Способ получения комбретастатинов
Номер патента: 7965
Опубликовано: 27.02.2007
Авторы: Казимир Жан-Поль, Лавинь Мишель, Малежонок Ирина, Мютти Стефан







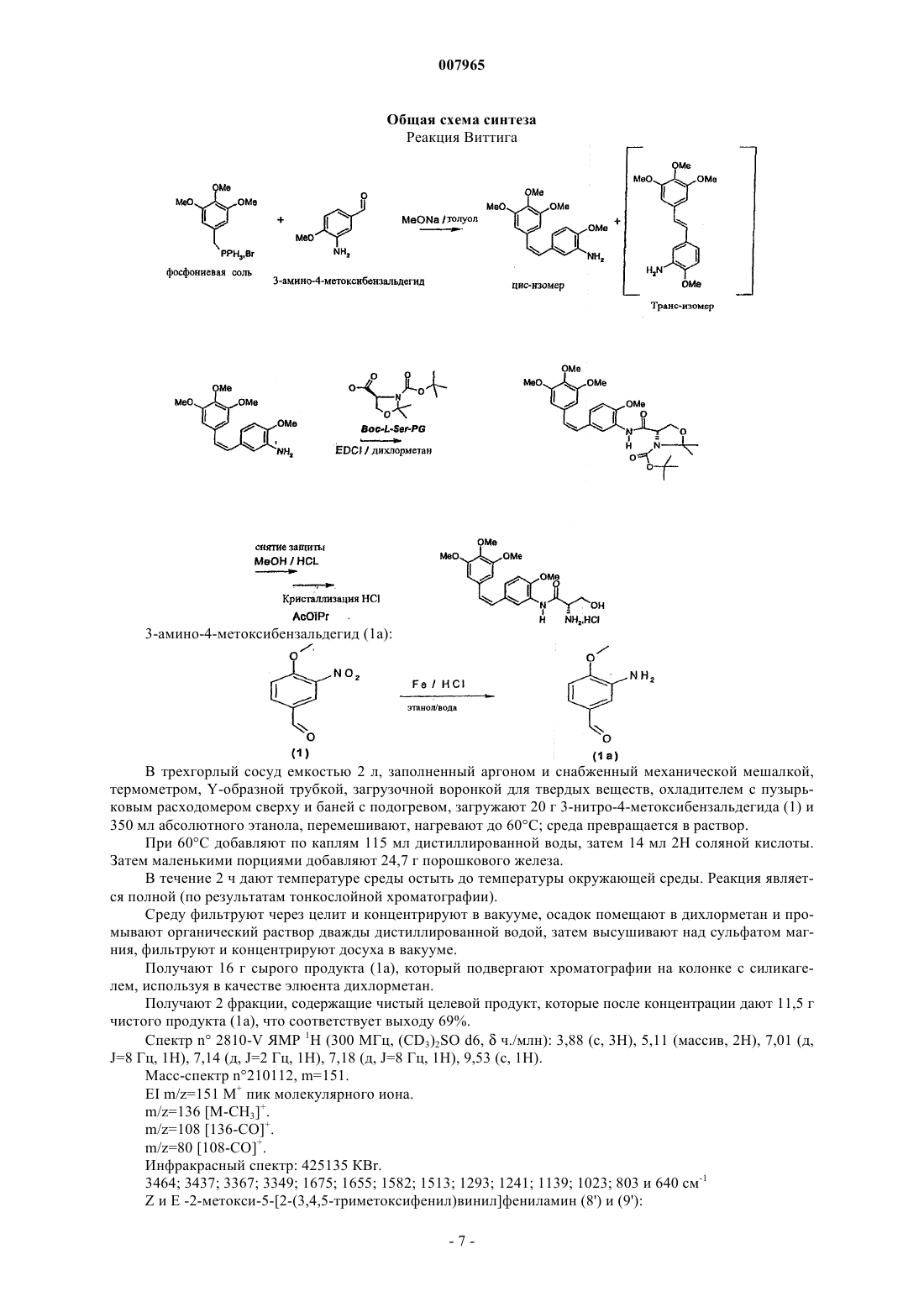

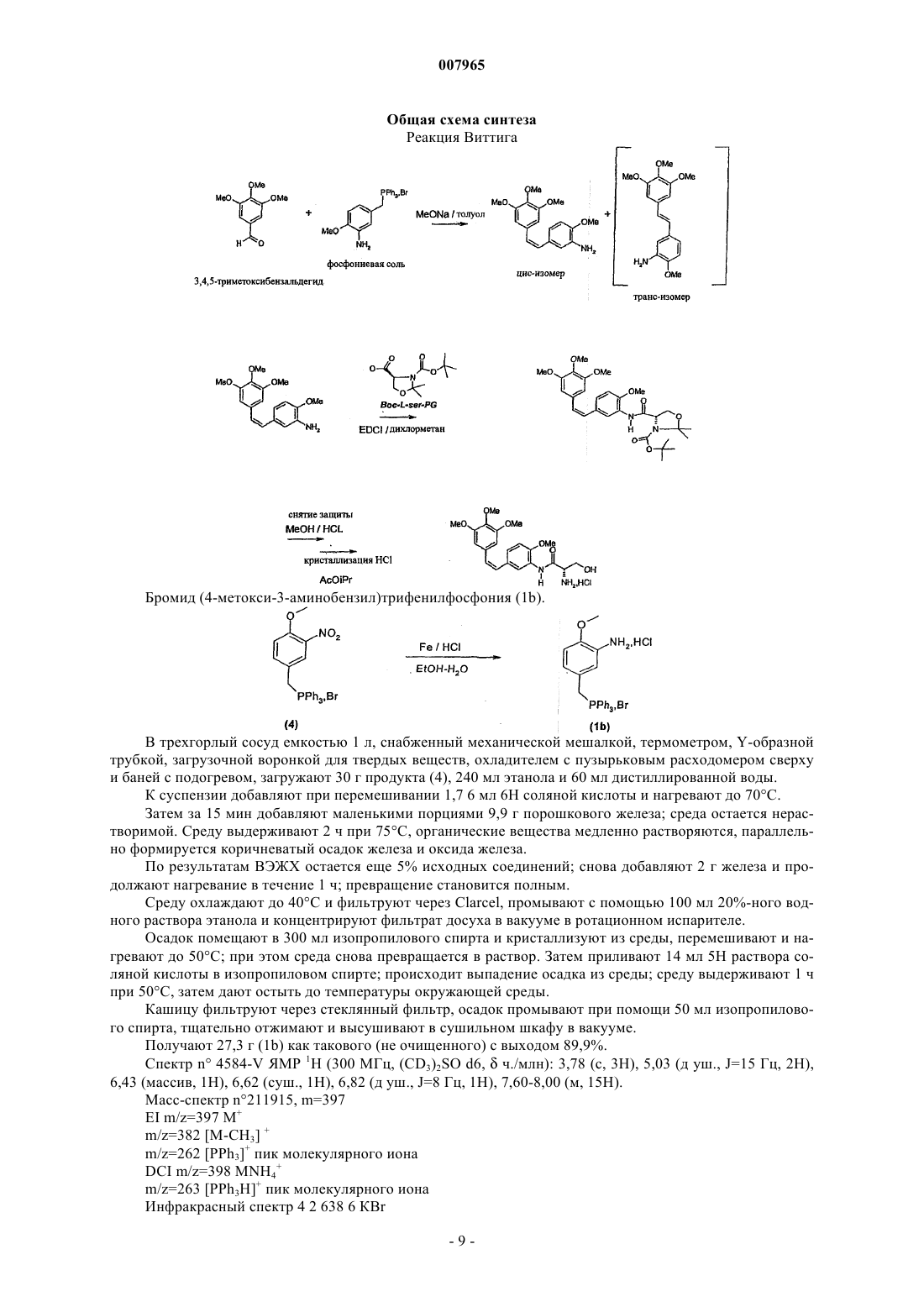



Формула / Реферат
1. Способ получения комбретастатинов следующей общей формулы (I)

в которой А обозначает аминогруппу, отличающийся тем, что конденсируют соль (3,4,5-триметоксибензил)трифенилфосфония с 3-амино-4-метоксибензальдегидом или соль (3-амино-4-метоксибензил)трифенилфосфония с 3,4,5-триметоксибензальдегидом.
2. Способ по п.1, отличающийся тем, что конденсируют соль (3,4,5-триметоксибензил)трифенилфосфония с 3-амино-4-метоксибензальдегидом.
3. Способ по п.1 или 2, отличающийся тем, что реакцию проводят предпочтительно в присутствии основания, выбранного из трет-бутилата калия, трет-амилата натрия, гидрида натрия, бутиллития, литийдиизопропиламина, метилата натрия, карбоната калия или щелочных производных гексаметилдисиланов.
4. Способ по п.1 или 2, отличающийся тем, что используют метилат натрия.
5. Способ по п.1 или 2, отличающийся тем, что реакционный растворитель, инертный в отношении реакции, выбирают из простых эфиров (ТГФ), полярных апротонных растворителей (ацетонитрил, NMP, DMF, DMSO...), спиртов, ароматических растворителей или воды.
6. Способ по п.4, отличающийся тем, что температура реакции предпочтительно составляет от 0 до 10шС.
7. Способ получения соединений формулы (I) в форме соли серина путем соединения производных формулы (IIа)

с циклическим производным защищенного серина формулы (IIb)

где PG обозначает защитную группу аминогруппы, с получением нового промежуточного соединения следующей общей формулы

которое затем подвергают снятию защиты.
8. Промежуточное соединение, образующееся в результате соединения аминокомбретастатина и циклического производного защищенного серина, отличающееся тем, что оно соответствует следующей формуле:

в которой PG обозначает защитную группу.
9. Промежуточное соединение по п.8, отличающееся тем, что PG обозначает защитную группу, выбранную из следующих групп: трет.-бутоксикарбонил, бензилоксикарбонил (CBZ) или 9-флуоренилметилоксикарбонил (FMOC).
Текст
007965 Настоящее изобретение относится к новому способу получения комбретастатинов и их производных. Под комбретастатинами, или производными стильбена, понимают производные следующей общей формулы в которой А обозначает гидроксильную группу или аминогруппу, а также их фармацевтически приемлемые соли. Из этих солей можно назвать гидрохлорид, ацетат, фосфат, метансульфонат. Когда соединение или А обозначает аминогруппу, она может быть также сопряжена с аминокислотами с образованием амидов,а также их фармацевтически приемлемых солей. Синтез производных стильбена, или комбретастатинов, которые могут находиться в форме фармацевтически приемлемой соли, а также содержащие их фармацевтические композиции описаны в патентах US 4996237; US 5525632; US 5731353 и US 5674906. В этих патентах описаны комбретастатины, а также их метаболиты и описана их онкологическая активность in vitro. Согласно этим патентам, комбретастатины получают из солей (3,4,5-триметоксибензил)трифенилфосфония, который конденсируют с 3-нитро- или 3-гидрокси-(где гидроксильная группа защищена) 4 метоксибензальдегидом в присутствии гидрида натрия или производных лития, после чего полученное производное, если оно является нитропроизводным, восстанавливают в присутствии цинка. Затем получают изомер в цис-конфигурации действием света или хроматографическим разделением смеси. Определены различные пути осуществления указанного способа получения комбрестатинов. Способ по пути V 01 получения производных формулы (I), в которых А обозначает аминогруппу, который представляет собой усовершенствованный способ, описанный в вышеперечисленных патентах, заключается в том, что после конденсации по Виттигу в присутствии бромида или хлорида (3,4,5 триметоксибензил)трифенилфосфония и 3-нитро-4-метоксибензальдегида проводят восстановление в присутствии железа вместо цинка, используемого в известном способе, что позволяет получить общий выход реакции, по отношению к использованному альдегиду, составляющий 60% (выход по отношению к использованному альдегиду в патенте US 5525632 составляет от 21 до 33%). Способ по пути V 02 заключается в конденсации 3,4,5-триметоксибензальдегида с бромидом или хлоридом (4-метокси-3-нитробензил)трифенилфосфония. В обоих способах по пути V 01 и V 02 реакцию проводят в присутствии основания, выбранного, в частности, из трет.-бутилата калия, трет.-амилата натрия, гидрида натрия, бутиллития, литийдиизопропиламина (LDA), метилата натрия, карбоната калия или щелочных производных гексаметилдисиланов. Эту реакцию осуществляют в различных растворителях, таких как простые эфиры (ТГФ), полярные апротонные растворители (ацетонитрил, NMP, DMF, DMSO), спирты, ароматические растворители или вода, при температуре, выбранной специалистом в зависимости от используемого основания и используемого растворителя. Эта реакция, в том, что касается способа по пути V 02, описана, в частности, в публикации K.G.Piney, Bioorg. Med. Chem. 8(2000) 2417-2425. 2-Метокси-5-[2-(3,4,5-триметоксифенил)винил]нитрофенил восстанавливают согласно улучшенному способу по изобретению действием железа. Предпочтительно используют избыток железа, если требуется полное превращение исходного продукта. Этот избыток предпочтительно превышает 2 эквивалента на моль исходного нитропроизводного. Было доказано, что та же стадия, осуществленная в присутствии цинка в уксусной кислоте, традиционном растворителе, используемом при восстановлении с помощью цинка, не позволяет осуществить полную реакцию (в патенте US 5525632 выход реакции восстановления, осуществляемой на чистом изомере Z, составляет от 46 до 66%) и, с другой стороны, используемые количества цинка непомерно велики, что приводит к большому объему промышленных отходов; кроме того, такой способ приводит к образованию большого количества азо-соединений, происходящих от слияния образуемого амино и нитрозо, являющегося промежуточным продуктом реакции восстановления. Восстановление за счет водорода, образуемого формиатом аммония в присутствии классических катализаторов, таких как палладий или платина, приводит к сильной изомеризации двойной связи с образованием нежелательного изомера Е, а также к частичному насыщению двойной связи. В вышеуказанной публикации Piney описано восстановление с помощью гидросульфита натрия чистого изомера Z нитро, полученного хроматографией и перекристаллизацией, с получением изомера Z амино с выходом, составляющим всего-навсего 37%. Гидрирования молекулярным водородом, катализируемые платиной или палладием, редко оказываются полными и, что хуже, приводят к насыщению этиленовой двойной связи. Настоящее изобретение относится к способу получения комбрестатинов или их производных, позволяющий избежать стадии промежуточного восстановления, которая является обязательной при ис-1 007965 пользовании нитро-производного в качестве исходного продукта. Действительно, значительно более экономичным является первый метод осуществления этого второго способа, в котором конденсируют бромид или хлорид (3,4,5-триметоксибензил)трифенилфосфония с 3-амино-4-метоксибензальдегидом,или второй метод осуществления этого второго способа, в котором конденсируют 3,4,5 триметоксибензальдегид с солью (3-амино-4-метоксибензил)трифенилфосфония. Этот способ согласно обоим вариантам требует на одну стадию меньше из числа таких, на которых используют продукты CMR (канцерогены, мутагены, соединения, токсичные в отношении репродуктивной функции) по сравнению с вышеописанными способами по пути V 01 и V 02, что в промышленном масштабе представляется ощутимым преимуществом с точки зрения безопасности и производственных затрат. Согласно первому варианту способа по пути V 03 осуществления изобретения вводят в реакцию соль (3,4,5-триметоксибензил)трифенилфосфония и 3-амино-4-метоксибензальдегид, причем реакцию осуществляют предпочтительно в присутствии основания, выбранного, в частности, из трет.-бутилата калия, трет.-амилата натрия, гидрида натрия, бутиллития, LDA, метилата натрия, карбоната калия или щелочных производных гексаметилдисиланов. Предпочтительно используют метилат натрия. Эту реакцию проводят в различных растворителях, таких как простые эфиры (ТГФ), полярные апротонные растворители (ацетонитрил, NMP, DMF, DMSO), спирты, ароматические растворители или вода, при температуре, выбираемой специалистом в зависимости от используемого основания и используемого растворителя. Температура реакции выбирается специалистом в зависимости от используемого основания; при использовании метилата она составляет предпочтительно от 0 до 10 С. По завершении реакции используемое основание нейтрализуют водным раствором кислоты, органическую фазу промывают, концентрируют и, в результате хроматографии сырого концентрата, получают целевой продукт. Согласно способу по пути V 04 осуществления изобретения, в котором вводят в реакцию соль (3 амино-4-метоксибензил)трифенилфосфония и 3,4,5-триметоксибензальдегид, реакцию предпочтительно осуществляют в присутствии органического основания, выбранного, в частности, из трет.-бутилата калия, трет.-амилата натрия, гидрида натрия, бутиллития, LDA, метилата натрия, карбоната калия или щелочных производных гексаметилдисиланов. Предпочтительно используют метилат натрия. Эту реакцию проводят в различных растворителях, таких как простые эфиры (ТГФ), полярные апротонные растворители (ацетонитрил, NMP, DMF, DMSO), спирты, ароматические растворители или вода, при температуре, выбираемой специалистом в зависимости от используемого основания и используемого растворителя. Температура реакции выбирается специалистом в зависимости от используемого основания; при использовании метилата она составляет предпочтительно от 0 до 10 С. По завершении реакции используемое основание нейтрализуют водным раствором кислоты, органическую фазу промывают, концентрируют и, в результате хроматографии сырого концентрата, получают целевой продукт. Производное, получаемое согласно способу по изобретению по пути V 03 или V 04 или на второй стадии способа по пути V 01 или V 02, имеет следующую формулу: Когда требуется соединить серии с соединением формулы (IIа), предпочтительно использовать L-серин,дважды защищенный по атому азота серина и по гидроксильной группе и имеющий общую формулу (IIb) где PG обозначает защитную группу аминогруппы, известную любому специалисту, с получением нового промежуточного продукта следующей общей формулы: которое затем расщепляют, предпочтительно одновременно с раскрытием цикла путем кислотного гидролиза, в реакции удаления защитной группы, известной любому специалисту. Предпочтительно-2 007965 группа PG в формулах (IIb) или (III) представляет собой защитную группу, выбранную из следующих групп: трет.-бутоксикарбонил, бензилоксикарбонил (CBZ) или 9-флуоренилметилоксикарбонил (FMOC). Соединение вышеприведенной формулы (III) является новым и заявлено как таковое. Конденсацию предпочтительно осуществляют в присутствии EDCI (хлорида 1-этил-3-(3 диметиламинопропил)карбодиимида) или в присутствии DCC (дициклогексилкарбодиимида) и НОВТ(гидроксибензотриазола), или в присутствии DCC (дициклогексилкарбодиимида) и HOSU (Nгидроксисукцинимида), или, наконец, в присутствии TOTU (O-[(этоксикарбонил)цианометиленамино]N,N,N',N'-тетраметилуронийтетрафторбората) или HBTU (O-бензотриазол-1-ил-N,N,N',N'-тетраметилуронийгексафторфосфата) или N,N-карбонилдиимидазола. Реакцию предпочтительно проводят в растворителе, инертном по отношению к реакции, выбранном, в частности, из полярных апротонных растворителей, таких как ацетонитрил, диметилформамид, тетрагидрофуран, или хлорсодержащих алифатических растворителей, таких как дихлорметан, или из сложных эфиров. Связывание с производным формулы (IIа) может быть также осуществлено действием смешанного ангидрида, синтезированного in situ на основе хлорформиата или хлорангидрида карбоновой кислоты,например пивалоила, и дважды защищенного L-серина формулы (IIb) в присутствии третичного основания типа NMM (N-метилморфолина) в различных растворителях, инертных по отношению к реакции: сложных эфирах, простых эфирах, хлорсодержащих растворителях, ацетонитриле и т.д. Смешанный ангидрид получают предпочтительно при температуре от 0 до 10 С, затем реакцию проводят при температуре окружающей среды. По завершении реакции реакционную среду гидролизуют с помощью водного раствора,затем среду декантируют и полученную органическую фазу промывают гидроксилированным основанием. Снятие двойной защиты с соединения формулы (III) осуществляют действием органической или неорганической кислоты; предпочтительно, используют концентрированный водный раствор соляной кислоты в спиртовой среде. Согласно наиболее предпочтительному варианту осуществления изобретения температура реакции составляет от 50 до 70 С. Изобретение будет более полно раскрыто с помощью следующих примеров, которые не должны считаться имеющими ограничительный характер. Состав смесей, последовательность и ход реакций, а также выход не выделенных продуктов/промежуточных соединений и их титры определяли путем ВЭЖХ (высокоэффективной жидкостной хроматографии). Пример 1. Cпособ по пути V 02 согласно изобретению. Гидрохлорид (Z)-N-[2-метокси-5-[2-(3,4,5-(триметоксифенил)винил]фенил]-L-серинамида. Общая схема синтеза Реакция Виттига Новый способ, т.н. обратный Виттиг, исходя из бромида (4-метокси-3-нитробензил)трифенилфосфония и 3,4,5-триметоксибензальдегида позволяет получить смесь изомеров Z и Е 2-метокси-5-[2-3 007965(3,4,5-триметоксифенил)винил]нитрофенила с отношением Z/E=75/25. Это отношение отражает достаточно высокую долю изомера Z для того, чтобы смесь Z/E можно было непосредственно подвергнуть восстановлению с получением путем кристаллизации гидрохлорида изомера Z амино с титров ВЭЖХ 97% (внутренний стандарт). Получение бромида (4-метокси-3-нитробензил)трифенилфосфония (4) осуществляют согласно следующему примеру: 3-Нитро-4-метоксибензиловый спирт (2). В трехгорлый сосуд емкостью 2 л, снабженный механической мешалкой, термометром, Y-образной трубкой, загрузочной воронкой для твердых веществ и охладителем с пузырьковым расходомером сверху, загружают 90,5 г 3-нитро-4-метоксибензальдегида (1), затем 450 мл ТГФ и 90 мл этанола. Полученный бледно-желтый раствор охлаждают до 10 С, затем вносят 10 г боргидрида натрия в течение 40 мин при 10/15 С (эта реакция является сильно экзотермичной и температуру необходимо поддерживать при помощи ледяной бани с ацетоном); в конце добавления раствор меняет цвет с коричневого на цвет морской волны. Раствор перемешивают в течение 30 мин при 10 С, окончание реакции отслеживают при помощи тонкослойной хроматографии и перемешивают еще 1 ч при 10 С, затем температуре дают подняться до температуры окружающей среды. Загрузочную воронку заменяют изобарной капельницей емкостью 500 мл, через которую по каплям приливают 300 мл дистиллированной воды в течение 30 мин, поддерживая температуру среды 20 С. В начале приливания наблюдают выделение газа. Среду концентрируют до 2/3 в ротационном испарителе (50 С/20 ммНg); в водном концентрате кристаллизуется продукт белого цвета в форме блоков. Охлажденную водную фазу экстрагируют при помощи 250 мл, затем 150 мл дихлорметана, объединенные органические фазы промывают 250 мл дистиллированной воды, затем высушивают над сульфатом магния. После фильтрации хлорметиленовый раствор используют таким какой он есть в следующей реакции бромирования. Выход этой стадии считают равным 100%. Примечание: спирт (2) является промышленным, но малодоступным продуктом. 3-Нитро-4-метоксибромбензил (3). В трехгорлый сосуд емкостью 1 л, снабженный механической мешалкой, термометром, Y-образной трубкой, капельницей и охладителем с пузырьковым расходомером сверху, загружают хлорметиленовый раствор 3-нитро-4-метоксибензилового спирта (2) и добавляют 100 мл дихлорметана. Раствор при перемешивании охлаждают до 5 С, затем приливают по каплям, поддерживая температуру среды равной 5 С,135,4 г трибромида фосфора. Раствор перемешивают в течение 1 ч 30 мин при 5 С; окончание реакции отслеживают при помощи тонкослойной хроматографии, затем приливают по каплям, поддерживая температуру 15 С, 250 мл насыщенного раствора гидрокарбоната натрия; в процессе добавления с небольшой задержкой происходит очень интенсивное выделение газа. Среду декантируют в колбе и промывают последовательно при помощи 250 мл дистиллированной воды и 200 мл насыщенного раствора гидрокарбоната натрия. Органическую фазу высушивают над сульфатом магния, фильтруют и концентрируют в ротационном испарителе (50 С/20 ммНg). Получают 119 г твердого вещества в форме ворсистых игл желто-зеленого цвета с химическим выходом на 2 стадиях 97%. Примечание: этот продукт (3) может также быть получен по следующей схеме, описанной в публикации K.G. Piney et al. Bioorg. Med. Chem. 8(2000) 2417-2425. Бромид (3-Нитро-4-метоксибензил)трифенилфосфония (4). В трехгорлый сосуд емкостью 2 л, снабженный механической мешалкой, термометром, Y-образной трубкой, загрузочной воронкой для твердых веществ и охладителем с пузырьковым расходомером сверху, загружают 119 г 3-нитро-4-метоксибромбензила (3) на перемешиваемый слой 1000 мл толуола; при легком нагревании до 25 С среда становится раствором. Добавляют 126,5 г трифенилфосфина, получен-4 007965 ный раствор постепенно нагревают до 60 С; начиная с температуры 30 С, формируется осадок. Среду выдерживают в течение 4 ч при 60/65 С, затем охлаждают до 30 С, фильтруют через стеклянный фильтр,промывают на фильтре 2 раза с помощью 300 мл толуола, отжимают и высушивают в сушильном шкафу В трехгорлый сосуд емкостью 2 л, снабженный механической мешалкой, термометром, Y-образной трубкой, капельницей и охладителем с пузырьковым расходомером сверху, загружают при 20 С и в атмосфере азота 54,7 г 3,4,5-триметоксибензальдегида (5), 148,6 г бромида (4-метокси-3-нитробензил)трифенилфосфония (4) и 1300 мл толуола. Суспензию при перемешивании охлаждают до 5 С на ледяной бане, затем за 40 мин при 5 С приливают 63,2 г 25%-ного (по массе) раствора метилата натрия в метаноле. По мере приливания цвет суспензии меняется с грязно-белого на желтый и далее на коричневый. Перемешивают 1 ч при 5 С, окончание реакции отслеживают с помощью ВЭЖХ (общее потребление альдегида), после чего добавляют 3 г (0,05 моль) уксусной кислоты. Суспензию нагревают до 40 С и поддерживают 30 мин при 40 С; при этой температуре нерастворимыми остаются только соли; среду фильтруют при 40 С через стеклянный фильтр 3 и соли промывают на фильтре 3 раза с помощью 100 мл толуола. Фильтрат загружают обратно в сосуд с 250 мл дистиллированной воды, эту двухфазную смесь перемешивают 20 мин при 40 С, затем декантируют в колбе. Фазу, содержащую толуол, промывают дважды с 250 мл дистиллированной воды, затем концентрируют досуха в ротационном испарителе. Осадок помещают в 600 мл изопропилового спирта и 12 мл толуола с температурой 40 С, начинается кристаллизация, при медленном перемешивании дают температуре опуститься до комнатной и оставляют до следующего утра. Суспензию при перемешивании охлаждают и выдерживают 1 ч при 5 С, затем фильтруют через стеклянный фильтр, осадок промывают дважды с 125 мл изопропилового спирта, отжимают и высушивают в сушильном шкафу в вакууме (35 С/30 ммНg/18 ч). Получают 91,7 г смеси изомеров Z и Е (6) и (7) в соотношении Z/E=75/25 (ВЭЖХ внутренний стандарт) и с выходом 95%. Синтез описан в публикации (используемый растворитель: дихлорметан: используемое основание:NaH) K.G. Piney et al. Bioorg. Med. Chem. 8(2000) 2417-2425. Примечание: были испробованы разнообразные рабочие условия, такие как растворители: ТГФ, ацетонитрил, метанол и другие спирты, дихлорметан, NMP, DMF, DMS0 и т.д.; основания: трет.-бутилат калия, трет.-амилат натрия, гидроксид натрия, NaH, Buli/LDA, карбонат калия и т.д.; температуры: от -10 С до температуры кипения некоторых растворителей. Гидрохлорид Z-2-метокси-5-[2-(3,4,5-триметоксифенил)винил]фениламина (8). В трехгорлый сосуд емкостью 2 л, снабженный механической мешалкой, термометром, Y-образной трубкой, загрузочной воронкой для твердых веществ, охладителем с пузырьковым расходомером сверху и баней с подогревом, загружают при 20 С и в атмосфере азота 80 г смеси Z и Е 75/25 2-метокси-5-[2(3,4,5-триметоксифенил)винил]нитрофенила (6) и (7), 640 мл абсолютного этанола и 160 мл дистиллированной воды. Быстро перемешивают и нагревают на масляной бане, при 50 С к суспензии добавляют 7,8 мл 6 Н соляной кислоты, затем температуру среды повышают до 772 С; среда становится практически полностью растворима. В течение 5 мин порциями добавляют 52 г порошкового железа. Начиная с первого добавления среда превращается в раствор, затем на стенках сосуда формируется черноватый осадок. Среду выдерживают 2 ч при 772 С, с помощью ВЭЖХ контролируют исчезновение исходных нитропроизводных (6) и(7). Среде дают остыть до 40 С и фильтруют через стеклянный фильтр с фильтрующим материалом Clarcel, осадок промывают дважды с 160 мл смеси этанол/вода 80/20. Фильтрат, маточные и промывочные растворы концентрируют в ротационном испарителе; с момента исчезновения азеотропа начинается осаждение масла в остаточной водной фазе. Эту водную фазу экстрагируют в колбе дважды с помощью 300 мл дихлорметана, затем промывают органическую фазу дважды с 300 мл полунасыщенного водного раствора хлорида натрия и с 300 мл дистиллированной воды. Органическую фазу концентрируют досуха в ротационном испарителе; получают 76 г масла,имеющего соотношение Z/E=80/20 согласно данным ВЭЖХ. Это масло растворяют в 591 мл метанола и переливают в сосуд емкостью 1 л при перемешивании, затем добавляют 100 мл 2,32 Н раствора соляной кислоты в метаноле, запускают реакцию и оставляют на ночь при перемешивании для осаждения. Количество метанол + раствор соляной кислоты в метаноле таково, что конечная концентрация изомера Z (определенная с помощью ВЭЖХ) составляет 8,8% вес./об. На следующее утро среду фильтруют через стеклянный фильтр; высушенный осадок имеет массу 8,2 г и состоит исключительно из изомера Е (ВЭЖХ). Фильтрат (693 г) с соотношением Z/E = 86/14 (ВЭЖХ, внутренний стандарт) концентрируют вполовину в ротационном испарителе, добавляют к 347 г концентрата 400 мл ацетонитрила и снова концентрируют до получения массы концентрата 347 г. Добавляют 1000 мл ацетонитрила и концентрируют до начала кристаллизации, затем концентрат переливают в сосуд емкостью 4 л, содержащий 1500 мл ацетонитрила, при перемешивании; при 60 С наблюдается обильное выпадение осадка из среды. Среду поддерживают 2,5 ч при 60 С и при перемешивании, дают остыть до 30 С приблизительно за 1 ч, кашицу фильтруют через стеклянный фильтр, изомер Е (9) растворен в фильтрате, осадок промывают дважды с 200 мл ацетонитрила и высушивают в сушильном шкафу (35 С/30 ммНg/18 ч). Получают 45,7 г Z-2-метокси-5[2-(3,4,5-триметоксифенил)винил]фениламина (8) с титром по ВЭЖХ, внутренним стандаром, составляющим 97%, и выходом тель-кель 56%, что соответствует выходу изомера Z полученного по отношению к изомеру Z исходному 72%. Пример 2. Синтез согласно способу по пути V 03 по изобретению. Преимуществом способа по пути V 03 по сравнению со способом по пути V 02, называемым обратный Виттиг, является проведение реакции Виттига между уже восстановленным продуктом, аминоальдегидом (1 а) и фосфонием (2 а) и, следовательно, исключение химической стадии с использованием продуктов CMR. Гидрохлорид (Z)-N-[2-метокси-5-[2-(3,4,5-триметоксифенил)винил]фенил]-L-серинамида.-6 007965 Общая схема синтеза Реакция Виттига В трехгорлый сосуд емкостью 2 л, заполненный аргоном и снабженный механической мешалкой,термометром, Y-образной трубкой, загрузочной воронкой для твердых веществ, охладителем с пузырьковым расходомером сверху и баней с подогревом, загружают 20 г 3-нитро-4-метоксибензальдегида (1) и 350 мл абсолютного этанола, перемешивают, нагревают до 60 С; среда превращается в раствор. При 60 С добавляют по каплям 115 мл дистиллированной воды, затем 14 мл 2 Н соляной кислоты. Затем маленькими порциями добавляют 24,7 г порошкового железа. В течение 2 ч дают температуре среды остыть до температуры окружающей среды. Реакция является полной (по результатам тонкослойной хроматографии). Среду фильтруют через целит и концентрируют в вакууме, осадок помещают в дихлорметан и промывают органический раствор дважды дистиллированной водой, затем высушивают над сульфатом магния, фильтруют и концентрируют досуха в вакууме. Получают 16 г сырого продукта (1 а), который подвергают хроматографии на колонке с силикагелем, используя в качестве элюента дихлорметан. Получают 2 фракции, содержащие чистый целевой продукт, которые после концентрации дают 11,5 г чистого продукта (1 а), что соответствует выходу 69%. Спектр n 2810-V ЯМР 1 Н (300 МГц, (CD3)2SO d6,ч./млн): 3,88 (с, 3 Н), 5,11 (массив, 2 Н), 7,01 (д,J=8 Гц, 1 Н), 7,14 (д, J=2 Гц, 1 Н), 7,18 (д, J=8 Гц, 1 Н), 9,53 (с, 1 Н). Масс-спектр n210112, m=151. Примечание: фосфоний (2 а) является сырьем, описанным в патенте Ajinomoto Co., Ltd US 5525632 и WO 01/12579 A2. В трехгорлый сосуд емкостью 250 мл, заполненный азотом и снабженный магнитной мешалкой,термометром, Y-образной трубкой, капельницей, охладителем с пузырьковым расходомером сверху и охлаждаемой баней, загружают 8,0 г фосфония (2 а), затем 2,20 г аминобензальдегида (1 а) и 100 мл толуола. Суспензию охлаждают при перемешивании до 5 С и приливают за 15 мин 3,51 мл 25%-ого (по весу) раствора метилата натрия в метаноле. После выдерживания в течение 2,5 ч при 5 С реакция остается неполной (коэффициент превращения: 45%), но далее не развивается (ВЭЖХ) и отношение Z/E составляет 61/39. Приливают 0,2 мл уксусной кислоты, разбавленной 50 мл воды, повышают температуру до 13 С, перемешивают 30 мин, затем переливают в колбу, концентрируют органическую фазу в вакууме в ротационном испарителе и получают 8 г масла желтого цвета. По результатам ВЭЖХ, это масло содержит исходный альдегид, фосфиноксид и целевую смесь Z/E с соотношением 61/39. Масло хроматографируют на колонке с силикагелем (40 частей маc./маc.), используя в качестве элюента смесь циклогексан/этилацетат/триэтиламин (50/50/2). 2 серии фракций объединяют и концентрируют досуха, первый сухой экстракт массой 360 мг содержит 93% изомера Z + неидентифицированные примеси, второй, массой 2,09 г, содержит исходный альдегид и смесь Z/E, составляющих 39 и 37,5% по результатам ВЭЖХ (внутренний стандарт). Итоговое количество изомера Z (8'), определяемое с помощью ВЭЖХ (внутренний стандарт), составляет 1,15 г на 2,20 г использованного альдегида, что соответствует выходу 24%. Пример 3. Синтез согласно способу по пути V 03 по изобретению. Как и для пути V 02, преимуществом пути V 03 по сравнению с первым способом по пути V 02, называемым обратный Виттиг, является проведение реакции Виттига между уже восстановленным продуктом, бромидом (3-амино-4-метоксибензил)трифенилфосфонием (1b) и 3,4,5-триметоксибензальдегидом (5) и, следовательно, отсутствие химической стадии с использованием продуктов CMR. Гидрохлорид (Z)-N-[2-метокси-5-[2-(3,4,5-триметоксифенил)винил]фенил]-L-серинамида.-8 007965 Общая схема синтеза Реакция Виттига В трехгорлый сосуд емкостью 1 л, снабженный механической мешалкой, термометром, Y-образной трубкой, загрузочной воронкой для твердых веществ, охладителем с пузырьковым расходомером сверху и баней с подогревом, загружают 30 г продукта (4), 240 мл этанола и 60 мл дистиллированной воды. К суспензии добавляют при перемешивании 1,7 6 мл 6 Н соляной кислоты и нагревают до 70 С. Затем за 15 мин добавляют маленькими порциями 9,9 г порошкового железа; среда остается нерастворимой. Среду выдерживают 2 ч при 75 С, органические вещества медленно растворяются, параллельно формируется коричневатый осадок железа и оксида железа. По результатам ВЭЖХ остается еще 5% исходных соединений; снова добавляют 2 г железа и продолжают нагревание в течение 1 ч; превращение становится полным. Среду охлаждают до 40 С и фильтруют через Clarcel, промывают с помощью 100 мл 20%-ного водного раствора этанола и концентрируют фильтрат досуха в вакууме в ротационном испарителе. Осадок помещают в 300 мл изопропилового спирта и кристаллизуют из среды, перемешивают и нагревают до 50 С; при этом среда снова превращается в раствор. Затем приливают 14 мл 5 Н раствора соляной кислоты в изопропиловом спирте; происходит выпадение осадка из среды; среду выдерживают 1 ч при 50 С, затем дают остыть до температуры окружающей среды. Кашицу фильтруют через стеклянный фильтр, осадок промывают при помощи 50 мл изопропилового спирта, тщательно отжимают и высушивают в сушильном шкафу в вакууме. Получают 27,3 г (1b) как такового (не очищенного) с выходом 89,9%. Спектр n 4584-V ЯМР 1H (300 МГц, (CD3)2SO d6,ч./млн): 3,78 (с, 3 Н), 5,03 (д уш., J=15 Гц, 2 Н),6,43 (массив, 1 Н), 6,62 (суш., 1 Н), 6,82 (д уш., J=8 Гц, 1 Н), 7,60-8,00 (м, 15 Н). Масс-спектр n211915, m=397m/z=263 [PPh3H]+ пик молекулярного иона Инфракрасный спектр 4 2 638 6 КВr В трехгорлый сосуд емкостью 250 мл, заполненный азотом и снабженный магнитной мешалкой,термометром, Y-образной трубкой, капельницей, охладителем с пузырьковым расходомером сверху и охлаждаемой баней, загружают 11,02 г продукта (1b), 4 г продукта (5) и 70 мл толуола. Суспензию охлаждают при перемешивании до 5 С и приливают за 15 мин 4,92 мл 25%-ного (по массе) раствора метилата натрия в метаноле. Суспензию перемешивают в течение 2,5 ч при 5 С, затем приливают 0,2 мл уксусной кислоты, разбавленной 50 мл воды, повышают температуру до 14 С; среда становится очень густой, ее разбавляют 10 мл толуола и 10 мл воды; остается нерастворимая часть коричневого цвета. Смесь фильтруют через Clarcel, осадок промывают трижды при помощи 50 мл толуола (промывочные растворы содержат почти исключительно исходный альдегид и их не соединяют с двухфазным фильтратом), прозрачный фильтрат (рН 12) переливают в колбу и органическую фазу концентрируют досуха в вакууме при 40 С; отношение Z/E, определенное с помощью ВЭЖХ, составляет 43/57. Полученное масло коричневого цвета подвергают хроматографии на колонке с силикагелем (100 частей вес./вес.), используя в качестве элюента смесь циклогексан/этилацетат/триэтиламин (50/50/2). Две серии фракций объединяют и концентрируют досуха; первый сухой экстракт массой 1,1 г содержит 14% изомера Е и 59% Z, второй, массой 1,08 г, содержит 86% изомера Е и 7% Z. Итоговое количество изомера Z (8'), определенное при помощи ВЭЖХ (внутренний стандарт), составляет 0,725 г на 4 г использованного альдегида, что соответствует как таковому выходу 11,3%. трет.-Бутиловый эфир 2-4-2-метокси-5-[2-(3,4,5-триметоксифенил)винил]фенилкарбамоил-2,2 диметилоксазолидин-3-карбоновой кислоты (11). Высвобождение основания (8') из гидрохлорида (8). В колбу Эрленмейера емкостью 1 л загружают 44 г продукта (8), 16 г гидрокарбоната натрия, затем 200 мл дистиллированной воды и 375 мл дихлорметана. Перемешивают в течение 20 мин при температуре окружающей среды и получают две прозрачные фазы. Органическую фазу отделяют декантацией, высушивают над сульфатом натрия, затем фильтруют. Получают приблизительно 400 мл хлорметиленового раствора, содержащего продукт (8'). Получение 3-трет.-бутилового эфира 2,2-диметилоксазолидин-3,4-дикарбоновой кислоты (10). Этот продукт, хотя и выпускается серийно, встречается очень редко; его получали омылением литином его метилового сложного эфира согласно J. ORG. CHEM. 63(12) Р. 3983(1998). Спектр ЯМР 1H (300 МГц, (CD3)2SO d6,ч./млн): 1,38 (с, 1 Н), 1,45 (с, 9 Н), 1,55 (с, 3 Н), 3,95 (м, 1 Н),4,16 (м, 1 Н), 4,31 (м, 1 Н), 12,50-13,10 (массив уш., 1 Н). Масс-спектр: n213135, m=245- 10007965 Соединение В трехгорлый сосуд емкостью 2 л, снабженный механической мешалкой, термометром, Y-образной трубкой, загрузочной воронкой для твердых веществ, охладителем с пузырьковым расходомером сверху и ледяной баней, загружают раствор продукта (8'), добавляют 600 мл дихлорметана и охлаждают при перемешивании. При 5 С добавляют 42,9 г 3-трет.-бутилового эфира 2,2-диметилоксазолидин-3,4-дикарбоновой кислоты (10), который растворяется, затем добавляют порциями при температуре от 5 до 10 С 48 г гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (EDCI). Среде дают медленно нагреться до температуры окружающей среды в процессе таяния льда в водяной бане в течение ночи. На следующее утро добавляют 330 мл дистиллированной воды и интенсивно перемешивают. В течение 30 мин среда становится мутной (гидролиз EDCI); перемешивание продолжают еще 30 мин. Среду переливают в колбу и промывают органическую фазу последовательно дважды при помощи 280 мл 0,55 Н гидроксида натрия, затем при помощи 300 мл дистиллированной воды. Органическую фазу концентрируют досуха в ротационном испарителе (50 С/50 ммНg). Получают 79,4 г клейкого масла (11), твердеющего при 20 С, с выходом по весу по отношению к использованному продукту (8) 117%. Спектр n=5 578-V 1 Н ЯМР (400 МГц, (CD3)2SO d6, при температуре 373 К,ч./млн): 1,41 (с, 9 Н), 1,53 (с, 3 Н), 1,64 (с,3 Н), 3,64 (с, 6 Н), 3,71 (с, 3 Н), 3,86 (с, 3 Н), 3,99 (д, J=9 и 3 Гц, 1 Н), 4,19 (дд, J=9 и 7 Гц, 1 Н), 4,52 (дд, J=7 и 3 Гц, 1 Н), б,48 (д, J=12,5 Гц, 1 Н), 6,55 (д, J=12,5 Гц, 1 Н), 6,58 (с, 2 Н), 7,02 (м, 2 Н) , 8,13 (с уш., 1 Н), 8,82 (с уш., 1 Н). Масс-спектр n213565, m=542m/z=443 [MH-BOC]+ Инфракрасный спектр 425857 ССl4 3409; 2982; 2938; 2837; 1712; 1698; 1534; 1363; 1249; 1133; 1092 и 851 см-1 Были использованы другие условия связывания, такие как смешанный ангидрид (пивалоилхлорид/(10;EDCI, HCl в дихлорметане дает наилучший результат. Гидрохлорид (Z)-N-[2-метокси-5-[2-(3,4,5-триметоксифенил)винил]фенил]-L-серинамида. В трехгорлый сосуд емкостью 1 л, снабженный механической мешалкой, термометром, Y-образной трубкой, охладителем с пузырьковым расходомером сверху и баней с подогревом, загружают при 20 С 61,8 г продукта (11) в растворе в 54 мл метанола, добавляют 150 мл изопропилацетата, 99 мл 2,3 Н раствора соляной кислоты в метаноле и 8,2 мл дистиллированной воды. Перемешивают и нагревают до 60 С в течение 3 ч. Раствор охлаждают до 40 С и очищают фильтрацией через стеклянный фильтр n 4, затем промывают фильтр 40 мл метанола. Фильтрат снова помещают в трехгорлый сосуд при перемешивании и добавляют 194 мл изопропилацетата, нагревают до 40 С, добавляют 0,2 г продукта (12), затем по каплям приливают в течение 1 ч 194 мл изопропилацетата; в процессе приливания среда медленно кристаллизуется. Среде дают остыть до комнатной температуры, затем охлаждают до 5 С и оставляют при этой температуре в течение ночи. На следующее утро кашицу фильтруют через стеклянный фильтр, осадок отжимают и промывают 4 раза по 50 мл изопропилацетата, тщательно отжимают, затем высушивают в сушильном шкафу до постоянной массы (35 С/10 ммНg). Получают 28 г продукта (12) с выходом за 2 стадии (соединение, затем снятие защиты) 56% и титром по ВЭЖХ (внутренний стандарт) 98%. Этот показатель соответствует общему выходу как таковому для синтеза, осуществленного согласно первому способу по пути V 02, составляющему 30% (полученный продукт (12) по отношению к ис- 11007965 пользованному продукту (5. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения комбретастатинов следующей общей формулы (I) в которой А обозначает аминогруппу, отличающийся тем, что конденсируют соль (3,4,5 триметоксибензил)трифенилфосфония с 3-амино-4-метоксибензальдегидом или соль (3-амино-4 метоксибензил)трифенилфосфония с 3,4,5-триметоксибензальдегидом. 2. Способ по п.1,отличающийся тем,что конденсируют соль(3,4,5 триметоксибензил)трифенилфосфония с 3-амино-4-метоксибензальдегидом. 3. Способ по п.1 или 2, отличающийся тем, что реакцию проводят предпочтительно в присутствии основания, выбранного из трет-бутилата калия, трет-амилата натрия, гидрида натрия, бутиллития, литийдиизопропиламина, метилата натрия, карбоната калия или щелочных производных гексаметилдисиланов. 4. Способ по п.1 или 2, отличающийся тем, что используют метилат натрия. 5. Способ по п.1 или 2, отличающийся тем, что реакционный растворитель, инертный в отношении реакции, выбирают из простых эфиров (ТГФ), полярных апротонных растворителей (ацетонитрил, NMP,DMF, DMSO), спиртов, ароматических растворителей или воды. 6. Способ по п.4, отличающийся тем, что температура реакции предпочтительно составляет от 0 до 10 С. 7. Способ получения соединений формулы (I) в форме соли серина путем соединения производных формулы (IIа) с циклическим производным защищенного серина формулы (IIb) где PG обозначает защитную группу аминогруппы, с получением нового промежуточного соединения следующей общей формулы которое затем подвергают снятию защиты. 8. Промежуточное соединение, образующееся в результате соединения аминокомбретастатина и циклического производного защищенного серина, отличающееся тем, что оно соответствует следующей формуле: в которой PG обозначает защитную группу. 9. Промежуточное соединение по п.8, отличающееся тем, что PG обозначает защитную группу, выбранную из следующих групп: трет.-бутоксикарбонил, бензилоксикарбонил (CBZ) или 9 флуоренилметилоксикарбонил (FMOC). Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6
МПК / Метки
МПК: C07D 263/06, C07C 213/02
Метки: способ, получения, комбретастатинов
Код ссылки
<a href="https://eas.patents.su/13-7965-sposob-polucheniya-kombretastatinov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения комбретастатинов</a>
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Мазюри Алан, Дельтиль Мишель, Бонне Алан
МПК: C07H 17/08
Метки: биологически, активных, получения, способ, 5-0-дезозаминил-6-0-метилэритронолида, производные, продуктов, применение
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Ларкин Джон Патрик, Крок Вероник, Колладан Колетт, Руссель Патрик
МПК: C07D 487/04
Метки: 1,2-а, кислоты, производные, способ, октагидро-6, терапевтически, соединений, получения, 10-диоксо-6н-пиридазино, применение, 1,2, активных, диазепин-1-карбоновой
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Способ получения защищенного 4-аминометилпирролидин-3-она, промежуточные соединения и способ получения 3-аминометил-4-алкоксииминопирролидина

Номер патента: 2499
Опубликовано: 27.06.2002
Авторы: Ким Вон Суп, Моон Кванг Юл, Ли Тае Хи, Чанг Джей Хиок
МПК: C07D 207/24
Метки: 4-аминометилпирролидин-3-она, 3-аминометил-4-алкоксииминопирролидина, получения, соединения, защищенного, способ, промежуточные
Формула / Реферат:
1. Способ получения соединения формулы (1) в которой Р1 и Р2 обозначают защитные группы; включающий а) восстановление соединения формулы (5) где Р1 такой, как определено для формулы (1); в присутствии катализатора никель Ренея в растворителе в атмосфере водорода в присутствии одной или более добавок с получением соединения формулы (6) где Р1 такой, как определено для формулы (1); b) защиту аминогруппы в присутствии одного или более оснований...
Способ получения карведилола

Номер патента: 7393
Опубликовано: 27.10.2006
Авторы: Герцек Рихард, Прокса Богумил, Шкода Алойз
МПК: A61P 9/00, C07D 209/88
Метки: получения, карведилола, способ
Формула / Реферат:
1. Способ получения карведилола, отличающийся тем, что включает взаимодействие 4-(оксиран-2-илметокси)-9Н-карбазола с солью 2-(2-метоксифенокси)этиламина в количестве от 2,0 до 5,0 эквивалентов относительно исходного карбазола, где указанная соль может содержать от 0 до 10% воды, в присутствии основания, которое представляет собой карбонат щелочного металла или щелочно-земельного металла, которое добавляют в количестве от 2,0 до 5,0 эквивалентов...
Промежуточное соединение для получения паклитаксела и способ получения промежуточного соединения

Номер патента: 678
Опубликовано: 28.02.2000
Авторы: Систи Николас Дж., Чандер Мадхави С., Свинделл Чарльз С.
МПК: C07D 305/14
Метки: соединения, способ, соединение, получения, паклитаксела, промежуточного, промежуточное
Формула / Реферат:
1. Промежуточное соединение для получения паклитаксела, где промежуточное соединение имеет общую формулу где P1 представляет гидрирующуюся бензильную защитную группу. 2. Промежуточное соединение паклитаксела по п.1, где гидрирующуюся бензильную защитную группу выбирают из группы, включающей бензилоксиметил и бензил. 3. Способ получения промежуточного соединения для получения паклитаксела, где промежуточное соединение имеет общую формулу ...
Предыдущий патент: Способ получения эхинокандиновых соединений
Следующий патент: Пиридиноилпиперидины как агонисты 5-нт1f
Случайный патент: Способы замедления отверждения чувствительных к влаге отверждаемых эластомеров