Кристаллическая форма трипептидного кетоэпоксидного соединения и способ его получения











Формула / Реферат
1. Способ получения кристаллической формы соединения формулы (II)

где кристаллическая форма имеет величины 2θ 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48,
включающий
(i) получение раствора соединения формулы (II) в органическом растворителе;
(ii) доведение раствора до сверхнасыщения, чтобы вызвать образование кристаллов; и
(iii) выделение кристаллов.
2. Способ по п.1, где органический растворитель выбран из ацетонитрила, этилацетата, гептанов, гексанов, изопропилацетата, метанола, метилэтилкетона, тетрагидрофурана, толуола и воды или любой их комбинации.
3. Способ по п.2, где органический растворитель выбран из ацетонитрила, гептанов, гексанов, метанола, тетрагидрофурана и толуола.
4. Способ по п.3, где органический растворитель выбран из гексанов, тетрагидрофурана и толуола.
5. Способ по любому из пп.1-4, где доведение раствора до сверхнасыщения включает добавление антирастворителя, предоставление возможности раствору охладиться, уменьшение объема раствора или любую их комбинацию.
6. Способ по п.5, где доведение раствора до сверхнасыщения включает добавление антирастворителя, охлаждение раствора до температуры окружающей среды и уменьшение объема раствора.
7. Способ по п.5, где антирастворитель добавляется медленно.
8. Способ по п.5, где антирастворитель выбран из гексанов, толуола и воды.
9. Способ по п.5, где уменьшение объема осуществляется выпариванием.
10. Способ по любому из пп.1-9, дополнительно включающий внесение затравки в раствор.
11. Способ по п.10, дополнительно включающий промывание кристаллов.
12. Способ по п.11, где промывание включает промывание жидкостью, выбранной из ацетонитрила, гептанов, гексанов, метанола, тетрагидрофурана, толуола, воды или их комбинации.
13. Способ по п.12, где промывание включает промывание гексанами или гептанами.
14. Способ по любому из пп.1-13, где выделение кристаллов включает фильтрацию кристаллов.
15. Способ по любому из пп.1-14, дополнительно включающий сушку кристаллов при пониженном давлении.
16. Кристаллическая форма соединения, имеющего структуру формулы (II)

где кристаллическая форма имеет величины 2θ 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48.
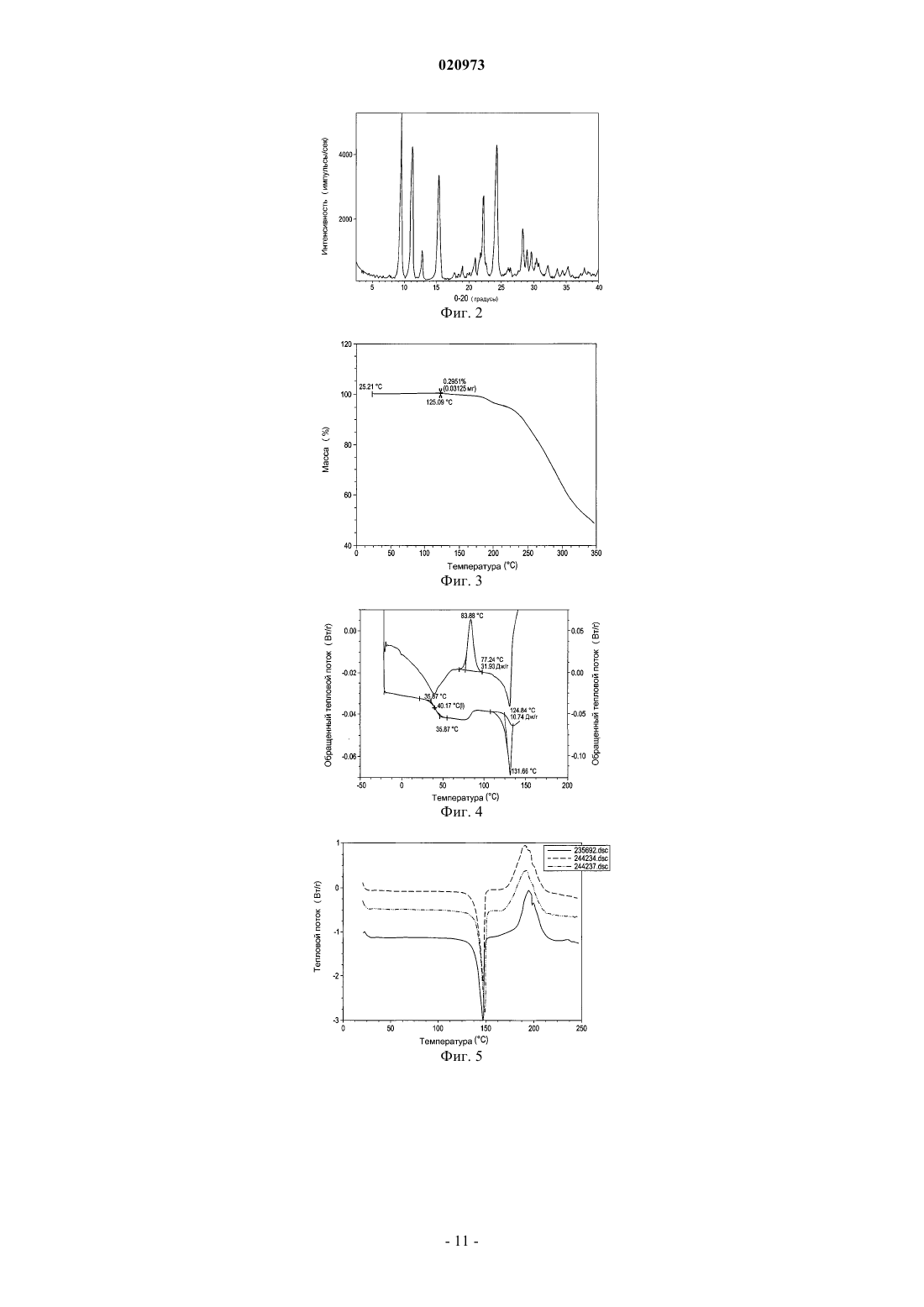
17. Кристаллическая форма соединения по п.16, имеющая термограмму дифференциальной сканирующей калориметрии, как показано на фиг. 1.
18. Кристаллическая форма соединения по п.16, имеющая точку плавления от 140 до 155°С.
19. Кристаллическая форма соединения по п.16, имеющая точку плавления от 145 до 150°С.
20. Кристаллическое соединение по п.16, имеющая порошковую рентгенограмму, как показано на фиг. 2.
21. Способ получения кристаллической формы соединения формулы (II)

где кристаллическая форма имеет величины 2θ 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48,
включающий (i) взаимодействие соединения формулы (III)

где X представляет любой подходящий противоион, с соединением формулы (IV) в органическом растворителе

(ii) получение раствора соединения формулы (II) в органическом растворителе;
(iii) доведение раствора до сверхнасыщения для обеспечения возможности образования кристаллов;
(iv) отделение кристаллов для получения кристаллической формы соединения формулы (II).

Текст
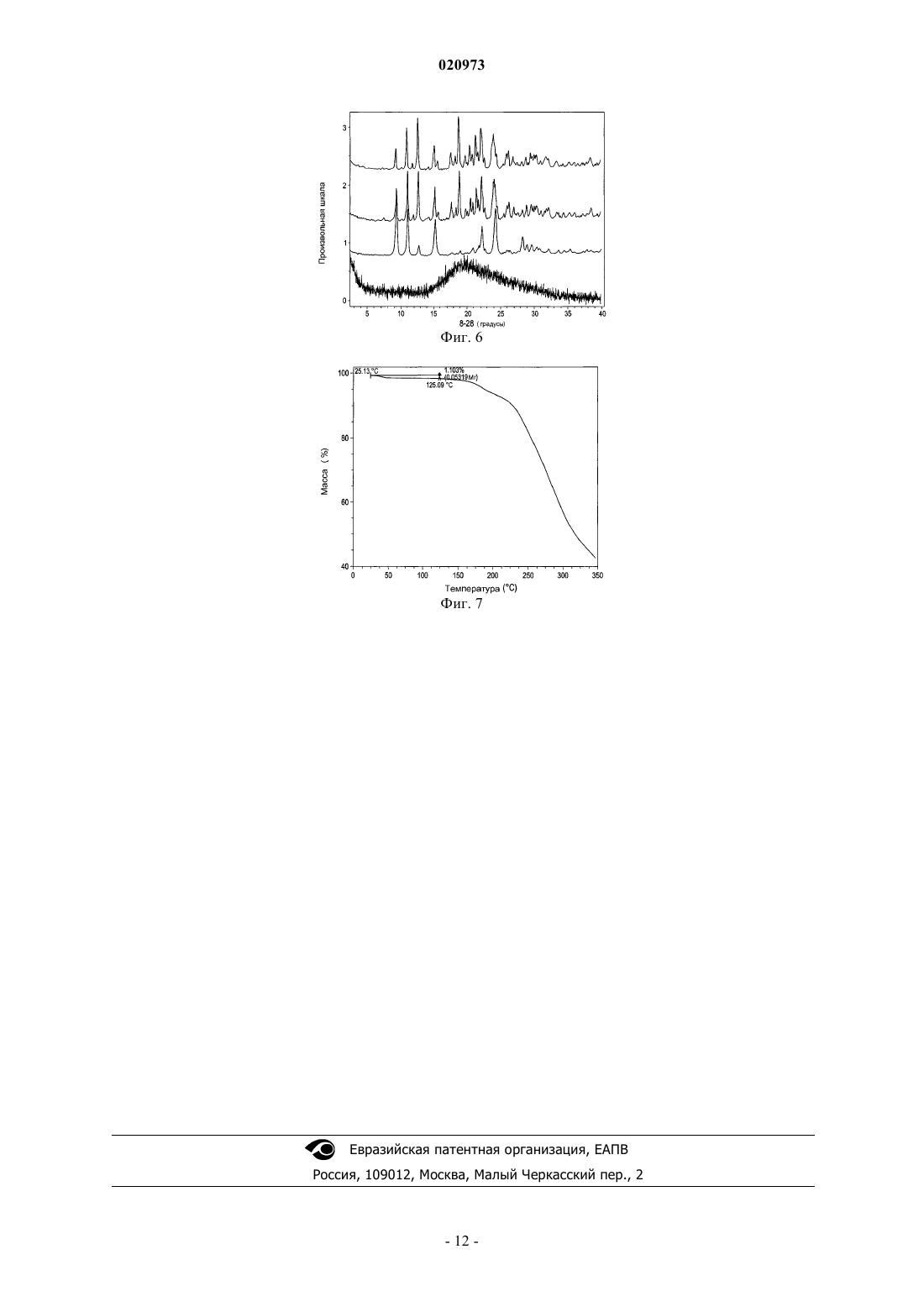
КРИСТАЛЛИЧЕСКАЯ ФОРМА ТРИПЕПТИДНОГО КЕТОЭПОКСИДНОГО СОЕДИНЕНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ Изобретение относится к кристаллическому трипептидному кетоэпоксидному соединению формулы (II) Родственные заявки Данная заявка испрашивает приоритет предварительной заявки на патент США под серийным 61/162196, поданной 20 марта 2009 г., и предварительной заявки на патент США под серийным 61/180561, поданной 22 мая 2009 г. Описания указанных выше заявок полностью включены в настоящую заявку путем ссылки. Предпосылки изобретения В эукариотах распад белка преимущественно опосредуется по убихитиновому пути, в котором являющиеся мишенью для распада белки легируются к полипептиду из 76 аминокислот убихитину. После нацеливания убихитинированные белки затем служат в качестве субстратов для протеасомы 26S, мультикаталитической протеазы, которая расщепляет белки на короткие пептиды посредством действия трех основных протеолитических активностей. Хотя опосредованный протеасомой распад выполняет общую функцию во внутриклеточном белковом обороте, он также играет ключевую роль во многих процессах,таких как представление антигенов класса I главного комплекса гистосовместимости (МНС), апоптоз,регуляция роста клеток, активация NF-KB (транскрипционного фактора В), процессинг антигенов и трансдукция провоспалительных сигналов. Протеасома 20S представляет собой мультикаталитический протеазный комплекс с молекулярной массой 700 кДа, состоящий из 28 субъединиц, организованных в четыре кольца. В дрожжах и других эукариотах 7 различныхсубъединиц образуют внешние кольца, и 7 различныхсубъединиц содержат внутренние кольца,субъединицы служат в качестве сайтов связывания для регуляторных комплексов 19S (РА 700) и HIS (PA28), а также в качестве физического барьера для внутренней протеолитической камеры, образованной двумя кольцамисубъединиц. Таким образом, считается, что протеасома существует в виде частицы 26S (протеасомы 26S). Эксперименты in vivo показали, что ингибирование формы 20S протеасомы может легко коррелироваться с ингибированием протеасомы 26S. Отщепление аминоконцевых пропоследовательностейсубъединиц во время образования частицы обнажает аминоконцевые остатки треонина, которые служат в качестве каталитических нуклеофитов. Таким образом, субъединицы, ответственные за каталитическую активность в протеасомах, обладают аминоконцевым нуклеофильным остатком, и эти субъединицы относятся к семейству гидролаз N-концевого нуклеофила (Ntn)(где нуклеофильный N-концевой остаток представляет собой, например, Cys, Ser, Thr и другие нуклеофильные части). Это семейство включает, например, пенициллин G ацилазу (PGA), пенициллин V ацилазу (PVA), глутамин PRPP амидотрансферазу (GAT) и бактериальную глиукуозиласпарагиназу. В дополнение к повсеместно экспрессированнымсубъединицам, высшие позвоночные также обладают тремя индуцируемыми интерфероном-субъединицами (LMP7, LMP2 и MECL1), которые замещают их нормальные копии, соответственно, 5, 1 и 7, таким образом, изменяя каталитическую активность протеасомы. Посредством использования различных пептидных субстратов, три основных вида протеолитической активности были определены для 20S протеасомы эукариота: активность, подобная химотрипсину(CT-L), которая отщепляет после крупных гидрофобных остатков; подобная трипсину активность (T-L),которая отщепляет после основных остатков; и активность, гидролизующая пептидилглутамил-пептид(PGPH), которая отщепляет после кислотных остатков. Два дополнительных менее охарактеризованных вида активности также приписывали протеасоме: активность BrAAP, которая отщепляет после разветвленноцепочечных аминокислот; и активность SNAAP, которая отщепляет после мелких нейтральных аминокислот. Представляется, что различные каталитические сайты вносят вклад в основные виды протеолитической активности протеасомы, поскольку ингибиторы, точечные мутации всубъединицах и обмен индуцирующихинтерферонсубъединиц в различных степенях изменяют указанные виды активности. Необходимы улучшенные способы получения ингибитора протеасом. Краткое изложение сущности изобретения Изобретение относится к способу получения кристаллической формы соединения формулы (II) где кристаллическая форма имеет величины 2 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48,включающему (i) получение раствора соединения формулы (II) в органическом растворителе; (ii) доведение раствора до сверхнасыщения, чтобы вызвать образование кристаллов; и (iii) выделение кристаллов. Органический растворитель выбран из ацетонитрила, этилацетата, гептанов, гексанов, изопропилацетата, метанола, метилэтилкетона, тетрагидрофурана, толуола и воды или любой их комбинации. В одном из вариантов осуществления способа органический растворитель выбран из ацетонитрила, гептанов, гексанов, метанола, тетрагидрофурана и толуола. В другом варианте осуществления способа органический растворитель выбран из гексанов, тетрагидрофурана и толуола. В заявленном способе доведение раствора до сверхнасыщения включает добавление антирастворителя, предоставление возможности раствору охладиться, уменьшение объема раствора или любую их комбинацию. В заявленном способе доведение раствора до сверхнасыщения включает добавление антирастворителя, охлаждение раствора до температуры окружающей среды и уменьшение объема раствора. В заявленном способе антирастворитель добавляется медленно и антирастворитель выбран из гексанов, толуола и воды. Уменьшение объема в способе осуществляется выпариванием. Заявленный способ дополнительно включает внесение затравки в раствор и промывание кристаллов, при этом промывание включает промывание жидкостью, выбранной из ацетонитрила, гептанов, гексанов, метанола, тетрагидрофурана, толуола, воды или их комбинации. В одном из вариантов осуществления изобретения промывание включает промывание гексанами или гептанами. Выделение кристаллов в заявленном способе включает фильтрацию кристаллов, а также дополнительно включает сушку кристаллов при пониженном давлении. Изобретение также относится к кристаллической форме соединения, имеющего структуру формулы где кристаллическая форма имеет величины 2 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48. Заявленная кристаллическая форма имеет термограмму дифференциальной сканирующей калориметрии, как показано на фиг. 1, и точку плавления от 140 до 155 С. Кристаллическое соединение имеет порошковую рентгенограмму, как показано на фиг. 2. Изобретение также относится к способу получения кристаллической формы соединения формулы где кристаллическая форма имеет величины 2 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48,включающему (i) взаимодействие соединения формулы (III) где X представляет любой подходящий противоион, с соединением формулы (IV) в органическом растворителе(ii) получение раствора соединения формулы (II) в органическом растворителе; (iii) доведение раствора до сверхнасыщения для обеспечения возможности образования кристаллов; и (iv) отделение кристаллов для получения кристаллической формы соединения формулы (II). Краткое описание чертежей На фиг. 1 показана DSC (дифференциальная сканирующая калориметрическая) термограмма кристаллического соединения 1. На фиг. 2 показан тип XRPD (порошковой рентгеновской дифракции) кристаллического соединения 1. На фиг. 3 показана TG (термограмма) кристаллического соединения 1. На фиг. 4 показаны модулированные термограммы аморфного соединения 1, обращенного теплового потока (нижняя кривая) и необращенного теплового потока (верхняя кривая). На фиг. 5 показано сравнение DSC термограмм кристаллического соединения 1, полученного в соответствии с примером 2 (средняя кривая), примером 3 (верхняя кривая) и примером 4 (нижняя кривая). На фиг. 6 показан тип XRPD аморфного соединения 1, полученного в соответствии с примером 1(верхняя кривая), по сравнению с типами XRPD кристаллического соединения 1, полученного в соответствии с примером 2 (верхняя кривая), примером 3 (2-я кривая снизу) и примером 4 (2-я кривая сверху). На фиг. 7 показана TG термограмма аморфного соединения 1. Подробное описание изобретения В определенных вариантах осуществления изобретение относится к кристаллическому соединению формулы (II) В определенных вариантах осуществления изобретение относится к способу получения кристаллического соединения формулы (II), включающему (i) получение аморфного соединения, например, в соответствии с заявкой на патент США 11/595804; (ii) растворение аморфного соединения в органическом растворителе; (iii) доведение раствора до сверхнасыщения; (iv) отделение кристаллов, например, фильтрацией кристаллов, декантацией жидкости из кристаллов или другой подходящей методикой отделения; и (v) промывание кристаллов. В определенных вариантах осуществления получение, кроме того, включает индукцию кристаллизации. В определенных вариантах осуществления получение, кроме того,включает сушку, предпочтительно при пониженном давлении, например, в условиях вакуума. В определенных вариантах осуществления аморфное соединение может быть растворено в растворителе, выбранном из ацетонитрила, этилацетата, гептанов, гексанов, изопропилацетата, метанола, метилэтилкетона, тетрагидрофурана, толуола и воды или любой их комбинации. В определенных вариантах осуществления аморфное соединение формулы (II) может быть растворено в органическом растворителе,выбранном из ацетонитрила, гептанов, гексанов, метанола, тетрагидрофурана и толуола или любой их комбинации. В определенных предпочтительных вариантах осуществления органический растворитель представляет собой гексан, тетрагидрофуран и толуол. В определенных вариантах осуществления доведение раствора до сверхнасыщения включает медленное добавление антирастворителя, такого как вода, гептаны, гексаны или другая полярная или неполярная жидкость, смешиваемая с органическим растворителем, предоставление возможности раствору охладиться (с осаждением или без осаждения раствора), уменьшение объема раствора или любую их комбинацию. В определенных вариантах осуществления доведение раствора до сверхнасыщения включает добавление антирастворителя, охлаждение раствора до окружающей температуры или ниже и уменьшение объема раствора, например, выпариванием растворителя из раствора. В определенных вариантах осуществления предоставление раствору возможности охладиться может быть пассивным (например, предоставление раствору возможности отстояться при окружающей температуре) или активным(например, охлаждением раствора на ледяной бане или морозильнике). В определенных вариантах осуществления способ, кроме того, включает индукцию осаждения или кристаллизации. В определенных вариантах осуществления индукция осаждения или кристаллизации включает вторичную нуклеацию, где нуклеация происходит в присутствии кристаллов затравки или при взаимодействиях с окружающей средой (стенками кристаллизатора, перемешивающими импеллерами,обработкой ультразвуком и т.д.). В определенных вариантах осуществления промывание кристаллов включает промывание жидкостью, выбранной из ацетонитрила, гептанов, гексанов, метанола, тетрагидрофурана, толуола, воды или их комбинации. В определенных вариантах осуществления промывание кристаллов включает промывание кристаллического соединения формулы (II) гексанами или гептанами. В определенных вариантах осуществления точка плавления кристаллического соединения формулы(II) находится в диапазоне от примерно 140 до примерно 155 С, от примерно 145 до примерно 150 С. В определенных вариантах осуществления DSC кристаллического соединения формулы (II) имеет резкий эндотермический максимум при температуре примерно 147 С, например, в результате плавления и разрушения кристаллической формы, как показано на фиг. 1. В определенных вариантах осуществления тип порошковой рентгеновской дифракции кристаллического соединения формулы (II) представляет собой (-2) 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48, как показано на фиг. 2. В определенных вариантах осуществления TG термограмма кристаллического соединения формулы(II) проявляет потерю массы от 0,0 до 0,3% в температурном диапазоне от 25 до 125 С, как показано на фиг. 3. В определенных вариантах осуществления кристаллическое соединение формулы (II) не сольватировано (например, кристаллическая решетка не содержит молекул растворителя). В определенных вариантах осуществления кристаллическое соединение формулы (II) является сольватированным. В определенных вариантах осуществления изобретение относится к способу получения кристаллического соединения формулы (II) включающему (i) взаимодействие соединения формулы (III) где X представляет любой подходящий противоион, с соединением формулы (IV) в органическом растворителе(ii) получение раствора соединения формулы (II) в органическом растворителе; (iii) доведение раствора до сверхнасыщения для обеспечения возможности образования кристаллов; и (iv) отделение кристаллов для получения кристаллического соединения формулы (II), например, фильтрацией кристаллов,декантацией или любой другой подходящей методикой отделения. В определенных вариантах осуществления получение, кроме того, включает индукцию кристаллизации. В определенных вариантах осуществления получение, кроме того, включает промывание кристаллов, например, растворителем или жидкостью, не являющейся растворителем. В определенных вариантах осуществления получение, кроме того, включает сушку, предпочтительно при пониженном давлении, например, в вакууме. В определенных вариантах осуществления X представляет противоион, выбранный из гидробромида, гидрохлорида, сульфата, фосфата, нитрата, ацетата, трифторацетата, цитрата, метансульфоната, валерата, олеата, пальмитата, стеарата, лаурата, бензоата, лактата, сукцината, тосилата, малоната, малеата,фумарата, сукцината, тартрата, месилата, 2-гидроксиэтансульфоната и тому подобных (см., например,публикацию Berge et al. (1977) Pharmaceutical Salts, J. Pharm. Sci. 66: 1-19.) В определенных вариантах осуществления X выбран из трифторацетата, метансульфоната, толуолсульфоната, ацетата, хлорида и бромида, предпочтительно трифторацетата. В определенных вариантах осуществления органический растворитель выбран из ацетонитрила,этилацетата, гептанов, гексанов, изопропилацетата, метанола, метилэтилкетона, тетрагидрофурана, толуола и воды или любой их комбинации. В определенных вариантах осуществления аморфное соединение формулы (II) может быть растворено в органическом растворителе, выбранном из ацетонитрила, гептанов, гексанов, метанола, тетрагидрофурана и толуола или любой их комбинации. В определенных вариантах осуществления органический ацетонитрил, предпочтительно ацетонитрил или толуол. В определенных вариантах осуществления получение, кроме того, включает промывание кристаллов формулы (II). В определенных вариантах осуществления промывание кристаллов включает промывание жидкостью, выбранной из антирастворителя, ацетонитрила, гептанов, гексанов, метанола, тетрагидрофурана, толуола, воды или их комбинации. В определенных вариантах осуществления кристаллы промываются комбинацией антирастворителя и органического растворителя. В определенных вариантах осуществления антирастворитель представляет собой воду, тогда как в других вариантах осуществления,он представляет собой щелочной растворитель, такой как гексан или пентан, или ароматический углево-4 020973 дородный растворитель, такой как бензол, толуол или ксилол. В определенных вариантах осуществления получение, кроме того, включает сушку кристаллов обоих соединений формулы (II), предпочтительно при пониженном давлении, например, в вакууме. Виды применения кристаллических трипептидных эпоксикетонов Организованный распад белка играет решающую роль в поддержании нормальных клеточных функций, и протеасома является неотъемлемой частью процесса распада белка. Протеасома регулирует уровни белков, которые важны для развития клеточного цикла и апоптоза в нормальных и злокачественных клетках, например циклинов, каспаз, BCL2 и nF-kB (Kumatori et al., Proc. Natl. Acad. Sci. USA (1990) 87:7071-7075; Almond et al., Leukemia (2002) 16: 433-443). Таким образом, неудивительно, что ингибирование активности протеасомы может внедряться в терапевтические способы для лечения различных патологических состояний, таких как злокачественные, не злокачественные и аутоиммунные заболевания, в зависимости от вовлеченных в процесс клеток. Модели и in vitro, и in vivo показали, что злокачественные клетки, в целом, восприимчивы к ингибированию протеасомы. Действительно, ингибирование протеасомы также было обосновано в качестве терапевтической стратегии для лечения множественной меланомы. Это частично могло быть связано с зависимостью обладающих высокой пролиферативной способностью злокачественных клеток от системы протеасомы для быстрого удаления белков (Rolfe et al., J. Mol. Med. (1997) 75:5-17; Adams, Nature (2004) 4: 349-360). Кристаллическое соединение формулы (II) согласно настоящему изобретению используется для лечения аутоиммунных заболеваний у млекопитающего, включающего введение терапевтически эффективного количества описанных в настоящей заявке соединения или композиции, включающей указанное соединение формулы (II). Кристаллическое соединение формулы (II) согласно настоящему изобретению может использоваться в способе применения соединения в качестве иммуномодулирующего средства для ингибирования или изменения представления антигена в клетке, включающем воздействие на клетку (или введение индивиду) соединения, описанного в настоящем изобретении, в способе лечения заболеваний трансплантата или связанных с ним, таких как болезнь трансплантат против хозяина или болезнь хозяин против трансплантата у млекопитающего, включающем введение терапевтически эффективного количества соединения, описанного в настоящем изобретении. Заявленная кристаллическая форма соединения (II) используется в способах воздействия на зависимую от протеасомы регуляцию онкопротеинов и способах лечения или ингибирования роста злокачественного новообразования и в способах лечения связанного с р 53 апоптоза. Кристаллическая форма соединения формулы (II) может использоваться для лечения нейродегенеративных заболеваний и состояний. Было также продемонстрировано, что ингибиторы, которые связываются с протеасомой 20S, стимулируют формирование костной ткани в органных культурах костей. Кроме того, когда такие ингибиторы вводились системно мышам, то определенные ингибиторы протеасомы увеличивали объем костей и скорость образования костной ткани более чем на 70% (Garrett, I. R. et al., J. Clin. Invest. (2003) 111: 1771-1782), поэтому свидетельствуя о том, что убихитин-протеасомный механизм регулирует дифференциацию остеобластов и формирование костной ткани. Поэтому, описанные соединение формулы (II) может использоваться при лечении и/или профилактике заболеваний, связанных с потерей костной ткани,таких как остеопороз. Таким образом, кристаллическое соединение формулы (II) может использоваться в способе лечения заболевания или состояния, выбранного из онкологического заболевания, аутоиммунного заболевания,состояния, связанного с трансплантатом, нейродегенеративного заболевания, состояния, связанного с фиброзом, состояний, связанных с ишемией, инфекции (вирусной, паразитарной или прокариотической) и заболеваний, связанных с потерей костной ткани, включающем введение описанных в настоящей заявке соединения или композиции. Введение кристаллических трипептидных эпоксикетонов Кристаллическое соединение формулы (II), полученное, как описано в настоящей заявке, могут вводиться в различных формах, в зависимости от подлежащего лечению расстройства и возраста, состояния и массы тела пациента, как хорошо известно в данной области. Количество активного ингредиента, которое может комбинироваться с материалом носителя для получения одной лекарственной формы, в целом составляет количество соединения, которое обеспечивает терапевтический эффект. Используемый в настоящем описании термин ингибитор предназначен для описания соединения,которое блокирует или снижает активность фермента (например, ингибирование протеолитического расщепления стандартных флуорогенных пептидных субстратов, таких как suc-LLVY-AMC, Box-LLRAMC и Z-LLE-AMC, ингибирование различных видов каталитической активности протеасомы 20S). Ингибитор может действовать конкурентным или неконкурентным ингибированием. Ингибитор может связываться обратимо или необратимо, и поэтому термин включает соединения, которые являются суицидальными субстратами фермента. Ингибитор может модифицировать один или более сайтов на активном сайте фермента или около него, или он может вызвать конформационное изменение в других участках фермента. Используемый в настоящем описании термин протеасома предназначен для включения иммуно- и конститутивных протеасом. Используемый в настоящем описании термин по существу, чистый относится к кристаллическому полиморфу, чистота которого составляет более чем 90%, означая, что он содержит менее чем 10% любого другого соединения, включая соответствующее аморфное соединение. Предпочтительно кристаллический полиморф имеет чистоту более чем 95%, или даже чистоту более чем 98%. Примеры Пример 1 Синтез соединения 1 Синтез соединения (А) К раствору простого метилового эфира N-Boc-серина (43,8 г, 200 ммоль), триэтиламина (26,5 г, 260 ммоль) и 4-(диметиламино)пиридина в дихлорметане (1,2 л) при 0 С добавляли раствор бензилхлорформиата (41 г, 240 ммоль) в дихлорметане (250 мл) в течение 30 мин. Полученную смесь перемешивали при такой же температуре в течение еще 3 ч. Добавляли насыщенный водный раствор бикарбоната натрия(200 мл), и органический слой отделяли, остаточную смесь экстрагировали дихлорметаном (2400 мл). Комбинированные органические слои промывали насыщенным водным бикарбонатом натрия (200 мл) и рассолом (200 мл), сушили над сульфатом натрия и фильтровали через Целит-545. Растворители удаляли при пониженном давлении, и остаток очищали флэш-хроматографией (силикагель, гексан и этилацетат). Соединение (А) (54 г) выделяли и характеризовали LC/MS (жидкостной хроматографией/массспектрометрией) (LRMS(МН) m/z: 310,16). Синтез соединения (В) К раствору соединения (А) (54 г) в дихлорметане (200 мл) при 0 С добавляли трифторуксусную кислоту (200 мл) в течение 10 мин, и полученную смесь перемешивали при такой же температуре еще в течение 3 ч. Растворители удаляли при пониженном давлении, и остаток помещали в высокий вакуум на ночь, получая соль TFA (трифторуксусной кислоты) соединения (В), которое характеризовали LC/MS(LRMS (MH) m/z: 210,11). Синтез соединения (С) К раствору соединения (В) (43,8 г, 200 ммоль), простого метилового эфира N-Boc-серина (36,7 г,167 ммоль), НОВТ (гидроксибензотриазола) (27 г, 200 ммоль) и HBTU (1 Н-бензотриазолия) (71,4 г, 200 ммоль) в тетрагидрофуране (1,2 л) при 0 С добавляли раствор N,N-диэтилизопропиламин (75 г, 600 ммоль) в тетрагидрофуране (250 мл) в течение 10 мин, и рН полученной смеси составил 8. Смесь перемешивали при комнатной температуре еще в течение 5 ч. Большую часть растворителя удаляли при пониженном давлении при комнатной температуре и разбавляли насыщенным водным раствором бикарбоната натрия (400 мл). Затем его экстрагировали этилацетатом (3400 мл), промывали бикарбонатом натрия (100 мл) и рассолом (100 мл). Объединенные органические слои сушили над сульфатом натрия и фильтровали через Целит-545. Растворители удаляли при пониженном давлении. И остаток очищали флэш-хроматографией (силикагель, гексан и этилацетат). Соединение (С) (65 г) выделяли и характеризовали LC/MS (LRMS (MH) m/z: 411,21). Синтез соединения (D) К раствору соединения (С) (18 г) в дихлорметане (100 мл) при 0 С добавляли трифторуксусную кислоту (80 мл) в течение 5 мин, и полученную смесь перемешивали при той же температуре в течение еще 3 ч. Растворители удаляли при пониженном давлении, и остаток помещали в высокий вакуум на ночь,получая соль TFA промежуточного соединения (D), которое характеризовали LC/MS (LRMS (MH) m/z: 311,15). Синтез соединения (Е) К раствору этил 2-метилтиазол-5-карбоксилата (15 г, 88 ммоль) в тетрагидрофуране (50 мл) при 0 С добавляли водный раствор гидроксида натрия (5N, 50 мл) в течение 10 мин, и полученный раствор перемешивали при комнатной температуре в течение еще 2 ч. Его затем подкисляли хлористо-водородной кислотой (2N) до рН 1 и экстрагировали тетрагидрофураном (3100 мл). Объединенные органические слои промывали рассолом (30 мл) и сушили над сульфатом натрия. Большую часть растворителей удаляли при пониженном давлении и остаток лиофилизировали для получения соединения (Е) (14 г). Синтез соединения (F) К раствору соединения (D) (41 ммоль) и 2-метилтиазол-5-карбоновой кислоты (Е) (6,0 г, 42 ммоль),НОВТ (7,9 г, 50 ммоль) и HBTU (18,0 г, 50 ммоль) в тетрагидрофуране (800 мл) при 0 С добавляли раствор N,N-диэтилизопропиламина (50 г) в тетрагидрофуране (200 мл) в течение 5 мин до тех пор, пока его рН не достигнет уровня приблизительно 8,5. Полученную смесь перемешивали при такой же температуре в течение ночи. Затем ее гасили насыщенным водным раствором бикарбоната натрия (200 мл), и большую часть растворителей удаляли при пониженном давлении. Остаточную смесь экстрагировали этилацетатом (3400 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (200 мл) и рассолом (100 мл), сушили над сульфатом натрия и фильтровали через Целит-545. Растворители удаляли при пониженном давлении,и остаток очищали флэш-хроматографией (силикагель, этилацетат с 2% метанолом). Соединение (F)(17,1 г) выделяли и характеризовали LC/MS (LRMS (МН) m/z: 436,15). Синтез соединения (G) К раствору соединения (F) (17,1 г, 95 ммоль) в метаноле (300 мл) добавляли 10% Pd/C (3 г). Полученной смеси давали возможность перемешиваться при давлении 1 атмосферы водорода в течение 48 ч. Смесь фильтровали через Целит 545, и фильтровальную лепешку промывали метанолом (200 мл). Органические слои концентрировали при пониженном давлении, и остаток помещали в высокий вакуум для получения соединения (G), которое характеризовали LC/MS (LRMS (МН) m/z: 346,1). Синтез соединения (Н)N-Boc фенилаланинкетоэпоксид (140 мг, 0,46 ммоль) разбавляли DCM (дихлорметаном) (2 мл) и охлаждали до 0 С. К данному раствору добавляли трифторуксусную кислоту (6 мл). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 1 ч, и в это время TLC (тонкослойная хроматография) показала полное расходование исходного материала. Полученный раствор концентрировали при пониженном давлении и помещали в высокий вакуум для получения соли TFA Соединения (Н). Синтез соединения 1 К раствору указанного выше соединения (Н) (131 мг, 0,38 ммоль) и (J) (0,46 ммоль), НОВТ (75 мг,0,48 ммоль) и HBTU (171 мг, 0,48 ммоль) в тетрагидрофуране (20 мл) и N,N-диметилформамиде (10 мл) при 0 С по каплям добавляли N,N-диэтилизопропиламин (1 мл). Смесь перемешивали при такой же температуре еще в течение 5 ч. Затем ее гасили насыщенным водным раствором бикарбоната натрия (20 мл),и большую часть растворителей удаляли при пониженном давлении. Затем остаточную смесь экстрагировали этилацетатом (340 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (20 мл) и рассолом (10 мл), сушили над сульфатом натрия и фильтровали через Целит-545. Растворители удаляли при пониженном давлении, и остаток очищали HPLC (ВЭЖХ)(0,02 М водный ацетат аммония и ацетонитрил (66/34) для получения соединения 1 (92 мг), которое лиофилизировали и характеризовали LC/MS (LRMS (MH) m/z: 533,2). Пример 2 Аморфное соединение 1 (50 мг) растворяли в ацетонитриле (1 мл), затем добавляли деионизированную воду (2 мл), и раствор доводили до сверхнасыщения медленным выпариванием 1 мл в течение примерно 1-2 недель. Полученные кристаллы фильтровали, промывали 1 мл раствора ацетонитрил-вода в соотношении 1:2 и сушили в вакууме в течение 12 ч для получения кристаллического полиморфа соединения 1 (25 мг) с точкой плавления 148 С. Характерная кривая DSC образца, зарегистрированная дифференциальным сканирующим калориметром ТА Instruments Differential Scanning Calorimeter 2920 при скорости нагревания 10 С/мин, показана на фиг. 1. Пример 3 Аморфное соединение 1 (611 мг) растворяли в тетрагидрофуране (5 мл) с последующим добавлением гексанов (5 мл), и в раствор вносили затравку кристаллического полиморфа соединения 1, полученного в примере 2, и раствор доводили до сверхнасыщения медленным выпариванием 5 мл в течение примерно 17 ч. Полученные кристаллы фильтровали, промывали 1 мл тетрагидрофурана-гексанов в соотно-7 020973 шении 1:1 и сушили в вакууме в течение 12 ч для получения кристаллического полиморфа соединения 1(150 мг) с точкой плавления 147 С. Пример 4 Аморфное соединение 1 (176 мг) растворяли в тетрагидрофуране (5 мл), затем добавляли толуол (25 мл). В раствор вносили затравку кристаллического полиморфа Соединения 1, полученного в примере 2, и раствор доводили до сверхнасыщения медленным выпариванием 20 мл в течение примерно 2 дней. Полученные кристаллы фильтровали, промывали 15 мл толуола и сушили в вакууме в течение 12 ч для получения кристаллического полиморфа соединения 1 (88 мг) с точкой плавления 149 С. Пример 5 Аморфное соединение 1 (312 мг) растворяли в толуоле (50 мл), нагретом до примерно 100 С для завершения растворения, затем добавляли гексаны (50 мл), и в раствор вносили затравку кристаллического полиморфа соединения I, полученного в примере 2, и раствор доводили до сверхнасыщения медленным выпариванием 60 мл в течение примерно 2 дней. Полученные кристаллы фильтровали, промывали 10 мл толуола и сушили в вакууме в течение 12 ч для получения кристаллического полиморфа соединения 1(156 мг) с точкой плавления 149 С. Пример 6 Аморфное соединение 1 (1,4 г) растворяли в толуоле (25 мл), нагретом до 50 С для завершения растворения, затем раствор доводили до сверхнасыщения охлаждением до 22 С и предоставлением соединению возможности кристаллизоваться в течение 12 ч. Полученные кристаллы фильтровали, промывали 5 мл гексанов и сушили в вакууме в течение 12 ч для получения кристаллического полиморфа соединения 1 (0,94 г) с точкой плавления 149 С. Пример 7 Синтез соединения 1 Синтез соединения (Н)N-Boc фенилаланинкетоэпоксид (1,0 эквивалент) растворяли в DCM (3 л/кг N-Boc фенилаланинкетоэпоксида) в 3-горловой колбе с круглым дном в инертной атмосфере, и раствор охлаждали на ледяной бане. Затем добавляли TFA (5,0 эквивалентов) со скоростью для поддержания внутренней температуры ниже 10 С. Затем реакционную смесь согревали приблизительно до 20 С и перемешивали в течение 1-3 ч. Затем к реакционной смеси добавляли МТВЕ (простой метил-трет-бутиловый эфир) (3,6 л/кг N-Вос фенилаланинкетоэпоксида) при поддержании температуры смеси ниже 25 С. Затем добавляли гептан(26,4 л/кг N-Boc фенилаланинкетоэпоксида), реакционную смесь охлаждали до температуры от -5 до 0 С в течение 2-3 ч для обеспечения возможности кристаллизации соединения (Н). Белое твердое вещество фильтровали и промывали гептаном (3 л/кг N-Boc фенилаланинкетоэпоксида). Белое твердое вещество затем сушили в вакууме в течение 12 ч при 22 С. Полученный выход составил 86%, при ВЭЖХ чистоте 99,4%. Синтез соединения 1 Соединение (Н) (1,2 эквивалента), соединение (G) (1,0 эквивалент), HBTU (1,2 эквивалента), НОВТ(1,2 эквивалента) и N-метилпирролидинон (8 л/кг соединения (G добавляли в сухую колбу при инертной температуре, и смесь перемешивали при 23 С для завершения растворения. Затем реакционную смесь охлаждали до температуры от -5 до 0 С, и диизопропилэтиламин (2,1 эквивалента) добавляли в течение 15 мин, в то же время, поддерживая внутреннюю температуру менее чем 0 С. Реакционную смесь перемешивали при 0 С в течение 12 ч. Неочищенное соединение 1 осаждали выливанием реакционной смеси на 8% бикарбонат натрия (40 л/кг соединения (G и суспензию неочищенного соединения 1 перемешивали в течение 12 ч при 2025 С, с последующим перемешиванием при 0-5 С в течение 1 ч. Белое твердое вещество фильтровали и промывали водой (5 л/кг соединения (G) ). Затем белое твердое вещество ресуспендировали в воде (15 л/кг) в течение 3 ч при 20-25 С, фильтровали и промывали водой (5 л/кг соединения (G и изопропилацетатом (22 л/кг соединения (G. Белое твердое вещество сушили в вакууме при 45 С до постоянной массы. Выход неочищенного соединения 1 составил 65%, при ВЭЖХ чистоте 97,2%. Неочищенное соединение 1 полностью растворяли в изопропилацетате (20 л/кг неочищенного соединения 1) перемешиванием и нагреванием при 85 С. Затем раствор в горячем состоянии фильтровали для удаления любого материала в виде частиц, и раствор вновь нагревали до 85 С с получением прозрачного раствора. Прозрачному раствору давали возможность охладиться со скоростью 10 С в ч до 20 С, когда происходила существенная кристаллизация соединения 1. Суспензию перемешивали при 20 С в течение 6 ч с последующим перемешиванием при 0-5 С в течение минимум 2 ч и фильтрацией и промыванием изопропилацетатом (1 л/кг неочищенного соединения 1). Очищенное соединение 1 сушили в вакууме при 45 С в течение минимум 24 ч до постоянной массы. Выход соединения 1 составил 87%,при ВЭЖХ чистоте 97,2%. Пример 8 Синтез соединения 1 Соединение (Н) (1,1 эквивалента), соединение (G) (1,0 эквивалент), HBTU (1,5 эквивалента), НОВТ перемешивали при 23 С для завершения растворения. Затем реакционную смесь охлаждали до температуры от -5 до 0 С, и добавляли диизопропилэтиламин (2,1 эквивалента) в течение 15 мин, в то же время,поддерживая внутреннюю реакционную температуру менее чем 0 С. Затем реакционную смесь перемешивали при 0 С в течение 3 ч. Реакционную смесь гасили добавлением предварительно охлажденным насыщенным раствором бикарбоната натрия (94 л/кг соединения (G, в то же время, поддерживая внутреннюю температуру на уровне менее 10 С. Содержимое затем переносили в делительную воронку. Смесь экстрагировали этилацетатом (24 л/кг соединения (G, и органический слой промывали насыщенным раствором бикарбоната натрия (12 л/кг соединения (G и насыщенным раствором хлорида натрия (12 л/кг соединения (G. Органический слой концентрировали при пониженном давлении при температуре бани менее чем 30 С до 15 л/кг соединения (G), с последующей совместной перегонкой с изопропилацетатом (224 л/кгPR-022). Конечный объем доводили до 82 л/кг соединения (G) изопропилацетатом перед нагреванием до 60 С с получением прозрачного раствора. Смеси прозрачного раствора давали возможность охладиться до 50 С перед добавлением кристаллов затравки. Раствору давали возможность охладиться до 20 С, когда происходила существенная кристаллизация соединения 1. Суспензию перемешивали при 0 С в течение 12 ч перед фильтрацией и промыванием изопропил ацетатом (2 л/кг соединения 1). Соединение 1 сушили в вакууме при 20 С в течение 12 ч до постоянной массы. Выход соединения 1 составил 48%, при ВЭЖХ чистоте 97,4%. Эквиваленты Специалистам в данной области будет понятно или они смогут определить не более чем обычным экспериментированием многочисленные эквиваленты соединений и способы их применения, описанные в настоящей заявке. Считается, что такие эквиваленты находятся в пределах объема настоящего изобретения и охватываются следующей формулой изобретения. Все приведенные выше ссылки и публикации включены в настоящее описание путем ссылки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения кристаллической формы соединения формулы (II) где кристаллическая форма имеет величины 2 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48,включающий(i) получение раствора соединения формулы (II) в органическом растворителе;(ii) доведение раствора до сверхнасыщения, чтобы вызвать образование кристаллов; и(iii) выделение кристаллов. 2. Способ по п.1, где органический растворитель выбран из ацетонитрила, этилацетата, гептанов,гексанов, изопропилацетата, метанола, метилэтилкетона, тетрагидрофурана, толуола и воды или любой их комбинации. 3. Способ по п.2, где органический растворитель выбран из ацетонитрила, гептанов, гексанов, метанола, тетрагидрофурана и толуола. 4. Способ по п.3, где органический растворитель выбран из гексанов, тетрагидрофурана и толуола. 5. Способ по любому из пп.1-4, где доведение раствора до сверхнасыщения включает добавление антирастворителя, предоставление возможности раствору охладиться, уменьшение объема раствора или любую их комбинацию. 6. Способ по п.5, где доведение раствора до сверхнасыщения включает добавление антирастворителя, охлаждение раствора до температуры окружающей среды и уменьшение объема раствора. 7. Способ по п.5, где антирастворитель добавляется медленно. 8. Способ по п.5, где антирастворитель выбран из гексанов, толуола и воды. 9. Способ по п.5, где уменьшение объема осуществляется выпариванием. 10. Способ по любому из пп.1-9, дополнительно включающий внесение затравки в раствор. 11. Способ по п.10, дополнительно включающий промывание кристаллов. 12. Способ по п.11, где промывание включает промывание жидкостью, выбранной из ацетонитрила,гептанов, гексанов, метанола, тетрагидрофурана, толуола, воды или их комбинации. 13. Способ по п.12, где промывание включает промывание гексанами или гептанами. 14. Способ по любому из пп.1-13, где выделение кристаллов включает фильтрацию кристаллов. 15. Способ по любому из пп.1-14, дополнительно включающий сушку кристаллов при пониженном давлении. 16. Кристаллическая форма соединения, имеющего структуру формулы (II) где кристаллическая форма имеет величины 2 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48. 17. Кристаллическая форма соединения по п.16, имеющая термограмму дифференциальной сканирующей калориметрии, как показано на фиг. 1. 18. Кристаллическая форма соединения по п.16, имеющая точку плавления от 140 до 155 С. 19. Кристаллическая форма соединения по п.16, имеющая точку плавления от 145 до 150 С. 20. Кристаллическое соединение по п.16, имеющая порошковую рентгенограмму, как показано на фиг. 2. 21. Способ получения кристаллической формы соединения формулы (II) где кристаллическая форма имеет величины 2 8,94; 9,39; 9,76; 10,60; 11,09; 12,74; 15,27; 17,74; 18,96; 20,58; 20,88; 21,58; 21,78; 22,25; 22,80; 24,25; 24,66; 26,04; 26,44; 28,32; 28,96; 29,65; 30,22; 30,46; 30,78; 32,17; 33,65; 34,49; 35,08; 35,33; 37,85; 38,48,включающий (i) взаимодействие соединения формулы (III) где X представляет любой подходящий противоион, с соединением формулы (IV) в органическом растворителе(ii) получение раствора соединения формулы (II) в органическом растворителе;(iii) доведение раствора до сверхнасыщения для обеспечения возможности образования кристаллов;(iv) отделение кристаллов для получения кристаллической формы соединения формулы (II).
МПК / Метки
МПК: C07K 5/08, A61K 38/55, C07D 277/56
Метки: трипептидного, получения, кристаллическая, соединения, способ, кетоэпоксидного, форма
Код ссылки
<a href="https://eas.patents.su/13-20973-kristallicheskaya-forma-tripeptidnogo-ketoepoksidnogo-soedineniya-i-sposob-ego-polucheniya.html" rel="bookmark" title="База патентов Евразийского Союза">Кристаллическая форма трипептидного кетоэпоксидного соединения и способ его получения</a>
Кристаллическая форма хинолинон-карбоксамидного соединения

Номер патента: 12115
Опубликовано: 28.08.2009
Авторы: Дженов Дэниэл, Тернер С.Дерек, Фазери Пол Р., Голдблум Адам, Чао Роберт
МПК: C07D 451/04, A61P 1/00, A61K 31/40...
Метки: соединения, форма, хинолинон-карбоксамидного, кристаллическая
Формула / Реферат:
1. Кристаллическая солевая форма, представляющая собой кристаллический гидрохлорид {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3,2,1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты или сольват этой соли. 2. Кристаллическая солевая форма по п.1, которая представляет собой кристаллический гидрохлорид. 3. Кристаллическая солевая форма по п.2, которая характеризуется порошковой...
Стабильная негигроскопичная кристаллическая форма n-[n-n-(4-(пиперидин-4-ил)бутаноил)-n-этилглицилового] соединения

Номер патента: 1362
Опубликовано: 26.02.2001
Авторы: Пауэрс Мэттью Р., Кубиак Грегори Г., Ванасс Бенуа Дж., Лиу Роберт С., Родригез Вальтер, Вемури Нарасимха М., Виндиш Винсент, Шербайн Джеймс П., Салазар Дайан К., Менсел Джеймс Дж., Толедо-Веласкез Дэвид, Гардетто Энтони Дж., Вудвард Рик Г., Хшан Зофия Дж., Следески Адам В.
МПК: A61K 31/445, C07D 211/06, A61P 7/02...
Метки: форма, соединения, кристаллическая, негигроскопичная, стабильная, n-[n-n-(4-(пиперидин-4-ил)бутаноил)-n-этилглицилового
Формула / Реферат:
1. Соединение, представляющее N-(N-трет-бутоксикарбонил-N-этилглицил)-(L)-аспарагиновой кислоты b-бензиловый сложный эфир. 2. Соединение, представляющее N-[N-[N-[4-[N-бензилоксикарбонилпиперидин-4-ил]бутаноил]-N-этилглицил]-(L)-аспартил] b-бензиловый сложный эфир]-(L)-b-циклогексилаланинамид. 3. Соединение, представляющее 4-(4-пиперидин)бутилиденилкарбоновую кислоту. 4. Соединение, представляющее...
Новая кристаллическая форма iv агомелатина, способ её получения и фармацевтические композиции, которые её содержат

Номер патента: 10867
Опубликовано: 30.12.2008
Авторы: Линоль Жюли, Суви Жан-Клод, Кокерель Жерар
МПК: C07C 233/18, A61K 31/165, C07C 231/00...
Метки: которые, форма, получения, содержат, агомелатина, кристаллическая, новая, композиции, фармацевтические, способ
Формула / Реферат:
1. Кристаллическая форма IV агомелатина формулы (I) которая характеризуется следующей рентгеновской дифракционной порошкограммой, измеренной с помощью дифрактометра (медный антикатод) и выраженной в виде межплоскостного расстояния d, брэгговского угла 2q, интенсивности и относительной интенсивности (выраженной в процентах наиболее интенсивного луча) 2. Способ получения кристаллической формы IV соединения формулы (I), определенной в п.1,...
Новая кристаллическая форма v агомелатина, способ её получения и фармацевтические композиции, которые её содержат

Номер патента: 11030
Опубликовано: 30.12.2008
Авторы: Суви Жан-Клод, Кокерель Жерар, Линоль Жюли
МПК: C07C 231/00, C07C 233/18, A61K 31/165...
Метки: форма, композиции, которые, фармацевтические, новая, получения, агомелатина, способ, содержат, кристаллическая
Формула / Реферат:
1. Кристаллическая форма V агомелатина формулы (I) которая характеризуется следующей рентгеновской порошковой дифрактограммой, измеренной с помощью дифрактометра (медный антикатод) и выраженной в виде межплоскостного расстояния d, брэгговского угла 2q, интенсивности и относительной интенсивности (выраженной в % наиболее интенсивного луча) 2. Способ получения кристаллической формы V соединения формулы (I) в соответствии с п.1, который...
Новая кристаллическая форма iii агомелатина, способ её получения и фармацевтические композиции, которые её содержат

Номер патента: 11031
Опубликовано: 30.12.2008
Авторы: Линоль Жюли, Кокерель Жерар, Суви Жан-Клод
МПК: C07C 231/00, C07C 233/18, A61K 31/165...
Метки: получения, композиции, содержат, агомелатина, фармацевтические, которые, кристаллическая, способ, форма, новая
Формула / Реферат:
1. Кристаллическая форма III агомелатина формулы (I) которая характеризуется следующей рентгеновской порошковой дифрактограммой, измеренной с помощью дифрактометра (медный антикатод) и выраженной в виде межплоскостного расстояния d, брэгговского угла 2q, интенсивности и относительной интенсивности (выраженной в процентах наиболее интенсивного луча): 2. Способ получения кристаллической формы III соединения формулы (I) в соответствии с п.1,...
Предыдущий патент: Ручной аэрозольный распылитель
Следующий патент: Аминоэфирные производные алкалоидов и их медицинские композиции
Случайный патент: Способ удаления карбонилов металлов из потоков газа и сорбент карбонила металла