Оптически активное производное фенилпиримидина, способ его получения и его применение в качестве анальгетика.
















Формула / Реферат
1. Пиримидин формулы (I)

или его соли присоединения к кислотам.
2. Соль по п.1, отличающаяся тем, что она является фармацевтически приемлемой солью присоединения к кислоте.
3. Соль по п.1, отличающаяся тем, что она является сульфатом, фосфатом или изэтионатом.
4. Соль по п.1, отличающаяся тем, что она является гидрохлоридом или метансульфонатом.
5. Способ получения пиримидина формулы (I) по п.1 или его соли присоединения к кислоте, при котором:
(1) рацемический 2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидин разделяют с помощью подходящей хиральной кислоты и полученную соль перекристаллизовывают с получением соли, которая состоит практически только из соли R(-)-2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина; и
(2) при желании перекристаллизованную соль превращают в свободное основание или другую соль присоединения к кислоте.
6. Способ получения пиримидина формулы (I) по п.1 или его соли присоединения к кислоте, при котором:
(а) рацемический 2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидин разделяют с помощью соответствующей хиральной кислоты и полученную соль перекристаллизовывают с получением соли, которая состоит практически только из соли (-)-2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидина;
(б) при желании перекристаллизованную соль превращают в свободное основание или другую соль;
(в) перекристаллизованную соль со стадии (а) или свободное основание или указанную другую соль со стадии (б) фторируют в условиях, при которых не происходит рацемизации (-)-2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметил пиримидина или полученного (-)-2,4-диамино-5-(2,3-дихлорфенил)-6-фторметил пиримидина; и
(г) при желании полученное фторированное соединение превращают в свободное основание или в его соль присоединения к кислоте.
7. Фармацевтический препарат, содержащий в качестве активного ингредиента пиримидин формулы (I) по п.1 или его фармацевтически приемлемую соль присоединения к кислоте и фармацевтически приемлемый носитель или разбавитель.
8. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли присоединения к кислоте в качестве терапевтического средства.
9. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при получении анальгетика.
10. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при получении противосудорожного средства.
11. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при получении лекарственного средства при лечении синдрома раздражённого кишечника.
12. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при получении лекарственного средства для лечения биполярного расстройства.
13. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при получении лекарственного средства для предупреждения или снижения зависимости или толерантности к вызывающему зависимость агенту.
14. Пиримидин формулы (III)

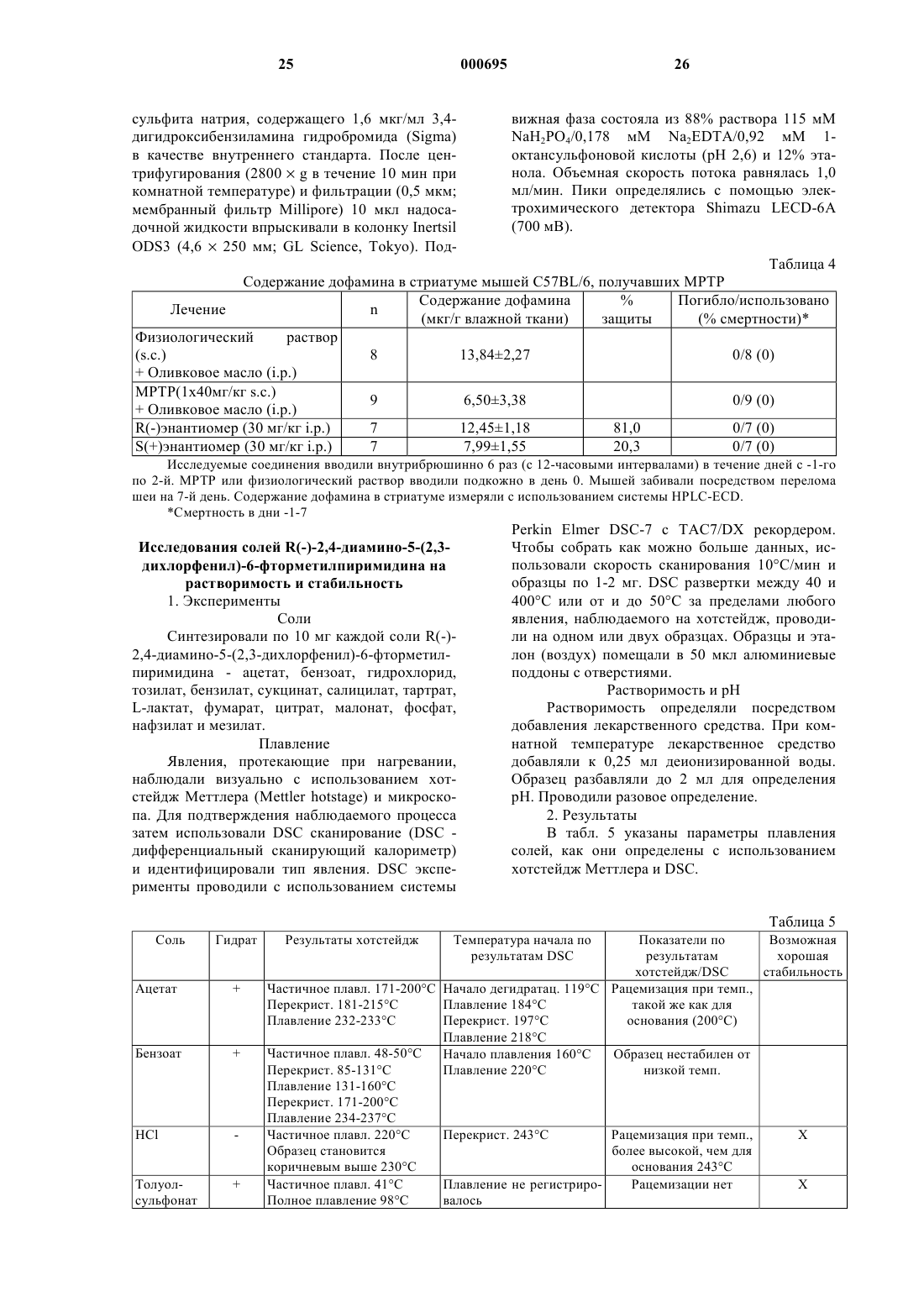
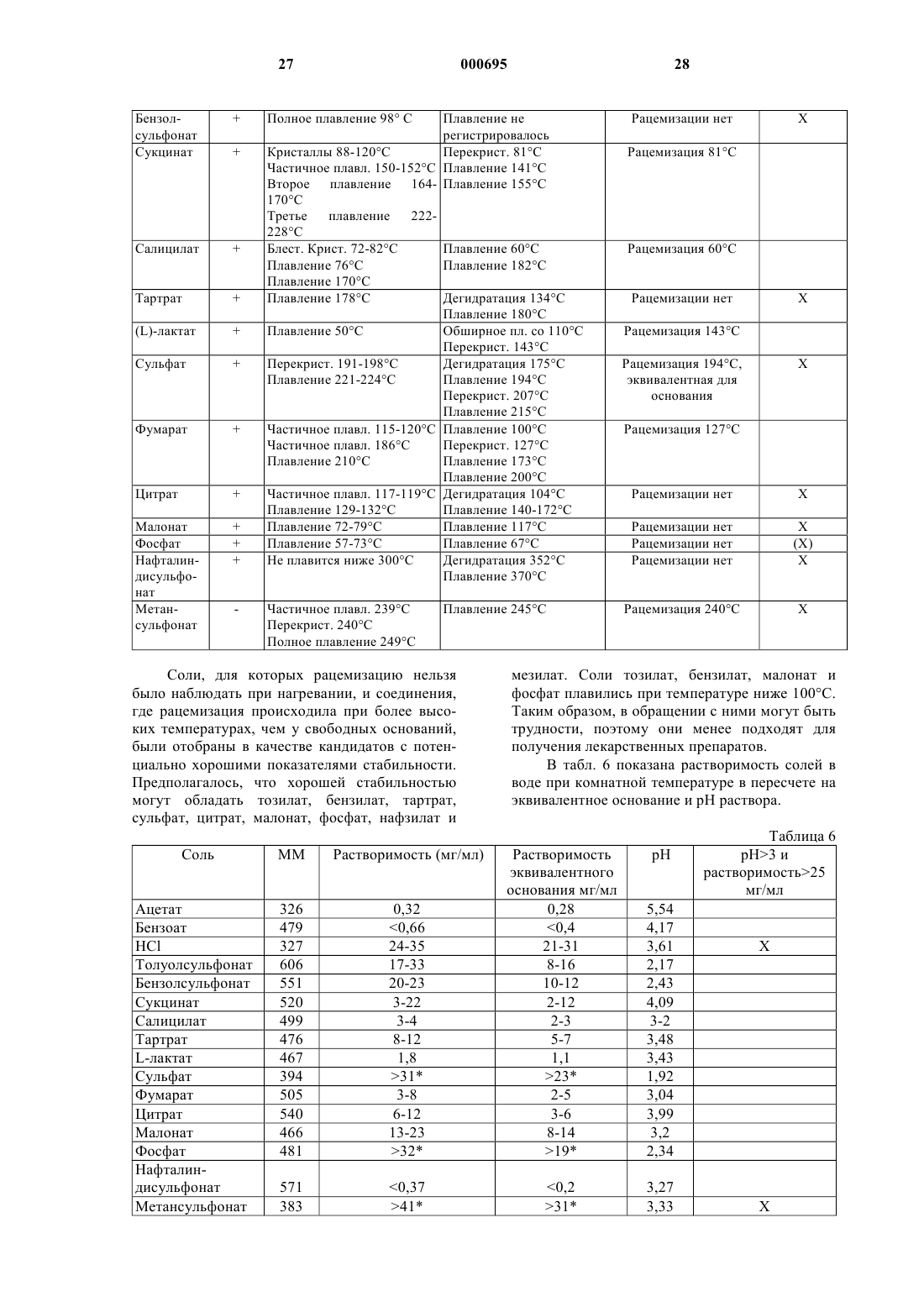
Текст
1 Настоящее изобретение относится к соединению пиримидина, его получению, содержащим его фармацевтическим препаратам и его применению в терапии. Известна[ЕР-А-21121] группа 3,5 диамино-6-(замещенный фенил)-1,2,4-триазинов, которые активны при лечении расстройств центральной нервной системы (ЦНС), например при лечении эпилепсии. Одним таким триазином является 3,5-диамино-6-(2,3-дихлорфенил)1,2,4-триазин, который иначе называется ламотригин. Известны [ЕР-0372934-А] соединения пиримидина, полезные при лечении расстройств ЦНС. Пример 18 из ЕР-0372934-А раскрывает 2,4-диамино-5-(2,3-дихлорфенил)-6-фторметил пиримидин. Согласно настоящему изобретению предложен пиримидин формулы (I) и его соли присоединения кислот. Пиримидин формулы (I) представляет собойR(-)-2,4-диамино-5-(2,3-дихлорфенил)-6 фторметилпиримидин. Он практически свободен от S(+)-энантиомера, S(+)-2,4-диамино-5(2,3-дихлорфенил)-6-фторметилпиримидина.R(-)-энантиомер по изобретению имеет более желательные свойства, чем ламотригин: он менее активен против дигидрофолатредуктазы(DHFR) и более активен в аналгетических и противосудорожных тестах. Он также имеет более желательные свойства, чем S(+)-энантиомер. Так,- R(-)-энантиомер имеет фармакокинетические показатели лучше, чем S(+)-энантиомер,например он не так быстро метаболизируется и поэтому имеет более длительный период полураспада (продолжительность действия);- R(-)-энантиомер проявляет более высокую аналгетическую активность по сравнению с- R(-)-энантиомер проявляет более высокую противосудорожную активность по сравнению с S(+)-энантиомером; и- R(-)-энантиомер проявляет меньшую активность против DHFR, чем S(+)-энантиомер. 2 Удивительно, что R(-)-энантиомер лучше,чем S(+)-энантиомер во всех этих отношениях.R(-)-энантиомер может быть приготовлен практически чистым. Так, соотношение R(-)энантиомер : S(+)-энантиомер может быть, по меньшей мере, 94:6, таким как, по меньшей мере, 98:2 или, по меньшей мере, 99:1. Предпочтительно предоставляетсяR(-)-энантиомер и его соли присоединения кислоты могут быть получены в соответствии с изобретением первым способом, при котором 1) рацемический 2,4-диамино-5-(2,3 дихлорфенил)-6-фторметилпиримидин разделяют с помощью подходящей хиральной кислоты и полученную cоль перекристаллизовывают таким образом, чтобы получить соль, которая состоит практически только из соли с R(-)-2,4 диамино-5-(2,3-дихлорфенил-6-фторметилпиримидином; и 2) при желании, перекристаллизованную соль превращают в свободное основание или другую соль присоединения кислоты. Стадию разделения (1) осуществляют с помощью подходящей хиральной кислоты в подходящем растворителе. Предпочтительно кислота представляет собой (-)-дибензоил-Lвинную кислоту. Другие подходящие кислоты могут быть определены опытным путм. Предпочтительно растворитель представляет собой этанол. Опять же другие подходящие растворители могут быть определены опытным путм. Полученная в результате соль, которая может быть выделена, состоит, главным образом, из R(-)-2,4-диамино-5-(2,3-дихлорфенил)-6 фторметилпиримидина. Может присутствовать незначительная доля соли с S(+)-энантиомером. Доля соли с R(-)-энантиомером может быть увеличена проведением одной или более чем одной, например двух или трх перекристаллизаций на стадии (1). Наконец, кристаллическая соль, полученная в результате разделения, может быть растворена в растворителе. Это может быть осуществлено путем нагревания. Из полученного раствора соль перекристаллизовывают. Это может быть осуществлено, если раствору дать возможность охладиться. Растворителем может быть этанол. Долю соли с R(-)-энантиомером можно таким образом увеличивать до тех пор, пока раствор не станет практически чистым, т.е. до тех пор, пока не будет присутствовать практически только соль с R(-)-энантиомером. Маточная жидкость со стадии разделения и маточные жидкости с каждой стадии перекристаллизации обогащаются S(+)-энантиомером. Одна или более чем одна из этих жидкостей или объединнные жидкости могут быть обработаны основанием, таким как гидроксид натрия,для удаления остаточной хиральной кислоты и,тем самым, с получением свободного основа 3 ния. Полученное свободное основание может быть высушено. Свободное основание, обогащнное S(+)энантиомером, затем может быть превращено в рацемат. Это может быть осуществлено путем нагревания с обратным холодильником в растворителе, таком как толуол, например в течение от 12 до 48 ч. Рацемат, полученный таким образом, затем может быть рециклизован на стадию (1) настоящего способа. Таким образом можно увеличить выход. Соль, полученная на стадии (1), представляет собой соль хиральной кислоты, использованной для разделения, и R(-)-энантиомера,практически свободного от S(+)-энантиомера. Эта соль может быть превращена в свободное основание или другую соль присоединения кислоты в соответствии со стадией (2) настоящего способа. Соль хиральной кислоты и R(-)энантиомера могут, таким образом, быть обработаны в растворе основанием, таким как гидроксид натрия, с получением свободного основания. Свободное основание затем само может быть превращено в его соль присоединения кислоты. Подходящие соли присоединения кислот,которые могут быть образованы на стадии (2),включают в себя соли, образованные как с органическими, так и с неорганическими кислотами. Такие соли присоединения кислот обычно должны быть фармацевтически приемлемыми. Так, подходящие соли включают в себя соли,образованные с хлористоводородной, бромистоводородной, серной, лимонной, виннокаменной,фосфорной, молочной, пировиноградной, уксусной, янтарной, фумаровой, малеиновой, щавелевоуксусной, метансульфоновой, этансульфоновой, р-толуолсульфоновой, бензолсульфоновой и изэтионовой кислотами. Эти соли могут быть получены взаимодействием свободного основания с соответствующей кислотой. Предпочтительными солями являются гидрохлорид, сульфат, фосфат, метансульфонат и изэтионат. Гидрохлорид и метансульфонат являются, в частности, подходящими для внутривенного введения.R(-)-энантиомер и его соли присоединения кислот альтернативно могут быть получены,согласно данному изобретению, посредством второго способа, при котором(а) рацемический 2,4-диамино-5-(2,3 дихлорфенил)-6-гидроксиметилпиримидин разделяют с помощью подходящей хиральной кислоты и полученную соль перекристаллизовывают таким образом, чтобы получить соль, которая состоит практически только из соли с (-)2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидином;(б) при желании, перекристаллизованную соль превращают в свободное основание или другую соль;(в) перекристаллизованную соль из стадии(а) или свободное основание или указанную другую соль из стадии (б) фторируют в условиях, при которых не происходит рацемизации (-)2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидина или полученного R(-)-2,4 диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина; и(г) при желании, полученное фторированное соединение превращают в свободное основание или соль присоединения кислоты, когда как подходит. Стадию разделения (а) осуществляют с помощью подходящей хиральной кислоты в подходящем растворителе. Предпочтительно кислота представляет собой (+)-ди-п-толуоил-Dвинную кислоту. Другие подходящие кислоты могут быть определены опытным путм. Предпочтительно растворитель представляет собой этанол. Опять же другие подходящие растворители могут быть определены опытным путем. Полученная соль, которую можно выделить, состоит, главным образом, из соли с (-)2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидином. Может присутствовать незначительная доля соли с (+)-энантиомером. Доля соли с (-)-энантиомером может быть увеличена проведением одной или более чем одной, например двух или трх, перекристаллизации на стадии (а) данного процесса. Кристаллическая соль, полученная в результате разделения, следовательно, может быть растворена в растворителе. Это может быть осуществлено путем подогревания. Соль перекристаллизовывают из полученного раствора. Это может быть осуществлено, если раствору дать возможность остыть. Растворителем может быть этанол. Долю соли с (-)-энантиомером можно таким образом увеличивать до тех пор,пока соль не станет практически чистой, т.е. до тех пор, пока не будет присутствовать практически только соль с (-)-энантиомером. Маточная жидкость со стадии разделения и маточные жидкости с каждой стадии перекристаллизации обогащаются (+)-энантиомером. Одна или более чем одна из этих жидкостей или объединнные жидкости могут быть обработаны основанием, таким как гидроксид натрия,чтобы удалить остаточную хиральную кислоту и, тем самым, получить свободное основание. Полученное свободное основание может быть высушено. Свободное основание, обогащнное S(+)энантиомером, затем может быть превращено в рацемат. Это может быть осуществлено путем нагревания с обратным холодильником в растворителе, таком как толуол, например в течение от 12 до 48 ч. Рацемат, полученный таким образом, затем может быть рециклизован на стадию (а) настоящего способа. Таким образом можно увеличить выход (-)-энантиомера. 5 Соль, полученная в результате этих процедур, представляет собой соль хиральной кислоты, использованной для разделения, и (-)энантиомера 2,4-диамино-5-(2,3-дихлорфенил)6-гидроксиметилпиримидина. Соль хиральной кислоты и(-)энантиомера, практически свободная от (+)энантиомера, может быть превращена в свободное основание или другую соль в соответствии со стадией (б) настоящего способа. Соль хиральной кислоты можно, таким образом, обработать в растворе основанием, таким как гидроксид натрия, с получением свободного основания. Фторирование(-)-2,4-диамино-5-(2,3 дихлорфенил)-6-гидроксиметилпиримидина осуществляют на стадии (в). (-)-Энантиомер может присутствовать либо в форме соли, либо как свободное основание. В любом случае (-)энантиомер практически свободен от (+)энантиомера. Поэтому в результате получают практически только R(-)-2,4-диамино-5-(2,3 дихлорфенил)-6-гидроксиметилпиримидин. Фторирование проводят в условиях, при которых не происходит рацемизации 6 гидроксиметил и фторметил (-)-энантиомеров. Температура должна, таким образом, быть ниже чем 80 С, например ниже чем 50 С. Фторирование может быть осуществлено, например, взаимодействием(-)2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидина с трифторидом диэтиламиносеры (DAST). Это взаимодействие может быть осуществлено в дихлорметане при 78 С. Раствор затем перемешивают, пока происходит нагревание до 10 С, в течение четырх с половиной часов с получением (-)2,4 диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина. Когда как подходит, полученное фторированное соединение может быть превращено в свободное основание или его соль присоединения кислоты. Подходящие соли присоединения кислот указаны выше. Эти соли могут быть получены путем обработки R(-)-энантиомера со 000695 6 ответствующей кислотой в форме свободного основания. Первый способ в соответствии с данным изобретением начинают с рацемического 2,4 диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина. Он может быть получен двумя способами. Способ 1 2,3-Дихлорбензальдегид чисто восстанавливают боргидридом натрия, например в смеси толуол/метанол. После разложения избытка боргидрида полученную суспензию 2,3 дихлорбензилового спирта обрабатывают метансульфонилхлоридом с образованием метансульфоната, который водным цианидом калия в присутствии катализатора фазового переноса превращают непосредственно в 2,3 дихлорфенилацетонитрил. Конденсация по Клайзену между 2,3 дихлорфенилацетонитрилом и этилфторацетатом в присутствии метоксида натрия в метаноле приводит к образованию енолята натрия. Регулирование рН приводит к образованию неочищенного 2-(2,3-дихлорфенил)-4-фтор-3 гидрокси-2-бутеннитрила. Алкилирование 2-(2,3-дихлорфенил)-4 фтор-3-гидрокси-2-бутеннитрила может быть соответствующим образом осуществлено с использованием этилйодида в диметилформамиде в присутствии карбоната калия с получением неочищенного 2-(2,3-дихлорфенил)-3-этокси-4 фтор-3-гидрокси-2-бутеннитрила. Сочетание неочищенного 2-(2,3 дихлорфенил)-3-этокси-4-фтор-3-гидрокси-2 бутеннитрила с гидрохлоридом гуанидина в присутствии метоксида натрия в метаноле приводит к образованию рацемического 2,4 диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина. Способ 2 2,3-Дихлорбензальдегид чисто восстанавливают с использованием щелочного раствора боргидрида натрия в метаноле с образованием 2,3-дихлорбензилового спирта. Обработка 2,3-дихлорбензилового спирта метансульфонилхлоридом в толуоле приводит к образованию метансульфоната, который водным цианидом калия в присутствии катализатора фазового переноса превращают непосредственно в 2,3-дихлорфенилацетонитрил. Конденсация по Клайзену между 2,3 дихлорфенилацетонитрилом и этилдиэтоксиацетатом в диметоксиэтане в присутствии третбутоксида натрия приводит к образованию енолята натрия. Алкилирование енолята натрия осуществляют с использованием этилйодида до получения неочищенного 2-(2,3-дихлорфенил)3,4,4-триэтокси-бут-2-еннитрила. Сочетание 2-(2,3-дихлорфенил)-3,4,4 триэтокси-бут-2-еннитрила с гидрохлоридом гуанидина в присутствии этоксида натрия в этаноле приводит к образованию 2,4-диамино-5 7(2,3-дихлорфенил)-6-диэтоксиметилпиримидина. Гидролиз 2,4-диамино-5-(2,3 дихлорфенил)-6-диэтоксиметилпиримидина в водной хлористоводородной кислоте при 90 С после охлаждения и нейтрализации приводит к образованию 2,4-диамино-5-(2,3-дихлорфенил)6-формилпиримидина. Восстановление 2,4-диамино-5-(2,3 дихлорфенил)-6-формилпиримидина боргидридом натрия в этаноле приводит к образованию 2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидина. Фторирование рацемического 2,4 диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидина может быть эффективным при использовании трифторида диэтиламиносеры(DAST). Фторирование может быть проведено в дихлорметане вначале при 78 С с последующим прогреванием до 10 С в течение четырх с половиной часов с образованием 2,4-диамино 5-(2,3-дихлорфенил)-6-фторметилпиримидина. Второй способ согласно данному изобретению начинают с рацемического 2,4-диамино 5-(2,3-дихлорфенил)-6-гидроксиметилпиримидина. Он может быть получен, как описано в способе 2. Соединение формулы (I) и его фармацевтически приемлемые соли присоединения кислот полезны в качестве анальгетиков. Следовательно, они полезны при лечении или предупреждении боли. Их можно использовать для улучшения состояния субъекта, как правило человека, страдающего от боли. Их можно использовать для облегчения боли у субъекта. Так,соединение формулы (I) и его фармацевтически приемлемые соли присоединения кислот можно использовать в качестве упреждающих анальгетиков при послеоперационной боли; для лечения острой боли, например послеоперационной боли, такой как удаление зуба; и для лечения хронической боли, такой как хроническая воспалительная боль, невропатическая боль и боль при раке. Невропатическая боль, как описано здесь, может включать, например, невропатию при СПИДе, невралгию при опоясывающем лишае, диабетическую невропатию и невралгию тройничного нерва. Соединение формулы (I) можно также использовать при лечении или предупреждении боли, связанной с мигренью. Соединение формулы (I) и его фармацевтически приемлемые соли присоединения кислот пригодны также при лечении функциональных расстройств пищеварительного тракта,которые включают в себя неязвенную диспепсию, несердечную боль в груди и синдром раздражнного кишечника. Синдром раздражнного кишечника представляет собой расстройство желудочно-кишечного тракта, которое характеризуется наличием боли в животе и изменнными свойствами кишечника в отсутствии доказа 000695 8 тельств органического заболевания. Соединение формулы (I) и его фармацевтически приемлемые соли присоединения кислот можно применять, таким образом, для облегчения боли, связанной с синдромом раздражнного кишечника. Состояние пациента, страдающего от синдрома раздражнного кишечника, таким образом, может быть улучшено. Соединение формулы (I) и его фармацевтически приемлемые соли присоединения кислот пригодны также в качестве противосудорожных средств. Следовательно, они полезны при лечении эпилепсии. Их можно применять для улучшения состояния субъекта, как правило человека, страдающего от эпилепсии. Их можно применять для облегчения симптомов эпилепсии у субъекта. Соединение формулы (I) и его фармацевтически приемлемые соли присоединения кислот полезны также при лечении биполярного расстройства, альтернативно известного как маниакальная депрессия. Можно лечить тип I или II биполярного расстройства. Соединение формулы (I) и его фармацевтически приемлемые соли присоединения кислот можно, таким образом, применять для улучшения состояния пациентов, страдающих биполярным расстройством. Их можно применять для облегчения симптомов биполярного расстройства у субъекта. Соединение формулы (I) можно также применять при лечении однополярной депрессии. Далее, соединение формулы (I) и его фармацевтически приемлемые соли присоединения кислот пригодны также для предупреждения или снижения зависимости от или предупреждения или снижения толерантности или обратной толерантности к агентам, вызывающим зависимость. Примеры вызывающих зависимость агентов включают в себя опиоиды (например морфин), депрессанты ЦНС (например этанол),психостимуляторы (например кокаин) и никотин. Соединение формулы (I) и его фармацевтически приемлемые соли присоединения кислот могут быть полезны при лечении нейродегенеративных заболеваний, таких как болезнь Альцгеймера, ALS (амиотрофический боковой склероз), двигательные нейронные заболевания и, в частности, болезнь Паркинсона. Соединения формулы (I) также можно применять при лечении нейродегенерации вследствие удара,травматического повреждения мозга или т.п. Поэтому далее настоящим изобретением предусмотрено применение соединения формулы (I) в производстве лекарственных средств для применения при лечении расстройств по существу, как описано выше. Далее, настоящее изобретение включает в себя способ лечения пациентов, страдающих от или подверженных расстройствам по существу, как описано выше,при котором пациенту вводят терапевтически 9 эффективное количество соединения формулы(I). Точное количество соединения формулы(I) или его соли, вводимое субъекту, в частности человеку, должно быть на ответственности лечащего врача. Однако применяемая доза будет зависеть от ряда факторов, включая возраст и пол пациента, точное состояние и его тяжесть и способ введения. Соединение формулы (I) и его соли можно вводить в дозе от 0,1 до 30 мг/кг веса тела в день в расчете на свободное основание. Интервал дозы для взрослого человека обычно составляет от 8 до 2400 мг/день, предпочтительно от 35 до 1050 мг/день в расчете на свободное основание. Хотя соединение формулы (I) или его фармацевтически приемлемую соль присоединения кислоты возможно вводить в виде исходного химического вещества, предпочтительно вводить его в форме фармацевтического препарата. Препараты настоящего изобретения содержат соединение формулы (I) или его фармацевтически приемлемую соль вместе с одним или более чем одним приемлемым носителем или разбавителем и возможно другим терапевтическим ингредиентом. Носитель(и) должен быть "приемлемым" в смысле совместимости с другими ингредиентами препарата и не вредным для реципиента. Препараты включают в себя препараты,подходящие для перорального, парентерального(включая подкожное, внутрикожное, внутриоболочечное, внутримышечное и внутривенное), ректального и местного (включая кожное,буккальное и подъязычное) введения, хотя наиболее подходящий способ может зависеть от,например, состояния и заболевания реципиента. Препараты могут быть приготовлены в форме стандартной дозы и могут быть получены любым из хорошо известных в фармации способом. Все способы включают стадию приведения в контакт соединения по формуле (I) или его фармацевтически приемлемой соли присоединения кислоты ("активный ингредиент") с носителем, который состоит из одного или более чем одного дополнительного ингредиента. В общем,препараты получают приведением к однородному и тесному контакту активного ингредиента с жидкими носителями или тонко измельченными тврдыми носителями или теми и другими, а затем, при необходимости, формованием продукта в целевой препарат. Препараты по настоящему изобретению,подходящие для перорального введения, могут быть приготовлены в виде отдельных стандартных доз, таких как капсулы, облатки или таблетки, каждая из которых содержит предопределнное количество активного ингредиента: в виде порошка или гранул; в виде раствора или суспензии в водном или неводном разбавителе; или в виде эмульсии масло-в-воде или эмульсии 10 вода-в-масле. Активный ингредиент может быть также представлен в виде большой пилюли,электуария или пасты. Таблетка может быть спрессована или отлита в форму, возможно с одним или более чем одним вспомогательным ингредиентом. Прессованные таблетки могут быть получены прессованием в соответствующем аппарате активного ингредиента в сыпучей форме, такой как порошок или гранулы, возможно смешанного со связывающим веществом, смазывающим веществом, инертным разбавителем, поверхностно активным или диспергирующим агентом. Формованные таблетки могут быть получены формованием в соответствующем аппарате смеси увлажннного порошкообразного соединения с инертным жидким разбавителем. Таблетки возможно могут быть покрыты оболочкой или на них могут быть сделаны метки и могут быть получены таким образом, чтобы обеспечить медленное или регулируемое высвобождение активного ингредиента. Препараты для парентерального введения включают в себя водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворнные вещества, которые делают препарат изотоническим с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители. Препараты могут быть представлены в сосудах для одноразовой или многоразовой дозы, например запаянных ампулах или флаконах, и могут хранится лиофилизированными (высушенными вымораживанием), требующими только добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед применением. Без подготовки растворы и суспензии могут быть приготовлены из стерильных порошков, гранул и таблеток вида, описанного ранее. Препараты для ректального введения могут быть представлены в форме суппозиториев с обычными носителями, такими как масло какао,тврдый жир или полиэтиленгликоль. Препараты для местного введения в рот,например трансбуккально или подъязычно,включают в себя лепешки, содержащие активный ингредиент в основе из корригента, такого как сахароза и акация или трагакант, и пастилки, содержащие активный ингредиент в основе,такой как желатин и глицерин или сахароза и акация. В дополнение к ингредиентам, в частности упомянутым выше, препараты могут содержать другие агенты, традиционные в данной области,имеющие отношение к типу препарата, о котором идет речь, например агенты, подходящие для перорального введения могут включать агенты, придающие вкус и аромат. Предпочтительными препаратами в форме стандартной дозы являются препараты, содер 11 жащие эффективную суточную дозу активного ингредиента, как сказано выше, или ее соответствующую долю. Подходящей суточной дозой может быть 5-500 мг, более подходящей 10-250 мг и наиболее подходящей 20-200 мг в расчете на свободное основание. Следующие примеры иллюстрируют изобретение. Предоставлены эталонные примеры. Эталонный пример 1. Синтез рацемического (+/-)-2,4-диамино-5-(2,3-дихлорфенил)6-фторметилпиримидина. 1. Получение 2,3-дихлорфенилацетонитрила. К суспензии из 2,3-дихлорбенальдегида(40 л) добавляют боргидрид натрия (2,59 кг, 68,6 моль) порциями за период времени 1 ч. Смесь перемешивают в течение 30 мин, затем обрабатывают ацетоном (20 л). После разложения избытка боргидрида добавляют воду (80 л). Толуол (54 л) добавляют в толуольную фазу и суспензию подогревают до 422 С, чтобы получить раствор, который затем разделяют. Органическую фазу дистиллируют с удалением 54 л азеотропа и таким образом способствуют удалению воды, ацетона и изопропилового спирта. Полученный раствор 2,3 дихлорбензилового спирта в толуоле охлаждают. К полученной суспензии добавляют триэтиламин (27,8 кг, 274,3 моля), затем метансульфонилхлорид (31,4 кг, 274,3 моля) в течение 1,5 ч,поддерживая температуру 02 С. Смесь перемешивают в течение 1 ч, затем в суспензию загружают воду (100 л) и смесь энергично перемешивают, затем разделяют. К метансульфонату в толуольной фазе добавляют гидросульфат тетрабутиламмония (15,6 кг, 45,8 моля) и водный раствор цианида калия(22,4 кг, 342 моля) в воде (70 л) в течение 40 мин. Смесь из двух фаз перемешивают в течение ночи, разделяют и органическую фазу промывают водой (70 л). Толуольную фазу дистиллируют с удалением 130 кг толуола в присутствии древесного угля (2,8 кг) и дикалита (2,8 кг). В остаток загружают петролейный эфир 60/80(300 л), смесь фильтруют горячей и кристаллизуют под вакуумом с получением 2,3 дихлорфенилацетонитрила (30 кг, выход 72%). 2. Получение 2-(2,3-дихлорфенил)-3 этокси-4-фтор-2-бутеннитрила. В суспензию из 2,3-дихлорфенилацетонитрила (45 кг, 241,9 моля) в метаноле (90 л) загружают 30 мас.%/мас. раствор метоксида натрия в метаноле (113,5 кг, 630,6 моля), затем этилфторацетат (29,7 кг, 280,1 моля). Реакционную смесь перемешивают в течение ночи и продукт осаждают из водной хлористоводородной кислоты (63,7 кг, 648 моля) в воде (350 л). Суспензию фильтруют и тврдое вещество растворяют в этилацетате и промывают рассолом. 12 Этилацетат (100 л) удаляют посредством вакуумной дистилляции. Добавляют диметилформамид (DMF) (70 л) и дистилляцию продолжают до удаления оставшегося этилацетата. К полученному енолу в DMF добавляют карбонат калия (20 кг, 145 молей) в течение 10 мин. Алкилирование енолята калия осуществляют с использованием этилйодида (37,7 кг,241,9 моля) при 70 С в течение 11/4 ч. Реакционную смесь разделяют между толуолом (140 л) и водой (75 л) и толуольную фазу промывают водой (50 л). Толуол (75 л) удаляют дистилляцией с получением неочищенного продукта в виде раствора в толуоле. 3. Получение рацемического (+/-)-2,4 диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина. К гидрохлориду гуанидина (25,4 кг, 266 молей) в метаноле (60 л) добавляют 30 маc.%/маc. раствор метоксида натрия в метаноле (49,2 кг, 273,3 моля). Суспензию нагревают до 552 С. Добавляют раствор 2-(2,3 дихлорфенил)-3-этокси-4-фтор-2-бутеннитрила в толуоле в течение 45 мин и полученную смесь кипятят с обратным холодильником в течение 4 ч, охлаждают, затем выливают в воду (230 л) для быстрого охлаждения. Тврдый осадок промывают 5 порциями метанола (25 л) с получением рацемата в виде тврдого белого вещества(26,3 кг, выход 38% в расчете на 2,3 дихлорфенилацетонитрил). Эталонный прмер 2. Альтернативный синтез рацемического (+/-)-2.4-диамино-5(2,3-дихлорфенил)-6-фторметилпиримидина. 1. Получение 2,3-дихлорбензилового спирта. К 2,3-дихлорбензальдегиду (500 г, 2,85 моля) в метаноле (3,5 л) добавляют щелочной раствор боргидрида натрия (113,5 г, 2,975 моля) в 0,2 н растворе гидроксида натрия в течение 1 ч. Через 2 ч реакционную смесь быстро охлаждают водой (3,7 л) и рН доводят до рН 6, используя ледяную уксусную кислоту (125 мл). Фильтрация приводит к получению 2,3 дихлорбензилового спирта в виде белого тврдого вещества (467 г, выход 92%). 2. Получение 2,3-дихлорфенилацетонитрила. К 2,3-дихлорбензиловому спирту (470,5 г,2,658 моля) в толуоле (1,97 л) добавляют триэтиламин (322,8 г, 3,19 моля) и диметиламинопиридин (16,23 г, 0,13 моля). Добавляют метансульфонилхлорид (365,4 г, 3,19 моля) в течение 1 ч. Через 2 ч толуольный раствор промывают водой. К метансульфонату в толуоле добавляют раствор гидросульфата тетрабутиламмония(180,5 г, 0,53 моля) в воде (641 мл), затем водный раствор цианида калия (259,6 г, 3,987 моля) в воде (712 мл). Двухфазную реакционную смесь перемешивают в течение ночи, разделяют и органическую фазу промывают водой (1069 13 мл). Толуол удаляют под вакуумом и продукт осаждают из петролейного эфира 60/80 (1069 мл), фильтруют и промывают петролейным эфиром 60/80 (356 мл) с получением неочищенного 2,3-дихлорфенилацетонитрила (406 г, выход 83%). 3. Получение 2-(2,3-дихлорфенил)-3,4,4 триэтокси-бут-2-енитрила. К 2,3-дихлорфенилацетонитрилу (100 г,0,54 моля) в диметоксиэтане (750 мл) и этилдиэтоксиацетате (142 г, 0,81 моля) добавляют трет-бутоксид калия за 1 раз. Смесь кипятят с обратным холодильником в течение 4,5 ч, охлаждают, затем добавляют этилйодид (169,8 г,1,08 моля), а затем нагревают при 65 С в течение ночи. Смесь охлаждают и концентрируют с получением остатка, который парционируют между водой (1,5 л) и этилацетатом (1 л). Водную фазу экстрагируют этилацетатом (1 л), а объединнную органическую фазу промывают водой (500 мл), сушат над MgSO4 и выпаривают в вакууме с получением целевого енольного эфира в виде масла, которое используют без дальнейшей очистки. 4. Получение 2,4-диамино-5-(2,3 дихлорфенил)-6-диэтоксиметилпиримидина. К гидрохлориду гуанидина (308,1 г, 3,24 моля) добавляют этоксид натрия в этаноле (1,15 кг, 3,54 моля) и этанол (3 л). К полученной смеси добавляют неочищенный енольный эфир(664 г, 1,62 моля) и дополнительную порцию этанола (1,85 л). После 2 ч при комнатной температуре смесь нагревают до 65 С в течение ночи, концентрируют до остатка, а затем быстро охлаждают в воде (5 л). Осадок отфильтровывают, промывают водой (1 л) и парционируют между тплым этилацетатом (9 л) и водой (1 л). Органическую фазу охлаждают и фильтруют с получением диэтоксиметилпиримидина (207 г). Маточную жидкость концентрируют до остатка,который перекристаллизовывают из изопропилового спирта (2,5 л) с получением ещ 159 г(232 мл) в воде (6,5 л) добавляют диэтоксиметилпиримидин (315 г, 0,88 моля). Смесь нагревают до 90 С в течение 2 ч и охлаждают, затем нейтрализуют с получением 2,4-диамино-5-(2,3 дихлорфенил)-6-формилпиримидина в виде олигомерного производного (218 г, выход 87%). 6. Получение рацемического (+/-)-2,4 диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидина. Способ А К суспензии из формилпиримидина (64 г,0,23 моля) в этаноле (343 мл) добавляют боргидрид натрия (3,4 г, 0,09 моля). По окончании реакции, что определяют тонкослойной хроматографией (TLC), добавляют этилацетат (262 14 мл) и смесь перемешивают в течение ночи,фильтруют и промывают этанолом. Тврдое вещество суспендируют в воде (2 л), фильтруют, промывают водой (1 л) и сушат с получением кремообразного тврдого вещества(43,8 г, 68%). Вторые порции получают концентрированием этанольного фильтрата с получением остатка, который суспендируют в этилацетате (5 объмов) с получением целевого продукта (4,3 г, 6,6%). Общий выход 48,14 г, 75%. Способ В К суспензии из формилпиримидина (52,3 г,0,18 моля) в этаноле (250 мл) добавляют боргидрид натрия (5 г, 0,13 моля). Полученную суспензию перемешивают при комнатной температуре до окончания реакции, что определяют соответствующей аналитической методикой(TLC), затем добавляют воду (750 мл). Суспензию фильтруют, промывают водой (3250 мл) и сушат с получением целевого продукта (40,8 г,выход 78%). 7. Получение рацемического (+/-)-2,4 диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина. Рацемический гидроксиметилпиримидин(1,25 л) до 78 С. За один раз добавляют трифторид диэтиламиносеры (DAST) (291,67 г, 2193 ммоля). Полученную смесь перемешивают при 78 С в течение 1 ч, затем подогревают до 10 С и при этой температуре перемешивают в течение 4,5 ч. Насыщенный раствор бикарбоната натрия (3,5 л) добавляют в течение 90 мин до рН 7. Водную и органическую фазы декантируют с органического осадка, разделяют, и водную фазу экстрагируют этилацетатом (21,5 л). Органические фазы объединяют и промывают рассолом, сушат над Na2SO4 и MgSO4, фильтруют и концентрируют с получением жлтого тврдого вещества, которое объединяют с оранжевым осадком и растирают с метанолом с получением целевого продукта в виде белого тврдого вещества. Последующие порции получают после концентрирования метанольных жидкостей (110 г, 87%). Эталонный пример 3. Разделение с использованием хиральных кислот. 1. Основной способ. 10-4 моля хиральной кислоты смешивают с-4 10 моля рацемического 2,4-диамино-5-(2,3 дихлорфенил)-6-фторметилпиримидина или рацемического 2,4-диамино-5-(2,3 дихлорфенил)-6-гидроксиметилпиримидина. К смеси добавляют 1 мл абсолютного этанола. Смесь подогревают до растворения тврдых веществ, а затем кристаллизуют. Декантирование и промывка приводят к получению солей,которые затем анализируют хиральной HPLC(R-2,2,2-трифтор-9 антрилэтанол). Тестируют следующие хиральные кислоты: 1. (+)-Дибензоил-D-винной кислоты моногидрат. 2. (+)-Ди-п-толуол-D-винная кислота. 3. (-)-Дибензоил-L-винной кислоты моногидрат. 4. (-)-Ди-р-толуол-L-винная кислота. 5. (S)(+)0'-ацетилминдальная кислота. 6. 1R(-)-камфар-10-сульфоновая кислота. 7. R(-)-миндальная кислота. 8. S(+)-миндальная кислота. 9. 1R, 3R, 4R, 5R(-)-хинная кислота. 10. L(-)-яблочная кислота. 11. L(+)-винная кислота. 12. (+)-Винная кислота (правовращающая). 13. 1R, 3S(+)-камфарная кислота. 14. L(-)-винная кислота. 15. (1S)(+)-3-бромкамфар-10-сульфоновой кислоты моногидрат. 16. S(+)-1,1-бинафтил 2,2'-диилгидрофосфат. 17.R(-)-1,1-бинафтил 2,2'-диилгидрофосфат. 18. D(+)-яблочная кислота. 19. (1S)(+)-камфар-10-сульфоновая кислота. 20. 2,3:4,6-Ди-0,0-изопропилиден-2-кето-Lглионовой кислоты моногидрат. 2. 2,4-Диамино-5-(2,3-дихлорфенил)-6 фторметилпиримидин. Кристаллизуют пятнадцать солей из двадцати использованных кислот. Только (-)дибензоил-L-винная кислота и (+)-дибензоил-Dвинная кислота приводят к разделению, причем первая увеличивает соотношениеR(-)энантиомер : S(+)-энантиомер. 3. 2,4-Диамино-5-(2,3-дихлорфенил)-6 гидроксиметилпиримидин. Кристаллизуют одиннадцать солей из двадцати использованных кислот. Из них (+)-ди-птолуоил-D-виннокаменная кислота приводит к повышенному соотношению R(-)-энантиомера :S(+)-энантиомер. 4. Растворители. Растворители, такие как бутанон, ацетон,метанол и этилацетат, также могут быть использованы, чтобы способствовать разделению. Кроме того, растворители, такие как изопропиловый спирт, н-бутанол и смеси воды с либо метанолом, ацетоном, либо этанолом могут быть использованы, чтобы способствовать разделению (+/-)-2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина. Пример 1. Получение R(-)-2,4-диамино 5-(2,3-дихлорфенил)-6-фторметилпиримидина посредством разделения в небольших масштабах. 1. К рацемическому (+/-)-2,4-диамино-5(2,3-дихлорфенил)-6-фторметилпиримидину 16 винную кислоту. Н 2O (1,0490 г). Добавляют абсолютный этанол (27,7 мл), смесь подогревают и полученный раствор оставляют стоять на ночь. Затем маточную жидкость декантируют с образовавшегося белого кристаллического тврдого вещества. Твердое вещество сушат в вакуумной печи при 50 С в течение ночи. Выход полученного кристаллического вещества (0,9534 г) составляет примерно 52%. СоотношениеR(-)-2,4-диамино-5-(2,3 дихлорфенил)-6-фторметилпиримидин ("R(-)энантиомер") : S(+)-2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидин ("S(+)-энантиомер") составляет 81:19. 2. Кристаллический материал (0,8796 г),полученный на начальной стадии разделения 1 растворяют при подогревании в абсолютном этаноле (36 мл). Раствор оставляют охлаждаться на ночь. Маточную жидкость декантируют. Белое кристаллическое вещество сушат в вакуумной печи при 50 С в течение ночи; выход(0,5227 г) из стадии 2 растворяют при подогревании в абсолютном этаноле (25 мл). Раствор оставляют охлаждаться на ночь. Маточную жидкость декантируют. Оставшееся белое кристаллическое вещество промывают этанолом (1 мл) и сушат в вакуумной печи при 50 С в течение ночи; выход (0,397 г) 76%. СоотношениеR(-)-энантиомер : S(+)-энантиомер составляет 99,84:0,2. 4. Кристаллическую соль из стадии 3 затем превращают в основание 2 М раствором NaOH. Так, к соли добавляют дистиллированную воду. Полученную суспензию перемешивают при комнатной температуре. Затем добавляют 2 М раствор NaOH до тех пор, пока не установится рН 12. Полученную суспензию оставляют стоять в течение 1 ч. Затем тврдое вещество отфильтровывают и промывают водой. Влажное тврдое вещество сушат при 50 С в вакууме с получением белого тврдого вещества. Пример 2. Получение R(-)-2,4-диамино 5-(2,3-дихлорфенил)-6-фторметилпиримидина посредством разделения в больших масштабах. 1. К рацемическому (+/-)-2,4-диамино-5(2,3-дихлорфенил)-6-фторметилпиримидину(78,83 г) в колбе добавляют (-)-дибензоил-Lвинную кислоту, Н 2 О (103,27 г), затем добавляют абсолютный этанол (2727 мл). Смесь нагревают с обратным холодильником до полного растворения тврдого вещества. Раствор оставляют на 18 ч охлаждаться при комнатной температуре. Образовавшееся белое тврдое вещество отфильтровывают и сушат в вакууме в течение 3 ч при 50 С. Высушенное тврдое вещество дважды перекристаллизовывают из абсолютного этанола (21500 мл). Полученное белое кристаллическое вещество сушат в вакууме в 17 течение 6 ч при 50 С. Соотношение R(-)энантиомера : S(+)-энантиомер в полученном высушенном кристаллическом веществе (22 г) составляет 99:1. 2. Маточные жидкости от перекристаллизации концентрируют в вакууме, а затем обрабатывают 2 М NaOH (водный раствор), чтобы превратить соль в основание. Так, к соли (98 г) добавляют воду (100 мл), затем 2 М растворNaOH (250 мл) порциями по 50 мл, при этом суспензию энергично перемешивают. Суспензию выдерживают при рН 12 в течение 2 ч. Белое тврдое вещество отфильтровывают и промывают водой (550 мл), пока не установится рН 7. Тврдое вещество сушат в вакууме в течение 4 ч при 50 С с получением свободного основания (39 г). Соотношение R(-)-энантиомера :S(+)-энантиомер в высушенном свободном основании составляет 30:70. 3. Свободное основание, обогащнноеS(+)-энантиомером затем рециклизуют в рацемат. Так, к свободному основанию (39 г) добавляют толуол. Смесь нагревают с обратным холодильником в течение 24 ч, а затем охлаждают до комнатной температуры. Коричневое тврдое вещество отфильтровывают, сушат в вакууме в течение 3 ч при 50 С. Соотношение R(-)энантиомер : S(+)-энантиомер в полученном высушенном веществе (33 г) составляет 50:50. 4. Рацемат затем возвращают на стадию 1,чтобы получить больше R(-)-энантиомера с энантиомерной чистотой 99%. Объединенные соли затем превращают в основание 2 М раствором NaOH. Так, к солям (86,6 г) добавляют дистиллированную воду (250 мл) и суспензию перемешивают при комнатной температуре. Затем добавляют 2 М NaOH (154 мл) порциями по 50 мл и затем две порции по 2 мл, пока не установится рН 12. Полученную суспензию оставляют на один час, а затем твердое вещество отфильтровывают и промывают водой (7100 мл). Влажное тврдое вещество сушат в вакууме при 50 С с получением в этом прогоне темножелтого тврдого вещества (37,9 г). Другие прогоны, однако, дают белое тврдое вещество. СоотношениеS(+)энантиомер в высушенном веществе составляет 99,7:0,3. Химическая чистота = 99,2%. Пример 3. Получение R(-)-2,4-диамино 5-(2,3-дихлорфенил)-6-фторметилпиримидина из (-)-2,4-диамино-5-(2,3-дихлорфенил)-6 гидроксиметилпиримидина. 1. Получение 18 холодильником в 60 мл этанола до полного растворения. Затем реакционную смесь оставляют охлаждаться при комнатной температуре в течение ночи. Образовавшееся тврдое вещество затем отфильтровывают и сушат в вакууме при 50 С. Соотношение(-)2,4-диамино-5-(2,3 дихлорфенил)-6-гидроксиметилпиримидин ("(-)энантиомер") : (+)2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидин ("(+)-энантиомер") в полученном высушенном веществе (3,12 г, как определено ЯМР анализом) составляет 82:18. 2,5 г вышеуказанного вещества растворяют затем в минимальном количестве этанола (60 мл). Этанольный раствор оставляют охлаждаться на ночь и фильтруют с получением 1,74 г хиральной соли (выделение 70%) после сушки в вакууме при 50 С в течение 14 ч. Соотношение(-)-энантиомер : (+)-энантиомер в высушенном веществе составляет 95:5. 1,5 г 95:5, (-):(+), вещества перекристаллизовывают снова из минимального количества этанола (60 мл). Этанольный раствор оставляют стоять в течение ночи, затем фильтруют и полученное тврдое вещество сушат в вакууме при 50 С в течение 5,5 ч. Выход полученного кристаллического вещества (1,19 г) составляет 80%. Соотношение (-)-энантиомер : (+)-энантиомер составляет 98:2 по данным 1H NMR (DCI, метилциклодекстрин в качестве растворителя) 99,8:0,2 по данным хиральной HPLC на колонке Daicel Chirapak AD (2504,6 мм нержавеющая сталь), подвижная фаза гексан:пропан 2 ол 650:350; температура окружающей среды; детектирование УФ при 254 нм; впрыскивается 20 мкл кристаллического вещества в 20 мл этанола; объмная скорость потока 1,0 мл/мин; ослабление 0,05. 2. Получение R(-)-2,4-диамино-5-(2,3 дихлорфенил)-6-фторметилпиримидина.(-)-Энантиомер (0,13 г, 0,00046 моля), полученный на стадии 1, охлаждают в дихлорметане (22 мл) до температуры ниже 50 С. К суспензии добавляют трифторид диэтиламиносеры (DAST) (0,153 г, 0,00115 моля). Через 1 ч реакционную смесь нагревают до 10 С. Через 40 мин полученный оранжевый раствор охлаждают до 50 С, затем добавляют насыщенный раствор бикарбоната натрия (1,6 мл). Вс вместе экстрагируют этилацетатом и объединнные экстракты промывают водой, насыщенным рассолом и сушат над MgSO4. Фильтрат концентрируют с получением не совсем белого продукта, затем растирают с петролейным эфиром 60/80 (80 мг, выход 61%): 99,6% R-(-)энантиомера и 0,4% S-(+)-энантиомера. 19 Пример 4. Получение изэтионата R(-)2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина. Ионообменную смолу AG8 (50 меш) вначале превращают из хлоридной формы в форму изэтионата элюированием водным раствором изэтионата натрия. После промывания водой колонку элюируют разбавленной HCl с получением изэтионовой кислоты в виде водного раствора, которую затем титруют раствором гидроксида натрия. 0,46 М раствор изэтионовой кислоты (11,35 мл, 1,0 экв) добавляют к суспензии R(-)-2,4 диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина (1,5 г, 5,22 ммоль) в воде (100 мл). Раствор затем фильтруют и сушат вымораживанием с получением продукта в виде кремообразного тврдого вещества. Выход 2,1 г (89%)R(-)-2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина в сухом эфире (21 мл). Полученную смесь перемешивают при комнатной температуре в течение 2 ч. Суспензию фильтруют, хорошо промывают сухим эфиром (5 мл),досуха отсасывают и сушат под вакуумом при комнатной температуре. Выход 0,911 г (93%) Т. пл. 245-247 С Пример 6. Получение моногидрохлоридаR(-)-2,4-диамино-5-(2,3-дихлорфенил)-6 фторметилпиримидин (0,70 г, 0,0024 моля) суспендируют в эфирном растворе хлористоводородной кислоты (5,60 мл) и перемешивают при комнатной температуре в течение 2 ч. Суспензию фильтруют, хорошо промывают сухим эфиром (210 мл), досуха отсасывают и сушат под вакуумом при комнатной температуре с получением белого тврдого вещества. Выход 0,773 г (98%) Т. пл. 232-235 С Свойства (-)-2,4-диамино-5-(2,3 дихлорфенил)-6-гидроксиметилпиримидина Физическое состояние: белое тврдое вещество. Температура плавления: 179-181 С Эмпирическая формула: C11H10Cl2N4O Молекулярная масса: 331,20 Оптическое вращение:(t), 1H, 5'): 7,23 (dd, 1H, 6'); 6,08 (синглет (s), 2 Н, 2-NH2): 5,83 (s, 2H, 4-NH2); 4,55 (t, 1H, ОН); 3,85 (t, 2H, СН 2) Свойства R(-)-2,4-диамино-5-(2,3 дихлорфенил)-6-фторметилпиримидина 1. Химические/физико-химические свойства Физическое состояние: белое тврдое вещество Температура плавления: 215-216 С Эмпирическая формула: С 11 Н 9 Сl2FN4 Молекулярная масса: 287,13 Оптическое вращение:(квартет (q), 1H, CH2F); 4,64 (q, 1H, CH2F) 2. Активность против дигидрофолатредуктазы (DHFR) АктивностьS(+)-энантиомера и ламотригина против DHFR печени крысы была исследована с помощью спектрофотометрического анализа. Исследование было модификацией описанного в Biochemical Pharmacology 20, 561-574, 1971. Были получены следующие результаты:S(+)-энантиомер - 33%-ное ингибирование при 100 мкМ Ламотригин: IС 50 = 119,6 мкМ. 3. Ингибирование высвобождения глютамата ВоздействиеR(-)-энантиомера,S(+)энантиомера и ламотригина на вызванное вератрином высвобождение глютамата из срезов мозга крыс было исследовано согласно протоколу, описанному в Epilepsia 27, 490-497, 1986. Были получены следующие результаты:S(+)-энантиомер - 0%-ное ингибирование при 10 мкМ Ламотригин: IC50 = 21 мкМ. 4. Активность против вольтаж-зависимых натриевых каналов Рекомбинантные каналы крысы типа IIА Воздействие R(-)-энантиомера по сравнению с ламотригином на натриевые каналы крысы типа IIА, стабильно выраженные в клетках яичников китайского хомяка, было исследовано с использованием техники внутриклеточной записи. Как R(-)-энантиомер (1-500 мкМ), так и ламотригин продуцировали ингибирование то 21 ков Na+ зависимым от концентрации и напряжения образом. IC50 при двух различных потенциалах покоя (Vh) были следующими:R(-)-энантиомер: 18 мкМ при Vh=-60 мВ 160 мкМ при Vh=-90 мВ Ламотригин: 98 мкМ при Vh=-60 мВ 413 мкМ при Vh=-90 мВ. Нативные каналы а). Культура нейронов стриатума крысы Воздействие R(-)-энантиомера на нативные каналы культуры нейронов стриатума крысы было исследовано с использованием техники внутриклеточной записи. Соединение продуцировало зависимое от концентрации и напряжения ингибирование токов Na+. IC50 при потенциале покоя (Vh) -60 мВ составила около 8 мМ,по сравнению с гораздо меньшей эффективностью при Vh= -90 мВ. Ингибирование, продуцируемое 10-30 мкМ R(-)-энантиомера, практически исчезало при гиперполяризации клеток доVh= -120 мВ. б). Культура нейронов гиппокампа зародыша крысы ВоздействиеR(-)-энантиомера,S(+)энантиомера и ламотригина на натриевые токи цельных клеток культуры нейронов гиппокампа зародыша крысы было исследовано с использованием клемм-электродов. Токи натрия вызывали применением диполяризующей пульсации длительностью 20 мс, снижая таким образом мембранный потенциал до -10 мВ с потенциала покоя -60 мВ. Все три соединения показали зависимое от концентрации снижение пикового тока натрия со следующими IC50:S(+)-энантиомер: 20 мкМ Ламотригин: 16 мкМ 5. Анальгетическая активность Воздействие на развитие острой гипералгезии,вызванной PGE2 (простагландин Е 2) За 1 ч до подошвенной инъекции PGE2 (100 нг) крысам давали перорально R(-)-энантиомер,S(+)-энантиомер и ламотригин. Через три часа Промежуток времениS(+)-энантиомер. В дополнение к этому, соль присоединения изэтионат R(-)-энантиомера (в пересчете на основание) была столь же активна,как и основание R(-)-энантиомера при интрапе 22 после инъекции PGE2 измеряли реакцию на сдавливание лапы. В то же время посредством наблюдения за помещенными на площадку крысами оценивали атаксию. Результаты приведены ниже в табл. 1. Атаксия представлена в виде соотношения между атаксией и анальгетическими ED50, p.o., n=5. Соединение Воздействие на развившуюся острую гипералгезию, вызванную PGE2 Через 2 ч после подошвенной инъекцииPGE2 (100 нг), когда гипералгезия уже развилась, крысам давали внутрь R(-)-энантиомер. Через три часа после инъекции PGE2 измеряли время реакции на сдавливание лапы. Анальгетическая ED50 и 95% границы достоверности составили 3,4 (3,1-3,7) мг/кг. 6.Противосудорожная активность Тест максимального электрошока В этой модели судорог используются ушные электроды-клипсы. Она чувствительна к антиэпилептическим агентам, используемым клинически для контроля клонико-тоническихR(-)-энантиомера,S(+)энантиомера и ламотригина на крыс при интраперитонеальном (i.p.) введении исследовали через различные промежутки времени после инъекции. Значения ED50, приведенные ниже в табл. 2, представляют собой дозы, предотвращающие разгибание задних конечностей у 50% животных. ритонеальном введении (ED50 через 2 ч: 1,8 и 2,5 мг/кг соответственно; р 0,05). В отдельной серии экспериментов было показано, что период полураспада (t1/2) R(-)энантиомера у крыс-самцов составляет 5,4 ч по сравнению с t1/2 S(+)-энантиомера, равным 3,1 ч. б). Различные способы введения Действие R(-)-энантиомера и ламотригина на мышей было исследовано через 1 ч после введения различными способами. Результаты приведены ниже в табл. 3. Соединение В отдельном исследовании действие R(-)энантиомера изэтионата на крыс было исследовано через 1 ч после внутривенного введения препарата. Использовали более сильный (200 мА) по сравнению с другими исследованиями(20 мА) ток. ED50 соли R(-)-энантиомера (в пересчете на основание) составила 1,5 мг/кг (ED50 ламотригина - 2,5 мг/кг). Приведенные результаты демонстрируют,что R(-)-энантиомер является мощным антиконвульсантом с приблизительно одинаковой активностью при различных способах введения, в 2-3 раза более активным, чем ламотригин в тесте максимального электрошока у крыс и мышей.R(-)-энантиомер обладает длительным действием и эффективен при всех способах введения. 7. Синдром раздраженного кишечника Использовали крыс-самцов породы Listar весом 100-150 г. Крыс сенсибилизировали посредством интраперитонеального введения (1 мл на крысу) раствора, содержащего яичный альбумин (10 мкг/мл), коклюшную вакцину (1 мг/мл) и гидроксид алюминия (10 мг/мл). Контрольным животным вводили физиологический раствор. Через семь дней крыс анестезировали изофураном и обнажали наружную косую мышцу. В мышцу имплантировали два нихромовых провода, которые выводили наружу на холке,кожу ушивали и животным позволяли пробудиться. Через шесть дней после этого животным не давали пищи в течение ночи. На следующий день крыс анестезировали и толстую кишку промывали физиологическим раствором. Латексный баллон длиной 4 см, привязанный к канюле Portex соединяли с устройством для раздувания, а нихромовые электроды на холке подсоединяли к головному каскаду. Электрическую активность наружных косых мышц регистрировали с помощью системы сбора данных ("спайк 2"), подсчитывающей число электрических спайков. Были построены кривые реакции сенсибилизированных и контрольных животных на давление (10-100 мм рт. ст.). Баллон раздували до каждого заданного давления в течение 1 мин, после чего следовал отдых в течение 5 мин. 24 Рассчитывали среднее количество спайков при каждом уровне давления у контрольных животных, сенсибилизированных животных и сенсибилизированных животных, получавшихR(-)-энантиомер. R(-)-энантиомер или разбавитель (0,25% метилцеллюлоза) вводили внутрь (5 мл/кг) за 60 мин до начала исследования кривой реакции на давление. Результаты У нормальных крыс растяжение толстой кишки вызывало обусловленное давлением возрастание электрической активности абдоминальной мускулатуры (при давлении, превышающем приблизительно 40 мм рт. ст.). После сенсибилизации крыс яичным альбумином отмечались выраженное возрастание электрической активности данной группы мышц при заданном растяжении (давлении), а также снижение порога подобной активности (приблизительно 20 мм рт. ст.). R(-)-энантиомер в дозе 10 мг/кг внутрь полностью купировал вызванные яичным альбумином изменения. Приведенные результаты свидетельствуют,что при обусловленных гиперчувствительностью состояниях, таких, как синдром раздраженного кишечника, R(-)-энантиомер должен быть эффективен для купирования гиперчувствительности и уменьшения подобным образом боли и нарушений моторики, связанных с синдромом раздраженного кишечника. 8. Модель нейротоксичности, вызванной МРТР Животные и лечение Шестинедельных самцов-мышей C57BL/6(Japan SLC Co., Hamamatu) содержали по десять в клетке в помещении с контролируемой температурой, в условиях чередования света и темноты по 12 ч и свободного доступа к пище и воде. Мышам вводили интраперитонеально R(-)энантиомер и S(+)-энантиомер (30 мг/кг) в оливковом масле, начиная за 12 ч до инъекции МРТР, а затем еще 5 раз каждые 12 ч. Контрольным мышам вводили только оливковое масло. Мышам вводили подкожно MPTPHCl (из рассчета 40 мг свободного основания на кг; Research Biochemicals) в физиологическом растворе. Контрольным мышам вводили только физиологический раствор. Измерение содержания дофамина в стриатуме Содержание дофамина в стриатуме исследовали с помощью HPLC с электрохимическим детектированием (J.C. Garcia: Journal of Chromatography В. 656 (1994) 77-80). Через семь дней после инъекции МРТР мышей (7,9 на группу) забивали, после чего стриатум вырезали, немедленно замораживали и хранили при температуре 80 С до исследования. В день исследования образцы ткани разрушали ультразвуком в 10 объемах (мас./об.) смеси 0,1 М перхлорной кислоты/1,9 мМ гидро 25 сульфита натрия, содержащего 1,6 мкг/мл 3,4 дигидроксибензиламина гидробромида (Sigma) в качестве внутреннего стандарта. После центрифугирования (2800g в течение 10 мин при комнатной температуре) и фильтрации (0,5 мкм; мембранный фильтр Millipore) 10 мкл надосадочной жидкости впрыскивали в колонку InertsilODS3 (4,6250 мм; GL Science, Tokyo). Под 26 вижная фаза состояла из 88% раствора 115 мМNaH2PO4/0,178 мМ Na2EDTA/0,92 мМ 1 октансульфоновой кислоты (рН 2,6) и 12% этанола. Объемная скорость потока равнялась 1,0 мл/мин. Пики определялись с помощью электрохимического детектора Shimazu LECD-6A Исследуемые соединения вводили внутрибрюшинно 6 раз (с 12-часовыми интервалами) в течение дней с -1-го по 2-й. МРТР или физиологический раствор вводили подкожно в день 0. Мышей забивали посредством перелома шеи на 7-й день. Содержание дофамина в стриатуме измеряли с использованием системы HPLC-ECD. Смертность в дни -1-7 Исследования солей R(-)-2,4-диамино-5-(2,3 дихлорфенил)-6-фторметилпиримидина на растворимость и стабильность 1. Эксперименты Соли Синтезировали по 10 мг каждой соли R(-)2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина - ацетат, бензоат, гидрохлорид,тозилат, бензилат, сукцинат, салицилат, тартрат,L-лактат, фумарат, цитрат, малонат, фосфат,нафзилат и мезилат. Плавление Явления, протекающие при нагревании,наблюдали визуально с использованием хотстейдж Меттлера (Mettler hotstage) и микроскопа. Для подтверждения наблюдаемого процесса затем использовали DSC сканирование (DSC дифференциальный сканирующий калориметр) и идентифицировали тип явления. DSC эксперименты проводили с использованием системыPerkin Elmer DSC-7 с TAC7/DX рекордером. Чтобы собрать как можно больше данных, использовали скорость сканирования 10 С/мин и образцы по 1-2 мг. DSC развертки между 40 и 400 С или от и до 50 С за пределами любого явления, наблюдаемого на хотстейдж, проводили на одном или двух образцах. Образцы и эталон (воздух) помещали в 50 мкл алюминиевые поддоны с отверстиями. Растворимость и рН Растворимость определяли посредством добавления лекарственного средства. При комнатной температуре лекарственное средство добавляли к 0,25 мл деионизированной воды. Образец разбавляли до 2 мл для определения рН. Проводили разовое определение. 2. Результаты В табл. 5 указаны параметры плавления солей, как они определены с использованием хотстейдж Меттлера и DSC. Таблица 5 Температура начала по результатам DSC Показатели по Возможная результатам хорошая хотстейдж/DSC стабильность Частичное плавл. 171-200 С Начало дегидратац. 119 С Рацемизация при темп.,Перекрист. 181-215 С Плавление 184 С такой же как для Плавление 232-233 С Перекрист. 197 С основания (200 С) Плавление 218 С Частичное плавл. 48-50 С Начало плавления 160 С Образец нестабилен от Перекрист. 85-131 С Плавление 220 С низкой темп. Плавление 131-160 С Перекрист. 171-200 С Плавление 234-237 С Частичное плавл. 220 С Перекрист. 243 С Рацемизация при темп.,Х Образец становится более высокой, чем для коричневым выше 230 С основания 243 С Частичное плавл. 41 С Плавление не регистрироРацемизации нет Х Полное плавление 98 С валось Малонат Фосфат Нафталиндисульфонат Метансульфонат Плавление не регистрировалось Кристаллы 88-120 С Перекрист. 81 С Частичное плавл. 150-152 С Плавление 141 С Второе плавление 164- Плавление 155 С 170 С Третье плавление 222228 С Блест. Крист. 72-82 С Плавление 60 С Плавление 76 С Плавление 182 С Плавление 170 С Плавление 178 С Дегидратация 134 С Плавление 180 С Плавление 50 С Обширное пл. со 110 С Перекрист. 143 С Перекрист. 191-198 С Дегидратация 175 С Плавление 221-224 С Плавление 194 С Перекрист. 207 С Плавление 215 С Частичное плавл. 115-120 С Плавление 100 С Частичное плавл. 186 С Перекрист. 127 С Плавление 210 С Плавление 173 С Плавление 200 С Частичное плавл. 117-119 С Дегидратация 104 С Плавление 129-132 С Плавление 140-172 С Плавление 72-79 С Плавление 117 С Плавление 57-73 С Плавление 67 С Не плавится ниже 300 С Дегидратация 352 С Плавление 370 С Частичное плавл. 239 С Перекрист. 240 С Полное плавление 249 С Соли, для которых рацемизацию нельзя было наблюдать при нагревании, и соединения,где рацемизация происходила при более высоких температурах, чем у свободных оснований,были отобраны в качестве кандидатов с потенциально хорошими показателями стабильности. Предполагалось, что хорошей стабильностью могут обладать тозилат, бензилат, тартрат,сульфат, цитрат, малонат, фосфат, нафзилат и Соль Ацетат Бензоат НСl Толуолсульфонат Бензолсульфонат Сукцинат Салицилат ТартратL-лактат Сульфат Фумарат Цитрат Малонат Фосфат Нафталиндисульфонат Метансульфонат Рацемизации нет Рацемизации нет Рацемизации нет мезилат. Соли тозилат, бензилат, малонат и фосфат плавились при температуре ниже 100 С. Таким образом, в обращении с ними могут быть трудности, поэтому они менее подходят для получения лекарственных препаратов. В табл. 6 показана растворимость солей в воде при комнатной температуре в пересчете на эквивалентное основание и рН раствора. рН Растворимость эквивалентного основания мг/мл 0,28 0,4 21-31 8-16 10-12 2-12 2-3 5-7 1,1 23 2-5 3-6 8-14 19 Табл. 6 показывает, что растворимость четырх солей (ацетата, бензоата, L-лактата и нафзилата) была менее чем 1 мг/мл. Следовательно, скорее всего они не подходят для перорального и внутривенного применения. Пять солей(HCl, сульфат, фосфат, мезилат и изэтионат) имели растворимость эквивалентного основания свыше или около 25 мг/мл, но только две из них также имели рН в растворе свыше 3. Раствор для инъекций должен иметь рН свыше 3, чтобы исключить побочные действия в месте инъекции. Исходя из этих опытов для препаратов внутривенного введения рекомендуются солиHCl и мезилаты. Примеры фармацевтических препаратов 1. Таблетки Таблетка 1 Кукурузный крахмал Поливинилпирролидон Стеарат магния= содержание на таблеткуR(-)-энантиомер смешивают с лактозой и крахмалом и гранулируют с раствором поливинилпирролидона в воде. Полученные гранулы сушат, смешивают со стеаратом магния и прессуют в таблетки. Таблетка 2 Для приготовления таблеток, содержащихR(-) энантиомер Лактоза Гидроксипропилцеллюлоза Микрокристаллическая целлюлоза Повидон Стеарат магния Очищенная вода Давление прессования Количество на таблетку (мг) 35,0 50,0 170,2 155,2R(-)-энантиомер, лактозу, гидроксипропилцеллюлозу и микрокристаллическую целлюлозу смешивают с образованием сухой порошкообразной смеси. Повидон растворяют в очищенной воде. Раствор повидона добавляют к сухой порошкообразной смеси, содержащейR(-)-энантиомер, чтобы получить влажную массу с консистенцией, годной для гранулирования. Полученную влажную массу пропускают через сито, гранулы сушат и просеивают. Добавляют стеарат магния, смешивают и прессуют. 2. Инъекции Инъекция 1 Соль метансульфоновой кислоты R(-)энантиомера растворяют в стерильной воде для инъекций. Препарат для внутривенного введения II Соль метансульфоновой кислотыR(-)-энантиомера 200 г Стерильный, свободный от пирогена фосфатный буфер (рН 9,0) до 10 мл Соль метансульфоновой кислоты растворяют в большей части фосфатного буфера при 35-40 С, затем доводят до полного объма и фильтруют через стерильный микропористый фильтр в стерильные стеклянные флаконы на 10 мл (тип 1), которые затем закупоривают и закрывают (в расчете на основание). В следующих примерах активным ингредиентом может быть R(-)-энантиомер или его фармацевтически приемлемая соль присоединения кислоты (в расчете на основание). 3. Препараты в форме капсул Препарат в форме капсул I Препарат I может быть получен смешиванием ингредиентов и заполнением тврдых желатиновых капсул, состоящих из 2-х частей,полученной смесью. мг/капсула(в) Крахмал гликолата натрия 25 Капсулы могут быть приготовлены плавлением Макрогеля 4000 ВР, диспергированием активного ингредиента в расплаве и заполнением им тврдых желатиновых капсул, состоящих из 2-х частей. Препарат в форме капсул III (капсулы с регулируемым высвобождением) Активный ингредиент Микрокристаллическая целлюлоза Лактоза В.Р. Этилцеллюлоза 513 Препарат в форме капсул с регулируемым высвобождением может быть приготовлен экструдированием смешанных ингредиентов (а)-(г) с использованием экструдера, придания им сферической формы и сушкой экструдата. Высушенные шарики покрывают этилцеллюлозой (г) в качестве мембраны регулируемого высвобождения и заполняют в тврдые желатиновые капсулы, состоящие из 2-х частей. 4. Препарат в виде сиропа Активный ингредиент 0,2500 г Раствор сорбита 1,5000 г Глицерин 1,0000 г Бензоат натрия 0,0050 г Вкусовая добавка 0,0125 мл Очищенная вода q.s. до 5,0 мл Бензоат натрия растворяют в порции очищенной воды и добавляют раствор сорбита. Добавляют и растворяют активный ингредиент. Полученный раствор смешивают с глицерином и вкусовой добавкой, а затем доводят до требуемого объма очищенной водой. 5. Препарат в форме суппозиториев мг/суппозиторий Активный ингредиент Активный ингредиент используют в виде порошка,где, по меньшей мере, 90% частиц имеют диаметр 63 мкм или менее. Одну пятую часть Witepsol H15 расплавляют в чашке с паровой рубашкой при 45 С максимум. Активный ингредиент просеивают через сито 200 мкм и добавляют в расплавленную основу при перемешивании, используя Silverson, снабженный режущей головкой, до тех пор, пока не получают стабильную однородную дисперсию. Поддерживая температуру микстуры 45 С, в суспензию добавляют оставшийсяWitepsol H15 и перемешивают до получения гомогенной смеси. При непрерывном перемешивании всю суспензию пропускают через сито из нержавеющей стали в 250 мкм и дают ей 32 возможность охладиться до 40 С. При температуре 38-40 С аликвоты смеси по 2,02 г заполняют в подходящие пластиковые формы и дают возможность суппозиториям охладиться до комнатной температуры. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Пиримидин формулы (I) или его соли присоединения к кислотам. 2. Соль по п.1, отличающаяся тем, что она является фармацевтически приемлемой солью присоединения к кислоте. 3. Соль по п.1, отличающаяся тем, что она является сульфатом, фосфатом или изэтионатом. 4. Соль по п.1, отличающаяся тем, что она является гидрохлоридом или метансульфонатом. 5. Способ получения пиримидина формулы (I) по п.1 или его соли присоединения к кислоте, при котором(1) рацемический 2,4-диамино-5-(2,3 дихлорфенил)-6-фторметилпиримидин разделяют с помощью подходящей хиральной кислоты и полученную соль перекристаллизовывают с получением соли, которая состоит практически только из соли R(-)-2,4-диамино-5-(2,3 дихлорфенил)-6-фторметилпиримидина; и(2) при желании перекристаллизованную соль превращают в свободное основание или другую соль присоединения к кислоте. 6. Способ получения пиримидина формулы (I) по п.1 или его соли присоединения к кислоте, при котором(а) рацемический 2,4-диамино-5-(2,3 дихлорфенил)-6-гидроксиметилпиримидин разделяют с помощью соответствующей хиральной кислоты и полученную соль перекристаллизовывают с получением соли, которая состоит практически только из соли (-)-2,4-диамино-5(2,3-дихлорфенил)-6-гидроксиметилпиримидина;(б) при желании перекристаллизованную соль превращают в свободное основание или другую соль;(в) перекристаллизованную соль со стадии(а) или свободное основание или указанную другую соль со стадии (б) фторируют в условиях, при которых не происходит рацемизации (-)2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидина или полученного (-)-2,4 диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина; и(г) при желании полученное фторированное соединение превращают в свободное основание или в его соль присоединения к кислоте. 7. Фармацевтический препарат, содержащий в качестве активного ингредиента пиримидин формулы (I) по п.1 или его фармацевтически приемлемую соль присоединения к кислоте и фармацевтически приемлемый носитель или разбавитель. 8. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли присоединения к кислоте в качестве терапевтического средства. 9. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при получении анальгетика. 10. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при получении противосудорожного средства. 11. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при полу 34 чении лекарственного средства при лечении синдрома раздражнного кишечника. 12. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при получении лекарственного средства для лечения биполярного расстройства. 13. Применение пиримидина формулы (I) по п.1 или его фармацевтически приемлемой соли в качестве активного вещества при получении лекарственного средства для предупреждения или снижения зависимости или толерантности к вызывающему зависимость агенту. 14. Пиримидин формулы (III)
МПК / Метки
МПК: A61K 31/505, C07D 239/48
Метки: производное, способ, получения, анальгетика, фенилпиримидина, активное, применение, оптически, качестве
Код ссылки
<a href="https://eas.patents.su/18-695-opticheski-aktivnoe-proizvodnoe-fenilpirimidina-sposob-ego-polucheniya-i-ego-primenenie-v-kachestve-analgetika.html" rel="bookmark" title="База патентов Евразийского Союза">Оптически активное производное фенилпиримидина, способ его получения и его применение в качестве анальгетика.</a>
Трициклические соединения, способ их получения, способы получения оптически активных или рацемических производных колхицина и тиохолкицина с использованием трициклических соединений и промежуточныепродукты синтеза

Номер патента: 93
Опубликовано: 25.06.1998
Авторы: Шаппер Бернадетт, Пронин Дидье, Миддендорп Мишель, Брион Франсис, Мазюри Алан, Тороманофф Эдмон, Мари Кристиан, Диолез Кристиан
МПК: C07C 43/21, C07D 317/44
Метки: колхицина, производных, тиохолкицина, способ, получения, соединений, использованием, способы, соединения, оптически, трициклические, активных, промежуточныепродукты, трициклических, синтеза, рацемических
Формула / Реферат:
1. Трициклические соединения общей формулы I в которой либо а) оба R1 и R2 представляют собой алкильную группу, a R3 представляет собой атом водорода или группу A-SO2-, либо б) оба R2 и R3 представляют собой атом водорода или алкил, a R1 представляет собой группу A-SO2-, либо в) все три: R1, R2 и R3 представляют собой атом водорода или все три представляют собой алкил, либо г) R1 представляет собой группу А-SO2- или атом водорода, a...
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Мазюри Алан, Бонне Алан, Дельтиль Мишель
МПК: C07H 17/08
Метки: применение, активных, биологически, продуктов, получения, 5-0-дезозаминил-6-0-метилэритронолида, производные, способ
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Производные полипирролкарбоксамидонафталина, способ их получения и их применение

Номер патента: 6
Опубликовано: 30.12.1997
Авторы: Ломбарди Борджиа Андреа, Чомеи Марина, Монджелли Никола, Бьясоли Джиованни, Анджелуччи Францеско, Пезенци Энрико
МПК: C07H 15/252, C07D 207/34, A61K 31/40...
Метки: применение, полипирролкарбоксамидонафталина, способ, получения, производные
Формула / Реферат:
1. Соединение формулы (II): где R является кислотной группой; m - целое число от 1 до 3; n - ноль или целое число от 1 до 3; А представляет собой ферментативно гидролизуемый спейсер; Х является биологически активным соединением; или его фармацевтически приемлемые соли. 2. Соединение формулы (II) по п.1, где R является кислотной группой, выбранной из сульфоновой, карбоксильной и фосфоновой кислотных групп. 3. Соединение формулы (II)...
Трициклические диазепины в качестве антагонистов вазопрессина, способ их получения и способ лечения с использованием трициклических диазепинов

Номер патента: 56
Опубликовано: 30.04.1998
Авторы: Олбрайт Джей Дональд, Гросу Джордж Теодор, Венкатесан Аранапакан Мудумбай
МПК: A61K 31/55, C07D 487/04
Метки: вазопрессина, получения, антагонистов, использованием, лечения, диазепинов, трициклические, способ, диазепины, трициклических, качестве
Формула / Реферат:
1. Соединение, выбранное среди соединений с общей формулой I: где Y является радикалом, выбранным из -(СН2)n-, где n является целым числом, 0 или 1, и А-В является радикалом, выбранным из где m является целым числом от 1 до 2; и радикал: представляет: (1) фенил или замещенный фенил, необязательно замещенный одним или двумя заместителями, выбранными из (C1-С3) низшего алкила, галогена, амина, (C1-С3) низшего алкокси или (C1-С3)...
Твердое синтетическое топливо в форме поленьев, используемое в качестве заменителя древесины, и способ его получения

Номер патента: 41
Опубликовано: 26.02.1998
Авторы: Пеллегрини Леонардо, Гарибальди Пьерпаоло
МПК: C10L 5/40
Метки: способ, древесины, форме, получения, поленьев, синтетическое, используемое, качестве, топливо, твердое, заменителя
Формула / Реферат:
1. Твердое синтетическое топливо в форме поленьев, используемое в качестве заменителя древесины, отличающееся тем, что оно получено способом, заключающимся в том, что смешивают в расплавленном состоянии: а) термопластичную смолу с высокой молекулярной массой, плотностью от 0,8 до 1,0 г/см3 и точкой плавления, равной или меньшей 230°С, в количестве от 70 до 97% по массе, б) смолу с низкой молекулярной массой в количестве от 3 до 30% по массе, ...
Предыдущий патент: Способ очистки эритропоэтина
Следующий патент: Способ получения 2н-1-бензопиранов
Случайный патент: Способы стимуляции и ингибирования роста в-клеток, продуцирования иммуноглобулинов и коррекции нарушений, связанных с baff-лигандом, у животного