раствору соединения В (572 г, 2,18 моль) в смеси CH3CN-АсОН (3,6 л, 4:1) добавляли пропан-2 амин (646 г, 5,0 экв.). Реакционную смесь нагревали при температуре обратной конденсации в течение 24 ч и затем охлаждали до комнатной температуры, и растворитель удаляли при пониженном давлении. Осадок растворяли в воде (2,8 л) и добавляли 1 н водный раствор NaOH до достижения рН 8,0. Осадок собирали посредством фильтрования, и фильтрат экстрагировали ЭА (500 мл 3). Объединенные органические слои сушили над безводным Na2SO4 и затем концентрировали до достижения объема 150 мл. К указанной смеси при 0 С медленно добавляли ПЭ (400 мл), и полученную суспензию фильтровали. Объединенное твердое вещество перекристаллизовывали из ЭА-ПЭ с получением соединения С (253 г, выход 57%) в виде твердого вещества не совсем белого цвета. 1(bs, 2 Н), 1,45 (d, J=6,8 Гц, 6 Н). МС (ИЭР+) m/z: 204 (М+1)+. Соединение С является основным промежуточным соединением для синтеза соединения формулы (I). Таким образом, задачей настоящего изобретения также является получение промежуточного соединения С его солей или защищенных форм для получения соединения формулы (I). Пример соли соединения С представляет собой соль присоединения HCl. Пример защищенной формы соединения С представляет собой карбаматное соединение, такое как соединение, полученное с помощью Cbz-Cl. Защитные группы,способы их получения и применения описаны в источнике Peter G.M. Wuts, Theodora W. Greene, Protective Groups in Organic Chemistry, 2nd edition, 1991, Wiley and Sons, Publishers. Схема 2 Получение соединения формулы (I) продолжали следующим образом: Формула (I) Соединение 6 представляет собой основное промежуточное соединение для синтеза соединения формулы (I). Таким образом, задачей настоящего изобретения также является получение промежуточного соединения 6 его солей или защищенных форм для получения соединения формулы (I). Пример соли соединения 6 представляет собой соль присоединения HCl. Пример защищенной формы соединения 6 представляет собой сложный эфир (например, метиловые, этиловые или бензиловые сложные эфиры) или карбаматное соединение, такое как соединение, полученное с помощью Cbz-Cl. Защитные группы, способы их получения и применения описаны в источнике Peter G.M. Wuts and Theodora W. Greene, Protective Groups inOrganic Chemistry, 2nd edition, 1991, Wiley and Sons, Publishers. Этап 1. Получение 5-амино-2-фтор-4-метилбензонитрила - соединения (2). Исходное соединение 5-бром-4-фтор-2-метиланилин (1) (20 г, 98 ммоль) растворяли в безводном 1 метилпирролидоне (100 мл) и добавляли цианид меди (I) (17,6 г, 196 ммоль). Реакционную смесь нагревали до 180 С в течение 3 ч, охлаждали до комнатной температуры и добавляли воду (300 мл) и концентрированный раствор гидроксида аммония (300 мл). Смесь перемешивали в течение 30 мин и экстрагировали ЭА (3200 мл). Объединенные экстракты сушили над сульфатом магния и растворитель удаляли при пониженном давлении. Маслянистый осадок промывали гексаном (2100 мл), и твердое вещество растворяли в дихлорметане и наносили на колонку, заполненную силикагелем. В результате элюирования в градиенте от 0 до 25% ЭА в гексане получали 5-амино-2-фтор-4-метилбензонитрил (10,06 г, 67,1 ммоль). ЖХ/МС (m/z:151 M+1). Этап 2. Получение 5-(2-циклопропил-2-оксоэтиламино)-2-фтор-4-метилбензонитрила - соединения(3). 5-Амино-2-фтор-4-метилбензонитрил (12 г, 80 ммоль) растворяли в безводном N,Nдиметилформамиде (160 мл) в атмосфере азота и добавляли при перемешивании карбонат калия (13,27 г,96 ммоль) и йодид калия (14,61 г, 88 ммоль) в виде твердого вещества. Реакционную смесь перемешивали в течение 5 мин при комнатной температуре и затем добавляли бромметилциклопропилкетон (20,24 мл, 180 ммоль). Реакционную смесь нагревали до 60 С в течение 3 ч, и затем растворители удаляли при пониженном давлении. Осадок растворяли в ЭА (400 мл) и промывали 400 мл воды. Органический слой сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Осадок повторно растворяли в минимальном количестве ЭА и добавляли гексан для достижения отношения гексан:ЭА в растворе 3:1 по объему. Продукт высаживали из раствора и собирали посредством фильтрования с получением 5-(2-циклопропил-2-оксоэтиламино)-2-фтор-4-метилбензонитрила (14,19 г, 61,2 ммоль). ЖХ/МС- соединения (4). 5-(2-Циклопропил-2-оксоэтиламино)-2-фтор-4-метилбензонитрил (14,19 г, 61,2 ммоль) растворяли в ледяной уксусной кислоте (300 мл). Добавляли при перемешивании тиоцианат натрия (11,9 г, 122,4 ммоль) в виде твердого вещества. Реакционную смесь нагревали до 110 С в течение 4 ч, затем растворитель удаляли при пониженном давлении. Осадок отбирали в дихлорметане (200 мл) и промывали 200 мл воды. Водный экстракт экстрагировали дополнительной порцией дихлорметана (2200 мл), органические экстракты объединяли и сушили над сульфатом магния. Растворитель удаляли при пониженном давлении, и полученный маслянистый осадок повторно растворяли в ЭА (50 мл) и добавляли 150 мл гексанов. Образовывался темный слой, и в колбу помещали магнитную мешалку. Интенсивное перемешивание приводило к осаждение продукта в виде твердого вещества персикового цвета. Продукт собирали посредством фильтрования с получением 5-(4-циклопропил-2-меркапто-1 Н-имидазол-1-ил)-2-фтор-4 метилбензонитрила (14,26 г, 52,23 ммоль). Аналитическая ЖХ/МС (m/z: 274, M+1). Этап 4. Получение 5-(4-циклопропил-1 Н-имидазол-1-ил)-2-фтор-4-метилбензонитрила - соединения(5). В трехгорлую круглодонную колбу на 500 мл наливали уксусную кислоту (96 мл), воду (19 мл) и перекись водорода (30%, 7,47 мл, 65,88 ммоль). Смесь нагревали до 45 С при перемешивании в атмосфере азота и следили за внутренней температурой. Затем добавляли небольшими порциями 5-(4 циклопропил-2-меркапто-1 Н-имидазол-1-ил)-2-фтор-4-метилбензонитрил (6,00 г, 21,96 ммоль) в виде твердого вещества в течение 30 мин при поддержании внутренней температуры ниже 55 С. После завершения добавления тиоимидазола реакционную смесь перемешивали в течение 30 мин при температуре 45 С и затем охлаждали до комнатной температуры и медленно добавляли 20 мас.% раствор сульфата натрия в воде (6 мл). Смесь перемешивали в течение 30 мин, и растворители удаляли при пониженном давлении. Осадок суспендировали в 250 мл воды и добавляли 4 н водный раствор гидроксида аммония для доведения рН до 10. Смесь экстрагировали дихлорметаном (3200 мл), органические слои объединяли, сушили над сульфатом магния и удаляли растворитель при пониженном давлении. Осадок растворяли в 20 мл ЭА и добавляли 80 мл гексанов при перемешивании. Растворители сливали, оставляя маслянистый осадок. Указанный процесс повторяли и получали продукт, 5-(4-циклопропил-1 Н-имидазол-1 ил)-2-фтор-4-метилбензонитрил, в виде вязкого масла (5,14 г, 21,33 ммоль). Аналитическая ЖХ/МС (m/z: 242, M+1). Этап 5. Получение гидрохлорида 5-(4-циклопропил-1 Н-имидазол-1-ил)-2-фтор-4-метилбензойной кислоты (6). 5-(4-Циклопропил-1 Н-имидазол-1-ил)-2-фтор-4-метилбензонитрил (11,21 г, 46,50 ммоль) помещали в круглодонную колбу, снабженную обратным конденсатором, и суспендировали в 38%-ном растворе соляной кислоты (200 мл). Смесь нагревали до 100 С в течение 4,5 ч и затем охлаждали до комнатной температуры. Растворитель удаляли при пониженном давлении с получением твердого вещества розового цвета, к которому добавляли 100 мл ЭА. Твердый продукт собирали посредством фильтрования и промывали 3100 мл ЭА. К полученному твердому продукту добавляли 100 мл 10%-ного метанола в дихлорметане, смесь перемешивали и собирали фильтрат. Указанную процедуру повторяли с использованием 2 дополнительных порций по 200 мл 10%-ного раствора метанола в дихлорметане. Фильтраты объединяли, и растворитель удаляли при пониженном давлении с получением неочищенного гидрохлорида 5-(4-циклопропил-1 Н-имидазол-1-ил)-2-фтор-4-метилбензойной кислоты. Дополнительную очистку не проводили (11,13 г, 37,54 ммоль). Аналитическая ЖХ/МС (m/z: 261, M+1). Этап 6. Получение 5-(4-циклопропил-1 Н-имидазол-1-ил)-2-фтор-N-(6-(4-изопропил-4 Н-1,2,4 триазол-3-ил)пиридин-2-ил)-4-метилбензамида - формулы (I). Гидрохлорид 5-(4-циклопропил-1 Н-имидазол-1-ил)-2-фтор-4-метилбензойной кислоты (1,5 г, 5,07 ммоль) суспендировали в безводном 1,2-дихлорметане (25 мл) при комнатной температуре. Добавляли оксалилхлорид (0,575 мл, 6,59 ммоль) при перемешивании в атмосфере азота с последующим добавлением N,N-диметилформамида (0,044 мл, 0,507 ммоль). Смесь перемешивали в течение 4 ч при комнатной температуре и затем растворитель удаляли при пониженном давлении. Осадок растворяли в 25 мл безводного дихлорметана. Быстро добавляли 6-(4-изопропил-4 Н-1,2,4-триазол-3-ил)пиридин-2-амин (1,13 г,5,58 ммоль) (соединение С) и 4-диметиламинопиридин (0,62 г, 5,07 ммоль) при перемешивании в атмосфере азота. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре и добавляли водный насыщенный раствор NaHCO3 (15 мл). Смесь перемешивали в течение 10 мин, и слои разделяли и водный слой промывали 120 мл дихлорметана. Объединенные органические слои сушили (MgSO4),фильтровали и концентрировали. Осадок растворяли в минимальном количестве CH3CN и медленно добавляли воду до тех пор, пока твердое вещество не осаждалось из смеси. Указанное твердое вещество собирали посредством фильтрования и сушили с получением 5-(4-циклопропил-1 Н-имидазол-1-ил)-2 фтор-N-(6-(4-изопропил-4 Н-1,2,4-триазол-3-ил)пиридин-2-ил)-4-метилбензамида с чистотой -96% (1,28 г,2,88 ммоль). Аналитическая ЖХ/МС (m/z: 446, M+1). Материал дополнительно очищали с помощью ОФВЭЖХ (обращенно-фазовой ВЭЖХ) с получением образца в виде соли HCl, чистого для анализа.C24H24FN7O-HCl. 446,2 (М+1). 1H-ЯМР (ДМСО):11,12 (s, 1H), 9,41 (s, 1H), 9,32 (s, 1H), 8,20 (d,J=8,4 Гц, 1H), 8,07 (t, J=8,4 Гц, 1H), 7,95 (d, J=6,4 Гц, 1H), 7,92 (d, J=7,6 Гц, 1H), 7,79 (s, 1H), 7,59 (d,J=10,4 Гц, 1H), 5,72 (sept, J=6,8 Гц, 1H), 2,29 (s, 3H), 2,00-2,05 (m, 1H), 1,44 (d, J=6,8 Гц, 6 Н), 1,01-1,06 (m,2H), 0,85-0,89 (m, 2H). Биологические анализы. Анализ киназной активности ASK1 (регулирующей апоптотические сигналы киназы 1) с помощьюTR-FRET (биохимическая IC50). Способность соединений ингибировать киназную активность ASK1 определяли с использованием метода резонансного переноса энергии флуоресценции с временным разрешением [TR-FRET], в котором в качестве белкового субстрата используется биотинилированный основный миелиновый белок [биотинМВР]. Устройство регулирования подачи жидкости Beckman Biomek FX использовали для нанесения в малообъемные 384-луночные полипропиленовые планшеты [Nunc, 267460] соединения в концентрации 2 мкл/лунку в 2,44%-ном водном растворе ДМСО с получением конечной концентрации соединения,составляющей между 100 мкМ и 0,5 нМ, для анализа киназной активности. Прибор Equator (Deerac Fluidics) использовали для нанесения 0,667 нг/мкл в объеме 3 мкл/лунку [Upstate Biotechnologies, 14-606,или эквивалентный белок собственного производства] и 0,1665 нг/мл биотин-МВР [Upstate Biotechnologies, 13-111] в буфере (85 мМ MOPS, pH 7,0, 8,5 мМ Mg-ацетат, 5% глицерин, 0,085% NP-40, 1,7 мМ ДТТ и 1,7 мг/мл БСА) в планшеты, содержащие нанесенные соединения. Фермент предварительно инкубировали с соединением в течение 20 мин до инициации киназной реакции путем добавления 5 мкл/лунку 300 мкМ АТФ в буфере (50 мМ MOPS, pH 7,0, 5 мМ Mg-ацетат, 1 мМ ДТТ, 5% ДМСО) с помощью прибора Equator (Deerac Fluidics). Киназным реакциям позволяли протекать в течение 20 мин при комнатной температуре и затем останавливали путем добавления 5 мкл/лунку 25 мМ ЭДТА с использованием прибора Equator (Deerac Fluidics). Затем использовали устройство Biomek FX для переноса каждой завершенной киназной реакции в объеме 1 мкл/лунку в лунки белого полистиролового планшета OptiPlate-1536 [PerkinElmer, 6004299], который содержал реагенты для выявления, по 5 мкл/лунку (1,11 нМ меченного Eu-W1024 антитела против фосфотреонина [PerkinElmer,AD0094] и 55,56 нМ аллофикоцианина стрептавидина [PerkinElmer, CR130-100] в 1 буфере для выявления LANCE [PerkinElmer, CR97-100]). Сигнал TR-FRET затем считывали на планшетном ридере Perkin Elmer Envision после инкубации планшетов при комнатной температуре в течение 2 ч. Лунки, содержащие положительный контроль, соответствующий 100% ингибированию, были получены путем изменения порядка добавления растворов ЭДТА и АТФ, описанных выше. Указанные лунки и лунки, соответствующие 0% ингибированию, содержащие капли 2,44% ДМСО в начале анализа использовали для расчета % ингибирования для исследуемых соединений. Результат.IC50 соединения формулы (I) для ингибирования ASK1 составляла 3,0 нМ. Указанные данные дают основания полагать, что соединение формулы (I) является активным ингибитором ASK1 в присутствии конкурентного лиганда АТФ. В усовершенствованном варианте указанного выше анализа ингибирующую активность соединения согласно изобретению в отношении ASK1 оценивали с использованием анализа ASK1 с помощью методаTR-FRET, определяющего количество фосфата, перенесенного на пептидный субстрат от АТФ. Материалы и способы. Реагенты. Дефосфорилированную рекомбинантную человеческую киназу ASK1 получали из компании GileadSciences. Низкомолекулярный ингибитор киназы стауроспорин (каталожныйS6942) и дитиотреитол(ДТТ, каталожный 43815-5G) были получены из компании Sigma Chemicals (Сент-Луис, Миссури). АТФ (каталожный 7724) был получен из компании Affymetrix (Санта-Клара, Калифорния), и соединение формулы (I) было получено было из компании Gilead Sciences. Набор для анализа методом гомогенной разрешенной во времени флуоресценции KinEASE-STK S3 получали из компании Cisbio (Бедфорд,Массочусетс). Все другие реагенты представляли собой коммерчески доступные реагенты высшего качества. Анализы. Анализ позволяет измерять уровень фосфорилирования биотинилированого пептидного субстрата киназой ASK1 путем выявления с помощью анализа методом гомогенной разрешенной во времени флуоресценции, HTRF (6,1). Указанный анализ представляет собой конкурентный иммунологический анализ резонансного переноса энергии флуоресценции с временным разрешением (TR-FRET), основанный на руководстве для анализа HTRF KinEASE-STK от Cisbio (6,1). Исследуемое соединение, 1 мкМ пептидного субстрата STK3, 4 нМ киназы ASK1 инкубировали с 10 мМ МОР буфером, рН 7,0, содержащим 10 мМ Mg-ацетат, 0,025% NP-40, 1 мМ ДТТ, 0,05% БСА и 1,5% глицерина, в течение 30 мин, затем добавляли 100 мкМ АТФ для начала киназной реакции и инкубировали в течение 3 ч. Добавляли пептидное антитело, меченное IX Eu3+ криптатным буфером, содержащим 10 мМ ЭДТА и 125 нМ стрептавидинXL665, для остановки реакции, и фосфорилированный пептидный субстрат выявляли с использованием ридера для нескольких меток Envision 2103 от компании PerkinElmer. Флуоресценцию измеряли при 615 нм (криптат) и 665 нм (XL665) и отношение 665 нм/615 нм рассчитывали для каждой лунки. Полученный в результате уровень TR-FRET (отношение 665 нм/615 нм) пропорционален степени фосфорилирования. В указанных условиях анализа степень фосфорилирования пептидного субстрата линейно изменялась в зависимости от времени и концентрации фермента. С помощью аналитической системы были получены согласующиеся результаты в отношении Km и специфичной активности для фермента. В случае экспери- 10024746 ментов по ингибированию (значение IC50) значения активности были представлены для постоянных концентраций АТФ, пептида и нескольких фиксированных концентраций ингибитора. Стауроспорин, неселективный киназный ингибитор, использовали в качестве положительного контроля. Все данные по ферментативной активности показаны как среднее четырех повторов измерений. Анализ данных. Значения IC50 рассчитывали с помощью следующего уравнения: у=предел/1+(х/IC50)s+исходное значение,где х и у представляют собой концентрацию ингибитора и ферментативную активность соответственно. Ферментативную активность выражали как количество фосфата, включенного в пептидный субстрат из АТФ. Предел представляет собой максимальное значение у (отсутствие ингибитора, контроль ДМСО) и s представляет собой фактор наклона кривой (6,2). Результаты.IC50 соединения формулы (I) составляла 3,2 нМ при указанных условиях проведения анализа. Данные показали, что соединение формулы (I) является активным ингибитором рецептора ASK-1 1. Клеточный анализ ASK1 (регулирующей апоптотические сигналы киназы 1) на клетках линии 293(клеточная EC50). Клеточную активность соединений оценивали на клетках, стабильно экспрессирующих АР 1:репортерную конструкцию люциферазы (клетки 293/AP1-Luc, Panomics Inc., 6519 Dumbarton Circle,Фримонт, Калифорния). Клетки инфицировали аденовирусом, экспрессирующим активную киназу ASK1(631-1381 кДНК ASK1 крысы), которая активирует транскрипционный фактор АР-1 и повышает экспрессию люциферазы. Ингибиторы ASK1 снижают ферментативную активность ASK1 и, таким образом,снижают активность транскрипционного фактора АР-1 и экспрессию люциферазы. 1. Материалы, необходимые для осуществления указанного протокола 2. Исходные материалы. Листок-вкладыш для стабильной клеточной линии 293/AP1-Luc, Panomics. Листок-вкладыш для системы анализа люциферазы Steady-Glo, Promega. 3. Необходимые среды. Полная питательная среда "ППС" 1% ПСГ 4. Способы. Поддержание. 293/AP1-Luc. Клетки линии 293/АР 1 поддерживали в соответствии с инструкциями производителя; при 80% монослоя клетки собирали во флаконы Т 150, как следует далее: Отбирали среду, осторожно промывали 12 мл стерильного D-ФСБ, жидкость отбирали. Добавляли 5 мл раствора трипсин-ЭДТА,осторожно наклоняли флакон, чтобы покрыть его поверхность, и инкубировали в течение 5 мин при 37 С. Флакон не закрывали крышкой; добавляли 5 мл ППС, промывали флакон 4 х суспензией клеток,переносили в коническую пробирку на 50 мл, центрифугировали в течение 5 мин при 1200 об/мин. Отбирали среду от клеточного осадка, добавляли 20-30 мл ППС, ресуспендировали осадок 6 раз путем пипетирования, пропускали через клеточный фильтр, чтобы разбить кластеры (при необходимости),и считали клетки с помощью гемоцитометра. 1 день анализа. Собирали клетки, как описано выше, за исключением ресуспендирования клеточного осадка. Клетки считали и разводили до концентрации 1,5105 клеток на мл; добавляли аденовирус для достижения отношения 5 инфекционных единиц на клетку. Отбирали (20-30 мл) и помещали клетки на покрытые поли-D-лизином 384-луночные планшеты Greiner в концентрации 1,2104 клеток на лунку с помощью прибора uFill, BioTek (80 мкл на лунку). Незамедлительно в планшеты вводили серии доз соединения по 0,4 мкл (в 100% ДМСО) и инкубировали 24 ч в инкубаторе с увлажнением (37 С, 5% СО 2). 2 день анализа. Планшеты обрабатывали (в соответствии с инструкциями производителя) как следует далее. Планшеты помещали в ламинарный бокс и оставляли без крышки на 30 мин при комнатной температуре для охлаждения. Удаляли 60 мкл АС из анализируемых лунок. Добавляли по 20 мкл на лунку субстрата Steady-Glo, Firefly, оставляли на 10-20 мин при комнатной температуре. Дно аналитического планшета покрывали белой фоновой лентой. Данные получали с помощью флуоресцентного планшетного ридера. Лунки с положительным контролем, соответствующим 100% ингибированию, получали путем инфицирования клеток аденовирусом, экспрессирующим мутант ASK1 с инактивированной каталитической активностью, содержащий замену лизина на аргинин в положении остатка 709. Результат.EC50 соединения формулы (I) составляла 2,0 нМ. Определение Kd. Анализы киназной активности. Штаммы фага Т 7 с киназной меткой получали в хозяине Е. coli, полученном из штамма BL21. Е.coli растили до фазы логарифмического роста и инфицировали фагом Т 7 и инкубировали при перемешивании при температуре 32 С до лизирования. Лизаты центрифугировали и фильтровали для удаления клеточных остатков. Остальные киназы продуцировали в клетках линии НЕК-293 и затем метили ДНК для выявления с помощью количественной ПЦР. Покрытые стрептавидином магнитные шарики обрабатывали биотинилированными низкомолекулярными лигандами в течение 30 мин при комнатной температуре для получения смол аффиности для анализа киназной активности. Покрытые лигандом гранулы блокировали с помощью избытка биотина и промывали блокирующим буфером (SeaBlock, Pierce), 1% БСА (бычий сывороточный альбумин), 0,05% Твин 20, 1 мМ ДТТ(дитиотреитол) для удаления несвязанного лиганда и снижения неспецифического связывания. Реакции связывания проводили путем объединения киназ, связанных с лигандом гранул для анализа аффинности и исследуемых соединений в 1 х связывающем буфере (20% SeaBlock, 0,17 ФСБ, 0,05% Твин 20, 6 мМ ДТТ). Все реакции проводили в полистироловых 96-луночных планшетах в конечном объеме 0,135 мл. Аналитические планшеты инкубировали при комнатной температуре при перемешивании в течение 1 ч и гранулы для анализа аффинности промывали промывочным буфером (1 ФСБ, 0,05% Твин 20). Затем гранулы ресуспендировали в элюирующем буфере (1 ФСБ, 0,05% Твин 20, 0,5 мкМ небиотинилированный лиганд для анализа аффинности) и инкубировали при комнатной температуре при перемешивании в течение 30 мин. Концентрацию киназы в элюатах измеряли с помощью количественной ПЦР. Константы связывания (Kd) рассчитывали с помощью стандартной кривой зависимости эффекта от дозы с использованием уравнения Хилла. Результат.Kd для соединения формулы (I) составляла 0,24 нМ. Указанные данные дают основания полагать,что соединение формулы (I) активно связывается с рецептором ASK1 в отсутствии АТФ. Определение процентного количества соединения, связанного с плазмой крови. Схема эксперимента. В указанных экспериментах использовали 1 мл тефлоновых диализных ячеек от компании HarvardApparatus (Холлистон, Массачусетс, США). До начала проведения исследования диализную мембрану смачивали в течение примерно одного часа в 0,133 М фосфатном буфере, рН 7,4. Соединение в малой концентрации 2 мкМ вводили в 1 мл плазмы или 1 мл клеточной питательной среды. Общий объем жид- 12024746 кости с каждой стороны ячейки составлял 1 мл. Через 3 ч инкубации в равновесном состоянии на водяной бане при 37 С аликвоты образцов из каждой стороны ячейки помещали в подходящие пробирки,содержащие либо 1 мл человеческой плазмы (клеточная питательная среда), либо буфер. Пробирки с образцом взвешивали и данные фиксировали. Отбирали аликвоты по 100 мкл и добавляли к 400 мкл останавливающего реакцию раствора (50% метанол, 25% ацетонитрил, 25% вода и внутренний стандарт). Образцы перемешивали на вортексе и центрифугировали в течение 15 мин при 12000 G. 200 мкл супернатанта отбирали и помещали в новый 96-луночный планшет. Добавляли дополнительные 200 мкл смесиACN:вода, 1:1. Затем содержимое планшета перемешивали на вортексе и подвергали ЖХ-МС анализу. Процент несвязанного анализируемого соединения в плазме рассчитывали с использованием следующего уравнения:% несвязанного соединения=100(Cf /Ct),где Cf и Ct представляют собой концентрации в буфере и плазме после диализа соответственно. Результаты. Процент несвязанного соединения формулы (I), измеренного в плазме крови человека, составлял 11,94 %. Определение отношения оттока для клеток САСО-2. Экспериментальная часть. Клетки линии Сасо-2 поддерживали в модифицированной Дальбеко среде Игла (DMEM) с пируватом натрия, Glutamax с добавлением 1%-ного раствора пенициллин/стрептомицин, 1% NEAA и 10%-ной эмбриональной бычьей сыворотки в инкубаторе, для которого были установлены температура 37 С,влажность 90% и содержание СО 2 5%. Клетки Сасо-2 на 62-72 пассаже высевали в концентрации 2100 клеток/лунку и растили до монослоя в течение по меньшей мере 21 дня на 24-луночных PET (полиэтилен-терефталатных) планшетах (BD Biosciences). Акцепторная лунка содержала HBSS-буфер (10 мМHEPES, 15 мМ глюкоза с рН, доведенным до рН 6,5) с добавлением 1% БСА, рН, доведенный до рН 7,4. После предварительного уравновешивания буфером переноса считывали значения TEER для исследования целостности исследуемой мембраны. Добавляли буферы, содержащие исследуемые соединения, и отбирали 100 мкл раствора через 1 и 2 ч из акцепторных камер. Удаленный буфер замещали на свежий буфер, и все расчеты корректировали с учетом удаленного материала. Эксперимент проводили в двух повторностях. Все образцы сразу собирали в 400 мкл 100% ацетонитриловой кислоты для осаждения белка и стабилизации исследуемых соединений. Дозу вводили к клеткам с апикальной или базолатеральной стороны для определения прямой (от А к В) и обратной (от В к А) проницаемости. Проницаемость через бесклеточный трансвелл также определяли как меру клеточной проницаемости через мембрану и неспецифического связывания. Для исследования неспецифического связывания и нестабильности соединения определяли процентное восстановление. Образцы анализировали с помощью ЖХ/МС/МС. Очевидную проницаемость, Papp, и % восстановление рассчитывали, как следует далее:% восстановление=100VrR120)+(VdD120/(VdD0) где dR/dt представляет собой снижение суммарной концентрации в акцепторной камере в зависимости от времени, мкМ/с, определенное на основе концентраций акцептора, измеренных через 60 и 120 мин.Vr и Vd представляют собой объем в акцепторной и донорной камере в см 3 соответственно. А представляет собой площадь клеточного монослоя (0,33 см 2).D0 и D120 представляют собой измеренную концентрацию в донорной камере в начале и конце эксперимента соответственно.R120 представляет собой концентрацию в акцепторной камере в конце эксперимента (120 мин). Классификация абсорбции и оттока Результат. Наблюдали, что значение АВ соединения формулы (I) для клеток САСО составляло 27; и значение ВА для клеток САСО составляло 35, что приводило к значению отношения оттока (ВА)/(АВ) 1,3. Определение метаболической стабильности в микросомальной фракции печени. Экспериментальная часть. Метаболическую стабильность оценивали с использованием кофакторов для окислительного метаболизма (NADPH) и конъюгации (UDP-глюкуроновая кислота (UDPGA. По две аликвоты соединения формулы (I) (3 мкл 0,5 мМ исходного раствора ДМСО) или метаболически стабильных стандартов (буспирон) добавляли к исходному раствору микросом, разведенному калий-фосфатным буфером, рН 7,4,для получения концентрации белка 1,0 мг/ мл, и содержащим аламетицин в качестве пермеабилизирующего агента. Метаболические реакции инициировали путем добавления воспроизводящей системыNADPH и кофактора UDPGA. Конечный состав каждой реакции был следующим: 3 мкМ исследуемого соединения, 1 мг микросомального белка/мл, 5 мМ UDPGA, 23,4 мкг/мл аламетицина, 1,25 мМ NADP,3,3 мМ глюкозо-6-фосфата, 0,4 ед/ мл глюкозо-6-фосфатдегидрогеназы а и 3,3 мМ MgCl2 в 50 мМ калийфосфатном буфере, рН 7,4. Через 0, 2, 5, 10, 15, 30, 45 и 60 мин аликвоты реакционной смеси по 25 мкл переносили в планшеты, содержащие 250 мкл IS/Q (останавливающего раствора, содержащего внутренний стандарт). После остановки реакций планшеты центрифугировали при 3000g в течение 30 мин и аликвоты по 10 мкл супернатанта анализировали с использованием ЖХ/МС для получения отношений площади пика анализируемого соединения/внутреннего стандарта. Метаболическую стабильность в микросомальных фракциях печени определяли путем измерения скорости исчезновения соединения формулы (I). Данные (% сохранения исходного соединения) наносили на полулогарифмическую шкалу и приводили с помощью экспоненциального приближенияCt=С 0 е-Kt и Т 1/2=ln2/K,где Ct - % оставшегося исходного вещества на момент времени=t; С 0 - % оставшегося исходного вещества на момент времени=0;K - константа скорости выведения первого порядка (ч-1); Т 1/2 - время полувыведения in vitro (ч). Прогнозируемый печеночный клиренс рассчитывали, как следует далее (ссылка 1):CLh=(CLint Qh)/(CLint+Qh),где CLh - прогнозируемый печеночный клиренс (л/ч/кг массы тела);CLint - внутренний печеночный клиренс (л/ч/кг массы тела);YP - выход микросомального белка (мг белка/кг массы тела);Qh - печеночный кровоток (л/ч/кг массы тела). Затем рассчитывали прогнозируемое печеночное выведение путем сравнения прогнозируемого печеночного клиренса с печеночным кровотоком. Соединение считали стабильным, если снижение концентрации субстрата было 10% в процессе инкубации (соответствует экстраполированному периоду полувыведения 395 мин в микросомальных фракциях и 39,5 ч в гепатоцитах). Значения, используемые для расчета прогнозируемого печеночного клиренса, показаны в таблицах ниже. Таблица 1 Значения, полученные из анализа микросомальной стабильности,используемые для расчета прогнозируемого печеночного клиренса Результат. Прогнозируемый печеночный клиренс у человека, определенный на основании экспериментов на микросомальных фракциях in vitro, составлял 0,1 л/ч/кг. Определение CL и Vss у крыс для исследуемого соединения. Фармакокинетика исследуемых соединений после ВВ инфузии дозы 1 мг/кг и пероральной дозы 5,0 мг/кг у крыс. Исследуемое изделие и лекарственная форма. Для ВВ введения исследуемое соединение готовили в смеси 60:40 PEG 400:вода с 1 эквивалентомHCl в концентрации 0,5 мг/ мл. Лекарственная форма представляла собой раствор. Для ПО (перорального) введения исследуемое соединение готовили в смеси 5/75/10/10 этанол/ПГ/солютол/вода в концентрации 2,5 мг/ мл. Лекарственная форма представляла собой раствор. Используемые животные. Каждая из групп внутривенного (ВВ) и перорального (ПО) введения дозы состояла из 3 самцов крыс линии SD. На момент введения дозы масса животных, в целом, составляла между 0,317 и 0,355 кг. Животных не кормили в течение ночи до введения дозы и в течение до 4 ч после введения дозы. Дозирование. Для группы ВВ инфузии исследуемое соединение вводили путем инфузии в течение 30 мин. Скорость инфузии регулировали в соответствии с массой тела каждого животного для доставки дозы 1 мг/кг в объеме 2 мл/кг. Для группы перорального введения дозы исследуемое изделие вводили путем принудительного кормления в объеме 2 мл/кг для дозы 5,0 мг/кг. Сбор образца. Серии образцов венозной крови (примерно по 0,4 мл каждый) отбирали у каждого животного в определенные точки времени после введения дозы. Образцы крови собирали в ампулы VacutainertM (Becton-Disckinson Corp, Нью-Джерси, США), содержащие ЭДТА в качестве антикоагулянта, и быстро подвергали центрифугированию на льду для получения плазмы. Определение концентрации соединения формулы (I) в плазме крови. Для измерения концентрации исследуемого соединения в плазме крови использовали метод ЖХ/МС/МС. Расчеты. Некомпартментный фармакокинетический анализ проводили на данных концентрация в плазме время. Результаты. Для соединение формулы (I) CL составляла 0,09 л/ч/кг; пероральная биодоступность составляла 75%; t1/2 составлял 5,07 ч и Vss составлял 0,55 л/кг у крыс. Анализ ингибирования Сур. Цель: оценить потенциальную способность исследуемого соединения ингибировать основные изоформы цитохрома Р 450, CYP1A, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 и CYP3A4 (2 субстрата). Определение IC50 для ингибирования цитохрома Р 450 (8 изоформ, 9 субстратов). Краткое описание протокола. Исследуемое соединение (0,1-25 мкМ) инкубировали с микросомами печени человека и NADPH в присутствии маркерного субстрата, специфичного для изоформы цитохрома Р 450. В случае реакций,специфичных для CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 и CYP3A4, за метаболитами следили с помощью масс-спектрометрии. За активностью CYP1A следили путем измерения образующегося флуоресцентного метаболита. Снижение образования метаболита по сравнению с носителем в качестве контроля использовали для расчета значения IC50 (концентрация исследуемого соединения, которая обеспечивает 50% ингибирование). Требования к проведению анализа. 500 мкл 10 мМ раствора исследуемого соединения в ДМСО. Ход эксперимента. Ингибирование CYP1A. Шесть концентраций исследуемого соединения (0,1, 0,25, 1, 2,5, 10, 25 мкМ в ДМСО; конечная концентрация ДМСО=0,3%) инкубировали с микросомами печени человека (0,25 мг/ мл) и NADPH (1 мМ) в присутствии маркерного субстрата этоксирезоруфина (0,5 мкМ) в течение 5 мин при 37 С. Селективный ингибитор CYP1A, альфа-нафтофлавон, использовали в скрининговом анализе вместе с исследуемыми соединениями в качестве положительного контроля. Ингибирование CYP2B6. Шесть концентраций исследуемого соединения (0,1, 0,25, 1, 2,5, 10, 25 мкМ в ДМСО; конечная концентрация ДМСО=0,3%) инкубировали с микросомами печени человека (0,1 мг/ мл) и NADPH (1 мМ) в присутствии маркерного субстрата бупропиона (ПО мкМ) в течение 5 мин при 37 С. Селективный ингибитор CYP2B6, тиклопидин, использовали в скрининговом анализе вместе с исследуемыми соединениями в качестве положительного контроля. Ингибирование CYP2C8. Исследуемое соединение в шести концентрациях (0,1, 0,25, 1, 2,5, 10, 25 мкМ в ДМСО; конечная концентрация ДМСО=0,3%) инкубировали с микросомами печени человека (0,25 мг/ мл) и NADPH (1 мМ) в присутствии маркерного субстрата паклитаксела (7,5 мкМ) в течение 30 мин при 37 С. Селективный ингибитор CYP2C8, монтелукаст, использовали в скрининговом анализе вместе с исследуемыми соединениями в качестве положительного контроля. Ингибирование CYP2C9. Исследуемое соединение в шести концентрациях (0,1, 0,25, 1, 2,5, 10, 25 мкМ в ДМСО; конечная концентрация ДМСО=0,25%) инкубировали с микросомами печени человека (1 мг/ мл) и NADPH (1 мМ) в присутствии маркерного субстрата толбутамида (120 мкМ) в течение 60 мин при 37 С. Селективный ингибитор CYP2C9, сульфафеназол, использовали в скрининговом анализе вместе с исследуемыми соединениями в качестве положительного контроля. Ингибирование CYP2C19. Исследуемое соединение в шести концентрациях (0,1, 0,25, 1, 2,5, 10, 25 мкМ в ДМСО; конечная концентрация ДМСО=0,25%) инкубировали с микросомами печени человека (0,5 мг/ мл) и NADPH (1 мМ) в присутствии маркерного субстрата мефенитоина (25 мкМ) в течение 60 мин при 37 С. Селективный ингибитор CYP2C19, транилципромин, использовали в скрининговом анализе вместе с исследуемыми соединениями в качестве положительного контроля. Ингибирование CYP2D6. Исследуемое соединение в шести концентрациях (0,1, 0,25, 1, 2,5, 10, 25 мкМ в ДМСО; конечная концентрация ДМСО=0,25%) инкубировали с микросомами печени человека (0,5 мг/ мл) и NADPH (1 мМ) в присутствии маркерного субстрата декстрометорфаном (5 мкМ) в течение 5 мин при 37 С. Селективный ингибитор CYP2D6, хинидин, использовали в скрининговом анализе вместе с исследуемыми соединениями в качестве положительного контроля. Ингибирование CYP3 А 4 (мидазолам). Исследуемое соединение в шести концентрациях (0,1, 0,25, 1, 2,5, 10, 25 мкМ в ДМСО; конечная концентрация ДМСО=0,26%) инкубировали с микросомами печени человека (0,1 мг/ мл) и NADPH (1 мМ) в присутствии маркерного субстрата мидазолама (2,5 мкМ) в течение 5 мин при 37 С. Селективный ингибитор CYP3A4, кетоконазол, использовали в скрининговом анализе вместе с исследуемыми соединениями в качестве положительного контроля. Ингибирование CYP3 А 4 (тестостерон). Исследуемое соединение в шести концентрациях (0,1, 0,25, 1, 2,5, 10, 25 мкМ в ДМСО; конечная концентрация ДМСО=0,275%) инкубировали с микросомами печени человека (0,5 мг/ мл) и NADPH (1 мМ) в присутствии маркерного субстрата тестостерона (50 мкМ) в течение 5 мин при 37 С. СелективныйCYP3A4 ингибитор, кетоконазол, использовали в скрининговом анализе вместе с исследуемыми соединениями в качестве положительного контроля. В случае инкубации с CYP1 А реакции останавливали метанолом и за образованием метаболита, резоруфина следили с помощью флуоресценции (длина волны возбуждения=535 нм, длина волны испускания=595 нм). В случае инкубации с CYP2B6, CYP2C9, CYP2C19, CYP2D6 и CYP3A4 реакции останавливали с помощью метанола. Образцы затем центрифугировали и супернатанты объединяли для одновременного анализа 4-гидрокситолбутамида, 4-гидроксимефенитоина, декстрарфана и 1 гидроксимидазолама с помощью ЖХ-МС/МС. Гидроксибупропион, 6-гидроксипаклитаксел и 6 гидрокситестостерон анализировали раздельно с помощью ЖХ-МС/МС. К конечному образцу до начала проведения анализа добавляли муравьиную кислоту в деионизированной воде (конечная концентрация=0,1%), содержащую внутренний стандарт. Снижение образования метаболитов по сравнению с носителем в качестве контроля использовали для расчета значения IC50 (концентрации исследуемого соединения, которая обеспечивает 50% ингибирование). Результаты. Общая схема исследования для модели фиброза почек, вызванного односторонней обструкцией мочеточника (ООМ), у крыс. Самцов крыс линии Sprague-Dawley обеспечивали нормальным кормом, содержали в стандартных условиях и позволяли акклиматизироваться в течение по меньшей мере 7 дней до проведения операции. В начале исследования крыс делили на группы на основе массы тела и вводили (2 мл/кг перорально два раза в день) носитель, соединение в одной из четырех дозы (1, 3, 10 или 30 мг/кг) путем принудительного кормления. Крыс подвергали анестезии с помощью изофлурана на носовой конус и проводили лапарото- 16024746 мию. Крыс подвергали полной обструкции правого мочеточника (ООМ) с помощью стерилизованных нагревом инструментов и асептического хирургического метода. Крысам вводили 50 мкл пенициллина G(внутримышечно) непосредственно после операции. Крысам позволяли восстанавливаться в чистой клетке с подогревом до возвращения в нормальные условия вивария. Крысам вводили соединения в дозе,описанной выше, два раза в день (с 12-часовыми интервалами) в течение следующих друг за другом 7 дней. На 7 день после операции крыс подвергали анестезии с помощью изофлурана и собирали сыворотку, плазму и мочу. Затем животных усыпляли, извлекали почки и брали биопсии коркового вещества почки для морфологического, гистологического и биохимического анализа. Все ткани для биохимического анализа являлись быстрозамороженными в жидком азоте и хранились при -80 С, ткани для гистологического анализа фиксировали в 10%-ном нейтральном забуференном формалине. Фиброз почки оценивали путем измерения количества коллагена IV в почке с помощью метода ИФА и путем оценки накопления положительно окрашенных на альфа-гладкомышечный актин миофибробластов в почке с помощью иммуногистохимии. Для проведения первого анализа небольшую часть замороженного коркового вещества почки переносили и гомогенизировали в RIPA-буфере, затем центрифугировали при 14000g в течение 10 мин при 4 С. Супернатант собирали в предварительно охлажденные ампулы и определяли концентрацию белка. Эквивалентное количество общего белка подвергали ИФА-анализу на коллаген IV (Exocell) в соответствии с инструкциями производителя. Фиксированную формалином и залитую парафином ткань почки окрашивали на альфа-гладкомышечный актин, как описано ранее (Stambe et al, The Role of p38 Mitogen-Activated Protein Kinase Activation in Renal Fibrosis J AmSoc Nephrol 15: 370-379, 2004). Результаты. Было обнаружено, что соединение формулы (I) в дозах от 3 до 30 мг/кг значительно снижает индукцию коллагена IV в почке (фиг. 1) и накопление положительно окрашенных на альфагладкомышечный актин миофибробластов (фиг. 2). Сравнительные данные для соединения формулы (I) и соединений сравнения. В следующей таблице представлены сравнительные результаты для соединения формулы (I) и соединений сравнения А и В, описанных в патентной публикации США 2001/00095410 А 1, опубликованной 13 января 2011 г. Необходимо отметить, что эксперименты, сравнение результатов которых приведено ниже, проводили в сходных условиях, но необязательно одновременно. Таблица( ) значения в круглых скобках показывают, во сколько раз соединение формулы (I) имеет преимущество по сравнению с указанным соединением для указанного параметра. Из описанных выше сравнительных данных можно вывести следующее: IC50 соединения формулы(I) сопоставима с IC50 соединения А. Функциональная IC50 соединения формулы (I) сопоставима с IC50 соединений А и В. Скорректированная с учетом связывания с белком IC50 соединения формулы (I) в 4 раза ниже по сравнению с соединением А и в 33 раз ниже по сравнению с соединением В. Соединение формулы (I) является более слабым ингибитором сур 3 А 4 по сравнению с соединениями А и В. Значение IC50 CYP3A4/ PBAdj.IC50 соединения формулы (I) в 43 раза выше по сравнению с соединением формулы А и в 92 раза выше по сравнению с соединением формулы В. Значение CL соединения формулы (I) у крыс в 2,7 раз ниже по сравнению с соединением формулы А и в 3,6 раз ниже по сравнению с соединением формулы В. Процентная биодоступность соединения формулы (I) у крыс в 6,8 раз выше по сравнению с соединением A и в 1,5 раз выше по сравнению с соединением В. Период полувыведения соединения формулы (I) у крыс в 8,6 раз больше по сравнению с соединением А и в 3,9 раз больше по сравнению с соединением В. Приведенные выше данные дают достаточные основания полагать, что соединение формулы (I) обладает неожиданными и предпочтительными свойства по сравнению с соединениями формулы А и В; и что указанное соединение формулы (I), вероятно, является лучшим кандидатом для дальнейшей разработки средств для лечения хронической почечной недостаточности, фиброза легких и/или почек и/или кардиоренальных заболеваний. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) представляющее собой 5-(4-циклопропил-1 Н-имидазол-1-ил)-N-(6-(4-изопропил-4 Н-1,2,4-триазол 3-ил)пиридин-2-ил)-2-фтор-4-метилбензамид, или его фармацевтически приемлемая соль. 2. Соединение формулы (I) представляющее собой 5-(4-циклопропил-1 Н-имидазол-1-ил)-2-фтор-N-(6-(4-изопропил-4 Н-1,2,4 триазол-3-ил)пиридин-2-ил)-4-метилбензамид. 3. Фармацевтическая композиция для лечения хронической почечной недостаточности, диабетической нефропатии, фиброза почек, фиброза печени, фиброза легких и диабетической почечной недостаточности, содержащая терапевтически эффективное количество соединения или соли по п.1 и фармацевтически приемлемый носитель. 4. Фармацевтическая композиция для лечения хронической почечной недостаточности, диабетической нефропатии, фиброза почек, фиброза печени, фиброза легких и диабетической почечной недостаточности, содержащая терапевтически эффективное количество соединения по п.2 и фармацевтически приемлемый носитель. 5. Способ лечения хронической почечной недостаточности, включающий введение терапевтически эффективного количества соединения по п.2 или его фармацевтически приемлемой соли нуждающемуся в этом пациенту. 6. Способ лечения диабетической нефропатии, включающий введение терапевтически эффективного количества соединения по п.2 или его фармацевтически приемлемой соли нуждающемуся в этом пациенту. 7. Способ лечения фиброза почек, фиброза печени или фиброза легких, включающий введение терапевтически эффективного количества соединения или соли по п.1 нуждающемуся в этом пациенту. 8. Способ лечения диабетического заболевания почек, включающий введение терапевтически эффективного количества соединения или соли по п.1 нуждающемуся в этом пациенту. 9. Применение соединения или соли по п.1 для получения лекарственного средства для лечения хронической почечной недостаточности. 10. Применение соединения или соли по п.1 для лечения диабетической нефропатии, фиброза почек, фиброза печени, фиброза легких и диабетической почечной недостаточности. 11. Промежуточное соединение формулы представляющее собой 2-амино-5-(4-изопропил-4 Н-1,2,4-триазол-3-ил)пиридин или его соль или защищенную форму. 12. Промежуточное соединение формулы представляющее собой 5-(4-циклопропил-1 Н-имидазол-1-ил)-2-фтор-4-метилбензойную кислоту,его соль или защищенную форму.
МПК / Метки
МПК: A61P 31/12, C07D 401/04, C07D 401/14, C07D 233/56, A61K 31/4164, A61K 31/4436
Метки: апоптотические, сигналы, регулирующей, ингибитор, киназы
Код ссылки
<a href="https://eas.patents.su/20-24746-ingibitor-reguliruyushhejj-apoptoticheskie-signaly-kinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибитор регулирующей апоптотические сигналы киназы</a>
Ингибитор p38 mapk-киназы

Номер патента: 19590
Опубликовано: 30.04.2014
Авторы: Уилльямс Джонатан Гарет, Чаррон Кэтрин Элизабет, Онионз Стюарт Томас, Рейппорт Уилльям Гарт, Ито Кадзухиро, Кинг-Андервуд Джон, Мюррей Питер Джон, Стронг Питер
МПК: A61P 11/00, A61K 31/415, A61P 29/00...
Метки: mapk-киназы, ингибитор
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль.2. Фармацевтическая композиция, содержащая соединение по п.1, в комбинации с одним или более фармацевтически приемлемыми разбавителями или носителями.3. Применение соединения формулы (I) по п.1 для получения лекарственного средства для лечения или предотвращения состояния, выбранного из COPD, включая хронический бронхит и эмфизему, астмы, педиатрической астмы, муковисцидоза,...
Ингибитор фосфоинозитид-3-киназы, содержащий цинксвязывающий фрагмент

Номер патента: 22434
Опубликовано: 30.12.2015
Авторы: Лай Чэнцзюн, Бао Жуди, Цянь Чангэн, Цай Сюн, Чжай Хайсяо
МПК: A61K 31/535
Метки: фосфоинозитид-3-киназы, ингибитор, содержащий, фрагмент, цинксвязывающий
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль,в котором R представляет собой водород или R1C(O)-, где R1 представляет собой С1-С24-алкил, С2-С24-алкенил или фенил.2. Соединение по п.1, в котором R1 представляет собой C1-С10-алкил или фенил.3. Соединение по п.2, в котором R1 представляет собой C1-С6-алкил.4. Соединение по п.1, в котором R представляет собой Н или ацетил.5. Соединение по п.1, представленное формулойили его...
Композиция, содержащая нестероидное противовоспалительное средство (nsaid) и ингибитор киназы egfr, для лечения или ингибирования полипов в ободочной кишке и рака ободочной и прямой кишки

Номер патента: 6876
Опубликовано: 28.04.2006
Авторы: Фрост Филип, Дискафани-Марро Каролин Мари
МПК: A61K 31/19, A61K 31/505, A61K 31/47...
Метки: средство, содержащая, кишке, egfr, композиция, ингибирования, ободочной, противовоспалительное, киназы, прямой, нестероидное, nsaid, кишки, лечения, ингибитор, рака, полипов
Формула / Реферат:
1. Способ лечения или ингибирования полипов в ободочной кишке у млекопитающего, нуждающегося в этом, который включает введение указанному млекопитающему эффективного количества NSAID и ингибитора киназы EGFR, где NSAID выбирают из группы, включающей ибупрофен, сулиндак, кетопрофен, фенопрофен, флурбипрофен, напроксен, тиапрофеновую кислоту, супрофен, этодолак, карпрофен, кетролак, пипрофен, индопрофен, целекоксиб, рофекоксиб, мобикокс и...
Композиции, включающие ингибитор абсорбции холестерина, ингибитор hmg-coa-редуктазы и стабилизирующий агент

Номер патента: 8888
Опубликовано: 31.08.2007
Авторы: Мур Уилльям Д., Петтс Кэтрин Р., Саклатвала Роберт, Фитцпатрик Шон, Зайлер Кристиан, Чо Винг-Ки Филип
МПК: A61K 31/365, A61K 31/35, A61K 31/22...
Метки: композиции, абсорбции, агент, hmg-coa-редуктазы, холестерина, стабилизирующий, включающие, ингибитор
Формула / Реферат:
1. Фармацевтическая композиция, включающая от 1 до 20 мас.% эзетимиба; от 1 до 80 мас.% симвастатина; от 0,01 до 2 мас.% бутилированного гидроксианизола (ВНА) и от 0,1 до 1,25 мас.% лимонной кислоты при условии, что композиция не содержит аскорбиновую кислоту. 2. Композиция по п.1, включающая от 1,25 до 10% эзетимиба и от 1 до 20% симвастатина. 3. Композиция по п.2, включающая от 5 до 10% симвастатина. 4. Композиция по п.1, включающая от 0,01 до...
Фармацевтическая композиция, включающая ингибитор sglt2, ингибитор дпп-iv и другой антидиабетический агент, и ее применение

Номер патента: 22349
Опубликовано: 30.12.2015
Авторы: Томас Лео, Гремплер Рольф, Симан Лео Джон, Марк Михаэль, Брёдль Ули, Айккельманн Петер
МПК: A61K 31/06, A61K 31/00, A61K 31/155...
Метки: включающая, dpp-iv, фармацевтическая, sglt2, композиция, антидиабетический, агент, ингибитор, применение
Формула / Реферат:
1. Фармацевтическая композиция, включающая:(а) ингибитор SGLT2 1-хлор-4-(β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензол,(б) ингибитор ДПП-IV линаглиптин или его фармацевтически приемлемую соль и(в) третий антидиабетический агент, отличающийся тем, что представляет собой метформин или его фармацевтически приемлемую соль.2. Фармацевтическая композиция по п.1, в которой третий антидиабетический агент представляет собой...
Предыдущий патент: Компонент катализатора для полимеризации олефинов
Следующий патент: Средство для лечения кокцидиозов у птиц и животных
Случайный патент: Способ и устройство для извлечения из анодного шлама селена и ценных металлов