Пуриновые соединения







Формула / Реферат
1. Соединение формулы

где R1 представляет собой Cl или СН3;
R2 представляет собой CºN, -CH2SO2CH3, -CONHCH3, -CH2NR4R5 или -CH2CºN;
R3 представляет собой C1-C2-алкил, C1-C2-фторалкил или C(O)CH3;
R4 представляет собой Н, С(О)СН3, СО2СН3 или SO2CH3;
R5 представляет собой Н или совместно с R4 образует пирролидин-2-он,
или его фармацевтически приемлемая соль.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 представляет собой Cl.
3. Соединение по п.1 или 2 или его фармацевтически приемлемая соль, где R2 представляет собой
-CH2SO2CH3, -CH2NR4R5 или -CH2CºN.
4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль, где R2 представляет собой
-CH2SO2CH3.
5. Соединение по любому из пп.1-4 или его фармацевтически приемлемая соль, где R3 представляет собой метил, 2-фторэтил или С(О)СН3.
6. Соединение по любому из пп.1-5 или его фармацевтически приемлемая соль, где R3 представляет собой метил.
7. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль, где R5 представляет собой Н и R4 представляет собой С(О)СН3 или СО2СН3.
8. Соединение по п.1, представляющее собой 8-(2-хлорфенил)-6-(4-метилпиперазин-1-ил)-2-метил-9-(2-метилсульфонилэтил)пурин, или его фармацевтически приемлемая соль.
9. Фармацевтическая композиция для лечения боли, содержащая соединение по любому из пп.1-8 или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель.
10. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемой соли для лечения боли.
11. Применение по п.10 для лечения остеоартритной боли или мигрени.
12. Способ лечения боли, включающий введение эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли человеку или животному, нуждающимся в этом.
13. Способ по п.12 для лечения остеоартритной боли или мигрени.
Текст
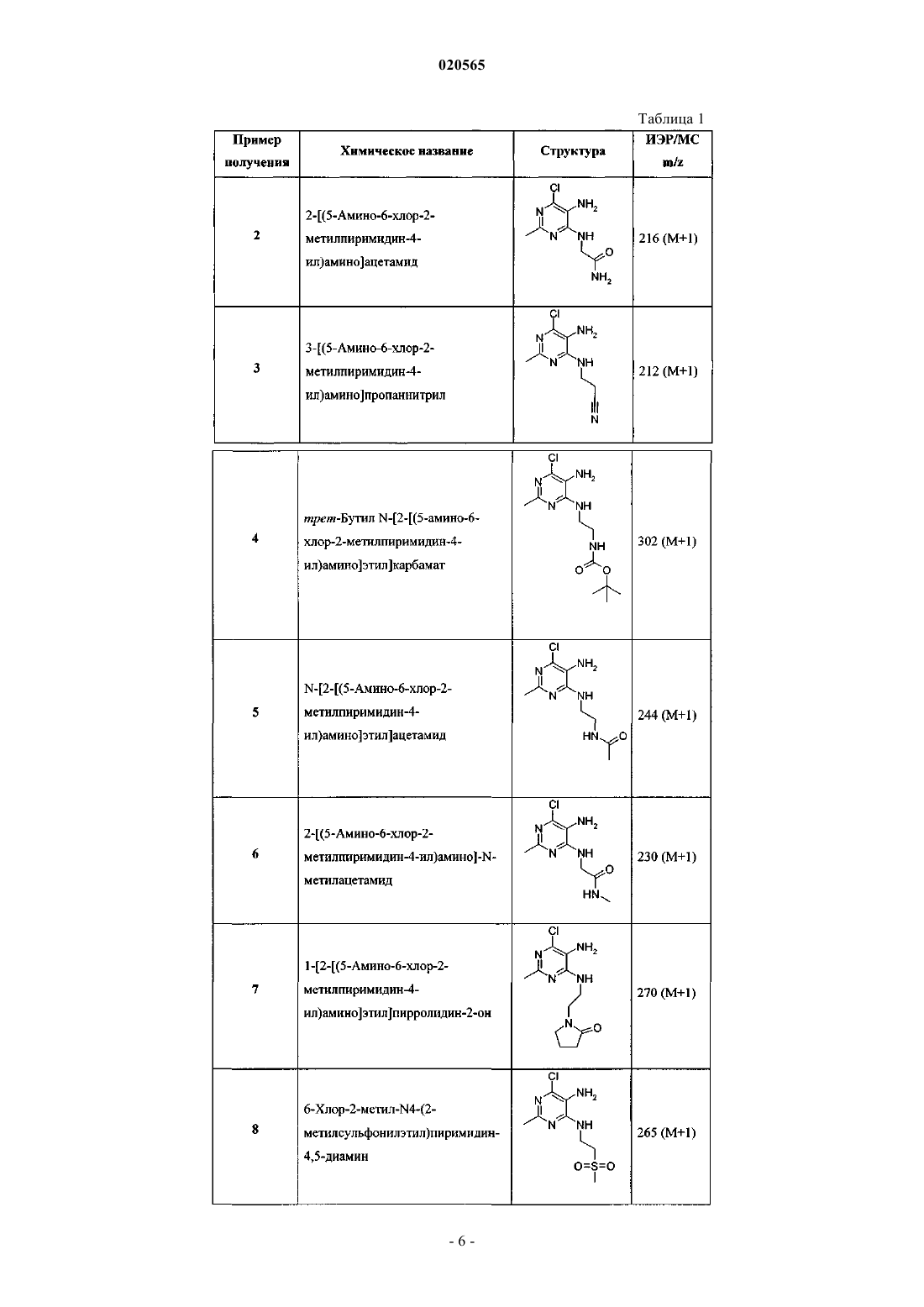
Изобретение относится к соединению формулы (I) и фармацевтическим композициям для лечения боли: Холлинсхед Шон Патрик (US) Лыу Т.Н. (RU)(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US)(GPCR). Рецепторы CB1 экспрессируются как в центральной, так и в периферической системах, тогда как рецепторы СВ 2 экспрессируются преимущественно в периферической системе, в основном в клетках и тканях иммунной системы. В недавнем обзоре, посвященном фармакологическому и терапевтическому потенциалу рецептора СВ 2 (Br. J. Pharmacol. (2008), 153, 319-334), СВ 2 идентифицирован в качестве терапевтической мишени для лечения боли, в частности воспалительной и невропатической боли. Агонисты СВ 2, в частности селективные в отношении СВ 2 агонисты, представляют собой мишень для лечения боли с ограничением побочных эффектов, опосредованных центральной нервной системой.WO 2004/037823 относится к пуриновым соединениям и их применению в качестве лигандов каннабиоидных рецепторов, в частности в качестве антагонистов рецептора СВ 1. В связи с побочными эффектами, связанными с существующими пероральными фармакологическими агентами, по-прежнему существует необходимость в разработке альтернативных способов терапии для лечения боли. Согласно настоящему изобретению предложено соединение формулыR5 представляет собой Н или совместно с R4 образует пирролидин-2-он,или его фармацевтически приемлемая соль. Было обнаружено, что соединения согласно настоящему изобретению являются агонистами рецептора СВ 2 in vitro. Некоторые соединения согласно настоящему изобретению обладают большей активностью по сравнению с существующими агонистами СВ 2. Некоторые соединения согласно настоящему изобретению представляют собой селективные агонисты СВ 2. Некоторые соединения согласно настоящему изобретению обладают большей селективностью в отношении СВ 2 по сравнению с существующими агонистами СВ 2. Согласно настоящему изобретению предложена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель. Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль, совместно с фармацевтически приемлемым разбавителем или носителем и, необязательно, одним или более терапевтическими ингредиентами. Согласно настоящему изобретению предложено соединение или его фармацевтически приемлемая соль для применения в терапии. Согласно настоящему изобретению предложено соединение или его фармацевтически приемлемая соль для применения для лечения боли, в частности остеоартритной боли или мигрени. Согласно другому аспекту настоящего изобретения предложено применение соединения или его фармацевтически приемлемой соли для получения лекарственного средства для лечения боли, в частности остеоартритной боли или мигрени. Согласно настоящему изобретению предложен способ лечения боли, включающий введение эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли человеку или животному, нуждающемуся в этом. Согласно настоящему изобретению также предложен способ лечения остеоартритной боли или мигрени, включающий введение эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли человеку или животному, нуждающимся в этом. Соединения согласно настоящему изобретению предпочтительно применять для лечения боли, в частности остеоартритной боли или мигрени. Также было установлено, что агонисты рецептора СВ 2 обладают терапевтическим потенциалом для лечения рассеянного склероза (Br. J. Pharmacol. (2008), 153, 216-225 и J. Bid. Chem. (2008), 283, 1332013329). Предпочтительные варианты реализации настоящего изобретения представляют собой соединения формулы или их фармацевтически приемлемые соли,где R2, R3, R4 и R5 являются такими, как определено выше. Некоторые классы соединений формулы (I) или (II) являются предпочтительными. В следующих пронумерованных группах описаны указанные предпочтительные классы: 1) R2 представляет собой -CH2SO2CH3, -CH2NR4R5 или -CH2CN; 2) R2 представляет собой -CH2SO2CH3; 3) R3 представляет собой метил, этил, 2-фторэтил или С(О)СН 3; 4) R3 представляет собой метил или этил; 5) R4 представляет собой С(О)СН 3 или СО 2 СН 3; 6) R5 представляет собой Н; 7) R5 представляет собой Н и R4 представляет собой С(О)СН 3 или СО 2 СН 3; 8) R2 представляет собой -CH2SO2CH3, -CH2NR4R5 или -CH2CN; R5 представляет собой Н и R4 представляет собой С(О)СН 3 или СО 2 СН 3; 9) R2 представляет собой -CH2SO2CH3, -CH2NR4R5 или -CH2CN; R5 представляет собой Н; R4 представляет собой С(О)СН 3 или СО 2 СН 3 и R3 представляет собой метил, этил, 2-фторэтил или С(О)СН 3; 10) R2 представляет собой -CH2SO2CH3; R3 представляет собой метил, этил, 2-фторэтил или С(О)СН 3; 11) R2 представляет собой -CH2SO2CH3; R3 представляет собой метил или этил. Фармацевтически приемлемые соли каждого соединения согласно настоящему изобретению находятся в рамках настоящего изобретения. Следует понимать, что если в настоящем изобретении группа охарактеризована как "определенная в настоящем изобретении" или "как определено в настоящем изобретении", то указанная группа включает первое из приведенных и самое широкое определение наряду со всеми без исключения конкретными определениями указанной группы. Выше и далее в описании изобретения следующие термины, если иное не указано, имеют следующие значения. В настоящем изобретении термин "C1-C2-алкил" относится к метилу или этилу. В настоящем изобретении термин "C1-C2-фторалкил" относится к C1-C2-алкильной группе, определенной в настоящем изобретении, где один или более атомов водорода заменены на фтор, и включает трифторметил, 2-фторэтил, 2,2-дифторэтил и 2,2,2 трифторэтил. Предпочтительно C1-C2-фторалкильная группа представляет собой 2-фторэтил. В настоящем изобретении термин "фармацевтически приемлемая соль" относится к солям соединений согласно настоящему изобретению, которые, по существу, являются не токсичными для живых организмов. Указанные соли и общая методика их получения хорошо известны в данной области техники. См., например, P. Stahl, et al., Handbook of Pharmaceutical Salts: Properties Selection and Use,(VCHA/Wiley-VCH, 2002) и J. Pharm. Sci. 66, 2-19 (1977). Предпочтительно фармацевтически приемлемые соли представляет собой гидрохлорид. Варианты реализации изобретения включают примеры, предложенные в настоящем изобретении, и,несмотря на то, что пример или его соль может быть представлен в одной хиральной или конформационной форме, дополнительные варианты реализации изобретения включают все другие стереоизомерные и/или конформационные формы описанных примеров, а также их фармацевтически приемлемых солей. В настоящем изобретении термин "селективные в отношении CB2 агонисты" или "селективность в отношении CB2" относится к соединениям, имеющим более высокую активность в отношении CB2 по сравнению с CB1. Предпочтительно соединения согласно настоящему изобретению обладают 50-кратной селективностью в отношении CB2. Более предпочтительно соединения согласно настоящему изобретению обладают 100-кратной селективностью в отношении CB2. Наиболее предпочтительно соединения согласно настоящему изобретению обладают 500-кратной селективностью в отношении CB2. Соединения согласно настоящему изобретению предпочтительно представлены в виде фармацевтических композиций, которые вводят различными путями. Предпочтительно указанные композиции представляют собой композиции для перорального введения. Указанные фармацевтические композиции и способы их получения хорошо известны в данной области техники. См., например, Remington: TheScience and Practice of Pharmacy (A. Gennaro, et al., eds., 19th ed., Mack Publishing Co., 1995). Следующие схемы, примеры получения и примеры приведены для лучшего описания практического осуществления настоящего изобретения. Подходящие условия реакций для этапов указанных схем,примеров получения и примеров известны в данной области техники, и соответствующие модификации условий реакций, включая замену растворителей и сопутствующих реагентов, находятся в пределах возможностей специалистов в данной области техники. Более того, специалистам в данной области техники ясно, что в некоторых обстоятельствах порядок введения фрагментов не имеет определяющего значения. Конкретный порядок этапов, необходимых для получения соединений формулы (I), зависит от конкретного синтезируемого соединения, исходного соединения и относительной подвижности замещаемых фрагментов, что очевидно специалисту в области химии. Специалисту в данной области техники ясно, что не все заместители совместимы с любыми условиями проведения реакций. Для таких соединений можно применять введение защитных групп или модифицирование на подходящем этапе синтеза с помощью методов, хорошо известных в данной области техники. Подходящие защитные группы включают защитные группы, описанные в источнике T.W. Greene,"Protective Groups in Organic Synthesis", John Wiley and Sons, New York, N.Y., 1991, далее называемом"Greene". Согласно указанному источнику описаны соответствующие условия "введения" и "удаления" подходящих защитных групп для использования специалистами в данной области техники. Промежуточные вещества и конечные продукты согласно настоящему изобретению при желании могут быть дополнительно очищены с помощью общих методик, таких как перекристаллизация или хроматография на твердых носителях, таких как силикагель или оксид алюминия. Названия соединений согласно настоящему изобретению получали с использованием программыSymyx Version 3,1.NET с названиями функциональных групп согласно IUPAC. Используемые в настоящем изобретении аббревиатуры имеют следующие определения:"солевой раствор" означает насыщенный водный раствор хлорида натрия;"БСА" означает бычий сывороточный альбумин; Соединение формулы (I) может быть получено в соответствии с реакциями, показанными на схеме 1. На 1 этапе 4,6-дихлор-2-метилпиримидин-5-иламин подвергали реакции с амином (1) в реакции замещения с получением диаминопиримидина (2). Реакция может протекать в присутствии подходящего основания, такого как триэтиламин или диизопропилэтиламин, в растворителе, таком как этанол или изопропанол, при повышенной температуре, такой как примерно от 90 до 160 С, предпочтительно в запечатанной пробирке. Альтернативно, реакцию можно проводить с использованием микроволнового излучения. На 2 этапе из диаминопиримидина (2) и бензальдегида (3) в присутствии кислотного катализатора,такого как хлорид железа на диоксиде кремния или п-толуолсульфоновая кислота, образовывался имин. Реакция проходит в подходящем растворителе, таком как 1,4-диоксан или толуол, при повышенной тем-3 020565 пературе, такой как примерно от 70 до 110 С. При отсутствии диоксида кремния можно добавлять молекулярные сита для удаления воды из реакционной смеси. После фильтрования для удаления твердого вещества и концентрирования можно проводить окислительную циклизацию имина в подходящем растворителе, таком как дихлорметан, в присутствии окислителя, такого как DDQ, при подходящей температуре, такой как примерно от -30 до 40 С, с получением 6-хлорпурина (4). На 3 этапе проводили реакцию замещения между 6-хлорпурином (4) и пиперазином (5) с получением соединения формулы (I). Реакция может протекать в присутствии подходящего основания, такого как триэтиламин или диизопропилэтиламин, в растворителе, таком как метанол, этанол или изопропанол,при повышенной температуре, такой как примерно от 50 до 100 С. Альтернативно, реакцию можно проводить с использованием микроволнового излучения. Специалистам в данной области техники ясно, что можно вводить подходящую защитную группу,такую как tBOC, для аминной функциональной группы, присутствующей на пиперазинильном фрагменте. После замещения на 3 этапе можно удалить защитную группу и ацилировать или алкилировать амин для получения дополнительных соединений формулы (I). Подобным образом, когда R2 содержит аминную функциональную группу, можно вводить подходящую защитную группу для амина, такую как группа tBOC, в приведенной выше последовательности. Затем можно удалять защитную группу из соединения и ацилировать или сульфонилировать указанное соединение для получения дополнительных соединений согласно изобретению. Кроме того некоторые функциональные группы можно подвергать дополнительным реакциям на различных этапах пути синтеза. Например, ацетамидное промежуточное соединение (где R2=C(O)NH2) можно превратить в нитрил с помощью фосфорилхлорида. Схема 2 На схеме 2 изображены способы получения соединений формулы (9). На 1 этапе 6-хлор-2-метил-N4-(2-метилсульфанилэтил)пиримидин-4,5-диамин (6) подвергали реакции с бензальдегидом (3), по существу, как описано выше для 2 этапа схемы 1, с получением алкилтиопурина (7) На 2 этапе алкилтиопурин (7) окисляли до алкилсульфонилпурина (8). Реакция протекала в присутствии подходящего окислителя, такого как МСРВА, в подходящем растворителе, таком как дихлорметан,при подходящей температуре, такой как примерно от 0 до 40 С. На 3 этапе проводили реакцию замещения между хлоралкилсульфонилпурином (8) и содержащим соответствующие заместители пиперазином (5) с получением пиперазинилпурина (9), по существу, как описано выше на 3 этапе схемы 1. На схеме 3 показан альтернативный способ синтеза соединений формулы (9). На 1 этапе 6-хлор-2-метил-N4-(2-метилсульфанилэтил)пиримидин-4,5-диамин (6) объединяли с бензальдегидом (3) и пиперазином (5) в подходящем растворителе, таком как анизол, при повышенной температуре, такой как примерно от 120 до 150 С, в течение примерно от 3 до 6 дней, с получением алкилтиопиперазинилпурина (10). На 2 этапе алкилтиопиперазинилпурин (10) окисляли до сульфона формулы (9). Реакцию проводили в подходящем растворителе, например в смеси ТГФ и МеОН, с использованием водного раствора пероксимоносульфата калия (Oxone) при подходящей температуре, такой как примерно от 0 до 60 С. Альтернативно, алкилтио может быть окислен с помощью МСРВА. Пример получения 1. 6-Хлор-2-метил-N4-(2-метилсульфанилэтил)пиримидин-4,5-диамин(40,05 г, 0,439 моль) и триэтиламина (53,3 г, 0,517 моль) в изопропаноле (500 мл) нагревали при 90 С в течение 30 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Осадок растворяли в дихлорметане (2 л) и промывали водой (2500 мл) и солевым раствором (500 мл). Органический слой сушили над сульфатом натрия, фильтровали и выпаривали с получением твердого вещества коричневого цвета. Осадок очищали на колонке, заполненной силикагелем, при использовании в качестве элюента этилацетата с получением указанного в заголовке соединения (56 г). ИЭР/МС m/z 233 (М+1). Диаминопиримидины в табл. 1 готовили, по существу, в соответствии со способом, описанным в примере получения 1, с использованием соответствующего амина и 4,6-дихлор-2-метилпиримидин-5 амина. Очищали с помощью хроматографии на силикагеле с использованием в качестве элюента смеси дихлорметан/метанол, этилацетат/гексан (пример получения 6) или ацетон/гексаны (пример получения 8). Смесь 6-хлор-2-метил-N4-(2-метилсульфанилэтил)пиримидин-4,5-диамина (1,63 г, 0,007 моль),2-хлорбензальдегида (1,96 г, 0,014 моль) и 15% FeCl3 на SiO2 (4,89 г) в 1,4-диоксане (15 мл) нагревали до 100 С в течение 16 ч. Реакционную смесь охлаждали и удаляли диоксид кремния посредством фильтрования через диатомовую землю. Фильтрат концентрировали при пониженном давлении с получением осадка. Указанный осадок растворяли в безводном дихлорметане (10 мл) и добавляли 2,3-дихлор-5,6 дициано-1,4-бензохинон (1,58 г, 0,007 моль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Указанную реакционную смесь разбавляли дихлорметаном и промывали 1 н. водным раствором гидроксида натрия, водой и солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением осадка. Указанный осадок очищали на колонке, заполненной силикагелем, при использовании в качестве элюента смесь этилацетаттексан (15:85) с получением указанного в заголовке соединения (1,9 г). ИЭР/МС m/z 353 (М+1). Фенилпурины в табл. 2 получали, по существу, в соответствии со способом, описанным в примере получения 9, с использованием соответствующих диаминопиримидина и бензальдегида. Очищали с помощью хроматографии на силикагеле с использованием в качестве элюента смеси этилацетат:гексан,дихлорметан:метанол или гексан:ацетон. Таблица 2 Смесь 2-[(5-амино-6-хлор-2-метилпиримидин-4-ил)амино]ацетамида (2,2 г, 0,01 моль),2-хлорбензальдегида (2,86 г, 0,02 моль), п-толуолсульфоновой кислоты (0,2 г) и молекулярных сит (1,0 г) в толуоле (50 мл) нагревали с обратным холодильником в течение 16 ч. Охлаждали и указанные сита удаляли посредством фильтрования через диатомовую землю. Фильтрат концентрировали при пониженном давлении с получением осадка. Осадок растворяли в безводном дихлорметане (50 мл) и добавляли 2,3-дихлор-5,6-дициано-1,4-бензохинон (2,3 г, 0,01 моль) при 0 С. Позволяли нагреваться до комнатной температуры и перемешивали в течение 2 ч. Реакционную смесь разбавляли дихлорметаном, промывали 1 н. водным раствором гидроксида натрия, водой и солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением осадка. Осадок очищали на колонке, заполненной силикагелем, при использовании в качестве элюента смеси дихлорметан:метанол(98:2) с получением указанного в заголовке соединения (0,3 г). ИЭР/МС m/z 336 (М+1). Фенилпурин в табл. 3 получали, по существу, в соответствии со способом, описанным в примере получения 15, с использованием соответствующих диаминопиримидина и 2-хлорбензальдегида. Таблица 3 Раствор 2-[6-хлор-8-(2-хлорфенил)-2-метилпурин-9-ил]ацетамида (0,3 г, 0,8 ммоль) и фосфорилхлорида (2,5 мл) нагревали при 110 С в течение 16 ч. Реакцию гасили водным раствором бикарбоната натрия и экстрагировали дихлорметаном. Органическую фазу промывали водой и солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (0,2 г). ИЭР/МС m/z 318 (M+l). Пример получения 18. 6-Хлор-8-(2-хлорфенил)-2-метил-9-(2-метилсульфонилэтил)пурин К раствору 6-хлор-8-(2-хлорфенил)-2-метил-9-(2-метилсульфанилэтил)пурина (1,98 г, 5,0 ммоль) в дихлорметане (15 мл) добавляли метахлорпербензойную кислоту (2,4 г, 0,014 моль) и нагревали с обратным холодильником в течение 6 ч. Реакционную смесь охлаждали, реакцию гасили насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением осадка. Осадок очищали на колонке, заполненной силикагелем, при использовании в качестве элюента смеси дихлорметан:метанол (98:2) с получением указанного в заголовке соединения (1,95 г). ИЭР/МС m/z 385 (М+1). Реакционную колбу наполняли N-трет-бутоксикарбонилпиперазином (1,600 г, 8,590 ммоль), карбонатом калия (3,56 г, 25,77 ммоль), йодидом натрия (катализатор) (10 мг, 66,7 мкмоль), 1,4-диоксаном(20 мл) и 1-бром-2-фторэтаном (704,0 мкл, 9,45 ммоль). Смесь нагревали с обратным холодильником при перемешивании в течение ночи. После завершения реакции полученную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный осадок распределяли между этилацетатом и водой. Органический слой отделяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением чистого сложного трет-бутилового эфира 4-(2-фторэтил)пиперазин-1-карбоновой кислоты.GC-MS m/z 232 (М). К перемешанному раствору сложного трет-бутилового эфира 4-(2-фторэтил)пиперазин-1 карбоновой кислоты (2,00 г, 8,61 ммоль) в безводном дихлорметане (60 мл) добавляли 4 н. раствор HCl в 1,4-диоксане (21,52 мл, 86,1 ммоль) при комнатной температуре в атмосфере азота. Перемешивали в течение ночи а атмосфере азота. Реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,78 г). ИЭР/МС m/z 133 (М+1). Пример 1. Гидрохлорид 8-(2-хлорфенил)-6-(4-метилпиперазин-1-ил)-2-метил-9-(2-метилсульфонилэтил)пурина Раствор 6-хлор-8-(2-хлорфенил)-2-метил-9-(2-метилсульфонилэтил)пурина (0,3 г, 0,0007 моль),N-метилпиперазина (0,08 г, 0,0008 моль) и триэтиламина (0,08 г, 0,0008 моль) в этаноле (15 мл) нагревали при 90 С в течение 16 ч. Реакционную смесь охлаждали и концентрировали при пониженном давлении. Осадок растворяли в безводном дихлорметане и промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором. Органический слой сушили над безводным сульфатом натрия,фильтровали и концентрировали с получением осадка. Указанный осадок очищали на колонке, заполненной силикагелем, с использованием в качестве элюента смеси дихлорметан:метанол (96:4) с получением 8-(2-хлорфенил)-6-(4-метилпиперазин-1-ил)-2-метил-9-(2-метилсульфонилэтил)пурина (0,24 г). ИЭР/МС m/z 449 (М+1). К раствору 8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(2-(метилсульфонил)этил)-9 Нпурина (0,24 г, 0,0005 моль) в эфире (4 мл) добавляли HCl (2,0 М раствор в эфире) (0,018 г, 0,0005 моль,1,0 экв.) при 0 С и перемешивали в течение 2 ч при комнатной температуре. Осадок фильтровали, промывали эфиром и сушили в вакууме с получением указанного в заголовке соединения (0,15 г) в виде твердого вещества белого цвета. ИЭР/МС m/z 449 (М+1). Фенилпиперазинилпурины в табл. 4 получали, по существу, в соответствии со способом, описанным в примере 1, с использованием содержащих соответствующие заместители пиперазина и 6 хлорпурина.N-метилпиперазин (14,76 мл, 132,81 ммоль) растворяли в метоксибензоле в круглодонной колбе на 2 л. Добавляли одной порцией 2-хлорбензальдегид (20,38 мл, 181,1 ммоль) и повышали температуру до 140 С и поддерживали при указанной температуре в течение 4 дней. Реакционную смесь охлаждали и концентрировали при пониженном давлении. Полученное масло разбавляли 2 н. водным раствором соля- 12020565 ной кислоты (200 мл) и промывали дихлорметаном (500 мл). Удаляли органический слой. Водный слой обрабатывали раствором гидроксида натрия до достижения рН 14. Экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения в виде масла коричневого цвета (43 г). ИЭР/МС m/z 417 (М+1). Пример 11. Гидрохлорид 8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(2-метилсульфонилэтил)пурина В растворе тетрагидрофурана (277 мл) и метанола (277 мл) растворяли 8-(2-хлорфенил)-2-метил-6(4-метилпиперазин-1-ил)-9-(2-метилсульфанилэтил)пурин (38,5 г, 92,3 ммоль). Готовили раствор пероксимоносульфата калия (Oxone) (79,5 г, 129,3 ммоль) в воде (554 мл). Указанный раствор пероксимоносульфата калия (300 мл) добавляли в течение 5 мин и перемешивали в течение 30 мин. Затем добавляли дополнительную порцию раствора пероксимоносульфата калия (150 мл, с последующим добавлением 50 мл через 30 мин). Реакционную смесь перемешивали в течение 30 мин после окончания добавления. Добавляли твердый метабисульфит натрия (49,1 г, 258,5 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Добавляли водный раствор бикарбоната натрия (400 мл) и экстрагировали этилацетатом (31 л). Объединенный органический слой сушили над безводным сульфатом натрия,фильтровали и концентрировали с получением осадка оранжевого цвета. Осадок очищали на колонке,заполненной силикагелем, с использованием в качестве элюента 5-50% этанола в смеси дихлорметан:гексан (1:1). Соответствующие фракции объединяли и выпаривали с получением твердого вещества. Указанное твердое вещество растирали с эфиром и сушили в вакууме с получением 8-(2-хлорфенил)-2 метил-6-(4-метилпиперазин-1-ил)-9-(2-метилсульфонилэтил)пурина (11,4 г). ИЭР/МС m/z 449 (М+1). 8-(2-Хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(2-метилсульфонилэтил)пурин(11,3 г,25,2 ммоль) суспендирвали в этаноле (150 мл) и добавляли 1 н. водный раствор соляной кислоты (6,29 мл,25,2 ммоль, 1 экв.). Смеси позволяли перемешиваться в течение ночи и концентрировали при пониженном давлении. Выпавшее в осадок твердое вещество собирали посредством фильтрования, промывали ацетоном и сушили в вакууме с получением указанного в заголовке соединения (10,6 г). ИЭР/МС m/z 449 (М+1). Пример 12. Гидрохлорид 2-[8-(2-хлорфенил)-6-(4-этилпиперазин-1-ил)-2-метилпурин-9-ил]этанамина К раствору трет-бутил-N-[2-[8-(2-хлорфенил)-6-(4-этилпиперазин-1-ил)-2-метилпурин-9 ил]этил]карбамата (0,6 г, 1,2 ммоль) в дихлорметане (3 мл) добавляли трифторуксусную кислоту (3 мл) при 0 С и перемешивали в течение 2 ч при комнатной температуре. Реакцию гасили насыщенным водным раствором бикарбоната натрия и экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением осадка. Указанный осадок очищали на колонке, заполненной силикагелем, с использованием в качестве элюента смеси дихлорметан:метанол (96:4) с получением 2-[8-(2-хлорфенил)-6-(4-этилпиперазин-1-ил)-2-метилпурин-9 ил]этанамина (0,3 г). ИЭР/МС m/z 400 (М+1). К смеси 2-[8-(2-хлорфенил)-6-(4-этилпиперазин-1-ил)-2-метилпурин-9-ил]этанамина (0,3 г,0,7 ммоль) в эфире (5 мл) добавляли HCl (2,0 М раствор в эфире) (0,027 г, 0,7 ммоль) при 0 С и перемешивали в течение 2 ч при комнатной температуре. Осадок собирали посредством фильтрования и промывали эфиром. Сушили в вакууме с получением указанного в заголовке соединения (0,25 г) в виде твердого вещества белого цвета. ИЭР/МС m/z 400 (М+1). метил-N-[2-[8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)пурин-9 К раствору трет-бутил-N-[2-[8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)пурин-9 ил]этил]карбамата (0,51 г, 0,001 моль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (5 мл) при температуре 0 С. Реакционной смеси позволяли нагреваться до комнатной температуры и перемешивали указанную смесь в течение 2 ч. Реакцию гасили насыщенным водным раствором бикарбоната натрия и затем реакционную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением 2-[8-(2-хлорфенил)-2-метил-6(4-метилпиперазин-1-ил)пурин-9-ил]этанамина (0,39 г). ИЭР/МС m/z 386 (М+1). К раствору 2-[8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)пурин-9-ил]этанамина (0,39 г,0,0010 моль) и пиридина (4,0 мл) в безводном дихлорметане (4 мл) добавляли метилхлорформиат (0,25 г,0,0027 моль) при 0 С. Полученной смеси позволяли охлаждаться до комнатной температуры и перемешивали указанную смесь в течение 2 ч. Реакцию гасили насыщенным водным раствором бикарбоната натрия и затем реакционную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением осадка. Указанный осадок очищали на колонке, заполненной силикагелем, с использованием в качестве элюента смеси дихлорметан:метанол (97:3) с получением метил-N-[2-[8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)пурин 9-ил]этил]карбамата (0,3 г). ИЭР/МС m/z 444 (М+1). К раствору метил-N-[2-[8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)пурин-9 ил]этил]карбамата (0,3 г, 0,0006 моль) в эфире (6 мл) добавляли HCl (2,0 М раствор в эфире) (0,024 г,0,0006 моль) при 0 С и перемешивали в течение 2 ч при комнатной температуре. Осадок фильтровали,промывали эфиром и сушили в вакууме с получением указанного в заголовке соединения (0,28 г) в виде твердого вещества белого цвета. ИЭР/МС m/z 444 (М+1). Примеры в табл. 5 получали, по существу, в соответствии со способами, описанными в примере 13,начиная с трет-бутил-N-[2-[8-(2-хлорфенил)-6-[4-(2-фторэтил)пиперазин-1-ил]-2-метилпурин-9 ил]этил]карбамата или трет-бутил-N-[2-[6-(4-ацетилпиперазин-1-ил)-8-(2-хлорфенил)-2-метилпурин-9 ил]этил]карбамата. Удаляли защитную группу и подвергали реакции с метилхлорформиатом, метансульфонилхлоридом или уксусным ангидридом. Исследование функциональной активности в отношении CB1 и СВ 2 in vitro. Приведенные в примерах соединения исследовали по принципу агонизма с использованием основанного на сцинтилляционном анализе сближения (SPA) анализа связывания GTP35S. Все компоненты для анализа готовили в аналитическом буфере, содержащем 20 мМ HEPES, 100 мМ NaCl, 5 мМ MgCl2,(pH 7,4, при комнатной температуре). Полулогарифмические разведения соединения готовили на аналитическом буфере, содержащем БСА (конечная концентрация 0,125%). Связывание GTP35S определяли в 96-луночных планшетах с использованием метода полного захвата мембран для анализа CB1 и модифицированного метода "захвата" антител, описанного ранее (DeLapp et al. J. Pharmacol. Exp. Ther. 289:946-955, 1999), для анализа СВ 2. Все инкубации проводили при комнатной температуре. СВ 1. Мембраны hCB1-CHO, GTP (конечная концентрация 1 мкМ) и сапонин (конечная концентрация 10 мкг/мл) добавляли в аналитический буфер и гомогенизировали. Разведенные соединения, GTP35S(конечная концентрация 500 нМ) и мембраны добавляли в аналитический планшет и инкубировали в течение 30 мин. Затем добавляли 1 мг/лунку SPA-микрогранул с агглютинином из проростков пшеницы и планшеты герметизировали, содержимое перемешивали на вортексе и инкубировали в течение допол- 15020565 нительного часа. Затем планшеты центрифугировали при 700g в течение 10 мин и анализировали каждую лунку по 1 мин с помощью сцинтилляционного счтчика.CB2-Sf9. Мембраны hCB2-Sf9 и GTP (конечная концентрация 1 мкМ) добавляли в аналитический буфер и гомогенизировали. Разведенные соединения и мембраны добавляли в аналитический планшет и предварительно инкубировали в течение 15 мин. Затем добавляли GTP35S (конечная концентрация 500 нМ) и инкубировали еще 35 мин. Затем добавляли смесь, содержащую детергент нонидет Р 40 (конечная концентрация 0,2%), антитела против Gi (конечное разведение 1:362) и добавляли 1,25 мг микрогранул для сцинтилляционного анализа сближения с антикроличьими антителами. Планшеты запечатывали и их содержимое перемешивали на вортексе и затем инкубировали еще в течение 2 ч, затем центрифугировали и анализировали, как в случае CB1. Для анализа данных сначала вычитали фон из значений для всех лунок. Относительную эффективность агониста определяли путем приведения показателей зависимости доза-эффект для агониста/обратного агониста к показателям, полученным для эффекта полного агониста (метанандамида). Данные анализировали с использованием 4-параметрической логистической сокращенной модели с помощью программы Activity Base и XLFit3. Все приведенные в примерах соединения анализировали, по существу, как описано выше, и было обнаружено, что относительное значение ЕС 50 каждого из указанных соединений для СВ 2 составляло 100 нМ. Относительное значение ЕС 50 примера 2 для CB2 составляло 17,2 нМ, и относительное значение ЕС 50 для CB1 составляло 5560 нМ. Относительное значение ЕС 50 примера 16 для CB2 составляло 13,5 нМ, и относительное значение ЕС 50 для CB1 составляло 100000 нМ. Таким образом, была показана активность соединений согласно настоящему изобретению в отношении СВ 2 in vitro. Более того, была показана селективность соединений согласно настоящему изобретению в отношении CB2 по сравнению с CB1 что обеспечивает ограничение вероятности возникновения побочных эффектов, опосредованных центральной нервной системой. Замещение 3H-CP55940 в человеческих и крысиных рецепторах СВ 2. Использовали методы, описанные Фелдером (Felder et al., Mol. Pharmaocol. 48:443-450, 1995), с незначительными модификациями. В частности, гомогенаты мембран из клеток, которые постоянно или временно экспрессируют человеческий или крысиный рецептор CB2, промывали посредством центрифугирования и разводили в буфере, содержащем 50 мМ трис-HCl (рН 7,4), 5 мМ MgCl2, 2,5 мМ ЭДТА и 0,1% БСА. Определяли специфическое связывание 3H-СР 55940 с 1 мкМ СР 55940. Способность соединений замещать 3H-СР 55940 в специфическом связывании анализировали в диапазоне концентраций при разведении в буфере, содержащем трис, MgCl2, ЭДТА, БСА, в присутствии 1% диметилсульфоксида,путем инкубирования при комнатной температуре в течение 90 мин в объеме 300 мкл. 96-луночный микропланшет с фильтрами, предварительно обработанный 0,5% поливинилпирролидоном, 0,1% полисорбатом 20 в воде, промывали три раза холодным трис-буфером. Реакционную смесь затем переносили в планшет с фильтром непосредственно перед завершением инкубации посредством быстрого фильтрования и трех промывок 200 мкл холодным трис-буфером. После высушивания планшета с фильтрами в каждую лунку добавляли сцинтилляционную жидкость Microscint 20, планшет герметизировали и анализировали для определения распада за 1 мин. Кривые замещения наносили на график и результирующие значения Ki определяли с использованием программы Graphpad Prism. Значение Ki в отношении примера 3 для человеческого рецептора составляет 27,8 нМ, и для крысиного рецептора значение Ki составляет 12,6 нМ. Значение Ki примера 2 для человеческого рецептора составляло 28,4 нМ, и значение Ki для крысиного рецептора составляло 48,7 нМ. Таким образом, было показано, что соединения согласно настоящему изобретению связываются как с человеческими, так и с крысиными рецепторами CB2 in vitro. Модель с использованием монойодацетата (MIA). Во всех исследованиях для измерения боли в модели MIA использовали самцов крыс породы Lewis,возраст которых составлял примерно 8 недель на момент проведения инъекции MIA. Крыс содержали в группах по 2 или 3 животных на клетку и поддерживали при постоянной температуре и на световом режиме 12 ч света/12 ч темноты. Животные имели свободный доступ к пище и воде в течение всего времени, за исключением периода сбора данных. В стандартной модели MIA в правое колено каждой крысы инъецировали 0,3 мг MIA в 50 мкл солевого раствора, а в левые колени инъецировали 50 мкл солевого раствора. Боль измеряли в различное время после инъекции MIA (в норме не ранее чем через 10 дней после инъекции MIA) с помощью теста недееспособности. С помощью указанного теста измеряли различие между весовой нагрузкой на задние лапы, в колени которых вводили MIA и солевой раствор, и каждое измерение представляло собой среднее 3 отдельных измерений, каждое из которых проводили в течение 1 с. Для исследований агонистов CB2 крыс распределяли случайным образом по группам дозирования(n=5 или 6) и затем однократно вводили дозу исследуемого соединения. Дозирование проводили с интервалом в 15 мин между крысами и измеряли боль в заранее определенное время после введения дозы(обычно через 2 ч) с помощью теста недееспособности. Исследования обычно проводили на 4 группах группе плацебо (1% карбоксиметилцеллюлоза в воде плюс 0,25% полисорбат 80) и 3 группах, включающих соединения, которые могут представлять собой группы, включающие разные соединения в одной дозе, или группы, включающие одно и то же соединение в 3 дозах. Результаты представляли в виде различия весовой нагрузки между коленями, в которые вводили солевой раствор, и коленями, в которые вводили MIA, и проводили статистическое сравнение животных, получавших плацебо, и животных, получавших соединение, для оценки влияния соединений на боль в колене в указанной модели. Соединение примера 1 исследовали, по существу, как описано выше, и было обнаружено, что указанное соединение в дозах 0,3 и 1 мг/кг приводило к уменьшению боли по сравнению с носителем. Пример 17 анализировали, по существу, как описано выше, и было обнаружено, что указанное соединение в дозах 0,1, 0,3 и 1 мг/кг приводит к уменьшению боли по сравнению с носителем. Таким образом, было показано, что соединения согласно настоящему изобретению подходят для лечении боли, в частности боли в суставах. Животная модель экстравазации белков плазмы (РРЕ) через твердую мозговую оболочку. Самцов крыс Sprague-Dawley (250-350 г), полученных из Harlan, подвергали анестезии пентобарбиталом натрия (65 мг/кг, внутрибрюшинно) и помещали на стереотаксическую рамку (David KopfInstruments), нож устанавливали на уровне -2,5 мм. После сагиттального разреза скальпа по средней линии в черепе были просверлены 2 пары билатеральных отверстий (3,2 мм постериально, 1,8 и 3,8 мм латерально, все координаты приведены относительно брегмы). Пары нержавеющих стальных стимулирующих электродов (Rhodes Medical Systems Inc), полностью изолированных за исключением их окончаний, вводили через отверстия в оба полушария на глубину 9,2 мм. Находили бедренную вену и вводили внутривенно исследуемое соединение в дозировке 1 мл/кг. Примерно через 8 мин после ввутривенного введения также вводили внутривенно комплекс изотиоцианат флуоресцеина и бычьего сывороточного альбумина (FITC-БСА) в дозе 20 мг/кг. FITC-БСА использовали в качестве маркера экстравазации белков. Через 10 мин после введения исследуемого соединения проводили электрическую стимуляцию левого тригеминального ганглия в течение 5 мин при силе тока 1,0 мА (5 Гц, продолжительность 5 мс) с помощью стимулятора Grass Instrument, модели S48, с блоком фотоэлектрической изоляции PSIU6 (Grass-Telefactor). Альтернативно, крысам, которые голодали в течение ночи, перорально вводили дозу исследуемого соединения в объеме 2 мл/кг посредством принудительного кормления. Примерно через 50 мин животных анестезировали и помещали на стереотаксическую рамку, как описано выше. Через 60 мин после перорального введения дозы животным вводили дозу FITC-БСА (20 мг/кг, внутривенно). Через 1 ч после перорального введения дозы животных подвергали стимуляции, как описано выше. Через 5 мин после стимуляции животных умерщвляли путем вымывания крови с помощью 40 мл физиологического раствора. Верхнюю часть черепа удаляли для облегчения сбора твердых мозговых оболочек. Образцы мембран отбирали из обоих полушарий, промывали водой и распределяли на предметных стеклах. После высыхания образцов ткани наносили раствор 70% глицерин/вода и покрывали покровным стеклом. Флуоресцентный микроскоп (Zeiss), снабженный дифракционным монохроматором и спектрофотометром, использовали для оценки количества комплекса краситель FITC-БСА в каждом образце. Использовали длину волны возбуждения примерно 490 нм и определяли интенсивность излучения при 535 нм. Микроскоп был снабжен механизированным столиком и соединен с персональным компьютером, что обеспечивало контролируемое компьютером движение столика с измерениями флуоресценции в 25 точках (шаг 500 мм) в каждом образце твердой мозговой оболочки. Среднее и стандартное отклонение измерений определяли с помощью компьютера. Экстравазация, вызванная электрической стимуляцией тригеминального ганглия, представляла собой ипсилатеральное явление (которое наблюдалось только на той стороне твердой мозговой оболочки, с которой проводили стимуляцию тригеминального ганглия). Это позволяло использовать другую (нестимулированную) половину твердой мозговой оболочки в качестве контроля. Рассчитывали отношение количества экстравазированных белков в твердой мозговой оболочке на стимулированной стороне по сравнению с количеством на нестимулированной стороне. Относительная экстравазация у животных,которым вводили только дозу солевого раствора, составляла приблизительно 2,0, в то время как соединение, которое эффективно предотвращает экстравазацию через твердую мозговую оболочку на стимулированной стороне, должно приводить к относительной экстравазации, составляющей приблизительно 1,0. Пример 1 анализировали, по существу, как описано выше, и было обнаружено, что при его применении в концентрации 10 мг/кг относительная экстравазация составляла 1,12 через 2 ч после введения пероральной дозы. Соединение примера 7 исследовали, по существу, как описано выше, и было обнаружено, что при его применении в концентрации 10 мг/кг относительная экстравазация составляла 1,18 через 2 ч после введения пероральной дозы. Таким образом, было показано, что соединения согласно настоящему изобретению подходят для применения для лечения боли, в частности мигрени.R5 представляет собой Н или совместно с R4 образует пирролидин-2-он,или его фармацевтически приемлемая соль. 2. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 представляет собой Cl. 3. Соединение по п.1 или 2 или его фармацевтически приемлемая соль, где R2 представляет собой-CH2SO2CH3, -CH2NR4R5 или -CH2CN. 4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль, где R2 представляет собой -CH2SO2CH3. 5. Соединение по любому из пп.1-4 или его фармацевтически приемлемая соль, где R3 представляет собой метил, 2-фторэтил или С(О)СН 3. 6. Соединение по любому из пп.1-5 или его фармацевтически приемлемая соль, где R3 представляет собой метил. 7. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль, где R5 представляет собой Н и R4 представляет собой С(О)СН 3 или СО 2 СН 3. 8. Соединение по п.1, представляющее собой 8-(2-хлорфенил)-6-(4-метилпиперазин-1-ил)-2-метил 9-(2-метилсульфонилэтил)пурин, или его фармацевтически приемлемая соль. 9. Фармацевтическая композиция для лечения боли, содержащая соединение по любому из пп.1-8 или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель. 10. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемой соли для лечения боли. 11. Применение по п.10 для лечения остеоартритной боли или мигрени. 12. Способ лечения боли, включающий введение эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли человеку или животному, нуждающимся в этом. 13. Способ по п.12 для лечения остеоартритной боли или мигрени.
МПК / Метки
МПК: C07D 473/34, A61P 25/06, A61K 31/52, A61P 29/00
Метки: соединения, пуриновые
Код ссылки
<a href="https://eas.patents.su/19-20565-purinovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Пуриновые соединения</a>
Пуриновые соединения

Номер патента: 18386
Опубликовано: 30.07.2013
Авторы: Холлиншед Шон Патрик, Эстлз Питер Чарльз, Тидуэлл Майкл Вэйд, Гвидетти Росселла
МПК: A61K 31/52, A61P 25/04, C07D 473/34...
Метки: пуриновые, соединения
Формула / Реферат:
1. Соединение формулыгде R1 выбран из Н, F, Cl, C1-C2-алкила, CF3, циклопропила, ОСН3, OCF3 и CN;R2 выбран из тетрагидрофуранила, тетрагидропиранила, метилового эфира азетидин-1-карбоновой кислоты и тетрагидротиофен-1,1-диоксида;R3 представляет собой Н или совместно с R4 образует конденсированный пирролидин-2-он;R4 выбран из C1-C2-алкила, C1-С2-фторалкила, циклопропила и СОСН3;R5 выбран из Н, СН3 и CF3;n равно 0 или 1;X1 и X3 независимо выбраны...
Пуриновые соединения

Номер патента: 20529
Опубликовано: 28.11.2014
Авторы: Холлиншед Шон Патрик, Гвидетти Росселла, Астлс Питер Чарльз, Сандерсон Адам Ян
МПК: A61K 31/52, A61P 25/02, C07D 487/04...
Метки: пуриновые, соединения
Формула / Реферат:
1. Соединение формулыгде R1 представляет собой Cl или СН3;R2 представляет собой ОН, ОСН3, СН2ОН или СН2ОСН3;R3 представляет собой Н или совместно с R4 образует конденсированный пирролидин-2-он;R4 представляет собой C1-C2 алкил, C1-C2 фторалкил, С(O)СН3 или CO2CH3;R5 представляет собой Н, СН3 или СН2ОСН3;R6 представляет собой Н, СН3 или совместно с R5 образует циклопропановое кольцо;n равно 0 или 1,или фармацевтически приемлемая соль указанного...
Пуриновые соединения и их применение в качестве лигандов каннабиноидных рецепторов

Номер патента: 8176
Опубликовано: 27.04.2007
Автор: Гриффит Дейвид Эндрю
МПК: A61P 3/04, A61K 31/52, C07D 239/48...
Метки: качестве, каннабиноидных, лигандов, пуриновые, рецепторов, соединения, применение
Формула / Реферат:
1. Соединение формулы (I) где А представляет собой возможно замещенный фенил или возможно замещенный гетероарил; В представляет собой возможно замещенный фенил или возможно замещенный гетероарил; R1 представляет собой водород, (С1-С4)алкил, галогенозамещенный (С1-С4)алкил или (С1-С4)алкокси; R4 представляет собой (1) группу, имеющую формулу (IA) или формулу (IB) где R4a представляет собой водород или (С1-С3)алкил; R4b и R4b', каждый...
Пуриновые производные

Номер патента: 4655
Опубликовано: 24.06.2004
Автор: Монахан Сандра Марина
МПК: C07H 19/167, A61K 31/70, A61P 11/00...
Метки: производные, пуриновые
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемые соль либо сольват, где R1 представляет собой водород или C1-C6алкил, возможно замещенный 1 или 2 заместителями, каждый из которых независимо выбран из фенила и нафтила, причем указанные фенил и нафтил возможно замещены C1-C6алкилом, C1-C6алкокси, галогено или циано; A представляет собой связь или C1-C3алкилен; R2 представляет собой (1) водород, C1-C6алкил, C3-C7циклоалкил, фенил или...
Пуриновые производные в качестве агонистов аденозиновых а1 рецепторов и способы их применения

Номер патента: 11826
Опубликовано: 30.06.2009
Авторы: Джагтап Пракаш, Сабо Чаба, Салзман Эндрю Л.
МПК: A61K 31/70
Метки: производные, способы, агонистов, рецепторов, пуриновые, применения, качестве, аденозиновых
Формула / Реферат:
1. Соединение, имеющее формулу или его фармацевтически приемлемая соль, где A представляет собой -CH2ONO2; В и С представляют собой -ОН; D представляет собой А и В представляют собой транс- в отношении друг к другу; В и С представляют собой цис- в отношении друг к другу; С и D представляют собой цис- или транс- в отношении друг к другу; R1 представляет собой тетрагидрофуранил, -C3-C8-моноциклический циклоалкил или -C8-C12-бициклический...
Предыдущий патент: Замещенные n-фенил-1-(4-пиридинил)-1н-пиразол-3-амины
Следующий патент: Спироциклические производные амидов
Случайный патент: Способ декорирования поверхности здания или сооружения (варианты)