Производные оксоаденина, конъюгированные с фосфо- или фосфонолипидами









Формула / Реферат
1. Соединение формулы (II)

где R1 выбран из С1-6алкила, С1-6алкиламино, C1-6алкокси, С3-6циклоалкил-С1-6алкила, С3-6циклоалкил-С1-6алкиламино, С3-6циклоалкил-С1-6алкокси, С1-6алкокси-С1-6алкила, С1-6алкокси-С1-6алкиламино или С1-6алкокси-С1-6алкокси;
n равно целому числу от 1 до 6;
X представляет собой СН или N;
Q представляет собой О, NH или ковалентную связь;
W представляет собой О или S;
m равно 0 или целому числу от 1 до 6;
R2 представляет собой Н или прямой или разветвленный C4-C24алкил или алкил-С(О);
R3 представляет собой прямой или разветвленный C4-C24алкил или алкил-С(О);
или его фармацевтически приемлемая соль.
2. Соединение формулы (IX)

где R1 выбран из C1-6алкила, C1-6алкиламино, С1-6алкокси, С1-6алкокси-С1-6алкиламино или С1-6алкокси-С1-6алкокси;
n равно 1 или 2;
X представляет собой СН или N;
Q представляет собой О, NH или ковалентную связь;
m равно 0, 1 или 2;
R2 представляет собой Н или прямой или разветвленный C4-C24алкил или алкил-С(О) и
R3 представляет собой прямой или разветвленный C4-C24алкил или алкил-С(О);
или его фармацевтически приемлемая соль.
3. Соединение по п.2,
где R1 представляет собой С1-6алкокси или С1-6алкоксиС1-6алкил;
Q представляет собой О или NH;
m равно 0, 1 или 2;
R2 представляет собой Н или прямой или разветвленный C4-C24алкил или алкил-С(О) и
R3 представляет собой прямой или разветвленный C4-C24алкил или алкил-С(О);
или его фармацевтически приемлемая соль.
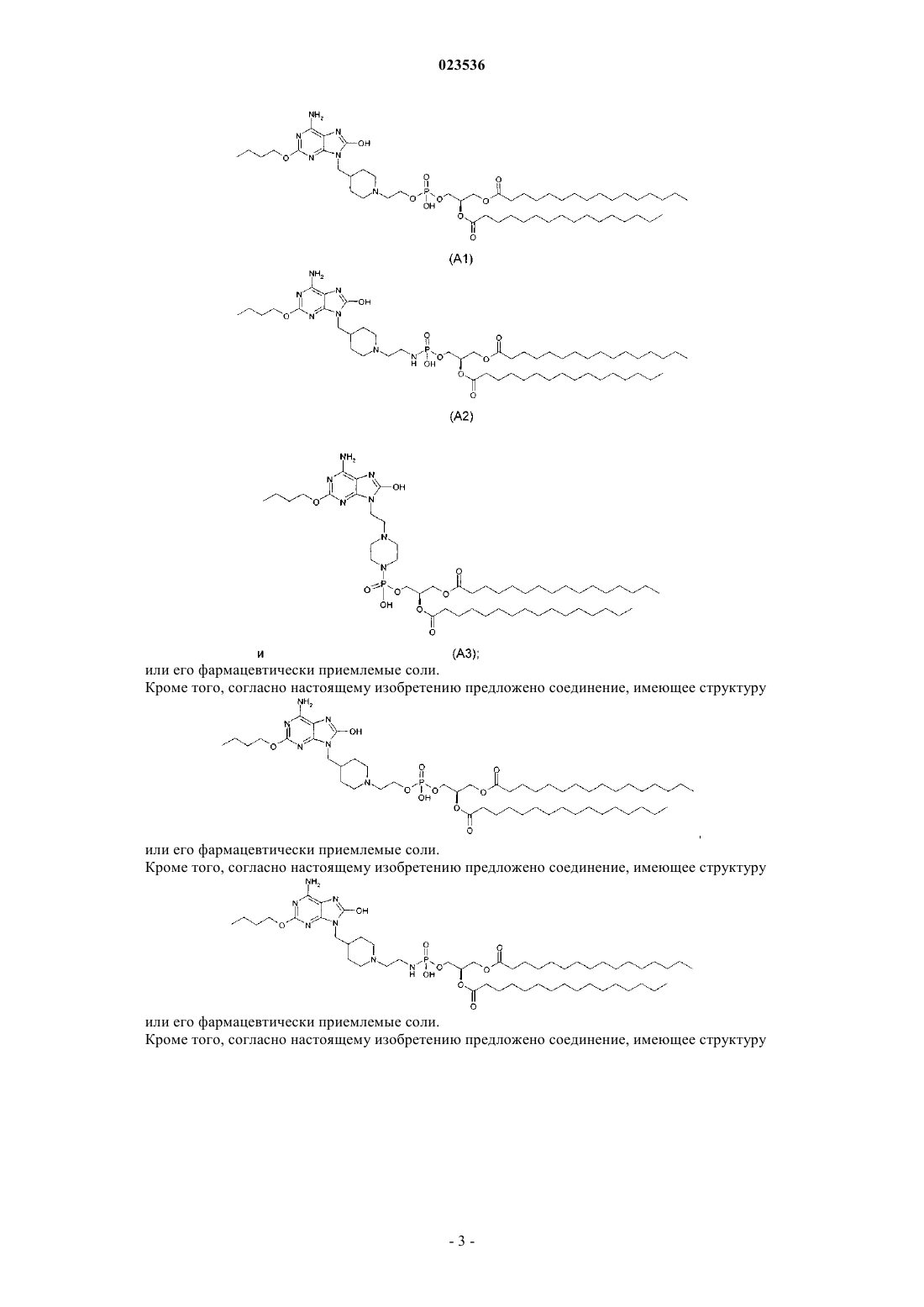
4. Соединение, выбранное из


или его фармацевтически приемлемая соль.
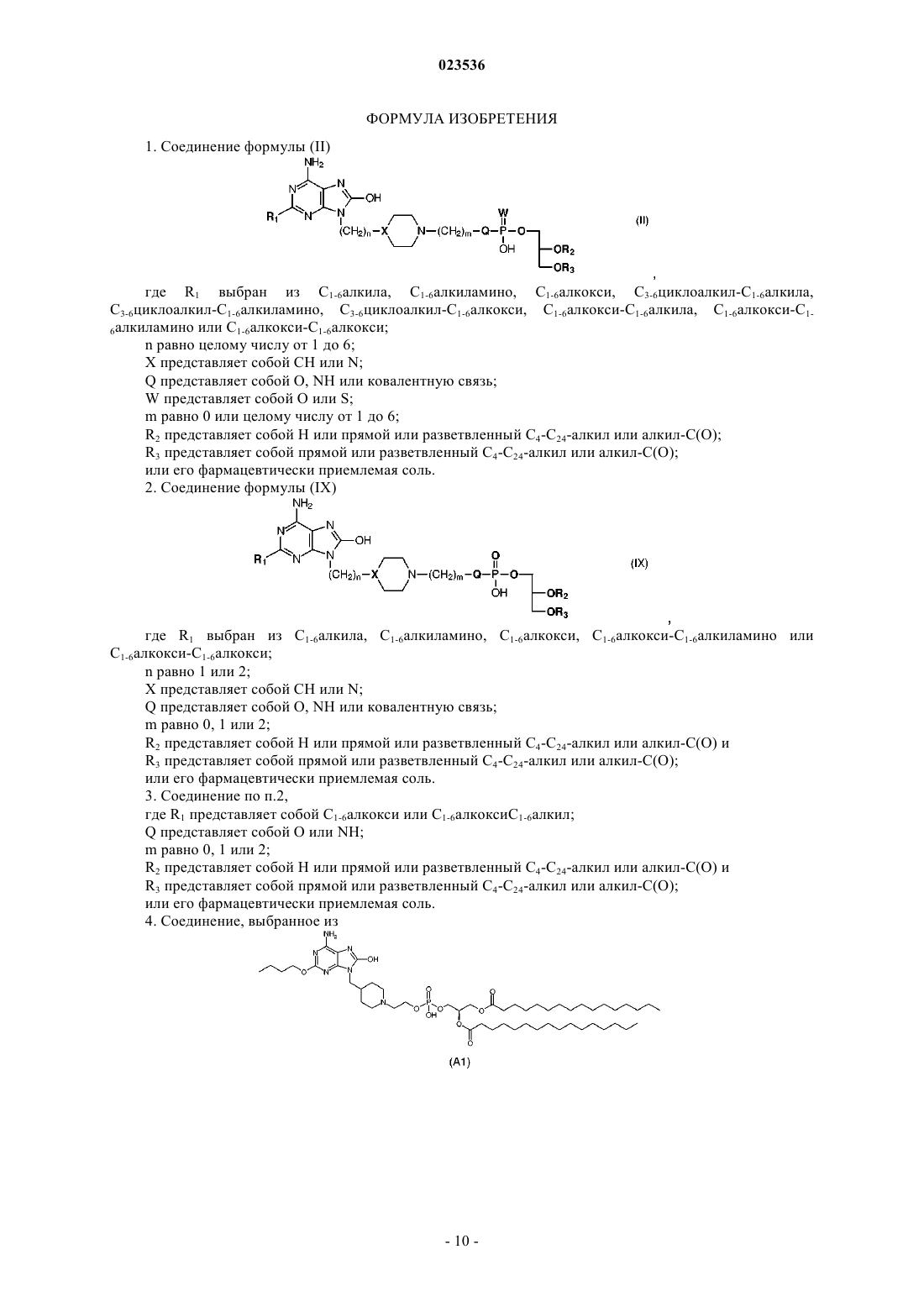
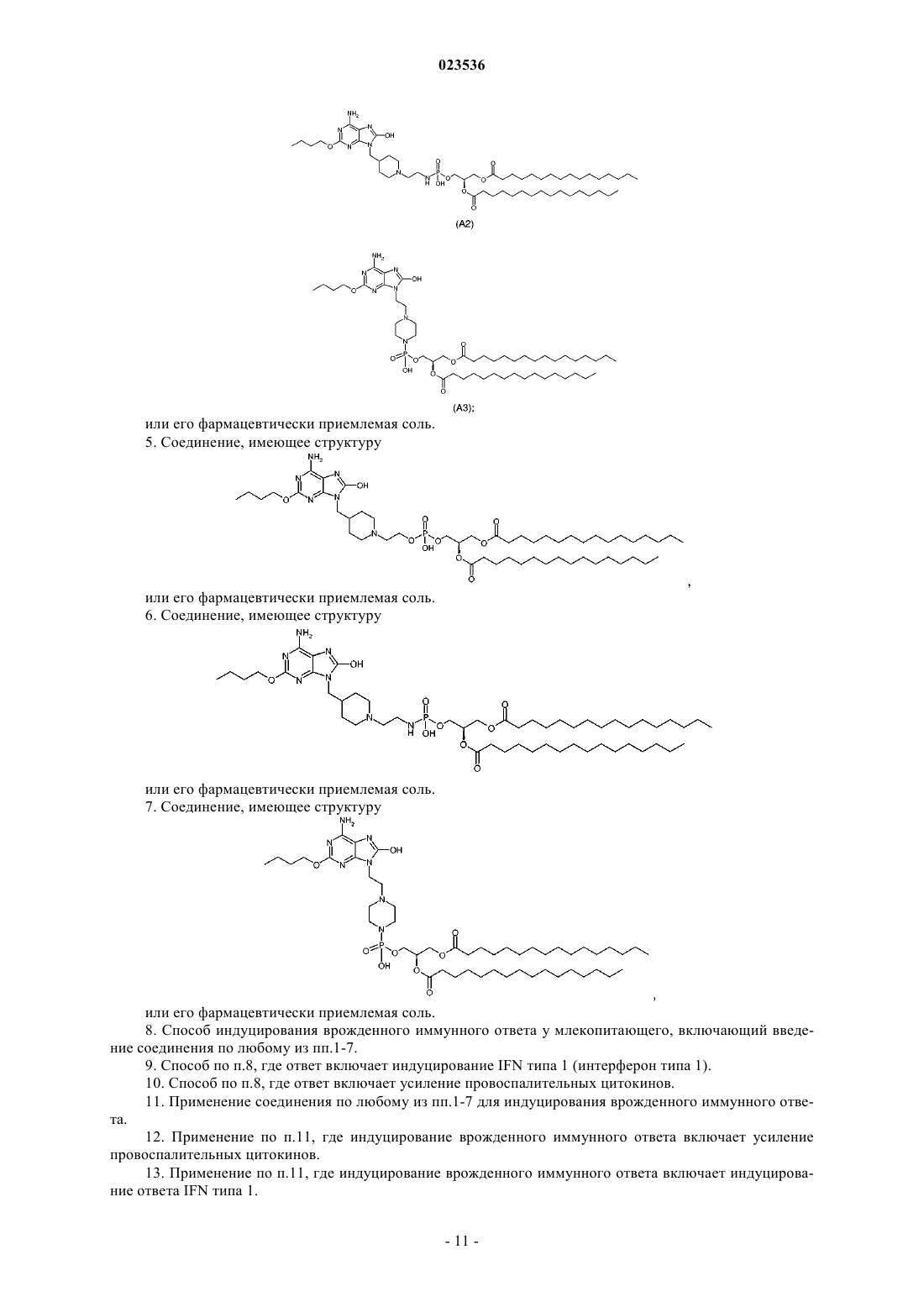
5. Соединение, имеющее структуру

или его фармацевтически приемлемая соль.
6. Соединение, имеющее структуру

или его фармацевтически приемлемая соль.
7. Соединение, имеющее структуру

или его фармацевтически приемлемая соль.
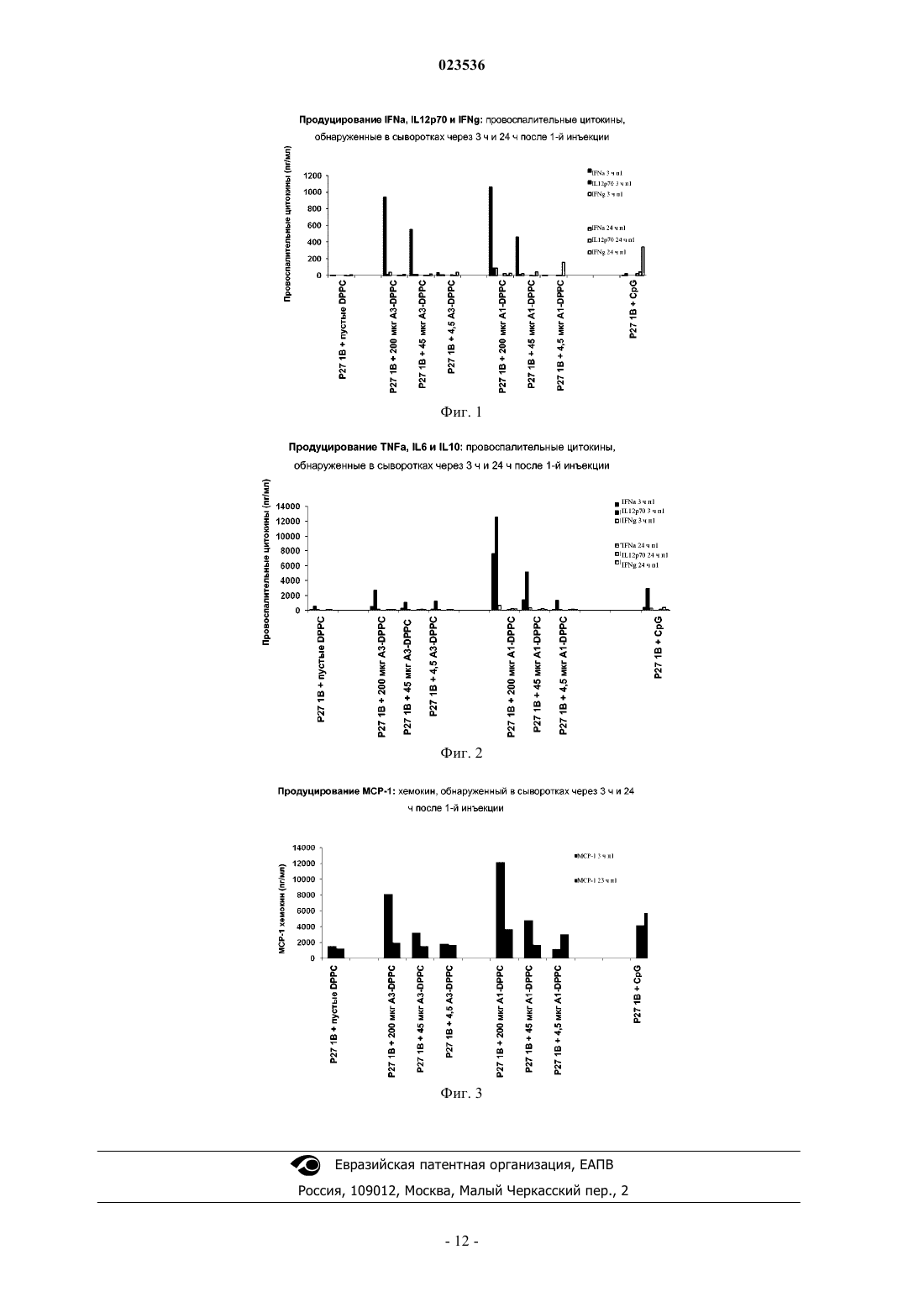
8. Способ индуцирования врожденного иммунного ответа у млекопитающего, включающий введение соединения по любому из пп.1-7.
9. Способ по п.8, где ответ включает индуцирование IFN типа 1 (интерферон типа 1).
10. Способ по п.8, где ответ включает усиление провоспалительных цитокинов.
11. Применение соединения по любому из пп.1-7 для индуцирования врожденного иммунного ответа.
12. Применение по п.11, где индуцирование врожденного иммунного ответа включает усиление провоспалительных цитокинов.
13. Применение по п.11, где индуцирование врожденного иммунного ответа включает индуцирование ответа IFN типа 1.

Текст
Новые производные оксоаденина, содержащие замещенную азотсодержащим гетероциклилом молекулу оксоаденина, ковалентно связанную с фосфо- или фосфонолипидом, формулы (II)(71)(73) Заявитель и патентовладелец: ГЛАКСОСМИТКЛАЙН БАЙОЛОДЖИКАЛС СА (BE) Предшествующий уровень техники Настоящее изобретение относится к новым соединениям-адъювантам, способам их получения, композициям, содержащим их, и к их применению в качестве вакцинных адъювантов. Очистка и упрощение микробиологических вакцин, а также использование синтетических и рекомбинантных субъединичных антигенов для улучшения технологичности и безопасности вакцин привели к снижению эффективности вакцин. Это стало причиной проведения исследований по совместному введению адъювантов с антигенами для усиления активности вакцин и усиления слабой иммуногенности синтетических и рекомбинантных эпитопов. Адъюванты представляют собой добавки, которые усиливают гуморальные и/или опосредованные клетками иммунные ответы на вакцинный антиген. Однако создание вакцинных адъювантов исторически было сопряжено с трудностями из-за сложной природы молекулярных механизмов, вовлеченных в функцию иммунной системы. Хотя давно известно, что добавление микробных компонентов усиливает адаптивные иммунные ответы, только недавно было показано, чтоtoll-подобные рецепторы (TLR) на клетках, вовлеченных в иммунный контроль, таких как эпителиальные и дендритные клетки, захватывают многие из этих микробных продуктов через так называемые "патогенассоциированные молекулярные паттерны" (или РАМР). Представляется, что многие вакцинные адъюванты и автономные иммуномодуляторы взаимодействуют с членами семейства TLR. Из 10 известных TLR, которые идентифицированы у людей, пять ассоциированы с распознаванием бактериальных компонентов (TLR 1, 2, 4, 5, 6), и другие четыре (TLR 3, 7, 8, 9) по всей вероятности ограничены цитоплазматическими компартментами и вовлечены в детекцию вирусной РНК (TLR 3, 7, 8) и неметилированной ДНК (TLR 9) (Iwasaki, A., Nat. Immunol. 2004, 5, 987). Активация TLR регулирует внутриклеточные сигнальные пути и приводит к экспрессии гена через взаимодействие с внутриклеточными адаптерными молекулами, такими как MyD88, TRIF, TIRAP и TRAM (Akira, S. Nat. Rev. Immunol. 2004, 4, 499; Takeda, K. Semin. Immunol. 2004, 16, 3). Эти адаптерные молекулы могут дифференцированно регулировать экспрессию воспалительных цитокинов/хемокинов и интерферонов типа I (IFN/),что может приводить к преимущественному усилению антиген-специфических гуморальных и опосредованных клетками иммунных ответов (Zughaier, S. Infect. Immun. 2005, 73, 2940). Гуморальный иммунитет является главной линией защиты от бактериальных патогенов, тогда как индуцирование цитотоксических Т-лимфоцитов (CTL) по всей вероятности является решающим для защитного иммунитета в случае вирусного заболевания и рака. В случае активации TLR7 и TLR8 идентифицировано несколько разных классов небольших молекул-миметиков природных (U- и/или G-богатых) лигандов вирусной онРНК (однонитевая РНК). Они включают некоторые антивирусные соединения, относящиеся к окисленным гуанозиновым метаболитам(оксогуанозины), которые взаимодействуют главным образом с TLR7 (Heil, F. Eur. J. Immunol. 2003, 33,2987; Hemmi, 2002), и производные аденина, которые связываются с TLR7 и/или TLR8. Иммуностимулирующую способность этих соединений приписывают TLR/MyD88-зависимым сигнальным путям и продуцированию цитокинов, включая IL-6 (интерлейкин-6) и интерфероны типа I (в частности интерферона-а) и типа II. Активация TLR7 или TLR8 приводит к положительной регуляции ко-стимулирующих молекул (например CD-40, CD-80, CD-86) и молекул МНС (главный комплекс гистосовместимости) класса I и II на дендритных клетках (DC). DC являются основными клетками иммунной системы, вовлеченными в захват и презентацию антигенов в Т-лимфоциты. Плазмацитоидные дендритные клетки(pDC), которые преимущественно экспрессируют TLR7, являются профессиональными интерферон-продуцирующими клетками, a mDC (миелоидные дендритные клетки) экспрессируют только TLR8. Активация TLR8 на mDC приводит к преимущественному продуцированию провоспалительных цитокинов,таких как IL-12, TNF- (фактор некроза опухоли альфа) и IFN-, и опосредованного клетками иммунитета (CMI). Было показано, что агонисты TLR7 более эффективны в выработке IFN- и INF-регулируемых цитокинов, а агонисты LR8, которые приводят к реверсии CD4+ регуляторной (Treg) клеточной функции, более эффективны в индуцировании провоспалительных цитокинов, таких как TNF- и IL-12, что свидетельствует о том, что активация TLR7 может быть более важной для антительных ответов (ответыTh2-типа), тогда как активация TLR8 должна индуцировать CMI или иммунные ответы Th1-типа(Gordon, J. Immunol. 2005, 1259). Один класс TLR-активных производных аденина, который привлек большое внимание, составляют оксоаденины. Оксоаденины обычно содержат гидроксильную группу в положении 8 аденинового кольца(часто показано в 8-кето/оксо таутомерной форме), различные заместители в положениях 2 и 9 и незамещенную ароматическую аминогруппу в положении 4. Как и в случае других IFN-индуцирующих производных аденина, таких как имидазохинолины, считается, что незамещенная ароматическая аминогруппа важна для IFN-индуцирующей активности. Было показано, что многие оксоаденины, которые вначале были разработаны для ослабления некоторых побочных эффектов, ассоциированных с имидазохинолинами, являются в значительной степени более сильнодействующими, чем прототипические имидазохинолины, такие как имиквимод и резиквимод, в отношении IFN-индуцирующей активности in vitro иin vivo, но не оказывают рвотного действия, основного клинического побочного эффекта имидазохинолинов. Например, оксоаденин SM360320 в настоящее время находится в предклинической разработке против HCV (вирус гепатита С), и он, как было показано, подавляет репликацию HCV в гепатоцитах человека путем индуцирования IFN типа I, а также через IFN-независимый механизм (Lee, PNAS 2006, 103,1828). Тем не менее, несмотря на то, что класс оксоаденинов по всей вероятности проявляет лучшие общие профили токсичности/биологической активности, чем имидазохинолины, введение SM360320 мышам различными путями приводит к системному болезненному ответу, опосредованному частично воспалительными цитокинами, высвобождаемыми тучными клетками (Hayashi, Am. J. Physiol. Regul. Integr. Сотр. Physiol. 2008, 295, R123). Фактически, во многих случаях большой иммунологический "отпечаток" агонистов TLR7 в общем привел к проблемам токсичности и к временному прекращению клинических испытаний (Strominger, Brain Res. Bull. 2001, 55, 445; Schmidt, Nat. Biotech. 2007, 25, 825). Поскольку большинство агонистов TLR7/8, находящихся в разработке в настоящее время, часто проявляют токсические свойства, являются нестабильными и/или имеют несущественные иммуностимулирующие эффекты, открытие и разработка эффективных и безопасных вакцинных адъювантов, которые активируют TLR7 и/или TLR8, важны для повышения безопасности и эффективности существующих и новых вакцин. Краткое изложение сущности изобретения Согласно настоящему изобретению предложено соединение формулы IIn равно целому числу от 1 до 6;X представляет собой СН или N;m равно 0 или целому числу от 1 до 6;R2 представляет собой Н или прямой или разветвленный C4-C24-алкил или алкил-С(О);R3 представляет собой прямой или разветвленный C4-C24-алкил или алкил-С(О); или его фармацевтически приемлемые соли. В другом варианте выполнения предложено соединение формулы (IX) 6 алкила, где R1 выбран из группы, состоящей из C1-6 алкила, C1-6 алкиламино, C1-6 алкокси,C1-6 алкокси-C1-6 алкиламино, C1-6 алкокси-C1-6 алкокси;n равно целому числу от 1 до 2;X представляет собой СН или N;m равно 0 или целому числу от 1 до 2;R2 представляет собой Н или прямой или разветвленный C4-C24-алкил или алкил-С(О); иR3 представляет собой прямой или разветвленный C4-C24-алкил или алкил-С(О); или его фармацевтически приемлемые соли. Предпочтительно в указанном соединении:m равно 0 или целому числу от 1 до 2;R2 представляет собой Н или прямой или разветвленный C4-C24-алкил или алкил-С(О); иR3 представляет собой прямой или разветвленный C4-C24-алкил или алкил-С(О); или его фармацевтически приемлемые соли. Кроме того, согласно настоящему изобретению предложено соединение, выбранное из группы, состоящей из или его фармацевтически приемлемые соли. Кроме того, согласно настоящему изобретению предложено соединение, имеющее структуру или его фармацевтически приемлемые соли. Кроме того, согласно настоящему изобретению предложено соединение, имеющее структуру или его фармацевтически приемлемые соли. Кроме того, согласно настоящему изобретению предложено соединение, имеющее структуру или его фармацевтически приемлемые соли. Согласно настоящему изобретению также предложен способ индуцирования врожденного иммунного ответа у млекопитающего, включающий введение любого из соединений, определенных выше. Предпочтительно, когда в указанном способе индуцирования ответ включает индуцирование IFN типа 1 (интерферон типа 1). Предпочтительно, когда в указанном способе индуцирования ответ включает усиление провоспалительных цитокинов. Кроме того, согласно настоящему изобретению предложено применение соединения по изобретению для индуцирования врожденного иммунного ответа. Предпочтительно, когда в указанном применении индуцирование врожденного иммунного ответа включает усиление провоспалительных цитокинов. Предпочтительно, когда в указанном применении индуцирование врожденного иммунного ответа включает индуцирование ответа IFN типа 1. Подробное описание репрезентативных воплощений В данном документе авторы изобретения описывают новые липидированные оксоаденины, содержащие замещенную азотсодержащим гетероциклилом молекулу оксоаденина, ковалентно связанную с фосфо- или фосфонолипидом, чтобы облегчать захват в иммунные клетки и усиливать активацию эндосомальных TLR7/8 и презентацию антигена, при введении их самих по себе или в депо-препарате с антигеном. Усиленные иммунные ответы с использованием соединений по данному изобретению возможно являются следствием прямого взаимодействия соединений по изобретению с эндосомальными TLR7 и/или TLR8 или другими молекулярными рецепторами и/или взаимодействия активного метаболита после ферментативного действия с TLR7 и/или TLR8 или другими молекулярными рецепторами. Показано, что соединения по изобретению являются индукторами интерферона- и других иммуностимулирующих цитокинов и могут иметь улучшенный профиль активности-токсичности по сравнению с известными индукторами цитокинов при использовании в качестве адъювантов для вакцинных антигенов в терапевтическом или профилактическом лечении инфекционных заболеваний и рака. Эти соединения также являются новыми per se. По всему тексту данной заявки сделаны ссылки на различные воплощения, относящиеся к соединениям, композициям и способам. Имеется в виду, что описанные различные воплощения предоставляют множество иллюстративных примеров, и их не следует рассматривать как описания альтернатив. Следует отметить, что описания различных воплощений, приведенные здесь, скорее могут быть перекрывающими объем. Воплощения, обсуждаемые в данном описании, являются только иллюстративными и не ограничивают объем настоящего изобретения. Следует иметь в виду, что использованная здесь терминология предназначена только для описания конкретных воплощений и не предназначена для ограничения объема настоящего изобретения. В данном описании изобретения и в прилагаемой формуле изобретения упоминается ряд терминов, которые будут определены как имеющие следующие значения."Алкил" относится к одновалентным насыщенным алифатическим углеводородным группам, имеющим от 1 до 14 атомов углерода и, в некоторых воплощениях, от 1 до 6 атомов углерода. "(Cx-Cy)алкил" относится к алкильным группам, имеющим от x до y атомов углерода. Этот термин охватывает, в качестве примера, линейные и разветвленные углеводородные группы, такие как метил (СН 3-), этил"Амино" относится к группе -NHR4, где R4 независимо выбран из водорода, алкила, алкенила, алкинила, арила, циклоалкила, гетероарила и гетероциклической группы."Циклоалкил" относится к насыщенный или частично насыщенный циклической группе из 3-14 атомов углерода и без кольцевых гетероатомов. Если не указано иное, номенклатура заместителей, которые в прямой форме не определены в данном описании, базируется на указании названия концевой части функциональной группы, затем соседней функциональной группы в направлении точке присоединения. Например, заместитель "арилалкилоксикарбонил" относится к группе (арил)-(алкил)-О-С(О)-. Понятно, что приведенные выше определения не предусматривают недопустимые конфигурации замещения (например метил, замещенный 5 группами фторо). Такие недопустимые конфигурации замещения общеизвестны специалистам. Соединения по данному изобретению являются молекулами-адъювантами, которые содержат молекулу замещенного азотсодержащим гетероциклилом оксоаденина, ковалентно связанную с фосфо- или фосфонолипидной группой. В одном варианте выполнения соединения по настоящему изобретению описаны общей формулойX равно СН или N;R2 равно H или прямой или разветвленный C4-C24-алкил или алкил-С(О);R3 равно прямой или разветвленный C4-C24-алкил или алкил-С(О); или его фармацевтически приемлемые соли. Соединения по данному изобретению получают из известных оксоадениновых промежуточных соединений формулы III, как показано в одном воплощении на схеме 1, путем (1) алкилирования соединения формулы III соответствующим образом защищенным азотсодержащим гетероциклилалкилбромидом формулы IV, (2) одновременного удаления N- и О-защитных групп с соединения формулы V в кислотных условиях, и (3) либо прямого фосфатидилирования N-гетероциклического заместителя соединения VI (m = 0, Q = простая связь) с использованием соединения формулы VII с получением соединений по данному изобретению формулы VIII, где m = 0, и Q = простая связь; либо, альтернативно,N-алкилирования соединения формулы VI N- или О-защищенным амино- или гидрокси-алкилбромидом с последующим удалением защитных групп и О- или N-фосфатидилированием соединением формулыVII с получением соединений по данному изобретению формулы VIII, где m = 2-6, и Q = О или NH. Схема 1 6 алкиламино, В одном дополнительном аспекте изобретение включает соединение формулы (IX)X равно СН или N;R2 равно Н или прямой или разветвленный C4-C24-алкил или алкил-С(О); иR3 равно прямой или разветвленный C4-C24-алкил или алкил-С(О); или его фармацевтически приемлемые соли. В еще одном аспекте данное изобретение включает соединение, имеющее структуру формулы (IX),где R1 представляет собой C1-6 алкокси,n равно 1-2;R2 равно Н или прямой или разветвленный C4-C24-алкил или алкил-С(О); иR3 равно прямой или разветвленный C4-C24-алкил или алкил-С(О); или его фармацевтически приемлемые соли. Пример 1 (соединение А 1). Получение 6-амино-2-бутокси-9-[N-(2-(1,2-дипальмитоил-sn-глицеро-3-фосфо)этил)-4 пиперидинилметил]-8-гидроксипурин (соединение (II), R1 = н-бутокси, n = 1, X = СН, Q = О, Z = О,W = О, m = 2, р = 1, R1 = R2 = н-C15H31CO)(1) Карбонат калия (0,65 г) добавляли к раствору 6-амино-2-бутокси-8-метоксипурина (0,55 г) в сухом N,N-диметилформамиде (DMF, 5,5 мл), и полученную реакционную смесь нагревали при 60 С в течение 1 ч. Добавляли 1,1-диметилэтил-4-(бромметил)-1-пиперидинкарбоксилат (0,5 г), используя дополнительное количество DMF (1,5 мл) для переноса остаточного бромида, и реакционную смесь перемешивали при 50 С в течение 3 ч. После перемешивания в течение 16 ч при комнатной температуре нагревание продолжали при 50 С в течение еще 5 ч до завершения реакции. Затем добавляли воду, и полученную смесь экстрагировали три раза этилацетатом (EtOAc). Объединенные органические экстракты промывали водой, сушили (Na2SO4) и концентрировали. После флэш-хроматографии на силикагеле с использованием МеОН-CHCl3 (градиентное элюирование; 1:992,5:97,5) получили 0,64 г (94%) 1,1 диметилэтил-4-[6-амино-2-бутокси-8-метокси-пурин-9-ил]метил-1-пиперидинкарбоксилата в виде желтого твердого вещества. 1 Н ЯМР (CDCl3):5.14 (s, 2H), 4.27(t, 2H), 4.11 (s, 3 Н), 3.81 (d, 2H), 2.65 (m, 2H), 2.03 (m, 1H), 1.77(2) К раствору соединения, полученного на стадии (1) выше (0,63 г), в МеОН (16 мл) добавляли 4,0 М HCl в диоксане (5,3 мл). После перемешивания в течение 4,5 ч при комнатной температуре реакционную смесь концентрировали и сушили в условиях глубокого вакуума. После флэш-хроматографии на силикагеле с использованием CHCl3-МеОН-Н 2 О (градиентное элюирование; 90:10:175:25:1) получили 0,404 г (85%) гидрохлорида 6-амино-2-бутокси-9-(4-пиперидинилметил)-8-гидроксипурина в виде белого твердого вещества. 1 Н ЯМР (CDCl3):4.84 (s, 5H), 4.27(t, 2H), 3.79 (d, 2H), 3.40 (d, 2H), 2.96 (t, 2H),2.21 (m, 1H), 1.92 (d, 2H), 1.75 (m, 2H), 1.60-1.47 (m,4 Н), 1.00 (t, 3 Н).(3) К раствору соединения, полученного на стадии (2) выше (1,24 г), в DMF (12,6 мл; 0,25 М) добавляли K2CO3 (1,74 г). Полученную смесь нагревали до 60 С в течение 1 ч, обрабатывали 2-бромэтокситрет-бутилдиметилсиланом (0,81 мл) и затем нагревали при 50 С в течение 18 ч. Охлажденную реакци-6 023536 онную смесь гасили водой, переносили в делительную воронку и экстрагировали три раза EtOAc. Объединенные органические слои сушили (Na2SO4) и концентрировали. После флэш-хроматографии на силикагеле получили 1,25 г (83%) 6-амино-2-бутокси-9-[N-(2-О-трет-бутилдиметилсилил)этил)-4 пиперидинилметил-]-8-гидроксипурина в виде не совсем белого твердого вещества. 1 Н ЯМР (CDCl3-CD3OD, 400 МГц):4.26 (m, 4 Н), 3.75 (m, 4 Н), 3.36 (m, 2 Н), 2.96 (m, 2 Н), 2.54 (t,2H), 1.92 (bs, 1H), 1.73 (m, 2 Н), 1.66 (m, 2 Н), 1.40-1.51 (m, 5 Н), 0.98 (t, 3 Н), 0.89 (m, 9 Н), 0.06 (s, 6H).(4) К раствору соединения, полученного на описанной выше стадии (3) (2,74 г), в МеОН (41 мл) добавляли 4,0 М HCl в диоксане (5,7 мл). После перемешивания в течение 0,5 ч при комнатной температуре реакционную смесь концентрировали и сушили в условиях глубокого вакуума. После флэшхроматографии на силикагеле с использованием CHCl3-МеОН-Н 2 О (градиентное элюирование; 90:10:175:25:1) получили 2,29 г (100%) гидрохлорида 6-амино-2-бутокси-9-(N-(2-гидроксиэтил)-4 пиперидинилметил)-8-гидроксипурина в виде белого твердого вещества. 1 Н ЯМР (CD3OD, 400 МГц):4.53 (t, 2 Н), 3.87 (m, 4 Н), 3.66 (d, 2 Н), 3.22 (m, 2 Н), 3.01 (t, 2 Н), 2.21(5) Раствор соединения, полученного на стадии (4) выше (51 мг), и 1,2-дипальмитоил-sn-глицерилгидрофосфоната (93 мг; соединение V1, R1 = R2 = пальмитоил) в пиридине (6,4 мл) обрабатывали пивалоилхлоридом (0,047 мл), и полученную реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Затем добавляли раствор йода (129 мг) в смеси пиридин-вода (19:1, об./об.), и реакционную смесь перемешивали при комнатной температуре в течение 1 ч, разбавляли CHCl3 и затем гасили добавлением 1 М Na2S2O5. Водный слой экстрагировали дважды CHCl3, и объединенные органические слои промывали 1 М триэтиламмонийборатом (рН 8). Органический слой сушили (Na2SO4) и концентрировали. После флэш-хроматографии на силикагеле с использованием CHCl3-MeOH-Et3N (градиентное элюирование; 90:10:175:25:1) получили 38 мг (30%) 6-амино-2-бутокси-9-[N-(2-(1,2-дипальмитоил-snглицеро-3-фосфо)этил-4-пиперидинилметил]-8-гидроксипурина в виде не совсем белого твердого вещества. 1 Н ЯМР (CDCl3-CD3OD, 400 МГц):5.17 (bs, 1H), 4.32 (dd, 1H), 4.20-4.09 (m, 5H), 3.98 (br t, 2H),3.69 (br d, 3 Н), 3.23 (br s, 1 Н), 1.86 (br s, 4H), 1.69 (m, 2H), 1.53 (br s, 4H), 1.42 (dd, 2H), 1.20 (m, 48H), 0.91(1) Способом, аналогичным способу, описанному в примере 1-(3), соединение, полученное, как описано в примере 1-(2) (107 мг), подвергали алкилированию в DMF (1,1 мл) 2-(третбутоксикарбониламино)этилбромидом (67 мг) в присутствии K2CO3 с получением 84 мг (67%) 1,1 диметилэтил 4-[6-амино-2-бутокси-8-метокси-пурин-9-ил]метил-1-пиперидинилэтиламинилкарбоксилата в виде не совсем белого твердого вещества. 1 Н ЯМР (CDCl3-CD3OD, 400 МГц):4.25 (t, 2H), 3.73 (d, 2H), 3.38 (m, 2 Н), 3.20 (m, 2 Н), 2.89 (m,2 Н), 2.44 (m, 2 Н), 1.97 (t, 3 Н), 1.75 (m, 2 Н), 1.66 (m, 2 Н), 1.42-1.51 (m, 11H), 0.97 (t, 3 Н).(2) Способом, аналогичным способу, описанному в примере 1-(4), с соединения, полученного на стадии (1) выше (83 мг), удаляли N-защитную группу с использованием 4 н. HCl в диоксане с получением 58 мг (74%) дигидрохлорида 6-амино-2-бутокси-9-[N-(2-аминоэтил)4-пиперидинилметил]-8 гидроксипурина в виде не совсем белого твердого вещества. 1 Н ЯМР (DMSO-d6, 400 МГц):4.15 (t, 2H), 3.58 (d, 2H), 2.88 (t, 2H), 2.81 (d, 2H), 2.46 (t, 2H), 1.91 (t,2 Н), 1.79 (m, 1 Н), 1.64 (m, 2 Н), 1.52 (m, 2 Н), 1.39 (dd, 2 Н), 1.28 (m, 2 Н), 0.91 (t, 3 Н).(0,126 мл), и полученную реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. Суспензию соединения, полученного на стадии (2) выше (37 мг), и триэтиламина (0,126 мл) в пиридине(2,3 мл) добавляли в реакционную смесь, затем добавляли йод (75 мг). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали и дважды выпаривали совместно с толуолом. После флэш-хроматографии на силикагеле с использованием CHCl3-MeOH-Et3N (градиентное элюирование; 90:10:175:25:1) получили 39 мг (18%) 6-амино-2-бутокси-9-[N-(2-(1,2-дипальмитоил-sn-7 023536 глицеро-3-фосфорамидо)этил-4-пиперидинилметил]-8-гидроксипурина в виде оранжевого стекловидного твердого вещества. Rf (CHCl3-CH3OH-H2O-NH4OH) = 0,68. Пример 3 (соединение A3). Получение 6-амино-2-бутокси-9-[1-(1,2-дипальмитоил-sn-глицеро-3-фосфорамидо)-4-пиперазинилэтил]-8-гидроксипурина (соединение (II), R1 = н-бутокси, n = 2, X = N, Q = простая связь, Z = О, W = О,m = 0, р = 1, R1 = R2 = н-C15H31CO)(2) Способом, аналогичным способу, описанному в примере 1-(2), с соединения, полученного на стадии (1) выше (124 мг), удаляли N-защитную группу с использованием 4,0 М HCl в диоксане с получением 87 мг (77%) гидрохлорида 6-амино-2-бутокси-9-(4-пиперазинилэтил)-8-гидроксипурина в виде не совсем белого твердого вещества. 1 Н ЯМР (CD3OD, 400 МГц)4.26 (t, 2 Н), 3.96 (m, 2 Н), 3.11 (m, 5 Н), 2.78 (m, 7 Н), 1.73 (m, 2 Н), 1.48(3) Способом, аналогичным способу, описанному в примере 2-(3), соединение, полученное на стадии (2) выше (288 мг), подвергали фосфатидилированию 1,2-дипальмитоил-sn-глицерилгидрофосфонатом (518 мг) с получением 313 мг (46%) 6-амино-2-бутокси-9-[1-(1,2-дипальмитоил-snглицеро-3-фосфорамидо)-4-пиперазинилэтил]-8-гидроксипурина в виде оранжево-коричневого твердого вещества. 1 Н ЯМР (CDCl3-CD3OD, 400 МГц):5.11 (br s, 1 Н), 4.28 (d, 1 Н), 4.12-4.01 (m, 5 Н), 3.76 (br s, 2 Н),3.60-2.60 (m, 11 Н), 2.21 (m, 4 Н), 1.65 (m, 2 Н), 1.49 (m, 4 Н), 1.39 (q, 2 Н), 1.17 (m, 48 Н), 0.88 (t, 3 Н), 0.81 (t,6H); MCBP: вычислено для [М-Н] C50H92N7O9P 964,6616; найдено 964,6600. Авторы изобретения оценивали способность лигандов TLR7 промотировать различные аспекты иммунного ответа у мышей. Врожденный иммунитет специфически исследовали, чтобы прийти к заключению, способны ли липидированные соединения-лиганды TLR7 индуцировать природные хемокины и провоспалительные цитокины. Этот врожденный иммунный ответ обеспечивает организм начальной,неспецифической защитой от патогенов. Считается, что среди этих врожденных хемокинов и цитокинов для программирования не подвергнутой никакому воздействию CD8 Т-клетки в отношении выживания,дифференцировки и развития памяти требуется IFN типа I. Исследования на мышах подтвердили, что величина и продолжительность воспаления регулирует скорость, при которой CD8+ Т-клетки приобретают характеристики памяти. Пример 4. Получение липосом TLR7-DPPC. 10,4 мл липидированного-TLR7-L А 1 в концентрации 10 мг/мл в хлороформе переносили в круглодонный стеклянный сосуд. В этот сосуд добавляли 18,4 мл DPPC (дипальмитоилфосфатидилхолин), растворенного в хлороформе в концентрации 100 мг/мл. 21,9 мл липидированного-TLR7-L соединения A3,растворенного в концентрации 5 мг/мл в хлороформе, переносили в круглодонный стеклянный сосуд. В этот сосуд добавляли 20 мл DPPC, растворенного в хлороформе в концентрации 100 мг/мл. Хлороформ выпаривали в роторном испарителе Buchi Rotavapor при медленном снижении давления. Полученную пленку затем высушивали по меньшей мере в течение ночи в эксикаторе под вакуумом. После этого липидную пленку медленно гидратировали на орбитальном шейкере (максимальная скорость: 250 об/мин),используя буфер 50 мМ РО 4-100 мМ NaCl, pH 7, предварительно нагретый при 60 С. После завершения ресуспендирования проводили стадию набухания для дополнительного увеличения гидратации фосфолипидов, которая заключается в инкубировании препарата при комнатной температуре. Затем суспензию предварительно гомогенизировали, используя роторно-статорный гомогенизатор (Х 620 CAT), в течение 12 мин при 8000 об/мин. Затем проводили микрофлюидизацию с использованием микрофлюидизатора (Model 110S-Serial 90181-Microfluidics Corporation) при 6 бар (630 Па), поддерживая в камере микрофлюидизации температуру примерно 40 С. Стерилизующую фильтрацию выполняли на фильтре PES (GD/X 0,2 мкм, 25 мм,Whatman). Пример 5. 20080672: SIV-p27 модель - DPPC-липосомы - A3 и А 1. Липидированные молекулы A3 и А 1 оценивали in vivo. SIV-p27-содержащие композиции, указанные в таблице, использовали для вакцинации 6-8-недельных самок мышей C57BL/6 (10/группа). Мышам делали две внутримышечные (в.м.) инъекции по 50 мкл с перерывом 14 суток и отбирали кровь в разные моменты времени после первичной вакцинации и ревакцинации, как иллюстрируется ниже:DPPC-липосомами. Итоговая таблица данных по составу композиций Содержание всех соединений приведено в мкг, если не указано иное.Дозы приведены в расчете на количество TLR7-L, использованного для изготовления липосом A1-DPPC и A3-DPPC. Реальные инъецируемые дозы могут быть ниже вследствие потери во время фильтрации. У группы сравнения AS01B и групп, иммунизированных TLR7-L-содержащими композициями, наблюдались разные профили цитокинов. Через короткое время после инъекции (3 ч) IFN-альфа обнаруживались доза-зависимым образом в сыворотках мышей, иммунизированных обоими TLR7-L, приготовленными в DPPC-липосоме (фиг. 1). Такой сывороточный IFN- врожденный ответ не наблюдался в случае, когда TLR7-L приготовлены вDOPS (диолеилфосфатидилсерин)/DOPC (диолеилфосфатидилхолин)-липосоме (данные не показаны). Уровни других воспалительных цитокинов, такие как TNF- и IL-6, были повышены, когда в композициях присутствовало соединение A3 или соединение А 1, причем А 1 было более эффективным, чемA3 (фиг. 2). Уровни хемокина МСР-1 также повышались в присутствии обоих соединений (фиг. 3). Все вместе эти данные показывают, что TLR7-L липидированные молекулы эффективны в индуцированииIFN типа I и в усилении ответов провоспалительных цитокинов, когда они объединены с препаратом на основе MPL/QS21.TLR7-L молекулы, использованные в данном изобретении, дополнительно не усиливали адаптивный Т-клеточный ответ по сравнению с группой сравнения AS01 (данные не показаны).n равно целому числу от 1 до 6;X представляет собой СН или N;m равно 0 или целому числу от 1 до 6;R2 представляет собой Н или прямой или разветвленный C4-C24-алкил или алкил-С(О);R3 представляет собой прямой или разветвленный C4-C24-алкил или алкил-С(О); или его фармацевтически приемлемая соль. 2. Соединение формулы (IX)X представляет собой СН или N;R2 представляет собой Н или прямой или разветвленный C4-C24-алкил или алкил-С(О) иR3 представляет собой прямой или разветвленный C4-C24-алкил или алкил-С(О); или его фармацевтически приемлемая соль. 3. Соединение по п.2,где R1 представляет собой С 1-6 алкокси или С 1-6 алкоксиС 1-6 алкил;R2 представляет собой Н или прямой или разветвленный C4-C24-алкил или алкил-С(О) иR3 представляет собой прямой или разветвленный C4-C24-алкил или алкил-С(О); или его фармацевтически приемлемая соль. 4. Соединение, выбранное из или его фармацевтически приемлемая соль. 5. Соединение, имеющее структуру или его фармацевтически приемлемая соль. 6. Соединение, имеющее структуру или его фармацевтически приемлемая соль. 7. Соединение, имеющее структуру или его фармацевтически приемлемая соль. 8. Способ индуцирования врожденного иммунного ответа у млекопитающего, включающий введение соединения по любому из пп.1-7. 9. Способ по п.8, где ответ включает индуцирование IFN типа 1 (интерферон типа 1). 10. Способ по п.8, где ответ включает усиление провоспалительных цитокинов. 11. Применение соединения по любому из пп.1-7 для индуцирования врожденного иммунного ответа. 12. Применение по п.11, где индуцирование врожденного иммунного ответа включает усиление провоспалительных цитокинов. 13. Применение по п.11, где индуцирование врожденного иммунного ответа включает индуцирование ответа IFN типа 1.
МПК / Метки
МПК: A01N 43/90, A61K 31/52, A61K 31/522
Метки: фосфо, фосфонолипидами, оксоаденина, производные, конъюгированные
Код ссылки
<a href="https://eas.patents.su/13-23536-proizvodnye-oksoadenina-konyugirovannye-s-fosfo-ili-fosfonolipidami.html" rel="bookmark" title="База патентов Евразийского Союза">Производные оксоаденина, конъюгированные с фосфо- или фосфонолипидами</a>
Конъюгированные цитокины, используемые для терапии опухолей

Номер патента: 7838
Опубликовано: 27.02.2007
Автор: Корти Анджело
МПК: A61K 38/19, A61K 38/21, A61K 31/70...
Метки: конъюгированные, опухолей, терапии, используемые, цитокины
Формула / Реферат:
1. Конъюгированный продукт, обладающий противоопухолевой активностью, включающий цитокин, выбранный из TNF или IFNg, и лиганд, содержащий NGR-мотив. 2. Конъюгированный продукт по п.1, где цитокин представляет собой TNFa или TNFb. 3. Конъюгированный продукт по пп.1-2, где лиганд представляет собой лиганд рецептора CD13, который выбирают из группы, включающей антитела или их активные фрагменты, пептиды или миметики пептидов. 4. Конъюгированный...
Фармацевтические композиции, содержащие производные 3-аминоазетидина, новые производные и способ их получения

Номер патента: 4649
Опубликовано: 24.06.2004
Авторы: Иттэнжер Огюстэн, Филош Брюно, Букерель Жан, Гризони Серж, Ашар Даниель, Майерс Майкл, Бушар Эрве
МПК: A61P 25/00, C07D 205/04, A61K 31/397...
Метки: способ, новые, фармацевтические, производные, композиции, 3-аминоазетидина, получения, содержащие
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы в которой R1 обозначает радикал -NHCOR4 или -N(R5)-Y-R6, Y обозначает CO или SO2, R2 и R3, одинаковые или разные, обозначают либо ароматический радикал, выбранный из фенила, нафтила и инденила, которые могут быть незамещенными или замещенными одним или несколькими заместителями: галоген, алкил, алкокси, формил, гидрокси, трифторметил, трифторметокси,...
Производные бензоксазина в качестве модуляторов 5-нт-6, способ их получения, фармацевтические композиции, содержащие эти производные, и их применение

Номер патента: 9982
Опубликовано: 28.04.2008
Авторы: Бергер Якоб, Чжао Шухай, Кларк Робин Дуглас
МПК: C07D 265/36, A61P 25/18, A61K 31/536...
Метки: бензоксазина, композиции, эти, способ, фармацевтические, модуляторов, получения, содержащие, производные, качестве, применение, 5-нт-6
Формула / Реферат:
1. Соединение формулы его фармацевтически приемлемая соль или пролекарство, где m означает целое число от 0 до 3; n означает 2 или 3; р означает 2; Y означает -S(O2)-; Z1 означает N; R1 и R2 означают водород; R3 означает фенил или нафтил, которые необязательно замещены галогеном, алкилом, алкокси, гидрокси, циано, пиперазинилом, метилсульфониламино, метилсульфонилом, аминокарбонилом, или означает хинолинил, изохинолинил, тиофенил,...
Производные 2,3-бензодиазепина и фармацевтические композиции, содержащие эти производные в качестве активного ингредиента

Номер патента: 5867
Опубликовано: 30.06.2005
Авторы: Сабо Геза, Грефф Зольтан, Леваи Дьердь, Раткаи Зольтан, Вег Миклош, Линг Иштван, Сенаши Габор, Харшинг Ласло Габор, Баркоци Йожеф, Гиглер Габор, Шимиг Дьюла, Мартонне Марко Бернадетт
МПК: A61P 25/00, A61K 31/551, C07D 243/02...
Метки: содержащие, качестве, эти, 2,3-бензодиазепина, производные, активного, композиции, фармацевтические, ингредиента
Формула / Реферат:
1. Производное 2,3-бензодиазепина формулы I где X - водород, хлор или метоксигруппа, Y - водород или галоген, Z - метил или хлор, R - C1-4 алкил или группа формулы -NR1R2, где R1 и R2, независимо, представляют собой водород, C1-4 алкил, C1-4 алкоксил или C3-6 циклоалкил, и его фармацевтически приемлемые соли с кислотами. 2. Производное 2,3-бензодиазепина по п.1, где X - хлор, Y - водород, хлор или бром, R - C1-4 алкил, Z - определен в п.1, и...
Полициклические производные аминокислот, содержащая их фармацевтическая композиция и способ лечения, использующий эти производные

Номер патента: 19879
Опубликовано: 30.07.2014
Авторы: Маринелли Бретт, Самала Лакшама, Цзинь Хайхун, Чжан Чэньминь, У Вэньсюэ, Чжан Хаймин, Ши Чжи-Цай, Ван Ин, Туноори Ашок, Девасагаярадж Арокиасами, Лю Цинюнь
МПК: A61K 31/4192, A61K 31/4965, A61K 31/53...
Метки: эти, производные, аминокислот, способ, использующий, содержащая, полициклические, лечения, композиция, фармацевтическая
Формула / Реферат:
1. Соединение формулыили его фармацевтически приемлемая соль или сольват, гдеА представляет собой необязательно замещенный фенил, нафтил или 5-6-членный гетероцикл, включающий 1-3 гетероатома, выбранных из О, N и S, причем необязательное замещение означает замещение одной или более группами С1-6-алкоксила, амино, циано, галогена, гидроксила или фенила, причем фенильная группа необязательно замещена одной или более группами С1-6-алкоксила, амино,...
Предыдущий патент: Композиция водного радиопротекторного фармацевтического раствора и способ предотвращения, снижения или исключения эффектов ионизирующей радиации
Следующий патент: Способ и система независимого управления транспортным средством
Случайный патент: Устройство для термозависимой цепной амплификации последовательностей - мишеней нуклеиновых кислот