Фармацевтические композиции на основе катехоламиновых производных и их применение
Номер патента: 18413
Опубликовано: 30.07.2013
Авторы: Ларсен Дженнифер, Банг-Андерсен Бенни, Мерк Нильс, Викстрем Хокан Вильхельм, Пюшль Аск, Йергенсен Мортен













Формула / Реферат
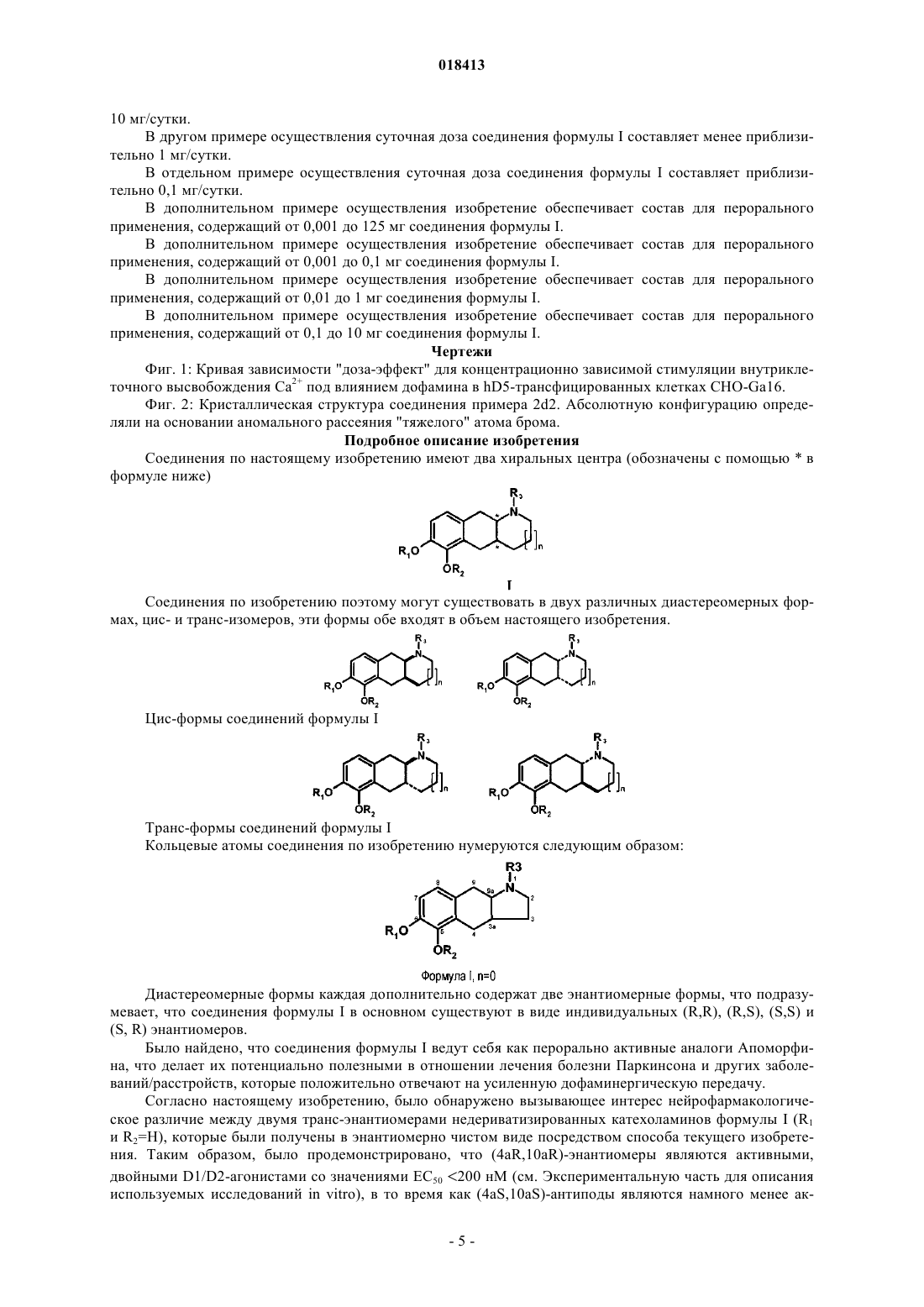
1. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы I и один или несколько фармацевтически приемлемых носителей, разбавителей и вспомогательных веществ, где соединение формулы I имеет следующую структуру:

где n=0,
R1 и R2 независимо выбирают из водорода, C1-6алканоила, фенилацетила или бензоила, R3 выбирают из группы, состоящей из водорода, метила, этила, н-пропила, циклопропила, циклобутила, аллила, пропаргила, гидроксиэтила, 3-фторпропила и 2-фторэтила, и его фармацевтически приемлемые кислотно-аддитивные соли.
2. Фармацевтическая композиция по п.1, где R3 выбирают из группы, состоящей из водорода, метила, этила, н-пропила, аллила и пропаргила.
3. Фармацевтическая композиция по любому из предшествующих пунктов, где R3 выбирают из группы, состоящей из циклопропила, циклобутила и гидроксиэтила.
4. Фармацевтическая композиция по любому из предшествующих пунктов, отличающаяся наличием, по существу, чистого транс-диастереоизомера соединения формулы I.
5. Фармацевтическая композиция по любому из пп.1-4, где по меньшей мере один из R1 и R2 является ацетилом.
6. Фармацевтическая композиция по любому из пп.1-4, где по меньшей мере один из R1 и R2 является пивалоилом.
7. Фармацевтическая композиция по любому из пп.1-4, где по меньшей мере один из R1 и R2 является бензоилом или фенилацетилом.
8. Фармацевтическая композиция по п.1, где соединение выбирают из
транс-1-метил-2,3,3а,4,9,9а-гексагидро-1H-бензо[f]индол-5,6-диола,
цис-1-метил-2,3,3а,4,9,9а-гексагидро-1H-бензо[f]индол-5,6-диола,
транс-1-н-пропил-2,3,3а,4,9,9а-гексагидро-1H-бензо[f]индол-5,6-диола,
цис-1-н-пропил-2,3,3а,4,9,9а-гексагидро-1H-бензо[f]индол-5,6-диола
или их фармацевтически приемлемой кислотно-аддитивной соли.
9. Фармацевтическая композиция по п.4, где R1 и R2 оба являются водородами и R3 выбирают из группы, состоящей из водорода, метила, этила и н-пропила.
10. Фармацевтическая композиция по п.4, где R1 и R2 оба являются C1-6алканоилом и R3 выбирают из группы, состоящей из водорода, метила, этила и н-пропила.
11. Применение фармацевтической композиции по любому из пп.1-10 или ее фармацевтически приемлемой кислотно-аддитивной соли для получения лекарственного средства для лечения нейродегенеративных расстройств у млекопитающего.
12. Применение фармацевтической композиции по п.11 для лечения болезни Паркинсона или болезни Хантингтона у млекопитающего.
13. Фармацевтически приемлемые соли соединения, где соединение выбирают из
транс-1-метил-2,3,3а,4,9,9а-гексагидро-1H-бензо[f]индол-5,6-диола,
цис-1-метил-2,3,3а,4,9,9а-гексагидро-1H-бензо[f]индол-5,6-диола,
транс-1-н-пропил-2,3,3а,4,9,9а-гексагидро-1H-бензо[f]индол-5,6-диола,
цис-1-н-пропил-2,3,3а,4,9,9а-гексагидро-1H-бензо[f]индол-5,6-диола.
Текст
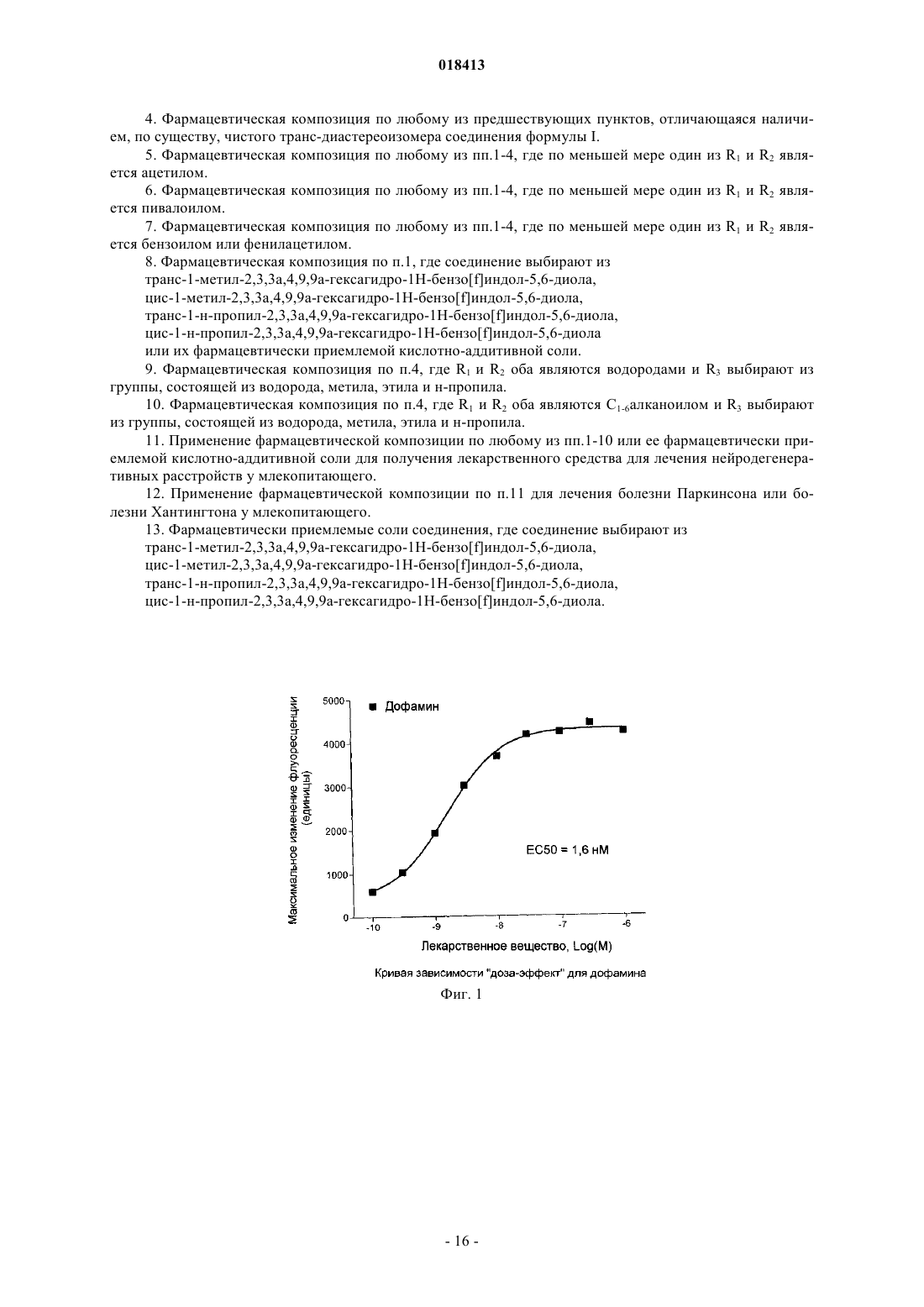
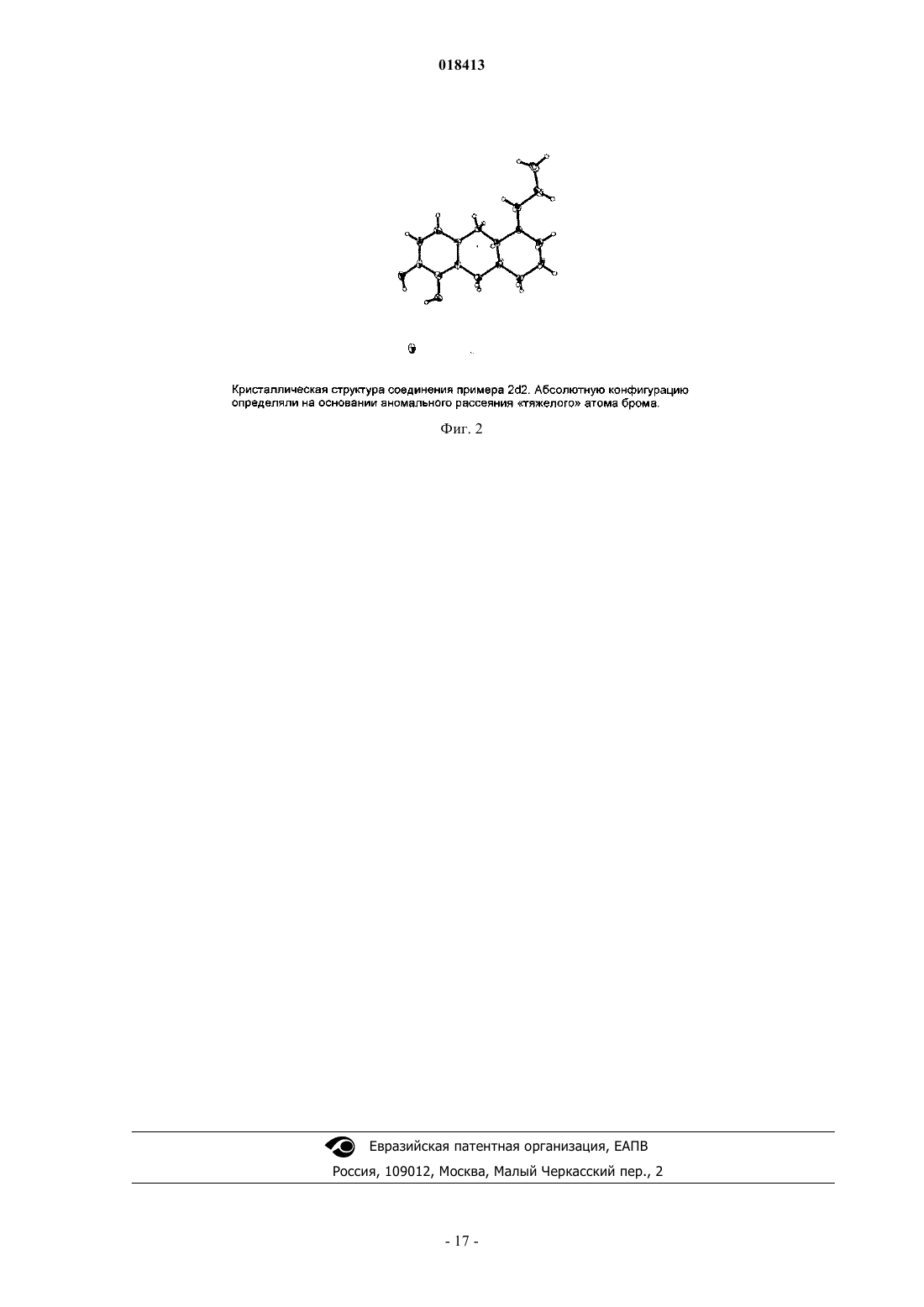
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ОСНОВЕ КАТЕХОЛАМИНОВЫХ ПРОИЗВОДНЫХ И ИХ ПРИМЕНЕНИЕ Настоящее изобретение относится к фармацевтическим композициям, содержащим терапевтически эффективное количество соединения формулы I и один или несколько фармацевтически приемлемых носителей, разбавителей и вспомогательных веществ, и к их применению в терапии. Йергенсен Мортен, Банг-Андерсен Бенни, Пюшль Аск, Мерк Нильс,Ларсен Дженнифер (DK), Викстрем Хокан Вильхельм (SE) Медведев В.Н. (RU) Область техники, к которой относится изобретение Настоящее изобретение относится к фармацевтическим композициям, содержащим катехоламиновые производные, и их применению в терапии. Уровень техники Такие нейродегенеративные заболевания, как болезнь Альцгеймера и Хантингтона приобретают все большее распространение со старением населения. Одним конкретным нейродегенеративным заболеванием, которое обычно развивается в возрасте между 50 и 80 годами, является болезнь Паркинсона (БП). БП представляет собой поражение мозга, которое характеризуется тремором и затрудненностью при ходьбе, движении и координации. Дофамин (ДА) является нейромедиатором, который используется в клетках мозга для передачи импульсов для контроля или регулирования сокращения периферических мышц. Полагают, что БП вызывается прогрессирующей дегенерацией дофаминергических нейронов в компактной части черной субстанции головного мозга. Дегенерация дофаминергических нейронов приводит к снижению количества ДА в головном мозге. Полагают, что данный процесс нарушает функции нервных клеток, так что импульсы не передаются должным образом, приводя к потере мышечного контроля и функции. В настоящее время не существует лечения, останавливающего прогрессирование БП. Лечение обычно направлено на контролирование симптомов БП, в первую очередь, путем замещения ДА на лево 3,4-дигидроксифенилаланин (L-ДОФА), который в результате метаболизма превращается в ДА, или путем введения химических агентов, которые стимулируют ДА-рецепторы. Данные рецепторы подразделяются на два обширных класса, рецепторы D1-типа и D2-типа. Первый класс подразделяется на D1- иD5-рецепторы, в то время как семейство D2-рецепторов состоит из D2-, D3- и D4-рецепторов. Известно, что некоторые гидроксилированные (фенолы или катехолы) фенилэтиламины (сами по себе или являющиеся частью полужесткой/жесткой кольцевой системы) обладают дофаминергической активностью, по меньшей мере, на моделях животных. Однако их клиническое применение ограничено,поскольку они обладают низкой или не обладают биологической доступностью при пероральном применении, вероятнее всего благодаря их высокому метаболизму первичного прохождения. Однако Апоморфин, который принадлежит к этому классу соединений, применяется в клинике при лечении БП, хотя и не доставляется перорально (обычно путем периодического подкожного введения или непрерывной инфузии в течение дня). В настоящее время проводится несколько клинических исследований альтернативной стратегии доставки для лечения Апоморфином при БП, например, лекарственных форм для интраназального и сублингвального введения. Однако данные усилия еще не привели к альтернативному варианту клинического лечения БП. Прямые агонисты ДА-рецепторов способны активировать ДА-ауторецепторы, а также постсинаптические ДА-рецепторы. По-видимому, преобладает влияние ауторецепторной стимуляции, когда, например, Апоморфин вводится в низких дозах, тогда как при более высоких дозах снижение переноса ДА перевешивается за счет увеличения стимуляции постсинаптических рецепторов. Антипсихотическое действие на человека низких доз, например, Апоморфина вероятно обусловлено ауторецепторной стимуляцией (для обсуждения данных клинических исследований см. Tamminga; J. Neurol. Trans., 109(3), 411L-ДОФА является эффективным лекарственным средством при БП (предшественник дофамина) с не очень хорошим ФК-профилем, приводящим к дискинезии и другим ответным изменениям. Селективные D2-агонисты (например Прамиксепол) реже вызывают дискинезии, но теряют эффективность на поздней стадии БП и с течением времени нуждаются в дополнении или замене на L-ДОФА. L-ДОФА и Апоморфин являются в настоящее время наиболее эффективными лекарственными средствами при БП и осуществляют стимулирование как D1-, так и D2-рецепторов. Как упоминалось выше, низкая биодоступность при пероральном применении катехоламинов препятствует их клиническому применению в качестве пероральных лекарственных средств. Родственные фенольные амины обладают аналогичной низкой биодоступностью при пероральном применении, ограничивающей их клиническое применение в качестве активных при пероральном применении лекарственных средств. Однако Ротиготин, который принадлежит к этому классу соединений, был недавно рекомендован в качестве нового лекарственного средства при БП, основанного на трансдермальной доставке. В случае Апоморфина, исследования на животных показали, что трансдермальная доставка или с помощью имплантатов может являться возможными способами введения. Однако, когда исследовали доставку Апоморфина из имплантов у обезьян [F. Bibbiani, L. С. Constantini, R. Patel, T.N. Chase Experimental Neurology 2005, 192, 73], было обнаружено, что в большинстве случаев животные должны были подвергаться лечению иммунодепрессантом Дексаметазоном для предотвращения местно раздражающего действия и других осложнений после операции по имплантации. Трансдермальная доставка Апоморфина также была связана с местным раздражением и окрашиванием кожи. Помимо БП другими заболеваниями, при которых может быть благоприятным усиление дофаминергической передачи, являются болезни старческого возраста, благодаря предотвращению брадикинезии и депрессии и улучшению психических функций, включая различные аспекты познавательной способности, как описано выше. Может иметь положительный эффект у пациентов в депрессии и может применяться при ожирении в качестве анорексигенного средства. Может способствовать улучшению при минимальной мозговой дисфункции (ММД), нарколепсии и вероятно негативных, позитивных, а также когнитивных симптомах шизофрении. Синдром беспокойных ног (СБН) и синдром периодических движений конечностями (СПДК) являются другими показателями, которые подвергаются клиническому лечению с помощью ДА-агонистов. Кроме того, также, по-видимому, улучшается состояние при импотенции и эректильной дисфункции посредством лечения с ДА-агонистами. Таким образом, улучшение сексуальных функций как у женщин, так и мужчин является другим возможным показанием для лечения с помощью ДА-агонистов после эректильной дисфункции (импотенции у мужчин) и сексуальная стимуляция, например, у женщин при менопаузе (стимуляция смазывания влагалища и эрекции клитора), вероятно, может быть достигнута посредством ДА-рецепторной стимуляции. В этой связи стоит отметить,что Апоморфин, когда дается сублингвально, применяется в клинике для улучшения состояния при эректильной дисфункции. Клинические исследования терапии L-ДОФА и D2-агонистом Прамипексолом при болезни Хантингтона показали многообещающие результаты; таким образом, лечение болезни Хантингтона является другим потенциальным применением соединений по настоящему изобретению. ДА вовлечен в регулирование сердечно-сосудистой и почечной системы, и соответственно почечная недостаточность и гипертензия могут считаться другими показаниями для соединений по изобретению. Альтернатива лекарственным формам катехоламинов для неперорального применения включает в себя применение пролекарств. Проблема, связанная с разработкой таких соединений для клинического применения, заключается в сложности, связанной с предсказанием превращения в сам катехоламин у человека. В литературе представлены различные пролекарства катехоламинов, представляющие собой сложные эфиры, такие как покрытые энтеросолюбильной оболочкой сложные эфиры NPA для дуоденальной доставки [см., например, Wikstrom, Dijkstra, Cremers, Ivo; WO 02100377] и D1-подобный агонист Адроголид (Adrogolide, АВТ-431; DAS-431, диацетильное пролекарство А-86929). Адроголид подвергается высокому метаболизму при первичном прохождении через печень у человека после перорального введения дозы и в результате имеет низкую биодоступность при пероральном введении (приблизительно 4%). У пациентов с БП введенный внутривенно (в/в) Адроголид обладает антипаркинсонической эффективностью, сравнимой с таковой для L-ДОФА [Giardina, Williams; CNS Drug Reviews, 7, 305 (2001)]. Альтернативный подход предусматривает "защиту" двух гидроксильных групп у катехола в виде соответствующего метилендиокси (МДО) ацеталя в виде ацеталя, полученного из отличного от формальдегида альдегида, или в виде кеталя, полученного из различных кетонов. О данном принципе получения пролекарства сообщалось для Апорфинов более 20 лет назад [Baldessarini, Ram, Neumeyer; Neuroropharmacology, 21(10), 953 (1982)]. Из этих возможных пролекарств для Апоморфина и родственных соединений только пролекарство, полученное из N-н-пропил-Апоморфина (NPA) и формальдегида, демонстрировало значительную эффективность на животных моделях БП. За следующие 25 лет данные результаты не привели к лекарственному средству для БП, основанному на МДО-защищенных Апоморфинах или родственных соединениях. Несмотря на многолетний интерес в данной области, очевидно, по-прежнему существует неудовлетворенная потребность относительно разработки эффективных, хорошо переносимых и перорально активных лекарственных средств для лечения БП. Смешанный D1-подобный/D2-подобный агонист, дающий длительную дофаминергическую стимуляцию, может удовлетворить такую неудовлетворенную потребность. Сущность изобретения Настоящее изобретение относится к фармацевтическим композициям на основе новых катехоламиновых производных, которые, как обнаружили авторы изобретения, могут являться подходящей заменой современным продаваемым препаратам для терапии нейродегенеративных заболеваний, таких как БП и болезнь Хантингтона, и препаратам для терапии по другим показаниям, обсуждаемым здесь, таким как,например, дискинетические расстройства, когнитивное расстройство и синдром беспокойных ног (СБН),и соединений, которые являются их in vivo метаболизируемыми пролекарствами. Когнитивное расстройство может происходить у нескольких групп пациентов, например пациентов с шизофренией, депрессией или психически больных и пациентов с синдромом дефицита внимания и гиперактивности (СДВГ), болезнью Паркинсона, умеренным когнитивным расстройством (УКР), деменцией, тревожностью, возрастным нарушением памяти, болезнью Альцгеймера или посттравматическим стрессовым расстройством и пациентов, принимающих бензодиазепины или трициклические антидепрессанты, и в ряду нейродегенеративных заболеваний помимо болезни Паркинсона и болезни Альцгеймера. Выражение "когнитивное расстройство" относится к затруднениям в отношении внимания, обучения, памяти и регуляторной функции (соответствующие реакции на внешние стимулы). Они могут включать в себя нарушение внимания, дезорганизованное мышление, замедленное мышление, затрудненность понимания, снижение концентрации, снижение способности решать проблему, снижение памяти, затрудненность в выражении мыслей и/или затрудненность в обобщении мыслей, чувств и поведения и торможении несоответствующих (неадекватных) мыслей, а также внимание и активность, речевое обучение и память, визуальное обучение и память, скорость обработки информации и социальное познание. Целью настоящего изобретения является обеспечение новых соединений, которые представляются собой как D1-подобными, так и D2-подобными агонистами и которые могут применяться при лечении неврологических и психиатрических заболеваний. Дополнительной целью настоящего изобретения является обеспечение новых соединений для перорального введения при лечении БП и других заболеваний или расстройств, которые положительно отвечают на усиленную дофаминергическую передачу. Таким образом, в одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I и один или несколько фармацевтически приемлемых носителей, разбавителей и вспомогательных веществ, где соединение формулы I имеет следующую структуру:R1 и R2 независимо выбирают из водорода, C1-6 алканоила, фенилацетила или бензоила;R3 выбирают из группы, состоящей из водорода, метила, этила, н-пропила, циклопропила, циклобутила, аллила, пропаргила, гидроксиэтила, 3-фторпропила и 2-фторэтила, и его фармацевтически приемлемые кислотно-аддитивные соли.C1-6 алканоильная группа подразумевает неразветвленную или разветвленную алканоильную группу, содержащую от 1 до 6 атомов углерода, примеры которой включают в себя формильную группу, ацетильную группу, пивалоильную группу и т.п. В конкретном примере осуществления изобретение относится к фармацевтической композиции на основе соединения формулы I, где R3 выбирают из группы, состоящей из водорода, метила, этила, нпропила, аллила и пропаргила. В другом конкретном примере осуществления изобретение относится к фармацевтической композиции на основе соединения формулы I, где R3 выбирают из группы, состоящей из циклопропила, циклобутила и гидроксиэтила. В конкретном примере осуществления изобретение относится к фармацевтической композиции на основе соединения формулы I, отличающейся наличием, по существу, чистого транс-диастереоизомера соединения формулы I. В другом примере осуществления изобретение относится к фармацевтической композиции на основе соединения формулы I, где по меньшей мере один из R1 и R2 является ацетилом. В примере осуществления изобретение относится к фармацевтической композиции на основе соединения формулы I, где по меньшей мере один из R1 и R2 является пивалоилом. В другом примере осуществления изобретение относится к фармацевтической композиции на основе соединения формулы I,где по меньшей мере один из R1 и R2 является бензоилом или фенилацетилом. В конкретном примере осуществления изобретение относится к фармацевтической композиции на основе соединения формулы I, где соединение выбирают из транс-1-метил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,цис-1-метил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,транс-1-н-пропил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,цис-1-н-пропил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,или их фармацевтически приемлемой кислотно-аддитивной соли. В конкретном примере осуществления изобретение относится к фармацевтической композиции на основе соединения формулы I, где R1 и R2 оба являются водородами и R3 выбирают из группы, состоящей из водорода, метила, этила и н-пропила. Изобретение дополнительно относится к фармацевтической композиции на основе соединения формулы I, где R1 и R2 оба являются C1-6 алканоилом и R3 выбирают из группы, состоящей из водорода,метила, этила и н-пропила. В дополнительном аспекте изобретение обеспечивает применение фармацевтической композиции на основе соединения формулы I или его фармацевтически приемлемой кислотно-аддитивной соли для получения лекарственного средства для лечения нейродегенеративных расстройств у млекопитающего. В еще одном аспекте изобретение обеспечивает применение фармацевтической композиции на основе соединения формулы I или его фармацевтически приемлемой кислотно-аддитивной соли для получения лекарственного средства для лечения болезни Паркинсона или болезни Хантингтона у млекопитающего. Соединение формулы I, либо в виде свободного основания, либо в виде фармацевтически приемлемой кислотно-аддитивной соли, либо в виде фармацевтической композиции, может вводиться любым приемлемым способом, например перорально, буккально, сублингвально, неперорально или парентерально, и соединение может быть представлено в виде любой приемлемой формы для такого введения,-3 018413 например перорально в форме таблеток, капсул, порошков, сиропов, растворов или дисперсий, неперорально в форме, например, трансдермальных пластырей или парентерально в форме дисперсий или растворов для инъекции. В одном примере осуществления соединение формулы I вводится в форме твердой фармацевтической субстанции, применимой в виде таблетки или капсулы. Соединения формулы I образуют фармацевтически приемлемые кислотно-аддитивные соли с широким кругом органических и неорганических кислот. Такие соли также являются частью данного изобретения. Фармацевтически приемлемые кислотно-аддитивные соли соединения формулы I образуются из фармацевтически приемлемой кислоты, что хорошо известно в данной области техники. Такие соли включают в себя фармацевтически приемлемые соли, перечисленные в Journal of Pharmaceutical Science,66, 2-19 (1977), и известны специалисту. Типичные неорганические кислоты, применяемые для образования таких солей, включают в себя хлорводородную, бромводородную, йодводородную, азотную, серную, фосфорную, гипофосфорную, метафосфорную, пирофосфорную и тому подобную. Соли, полученные из органических кислот, таких как алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановая и гидроксиалкандиовые кислоты, ароматические кислоты,алифатические и ароматические сульфоновые кислоты, могут также применяться. Такие фармацевтически приемлемые соли, таким образом, включают в себя хлорид, бромид, йодид, нитрат, ацетат, фенилацетат, трифторацетат, акрилат, аскорбат, бензоат, хлорбензоат, динитробензоат, гидроксибензоат, метоксибензоат, метилбензоат, о-ацетоксибензоат, изобутират, фенилбутират, -гидроксибутират, бутин 1,4-дикарбоксилат, гексин-1,4-дикарбоксилат, капрат, каприлат, циннамат, цитрат, формиат, фумарат,гликолят, гептаноат, гиппурат, лактат, малат, малеат, гидроксималеат, малонат, манделат, мезилат, никотинат, изоникотинат, оксалат, фталат, терефталат, пропиолат, пропионат, фенилпропионат, салицилат,себакат, сукцинат, суберат, бензолсульфонат, п-бромбензолсульфонат, хлорбензолсульфонат, этилсульфонат, 2-гидроксиэтилсульфонат, метилсульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, нафталин-1,5-сульфонат, п-толуолсульфонат, ксилолсульфонат, тартрат и тому подобное. Способы получения твердых фармацевтических препаратов также хорошо известны в данной области техники. Таблетки могут, таким образом, быть получены путем смешивания активного компонента с традиционными адъювантами, наполнителями и разбавителями и затем прессования смеси в подходящей таблеточной машине. Примеры адъювантов, наполнителей и разбавителей включают в себя микрокристаллическую целлюлозу, кукурузный крахмал, картофельный крахмал, лактозу, маннит, сорбит тальк, стеарат магния, желатин, лактозу, камеди и тому подобное. Может также применяться любой другой адъювант или добавка, такие как красители, ароматизатор, консерванты и так далее, при условии, что они совместимы с активными компонентами. В частности, таблетированная лекарственная форма по изобретению может быть получена прямым прессованием соединения формулы I в смеси с традиционными адъювантами и разбавителями. Альтернативно, для прессования в таблетки может использоваться полученный путем влажного гранулирования или путем гранулирования из расплава гранулят соединения формулы I, необязательно в смеси с традиционными адъювантами или наполнителями. Растворы соединения формулы I для инъекций могут быть получены путем растворения активного компонента и возможных добавок в части растворителя для инъекций, предпочтительно стерильной воде, доведения раствора до желаемого объема, стерилизации раствора и заполнения подходящих ампул или сосудов. Может быть добавлена любая подходящая добавка, традиционно применяемая в области техники, такая как регулирующие тоничность агенты, консерванты, антиоксиданты, повышающие растворимость агенты и так далее. Альтернативно, активный компонент, например, в виде свободного основания может быть растворен в усвояемом или неусвояемом масле, их смеси или подобном для получения депо-формы для внутримышечного введения, способной высвобождать активный компонент в течение длительного периода времени. Фармацевтические составы соединения формулы I, которые используются при трансдермальном применении, такие как трансдермальные пластыри, могут необязательно содержать активаторы проникновения, способствующие переносу активного компонента через кожу. В соответствии с другим аспектом, изобретение относится к фармацевтически приемлемым солям соединения, где соединение выбирают из транс-1-метил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,цис-1-метил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,транс-1-н-пропил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,цис-1-н-пропил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола. В конкретном примере осуществления изобретения млекопитающим является человек. Терапевтически эффективное количество соединения формулы I, рассчитываемое в виде суточной дозы соединения формулы (I) выше в форме свободного основания, целесообразно, составляет между 0,01 и 125 мг/сутки, более целесообразно, между 0,05 и 100 мг/сутки, например, предпочтительно между 0,1 и 50 мг/сутки. В конкретном примере осуществления суточная доза соединения формулы I составляет между 1 и 10 мг/сутки. В другом примере осуществления суточная доза соединения формулы I составляет менее приблизительно 1 мг/сутки. В отдельном примере осуществления суточная доза соединения формулы I составляет приблизительно 0,1 мг/сутки. В дополнительном примере осуществления изобретение обеспечивает состав для перорального применения, содержащий от 0,001 до 125 мг соединения формулы I. В дополнительном примере осуществления изобретение обеспечивает состав для перорального применения, содержащий от 0,001 до 0,1 мг соединения формулы I. В дополнительном примере осуществления изобретение обеспечивает состав для перорального применения, содержащий от 0,01 до 1 мг соединения формулы I. В дополнительном примере осуществления изобретение обеспечивает состав для перорального применения, содержащий от 0,1 до 10 мг соединения формулы I. Чертежи Фиг. 1: Кривая зависимости "доза-эффект" для концентрационно зависимой стимуляции внутриклеточного высвобождения Ca2+ под влиянием дофамина в hD5-трансфицированных клетках CHO-Ga16. Фиг. 2: Кристаллическая структура соединения примера 2d2. Абсолютную конфигурацию определяли на основании аномального рассеяния "тяжелого" атома брома. Подробное описание изобретения Соединения по настоящему изобретению имеют два хиральных центра (обозначены с помощьюв формуле ниже) Соединения по изобретению поэтому могут существовать в двух различных диастереомерных формах, цис- и транс-изомеров, эти формы обе входят в объем настоящего изобретения. Транс-формы соединений формулы I Кольцевые атомы соединения по изобретению нумеруются следующим образом: Диастереомерные формы каждая дополнительно содержат две энантиомерные формы, что подразумевает, что соединения формулы I в основном существуют в виде индивидуальных (R,R), (R,S), (S,S) и(S, R) энантиомеров. Было найдено, что соединения формулы I ведут себя как перорально активные аналоги Апоморфина, что делает их потенциально полезными в отношении лечения болезни Паркинсона и других заболеваний/расстройств, которые положительно отвечают на усиленную дофаминергическую передачу. Согласно настоящему изобретению, было обнаружено вызывающее интерес нейрофармакологическое различие между двумя транс-энантиомерами недериватизированных катехоламинов формулы I (R1 и R2=H), которые были получены в энантиомерно чистом виде посредством способа текущего изобретения. Таким образом, было продемонстрировано, что (4aR,10aR)-энантиомеры являются активными,двойными D1/D2-агонистами со значениями ЕС 50 200 нМ (см. Экспериментальную часть для описания используемых исследований in vitro), в то время как (4aS,10aS)-антиподы являются намного менее ак-5 018413 тивными D1-агонистами и демонстрируют единственно умеренно сильный D2-агонизм. Некоторые из (4aR,10aR)-энантиомеров соединений формулы I кроме того были протестированы наD5-аффинность и доказано, что являются очень активными D5-агонистами со значениями ЕС 50 10 нМ. Рацемические соединения формулы I, для которых n=1, R1 и R2=водородом и R3=водородом, метилом, этилом и н-пропилом, были ранее описаны [см., например, Cannon, Lee, Beres, Goldman; J. Heterocycl. Chem., 17, 1633 (1980)], и рассмотрена их дофаминергическая активность [см., например, Bradbury,Costall, Naylor; Neuropharmacology 23(9), 1025 (1984); Bradbury, Cannon, Costall, Naylor; Eur. J. Pharmacol. 105(1-2), 33 (1984)]. Сообщалось, что рацемическое соединение формулы I, для которого n=1, R1 иR2=водород и R3=этил, стимулирует как D1-, так и D2-рецепторы [Itoh, Goldman, Kebabain; Eur. J. Pharmacol., 108 (1), 99 (1985)]. Однако ни в одном из данных документов предшествующего уровня техники не рассматривается энантиоселективность соединений формулы I или разные селективности, полученные в случае in vitro в сравнении с in vivo. Как упомянуто ранее, соединение Апоморфин в настоящее время применяется в клинике при лечении БП. Апоморфин является смешанным D1-подобным/D2-подобным агонистом: Когда соединения по изобретению тестировали in vitro и in vivo на их действие в отношение D1 иD2 рецепторов, их фармакологические профили очень отличались от такового для Апоморфина (более подробно см. Экспериментальную часть). Было показано, что коэффициент селективности D1/D2 для модифицированных катехоламинов формулы I (R1 и R2=H) существенно изменяется при сравнении результатов измерений in vitro с in vivo. В случае исследований in vitro данные соединения являются значительно более активными в отношенииD2-рецепторов, чем в отношении D1-рецепторов (как правило, с коэффициентом 100). Однако коэффициент in vivo изменяется в сторону 2-10-кратной селективности. Таким образом, очевидно, что экстраполяция от данных in vitro до состояния in vivo не может быть проведена для соединения по изобретению. Как упомянуто ранее, доступная в настоящий момент информация поддерживает гипотезу, что D1 подобный агонист (будь то селективный в отношении каждого подтипа или смешанный D1/D5-агонист) может иметь значительное практическое применение в лечении когнитивного расстройства при, например, психозе, БП и болезни Альцгеймера (БА), и болезни Хантингтона. Это может быть так в случаеD1/D2-агонистов двойного действия, таких как соединения формулы I. С учетом сложившейся ситуации, в частности, в случае фармацевтического применения, подразумевается, что когда определяется соединение формулы (I), которое является по существу энантиомерно или диастереомерно чистым, то соединение является относительно стереохимически чистым, предпочтительно избыток энантиомера или диастереомера составляет по меньшей мере 60%, по меньшей мере 70%, и более предпочтительно по меньшей мере 80% (избыток энантомера 80% подразумевает, что соотношение, например, (4aR,10aR) к (4aS,10aS) составляет 90:10 в данной смеси), по меньшей мере 90%, по меньшей мере 96% или предпочтительно по меньшей мере 98%. Экспериментальная часть Общие методы. Аналитические ЖХ/МС-данные были получены на приборе РЕ Sciex API 150EX, снабженном фотоионизацией при атмосферном давлении (APPI) и ЖХ-системой Shimadzu LC-8A/SLC-10A. Чистоту определяли путем интегрирования кривых УФ (254 нм) и ELSD. МС-приборы были от PESciex (API),снабженные APPI-источником и работающие в режиме положительных ионов. Время удерживания в УФ-кривых (RT) выражали в мин. Раствор А получали из 0,05% ТФУ в воде, а раствор В получали из 0,035% ТФУ и 5% воды в ацетонитриле. Были использованы несколько различных методик: Методика 14: API 150EX и ЖХ-система Shimadzu LC8/SLC-10A. Колонка: С-18 4,630 мм, 3,5 мкм(Symmetry, Waters). Температура колонки: 60C. Градиент: обращенная фаза с образованием ионных пар. Скорость потока: 3,3 мл/мин. Объем введенной пробы: 10 мкл (1 мкл инъецируемый непосредственно в колонку). Градиент: 10% В в А до 100% В в течение 2,4 мин, затем 10% В в А в течение 0,4 мин. Общая продолжительность: 2,8 мин. Методика 314: API 150EX и ЖХ-система Shimadzu LC8/SLC-10A. Колонка: С-18 4,630 мм, 3,5 мкм(Symmetry, Waters). Температура колонки: комн. темп. Скорость потока 2 мл/мин. Объем введенной пробы 10 мкл. Градиент: 10% В в А в течение 4 мин, затем 100% В в течение 0,1 мин, затем 10% В в А в течение 0,9 мин. Общая продолжительность: 5,0 мин. Методика 23 SUN: API 150EX и ЖХ-система Shimadzu LC8/SLC-10 А. Колонка: С-18 4,630 мм, 3,5 мкм (Sunfire, Waters). Температура колонки: 40C. Градиент: обращенная фаза с образованием ионных пар. Скорость потока: 3,3 мл/мин. Объем введенной пробы: 15 мкл. Градиент: 10% В в А до 100% В в течение 2,4 мин, затем 10% В в А в течение 0,4 мин. Общая продолжительность: 2,8 мин. Препаративную очистку методом ЖХ/МС осуществляли на том же самом приборе с химической ионизацией при атмосферном давлении. Колонка: 5020 мм YMC ODS-A с размером частиц 5 мкм. Метод: элюирование в линейном градиенте с 80% А до 100% В в течение 7 мин и со скоростью потока 22,7 мл/мин. Сбор фракций осуществляли с помощью МС-детекции разделения потока. Реакции гидрирования осуществляли, используя либо стандартный шейкер Парра (Parr), либо прибор Endavour от Argonaut. Во всех случаях использовали низкое давление (давление водорода 1-5 бар(105-5105 Па). Термин "хроматография на силикагеле (EtOAc/гептан)" имеет следующее значение: подвергаемое очистке соединение обычно растворяли в небольшом количестве ДХМ и вносили в колонку, предварительно заполненную силикагелем, и элюировали смесью EtOAc и гептана, либо в изократическом режиме, либо с помощью градиента, такого как 0-100% EtOAc в гептане. Одним примером используемой колонки, заполняемой силикагелем, является "ISOLUTE SPE COLUMNS" [например, 20 г FLASH Si 70 мл от International sorbent technology]. Альтернативно, осуществляли классическую ручную хроматографическую очистку, используя силикагель [например, Machery-Nagel 60M; 0,04-0,063 мм, 230-400 меш], с осуществлением идентификации соединения путем анализа стандартным методом ТСХ на алюминиевых пластинах, предварительно покрытых силикагелем [например Merck 60 F254]. Соединения визуализировали путем освещения с использованием УФ-лампы (254 нм) или путем обжигания после погружения в раствор молибдата аммония (6,25 г) и сульфата церия (IV) (2,5 г) в 10% водной серной кислоте (250 мл). Микроволновое ускорение реакций осуществляли в герметично закрытых реакционных сосудах для микроволнового облучения. Эксперименты осуществляли на синтезаторе Smith от Personal Chemistry. Термин "лиофилизовали" относится к сублимационной сушке вещества с использованием прибораChrist Aplha 2-4 LSC от WWR International. Термины "сушили (Na2SO4)" и "сушили (MgSO4)" относится к удалению воды из органического слоя путем добавления сухого Na2SO4 или MgSO4, соответственно, с последующим перемешиванием в течение подходящего количества времени для обеспечения эффективного процесса сушки. Затем осадок удаляется путем фильтрования, и фильтрат обычно концентрируется в вакууме (см. ниже). Термин "концентрировали в вакууме" имеет следующее значение: летучие компоненты удаляются из смеси с помощью стандартного роторного испарителя при пониженном давлении. Термин "сушили в вакууме при 40C" относится к применению стандартного вакуум-сушильного шкафа при 40C, связанного с масляным насосом. Термин "сушили в вакууме" относится к процессу сушки, в котором вещество,которое подвергается сушке, помещают в сосуд, связанный непосредственно с масляным насосом, на период времени, достаточный для удаления летучих компонентов. Определения кристаллических структур рентгеноструктурным анализом осуществляли следующим образом. Кристалл соединений охлаждали до 120 К с применением системы охлаждения азотом Cryostream. Данные собирали на дифрактометре SMART Platform Siemens с двумерным чувствительным ПЗС-детектором. Структуры решали прямыми методами и уточняли методом наименьших квадратов плотно заполненной матрицы по F2 всех данных. Атомы водорода в структурах не могут быть обнаружены в разностных картах электронной плотности. Неводородные атомы уточняли в анизотропном приближении. Все атомы водорода помещали в рассчитанные позиции с применением модели "наездника" сO-H=0,84, C-H=0,99-1,00, N-H=0,92-0,93. Для всех атомов водорода фиксировали тепловые параметры зывая на то, что абсолютные структуры являются корректными. Используемыми для сбора данных, обработки данных и поглощения программами были SMART, SAINT и SADABS [см. также "SMART и"SHELXTL, Structure Determination Programs", Версия 6.12, Bruker Analytical X-Ray Instruments Inc., Мэдисон, США (2001)] использовали для решения структур и изображения молекул. Общие методы синтеза для структуры Маркуша 1 а Исходя из промежуточного соединения I, чей синтез описан здесь, конденсация с первичным амином R3NH2 дает соединение Маркуша 1 а-1 согласно условиям здесь для синтеза соединения 25 из промежуточного соединения I. Восстановление соединения Маркуша 1 а-1 с помощью ЛАГ дает соединение Маркуша 1 а-2, например, согласно условиям здесь для синтеза соединений 13 и 14. После разделения цис/транс-смеси, любой диастереомер может быть обработан 48% HBr или родственным реагентом для расщепления метоксигрупп, чтобы давать соединение Маркуша 1 а, например, согласно условиям, описанным здесь для синтеза примера 1a1. Далее взаимодействие соединения Маркуша 1 а с CH2ClBr или родственным реагентом в присутствии основания для получения соединения Маркуша 1a-МДО, например, согласно условиям, описанным здесь для синтеза примера 3b1. Полученное соединение Маркуша 1a-МДО может быть обратно превращено в соединение Маркуша 1 а путем обработки BCl3/(н-бутил)4NI или родственным реагентом. Соединение Маркуша 1 а может быть превращено в соединение Маркуша 1 а-ди(сложный эфир) путем обработки соответствующими хлорангидридами кислот в ТФУ для получения соединения Маркуша 1 а-ди(сложный эфир), например, как описано здесь для синтеза примера 4 а 1. Данное вещество может быть подвергнуто гидролизу до соединения Маркуша 1 а. Следующий раздел общих методов синтеза для получения промежуточных соединений будет представлен следующими конкретными примерами. Общая методика получения бензо[f]индольных катехоламинов Промежуточное соединение I, чей синтез описан здесь, вводят во взаимодействие с первичным амином и полученный енаминлактам затем восстанавливали аланом и затем боргидридом натрия. Это дает смесь цис/транс защищенных бензо[f]индольных катехоламинов. Данные диастереомеры разделяют,например, хроматографией на силикагеле [для примера близкого синтеза см.: Lin, Haadsma-Svensson,Phillips, Lahti, McCall, Piercey, Schreur, von Voigtlander, Smith, Chidester; J. Med. Chem., 36(8), 1069(1993)]. Защищенный катехоламин подвергали удалению защиты, например, путем обработки 48% HBr или BBr3. Получение промежуточного соединения I Раствор метилового эфира рацемической 5,6-диметокси-2-оксо-1,2,3,4-тетрагидронафталин-1 карбоновой кислоты (6,60 г) [соединение 1; полученное как описано у Taber, Neubert, Rheingold; J. Am.Chem. Soc, 124(42), 12416 (2002)] в ТГФ (25 мл) добавляли по каплям к раствору ЛДА (27 мл, 2 М в ТГФ/гептане/этилбензоле) в ТГФ (125 мл) при 0C. Раствор перемешивали при 0C в течение 1,5 ч. Добавляли аллилбромид (3,44 мл) и раствор перемешивали при комнатной температуре в течение ночи. Добавляли Et2O (300 мл) и 1 М HCl (300 мл) и слои разделяли. Органический слой промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали в вакууме. Оставшееся масло растворяли в ДМСО (25 мл) и добавляли воду (2,5 мл) и LiCl (1 г). Реакционную смесь перемешивали при 150C в течение 0,5 ч и затем охлаждали до комнатной температуры. Добавляли EtOAc (250 мл) и воду (250 мл) и слои разделяли. Водный слой экстрагировали EtOAc (125 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали в вакууме. Сырой продукт очищали хроматографией на силикагеле (EtOAc/гептан) для получения 2,55 г соединения 2 в виде белого твердого вещества. Рацемический 3'-аллил-5',6'-диметокси-3',4'-дигидро-1'Н-спиро 1,3]диоксолан-2,2'-нафталин] К перемешиваемому раствору соединения 2 (2,55 г) в ДХМ (45 мл) добавляли СН(ОСН 3)3 (4,53 мл),этиленгликоль (5,68 мл) и ПТСК (20 мг). Раствор перемешивали при комнатной температуре в течение 4,5 ч и затем гасили, добавляя насыщенный водный раствор NaHCO3 (45 мл). Органический слой промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали в вакууме. Сырой продукт очищали хроматографией на силикагеле (EtOAc/гептан) для получения 2,52 г соединения 3 в виде масла. Рацемический 3-аллил-5,6-диметокси-3,4-дигидро-1H-нафталин-2-он (промежуточное соединение I) течение дополнительных 5 мин. Добавляли раствор соединения 3 (14,8 г) в трет-бутиловом спирте (500 мл). Раствор перемешивали 3 ч и затем охлаждали на бане лед/вода. Добавляли по каплям гидросульфит натрия (38-40% водный раствор) в течение 0,5 ч. Добавляли ДХМ (1 л) и слои разделяли. Водный слой экстрагировали еще ДХМ (0,4 л) и объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали в вакууме с получением 11,3 г темного масла. Данное вещество растворяли в ацетонитриле (225 мл) и добавляли раствор AcCl (37 мл) в МеОН (190 мл). Раствор перемешивали 5 мин при комнатной температуре и затем держали при 4C в течение ночи, и затем перемешивали 2 ч при комнатной температуре. Добавляли воду (45 мл) и раствор перемешивали в течение 3 ч, после чего его концентрировали в вакууме. Сырой остаток очищали хроматографией на силикагеле К перемешиваемому раствору промежуточного соединения I (830 мг) в толуоле (7 мл) в реакционном сосуде для микроволнового облучения добавляли раствор метиламина (0,75 мл, 8 М в EtOH) и добавляли АсОН (0,34 мл). Реактор герметично закрывали и смесь нагревали при 120C в течение 15 мин микроволновым облучением. Раствор концентрировали в вакууме и остаток сушили в вакууме. Сырой продукт очищали хроматографией на силикагеле (EtOAc/гептан). Выход: 210 мг соединения 12 в виде масла. Рацемические транс- и цис-изомеры 5,6-диметокси-1-метил-2,3,3a,4,9,9 а-гексагидро-1H-бензо[f]индола К перемешиваемому раствору ЛАГ (3,9 мл, 1 М в ТГФ) при 0C добавляли AlCl3 (174 мг). Смеси позволяли нагреться до комнатной температуры и затем охлаждали до 0C снова. К данной смеси добавляли соединение 12 (200 мг), растворенное в ТГФ (4 мл), и смесь перемешивали при комнатной температуре 1 ч. Смесь охлаждали до 0C и затем гасили, добавляя влажный Na2SO4. Неорганические соли отфильтровывали и фильтрат концентрировали в вакууме. Остаток растворяли в 99% EtOH и добавлялиNaBH4 (146 мг), и раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили добавлением 2 М водным HCl (3 мл). Большую часть летучих компонентов удаляли путем концентрирования в вакууме и остаток экстрагировали Et2O. Органический слой экстрагировали более разбавленной HCl. Объединенные слои разбавленной HCl подщелачивали 9 М NaOH и затем экстрагировали Et2O. Органический слой промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали в вакууме. Сырую смесь очищали хроматографией на силикагеле (MeOH/EtOAc). Выход: 4 мг соединения 13 в виде масла (медленно элюируемый изомер) и 32 мг соединения 14 в виде масла (быстро элюируемый изомер). Получали из промежуточного соединения I (1,39 г) по методике, описанной для соединения 12, используя н-пропиламин вместо метиламина. Выход соединения 15: 0,69 г в виде твердого вещества. Рацемические транс- и цис-изомеры 5,6-диметокси-1-н-пропил-2,3,3a,4,9,9 а-гексагидро-1Hбензо[f]индола (соединения 16 и 17) Соединения 16 и 17 получали аналогичным методом как соединения 13 и 14 из соединения 15 (400 мг) вместо соединения 12. Неочищенную смесь продукта очищали хроматографией на силикагеле(MeOH/EtOAc). Выход: 55 мг соединения 16 в виде масла (медленно элюируемый изомер) и 40 мг соединения 17 в виде масла (быстро элюируемый изомер). Получение соединений по изобретению Раскрытое здесь изобретение дополнительно иллюстрируется следующими неограничивающими примерами: 1b1. Рацемический гранс-1-метил-2,3,3 а,4,9,9 а-гексагидро-1 Н-бензо[f]индол-5,6-диола трифторацетат Соединение 13 (4 мг) суспендировали в 48% HBr (1 мл) и нагревали до 155C в течение 0,5 ч в герметично закрытом реакционном сосуде для микроволнового облучения с помощью микроволнового излучения. Сырую смесь концентрировали в вакууме, и остаток очищали препаративной ЖХ/МС. Выход: 6 мг в виде белого твердого вещества. ЖХ/МС (методика 25): RT 0,52 мин, ELSD 94,1%, УФ 82,9%, МН+: 220,3. 1b2. Рацемический цис-1-метил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола трифторацетат Соединение 14 (32 мг) суспендировали в 48% HBr (1,5 мл) и нагревали до 155C в течение 0,5 ч в герметично закрытом реакционном сосуде для микроволнового облучения с помощью микроволнового облучения. Неочищенную смесь концентрировали в вакууме и остаток очищали препаративной ЖХ/МС. Выход: 23 мг в виде белого твердого вещества. ЖХ/МС (методика 25): RT 0,52 мин, ELSD 93,5%, УФ 92,7%, МН+: 220,2. Соединение 16 (55 мг) суспендировали в 48% HBr(2 мл) и нагревали до 155C в течение 0,5 ч в герметично закрытом реакционном сосуде для микроволнового облучения с помощью микроволнового облучения. Неочищенную смесь концентрировали в вакууме и остаток очищали препаративной ЖХ/МС. Выход: 30 мг в виде белого твердого вещества. ЖХ/МС (методика 25): RT 0,69 мин, ELSD 99,7%, УФ 97,9%, МН+: 248,2. 1d2. Рацемический цис-1-н-пропил-2,3,3 а,4,9,9 а-гексагидро-1 Н-бензо[f]индол-5,6-диола трифторацетат Соединение 17 (40 мг) суспендировали в 48% HBr (2 мл) и нагревали до 155C в течение 0,5 ч в герметично закрытом реакционном сосуде для микроволнового облучения с помощью микроволнового облучения. Неочищенную смесь концентрировали в вакууме и остаток очищали препаративной ЖХ/МС. Выход: 8 мг в виде белого твердого вещества. ЖХ/МС (методика 25): RT 0,69 мин, ELSD 99,1%, УФ 97,8%, МН+: 248,3. Аббревиатуры и список использованных химических реактивов Были использованы следующие аббревиатуры. В данном параграфе также перечислены использованные химические реактивы вместе с их коммерческим источником (не включено для стандартных растворителей).AcCl=ацетилхлорид (например, Aldrich 23,957-7), ACh=ацетилхолин, АсОН=уксусная кислота,БА=болезнь Альцгеймера, АРМВ (ADME)=абсорбция-распределение-метаболизм-выведение, Аллилбромид (например, Fluka 05870), AlCl3=алюминия хлорид (например, Aldrich 29,471-3), D=удельное оптическое вращение, BBr3=трибромид бора (использованный в виде раствора в ДХМ; Aldrich 17,893-4),Вос 2O=ангидрид Вос/ди-трет-бутилдикарбонат (например, Aldrich 19,913-3), насыщенный раствор соли=насыщенный водный раствор хлорида натрия, БСА=бычий сывороточный альбумин, (вторбутил)литий (использованный в виде раствора в циклогексане; например, Aldrich 19,559-6),цАМФ=циклический аденозинмонофосфат, целит=вспомогательный материал для фильтрования,CH2BrCl=бромхлорметан (Aldrich 13,526-7), CH3I=метилйодид/йодметан (например, Aldrich 28,956-6),клетки СНО=клетки яичника китайского хомячка, ClAcCl=хлорацетилхлорид (например, Aldrich 10,4493), Cs2CO3=карбонат цезия (Aldrich 441902), CuI=йодид меди(I) (Aldrich 215554), циклобутанон (например, Aldrich C9,600-1), циклопропилметилбромид/(бромметил)циклопропан (Aldrich 24,240-3),ДА=дофамин, D1=рецептор D1 дофамина, D2=рецептор D2 дофамина, D3=рецептор D3 дофамина,D4=рецептор D4 дофамина, D5=рецептор D5 дофамина, ДХМ=дихлорметан/метиленхлорид, 1,6-дибром 2-нафтол (например, Aldrich D4, 180-5), ДМФА=диметилформамид, ДМСО=диметилсульфоксид, LДОФА=лево-3,4-дигидроксифенилаланин, DOPAC=3,4-дигидроксифенилуксусная кислота (метаболит ДА), EC50=концентрация, требующаяся для индуцирования ответа, находящегося посередине между фоновым уровнем и максимальным ответом, для соединения, о котором идет речь, ELSD=детекция светорассеяния испаренного образца, Et3N=триэтиламин, Et2NH=диэтиламин, EtOAc=этилацетат, этил-2 хлорникотинат (например, ABCR AV20359), 99% EtOH=абсолютный этанол, этилмагнийбромид (используемый в виде 3 М раствора в Et2O; Aldrich 18,987-1), Et2O=диэтиловый эфир, [(1 этоксициклопропил)окси]триметилсилан (Aldrich 332739), этиленгликоль=1,2-этандиол, 35% Н 2 О 2=35% водный раствор перекиси водорода (например, Aldrich 34,988-7). FLIPR=флуоресцентный планшетный спектрофотометр, ФТС=фетальная телячья сыворотка, ч=часы, 48% HBr=48% водный раствор бромводорода, 18%/37% HCl=18%/37% водный раствор хлорводорода, 1 М HCl/2 М HCl=1 М/2 М водный раствор хлорводорода (если конкретно не указано иное в виде 2 М раствора в Et2O, который является коммерчески доступным, например Aldrich 45,518-0). ГМФА=гексаметилфосфортриамид, ГВК=гомованилиновая кислота (метаболит ДА), i=изо, ИБМК=3-изобутил-1-метилксантин, в.д.=внутренний диаметр, 1- 12018413(например, Aldrich 31,144-8), 1 М/9 М NaOH=1M/9M водный раствор гидроксида натрия, NaOMe=метилат натрия (используемый в виде приблизительно 5 М раствора в метаноле; например Aldrich 15,625-6).(0,02 М натрий фосфатный буфер с 0,15 М хлоридом натрия, pH доведено до 7,4). БП=болезнь Паркинсона. ПФК=предфронтальная кора. Pd/C=палладий на угле (например, Aldrich 20,569-9),Pd(OAc)2=ацетат палладия(II) (Alfa Aesar 010516), пиперониловый спирт (например, Aldrich P4,940-6). ФК=фармакокинетика, СПДК=синдром периодических движений конечностями, пропаргилхлорид (например, Aldrich 14,399-5), пропионовый альдегид (например, Aldrich 58,812-4), ПТСК=паратолуолсульфоновой кислоты гидрат(например,Aldrich 40,288-5),PivCl=пивалоилхлорид/триметилацетилхлорид (например, Aldrich Т 7,260-5), СБН=синдром беспокойных ног,к.т.=комнатная температура, RT=время удерживания, втор=вторичный, насыщ. NaHCO3 = насыщенный водный раствор гидрокарбоната натрия, насыщ. NH4Cl=насыщенный водный раствор хлорида аммония,ПК=подкожное, СФХ=сверхкритическая флеш-хроматография, металлический натрий (например, Aldrich 28,205-7). Трет = третичный, TBAI=йодид тетра-н-бутиламмония (например, Aldrich 14,077-5). ТФУ=трифторуксусная кислота, TFAA = трифторуксусной кислоты ангидрид, ТГФ=тетрагидрофуран(высушенный над молекулярными ситами 4), ТСХ=тонкослойная хроматография, CH(ОСН 3)3=триметилортоформиат (например, Aldrich 30,547-2), УФ=чистота по ультрафиолетовому излучению (если не указано другое при 254 нм). Фармакологические испытанияD1 цАМФ-тестирование Способность соединений к стимулированию или ингибированию D1-рецепторопосредованного образования цАМФ в клетках СНО, стабильно экспрессирующих рекомбинантный D1-рецептор человека,определяли следующим образом. Клетки высевали в 96-луночные планшеты при концентрации 11000 клеток/лунку за 3 дня до эксперимента. В день эксперимента клетки один раз промывали предварительно нагретым буфером G (1 мМ MgCl2, 0,9 мМ CaCl2, 1 мМ ИБМК (3-изобутил-1-метилксантин) в ФСБ(фосфатно-солевой буферный раствор и начинали тестирование путем добавления 100 мкл смеси 30 нМ А 68 930 и тестируемого соединения, разведенного в буфере G (антагонизм), или тестируемого соединения, разведенного в буфере G (агонизм). Клетки инкубировали в течение 20 мин при 37C и реакцию останавливали путем добавления 100 мкл буфера S (0,1 М HCl и 0,1 мМ CaCl2) и планшеты держали при 4C в течение 1 ч. Добавляли 68 мкл буфера N (0,15 М NaOH и 60 мМ NaOAc) и планшеты встряхивали в течение 10 мин. Переносили 60 мкл реакции в планшеты cAMP FlashPlate (DuPont NEN), содержащие 40 мкл 60 мМ ацетата натрия рН 6,2 и добавляли 100 мкл 1 С mix (50 мМ ацетат натрия рН 6,2, 0,1% азид натрия, 12 мМ CaCl2, 1% БСА (бычий сывороточный альбумин) и 0,1510-6 Ки (5550 Бк)/мл 125I-цАМФ). После 18 ч инкубирования при 4C планшеты промывали один раз и считывали в счетчике Wallac TriLux.D2 цАМФ-тестирование Способность соединений к стимулированию или ингибированию D2-рецепторопосредованного ингибирования образования цАМФ в клетках СНО, трансфецированный D2-рецептором человека, определяли следующим образом. Клетки высевали в 96-луночные планшеты при концентрации 8000 клеток/лунку за 3 дня до эксперимента. В день эксперимента клетки один раз промывали предварительно нагретым буфером G (1 мМ MgCl2, 0,9 мМ CaCl2, 1 мМ ИБМК в ФСБ) и начинали анализ путем добавления 100 мкл смеси 1 мкМ хинпирола, 10 мкМ форсколина и тестируемого соединения в буфере G (антагонизм) или 10 мкМ форсколина и тестируемого соединения в буфере G (агонизм). Клетки инкубировали 20 мин при 37C и реакцию останавливали путем добавления 100 мкл буфераS (0,1 М HCl и 0,1 мМ CaCl2) и планшеты держали при 4C в течение 1 ч. Добавляли 68 мкл буфера N(0,15 М NaOH и 60 мМ ацетата натрия) и планшеты встряхивали в течение 10 мин. Переносили 60 мкл реакции в планшеты cAMP FlashPlate (DuPont NEN), содержащие 40 мкл 60 мМ NaOAc pH 6,2, и добавляли 100 мкл 1 С mix (50 мМ ацетат натрия рН 6,2, 0,1% азид натрия, 12 мМ CaCl2, 1% БСА и 0,1510-6- 13018413 Ки (5550 Бк)/мл 125I-цАМФ). После 18 ч инкубирования при 4C планшеты промывали один раз и считывали в счетчике Wallac TriLux.D5-тестирование Концентрационно-зависимая стимуляция высвобождения внутриклеточного Ca2+ под влиянием дофамина в hD5-трансфецированных клетках CHO-Ga16. Клетки загружали фтор-4, красителем - индикатором кальция, в течение 1 ч. Кальциевый отклик (изменение флуоресценции) наблюдали с помощью(EC50) усредняли из двух ячеек для каждой экспериментальной точки и наносили на график с концентрациями лекарственного вещества (см. фиг. 1 для дофамина). Кривые концентрационных эффектов для агонистов строили при добавлении различных концентраций к различным лункам, используя флуоресцентный планшетный спектрофотометр (FLIPR) (Molecular Devices, Sunnyvale, CA). Кривые соответствовали сигмоидальному уравнению "доза-эффект" I=Imax/(1+ (EC50/[Агонист])n), где значение EC50 является концентрацией агониста, которая производила половину максимальной активации, и п представляет собой коэффициент Хилла. Подборы кривых осуществляли, используя программное обеспечение Graphpad Prism 4 (Сан-Диего, Калифорния).D1/D2 анализ Агонисты дофамина могут обладать активностью к либо D1-подобным рецепторам, D2-подобным рецепторам, либо обоим. Мы использовали ротационный отклик у крыс с односторонними 6-ОНДАпоражениями для оценки соединений на их способность стимулировать оба типа рецепторов и индуцировать вращение [Ungerstedt, Arbuthnott, Brain Res., 24, 485 (1970); Setler, Sarau, Zirkle, Saunders, Eur. J.Ungerstedt, Herrera-Marschitz, Jungnelius, Stahle, Tossman, Zetterstrom, в "Advances в Dopamine Research" (Kohsaka, Ed.), Pergamon Press, Oxford, p. 219 (1982)]. Эксперименты состояли из определения минимальной эффективной дозы (МЭД), чтобы индуцировать вращение в случае исследуемого соединения. После того, как МЭД было определено, второй эксперимент осуществляли, чтобы определить МЭД соединения для преодоления блокирования Немонапридом (МЭДнемонаприд). Немонаприд является D2 подобным антагонистом, который блокирует D2-подобный рецептор, поэтому любые наблюдаемые вращения могут зависеть от активности при D1-подобном рецепторе. В заключение, после того, как стала известна МЭДнемонаприд, проводили третий эксперимент, используя дозу МЭДнемонаприд и наблюдая эффектD1-подобного антагониста, только SCH 23390, D2-подобного антагониста, только Немонаприда и, в заключение, эффект комбинированного применения SCH 23390 и Немонаприда. Данный третий эксперимент подтверждал активность соединения в отношении обоих рецепторов, так как каждый антагонист в одиночку мог только частично ингибировать ротационный отклик, индуцируемый тестируемым соединением, в то время как применение комбинации полностью блокировало все вращения у крыс [Arnt,Hytell, Psychopharmacology, 85(3), 346 (1985); Sonsalla, Manzino, Heikkila, J. Pharmacol Exp. Ther., 247(1),180 (1988)]. Данную модель валидировали, используя Апоморфин в качестве контрольно-проверочного соединения для смешанных D1-подобный/D2-подобный агонистов. Модель для определения превосходства Апоморфин и L-ДОФА способны реверсировать нарушение подвижности на модели сильного истощения дофамина у мыши. Как Апоморфин, так и L-ДОФА стимулируют D1- и D2-подобные рецепторы дофамина. Прамипексол, агонист D2-подобных рецепторов, является неэффективным в данной модели. Несколько указанных здесь соединений были протестированы в данной модели и показали профиль,аналогичный Апоморфинину и L-ДОФА в том отношении, что они способны восстанавливать локомоцию у мышей. Таким образом, данные соединения "превосходят" другие соединения, такие как Прамипексол, мишенью которых являются только D2-подобные рецепторы. Модель дискинезии Исследовали дискинетический профиль нескольких соединений по изобретению, используя описанную в литературе животную модель [Lundblad, Andersson, Winkler, Kirik, Wierup, Cenci, Eur. J. Neurosci., 15(1), 120 (2002)]. В данной модели несколько соединений по изобретению приводили к меньшей дискинезии, чем L-ДОФА или Апоморфин у не подвергавшихся действию психотропных веществ животных. Несколько соединений по изобретению, кроме того, значительно больше уменьшали индуцированную L-ДОФА дискинезию, чем наблюдали при переводе животных с L-ДОФА на Прамипексол. Методы - культура клеток Конструкт экспрессии D5 человека (чD5) делали, используя модифицированный вектор pEXJ. Стабильная линия клеток, экспрессирующая промискуитетный белок Galpha16 G человека (СНО-Ga16), была куплена у Molecular Devices, Саннивейл, Калифорния. Клетки выращивали в среде HAMS F-12 (Invitrogen, Карлсбад, Калифорния), содержащей 10% ФТС (фетальная телячья сыворотка), 1% L-глутамина и 1% пенициллин/стрептомицин (П/С), при 37C в 5% СО 2. За 48 ч до тестирования клетки CHO-Ga16 кратковременно трансфецировали ДНК рецептора hD5, используя методику LIPOFECTAMINE Plus (Invitrogen, Карлсбад, Калифорния) и оставляли расти в течение 1 дня в сыворотке и П/С-свободной среде. За 24 ч до тестирования hD5-трансфецированные клетки CHO-Ga16 высевали с плотностью 10000 клеток на лунку в черные 384-луночные планшеты с прозрачным дном, предварительно обработанные поли-D- 14018413 лизином (Becton Dickinson, США). Затем клетки культивировали в среде для роста клеток HAMS F-12,содержащей 1,5% ФТС, 1% L-глутамина и 1% пенициллина/стрептомицина (П/С) при 37C в 5% СО 2. Методы - анализ активации внутриклеточного кальция Для определения концентрации свободного внутриклеточного кальция ( [Са 2+]i) культуральную среду заменяли свежеприготовленным загрузочным буфером. Загрузочный буфер содержит 1X HBSS(Invitrogen), 20 мМ HEPES (Sigma), 0,1% БСА (Sigma), 1,5 мкМ Фтор-4-АМ (Molecular Probes) и 2,5 мМ пробенецида (свежеприготовленный) (Sigma). Планшеты инкубировали в течение 1 ч при 37C и 5% CO2 и промывали три раза отмывочным буфером. Промывочный буфер содержит те же компоненты, как в загрузочном буфере за исключением Фтор-4-АМ. Затем клетки помещали в флуоресцентный планшетный спектрофотометр (FLIPR, Molecular Devices), чтобы контролировать флуоресценцию клеток до и после добавления различных соединений. Интересующие соединения разводили в промывочном буфере до 4 конечной концентрации и отбирали аликвоты в прозрачный круглодонный планшет. Возбуждали краситель при длине волны 488 нм,используя лазер на ионах аргона, и детектировали сигнал, используя эмиссию стандарта при 510-570 нм[Sullivan, Tucker, Dale, Methods Mol. Biol, 114, 125 (1999)]. Получали кривые концентрационного эффекта для агонистов путем добавления различных концентраций к различным лункам. Измеряли относительную флуоресценцию путем вычитания фоновой из максимальной флуоресценции после добавления лекарственного вещества. Затем данные собирали и анализировали с помощью программного обеспеченияFLIPR и GraphPad Prism 4. Антагонистическую активность соединений анализировали по ингибированию ими сигнала, вызываемого лигандами-агонистами. Клетки предварительно инкубировали с соединениями при увеличивающихся концентрациях и затем стимулировали с помощью агонистов, используя описанные выше методы. Анализ in vitro гепатоцитов. Замороженные собранные гепатоциты самца крысы (Sprague Dawley) и собранные гепатоциты человека у 10 доноров (мужчин и женщин) были куплены у Vitro Technologies Inc., BA, США. Клетки размораживали при 31C в водяной бане, живые клетки подсчитывали и высевали в общем количестве 100 мкл модифицированной по способу Дульбекко среды Игла (высокое содержание глюкозы) с 5 мМ буфера Hepes в 96-луночные планшеты с содержанием в каждой лунке 250000 и 500000 клеток/мл для гепатоцитов крысы и человека, соответственно. Инкубации начинали через 15 мин преинкубации и останавливали в моменты времени 0, 5, 15, 30 и 60 мин для гепатоцитов крыс и 0, 30, 60, 90 и 120 мин для гепатоцитов человека. Инкубации останавливали путем добавления равных объемов ледяного ацетонитрила,содержащего 10% 1 М HCl. После центрифугирования инъецировали 20 мкл супернатантов в ВЭЖХколонку Atlantis dC18 3 мкм, 1502,1 мм в.д. (Waters, Массачусетс, США). Подвижная фаза имела следующий состав: А: 5% ацетонитрила, 95% Н 2 О, 3,7 мл/л 25% водный NH3, 1,8 мл/л муравьиной кислоты. Подвижная фаза В: 100% ацетонитрил и 0,1% муравьиной кислоты. Скорость потока была 0,3 мл/мин. Градиент устанавливали от 0 до 75% В с 5 по 20 мин и элюат анализировали, используя массспектрометр Q-TOFmicro (Waters, Массачусетс, США). Образование продукта/метаболита подтверждали путем точного измерения масс и сравнения с синтезированным стандартом, дающим совпадающие времена удерживания. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы I и один или несколько фармацевтически приемлемых носителей, разбавителей и вспомогательных веществ, где соединение формулы I имеет следующую структуру: где n=0,R1 и R2 независимо выбирают из водорода, C1-6 алканоила, фенилацетила или бензоила, R3 выбирают из группы, состоящей из водорода, метила, этила, н-пропила, циклопропила, циклобутила, аллила, пропаргила, гидроксиэтила, 3-фторпропила и 2-фторэтила, и его фармацевтически приемлемые кислотноаддитивные соли. 2. Фармацевтическая композиция по п.1, где R3 выбирают из группы, состоящей из водорода, метила, этила, н-пропила, аллила и пропаргила. 3. Фармацевтическая композиция по любому из предшествующих пунктов, где R3 выбирают из группы, состоящей из циклопропила, циклобутила и гидроксиэтила. 4. Фармацевтическая композиция по любому из предшествующих пунктов, отличающаяся наличием, по существу, чистого транс-диастереоизомера соединения формулы I. 5. Фармацевтическая композиция по любому из пп.1-4, где по меньшей мере один из R1 и R2 является ацетилом. 6. Фармацевтическая композиция по любому из пп.1-4, где по меньшей мере один из R1 и R2 является пивалоилом. 7. Фармацевтическая композиция по любому из пп.1-4, где по меньшей мере один из R1 и R2 является бензоилом или фенилацетилом. 8. Фармацевтическая композиция по п.1, где соединение выбирают из транс-1-метил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,цис-1-метил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,транс-1-н-пропил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,цис-1-н-пропил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола или их фармацевтически приемлемой кислотно-аддитивной соли. 9. Фармацевтическая композиция по п.4, где R1 и R2 оба являются водородами и R3 выбирают из группы, состоящей из водорода, метила, этила и н-пропила. 10. Фармацевтическая композиция по п.4, где R1 и R2 оба являются C1-6 алканоилом и R3 выбирают из группы, состоящей из водорода, метила, этила и н-пропила. 11. Применение фармацевтической композиции по любому из пп.1-10 или ее фармацевтически приемлемой кислотно-аддитивной соли для получения лекарственного средства для лечения нейродегенеративных расстройств у млекопитающего. 12. Применение фармацевтической композиции по п.11 для лечения болезни Паркинсона или болезни Хантингтона у млекопитающего. 13. Фармацевтически приемлемые соли соединения, где соединение выбирают из транс-1-метил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,цис-1-метил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,транс-1-н-пропил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола,цис-1-н-пропил-2,3,3 а,4,9,9 а-гексагидро-1H-бензо[f]индол-5,6-диола.
МПК / Метки
МПК: C07D 209/60, A61P 25/00, C07D 221/08, A61K 31/473, A61K 31/4741
Метки: основе, применение, производных, катехоламиновых, композиции, фармацевтические
Код ссылки
<a href="https://eas.patents.su/18-18413-farmacevticheskie-kompozicii-na-osnove-kateholaminovyh-proizvodnyh-i-ih-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Фармацевтические композиции на основе катехоламиновых производных и их применение</a>
Фармацевтические композиции на основе производных азетидина

Номер патента: 6510
Опубликовано: 29.12.2005
Авторы: Бобино Валери, Коте Софи, Пераккия Мария-Тереза
МПК: A61K 47/14
Метки: основе, азетидина, композиции, фармацевтические, производных
Формула / Реферат:
1. Стабильная фармацевтическая композиция, содержащая по крайней мере одно производное азетидина общей формулы в которой Ar является ароматической или гетероароматической группой, возможно, замещенной одним или несколькими (C1-C4)алкилом, галогеном, NO2, CN, (C1-C4)алкокси или OH, возможно, в сочетании с другим действующим веществом, способным потенцировать действие производного азетидина общей формулы (Ia) или (Ib), в системе, содержащей не...
Соли производных гуанидина и фармацевтические композиции на их основе

Номер патента: 6558
Опубликовано: 24.02.2006
Автор: Амтманн Эберхард
МПК: A61K 31/155, C07C 281/18, A61P 35/00...
Метки: композиции, производных, фармацевтические, основе, соли, гуанидина
Формула / Реферат:
1. Соли производных гуанидина формулы причем X обозначает валентную связь, -CH2-NH-, -CH2-NH-NH- или -CH=N-NH- и R обозначает линейный или разветвленный C1-C30-алкильный, C3-C20-циклоалкильный, адамантильный, норборнильный, трициклодецильный, бензильный, фурильный, пиридильный, антрацильный, нафтильный, фенантрильный, перинафтильный или хинуклидинильный остаток, который может быть замещен одной или несколькими гидроксильными группами,...
Фармацевтические композиции на основе нейроактивного стероида и их применение

Номер патента: 13744
Опубликовано: 30.06.2010
Автор: Вудворд Ричард М.
МПК: A61K 31/58, A61K 9/16, A61K 9/20...
Метки: композиции, применение, нейроактивного, фармацевтические, основе, стероида
Формула / Реферат:
1. Фармацевтическая композиция, содержащая 3a-гидрокси-3b-метоксиметил-21-(1'-имидазолил)-5a-прегнан-20-он или его фармацевтически приемлемые соль или сольват и один или несколько фармацевтически приемлемых эксципиентов, отличающаяся тем, что она обеспечивает устойчивый уровень 3a-гидрокси-3b-метоксиметил-21-(1'-имидазолил)-5a-прегнан-20-она в плазме в пределах от примерно 5 до примерно 500 нг/мл в течение от примерно 12 до примерно 24 ч после...
Фармацевтическая композиция на основе 3,6-ангидрогалактопиранозы и/или ее производных, и/или растворимого сахарида, содержащего данное соединение, применение композиции и входящих в ее состав ингредиентов, пищевой продукт, содержащий указанную композицию, и применение пищевого продукта

Номер патента: 4148
Опубликовано: 26.02.2004
Авторы: Койама Нобуто, Йу Фу-Гонг, Сагава Хироаки, Нисийама Еидзи, Томинага Таканари, Икаи Катсусиге, Еноки Татсудзи, Сакаи Такеси, Като Икуносин
МПК: A01N 1/00, A23L 1/30, A61K 31/70...
Метки: продукта, входящих, растворимого, пищевого, основе, композиция, композиции, содержащего, продукт, композицию, сахарида, указанную, данное, ингредиентов, содержащий, 3,6-ангидрогалактопиранозы, пищевой, применение, соединение, производных, фармацевтическая, состав
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента по меньшей мере одно соединение, выбранное из группы, состоящей из 3,6-ангидрогалактопиранозы, представленной формулой I ее альдегида и гидрата и 2-O-метилированных производных 3,6-ангидрогалактопиранозы, ее альдегида и гидрата; и растворимого сахарида, содержащего данное соединение на восстанавливающем конце. 2. Фармацевтическая композиция по п.1, где сахарид...
Применение бактерицидных пиримидинов для профилактики передачи вич половым путём и фармацевтические композиции на их основе

Номер патента: 11298
Опубликовано: 27.02.2009
Авторы: Ван Рое Енс Марсель, Де Бетюн Мари-Пьер Т.М.М.Дж., Стоффелс Пол
МПК: A61K 31/506, A61K 31/535, A61K 31/505...
Метки: пиримидинов, бактерицидных, профилактики, половым, фармацевтические, применение, основе, композиции, вич, передачи, путём
Формула / Реферат:
1. Применение 4-[[4-[(2,4,6-триметилфенил)амино]-2-пиримидинил]амино]бензонитрила или его фармацевтически приемлемой аддитивной соли для получения лекарственного микробицидного средства местного применения для профилактики (предотвращения) предачи или заражения ВИЧ, где указанные передача или заражение происходит через половую связь или связанный с ней интимный контакт между партнерами. 2. Применение по п.1, где передача или заражение происходит...
Предыдущий патент: Производные изоксазола и их применение в качестве потенциирующих средств для метаботропных глутаматных рецепторов
Следующий патент: Ингибиторы seh и их применение
Случайный патент: Дозирующий ингалятор для пропионата флутиказона