Новые 1,2-диарилпиразолы, пригодные в качестве болеутоляющих и противовоспалительных средств
Номер патента: 13232
Опубликовано: 30.04.2010
Авторы: Фишер Янош, Лейбингер Янош, Киш-Варга Иштванне, Сабо Дьёрдь








Формула / Реферат
1. Соединения формулы (I)

где R1- атом водорода, С1-С5 - ацильная группа, бензоильная группа или группа R2-COOR3, Y - атом водорода или ион щелочного металла, R2- прямая или разветвленная С1-С4 алкилиденовая группа и R3 - атом водорода, алкильная группа С1-С4или ион щелочного металла, и/или их стереоизомеры, и/или диастереоизомеры, и/или фармацевтически приемлемые соли, и/или гидраты, и/или сольваты.
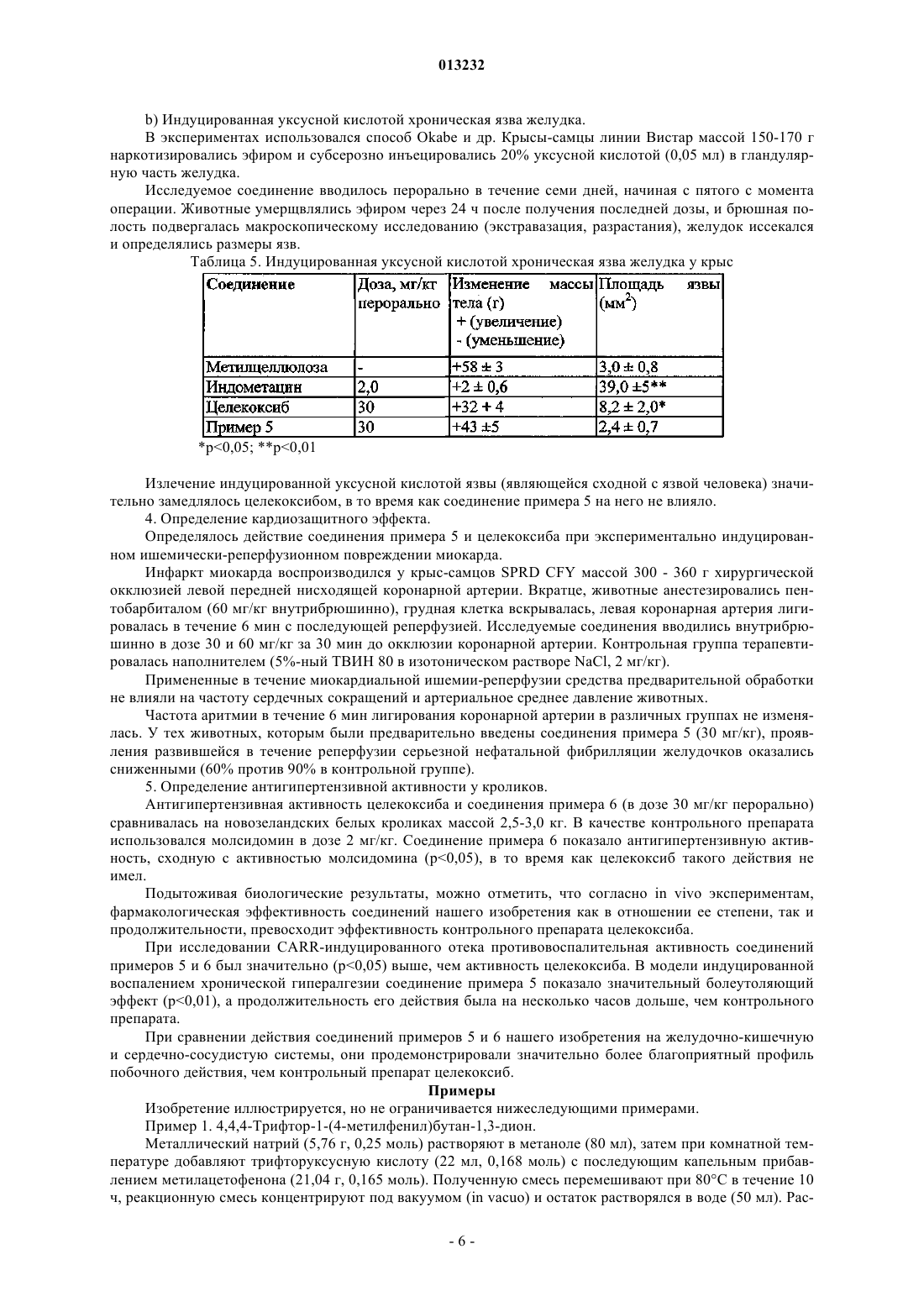
2. (R,S)-2-[4-(5-п-Метилфенил-3-трифторметилпиразол-1-ил)бензолсульфониламиноокси]пропионовая кислота.
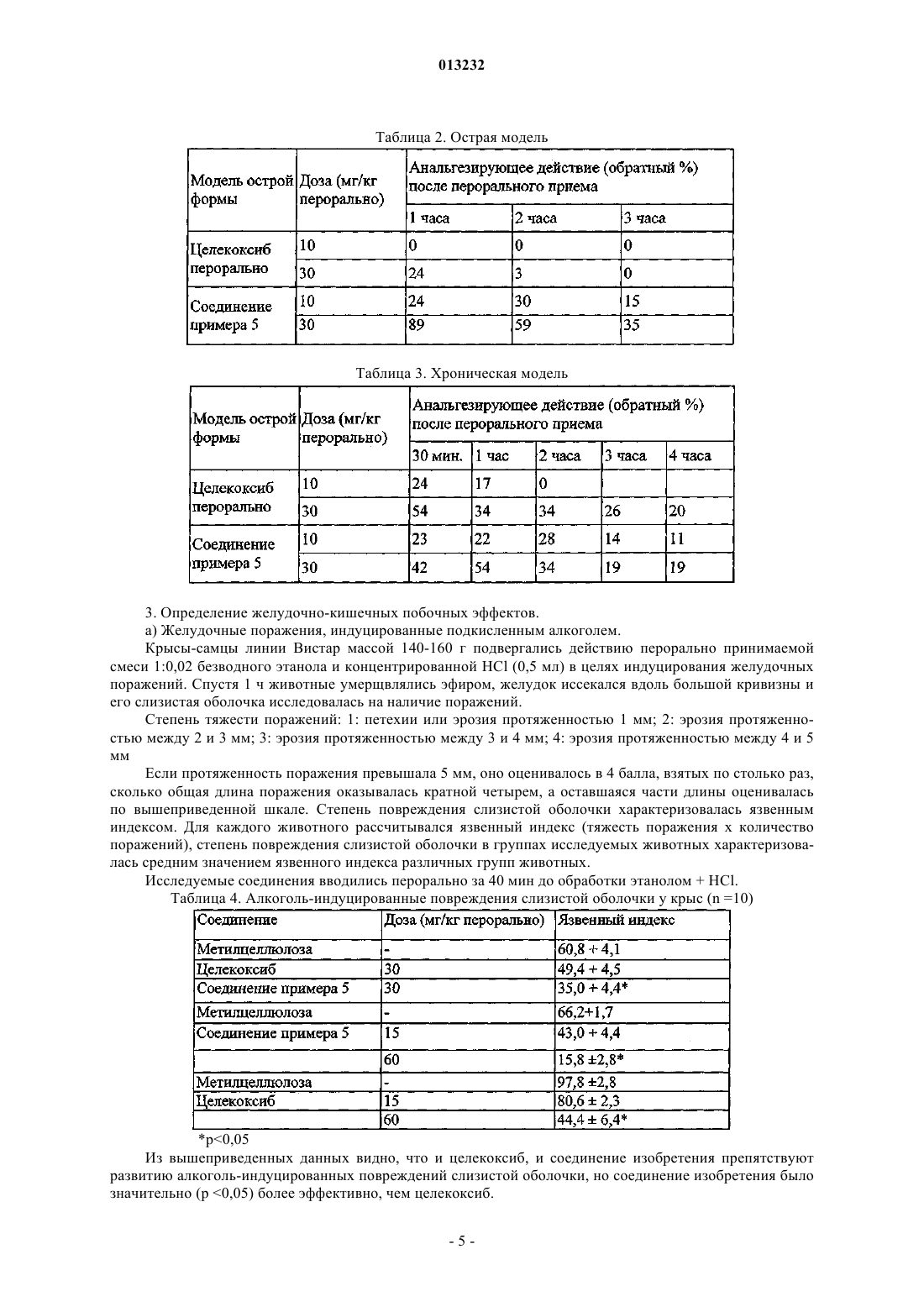
3. Моногидрат натриевой двузамещенной соли (R,S)-2-[4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)бензолсульфониламиноокси]пропионовой кислоты.
4. Способ синтеза соединений по п.п.1-3 и/или их геометрических изомеров, и/или стереоизомеров, и/или диастереомеров, и/или фармацевтически приемлемых солей, и/или гидратов, и/или сольватов, характеризующийся проведением реакции 4,4,4-трифтор-1-(4-метилфенил)бутан-1,3-диона с п-гидразин-бензолсульфоновой кислотой в инертном растворителе в присутствии соляной кислоты с последующим взаимодействием полученного соединения формулы (III) с пентахлоридом фосфора в инертном растворителе с образованием соединения формулы (II), которое взаимодействует
a) с гидроксиламином или
b) с соединением формулы NH2-O-R2-COOR3, в котором значения R2и R3 являются теми же самыми, что и упомянуты выше, или
c) с соединением формулы NH2-OCOR1, в котором значение R1является тем же самым, что и упомянуто выше,
для получения соединений формулы (I).
5. Фармацевтическая композиция, содержащая соединение по пп.1-3 и/или его стереоизомеры, и/или диастереомеры, и/или фармацевтически приемлемые соли, и/или гидраты, и/или сольваты, а также один или более фармацевтически приемлемый адъювант и/или вспомогательное вещество.
6. Применение соединения по пп.1-3 и/или его стереоизомеров, и/или диастереомеров, и/или фармацевтически приемлемых солей, и/или гидратов, и/или сольватов для производства фармацевтической композиции для терапии воспаления и связанных с воспалением нарушений.
7. Способ лечения воспалений и связанных с воспалениями нарушений, характеризующийся введением подвергаемым лечению млекопитающим, включая человека, эффективного количества/количеств соединения по пп.1-3 и/или его стереоизомеров, и/или диастереомеров, и/или фармацевтически приемлемых солей, и/или гидратов, и/или сольватов.
Текст
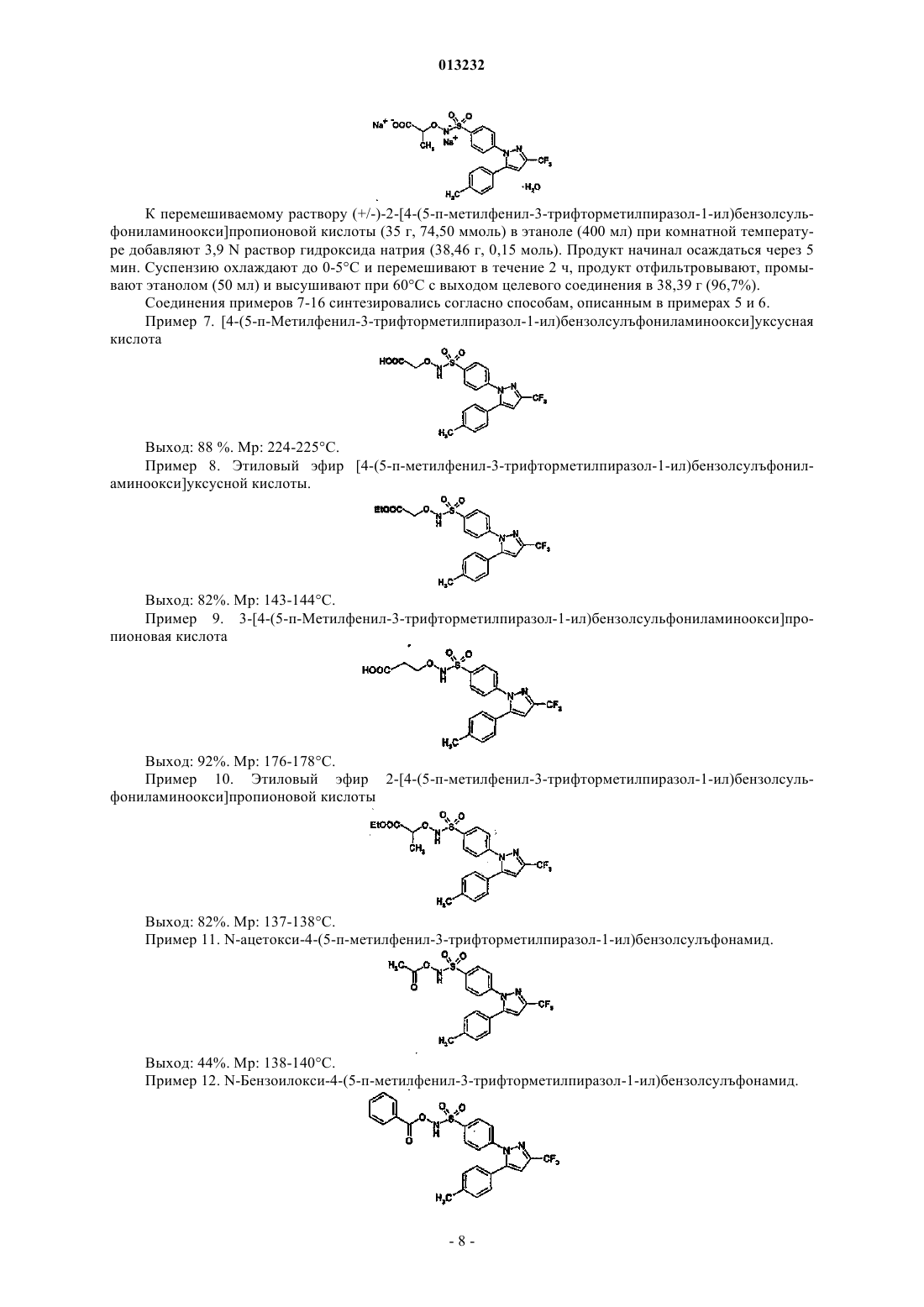
013232 Область техники, к которой относится изобретение Изобретение относится к новым соединениям химической формулы (I) и/или их стереоизомерам,и/или диастереомерам, и/или солям, и/или гидратам, и/или сольватам, которые являются пригодными для лечения боли, возникающей вследствие острого и хронического воспаления, а также хирургического вмешательства и дисменореи. Кроме того, изобретение относится к синтезу соединений химической формулы (I) и содержащих их фармацевтических композиций. Уровень техники Известно, что селективные ингибиторы фермента циклооксигеназы-2 (СОХ-2) обладают значительно более благоприятным профилем побочного действия на желудочно-кишечный тракт, чем традиционные нестероидные противовоспалительные средства, однако в том, что касается профиля побочного действия на сердечно-сосудистую систему, длительное применение как традиционных нестероидных противовоспалительных средств, так и коксибов может вызывать сердечно-сосудистые побочные эффекты(L.A.Garcia Rodriguez, Annals of the Rheumatic Diseases 2005, 64 (Suppl. Ill) page 40). В этом отношении наиболее предпочтителен целекоксиб, поскольку его побочное действие на сердечно-сосудистую систему менее выражено. Целекоксиб описан в европейском патенте ЕР 731795. В ходе его применения возникли некоторые проблемы. С одной стороны, целекоксиб не является эффективным для случаев всех пациентов, в связи с чем были начаты исследования с целью нахождения более эффективных аналогичных препаратов. С другой стороны, желательно уменьшить побочные желудочно-кишечные эффекты. Из литературы известно, что исследование было сосредоточено на усилении селективного ингибирования фермента СОХ 2 с целью уменьшения побочных желудочно-кишечных эффектов. Это поднимает вопрос о побочных сердечно-сосудистых эффектах, поскольку, согласно предыдущим наблюдениям, возрастание селективного ингибирования фермента СОХ-2 приводит к усилению побочных сердечно-сосудистых эффектов. Это было доказано так называемым исследованием VIGOR рофекоксиба - ингибитора фермента СОХ 2 первого поколения (Bombardier С., Laine L., Reicin A. et al. for the VIGOR Study Group. Comparison ofupper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N. Engl J. Med 343(21): 1520-1528, Nov. 2000.). Возможные причины этого явления описаны в исследовании D. Mukherjee (Mukherjee D., Mssen S.E., Topol E.J. Risk of cardiovascur events associated with selective COX-2 inhibitors. JAMA 2001; 286:954-959). Наша цель состояла в том, чтобы синтезировать аналогичные соединения, которые были бы более эффективны, чем целекоксиб, и имели бы меньше нежелательных побочных эффектов и, с другой стороны, чтобы их селективность в отношении СОХ-1/СОХ-2 оставалась неизменной. Сущность изобретения Неожиданно было найдено, что некоторые из производных целекоксиба являются более эффективными анальгетиками и противовоспалительными средствами, чем целекоксиб, кроме того, они не обладают in vitro ингибирующим действием в отношении СОХ-1 и СОХ-2 и профиль их желудочнокишечных побочных эффектов значительно более благоприятен, а также они обладают кардиозащитным действием. Настоящее изобретение относится к новым соединениям формулы (I) где R1 - атом водорода, C1-C5 - ацильная группа, бензоильная группа или группа R2-COOR3, Y атом водорода или ион щелочного металла, R2 - прямая или разветвленная алкилиденовая группа С 1-С 4 иR3 - атом водорода, С 1-С 4 алкил или ион щелочного металла, и/или их стереоизомерам, и/или диастереомерам, и/или фармацевтически приемлемым солям, и/или гидратам, и/или сольватам. Изобретение также относится к способу их синтеза, а также содержащей их фармацевтической композиции и использованию для лечения боли, воспаления и нарушений, связанных с воспалением. Подробное описание изобретения Настоящее изобретение относится к новым соединениям формулы (I)-1 013232 где R1 - атом водорода, С 1-С 5 - ацильная группа, бензоильная группа или группа R2-COOR3, Y атом водорода или ион щелочного металла, R2 - прямая или разветвленная С 4-С 4 алкилиденовая группа иR3 - атом водорода, алкильная группа С 1-С 4 или ион щелочного металла, и/или их стереоизомерам, и/или диастереомерам, и/или фармацевтически приемлемым солям, и/или гидратам, и/или сольватам.Y и R2 обозначают ион щелочного металла, предпочтительно натрия. Настоящее изобретение также относится к сольватам и гидратам соединений формулы (I). В тех случаях, когда соединения формулы (I) имеют хиральный центр, то, как стереоизомеры, так и их рацемическая смесь являются объектом настоящего изобретения. Изобретение также относится к способу синтеза соединений формулы (I) и химическому и фармацевтическому производству содержащих их фармацевтических композиций, а также к способам лечения и/или профилактики этими соединениями, которые подразумевают введение млекопитающему, включая человека, эффективного количества/количеств соединений формулы (I) настоящего изобретения как таковых или в виде медикаментов. Реакция Клайзена коммерчески доступного п-метилацетофенона и этилового эфира трифторуксусной кислоты дала хороший выход 4,4,4-трифтор-1-(4-метилфенил)-бутан-1,3-диона. Затем последний реагировал с коммерчески доступной п-гидразинбензолсульфоновой кислотой в инертном растворителе в присутствии соляной кислоты с образованием новой 4-(5-п-метилфенил)-3-трифторметилпиразол-1 ил)-бензолсульфоновой кислоты формулы (III) (выход более 80%) которая после перекристаллизации из инертного растворителя, предпочтительно из диизопропилового эфира, может быть получена в высокочистом состоянии в качестве ключевой субстанции. Новый интермедиат формулы (III) превращается в 4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)бензолсульфонилхлорид формулы (II) взаимодействием с пентахлоридом фосфора в инертном растворителе, предпочтительно в обезвоженном дихлорметане. Полученное соединение формулы (II) реагирует с гидроксиламином в инертном растворителе,предпочтительно в смеси воды и диоксана, давая соединение формулы (I), в которой R1 обозначает атом водорода. Реагент образуется in situ в реакционной смеси из гидроксиламин гидохлорида и ацетата натрия. Если соединение формулы (II) реагирует с аминооксикарбоновой кислотой в инертном растворителе, предпочтительно в диоксане, то с высоким выходом образуются такие продукты формулы (I), у которых R1 представлен группой R2-COOR3, где R2 - прямая или разветвленная C1-C4 алкилиденовая группа иR3 - атом водорода, или С 1-С 4 алкильная группа, или атом щелочного металла. При необходимости полученные соединения формулы (I) могут быть очищены перекристаллизацией из инертного растворителя,предпочтительно из толуола. Те соединения формулы (I), у которых значение R3 представлено водородным атомом, могут быть преобразованны в соли щелочного металла взаимодействием с раствором гидроксида щелочного металла, предпочтительно с двумя эквивалентами гидроксида натрия. Образующаяся двунатриевая соль может содержать в качестве сольвата гидрат, предпочтительно моногидрат. Если соединение формулы (II) реагирует с О-ацилированным гидроксиламином в инертном растворителе, предпочтительно в диоксане, то с высоким выходом образуются такие соединения формулы (I), у которых R1 представлен С 1-С 5 ацильной группой или бензоильной группой. Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединения формулы (I) в качестве активного ингредиента. Соединения формулы (I) могут использоваться при лечении воспаления и связанных с воспалением нарушений, например, в качестве анальгезирующее средства при лечении боли и цефалгии. Соединения формулы (I) и/или стереоизомеры, и/или их диастереомеры как таковые и/или их фармацевтически приемлемые соли, и/или гидраты, и/или сольваты могут использоваться в виде медикаментов, обычно как стандартные композиции. Также настоящее изобретение относится к фармацевтическим-2 013232 композициям, содержащим новое соединение формулы (I) и/или его стереоизомеры, и/или диастереомеры и/или фармацевтически приемлемые соли, и/или гидраты, и/или сольваты, а также один или более фармацевтически приемлемый адъювант и вспомогательное вещество. Соединения формулы (I) и/или их геометрические изомеры, и/или стереоизомеры, и/или диастереомеры, и/или фармацевтически приемлемые соли, и/или гидраты, и/или сольваты могут применяться любым подходящим способом, например, пероральным, парентеральным трансбуккальным, сублингвальным, назальным, ректальным или трансдермальным введением и в форме соответствующим образом адаптированных фармацевтических композиций. Фармацевтические композиции, содержащие соединения формулы (I) и/или их геометрические изомеры, и/или стереоизомеры, и/или диастереомеры, и/или фармацевтически приемлемые соли, и/или гидраты, и/или сольваты при пероральном назначении могут составляться в жидкой или твердой форме, например сиропов, суспензий или эмульсий, таблеток, капсул и жевательных таблеток. Жидкие рецептуры соединений формулы (I) и/или их геометрических изомеров, и/или стереоизомеров, и/или диастереомеров, и/или фармацевтически приемлемых солей, и/или гидратов, и/или сольватов,как правило, состоят из суспензии или раствора соединения формулы (I) в подходящем жидком носителе(-ях), например в водном растворителе, таком как вода, этиловый спирт или глицерин или в неводном растворителе, таком как полиэтиленгликоль или масло. Фармацевтическая композиция может также содержать суспендирующее вещество, консервирующие, ароматизирующие и окрашивающие вещества. Композиция в твердой форме - таблетки, может приготавливаться с использованием любого подходящего общеупотребительного на практике фармацевтического носителя(-й), такого как стеарат магния,крахмал, лактоза, сахароза, целлюлоза и т.д. Композиция в твердой форме - капсулы, может приготавливаться с использованием стандартных методов капсулирования. Например, используя стандартные носители, могут готовиться, а затем помещаться в твердую желатиновую капсулу содержащие активный ингредиент пеллеты; в альтернативном варианте, используя любой подходящий фармацевтический носитель(-и), например, водные растворы камеди, целлюлозу, силикаты или масла, может готовиться дисперсия или суспензия, которой затем заполняется мягкая желатиновая капсула. Типичные перентеральные композиции состоят из раствора или суспензии соединения формулы (I) и/или его геометрических изомеров, и/или стереоизомеров, и/или диастереомеров, и/или фармацевтически приемлемых солей, и/или гидратов, и/или сольватов в стерилизованном водном носителе или парентерально приемлемом масле, например, в полиэтиленгликоле, поливинилпирролидоне, лецитине, арахисовом масле или кунжутном масле. В альтернативном варианте раствор может быть лиофилизирован и затем непосредственно перед введением восстановлен подходящим растворителем. Биологическая оценка Исследования in vitro Согласно данным спектрофотометрии TMPD, соединения изобретения в концентрации 10 мкМ не обладали ингибирующим действием на фермент СОХ-2 (K. Gierse, S.D. Hauser, D.P. Creely, C.M. Koboldt,S.H. Rangwala, P.C. Isakson and K. Seibert, 1995: Expression and selective inhibition of the constitutive andinducible forms of human cyclo-oxygenase. Biochem. J. 305: 479-48) Анализы in vivo 1. Подавление каррагинан-индуцированного отека у крыс. Отек индуцировался подкожной инъекцией 50 мкл 1% суспензии каррагинана (CARR) в субплантарную область правой задней лапы самцов крыс линии Вистар (140-150 г). Введенный каррагинан вызывал воспаление лапы. Отек, то есть разница в объеме (в мл) инъецированный задней лапы до и после обработки, измерялся с помощью водяного вытеснительного плетизмометра (Ugo Basile, тип 7150). Обработанная лапа погружалась в камеру до тибиотарзального сустава и определялся объем вытесненной жидкости в качестве показателя степени воспаления. Степень воспаления (мл) = объем после обработки CARR (мл) - объем перед обработкой CARR(мл). Степень противовоспалительного действия в леченой группе (получавшей исследуемое средство) сравнивалась с величиной этого показателя в контрольной группе (получавшей лишь наполнитель). Исследуемое соединение и наполнитель вводились перорально с помощью тупоконечной игольной канюли за один час перед обработкой CARR. Объем обработанной лапы измерялся спустя 3 и 5 ч после обработки CARR и по следующей формуле рассчитывались процентные изменения в степени воспаления:% противовоспалительного действия = 100[контрольная группа (мл) - леченая группа (мл) / контрольная группа (мл)]. Целекоксиб и соединения изобретения изучались при пероральной дозе в 1-10 - 30 мг/кг (n = 12 животных в каждой группе). Процент противовоспалительного действия соединений определялся через 4 и 6 ч после пероральной обработки и рассчитывалось значение ED30. Результаты Подавление отека целекоксибом: сохранял действие через 4 ч после обработки с ED30 23 мг/кг. Через 6 ч после обработки противовоспалительное действие целекоксиба по сравнению с контрольной-3 013232 группой было ниже на 30%. Соединение примера 5: сохраняло действие через 4 и 6 ч после обработки сED30 5,7 мг/кг и ED30 8,4 мг/кг соответственно. Полученные результаты показывают, что соединение примера 5 значительно по сравнению с целекоксибом подавляло отек лапы в обеих исследуемых временных точках. Таблица 1 2. Каррагинан-индуцированная острая гипералгезия у крыс (модель Randall-Selitto). Отек (воспаление) индуцировался подкожной инъекцией суспензии каррагинана (CARR) в субплантарную область правой задней лапы самцов крыс линии Вистар массой 140-190 г (n = 8-12 животных в каждой группе). Ноцицептивные пороги воспаленной задней лапы после воздействия механических болезненных раздражителей определялись с помощью анальгезиметра (Ugo Basile, тип 37215, Италия). Прибор пригоден для измерения степени и латентности порога болевой реакции сенсибилизированной лапы после воздействия болезненных раздражителей. Анальгетики повышают низкий порог болевой реакции воспаленной лапы, а степень их антиноцицептивного действия выражается в обратных процентах. На лапу оказывалось возрастающее давление и пороговый момент отдергивания определялся в качестве первого признака болевой реакции (писк и/или борьба). Порог давления отображался в граммах. Усредненная величина показателей отдергивания задней лапы, полученных на не подвергавшейся обработке правой лапе, рассматривалась в качестве исходного показателя отдергивания задней лапы (в среднем 80-110 г) После определения исходного порога животные получали инъекцию каррагинана, вызывавшую интенсивное воспаление, связанное с гипералгезией. Пороговая величина механического воздействия определялась в различные моменты времени с целью установления степени и продолжительности гипералгезии. Максимальное снижение порога было определено через 2-3 ч после инъекции (порог болевой чувствительности воспаленной лапы 20-30 г, что соответствует снижению по сравнению с исходной величиной на 65-80%). Модель острой гипералгезии: животные получали терапию исследуемыми соединениями и целекоксибом (10 и 30 мг/кг перорально) спустя один час после обработки CARR (100 мкл 2% суспензии). Порог болевой чувствительности измерялся через 2 и 4 ч после терапии. Модель хронической гипералгезии: обширное воспаление и сниженный порог болевой чувствительности индуцировались повышенной дозой CARR. Порог болевой чувствительности воспаленной лапы измерялся спустя 24 часа после обработки CARR (150 мкл, 2% суспензия), затем животным давались исследуемое соединение и целекоксиб(30 мг/кг перорально). Порог болевой чувствительности измерялся через 30 мин и через 3 ч после терапии. Контрольные группы в обеих моделях после обработки CARR через те же временные промежутки,что и в случае исследуемых соединений, получали наполнитель перорально. В обеих моделях болеутоляющее действие соединений выражалось в обратных процентах от снижения порога болевой чувствительности. Обратный % = [среднее по леченой группе Txh (г) - леченая группа T0h (г)] / [среднее исходное по леченой группе T-1h (g) -леченая группа T0h (g)].T-1h = среднее порога болевой чувствительности лапы перед обработкой CARR в модели острой формы (г);T0h = среднее порога болевой чувствительности перед терапией спустя 1 ч после обработки CARR в модели острой формы (г);Txh = порог болевой чувствительности после обработки CARR в измеряемые моменты времени (1 ч, 2 ч, 3 ч) в модели острой формы (г). В обеих моделях (терапия острой и хронической форм) целекоксиб в дозе 30 мг/кг перорально показал частичный болеутоляющий эффект (24 и 54 обратных процента). В модели острой формы исследуемое соединение в дозе 30 мг/кг перорально показало почти полное обращение (90%), таким образом исследуемое соединение показало значительно лучший противовоспалительный/болеутоляющий эффект по сравнению с контрольным препаратом. В хронической модели исследуемый препарат показал сопоставимый с анальгезирующим действием целекоксиба эффект в 54 обратных процента. 3. Определение желудочно-кишечных побочных эффектов. а) Желудочные поражения, индуцированные подкисленным алкоголем. Крысы-самцы линии Вистар массой 140-160 г подвергались действию перорально принимаемой смеси 1:0,02 безводного этанола и концентрированной HCl (0,5 мл) в целях индуцирования желудочных поражений. Спустя 1 ч животные умерщвлялись эфиром, желудок иссекался вдоль большой кривизны и его слизистая оболочка исследовалась на наличие поражений. Степень тяжести поражений: 1: петехии или эрозия протяженностью 1 мм; 2: эрозия протяженностью между 2 и 3 мм; 3: эрозия протяженностью между 3 и 4 мм; 4: эрозия протяженностью между 4 и 5 мм Если протяженность поражения превышала 5 мм, оно оценивалось в 4 балла, взятых по столько раз,сколько общая длина поражения оказывалась кратной четырем, а оставшаяся части длины оценивалась по вышеприведенной шкале. Степень повреждения слизистой оболочки характеризовалась язвенным индексом. Для каждого животного рассчитывался язвенный индекс (тяжесть поражения х количество поражений), степень повреждения слизистой оболочки в группах исследуемых животных характеризовалась средним значением язвенного индекса различных групп животных. Исследуемые соединения вводились перорально за 40 мин до обработки этанолом + HCl. Таблица 4. Алкоголь-индуцированные повреждения слизистой оболочки у крыс (n =10) р 0,05 Из вышеприведенных данных видно, что и целекоксиб, и соединение изобретения препятствуют развитию алкоголь-индуцированных повреждений слизистой оболочки, но соединение изобретения было значительно (р 0,05) более эффективно, чем целекоксиб.b) Индуцированная уксусной кислотой хроническая язва желудка. В экспериментах использовался способ Okabe и др. Крысы-самцы линии Вистар массой 150-170 г наркотизировались эфиром и субсерозно инъецировались 20% уксусной кислотой (0,05 мл) в гландулярную часть желудка. Исследуемое соединение вводилось перорально в течение семи дней, начиная с пятого с момента операции. Животные умерщвлялись эфиром через 24 ч после получения последней дозы, и брюшная полость подвергалась макроскопическому исследованию (экстравазация, разрастания), желудок иссекался и определялись размеры язв. Таблица 5. Индуцированная уксусной кислотой хроническая язва желудка у крыс р 0,05; р 0,01 Излечение индуцированной уксусной кислотой язвы (являющейся сходной с язвой человека) значительно замедлялось целекоксибом, в то время как соединение примера 5 на него не влияло. 4. Определение кардиозащитного эффекта. Определялось действие соединения примера 5 и целекоксиба при экспериментально индуцированном ишемически-реперфузионном повреждении миокарда. Инфаркт миокарда воспроизводился у крыс-самцов SPRD CFY массой 300 - 360 г хирургической окклюзией левой передней нисходящей коронарной артерии. Вкратце, животные анестезировались пентобарбиталом (60 мг/кг внутрибрюшинно), грудная клетка вскрывалась, левая коронарная артерия лигировалась в течение 6 мин с последующей реперфузией. Исследуемые соединения вводились внутрибрюшинно в дозе 30 и 60 мг/кг за 30 мин до окклюзии коронарной артерии. Контрольная группа терапевтировалась наполнителем (5%-ный ТВИН 80 в изотоническом растворе NaCl, 2 мг/кг). Примененные в течение миокардиальной ишемии-реперфузии средства предварительной обработки не влияли на частоту сердечных сокращений и артериальное среднее давление животных. Частота аритмии в течение 6 мин лигирования коронарной артерии в различных группах не изменялась. У тех животных, которым были предварительно введены соединения примера 5 (30 мг/кг), проявления развившейся в течение реперфузии серьезной нефатальной фибрилляции желудочков оказались сниженными (60% против 90% в контрольной группе). 5. Определение антигипертензивной активности у кроликов. Антигипертензивная активность целекоксиба и соединения примера 6 (в дозе 30 мг/кг перорально) сравнивалась на новозеландских белых кроликах массой 2,5-3,0 кг. В качестве контрольного препарата использовался молсидомин в дозе 2 мг/кг. Соединение примера 6 показало антигипертензивную активность, сходную с активностью молсидомина (р 0,05), в то время как целекоксиб такого действия не имел. Подытоживая биологические результаты, можно отметить, что согласно in vivo экспериментам,фармакологическая эффективность соединений нашего изобретения как в отношении ее степени, так и продолжительности, превосходит эффективность контрольного препарата целекоксиба. При исследовании CARR-индуцированного отека противовоспалительная активность соединений примеров 5 и 6 был значительно (р 0,05) выше, чем активность целекоксиба. В модели индуцированной воспалением хронической гипералгезии соединение примера 5 показало значительный болеутоляющий эффект (р 0,01), а продолжительность его действия была на несколько часов дольше, чем контрольного препарата. При сравнении действия соединений примеров 5 и 6 нашего изобретения на желудочно-кишечную и сердечно-сосудистую системы, они продемонстрировали значительно более благоприятный профиль побочного действия, чем контрольный препарат целекоксиб. Примеры Изобретение иллюстрируется, но не ограничивается нижеследующими примерами. Пример 1. 4,4,4-Трифтор-1-(4-метилфенил)бутан-1,3-дион. Металлический натрий (5,76 г, 0,25 моль) растворяют в метаноле (80 мл), затем при комнатной температуре добавляют трифторуксусную кислоту (22 мл, 0,168 моль) с последующим капельным прибавлением метилацетофенона (21,04 г, 0,165 моль). Полученную смесь перемешивают при 80 С в течение 10 ч, реакционную смесь концентрируют под вакуумом (in vacuo) и остаток растворялся в воде (50 мл). Рас-6 013232 твор подкисляют добавлением 1N соляной кислоты (120 мл), экстрагируют этилацетатом (280 мл), осушают над MgSO4, отфильтровывают и концентрируют in vacuo с выходом целевого соединения в 34,60 г(95 %). Полученный кристаллический продукт используют на следующем этапе без дополнительной очистки. Пример 2. 4-(5-п-Метилфенил-3-трифторметилпиразол-1-ил)бензолсульфоновая кислота. К перемешиваемой суспензии п-гидразинбензолсульфоновой кислоты (42 г, 0,223 моль) в этаноле(450 мл) при комнатной температуре добавляют 6N соляную кислоту (74 мл, 0,446 моль) с последующим прибавлением 4,4,4-трифтор-1-(4-метилфенил)-бутан-1,3-диона (51,45 г, 0,223 моль). Полученную суспензию нагревают в колбе с обратным холодильником в течение 8 ч, затем концентрируют in vacuo. Остаток растворяют в воде (300 мл) и экстрагируют этилацетатом (2200 мл). Объединенные органические слои промывают водой (1100 мл) и насыщенным солевым раствором (1100 мл), высушивают надMgSO4, обесцвечивают, отфильтровывают и концентрируют in vacuo. Полученный кристаллический продукт перекристаллизовывают из диизопропилового эфира (300 мл) с выходом 70,12 г (82%) целевого соединения. Пример 3. 4-(5-п-Метилфенил-3-трифторметилпиразол-1-ил)бензолсульфонилхлорид. К перемешиваемой суспензии 4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)бензолсульфоновой кислоты (43 г, 0,112 моль) в дихлорэтане (250 мл) при комнатной температуре по частям добавляют пентахлорид фосфора (34,20 г, 0,168 моль) с последующим прибавлением N,N-диметилформамида (10 мл). Полученную таким образом суспензию нагревают в колбе с обратным холодильником в течение 8 ч, а затем концентрируют in vacuo. Остаток фракционируют между этилацетатом (150 мл) и водой (150 мл) и отделяют. Водную фазу экстрагируют этилацетатом (150 мл), объединенные органические слои высушивают над MgSO4, отфильтровывают и концентрируют. Полученное масло кристаллизуют из циклогексана (120 мл) с выходом целевого соединения в 33,40 г (74 %) в виде белых кристаллов. Мр: 97-98 С. Пример 4. N-гидрокси-4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)-бензолсульфонамид К перемешиваемой суспензии гидроксиламин гидрохлорида (2,74 г, 40 ммоль) в диоксане (25 мл) при комнатной температуре по каплям добавляют ацетат натрия (3,25 г, 40 ммоль) в воде (15 мл) с последующим прибавлением по каплям 4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)-бензолсульфонилхлорида (4,11 г, 10 ммоль) в диоксане (40 мл). Полученную реакционную смесь вливают в воду(100 мл), экстрагируют этилацетатом (250 мл), органические слои промывают водой (350 мл) и насыщенным солевым раствором (150 мл), высушивают над MgSO4, отфильтровывают и концентрируют invacuo. Полученное масло кристаллизуют из 75% водного этанола (75 мл) с выходом целевого соединения в 2,94 г (75 %) в виде белых кристаллов. Мр: 203 С. Пример 5. (R,S)-2-[4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)-бензолсулъфониламиноокси]пропионовая кислота К перемешиваемой суспензии гидохлорида 2-аминооксипропионовой кислоты (32,60 г, 0,23 моль) в диоксане (100 мл) при комнатной температуре по каплям добавляют ацетат натрия (18,86 г, 0,23 ммоль) в воде (100 мл) с последующим прибавлением по каплям 4-(5-п-метилфенил-3-трифторметилпиразол-1 ил)бензолсульфонилхлорида (38 г, 94,80 ммоль) в диоксане (100 мл). Полученную реакционную смесь перемешивают в течение 5 ч, а затем концентрируют in vacuo. Остаток растворяют в воде (200 мл), экстрагируют этилацетатом (2100 мл), объединенные органические слои промывают водой (450 мл) и насыщенным солевым раствором (150 мл), высушивают над MgSO4, отфильтровывают и концентрируютin vacuo. Полученное масло кристаллизуют из толуола (120 мл) с выходом целевого соединения в 37,94 г(85%) в виде белых кристаллов. Полученный таким образом продукт растворяют в толуоле (480 мл) и перемешивают при 0-5 С в течение 1 ч, осадившийся продукт отфильтровывают, промывают холодным толуолом и высушивают при комнатной температуре с выходом целевого соединения в 36,43 г (96%). Мр: 187-189 С, HPLC: 99,9%. Пример 6. Моногидрат натриевой двузамещенной соли (R,S)-2-[4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)-бензолсулъфониламиноокси]пропионовой кислоты. К перемешиваемому раствору (+/-)-2-[4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)бензолсульфониламиноокси]пропионовой кислоты (35 г, 74,50 ммоль) в этаноле (400 мл) при комнатной температуре добавляют 3,9 N раствор гидроксида натрия (38,46 г, 0,15 моль). Продукт начинал осаждаться через 5 мин. Суспензию охлаждают до 0-5 С и перемешивают в течение 2 ч, продукт отфильтровывают, промывают этанолом (50 мл) и высушивают при 60 С с выходом целевого соединения в 38,39 г (96,7%). Соединения примеров 7-16 синтезировались согласно способам, описанным в примерах 5 и 6. Пример 7. [4-(5-п-Метилфенил-3-трифторметилпиразол-1-ил)бензолсулъфониламиноокси]уксусная кислота Выход: 72%. Мр: 134-136 С. Пример 14 Тригидрат натриевой соли [4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)бензолсулъфониламиноокси]уксусной кислоты. Выход: 90%. Пример 15. Моногидрат натриевой двузамещенной соли 3-[4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)бензолсулъфониламиноокси]пропионовой кислоты Выход 87%. Пример 16. Моногидрат натриевой двузамещенной соли 2-метил-2-[4-(5-п-метилфенил-3 трифторметилпиразол-1-ил)бензолсульфониламиноокси]пропионовой кислоты(1,76 г, 10 ммоль). Смесь перемешивают при комнатной температуре в течение 1 дня, осадившиеся кристаллы отфильтровывают и промывают 91% этанолом с выходом продукта в 3,38 г (25%), рассчитанным на основе исходной рацемической смеси. Полученные таким образом кристаллы дважды перекристаллизовывают из 97% этанола с выходом 2,02 г (60%) белого кристаллического вещества. Диастереомерную соль суспендируют в этилацетат (30 мл) и добавляют 1N соляной кислоты (15 мл). Органический слой отделяют, высушивают над MgSO4, отфильтровывают и концентрируют invacuo. Полученное масло кристаллизуют из петролейного эфира с выходом 0,9 г (60%) целевого соединения. Мр: 158-160 С. []D = - 505,3 (с = 1, метанол). Пример 18. (+)-2-[4-(5-п-Метилфенил-3-трифторметилпиразол-1-ил)бензолсулъфониламиноокси] пропионовая кислота. Указанное соединение получалось согласно методу, описанному в примере 17 с использованием(+)-эфедрина в качестве расщепляющего агента и изопропанола в качестве растворителя. Выход: 0,62 г. Мр: 161-163 С. []D = + 457,6 (с = 1, метанол). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединения формулы (I)-9 013232 где R1 - атом водорода, С 1-С 5 - ацильная группа, бензоильная группа или группа R2-COOR3, Y атом водорода или ион щелочного металла, R2 - прямая или разветвленная С 1-С 4 алкилиденовая группа иR3 - атом водорода, алкильная группа С 1-С 4 или ион щелочного металла, и/или их стереоизомеры, и/или диастереоизомеры, и/или фармацевтически приемлемые соли, и/или гидраты, и/или сольваты. 2.(R,S)-2-[4-(5-п-Метилфенил-3-трифторметилпиразол-1-ил)бензолсульфониламиноокси]пропионовая кислота. 3. Моногидрат натриевой двузамещенной соли (R,S)-2-[4-(5-п-метилфенил-3-трифторметилпиразол-1-ил)бензолсульфониламиноокси]пропионовой кислоты. 4. Способ синтеза соединений по пп.1-3 и/или их геометрических изомеров, и/или стереоизомеров,и/или диастереомеров, и/или фармацевтически приемлемых солей, и/или гидратов, и/или сольватов, характеризующийся проведением реакции 4,4,4-трифтор-1-(4-метилфенил)бутан-1,3-диона с п-гидразинбензолсульфоновой кислотой в инертном растворителе в присутствии соляной кислоты с последующим взаимодействием полученного соединения формулы (III) с пентахлоридом фосфора в инертном растворителе с образованием соединения формулы (II), которое взаимодействуетb) с соединением формулы NH2-O-R2-COOR3, в котором значения R2 и R3 являются теми же самыми, что и упомянуты выше, илиc) с соединением формулы NH2-OCOR1, в котором значение R1 является тем же самым, что и упомянуто выше,для получения соединений формулы (I). 5. Фармацевтическая композиция, содержащая соединение по пп.1-3 и/или его стереоизомеры,и/или диастереомеры, и/или фармацевтически приемлемые соли, и/или гидраты, и/или сольваты, а также один или более фармацевтически приемлемый адъювант и/или вспомогательное вещество. 6. Применение соединения по пп.1-3 и/или его стереоизомеров, и/или диастереомеров, и/или фармацевтически приемлемых солей, и/или гидратов, и/или сольватов для производства фармацевтической композиции для терапии воспаления и связанных с воспалением нарушений. 7. Способ лечения воспалений и связанных с воспалениями нарушений, характеризующийся введением подвергаемым лечению млекопитающим, включая человека, эффективного количества/количеств соединения по пп.1-3 и/или его стереоизомеров, и/или диастереомеров, и/или фармацевтически приемлемых солей, и/или гидратов, и/или сольватов.
МПК / Метки
МПК: A61P 29/00, A61K 31/4155, C07D 231/12
Метки: качестве, 1,2-диарилпиразолы, новые, противовоспалительных, средств, болеутоляющих, пригодные
Код ссылки
<a href="https://eas.patents.su/11-13232-novye-12-diarilpirazoly-prigodnye-v-kachestve-boleutolyayushhih-i-protivovospalitelnyh-sredstv.html" rel="bookmark" title="База патентов Евразийского Союза">Новые 1,2-диарилпиразолы, пригодные в качестве болеутоляющих и противовоспалительных средств</a>
Имидазольные соединения, арил или гетероарил конденсированные, в качестве противовоспалительных и анальгетических средств

Номер патента: 5991
Опубликовано: 25.08.2005
Авторы: Окумура Есиюки, Синдзио Кацухиро, Унео Наоми, Накао Казунари, Нукуи Сейдзи, Хасизуме Есинобу, Мацумизу Мияко, Мияке Ерико, Танигути Кана, Като Томоки, Каваи Акиеси
МПК: A61K 31/4178, C07D 471/04, A61P 29/00...
Метки: анальгетических, арил, конденсированные, противовоспалительных, соединения, гетероарил, качестве, средств, имидазольные
Формула / Реферат:
1. Соединение, представленное следующей формулой или его фармацевтически приемлемые соли, где Y1, Y2, Y3 и Y4 независимо выбраны из N, CH и C(L); R1 представляет собой H, C1-8алкил, C2-8алкенил, C2-8алкинил, C3-7циклоалкил, C1-8алкокси, галогензамещенный C1-8алкокси, C1-8алкил-S(O)m-, Q1-, пирролидинил, пиперидил, оксопирролидинил, оксопиперидил, амино, моно- или ди-(C1-8алкил)амино, C1-4алкил-C(=O)-N(R3)- или C1-4алкил-S(O)m-N(R3)-, где...
Производные тетрагидронафталина, способ их получения и их применение в качестве противовоспалительных средств

Номер патента: 10186
Опубликовано: 30.06.2008
Авторы: Шэкке Хайке, Ревинкель Хартмунт, Бойерле Штефан, Менгель Анне, Бергер Маркус, Кроликевич Конрад, Шмеес Норберт, Скубалла Вернер, Ярох Штефан, Нгуен Дуй
МПК: A61K 31/416, A61K 31/404, A61K 31/4035...
Метки: качестве, тетрагидронафталина, получения, противовоспалительных, производные, применение, способ, средств
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 и R2, независимо друг от друга, представляют собой атом водорода, гидроксигруппу, атом галогена, необязательно замещенную С1-С10-алкильную группу, необязательно замещенную С1-С10-алкоксигруппу, C1-С10-алкилтиогруппу, C1-C5-перфторалкильную группу, цианогруппу, нитрогруппу, или R1 и R2 вместе представляют собой группу, выбранную из групп -О-(СН2)n-О-, -О-(СН2)n-СН2-, -О-СН=СН-, -(CH2)n+2-,...
Перегруппированные пентанолы, способ их получения и их применение в качестве противовоспалительных средств

Номер патента: 9958
Опубликовано: 28.04.2008
Авторы: Бергер Маркус, Скубалла Вернер, Менгель Анне, Шмес Норберт, Нгуен Дуй, Шэкке Хайке, Бойеле Штефан, Ярох Штефан, Ревинкель Хартмунт, Кроликевич Конрад
МПК: A61K 31/4035, A61K 31/416, A61K 31/352...
Метки: пентанолы, противовоспалительных, качестве, применение, перегруппированные, средств, способ, получения
Формула / Реферат:
1. Стереоизомеры общей формулы I в которой R1 представляет собой необязательно замещенный фенильный радикал, заместители которого выбраны из группы, включающей С1-С5-алкил, С1-С5-алкокси, галоген, гидроксил, трифторметил, R2 представляет собой моноциклическую или бициклическую, ароматическую, частично ароматическую или неароматическую кольцевую систему, которая содержит атомы азота или кислорода и необязательно замещена в одном или более местах...
Производные хинолина и изохинолина, замещённые в 5-м положении, способ их получения и их применение в качестве противовоспалительных средств

Номер патента: 13076
Опубликовано: 26.02.2010
Авторы: Ревинкель Хартмут, Петроф Орлин, Шмес Норберт, Скубалла Вернер, Ярох Штефан, Хюбнер Ян, Шнайдер Маттиас, Шэкке Хайке, Динтер Кристиан
МПК: A61P 29/00, C07C 69/68, A61K 31/47...
Метки: замещённые, производные, получения, положении, изохинолина, способ, хинолина, средств, качестве, применение, противовоспалительных
Формула / Реферат:
1. Соединения общей формулы (IIa) или (IIb)в которых R1 и R2 независимо друг от друга представляют собой атом водорода, С1-3-алкильную группу, атом галогена, C1-3-алкоксигруппу или гидроксигруппу,а также их рацематы или отдельно представленные стереоизомеры и их физиологически совместимые соли.2. Соединения общей формулы (IIa) или (IIb) по п.1, в которых R1и R2 независимо друг от друга представляют собой атом водорода, атом хлора, метильную...
N-ацильные производные аминокислот, способ их получения, фармацевтическая композиция и применение в качестве противоаллергических, противовоспалительных и гиполипидемических средств

Номер патента: 13057
Опубликовано: 26.02.2010
Авторы: Желтухина Галина Александровна, Небольсин Владимир Евгеньевич, Ковалева Виолетта Леонидовна, Кромова Татьяна Александровна
МПК: A61K 31/405, A61P 11/00, A61K 31/4172...
Метки: качестве, противоаллергических, композиция, фармацевтическая, противовоспалительных, способ, аминокислот, производные, n-ацильные, применение, гиполипидемических, получения, средств
Формула / Реферат:
1. N-Ацильные производные аминокислот общей формулы (I)где n равно 2 или 3 иR1 представляетR2=Н, -СН3, -С2Н5,и их фармацевтически приемлемые соли,при условии, что соединение общей формулы (I) не является Na-сукцинил-L-гистидином, сукцинил-L-триптофаном, сукцинил-D-триптофаном и сукцинил-D,L-триптофаном и его дикалиевой солью, Na-сукцинил-L-триптофаном метиловым эфиром, Na-глутарил-L-гистидином метиловым эфиром, Na-глутарил-L-триптофаном...
Предыдущий патент: Замещенные амидные производные в качестве ингибиторов протеинкиназы
Следующий патент: Антисептическая композиция для дезинфекции кожных покровов, способ ее приготовления и полупродукт для ее приготовления
Случайный патент: N-[(3s)пирролидин-3-ил]бензамидные производные в качестве ингибиторов обратного захвата моноаминов