Антагонисты мускариновых ацетилхолиновых рецепторов
Номер патента: 13689
Опубликовано: 30.06.2010
Авторы: Чжу Чунцзе, Вань Цзехун, Янь Хунсин, Палович Майкл Р., Буш-Петерсен Якоб













Формула / Реферат
1. Соединение, имеющее структуру II

в котором обозначенный атом Н находится в экзо-положении;
R2 и R3 независимо выбраны из арила или гетероарила, где арил представляет собой фенил или нафтил и гетероарил представляет собой 5-10-членную систему ароматических колец, в которой одно или более колец содержит один и более гетероатомов, выбранных из группы, состоящей из N, О или S;
R4 представляет собой -CN.
2. Соединение по п.1, где R2 и R3 оба являются арилом.
3. Соединение по п.1 или 2, где R2 и R3 оба являются фенилом.
4. Соединение по п.1, где R2 и R3 оба являются гетероарилом.
5. Соединение по п.1 или 4, где R2 и R3 оба являются тиенилом.
6. Соединение по п.1, выбранное из группы, состоящей из
3-((эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионитрила;
3-((эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дитиофен-2-ил-пропионитрила.
7. Соединение, которое представляет собой 3-((эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионитрил.
8. Фармацевтическая композиция, включающая соединение согласно пп.1-6 и фармацевтически приемлемый носитель или растворитель.
9. Фармацевтическая композиция, включающая соединение согласно п.7 и фармацевтически приемлемый носитель или растворитель.
Текст
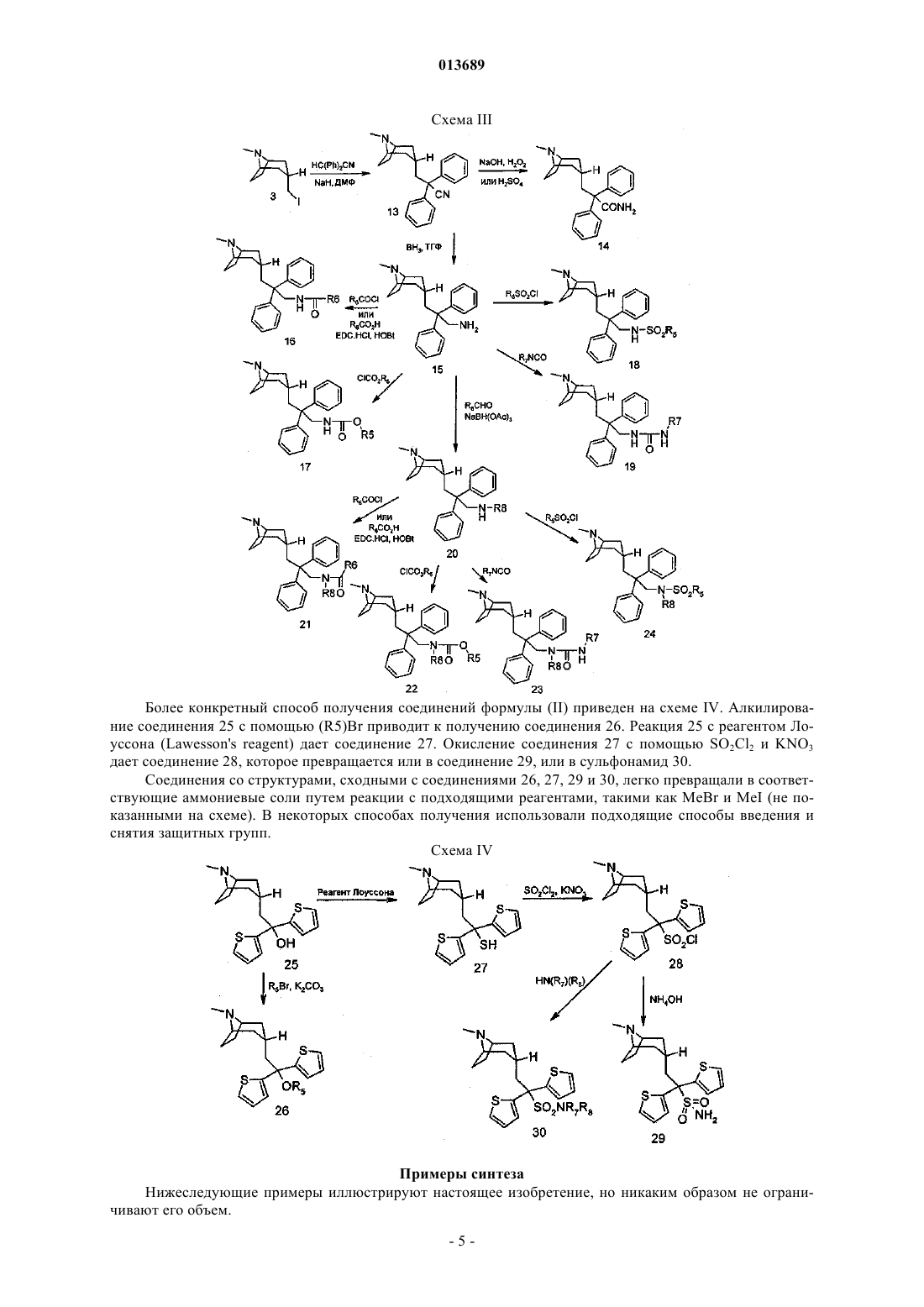
013689 Настоящее изобретение относится к новым производным соединений 8-азониабицикло[3,2,1]октанов, фармацевтическим композициям, способам их изготовления и их применению для лечения заболеваний, опосредованных М 3 мускариновыми ацетилхолиновыми рецепторами. Холинергические нейроны периферической и центральной нервной системы высвобождают ацетилхолин, который воздействует на множество различных биологических процессов путем взаимодействия с двумя главными классами ацетилхолиновых рецепторов, представленных никотиновыми и мускариновыми ацетилхолиновыми рецепторами. Мускариновые ацетилхолиновые рецепторы (mAChRs) принадлежат к суперсемейству рецепторов с иммобилизованным G-белком, имеющему семь трансмембранных доменов. Существует пять подтипов mAChRs, обозначаемых M1-M5, и каждый из подтипов представляет собой продукт отдельного гена. У каждого из этих пяти подтипов проявляются уникальные фармакологические свойства. Мускариновые ацетилхолиновые рецепторы широко распространены в органах позвоночных животных, и эти рецепторы могут являться медиаторами или торможения, или возбуждения. Например, в гладкой мускулатуре дыхательных путей, мочевого пузыря и желудочнокишечного тракта М 3 mAChRs вызывают реакции сокращения. Обзор этих данных можно прочитать в(Brown, 1989, 247). При некоторых различных патофизиологических состояниях отмечается дисфункция мускариновых ацетилхолиновых рецепторов. Например, состояние воспаления при астме и хронической обструктивной болезни легких (COPD) ведет к потере у М 2 мускариновых ацетилхолиновых ауторецепторов функции торможения парасимпатических нервов, иннервирующих гладкие мышцы легких, что вызывает увеличение высвобождения ацетилхолина с последующей вагусной стимуляцией. Такая дисфункция mAChRs приводит к повышенной реактивности дыхательных путей, вызванной усилением стимуляции М 3mAChRs (Costello, Evans, et al., 1999, 72) (Minette, Lammers, et al., 1989, 248). Аналогично, состояние воспаления желудочно-кишечного тракта при воспалительной болезни кишечника (IBD) приводит к М 3mAChRs-опосредованной гиперкинезии (Oprins, Meijer, et al., 2000, 245). Также выявлялось недержание мочи вследствие гиперсократимости, вызванной усилением стимуляции М 3 mAChRs (HegdeEglen,1999, 251). Таким образом, идентификация подтип-селективных антагонистов mAChRs может быть полезной для лечения этих mAChRs-опосредованных болезней. Несмотря на большое количество доказательств в поддержку использования антимускариновых рецепторов для лечения ряда клинических состояний, в клинике применяется относительно мало антимускариновых соединений. Таким образом, существует потребность в новых соединениях, способных вызывать блокаду М 3 mAChRs. Соединения, являющиеся ингибиторами связывания mAChRs, могут иметь эффект при состояниях, связанных с усилением стимуляции М 3 mAChRs, таких как астма, COPD, IBD и недержание мочи. Сущность изобретения Настоящее изобретение касается способа лечения заболевания, опосредованного мускариновыми ацетилхолиновыми рецепторами (mAChRs), в котором ацетилхолин связывается с М 3 mAChRs, при этом способ содержит введение эффективного количества соединения формулы (I) или формулы (II) [кроме соединения формулы (II), где R2 и R3 представляют собой 2-тиофен и R4 представляет собой-ОС(О)СН 3], или их фармацевтически приемлемой соли. Настоящее изобретение также относится к способу ингибирования связывания ацетилхолина его рецепторами у млекопитающих, нуждающихся в таком лечении, который включает введение вышеупомянутому млекопитающему эффективного количества соединения формулы (I) или формулы (II). Настоящее изобретение также относится к новым соединениям формулы (I) или формулы (II) и фармацевтическим композициям, содержащим соединения формулы (I) или формулы (II) и фармацевтический носитель или разбавитель. Соединения формулы (I) или формулы (II), используемые согласно изобретению, представлены следующей структурой: в которых обозначенный атом водорода Н находится в экзо-положении;R1- представляет собой анион, связанный с положительным зарядом атома N. R1- может представлять собой хлорид, бромид, йодид, сульфат, бензолсульфонат и толуолсульфонат, но не ограничивается вышеперечисленным;R2 и R3 независимо выбраны из группы, состоящей из прямой или разветвленной цепи групп низших алкилов (предпочтительно от 1 до 6 атомов углерода), циклоалкильных групп (имеющих от 5 до 6 атомов углерода), циклоалкил-алкила (имеющего 6-10 атомов углерода), гетероциклоалкила (имеющегоR7 и R8 независимо выбраны из группы, состоящей из Н, (C1-C6)алкила, (С 3-С 12)циклоалкила, (С 3 С 7)гетероциклоалкила, (C1-C6)алкил(С 3-С 12)циклоалкила, (C1-C6)алкил(С 3-С 7)гетероциклоалкила, (C1-C6) алкил-арила и (C1-C6)алкил-гетероарила. Подходящие фармацевтически приемлемые соли известны специалистам в данной области техники и включают основные соли неорганических и органических кислот, таких как соляная кислота, бромисто-водородная кислота, серная кислота, фосфорная кислота, метансульфокислота, этансульфокислота,уксусная кислота, яблочная кислота, винная кислота, лимонная кислота, молочная кислота, щавелевая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, бензойная кислота, салициловая кислота, фенилуксусная кислота и миндальная кислота. Кроме того, фармацевтически приемлемые соли соединений формулы (I) или формулы (II) могут также образовываться с фармацевтически приемлемым катионом. Подходящие фармацевтически приемлемые катионы известны специалистам в данной области техники и включают щелочные, щелочно-земельные, аммониевые и четверичные аммониевые катионы. Следующие термины, используемые в настоящем изобретении, означают"галогены" представляют собой все галогены, включая хлор, фтор, бром и йод;"C1-10 алкил" или "алкил" представляет собой функциональную группу в виде как прямой, так и разветвленной цепи, имеющей от 1 до 10 атомов углерода, если длина цепи не ограничена иначе, включающей метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил и т.п., но не ограниченной этим;"циклоалкил" приводится в настоящем изобретении для обозначения циклической функциональной группы, предпочтительно имеющей от 3 до 8 атомов углерода, включающей циклопропил, циклопентил,циклогексил, и т.п., но не ограничивается этим;"алкенил" приводится в настоящем изобретении для обозначения функциональной группы прямой или разветвленной цепи, имеющей от 2 до 10 атомов углерода, если длина цепи не ограничена иначе,включающей винил, 1-пропенил, 2-пропенил, 2-метил-1-пропенил, 1-бутенил, 2-бутенил и т.п., но не ограничивается этим;"арил" представляет собой фенил и нафтил;"гетероарил" (самостоятельно или в любой комбинации, такой как "гетероарилокси" или "гетероарилалкил") представляет собой 5-10-членную систему ароматических колец, в которой одно или более колец содержит один или более гетероатомов, выбранных из группы, состоящей из N, О или S, таких как пиррол, пиразол, фуран, тиофен, хинолин, изохинолин, хиназолинил, пиридин, пиримидин, оксазол, тетразол, тиазол, тиадиазол, триазол, имидазол или бензимидазол, но не ограничивается этим;"гетероциклический" (самостоятельно или в любой комбинации, такой как "гетероциклоалкил") представляет собой насыщенные или частично ненасыщенные 4-10-членные кольцевые системы, в которых одно или более колец содержит один или более гетероатомов, выбранных из группы, состоящей изN, О или S; таких как пирролидин, пиперидин, пиперазин, морфолин, тетрагидропиран, тиоморфолин или имидазолидин, но не ограничивается этим. Кроме того, сера может быть необязательно окислена до сульфона или сульфоксида;"арилалкил", или "гетероарилалкил", или "гетероциклоалкил" приводится в настоящем изобретении для обозначения C1-10 алкила, как указано выше, присоединенного к арильной, гетероарильной или гетероциклической функциональной группе, если не указано иное;"сульфинил" представляет собой оксид S(О) соответствующего сульфида, термин "тио" относится к сульфиду, и термин "сульфонил" относится к полностью окисленной функциональной группе S(O)2;"в которой две функциональные группы R1 (или две функциональные группы Y) могут вместе образовывать насыщенное или ненасыщенное 5- или 6-членное кольцо" приводится в настоящем изобретении для обозначения ароматической кольцевой системы, такой как нафталин, или обозначает фенильную функциональную группу, присоединенную к 6-членному частично насыщенному или ненасыщенному кольцу, такую как С 6-циклоалкенильную функциональную группу, то есть гексен, или С 5 циклоалкенильную функциональную группу, такую как циклопентен. Предпочтительные соединения, полезные для настоящего изобретения, включают[3,2,1]октан бромид. Более предпочтительные соединения настоящего изобретения включают[3,2,1]октан бромид. Способы получения Получение Получение соединений формулы (I) и формулы (II) можно осуществлять с применением способов синтеза, некоторые из которых показаны далее на схемах. Синтез, осуществленный по этим схемам,применяется для получения соединений формулы (I) и формулы (II), имеющих ряд различающихся R1,R2, R3 и R4, которые вступают в реакцию с заместителями, соответствующим образом защищенными,для достижения совместимости с реакциями настоящего изобретения. В этом случае последующее снятие защитных групп предоставляет соединениям возможность в целом раскрыть их природу. Ряд показанных схем содержат соединения только формулы (II), тем не менее, они приводятся лишь для иллюстрации. Схема I представляет общий способ получения. Синтез начинают с соединения 1. Восстановление с алюмогидридом лития (LAH) дает спирт 2. Замещение йодом дает соединение 3. Реакция связывания с анионом, полученная из HCR2(R3)(R4), дает затем соединение 4, которое легко превращается в аммониевую соль 5. Схема I Более конкретный способ получения, приводящий к соединению с формулой (II), приводится на схеме II. Алкилирование сложного эфира НС(Ph)2 СО 2 СН 3 соединением 3 дает соединение 6. Гидролиз соединения 6 приводит к образованию кислоты 7. Опосредованная 1,3-дициклогексилкарбодиимидом(DCC) конденсация кислоты с спиртом (R7)OH приводит к образованию затем сложного эфира 8. Конденсацией кислоты 7 с амином (R7)(R8)NH в подходящих условиях для связывания амида, хорошо из-3 013689 вестных специалистам в данной области техники, получают амид 9, такой как 1-(3-диметиламинопропил)3-этилкарбодиимид гидрохлорид (EDCHCl) и 1-гидроксибензотриазол гидрат (HOBt). Восстановлением полученного спирта 6 получают соединение 10. Реакция соединения 10 с хлорангидридом кислоты(R6)COCl или кислотой (R6)CO2H приводит к образованию сложного эфира 11. Алкилирование соединения 10 с подходящими реагентами, такими как (R5) бром, дает затем соединение 12. Соединения со структурами, сходными с соединениями 6, 7, 8, 9, 10, 11 и 12, превращали в соответствующие аммониевые соли путем реакции с подходящими реагентами, такими как MeBr и MeI (не показанными на схеме). В некоторых способах получения использовали способы введения и снятия защитных групп. Схема II Более конкретный способ получения соединений формулы (II) приведен на схеме III. Алкилирование HC(Ph)2CN с соединением 3 дает нитрил 13. Гидролиз соединения 13 как в присутствии оснований(например, NaOH и Н 2 О 2), так и присутствии кислот (например, H2SO4), дает амид 14. Восстановление соединения 13 приводит к получению амина 15, который преобразуется в амид 16, карбамид 17, сульфонамид 18 и мочевину 19. Конденсацией соединения 15 с альдегидом (R8)CH(O) с последующим восстановлением с помощью NaBH(OAc)3, получают амин 20, который легко превращается в амид 21, карбамид 22, мочевину 23 и сульфонамид 24. Соединения со структурами, сходными с соединениями 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 и 24,превращали в соответствующие аммониевые соли путем реакции с подходящими реагентами, такими какMeBr и MeI (не показанными на схеме). В некоторых способах получения использовали подходящие способы введения и снятия защитных групп. Более конкретный способ получения соединений формулы (II) приведен на схеме IV. Алкилирование соединения 25 с помощью (R5)Br приводит к получению соединения 26. Реакция 25 с реагентом Лоуссона (Lawesson's reagent) дает соединение 27. Окисление соединения 27 с помощью SO2Cl2 и KNO3 дает соединение 28, которое превращается или в соединение 29, или в сульфонамид 30. Соединения со структурами, сходными с соединениями 26, 27, 29 и 30, легко превращали в соответствующие аммониевые соли путем реакции с подходящими реагентами, такими как MeBr и MeI (не показанными на схеме). В некоторых способах получения использовали подходящие способы введения и снятия защитных групп. Схема IV Примеры синтеза Нижеследующие примеры иллюстрируют настоящее изобретение, но никаким образом не ограничивают его объем.(Эндо)-3-(2-метокси-2,2-дитиофен-2-ил-этил)-8,8-диметил-8-азонийбицикло[3,2,1]октан йодид. К раствору 2-(8-метил-8-азабицикло[3,2,1]окт-3-ил)-1,1-дитиофен-2-ил-этанола (полученного в соответствии с US2800482) (212 мг, 0,64 ммоль) в 5 мл метиленхлорида и йодметана (0,40 мл, 6,4 ммоль) добавляли 50%-ный водный гидроксид калия (0,25 мл, 3,2 ммоль) и тетрабутиламмония хлорид (5 мг, 3 мол.%). Реакционную смесь нагревали с обратным холодильником в течение 5 дней. Каждый день добавляли дополнительное количество 0,2 мл йодметана и 0,1 мл гидроксида калия. После этого реакционную смесь охлаждали до комнатной температуры, разводили метиленхлоридом и промывали водой. Водный слой экстрагировали с метиленхлоридом и объединенные органические слои промывали с солевым раствором, обезвоженным MgSO4, и выпаривали в вакууме. Сырой продукт перекристаллизовывали из метиленхлорида/этилацетата до получения 109 мг указанного в заголовке соединения со следующими показателями: LCMS (ES) жидкостная хроматография - масс-спектрометрия, отношение массы к заряду 3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионитрил. 2 а). Получение эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)метанола. Смесь 1,1-диметилэтил(эндо)-3-(гидроксиметил)-8-азабицикло[3,2,1]октан-8-карбоксилата (0,50 г,2,05 ммоль) и LiAlH4 (6,16 мл, 1,0 М в тетрагидрофуране (ТГФ), 6,16 ммоль) нагревали при 80 С в микроволновом реакторе в течение 60 мин. Затем раствор смешивали с насыщенным раствором Na2SO4,фильтровали через целит и выпаривали до получения указанного в заголовке соединения (0,31 г, 97%) со следующими показателями: LCMS (ES) m/z 156 (М+Н)+; 1 Н-ЯМР (CDCl3)1,28 (с, 1 Н), 1,59 (м, 4 Н), 1,90 (м, 1 Н), 2,13 (м, 4 Н), 2,32 (с, 3 Н), 3,17 (с, 2 Н), 3,59(д, 2 Н). 2b). Получение (эндо)-3-йодметил-8-метил-8-азабицикло[3,2,1]октана. Раствор йода (6,67 г, 25,8 ммоль) и эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)метанола (2,0 г,12,9 ммоль) в CH2Cl2 (120 мл) смешивали с PPh3 (на смоле, 8,6 г, 3 ммоль/г, 25,8 ммоль). Получаемую смесь перемешивали в течение 17 ч, фильтровали и выпаривали до получения указанного в заголовке соединения (2,63 г, 77%) со следующими показателями: LCMS (ES) m/z 266 (М+Н)+; 1 Н-ЯМР (CDCl3)2,05 (м, 4 Н), 2,39 (м, 3 Н), 2,79 (д, 3 Н), 2,98 (м, 2 Н), 3,45 (д, 2 Н), 3,81 (с, 2 Н). 2 с). Получение 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионитрил. Раствор (эндо)-3-йодметил-8-метил-8-азабицикло[3,2,1]октана (1,06 г, 4,0 ммоль) и Ph2CHCN (2,32 г, 12,0 ммоль) в диметилформамиде (ДМФ) (20 мл) смешивали с NaH (0,288 г, 12,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 60 мин. Фильтрацию и очистку осуществляли путем обращенно-фазной жидкостной хроматографии высокого разрешения ВЭЖХ (Gilson), с получением указанного в заголовке соединения (1,16 г, 93%) со следующими показателями: LCMS (ES) m/z 331 (М+Н)+; 1 Н-ЯМР (CDCl3)1,64 (м, 2 Н), 2,14 (м, 1 Н), 2,26 (м, 2 Н), 2,34 (м, 2 Н), 2,52 (м, 2 Н), 2,75 (м, 5 Н), 3,83(Эндо)-8-метил-3-(2,2,2-трифенил-этил)-8-азабицикло[3,2,1]октан. Раствор трифенилметана (0,276 г, 1,13 ммоль) в ТГФ (0,5 мл) смешивали с н-BuLi (0,706 мл, 1,6 М в гексане, 1,13 ммоль). Раствор перемешивали в течение 10 мин и добавляли раствором (эндо)-3-йодметил 8-метил-8-азабицикло[3,2,1]октана (100 мг, 0,377 ммоль) в ДМФ (1,0 мл). Смесь перемешивали при комнатной температуре в течение 60 мин, смешивали с Н 2 О (0,1 мл), выпаривали и фильтровали. Затем осуществляли очистку путем обращенно-фазной ВЭЖХ (Gilson.), с получением указанного в заголовке соединения (23,8 мг, 17%) со следующими показателями: LCMS (ES) m/z 382 (М+Н)+; 1 Н-ЯМР (CDCl3)1,07 (д, 2 Н), 2,12 (м, 1 Н), 2,22 (м, 4 Н), 2,31 (м, 2 Н), 2,65 (д, 3 Н), 2,97 (д, 2 Н), 3,63 3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионамид. Раствор 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионитрила (53 мг, 0,16 ммоль) в CH2Cl2 (0,25 мл) смешивали с H2SO4 (0,28 мл, 96%) и перемешивали при 40 С в течение 30 ч. Затем смесь выливали на лед, нейтрализовали NH3H2O, выделяли с помощью EtOAc и выпаривали. Полученный остаток растворяли в диметилсульфоксиде (ДМСО) и фильтровали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (17,2 мг,30%) со следующими показателями: масс-спектрометрия MS (ES) m/z 347 (М+Н)+; 1 3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионовая кислота. В раствор 2-[(3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил]-1,1-дифенилэтанола (100 мг, 1,56 ммоль) в НСООН (0,25 мл) быстро добавляли H2SO4 (2,73 мл, 90%) при 0 С. Реакционную пробирку немедленно закупоривали и сохраняли в холодильнике при -20 С в течение 7 дней. Раствор выливали на лед, нейтрализовали с NH3H2O, выделяли с помощью EtOAc и выпаривали. Полученный остаток растворяли в ДМСО и фильтровали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (52 мг, 48%) со следующими показателями: LCMS (ES)-7 013689 Раствор 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил]-2,2-дифенил-пропионитрила (310 мг, 0,938 ммоль) в ацетоне (6,0 мл) смешивали с MeBr (4,69 мл, 2,0 М в t-BuOMe, 0,938 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 60 мин и фильтровали. Твердый остаток промывали с ацетоном (23 мл) до получения указанного в заголовке соединения (333 мг, 83%) со следующими показателями: LCMS (ES) m/z 345 (М)+; 1(Эндо)-3-(2-циано-2,2-дифенил-этил)-8,8-диметил-8-азонийбицикло[3,2,1]октан йодид. Раствор 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионитрила (26,5 мг, 0,080 ммоль) в CH2Cl2 (0,5 мл) и MeCN (0,5 мл) перемешивали с MeI (0,125 мл, 2,00 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 ч, разбавляли ДМСО (0,3 мл) и выпаривали. Последующая очистка обращение-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (22,9 мг, 60%) со следующими показателями: LCMS (ES) m/z 345 (М)+; 1 Н-ЯМР (CDCl3)1,83 (д, 2 Н), 2,17 (м, 1 Н), 2,35 (м, 2 Н), 2,49 (м, 4 Н), 3,01 (д, 2 Н), 3,07 (с, 3 Н), 3,10 3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропан-1-ол. Смесь 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионовой кислоты (42,5 мг,0,122 ммоль) и LiAlH4 (0,488 мл, 1,0 М в ТГФ, 0,488 ммоль) нагревали в микроволновом реакторе при 100 С в течение 1 ч. Смесь разбавляли насыщенным раствором Na2SO4, фильтровали через целит и выпаривали. Полученный остаток растворяли в ДМСО и фильтровали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (29,1 мг, 71%) со следующими показателями: LCMS (ES) m/z 336 (М+Н)+; 1 Н-ЯМР (CDCl3)1,40 (д, 2 Н), 1,92 (м, 1 Н), 2,29 (м, 6 Н), 2,59 (м, 2 Н), 2,68 (д, 3 Н), 3,72 (с, 2 Н), 4,16(EDC) (49,5 мг, 0,258 ммоль), HOBt (3,2 мг, 0,024 ммоль) и (СН 3 СН 2)3N (0,232 мл, 1,65 ммоль). Смесь перемешивали при комнатной температуре в течение 60 ч и выпаривали. Полученный остаток растворяли в ДМСО и фильтровали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (29,8 мг, 30%) со следующими показателями: LCMS (ES) m/z 439 (М+Н)+; 1(Эндо)-3-(2-карбамоил-2,2-дифенил-этил)8,8-диметил-8-азонийбицикло[3,2,1]октан йодид. Указанное в заголовке соединение изготавливали из 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3 ил)-2,2-дифенил-пропионамида в соответствии со способом примера 7 (33% выхода) со следующими показателями: LCMS (ES) m/z 363 (М)+; 1 Н-ЯМР (CDCl3)1,49 (д, 2 Н), 1,95 (м, 1 Н), 2,25 (м, 2 Н), 2,42 (м, 4 Н), 2,84 (д, 2 Н), 3,17 (с, 3 Н), 3,23N-Бензил-3-[3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропил]мочевина. 11 а). 3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропиламин. Раствор 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионитрила (250 мг, 0,758 ммоль) в ТГФ (2,5 мл) смешивали с ВН 3 (2,53 мл, 1,5 M в ТГФ, 3,79 ммоль) при 0 С. Смесь перемешивали при комнатной температуре в течение 20 ч и разбавляли Н 2 О (1,0 мл). Затем раствор смешивали сK2CO3 (0,1 г) и перемешивали при комнатной температуре в течение 1 ч. Отделяли органические слои и водную часть экстрагировали с EtOAc (23 мл). Органические слои объединяли, обезвоживали с Na2SO4 и выпаривали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (159 мг, 63%) со следующими показателями: LCMS (ES) m/z 335(м, 4 Н), 7,33 (м, 2 Н), 7,43 (м, 4 Н). 11b). 1-Бензил-3-[3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропил]мочевина. Раствор 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропиламина (50,0 мг, 0,149 ммоль) в CH2Cl2 (2,0 мл) смешивали с PhCH2NCO (20,4 мкл, 0,164 ммоль) и (CH3CH2)3N (62,8 мкл, 0,447 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 ч и выпаривали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (13,0 мг, 19%) со следующими показателями: LCMS (ES) m/z 468 (M+Н)+; 1 Н-ЯМР (MeOD)1,24 (д, 2 Н), 1,94 (м, 3 Н), 2,25 (м, 4 Н), 2,49 (д, 2 Н), 2,67 (с, 3 Н), 3,62 (с, 2 Н), 3,97 1-Этил-3-[3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропил]мочевина. Титульное соединение изготавливали из 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2 дифенил-пропиламина и CH3CH2NCO в соответствии со способом примера 11 (45% выхода): LCMS (ES)N-[3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропил]ацетамид. Раствор 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропиламина (33,4 мг, 0,10 ммоль) в CH2Cl2 (0,5 мл) смешивали с Ас 2 О (18,9 мкл, 0,20 ммоль) и пиридином (16,2 мкл, 0,20 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч и выпаривали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (10,7 мг,29%) со следующими показателями: LCMS (ES) m/z 377 (М+Н)+; 1 Н-ЯМР (MeOD)1,26 (д, 2 Н), 1,82 (с, 3 Н), 1,96 (м, 3 Н), 2,26 (с, 4 Н), 2,53 (д, 2 Н), 2,67 (с, 3 Н), 3,66N-[3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропил]бензамид. Указанное в заголовке соединение изготавливали из 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3 ил)-2,2-дифенил-пропиламина и (PhCO)2O в соответствии со способом примера 13 (8% выхода) со следующими показателями: LCMS (ES) m/z 439 (М+Н)+; 1 Н-ЯМР (MeOD)1,28 (д, 2 Н), 2,00 (м, 3 Н), 2,24 (с, 4 Н), 2,59 (д, 2 Н), 2,67 (с, 3 Н), 3,65 (с, 2 Н), 4,21 3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дитиофен-2-ил-пропионитрил. Указанное в заголовке соединение изготавливали из(эндо)-3-йодметил-8-метил-8 азабицикло[3,2,1]октана и 2,2-дитиофен-2-ил-ацетонитрила в соответствии со способом примера 2 С (34% выхода) со следующими показателями: LCMS (ES) m/z 343 (М+Н)+; 1(Эндо)-3-(2-циано-2,2-дитиофен-2-ил-этил)-8,8-диметил-8-азонийбицикло[3,2,1]октан йодид. Указанное в заголовке соединение изготавливали из 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3 ил)-2,2-дитиофен-2-ил-пропионитрила в соответствии со способом примера 7 (43%) со следующими показателями: LCMS (ES) m/z 345 (М)+; 1N-[3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропил]бензолсульфонамид. Раствор 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропиламина (67,0 мг, 0,20 ммоль) в CH2Cl2 (2,0 мл) смешивали с PhSO2Cl (28,2 мкл, 0,22 ммоль) и (CH3CH2)3N (84,3 мкл, 0,60 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 ч и выпаривали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (51,5 мг, 54%) со следующими показателями: LCMS (ES) m/z 475 (М+Н)+; 1 Н-ЯМР (MeOD)1,39 (д, 2 Н), 2,01 (м, 3 Н), 2,30 (с, 4 Н), 2,69 (с, 5 Н), 3,60 (с, 2 Н), 3,68 (с, 2 Н), 7,12[3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропил]мочевина. К раствору 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропиламина (50,0 мг,0,149 ммоль) в CH2Cl2 (4,0 мл), добавляли CISO2NCO (31,2 мкл, 0,358 ммоль). Смесь перемешивали при комнатной температуре в течение 2 дней и выпаривали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (21,6 мг, 38%) со следующими показателями: LCMS (ES) m/z 378 (М+Н)+; 1 Н-ЯМР (MeOD)1,33 (д, 2 Н), 2,01 (м, 3 Н), 2,29 (с, 4 Н), 2,57 (м, 2 Н), 2,68 (с, 3 Н), 3,69 (с, 2 Н), 4,01N-[3-Эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропил]метансульфонамид. Указанное в заголовке соединение изготавливали из 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3 ил)-2,2-дифенил-пропиламина и MeSO2Cl в соответствии со способом примера 17 (28% выхода) со следующими показателями: LCMS (ES) m/z 413 (M+Н)+; 1 Н-ЯМР (MeOD)1,39 (д, 2 Н), 1,97 (м, 3 Н), 2,30 (с, 4 Н), 2,68 (с, 3 Н), 2,76 (с, 3 Н), 3,68 (с, 2 Н), 3,84(Эндо-3-2,2-дифенил-3-[(1-фенил-метаноил)амино]пропил-8,8-диметил-8-азонийбицикло[3,2,1] октан бромид. Раствор N-[3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропил]бензамида (29 мг,0,0683 ммоль) в CH2Cl2 (0,5 мл) и ацетоне (0,5 мл) смешивали с MeBr (0,342 мл, 2,0 М в tбутилметиловом эфире, 0,683 ммоль). Полученную смесь перемешивали при комнатной температуре в- 11013689 течение 3 ч и выпаривали. Последующая очистка обращенно-фазной ВЭЖХ (Gilson) обеспечивала получение указанного в заголовке соединения (19,6 мг, 64%) со следующими показателями: LCMS (ES) m/z 453 (М)+; 1(с, 2 Н), 7,30 (м, 6 Н), 7,39 (м, 6 Н), 7,50 (с, 3 Н). Биологические примеры Ингибирующее действие соединений M3 mAChRs настоящего изобретения определяли в следующих исследованиях in vitro и in vivo. Анализ ингибирования активации рецептора мобилизацией кальция Проводили мониторинг активированной рецептором мобилизации кальция, как ранее описано 10, для анализа экспрессированной в клетках СНО (клетках яичника китайского хомячка) стимуляции mAChRs. Клетки СНО с устойчивой экспрессией М 3 mAChRs помещали в 96-луночные планшеты с черными стенками и прозрачным дном. Через промежуток времени от 18 до 24 ч среду удаляли и заменяли на 100 мкл загрузочной среды (ЕМЕМ с солями Эрла, 0,1% бычий сывороточный альбумин (BSA) для радиоиммуноанализа (Sigma, St. Louis МО), и 4 мкл фтор-3-ацетоксиметилового сложноэфирного флуоресцентного индикаторного красителя (Fluo-3 AM, Molecular Probes, Eugene, OR) и помещали в инкубатор на 1 ч при 37 С. Затем содержащую краситель среду удаляли и заменяли на свежую среду (без Fluo-3 AM) и клетки помещали в инкубатор на 10 мин при 37 С. После этого клетки промывали 3 раза и помещали в инкубатор на 10 мин при 37 С в 100 мкл оценочного буфера (0,1% желатин (Sigma), 120 ммоль NaCl, 4,6 ммольKCl, 1 ммоль KH2PO4, 25 ммоль NaHCO3, 1,0 ммоль CaCl2, 1,1 ммоль MgCl2, 1,1 ммоль глюкозы, 20 ммоль HEPES (рН 7,4. Добавляли 50 мкл соединения (110-11 - 110-5 M на конец анализа) и планшеты помещали в инкубатор на 10 мин при 37 С. Затем планшеты помещали в планшет-ридер интенсивности флуоресцентного излучения (FLIPR, Molecular Probes), где содержащие краситель клетки подвергали возбуждению светом (488 нм) с помощью аргонного лазера мощностью в 6 Вт. Клетки активировали добавлением 50 мкл ацетилхолина со скоростью 50 мкл/с (0,1-10 нМ на конец анализа), приготовленном в буфере, содержащем 0,1% BSA. Мобилизацию кальция, отслеживаемую по изменению цитозольной концентрации кальция, измеряли по изменению интенсивности эмиссии в 566 нм. Изменение в интенсивности эмиссии непосредственно зависит от уровня содержания цитозольного кальция 11. Во всех 96 лунках излучение флуоресценции измеряли одновременно, используя охлажденную камеру прибора с зарядовой связью (ПЗС-камеру). Полученные данные отмечали точками каждую секунду. Это данные наносили на схему и анализировали с использованием программного обеспечения GraphPad PRISM. Бронхоспазм, индуцированный метахолином Ответную реакцию дыхательных путей на метахолин определяли у активных и свободно перемещающихся мышей BalbC в количестве (n=6 в каждой группе). Для измерения увеличенной паузы (Penh) использовали барометрическую плетизмографию, представляющую собой безразмерное измерение, показавшее зависимость от изменений в реактивности дыхательных путей, появляющегося во время метахолиновой провокации бронхов 12. Предварительно мышам интраназально вводили 50 мкл соединения(0,003-10 мкл на мышь) в 50 мкл носителя (10% ДМСО), и после чего их помещали в плетизмографическую камеру. В течение 10 мин мышам в камере давали успокоиться перед началом исходного Penh измерения, продолжающегося в течение 5 мин. Затем в течение 2 мин мышам вводили аэрозоль метахолина (10 мг/мл). Увеличенную паузу Penh начинали непрерывно регистрировать в течение 7 мин от начала введения аэрозоля и продолжали еще в течение 5 мин после введения. Данные по каждой мыши наносили на схему и анализировали с использованием программного обеспечения GraphPad PRISM. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, имеющее структуру II в котором обозначенный атом Н находится в экзо-положении;R2 и R3 независимо выбраны из арила или гетероарила, где арил представляет собой фенил или нафтил и гетероарил представляет собой 5-10-членную систему ароматических колец, в которой одно или более колец содержит один и более гетероатомов, выбранных из группы, состоящей из N, О или S;- 12013689 4. Соединение по п.1, где R2 и R3 оба являются гетероарилом. 5. Соединение по п.1 или 4, где R2 и R3 оба являются тиенилом. 6. Соединение по п.1, выбранное из группы, состоящей из 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дифенил-пропионитрила; 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2-дитиофен-2-ил-пропионитрила. 7. Соединение, которое представляет собой 3-эндо)-8-метил-8-азабицикло[3,2,1]окт-3-ил)-2,2 дифенил-пропионитрил. 8. Фармацевтическая композиция, включающая соединение согласно пп.1-6 и фармацевтически приемлемый носитель или растворитель. 9. Фармацевтическая композиция, включающая соединение согласно п.7 и фармацевтически приемлемый носитель или растворитель.
МПК / Метки
МПК: C07D 451/02, A61K 31/46
Метки: ацетилхолиновых, антагонисты, мускариновых, рецепторов
Код ссылки
<a href="https://eas.patents.su/14-13689-antagonisty-muskarinovyh-acetilholinovyh-receptorov.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты мускариновых ацетилхолиновых рецепторов</a>
Антагонисты мускариновых ацетилхолиновых рецепторов

Номер патента: 9899
Опубликовано: 28.04.2008
Авторы: Вань Цзехун, Буш-Петерсен Якоб, Палович Майкл Р., Янь Хунсин, Чжу Чунцзе
МПК: C07D 451/02, A61K 31/46
Метки: рецепторов, антагонисты, мускариновых, ацетилхолиновых
Формула / Реферат:
1. Соединение, которое является (эндо)-3-(2-циано-2,2-дифенилэтил)-8,8-диметил-8-азониабицикло [3,2,1]октана бромидом и (эндо)-3-(2-циано-2,2-дифенилэтил)-8,8-диметил-8-азониабицикло[3,2,1]октана йодидом. 2. Соединение по п.1, которое является (эндо)-3-(2-циано-2,2-дифенилэтил)-8,8-диметил-8-азониабицикло[3,2,1]октана бромидом. 3. Соединение по п.1, которое является (эндо)-3-(2-циано-2,2-дифенилэтил)-8,8-диметил-8-азониабицикло[3,2,1]октана...
Антагонисты мускариновых ацетилхолиновых рецепторов

Номер патента: 13435
Опубликовано: 30.04.2010
Авторы: Вань Цзехун, Палович Майкл Р., Буш-Петерсен Якоб, Янь Хунсин, Чжу Чунцзе
МПК: C07D 451/02, A61K 31/46
Метки: антагонисты, ацетилхолиновых, рецепторов, мускариновых
Формула / Реферат:
1. Соединение, имеющее структуру Iв котором обозначенный атом Н находится в экзо-положении;R1-представляет собой анион, связанный с положительным зарядом атома N;R2 и R3 независимо выбраны из арила или гетероарила, где арил представляет собой фенил или нафтил и гетероарил представляет собой 5-10-членную систему ароматических колец, в которой одно или более колец содержит один и более гетероатомов, выбранных из группы, состоящей из N, О или S;R4...
Азабициклопроизводные в качестве антагонистов мускариновых рецепторов

Номер патента: 7932
Опубликовано: 27.02.2007
Авторы: Силамкоти Арундутт Висванатан, Мехта Анита, Сарма Пакала Кумара Савитру, Салман Мохаммад, Дхармараджан Санкаранарейянан, Шетти Шанкар Джайрам, Чагх Анита, Кумар Нареш
МПК: C07D 401/12, A61K 31/403, C07D 209/52...
Метки: рецепторов, азабициклопроизводные, качестве, антагонистов, мускариновых
Формула / Реферат:
1. Соединения, имеющие структурную формулу I формула I и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры, где Ar представляет фенил; R1 представляет ОН, МеО и MeSO3; R2 представляет фенил, С4-С7 циклоалкил, 3,3-дифторциклопентил, 3-фторциклопентил; W представляет простую связь; X представляет NH, Nme, О или отсутствие атома; Y представляет простую связь; Z представляет NH, Nme,...
Карбоксамидные производные в качестве антагонистов мускариновых рецепторов

Номер патента: 13083
Опубликовано: 26.02.2010
Авторы: Глоссоп Пол Алан, Странг Росс Синклэр, Уотсон Кристин Энн Луис, Вуд Энтони, Мантелл Саймон Джон
МПК: A61K 31/397, A61K 31/4409, A61K 31/40...
Метки: производные, качестве, рецепторов, карбоксамидные, мускариновых, антагонистов
Формула / Реферат:
1. Соединение формулы (I)где R1 представляет собой CN или CONH2;А выбран изилигде * и ** представляют собой точки присоединения, причем ** означает связывание с кислородом,R2 и R3 представляют собой метил или, когда А представляет собой группу формулыR2 и R3 также могут образовывать вместе с атомом углерода, с которым они связаны, циклопентановое кольцо;р равно 0 или 1;А1 выбран иза) фенила, возможно замещенного 1, 2 или 3 группами, независимо...
Соединения мочевины, обладающие активностью антагонистов мускариновых рецепторов

Номер патента: 6437
Опубликовано: 29.12.2005
Авторы: Маммен Матай, Оуэр Дэвид
МПК: A61P 13/10, A61K 31/4468, C07D 211/58...
Метки: рецепторов, обладающие, антагонистов, мочевины, мускариновых, активностью, соединения
Формула / Реферат:
1. Соединение формулы где R46 означает C1-10алкил, C3-20циклоалкил или C1-40гетероцикл; R47 означает C1-10алкил, C6-20арил, HC(O)-, C1-10алкил-C(O)-, C3-20циклоалкил-C(O)-, C4-20циклоалкенил-C(O)-, C6-20арил-C(O)-, C1-15гетероарил-C(O)-, C1-40гетероциклил-C(O)-, C1-40гетероцикл или -COOR50, в котором R50 означает C1-10алкил; или R46 и R47 вместе с атомом азота, к которому они присоединены, образуют C1-40гетероцикл; X означает C3-20алкилен, где...