Способ получения азациклогексапетидов
Номер патента: 564
Опубликовано: 29.12.1999
Авторы: Бендер Дин Р., Белик Кевин М., Леонард Уильям, Блэк Регина М., Хьюджес Дэвид Л.






Формула / Реферат
1. Способ получения азациклопептидов формулы (I)

Послед. ь 1
где
R1 представляет собой CH2CH2NH2 или CH2CONH2;
RI представляет собой (C9-C21)-алкил, (C9-C21)-алкенил, (C1-C10)-алкоксифенил, (C1-С10)-алкоксинафтил или (C1-С10)-алкокситерфенил;
RII представляет собой Н, (C1-C4)-алкил, (С3-С4)-алкенил, (CH2)2-4ОН или (CH2)2-4NRIVRV;
RIII представляет собой Н, (C1-C4)-алкил, (С3-С4)-алкенил, (СН2)2-4OН; (CH2)2-4NRIVRV, или
RII и RIII, взятые вместе, представляют (СН2)4, (СН2)5, (СН2)2O(СН2)2 или (CH2)2NH(CH2)2;
RIV представляет собой Н или (C1-C4)-алкил;
RV представляет собой Н или (C1-C4)-алкил; или их фармацевтически приемлемых кислотно-аддитивных солей, отличающийся тем, что включает следующие стадии:
а) восстановление соединения II формулы

Послед. ь1
с использованием боранового комплекса или TiCl4/NaBH4, или ZrCl4/NaBH4
с образованием соединения III формулы

Послед. ь 1
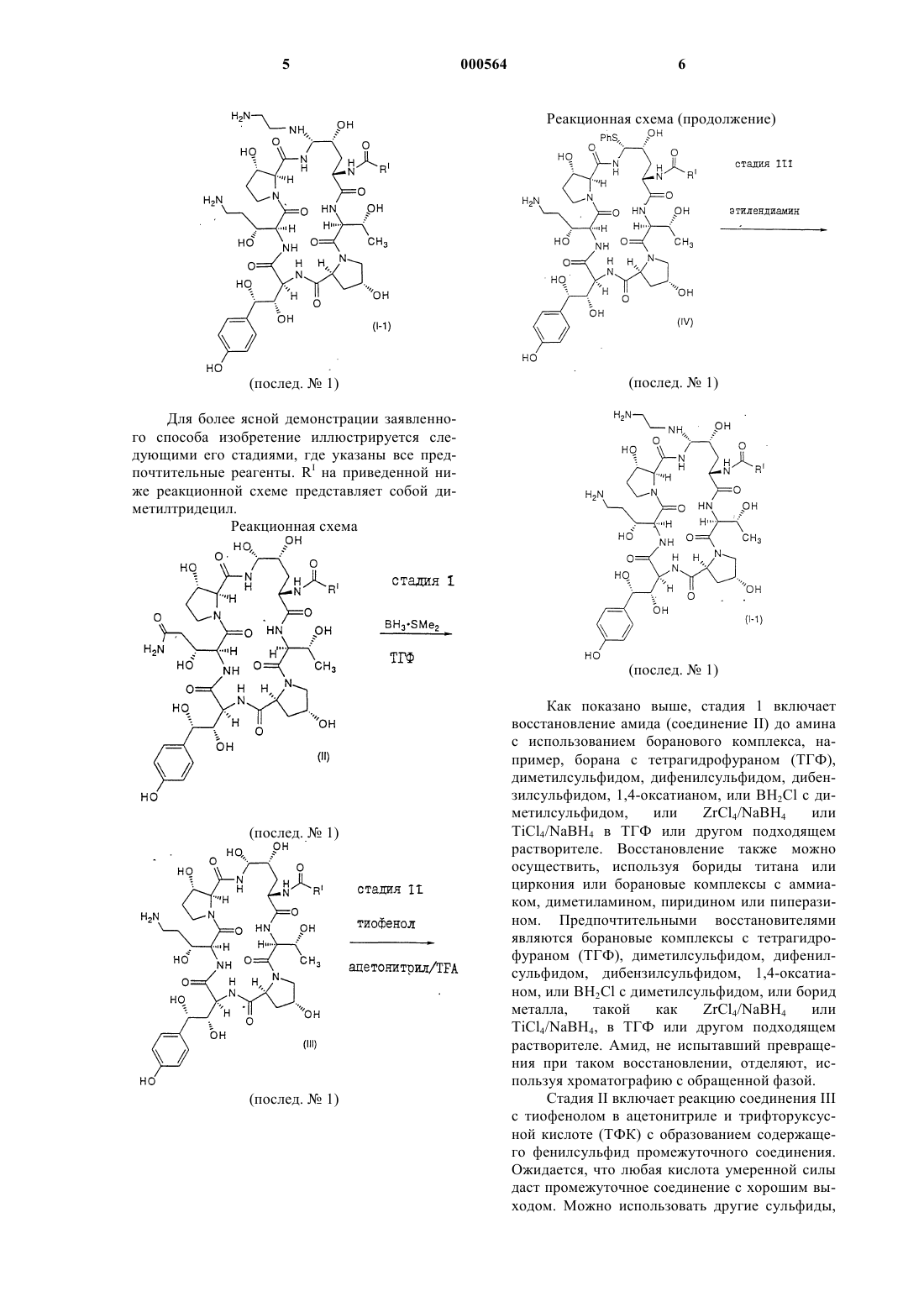
b) взаимодействие соединения III с тиофенолом в количестве 3-5 эквивалентов и в присутствии 5-25%-го раствора трифторуксусной кислоты в подходящем растворителе с образованием фенилсульфида IV формулы

Послед. ь 1
и
с) стереоселективное превращение соединения IV в соединение I путем замещения фенилтиогруппы в чистом этилендиамине или в растворе этилендиамина, в подходящем растворителе, при температуре от 10 до 40шС.
2. Способ по п.1, отличающийся тем, что борановый комплекс представляет собой комплекс борана с диметилсульфидом, дибензилсульфидом, дифенилсульфидом, тетрагидрофураном или 1,4-оксатианом, или BH2Cl с диметилсульфидом.
3. Способ по п.1, отличающийся тем, что в качестве растворителя на стадии b) используют ацетонитрил.
4. Способ по п.1, отличающийся тем, что растворитель на стадии с), выбирают из группы, включающей воду, метанол, этанол, тетрагидрофуран, изопропанол, трифторэтанол, ацетонитрил или дихлорметан.
5. Способ получения азациклопептидов формулы (I)

Послед. ь 1
где
R1 представляет собой CH2CH2NH2 или CH2CONH2;
RI представляет собой (C9-C21)-алкил, (C9-C21)-алкенил, (C1-C10)-алкоксифенил, (C1-С10)-алкоксинафтил или (C1-С10)-алкокситерфенил;
RII представляет собой Н, (C1-C4)-алкил, (С3-С4)-алкенил,
(СН2)2-4OН или (СН2)2-4NRIVRV;
RIII представляет собой Н, (C1-C4)-алкил, (С3-С4)-алкенил, (СН2)2-4OН; (СН2)2-4NRIVRV, или
RII и RIII, взятые вместе, представляют собой (СН2)4, (СН2)5, (СН2)2O(СН2)2 или (CH2)2NH(CH2)2;
RIV представляет собой Н или (C1-C4)-алкил;
RV представляет собой Н или (C1-C4)-алкил; или их фармацевтически приемлемых кислотно-аддитивных солей, отличающийся тем, что включает следующие стадии:
а) взаимодействие соединения II формулы

Послед.ь 1
с тиофенолом в подходящем растворителе с образованием фенилсульфида IV-а формулы

Послед. ь 1
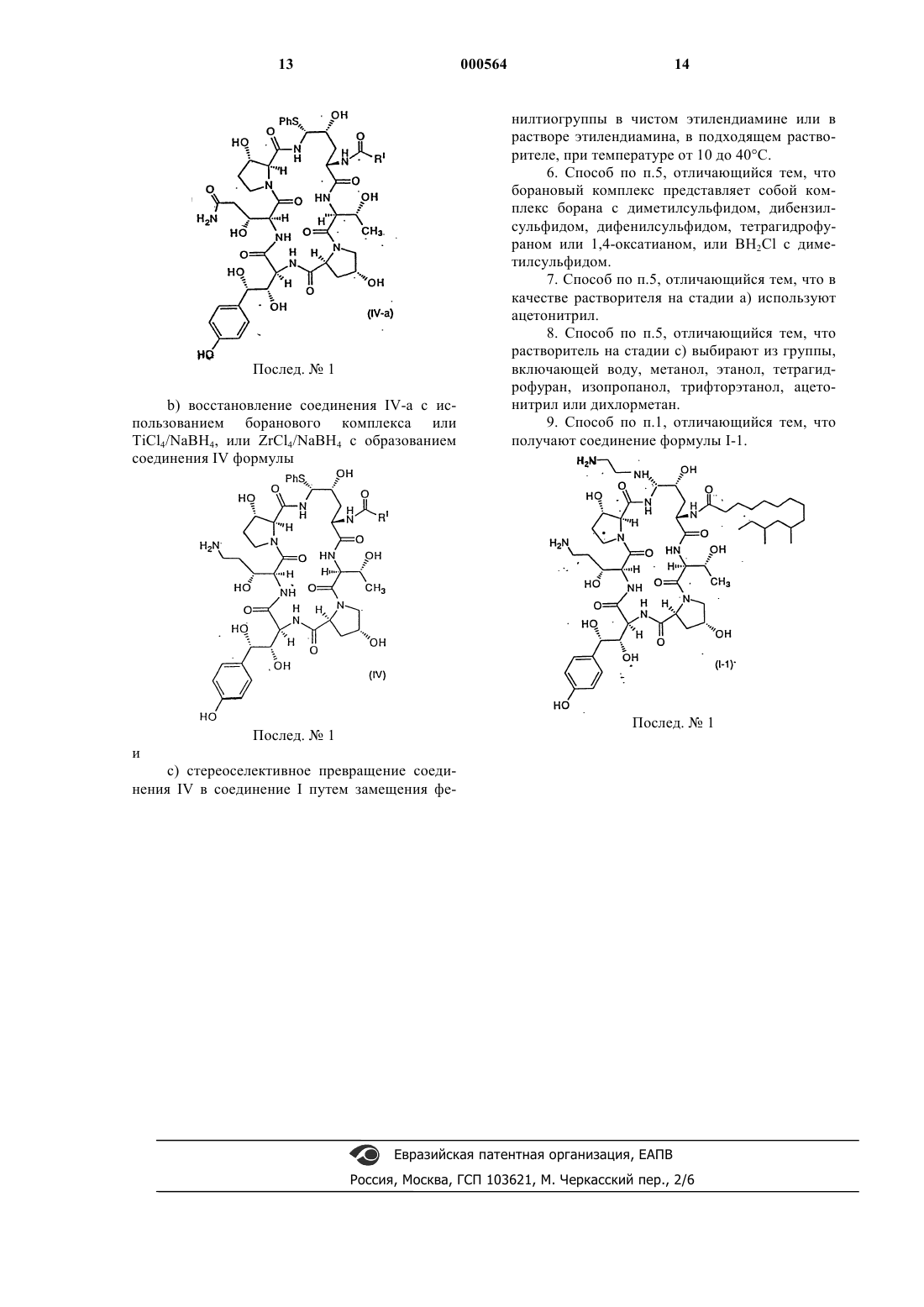
b) восстановление соединения IV-a с использованием боранового комплекса или TiCl4/NaBH4, или ZrCl4/NaBH4 с образованием соединения IV формулы

Послед. ь 1
и
с) стереоселективное превращение соединения IV в соединение I путем замещения фенилтиогруппы в чистом этилендиамине или в растворе этилендиамина, в подходящем растворителе, при температуре от 10 до 40шС.
6. Способ по п.5, отличающийся тем, что борановый комплекс представляет собой комплекс борана с диметилсульфидом, дибензилсульфидом, дифенилсульфидом, тетрагидрофураном или 1,4-оксатианом, или BH2Cl с диметилсульфидом.
7. Способ по п.5, отличающийся тем, что в качестве растворителя на стадии а) используют ацетонитрил.
8. Способ по п.5, отличающийся тем, что растворитель на стадии с) выбирают из группы, включающей воду, метанол, этанол, тетрагидрофуран, изопропанол, трифторэтанол, ацетонитрил или дихлорметан.
9. Способ по п.1, отличающийся тем, что получают соединение формулы I-1

Послед. ь 1
Текст
1 Предпосылки создания изобретения Настоящее изобретение относится к усовершенствованному способу получения некоторых азациклогексапептидов, описанных в патенте США 5378804, выданном 3 января 1995 г. Прежний синтез этих соединений предусматривал проведение пяти стадий и не обладал достаточной стереоселективностью или высоким выходом. Известные способы восстановления первичных амидов, такие как гидрирование,применение гидрида металла и электрохимическое восстановление, требуют форсированных режимов, несовместимых с другими амидами и функциональными группами в ряду пневмокандинов. Недостатком этих способов восстановления является отсутствие хемоселективности среди различных замещенных амидов. Новый способ, описанный в настоящей заявке, исключает две стадии и обеспечивает, в результате,более высокий выход и более легкий синтез аналогов указанных соединений. Краткое изложение сущности изобретения Настоящее изобретение относится к способу получения азациклогексапептидов формулыR1 представляет собой СН 2 СН 2NH2 илиRV представляет собой Н или (C1-C4)алкил; или их фармацевтически приемлемых кислотно-аддитивных солей. Установлено, что соединения, полученные заявленным способом, полезны для лечения грибковых инфекций и для лечения и предупреждения инфекций, вызванных Pneumocystiscarinii, которые часто обнаруживаются у паци 000564 2 ентов с ослабленным иммунитетом, например, у пациентов, страдающих от СПИДа. Подробное описание изобретения Настоящее изобретение относится к способу получения соединений формулы (I) путем стереоселективного, высокопродуктивного синтеза, который исключает две стадии из предшествующего способа их получения. По всему описанию и в прилагаемой формуле изобретения данная химическая формула или название будет охватывать все оптические и стереоизомеры, а также их рацемические смеси,когда такие изомеры и смеси существуют. Термин алкил подразумевает линейные,разветвленные или циклические углеводородные группы, например, метил, этил, н-пропил,изопропил, н-бутил, пентил, гексил, гептил,циклопентил, циклогексил, циклогексилметил и подобные группы. Термин циклоалкил относится к типу алкила, содержащему от 3 до 15 атомов углерода, без чередующихся или резонирующих двойных связей между атомами углерода. Термин алкенил относится к группам,таким как, например, винил, 1-пропен-2-ил, 1 бутен-4-ил, 2-бутен-4-ил, 1-пентен-5-ил, и подобным группам. Термин алкокси относится к линейным или разветвленным оксиалкильным группам,таким как, например, метокси, этокси, бутокси,гептокси, додецилокси, и подобным группам. Соединения настоящего изобретения, как правило, получают в виде смесей стереоизомерных форм, в которых обычно преобладает одна из них, причем специалист может подобрать условия для получения преимущественно нужного изомера. Соединения предпочтительной стереоизомерной формы, обозначаемой в настоящем изобретении как нормальная форма,являются соединениями, в которых группа в положении C-5-orn находится ниже плоскости в указанной позиции. Обозначение epi используется для тех соединений, в которых группа в положении C-5-orn находится выше этой плоскости. Положение C-5-orn определяется как углерод в положении 5 4-гидроксиорнитинового компонента. Фармацевтически приемлемыми солями,подходящими в качестве солей присоединения кислот, являются соли таких кислот, как хлористоводородная, бромистоводородная, фосфорная, серная, малеиновая, лимонная, уксусная,винная, янтарная, щавелевая, яблочная, глутаминовая и подобные кислоты, и включают соли других кислот, родственные с фармацевтически приемлемыми солями, перечисленными в Journal of Pharmaceutical Science, 66:2 (1977). В предпочтительном варианте осуществления изобретения способ настоящего изобретения включает стадии восстановления соединения II формулы(послед.1) для получения соединения III формулы(послед.1) которое затем превращают в соединение IV формулы(послед.1) которое стереоселективно превращают в соединение I посредством замещения фенилтиогруппы. В другом варианте осуществления изобретения способ включает стадии взаимодействия соединения II формулы(послед.1) которое стереоселективно превращают в соединение I посредством замещения фенилтиогруппы. Соединение II, в котором RI представляет собой диметилтридецил, можно получить путем культивирования Zalerion arboricola, ATCC 20868, в питательной среде, обогащенной маннитом в качестве основного источника углерода, как это описано в патенте США 5021341,выданном 4 июня 1991 г. Другие способы присоединения протеиновых цепей описаны в патентах США 5378804, 5514651 и 5516756. Предпочтительное соединение, полученное по способу изобретения, показано ниже: Для более ясной демонстрации заявленного способа изобретение иллюстрируется следующими его стадиями, где указаны все предпочтительные реагенты. RI на приведенной ниже реакционной схеме представляет собой диметилтридецил. Реакционная схема Как показано выше, стадия 1 включает восстановление амида (соединение II) до амина с использованием боранового комплекса, например, борана с тетрагидрофураном (ТГФ),диметилсульфидом, дифенилсульфидом, дибензилсульфидом, 1,4-оксатианом, или ВН 2 Сl с диметилсульфидом,илиTiCl4/NaBH4 в ТГФ или другом подходящем растворителе. Восстановление также можно осуществить, используя бориды титана или циркония или борановые комплексы с аммиаком, диметиламином, пиридином или пиперазином. Предпочтительными восстановителями являются борановые комплексы с тетрагидрофураном (ТГФ), диметилсульфидом, дифенилсульфидом, дибензилсульфидом, 1,4-оксатианом, или ВН 2 Сl с диметилсульфидом, или борид металла,такой какTiCl4/NaBH4, в ТГФ или другом подходящем растворителе. Амид, не испытавший превращения при таком восстановлении, отделяют, используя хроматографию с обращенной фазой. Стадия II включает реакцию соединения III с тиофенолом в ацетонитриле и трифторуксусной кислоте (ТФК) с образованием содержащего фенилсульфид промежуточного соединения. Ожидается, что любая кислота умеренной силы даст промежуточное соединение с хорошим выходом. Можно использовать другие сульфиды, 7 такие как 4-метокситиофенол, 2-меркапто-1 метилимидазол и 2-меркаптобензимидазол. Соединение III экстрагируют путем внесения разбавленного реакционного раствора в колонку для хроматографии с обращенной фазой С-18 с последующим элюированием метанолом. Количество используемой ТФК является критическим фактором для степени замещения,так же, как и для последующего образования нежелательного сульфида в гомотирозиновой части циклического пептида. Обнаружено, что 5-25% ТФК в ацетонитриле дает наилучший выход, а для времени необратимого процесса предпочтителен интервал, содержащий ТФК 715%. Не обнаружено, что количество воды относительно исходного вещества в реакционной смеси существенно влияет на выход. Количество тиофенола, используемого на этой стадии, также является критическим фактором для выхода конечного продукта. Наилучший выход обеспечивают 3-5 эквивалентов этого соединения. Установлено, что предпочтительными условиями для образования сульфида являются 5 эквивалентов тиофенола в 10% ТФК ацетонитриле при 0 С. При таких условиях после экстракции на твердой фазе достигают выхода 6570%. Стадия 3, включающая замещение фенилтиогруппы, осуществляется в обход прежнего пути, который проходил через образование промежуточного сульфона. Фенилсульфид реагирует в неразбавленном этилендиамине (1:3) при температуре окружающей среды с получением соединения 1-1 с выходом 95%. Реакция протекает при температуре от 10 до 40 С в течение 0,5-6,0 ч. Предпочтительно, чтобы реакция протекала при комнатной температуре в течение примерно 1,5 ч. Реакцию также можно проводить с использованием этилендиамина, растворенного в подходящем растворителе, таком как вода, метанол,этанол, изопропанол, тетрагидрофуран, трифторэтанол, дихлорэтан или ацетонитрил. Настоящее изобретение подробнее описывается в следующих примерах, в которых все количественные выражения в виде частей, а также соотношения и проценты являются весовыми, если на это нет других указаний. В приведенном примере R1 представляет собой диметилтридецил. Пример 1. а) Синтез и отделение соединения III от соединения II. Соединение II (15,9 г, с чистотой 89% по площади пика хроматограммы, 3,4 вес.% воды,0,0128 моль) добавляют к сухому ТГФ (0,64 л),и суспензию сушат до содержания воды менее 10 мол.% путем стекания флегмы через слой молекулярных сит ЗА. Добавляют дополнительное количество сухого ТГФ для восстановления 8 первоначального объема смеси, и суспензию охлаждают до температуры менее 4 С на бане лед/вода/метанол. Добавляют в течение десяти минут чистый ВН 3SМе 2 (10,91 г, 0,144 моль), и реакционную смесь выдерживают при 0-4 С. Развитие реакции контролируют методом ВЭЖХ до соотношения исходного вещества и конечного продукта 1:1, показывающего на окончание времени выдержки реакционной смеси (3,5 ч). Через 4 чсмесь охлаждают до -12 С и медленно гасят 2N НСl (0,036 л). Этот раствор разбавляют водой до 1,14 л. Выход соединения III по анализу составляет 6,60 г (47%). Погашенный раствор разбавляют до 4 л и загружают в колонку для хроматографии среднего давления с адсорбентом LiChroprep RP-C18(158 г). После загрузки колонку промывают 1,2 л воды, и амин элюируют 1,9 л смеси воды с ацетонитрилом (1:4 по объему), а затем 0,38 л смеси ацетонитрила с водой (1:3 по объему). Богатые продуктом фракции (более 80% площади пика) объединяют и разбавляют водой до раствора ацетонитрила в воде 1:7,3 по объему (всего 1,70 л). Эту смесь загружают в такую же колонку, какая описана выше, и колонку промывают 0,57 л воды. Нужное соединение элюируют 0,57 л метанола. Обогащенные фракции (более 85% площади пика) объединяют и концентрируют путем испарения на роторном испарителе и в условиях статического высокого вакуума, и получают 6,81 г (с чистотой 87 вес.%, 6,8 вес.% воды), содержащих 5,92 г гидрохлорида соединения III (где R1 является диметил-тридецилом) с выходом в чистом виде 43%.b) Получение фенилсульфидного соединения IV. Соединение III (5,80 г по анализу, 0,00533 моль) загружают в 0,23 л сухого ацетонитрила и охлаждают до -5 С, после чего добавляют тиофенол (3,10 г, 0,028 моль). Добавляют в течение 20 мин ТФК (36 г, 24,5 мл, 0,318 моль), чтобы поддержать температуру реакционной смеси ниже 0 С. Реакционную смесь выдерживают при температуре от -10 С до 0 С до тех пор,пока анализ с помощью ВЭЖХ не покажет менее 3% площади исходного вещества (3,75 ч). В это время медленно (1 ч) добавляют охлажденную воду (0,56 л), причем реакционную смесь охлаждают таким образом, чтобы ее температура оставалась ниже 5 С. Выход по анализу -и-фенилсульфидного аддукта в виде соли трифторуксусной кислоты составляет 4,82 г (71%). Этот раствор загружают на такую же колонку, какая описана в стадии а), и колонку промывают водой (0,57 л), затем адсорбированные органические соединения элюируют метанолом (0,50 л). Обогащенные фракции концентрируют с помощью испарения на роторном испарителе и в условиях статического высокого вакуума. Получают 7,20 г (с чистотой 57 вес.%, 9 5,1 вес.% воды) сырой соли трифторуксусной кислоты фенилсульфида в виде аморфного пенистого твердого вещества. Скорректированная стадия выделения дает 4,10 г (61%) фенилсульфида в виде смеси - и -аминальных диастереомеров 93:7. с) Превращение фенилсульфида в диамин(соединение 1-1). Сырой трифторметансульфонат (8,4 г сырого, чистотой 57 вес. %, 0,00377 моль) добавляют к этилендиамину (24 мл) при перемешивании при температуре окружающей среды. Получающийся в результате раствор перемешивают в течение 1,5 ч для завершения замещения, затем добавляют метанол (40 мл), а после него уксусную кислоту (45 мл), поддерживая температуру ниже 25 С путем охлаждения на ледяной бане и получая в результате густую суспензию. Добавляют воду (160 мл) для растворения суспензии,и водный слой экстрагируют путем осторожного встряхивания с гексаном (75 мл). Гексановый слой снова экстрагируют водой (40 мл), объединенный водный слой фильтруют через воронку с фильтром из плавленного стекла средней пористости, а затем очищают с помощью препаративной ВЭЖХ, используя колонку С-18 диаметром 50 мм, с использованием в качестве элюента смесь из 22% ацетонитрила и 78% 0,15%-ой водной уксусной кислоты. Обогащенную фракцию лиофилизуют, и получают 4,2 г (с чистотой 85 вес.%) соединения 1-1 в виде диацетата с выходом 78% на стадии получения в чистом виде.d) Кристаллизация соединения 1-1. Твердое вещество (2,3 г) растворяют в этаноле (25 мл), и затем добавляют воду (2,7 мл). Раствор пропускают через воронку с фильтром из плавленного стекла для удаления посторонних веществ. К полученному фильтрату добавляют уксусную кислоту (0,14 мл), а затем постепенно (1,75 ч) добавляют этилацетат (14 мл). В раствор вносят затравку, и затравочный слой оставляют для вызревания на 1 ч. Добавляют в течение 5 ч оставшуюся часть этилацетата (32 мл), и оставляют для вызревания еще на 1 ч. Твердое кристаллическое вещество собирают на воронке с фильтром из плавленного стекла и промывают раствором этанола с этилацетатом и водой (6 мл/9 мл/0,5 мл, соответственно). Сырой остаток на фильтре сушат в токе азота, и получают 1,91 г (1,75 г по анализу, извлечение 88%) диацетата соединения 1-1.RV представляет собой Н или (C1-C4)алкил; или их фармацевтически приемлемых кислотно-аддитивных солей, отличающийся тем, что включает следующие стадии: а) восстановление соединения II формулы 1. Способ получения азациклопептидов формулы (I) с использованием боранового комплекса илиb) взаимодействие соединения III с тиофенолом в количестве 3-5 эквивалентов и в присутствии 5-25%-го раствора трифторуксусной кислоты в подходящем растворителе с образованием фенилсульфида IV формулыRV представляет собой Н или (C1-C4)алкил; или их фармацевтически приемлемых кислотно-аддитивных солей, отличающийся тем, что включает следующие стадии: а) взаимодействие соединения II формулы и с) стереоселективное превращение соединения IV в соединение I путем замещения фенилтиогруппы в чистом этилендиамине или в растворе этилендиамина, в подходящем растворителе, при температуре от 10 до 40 С. 2. Способ по п.1, отличающийся тем, что борановый комплекс представляет собой комплекс борана с диметилсульфидом, дибензилсульфидом, дифенилсульфидом, тетрагидрофураном или 1,4-оксатианом, или BH2Cl с диметилсульфидом. 3. Способ по п.1, отличающийся тем, что в качестве растворителя на стадии b) используют ацетонитрил. 4. Способ по п.1, отличающийся тем, что растворитель на стадии с), выбирают из группы,включающей воду, метанол, этанол, тетрагидрофуран, изопропанол, трифторэтанол, ацетонитрил или дихлорметан. 5. Способ получения азациклопептидов формулы (I)b) восстановление соединения IV-a с использованием боранового комплекса или 14 нилтиогруппы в чистом этилендиамине или в растворе этилендиамина, в подходящем растворителе, при температуре от 10 до 40 С. 6. Способ по п.5, отличающийся тем, что борановый комплекс представляет собой комплекс борана с диметилсульфидом, дибензилсульфидом, дифенилсульфидом, тетрагидрофураном или 1,4-оксатианом, или BH2Cl с диметилсульфидом. 7. Способ по п.5, отличающийся тем, что в качестве растворителя на стадии а) используют ацетонитрил. 8. Способ по п.5, отличающийся тем, что растворитель на стадии с) выбирают из группы,включающей воду, метанол, этанол, тетрагидрофуран, изопропанол, трифторэтанол, ацетонитрил или дихлорметан. 9. Способ по п.1, отличающийся тем, что получают соединение формулы I-1. Послед.1 и с) стереоселективное превращение соединения IV в соединение I путем замещения фе Евразийская патентная организация, ЕАПВ Россия, Москва, ГСП 103621, М. Черкасский пер., 2/6
МПК / Метки
МПК: A61K 38/12, C07K 7/56
Метки: азациклогексапетидов, получения, способ
Код ссылки
<a href="https://eas.patents.su/8-564-sposob-polucheniya-azaciklogeksapetidov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения азациклогексапетидов</a>
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Ансель Жан-Эрик, Бьенейм Юг, Мейллян Пьер
МПК: A61K 31/355, C07C 39/19, B01J 31/24...
Метки: использованием, замещенных, cпособ, получения, способ, витамина, фенолов
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Трициклические соединения, способ их получения, способы получения оптически активных или рацемических производных колхицина и тиохолкицина с использованием трициклических соединений и промежуточныепродукты синтеза

Номер патента: 93
Опубликовано: 25.06.1998
Авторы: Пронин Дидье, Тороманофф Эдмон, Брион Франсис, Мазюри Алан, Шаппер Бернадетт, Диолез Кристиан, Мари Кристиан, Миддендорп Мишель
МПК: C07D 317/44, C07C 43/21
Метки: синтеза, рацемических, активных, трициклических, промежуточныепродукты, оптически, соединений, производных, использованием, способы, тиохолкицина, колхицина, способ, соединения, трициклические, получения
Формула / Реферат:
1. Трициклические соединения общей формулы I в которой либо а) оба R1 и R2 представляют собой алкильную группу, a R3 представляет собой атом водорода или группу A-SO2-, либо б) оба R2 и R3 представляют собой атом водорода или алкил, a R1 представляет собой группу A-SO2-, либо в) все три: R1, R2 и R3 представляют собой атом водорода или все три представляют собой алкил, либо г) R1 представляет собой группу А-SO2- или атом водорода, a...
Способ получения n-замещенных 3-гидроксипиразолов

Номер патента: 351
Опубликовано: 29.04.1999
Авторы: Кениг Хартман, Гётц Норберт, Клайн Ульрих, Эллер Карштен
МПК: C07D 231/22
Метки: способ, получения, 3-гидроксипиразолов, n-замещенных
Формула / Реферат:
1. Способ получения N-замещенных 3-гидроксипиразолов формулы I в которой R1 означает необязательно замещенный алкил, арил или гетероарил, а R2 и R3 означают водород, циано, галоген и необязательно замещенный алкил, арил или гетероарил, окислением пиразолидин-3-она формулы II отличающийся тем, что реакцию осуществляют с использованием кислорода воздуха в качестве окислителя в практически рН-нейтральной среде в присутствии...
Новые противосудорожные i-ar(алк)ил-имидазолин-2-оны, содержащие в 4-положении двузамещенный остаток амина, и способ их получения

Номер патента: 535
Опубликовано: 28.10.1999
Авторы: Гевальд Карл, Унферферт Клаус, Менцер Манфред, Ланкау Ханс-Йоахим, Шефер Харри
МПК: C07D 233/88, A61K 31/415
Метки: амина, содержащие, противосудорожные, способ, 4-положении, новые, двузамещенный, остаток, i-ar(алк)ил-имидазолин-2-оны, получения
Формула / Реферат:
1. Новые соединения общей формулы 1 где Х = водород, С1-С4-алкил, С1-С4-алкокси, трифторметил, галоген, R1 или R2 = С1-С4-алкил, циклоалкил, гетероалкил или R1 и R2 вместе обозначают группу алкилена с 2-6 атомами углерода, в которой группа -СН2- может быть замещена кислородом, азотом или серой. 2. Соединения по п.1, представляющие собой: 1-фенил-4-морфолино-имидазолин-2-он, 1-(4-метокси)-4-пиперидино-имидазолин-2-он, ...
Мезопористый алюмогель и способ его получения

Номер патента: 16
Опубликовано: 30.12.1997
Авторы: Миллини Роберто, Калемма Винченцо, Беллусси Джузеппе, Перателло Стефано
МПК: B01J 21/04, C04B 35/10, C01F 7/02...
Метки: способ, мезопористый, алюмогель, получения
Формула / Реферат:
1. Мезопористый гель, содержащий матрицу оксида алюминия, в которой могут быть гомогенно диспергированы один или более оксидов, выбранных из группы, включающей диоксид кремния, оксид бора, оксид фосфора, оксид металла группы VIII и/или VIB общей формулы МОx, при следующих молярных соотношениях между указанными оксидами и оксидом алюминия: SiO2/Al2O3 = 0-3,0 B2O3/Al2O3 = 0-4,0 P2O5/Al2O3 = 0-0,2 МОx/Аl2O3 = 0-0,2, и имеющий удельную...
Предыдущий патент: Способ получения пористых полиолефиновых частиц.
Следующий патент: Полимерный материал
Случайный патент: Модульные строительные элементы для возведения подпорной стенки и способ ее возведения