Способ очистки конденсированного производного пирролокарбазола
Номер патента: 21869
Опубликовано: 30.09.2015
Авторы: Пьясенза Ги, Оллвейн Шон П., Роз Себастьен, Грандери Арно







Формула / Реферат
1. Кристаллическая форма кислотного комплекса формулы (Ia)

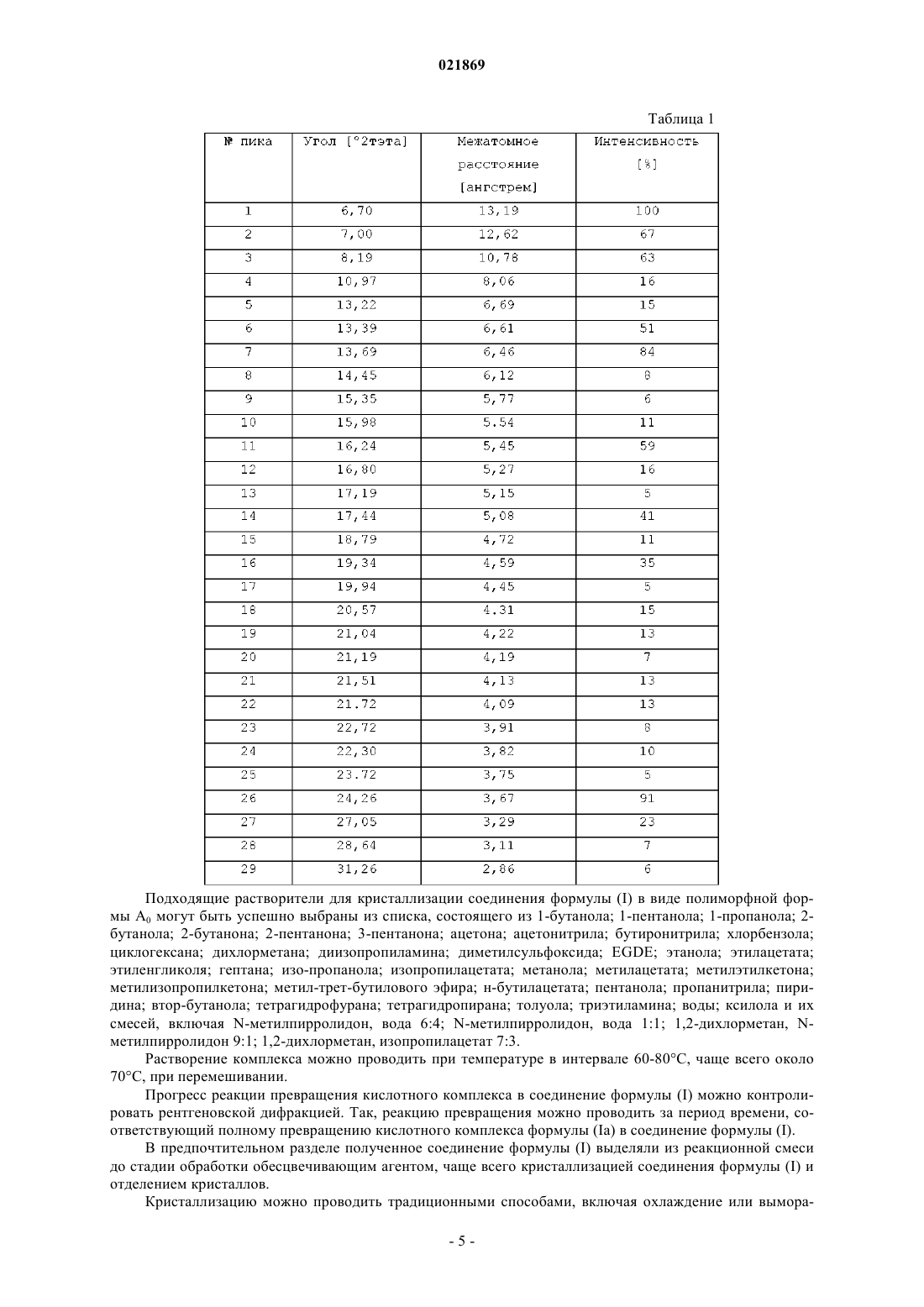
где R означает метил, характеризующийся рентгеновской порошковой дифрактограммой, содержащей один или несколько следующих пиков: 5,19±0,2 градуса 2-тэта; 6,17±0,2 градуса 2-тэта; 6,44±0,2 градуса 2-тэта; 14,36±0,2 градуса 2-тэта и 26,09±0,2 градуса 2-тэта, при измерениях с использованием Cu-Kα излучения.
2. Способ получения кислотного комплекса формулы (Ia) по п.1, содержащий:
i) взаимодействие соединения формулы (I) с уксусной кислотой

с получением кислотного комплекса формулы (Ia);
ii) кристаллизацию полученного кислотного комплекса формулы (Ia).
3. Способ по п.2, дополнительно включающий взаимодействие кислотного комплекса формулы (Ia) с обесцвечивающим агентом.
4. Способ по п.2 или 3, дополнительно включающий выделение кристаллизованного комплекса формулы (Ia).
5. Способ по п.3, где обесцвечивающий агент представляет собой активированный уголь, активированный паром или химически.
6. Способ по п.4, где выделенный кристаллизованный комплекс формулы (Ia) имеет чистоту по меньшей мере приблизительно 92%.
7. Способ по п.4, где выделенный кристаллизованный комплекс формулы (Ia) имеет чистоту по меньшей мере приблизительно 97%.
Текст
СПОСОБ ОЧИСТКИ КОНДЕНСИРОВАННОГО ПРОИЗВОДНОГО ПИРРОЛОКАРБАЗОЛА Изобретение описывает метод очистки конденсированного пирролокарбазольного соединения,известного как 11-изобутил-2-метил-8-(2-пиримидиниламино)-2,5,6,11,12,13-гексагидро-4 Ниндазоло[5,4-а]пирроло[3,4-с]карбазол-4-он, с использованием его кислотного комплекса. Изобретение описывает также кристаллическую форму кислотного комплекса. Область техники, к которой относится изобретение Настоящее изобретение предлагает способ очистки производного пироллокарбазола (соединение формулы (I) или соединение (I с использованием для этого кислотного комплекса. Настоящее изобретение описывает также кристаллическую форму кислотного комплекса формулы (Ia). Уровень техники Специальное соединение конденсированного пироллокарбазола, известное как 11-изобутил-2 метил-8-(2-пиримидиниламино)-2,5,6,11,12,13-гексагидро-4 Н-индазоло[5,4-а]пирроло-[3,4-с]карбазол-4 он, является потенциальным орально-активным TIE-2/VEG-R 40 ингибитором, обладающим противоопухолевой и противоангиогенной активностью, и представлен следующей формулой (I): Данное соединение обозначается здесь как соединение (I). Патент США 7169802 описывает соединение I и его применение. Особо описан способ получения этого соединения, соответствующий схеме: Тем не менее, авторы показали, что соединение (I) в соответствии с такой процедурой получается с низким выходом примерно 38% и низкой степенью чистоты. В частности, было показано, что полученное таким образом соединение формулы (I) содержало большое количество побочного продукта формулы (IV) Данный побочный продукт, образующийся в результате отщепления двух атомов водорода в индазолильном фрагменте соединения формулы (I) и, следовательно, ароматизации кольцевой системы, представлял особые трудности в отделении от соединения формулы (I). Несколько стадий очистки, чаще всего колоночной хроматографией, требуемых для получения соединения (I) с фармацевтически приемлемой чистотой, чаще всего более чем 95%, еще более понижали выход. Таким образом, возникла потребность улучшения процесса для получения соединения (I) из соединения (II), которое преодолело бы недостатки предыдущего способа и особенно позволило бы получить удовлетворительный выход и чистоту. Сущность изобретения Настоящее изобретение в одном разделе описывает кислотный комплекс соединения (I). Неожиданно заявители обнаружили, что кристаллизация такого комплекса позволяет удалить большинство загрязнений, особенно тех, которые трудно удалить по традиционным методикам, таким как хроматография, и, таким образом, получить высокий уровень чистоты. Кислотный комплекс соединения (I) в соответствии с изобретением делает последующую очистку соединения (I) легче и переводит процесс на индустриальную основу. В частности, он снижает потребность в больших объемах растворителя, требующегося обычно при очистке хроматографией. Другой аспект настоящего изобретения предлагает способ получения кислотного комплекса соединения (I) из соединения (II). Было удачно продемонстрировано, что применение основания на стадии нуклеофильного замещения позволяет повысить выход кислотного комплекса и, следовательно, соединения формулы (I), а также снизить конечные примеси. В частности, было показано, что присутствие основания не повышает разложения соединения (II). Другой аспект настоящего изобретения предлагает применение такого кислотного комплекса для очистки соединения (I) с тем, чтобы достичь чистоту более чем 95%. Другой аспект настоящего изобретения предлагает способ очистки соединения (I), в основном включающий обработку кислотного комплекса обесцвечивающим агентом для того, чтобы удалить побочный продукт формулы (IV). Еще одним предметом настоящего изобретения является получение кристаллической формы кислотного комплекса формулы (Ia). Эти и другие аспекты, тонкости и преимущества изобретения раскрываются далее в детальном описании. Описание чертежей Фиг. 1 представляет рентгеновскую порошковую дифрактограмму формы А 0 соединения (I); фиг. 2 представляет рентгеновскую порошковую дифрактограмму ацетатного кислотного комплекса соединения формулы (I); фиг. 3 представляет 1 Н ЯМР спектр ацетатного кислотного комплекса соединения формулы (I). Детальное описание изобретения Итак, в одном аспекте изобретение описывает кислотный комплекс соединения формулы (I) и указанный комплекс имеет формулу (Ia) где R означает С 1-С 8 алкил. Карбоновая кислота RCOOH может быть выбрана из списка, включающего уксусную кислоту, пропионовую кислоту, масляную кислоту, валерьяновую кислоту, капроевую кислоту, гептановую кислоту или октановую кислоту. В частном аспекте RCOOH означает уксусную кислоту. Следует понимать, что ацетатно-кислотный комплекс соединения (I) может быть описан как кислотный комплекс формулы (Ia), где R означает С 1 алкил. В следующем аспекте R означает С 2-С 8 алкил. В дополнительном аспекте изобретение описывает способ получения кислотного комплекса формулы (Ia), как здесь определено, включающий:i) взаимодействие соединения формулы (II) с соединением формулы (III) в присутствии основания в растворителеii) взаимодействие полученного соединения формулы (I) с кислотой формулы RCOOH и необязательноiii) выделение полученного кислотного комплекса формулы (Ia). Стадия i). В другом аспекте основанием является амин, в основном вторичный или третичный амин. В другом аспекте амин означает триалкиламин. В другом аспекте амин имеет формулу R1R2R3N, где R1 означаетC1-С 6 алкил, a R2 и R3 независимо выбирают из списка, содержащего Н и C1-С 6 алкил. Предпочтительно амин представлен триалкиламином, где R1, R2 и R3 независимо означают C1-С 6 алкил, чаще всего диизопропиламином или триэтиламином; триэтиламин наиболее предпочтителен. Показано, что присутствие основания повышает скорость реакции с повышением и выхода, и чистоты реакции главным образом за счет снижения количества побочных продуктов, в частности, тех, ко-2 021869 торые относятся к разложению соединений формулы (II). Далее было обнаружено, что основание не повышает количества побочного продукта формулы (IV). В следующем аспекте молярное соотношение основания к соединению формулы (II) варьируется от 1 до 2 и в основном составляет примерно 1,5 экв. В следующем аспекте молярное отношение соединения формулы (III) по отношению к соединению формулы (II) варьируется от 1 до 2, чаще всего составляет 1,5 экв. Нет специальных ограничений на применяемый растворитель, имея в виду, чтобы он не оказывал негативный эффект на реакцию или применяемые реагенты. Примеры подходящих растворителей включают полярные растворители, чаще всего спирты, особенно спирты с температурой кипения выше 100 С,такие как н-бутанол. Реакция может осуществляться в широком интервале температур и точное значение температуры реакции не имеет значения в изобретении. В общем случае реакционную смесь нагревают до кипения,чаще всего при температуре в интервале от 100 до 120 С. Время проведения реакции может также широко варьироваться в зависимости от многих факторов,главным образом температуры и природы реагентов. Мониторирование процесса методом ВЭЖХ показывает, что период от примерно 18 до 22 ч в среднем удовлетворителен. Стадия ii). В соответствии со стадией ii) соединение формулы (I) вводят в реакцию с карбоновой кислотой формулы RCOOH. В предпочтительном разделе карбоновую кислоту добавляют к реакционной смеси,полученной на стадии i). Предпочтительный объем RCOOH, добавляемой к реакционной смеси, варьируется от 1 до 20 об.,чаще всего от 5 до 15 об. по отношению к соединению формулы (II) или (I). Температура добавления кислоты RCOOH к реакционной смеси не принципиальна. Она может быть выбрана между точкой кипения и точкой плавления кислоты RCOOH, в частности в интервале от 60 до 120 С. Предпочтительно реакционную смесь, полученную на стадии i), охлаждают до примерно 75 С перед добавлением кислоты RCOOH. Реакционную смесь затем, как правило, нагревают до температуры от 60 до 80 С, чаще всего до 75 С, в течение периода времени от примерно 10 до 30 мин. Стадия iii). В дополнительном аспекте кислотный комплекс формулы (Ia) выделяют из реакционной смеси. В частном разделе стадия iii) включает: а) кристаллизацию полученного кислотного комплекса формулы (Ia) иb) выделение кристаллизованного комплекса формулы (Ia). Комплекс формулы (Ia) может быть кристаллизован из реакционной смеси традиционными способами, включая охлаждение или вымораживание, осаждение кристаллов, выпаривание части раствора или высаживание добавлением не растворяющего растворителя, такого как метил-трет-бутиловый эфир(МТБЭ). Предпочтительный раздел включает охлаждение реакционной смеси до примерно 20 С. В частности, реакционная смесь может быть быстро охлаждена стандартными методами охлаждения, обычно со скоростью охлаждения в интервале от -0,1 до -10 С/мин. Кристаллизованный комплекс формулы (Ia) может быть выделен традиционными способами, включая фильтрование или центрифугирование. Выделенные кристаллы кислотного комплекса могут быть затем промыты растворителем, например метил-трет-бутиловым эфиром (МТБЭ). В дополнительном разделе изобретение описывает кристаллическую форму кислотного комплекса формулы (Ia) где R означает С 1 алкил, охарактеризованную методом порошковой рентгеновской дифракции, с одним или более следующих пиков: 5,190,2 градуса 2-тэта; 6,170,2 градуса 2-тэта; 6,440,2 градуса 2 тэта; 14,360,2 градуса 2-тэта и 26,090,2 градуса 2-тэта при измерении с применением Cu-K излучения. В одном аспекте рентгеновская порошковая дифрактограма содержала пик 6,440,2 и один или более следующих пиков: 5,190,2 градуса 2-тэта; 6,170,2 градуса 2-тэта; 14,360,2 градуса 2-тэта и 26,090,2 градуса 2-тэта при измерении с использованием Cu-K излучения. В другом аспекте рентгеновская порошковая дифрактограмма содержала пики 6,440,2 градуса 2-тэта и 6,17 0,2 градуса 2-тэта и один или более следующих пиков: 5,190,2 градуса 2-тэта; 14,360,2 и 26,090,2 градуса тэта при измерении с использованием Cu-K излучения. В следующем аспекте рентгеновская порошковая дифрактограмма содержит пики 6,440,2 градуса 2-тэта; 6,170,2 градуса 2-тэта и 26,090,2 градуса тэта и один или более следующих пиков: 5,190,2 градуса 2-тэта и 14,360,2 градуса 2-тэта при измерении с использованием Cu-K излучения. В еще одном аспекте рентгеновская порошковая дифрактограмма содержит пики 5,190,2 градуса 2-тэта; 6,170,2 градуса 2-тэта; 6,440,2 градуса 2-тэта; 14,360,2 градуса 2-тэта и 26,090,2 градуса 2-тэта и один или более следующих пиков: 10,510,2 градуса 2-тэта; 15,840,2 градуса 2-тэта; 18,33 0,2 градуса 2-тэта; 20,690,2 градуса 2-тэта и 23,710,2 градуса 2-тэта при использовании для измерений Cu-K излучения. В другом аспекте кристаллический уксуснокислотный комплекс формулы (Ia) имеет рентгеновскую порошковую дифрактограму в основном такую, как указано на фиг. 2. В предпочтительном разделе кристаллическая форма кислотного комплекса формулы (Ia), где R означает С 1 алкил, имеет чистоту по крайней мере около 92%. В более предпочтительном разделе кристаллическая форма кислотного комплекса формулы (Ia), где R означает С 1 алкил, имеет чистоту по крайней мере около 97%. В наиболее предпочтительном разделе кристаллическая форма кислотного комплекса формулы (Ia), где R означает С 1 алкил, имеет чистоту по крайней мере около 99,5%. Преимущественно показано, что кристаллизация комплекса формулы (Ia) позволяет отделить большинство примесей, образованных на стадиях получения соединения формулы (I). Так, после кристаллизации конечный комплекс охарактеризован с чистотой в интервале от 92 до 99,5% или более. В частности, удаление большинства примесей в основном приводит к чистоте в интервале от 92 до 97%. Оставшиеся примеси - это главным образом побочный продукт формулы (IV), который кристаллизуется вместе с кислотным комплексом формулы (I). Дальнейшее удаление соединения формулы (IV) позволяет довести чистоту до уровня, равного или более чем 99,5%. В дополнительном аспекте изобретение предлагает кислотный комплекс формулы (Ia), полученный в соответствии с описанным здесь способом. В другом дополнительном аспекте изобретение предлагает использование кислотного комплекса формулы (Ia) для очистки или способ для очистки соответствующего соединения формулы (I) или его фармацевтически приемлемой соли. В предпочтительном разделе очищенное соединение формулы (I) имеет чистоту более 98%, предпочтительно более 99%. В дополнительном аспекте изобретение предлагает метод очистки соединения формулы (I), как это здесь определено, включающий:i) превращение кислотного комплекса формулы (Ia), как это здесь определено, в соответствующее соединение формулы (I);ii) взаимодействие полученного соединения формулы (I) с обесцвечивающим агентом и необязательноiii) выделение очищенного соединения формулы (I). В еще одном аспекте стадию превращения кислотного комплекса формулы (Ia) в соединение формулы (I) проводят высушиванием комплекса при температуре в интервале от 70 до 90 С, чаще всего при температуре около 80 С. Альтернативно, стадия превращения кислотного комплекса формулы (Ia) в соединение формулы (I) может быть проведена растворением комплекса формулы (Ia) в растворителе, чаще всего в растворителе,подходящем для кристаллизации соединения формулы (I), например, в виде полиморфной формы А 0. Полиморфная форма А 0 соединения (I) была предложена в международной патентной заявкеPCT/US2009/065099, содержание которой введено здесь в виде ссылки. Фиг. 1 представляет рентгеновскую порошковую дифрактограмму формы А 0 соединения (I). Она демонстрирует пики, представленные в табл. 1. Подходящие растворители для кристаллизации соединения формулы (I) в виде полиморфной формы А 0 могут быть успешно выбраны из списка, состоящего из 1-бутанола; 1-пентанола; 1-пропанола; 2 бутанола; 2-бутанона; 2-пентанона; 3-пентанона; ацетона; ацетонитрила; бутиронитрила; хлорбензола; циклогексана; дихлорметана; диизопропиламина; диметилсульфоксида; EGDE; этанола; этилацетата; этиленгликоля; гептана; изо-пропанола; изопропилацетата; метанола; метилацетата; метилэтилкетона; метилизопропилкетона; метил-трет-бутилового эфира; н-бутилацетата; пентанола; пропанитрила; пиридина; втор-бутанола; тетрагидрофурана; тетрагидропирана; толуола; триэтиламина; воды; ксилола и их смесей, включая N-метилпирролидон, вода 6:4; N-метилпирролидон, вода 1:1; 1,2-дихлорметан, Nметилпирролидон 9:1; 1,2-дихлорметан, изопропилацетат 7:3. Растворение комплекса можно проводить при температуре в интервале 60-80 С, чаще всего около 70 С, при перемешивании. Прогресс реакции превращения кислотного комплекса в соединение формулы (I) можно контролировать рентгеновской дифракцией. Так, реакцию превращения можно проводить за период времени, соответствующий полному превращению кислотного комплекса формулы (Ia) в соединение формулы (I). В предпочтительном разделе полученное соединение формулы (I) выделяли из реакционной смеси до стадии обработки обесцвечивающим агентом, чаще всего кристаллизацией соединения формулы (I) и отделением кристаллов. Кристаллизацию можно проводить традиционными способами, включая охлаждение или вымора-5 021869 живание, выпадение кристаллов, выпаривание части раствора или высаживание путем добавления не растворяющего растворителя. Предпочтительный раздел включает охлаждение реакционной смеси до примерно 10 С, быстро, по стандартным методам охлаждения, обычно в температурном интервале скорости охлаждения от -0,1 до-10 С/мин. Кристаллизованное соединение формулы (I) может быть выделено традиционными способами,включая фильтрование и центрифугирование. Выделенные кристаллы соединения формулы (I) могут затем быть промыты растворителем, например изопропилацетатом. Выделенный продукт может затем быть высушен в вакууме. В частном разделе стадия взаимодействия соединения формулы (I) с обесцвечивающим агентом проводится в растворителе, выбранном из списка, состоящего из дихлорметана, метанола, этанола или любого растворителя, способного растворять соединение (I), или любой их бинарной или трехкомпонентной смеси. В дальнейшем аспекте обесцвечивающим агентом является активированный уголь, например, активированный паром или химически. Примерами подходящих активированных углей являются те, которые предлагаются под торговыми наименованиями LSM, L3S, 3S, DARCO G60 для активированных паром углей и CPL, ENO PC, CAP SUPER для химически активированных углей; обычно от фирм Сеса или Norit. Предпочтительным углем является уголь ENO PC. Показано, что обесцвечивающий агент позволяет существенно с помощью адсорбции удалить остаточные побочные продукты, которые могут присутствовать в реакционной смеси вместе с соединением формулы (I) (или с кислотным комплексом формулы (Ia) соответственно), в частности побочный продукт формулы (IV). Очищенное соединение формулы (I) может затем быть выделено фильтрованием реакционной смеси и удалением растворителя в вакууме. Выделенное соединение формулы (I) может затем быть необязательно перекристаллизовано, особенно в полиморфную форму А 0. В определенном разделе соединение формулы (I) может быть растворено в изопропилацетате и затем охлаждено до температуры примерно 10-20 С, пока полностью не образуется полиморфная форма А 0. Кристаллы очищенного соединения формулы (I) могут затем быть выделены любыми удобными способами, особенно центрифугированием, и промыты растворителем, таким как изопропилцетат. В другом разделе изобретение предлагает способ очистки соединения формулы (I), как здесь определено, включающий следующие стадии:i) взаимодействие кислотного комплекса формулы (Ia), как здесь определено, с обесцвечивающим агентом в растворителе;ii) перевод полученного кислотного комплекса формулы (Ia) в соответствующее соединение формулы (I) и необязательноiii) выделение очищенного соединения формулы (I). Стадии обработки обесцвечивающим агентом (стадия i) и перевода кислотного комплекса формулы(Ia) в соединение формулы (I) (стадия ii) могут быть выполнены в соответствии с процедурами, описанными здесь. В дальнейшем аспекте полученное очищенное соединение формулы (I) далее вводят в реакцию с кислотой для получения кислотно-аддитивной соли, предпочтительно аддитивной соли монокислоты. В другом аспекте кислотой является пара-толуолсульфоновая кислота (ПТСК). Следующие термины и выражения, используемые здесь, имеют следующие значения. Как здесь используется, термин "около" относится к величинам от 10% от приведенного значения. Например, фраза "около 50 мг" включает 10% от 50 или от 45 до 55 мг. Термин "комплекс", как используется здесь, относится к нековалентно связанной ассоциации двух молекул кислоты RCOOH с одной молекулой соединения формулы (I). Как используется здесь, термин "алкил" относится к прямоцепочечной или разветвленной алкильной группе, имеющей от 1 до 8 атомов углерода, такой как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, неопентил, 1-этилпропил, 3-метилпентил, 2,2 диметилбутил, 2,3-диметилбутил, гексил, октил и т.д. К низшим алкильным группам, которые предпочтительны, относятся алкильные группы, как они определены выше, содержащие от 1 до 4 атомов углерода. Определение С 1-С 4 алкил относится к алкильному радикалу с 1-4 атомами углерода. Термин "растворитель", как используется здесь, означает вещество, обычно жидкое, которое способно полностью или частично растворять другое вещество, обычно твердое, и которое неактивно по отношению к исходным реагентам, промежуточным продуктам или продуктам при рассматриваемой температуре реакции, которая варьируется от точки растворения растворителем до точки кипения растворителя. Термин "нерастворяющий растворитель", как здесь используется, означает растворитель, в котором соединение в основном нерастворимо. Как используется здесь, термин "обесцвечивающий агент" относится к пористому или тонкоизмельченному углю, чаще всего активированному, с большой поверхностью, который может адсорбировать окрашенные примеси из жидкой реакционной смеси, чаще всего ароматические примеси. Как здесь используется, термин "объем" или "V", когда он применяется к соотношению, означает отношение литр/килограмм (л/кг). Примеры Другие специфические черты изобретения будут понятны из следующих описаний примеров. Эти примеры иллюстрируют изобретение, но не ограничивают его. Материалы и методы. Соединение (II) предоставлено Cephalon (обычная чистота 92% LCAP); оно может быть получено в соответствии с примером I-29 WO-2005/063763. Триэтиламин приобретен в SAFC (обычная чистота 99%). 2-Бромпиримидин приобретен в Acros(обычная чистоты 98%). Растворители приобретены в SDS Carlo Erba (обычная чистота: PPS степень). ВЭЖХ. Обращенно-фазный ВЭЖХ метод был разработан и применен для установления идентичности, проверки и чистоты соединения формулы (I) как лекарственной субстанции. Анализ осуществляют на колонке XTerra MS C18 (1504,6 мм, 5 мкм насыпка), используя 55-85% градиента органики в течение 27 мин и измеряя поглощение при 270 нм. Параметры анализа. Колонка: Xterra MS C18, 1504,6 мм, 5 мкм. Температура колонки: 30 С. Инжектируемый объем: 10 мкл. Детектирование: УФ, 270 нм. Скорость потока: 1,0 мл/мин. Время пробега: 27 мин. Подвижная фаза А:10 мМ водный ацетат аммония. Подвижная фаза В: 10 мМ ацетат аммония в смеси 50:50 ацетонитрил/метанол. Градиент Рентгеновская порошковая дифракция (XRPD). Спектр рентгеновской порошковой дифракции (XRPD) для ацетатного кислотного комплекса соединения формулы (I) получали, используя дифрактометр Rigaku Miniflex II с Cu-K излучением. Фиг. 2 представляет рентгеновскую порошковую дифрактограму ацетатного кислотного комплекса соединения формулы (I). Он представляет характерные XRPD пики, соответствующие табл. 2XRPD пики кристаллизованного ацетатного кислотного комплекса формулы (Ia) Ядерный магнитный резонанс (ЯМР). ЯМР спектры были получены на Bruker Avance AV-400 спектрометре при 400 МГц для 1 Н спектров и 100 МГц для 13 С спектров, используя CDCl3 в качестве растворителя. Фиг. 3 демонстрирует 1 Н ЯМР спектр ацетатного кислотного комплекса соединения формулы (I). Он представляет характерные пики, соответствующие табл. 3. Пики при 0,88 (соответствует соединению(I и при 2,13 (соответствует уксусной кислоте) устанавливают присутствие двух молекул уксусной кислоты на одну молекулу соединения формулы (I). Пример 1. Получение ацетатного кислотного комплекса формулы (Ia). В реактор помещали при примерно 20 С соединение формулы (II) (12,99 кг; 1 экв.) и бутанол-1 (130 л; 10 об. (V). Смесь перемешивали (80 об/мин) при 20 С 5 мин. Триэтиламин (6,82 л; 1,5 экв.) и 2 бромпиримидин (7,79 кг; 1,5 экв.) добавляли при 20 С. Затем реакционную смесь при перемешивании 100 об/мин нагревали при кипении (т.пл. 117 С) по крайней мере 20 ч (проверяя полноту протекания реакции методом ВЭЖХ, в случае необходимости продолжая кипячение). После охлаждения смеси до 60 С добавляли уксусную кислоту (195 л). Смесь нагревали до 75 С (исчезновение твердых частиц) и перемешивали 15 мин. Затем смесь охлаждали до 20 С (-0,3 С/мин) и перемешивали в течение 2 ч. Твердые осадки отделяли центрифугированием и промывали метил-трет-бутиловым эфиром (МТБЭ). Продукт высушивали в вакууме при 40 С, получая 17,9 кг уксуснокислого комплекса формулы (Ia) с выходом 92% и чистотой 96.5%. Пример 2. Превращение уксуснокислого комплекса в соединение формулы (I) посредством полиморфной трансформации. В реактор помещали при примерно 20 С уксуснокислый комплекс (пример 1) (9,02 кг; 1 экв.) и изопропилацетат (390 л; 40 V). Смесь перемешивали (80 об/мин) при 20 С в течение 15 мин. После нагревания до 70 С смесь перемешивали при 80 об/мин до полного образования полиморфной формы А 0 (проверка полноты реакции рентгеновским методом). Затем реакционную смесь охлаждали до 10 С и перемешивали по крайней мере 2 ч. Твердые осадки отделяли центрифугированием и промывали изопропилацетатом. Продукт высушивали в вакууме при 40 С, получая 6,11 кг неочищенного соединения (I) в форме А 0 (выход 85,6%). Пример 3. Обработка углем и полиморфная трансформация в очищенное соединение формулы (I). В реактор помещали при примерно 20 С неочищенное соединение формулы (I) в форме A0 (4,040 кг; 1 экв.), дихлорметан (222 л; 40 V) и этанол (56 л; 10 V). Смесь перемешивали (80 об/мин) при 20 С в течение 15 мин, чтобы получить абсолютно прозрачный раствор. Смесь очищали 50% вес./вес. кусочками активированного угля (21 кг; 49,5 вес./вес.). Затем жидкости фильтровали через 0,3-мкм фильтрующий катридж Cuno для удаления нерастворимых частиц (активированный уголь). Растворители выпаривали досуха в вакууме. К смеси добавляли изопропилацетат (265 л; 58 V) и 50 л азеотропной смеси выпаривали в вакууме. После охлаждения смеси до 20 С смесь перемешивали при 80 об/мин до полного образования полиморфной формы A0 (контролируя полноту реакции по рентген/ДСК, если необходимо нагревали до 70 С). Затем реакционную смесь охлаждали до 10 С. Выпавшие твердые осадки отделяли центрифугированием и промывали изопропилацетатом. Продукт сушили в вакууме при 40 С, получая соединение (I) в форме A0 (3,280 кг; выход 81,19%; чистота 99,2%). Пример 4. Получение кислотно-аддитивной соли соединения (I) с ПТСК (моно-тозилат). В реактор при примерно 20 С загружали соединение формулы (I) в форме A0 (6,075 кг; 1 экв.) и дихлорметан (92 л; 15 V). Смесь перемешивали (80 об/мин) при 20 С 15 мин. После охлаждения смеси до 10 С порциями добавляли пара-толуолсульфоновую кислоту (ПТСК, 2,417 кг; 1 экв.). Смесь перемешивали при 80 об/мин при 10 С 1 ч. Затем порциями через воронку с затвором добавляли МТБЭ (122 л; 15V). Смесь нагревали при 45 С по крайней мере 1 ч (контролируя полноту реакции по рентген/ДСК,если необходимо продолжая реакцию). После охлаждения смеси до 10 С выпавшие твердые осадки отделяли фильтрованием и промывали МТБЭ. Продукт сушили в вакууме при 40 С, получая аддитивную соль соединения формулы (I) с ПТСК (8,045 кг; выход 97,3%; чистота 99,1%). Пример 5. Получение соединения формулы (I) в виде свободного основания соединения формулы(I) из соединения формулы (II). В реактор загружали при примерно 20 С соединение формулы (II) (1 экв.) и бутанол-1 (10 V). Смесь перемешивали (80 об/мин) при 20 С в течение 5 мин. При 20 С добавляли триэтиламин (1,4 экв.) и 2 бромпиримидин (1,4 экв.). Затем смесь, перемешиваемую при 100 об/мин, нагревали до кипения (т.пл. 117 С) по крайней мере 20 ч (контролируя полноту реакции методом ВЭЖХ, если необходимо продолжая кипячение). После охлаждения смеси до 75 С добавляли уксусную кислоту (5 V). Смесь перемешивали при 75 С до исчезновения твердых частиц. Смесь затем охлаждали до 20 С (-0,3 С/мин). Выпавшие твердые осадки (влажное соединение (I)/ацетатный кислотный комплекс формулы (Ia отделяли центрифугированием и промывали метил-трет-бутиловым эфиром (МТБЭ). Продукт сушили в вакууме при 80 С, получая соединение (I) в виде свободного основания формулы (I). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Кристаллическая форма кислотного комплекса формулы (Ia) где R означает метил, характеризующийся рентгеновской порошковой дифрактограммой, содержащей один или несколько следующих пиков: 5,190,2 градуса 2-тэта; 6,170,2 градуса 2-тэта; 6,440,2 градуса 2-тэта; 14,360,2 градуса 2-тэта и 26,090,2 градуса 2-тэта, при измерениях с использованием CuK излучения. 2. Способ получения кислотного комплекса формулы (Ia) по п.1, содержащий:i) взаимодействие соединения формулы (I) с уксусной кислотой с получением кислотного комплекса формулы (Ia);ii) кристаллизацию полученного кислотного комплекса формулы (Ia). 3. Способ по п.2, дополнительно включающий взаимодействие кислотного комплекса формулы (Ia) с обесцвечивающим агентом. 4. Способ по п.2 или 3, дополнительно включающий выделение кристаллизованного комплекса формулы (Ia). 5. Способ по п.3, где обесцвечивающий агент представляет собой активированный уголь, активированный паром или химически. 6. Способ по п.4, где выделенный кристаллизованный комплекс формулы (Ia) имеет чистоту по меньшей мере приблизительно 92%. 7. Способ по п.4, где выделенный кристаллизованный комплекс формулы (Ia) имеет чистоту по меньшей мере приблизительно 97%.
МПК / Метки
МПК: A61K 31/506, C07D 487/14
Метки: производного, очистки, пирролокарбазола, конденсированного, способ
Код ссылки
<a href="https://eas.patents.su/13-21869-sposob-ochistki-kondensirovannogo-proizvodnogo-pirrolokarbazola.html" rel="bookmark" title="База патентов Евразийского Союза">Способ очистки конденсированного производного пирролокарбазола</a>
Способ получения и очистки n-алкилированного производного аспартама

Номер патента: 1948
Опубликовано: 22.10.2001
Автор: Пракаш Индра
МПК: A23L 1/236, C07K 5/075
Метки: производного, n-алкилированного, получения, способ, очистки, аспартама
Формула / Реферат:
1. Способ очистки 1-сложного метилового эфира N-[N-(3,3-диметилбутил)-L-a-аспартил]-L-фенилаланина формулы включающий стадии: (I) получения раствора 1-сложного метилового N-[N-(3,3-диметилбутил)-L-a-acпapтил]-L-фенилаланина в органическом растворителе; и (II) получения раствора вода/органический растворитель из раствора органического растворителя для осаждения 1-сложного метилового эфира N-[N-(3,3-диметилбутил)-L-a-аспартил]-L-фенилаланина...
Противоопухолевая композиция, содержащая синергическую комбинацию производного антрациклина и производного камптотецина

Номер патента: 3134
Опубликовано: 27.02.2003
Авторы: Сварато Антонино, Джерони Кристина, Карузо Микеле, Рипамонти Марина
МПК: A61P 35/00, A61K 31/704
Метки: содержащая, камптотецина, композиция, противоопухолевая, антрациклина, производного, комбинацию, синергическую
Формула / Реферат:
1. Композиция, содержащая антрациклин формул Ia или Ib и противоопухолевый ингибитор топоизомеразы-I в виде комбинированного препарата, предназначенного для одновременного, раздельного или последовательного введения для лечения опухолей. 2. Композиция по п.1, где ингибитором топоизомеразы-I является камптотецин, 9-аминокамптотецин, иринотекан (СРТ-11), топотекан, 7-этил-10-гидроксикамптотецин, GI-147211 или 9-нитрокамптотецин....
Азолидинонвинильные производные конденсированного бензола

Номер патента: 11807
Опубликовано: 30.06.2009
Авторы: Рюккле Томас, Цзян Сюйляан, Черч Деннис, Геллар Паскаль, Фаллоттон Таня
МПК: A61P 37/06, C07D 277/18, A61K 31/00...
Метки: производные, азолидинонвинильные, бензола, конденсированного
Формула / Реферат:
1. Применение соединения формулы (I) а также его геометрических изомеров, его оптически активных форм в виде энантиомеров, диастереомеров и его рацематов, а также его фармацевтически приемлемых солей и фармацевтически активных производных, где А означает 5-8-членную гетероциклическую или карбоциклическую группу, где упомянутая карбоциклическая группа может быть сконденсирована с арилом, гетероарилом, циклоалкилом или гетероциклоалкилом; X...
Способ получения производного тетразола

Номер патента: 4633
Опубликовано: 24.06.2004
Авторы: Хегедюш Иштван, Дойчне Юхас Ида, Фаркаш Йенёме, Цибула Ласло, Немеш Андраш, Балло Илдико, Крейдл Янош, Веркне Папп Эва, Надьне Багди Юдит, Петеньи Эндрене, Фишер Янош
МПК: C07D 403/10, A61P 9/12, A61K 31/41...
Метки: получения, производного, способ, тетразола
Формула / Реферат:
1. Способ синтеза лозартана калия формулы (I) имеющего химическое название 2-н-бутил-4-хлор-1-[(2'-(тетразол-5-ил)-1,1'-бифенил-4-ил)метил]имидазол-5-метанол калия, с использованием в качестве исходного соединения 2-н-бутил-4-хлор-1-[(2'-(2-трифенилметил-2H-тетразол-5-ил)-1,1'-бифенил-4-ил)метил]-1H-имидазол-5-метанол формулы (III) отличающийся тем, что проводят реакцию соединения формулы (III) в среде спирта формулы R-OH (VI), где R...
Устройство очистки, вкладыш и способ очистки жидкости

Номер патента: 10004
Опубликовано: 30.06.2008
Авторы: Хахманн Роберт, Коос Пауль, Коос Паулина
МПК: B01D 35/06, C02F 1/48, C02F 1/461...
Метки: способ, очистки, жидкости, вкладыш, устройство
Формула / Реферат:
1. Устройство очистки, предназначенное для очистки жидкости, такой как вода, предпочтительно питьевая вода, содержащее по меньшей мере один корпус (1), имеющий по меньшей мере один впуск (8), и по меньшей мере один выпуск (9) для жидкости, и по меньшей мере один вкладыш (10), содержащий по меньшей мере один фильтр (15), расположенный на траектории потока жидкости, причем устройство дополнительно содержит по меньшей мере одну гальваническую пару...
Предыдущий патент: Способ и система для управления связью в приграничной соте
Следующий патент: Устройство для ингаляции
Случайный патент: Сопло инжектора для впрыскивания реагентов в реактор