Синтез замещенных флуоренов
Номер патента: 15300
Опубликовано: 30.06.2011
Авторы: Воскобойников Александр Зельманович, Разави Аббас, Лебедев Артём Юрьевич, Асаченко Андрей Фёдорович






Формула / Реферат
1. Способ получения замещенных флуоренов, включающий стадии:
I) алкилирования дифенилметана по меньшей мере 2 экв. алкилирующего агента общей формулы

где X представляет собой галоген, выбранный из Cl, Br или I, и
Ra, Rb и Rc, каждый независимо, выбраны из алкила, арила, гетероарила, циклоалкила или алкенила, имеющего от 1 до 20 атомов углерода, при условии, что атом углерода каждого из Ra, Rb и Rc непосредственно связан с четвертичным атомом углерода,
в присутствии кислоты Льюиса с соответствующей операцией выделения;
II) бромирования продукта стадии I 2,01-2,05 экв. брома при температуре немного ниже комнатной температуры в присутствии кислоты Льюиса или металла, способного образовывать кислоту Льюиса в реакционной среде, с последующей операцией выделения, включающей удаление избытка брома с использованием подходящего реагента и перекристаллизацию после стандартной обработки;
III) взаимодействия продукта, полученного на стадии II, с восстановителем в присутствии катализатора, включающего лиганд общей формулы

где Rm, Rn, Rp и Rq, каждый независимо, выбраны из гидрокарбила, имеющего от 1 до 30 атомов углерода, или любого органического заместителя при условии, что галоген не может быть непосредственно связан с N, при этом любые два соседних R могут быть соединены с образованием кольца, и где Е являются одинаковыми или разными и каждый независимо выбран из С, N, Р или В;
и соединения любого металла группы 10, предпочтительно безводной соли любого металла группы 10, и возможно, в зависимости от типа и количества катализатора, в присутствии соли HCl, HBr или HI.
2. Способ по п.1, где в комплексе CXRaRbRc на стадии I X представляет собой хлор и Ra, Rb и Rc, каждый независимо, выбраны из алкила, имеющего от 1 до 6 атомов углерода.
3. Способ по п.2, где алкилирующий агент представляет собой t-BuCl.
4. Способ по любому из пп.1-3, где на стадии I кислота Льюиса представляет собой AlCl3.
5. Способ по любому из пп.1-4, где количество кислоты Льюиса составляет от 0,01 до 5 мас.% в расчете на массу всех реагентов на стадии I.
6. Способ по любому из пп.1-5, где на стадии II катализатор представляет собой Fe.
7. Способ по любому из пп.1-6, где в лиганде на стадии III заместители Rm и Rn, непосредственно связанные с азотом, являются стерически затрудненными.
8. Способ по любому из пп.1-7, где количество лиганда составляет от 0,01 до 20 мол.% включительно.
9. Способ по любому из пп.1-8, где восстановитель на стадии III представляет собой цинк в виде порошка.
10. Способ по любому из пп.1-9, где соединение металла группы 10 на стадии III представляет собой безводную соль Ni.
11. Способ по любому из пп.1-10, где возможная соль HCl, HBr или HI на стадии III представляет собой Et4NI, KBr, NaBr, KI, Nal или CsBr.
12. Способ по любому из пп.1-11, где конечный продукт представляет собой 3,6-ди-трет-бутилфлуорен.
Текст
Настоящее изобретение относится к экономичному способу получения дизамещенных флуоренов с высоким выходом. Лебедев Артм Юрьевич, Асаченко Андрей Фдорович, Воскобойников Александр Зельманович (RU), Разави Аббас (BE)(71)(73) Заявитель и патентовладелец: ТОТАЛ ПЕТРОКЕМИКАЛС РИСЕРЧ ФЕЛЮЙ (BE) 015300 Настоящее изобретение относится к синтезу замещенных флуоренов с высоким выходом. В общеизвестном способе получения 3,6-дизамещенных флуоренов в качестве исходного соединения использовали дорогостоящий 2,2'-дийод-4,4'-дизамещенный дифенилметан. 3,6-Дизамещенный флуорен получали путем взаимодействия примерно 0,5 экв. соли никеля, приблизительно 1 экв. крайне дорогостоящего и вредного для здоровья PPh3, по меньшей мере 1,5 экв. дорогостоящего Et4NI и по меньшей мере 1,5 экв. цинкового порошка. Взаимодействие проводили в тетрагидрофуране (ТГФ) при температуре примерно 70 С и в течение периода времени примерно 7 ч. Эта процедура была длительной,трудоемкой, дорогостоящей и опасной для здоровья. В документе известного уровня техники Синтез 3,6-ди-(трет-бутил)флуорена путем каталитического связывания над никелем арилгалогенидов, Gitendra С.Р., Gajewski J.J., In organic preparations andprocedures intl., vol. 30,2, 222-225, 1998, описан способ получения флуорена, исходя из дифенилметана с использованием дийодистых интермедиатов. Таким образом, существует необходимость в разработке способов получения замещенных флуоренов с высоким выходом. Задача настоящего изобретения заключается в разработке способа получения замещенных флуоренов. Задача настоящего изобретения также заключается в том, чтобы обеспечить получение указанных соединений с высоким выходом. Задача настоящего изобретения заключается также в разработке способа получения флуоренов, который является эффективным и экономичным. Соответственно, в настоящем изобретении предложен способ получения замещенных флуоренов,включающий стадии:I) алкилирования дифенилметана по меньшей мере 2 экв. алкилирующего агента общей формулы где X представляет собой галоген, выбранный из Cl, Br или I,Ra, Rb и Rc, каждый независимо, выбраны из алкила, арила, гетероарила, циклоалкила или алкенила,имеющего от 1 до 20 атомов углерода, при условии, что атом углерода каждого из Ra, Rb и Rc непосредственно связан с четвертичным атомом углерода,в присутствии кислоты Льюиса с соответствующей процедурой выделения;II) бромирования продукта стадии I 2,01-2,05 экв. брома при температуре немного ниже комнатной температуры в присутствии кислоты Льюиса или металла, способного образовывать кислоту Льюиса в реакционной среде, с последующей процедурой выделения, включающей удаление избытка брома с использованием подходящего реагента и перекристаллизацию после стандартной обработки;III) взаимодействия продукта, полученного на стадии II, с восстановителем в присутствии катализатора, включающего лиганд общей формулы где Rm, Rn, Rp и Rq, одинаковые или разные и каждый независимо, выбран из гидрокарбила, имеющего от 1 до 30 атомов углерода, или любого органического заместителя, при условии, что галоген не может быть непосредственно связан с N, где любые два соседних R могут быть соединены с образованием кольца и где Е являются одинаковыми или разными, и каждый независимо выбран из С, N, Р или В; и соединения любого металла группы 10, предпочтительно безводной соли металла группы 10, и возможно, в зависимости от типа и количества катализатора, в присутствии соли HCl, HBr или HI. Стадия I. Предпочтительно в соединении CXRaRbRc на стадии I X представляет собой хлор, и Ra, Rb и Rc, каждый независимо, представляют собой алкильные группы, имеющие от 1 до 6 атомов углерода. Более предпочтительно они являются одинаковыми, и еще более предпочтительно они являются одинаковыми и представляют собой трет-бутил (t-Bu). Альтернативным предпочтительным соединением является tBuCl. Наиболее предпочтительными положениями для заместителей у дифенилметана являются положения 4 и 4'. Небольшие заместители, такие как метильные группы, могут занимать положения 2 и 2', и поэтому они не являются предпочтительными. Предпочтительно используемая на стадии I кислота Льюиса может быть выбрана из AlX3, где X-1 015300 представляет собой Cl, Br, F, I, OR, CR3, NR2 или OC(O)R, где каждый R независимо может быть выбран из алкильной группы, имеющей от 1 до 6 атомов углерода. Более предпочтительно она представляет собой AlCl3. Количество кислоты Льюиса обычно может составлять от 0,01 до 5 мас.%, предпочтительно от 1 до 3 мас.% в расчете на массу замещенного дифенилметана на стадии I. Указанное соединение подвергают кристаллизации предпочтительно из изопропилового спирта или из гексанов, более предпочтительно из изопропилового спирта. На стадии очистки в конце стадии I перед перекристаллизацией промывка не требуется, поскольку в дизамещенном дифенилметановом соединении предпочтительно должна оставаться примесь алюминия. На самом деле, данные примеси алюминия(III) служат дополнительным катализатором на описанной ниже стадии бромирования (стадияII). Полученный на стадии I продукт предпочтительно представляет собой 4,4'-дизамещенный дифенилметан. Стадия II. Предпочтительным растворителем является CH2Cl2. Для получения чистого продукта с атомами Br, занимающими положения 2 и 2' дизамещенного соединения, количество брома, которое добавляют в систему, ограничивают узким диапазоном от 2,01 до 2,05 экв. по отношению к дизамещенному дифенилметану. Если добавлять бром в избытке, то он будет занимать другие положения и, возможно, сможет вытеснять заместители и препятствовать кристаллизации конечного продукта. Если добавлять бром в слишком малом количестве, то конечный продукт не будет кристаллизоваться. Предпочтительно катализатор представляет собой Fe. Катализатор помещают в реакционный сосуд с растворителем и дизамещенным дифенилметаном, и в этот сосуд, в котором поддерживают температуру ниже комнатной температуры, предпочтительно от 10 до 20 С, медленно добавляют бром в растворителе. Полученный продукт промывают раствором, который может поглощать Br2. Предпочтительно это раствор Na2SO3 в воде. Полученный продукт отделяют, сушат над осушителем, упаривают и подвергают кристаллизации из EtOH или гексанов, предпочтительно из EtOH. Полученный на стадии II продукт предпочтительно представляет собой 2,2'-дибром-4,4'дизамещенный дифенилметан. Стадия III. Продукт, полученный на стадии II, добавляют к апротонному растворителю, предпочтительно к эфирному растворителю, более предпочтительно к ТГФ. Лиганд может быть выбран из множества разнообразных соединений, но предпочтительно, чтобы заместители Rm и Rn, связанные непосредственно с атомами азота, были стерически затрудненными заместителями. Более предпочтительно они представляют собой вторичный или третичный гидрокарбил,имеющий по меньшей мере 3 и до 30 атомов углерода включительно. Предпочтительно оба Е являются одинаковыми и представляют собой атомы углерода. Количество лиганда составляет от 0,01 до 20 мол.% включительно. Восстановителем может быть органический восстановитель или цинк. Предпочтительно это цинк в виде порошка. В типичном случае используют от 1 до 2 экв. восстановителя по отношению к дифенилметановому соединению, предпочтительно 1,5 экв. Затем в систему добавляют соединение металла группы 10, а также соль HCl, HBr или HI. Предпочтительно соединение металла группы 10 представляет собой безводную соль Ni, Pd или Pt,более предпочтительно Ni. Более предпочтительно оно представляет собой безводный галогенид, такой как, например, NiCl2 или NiBr2. Наиболее предпочтительным соединением является NiCl2. Необходимое количество соединения металла группы 10 составляет от 0,01 до 20 мол.% включительно. Лиганд и соединение металла группы 10 в общем случае используют в одинаковых молярных количествах. Предпочтительная возможная соль HCl, HBr или HI может быть выбрана, например, из Et4NI, KBr,NaBr, KI, NaI, CsBr. Более предпочтительными из этих солей являются соли, у которых радиусы ионов(как катиона, так и аниона) по возможности являются наибольшими. Наиболее предпочтительной солью является Et4NI. Количество соли HCl, HBr или HI, необходимое в реакции, зависит от количества используемого катализатора и от природы используемой соли. Если количество используемого катализатора больше 10% мол., добавления соли не требуется. Реакцию проводят при кипячении с обратным холодильником при температуре в пределах от 50 до 100 С, предпочтительно при температуре приблизительно 70 С, и в течение периода времени от 1 до 3 ч,предпочтительно примерно 1,5 ч. Примеры Пример 1. Получение 4,4'-ди-трет-бутилдифенилметана. В колбу емкостью 500 мл к смеси 16,8 г (100 ммоль) дифенилметана и 21,3 г (230 ммоль) третбутилхлорида при комнатной температуре при энергичном перемешивании добавляли 67 мг (0,5 ммоль) безводного AlCl3, что вызывало немедленное выделение газообразного HCl. Цвет реакционной смеси менялся на красный. Через 5 мин добавляли вторую порцию 67 мг (0,5 ммоль) AlCl3. После 5 мин энергичного перемешивания реакционная смесь затвердевала. Спустя примерно 1 ч полученную твердую массу подвергали перекристаллизации из примерно 100 мл горячего изопропанола. Кристаллический продукт выпадал в осадок из этого раствора при температуре -30 С примерно через 2-3 ч. Его отделяли,промывали 50 мл холодного изопропанола и сушили в вакууме при пониженном давлении 10-20 мм Hg(1,33-2,67 кПа). Получили 19,6 г продукта с выходом 70%. Эту процедуру повторяли с выходом в пределах от 67 до 74%. Элементный анализ для C21H28. Вычислено: С 89,94; Н 10,06. Найдено: С 89,86; Н 10,03. 1 Н ЯМР (CDCl3):7.31 (d, J = 8.4 Гц, 4 Н), 7.13 (d, J = 8.4 Гц, 4 Н), 3.92 (s, 2 Н), 1.30 (s, 18H) 13 С(1 Н) ЯМР (CDCl3):148.7, 138.2, 128.5, 125.2, 41.0, 34.3, 31.4. Получение 2,2'-дибром-4,4'-ди-трет-бутилдифенилметана. В трехгорлую круглодонную колбу емкостью 250 мл, оснащенную термометром, капельной воронкой с обводной трубкой для выравнивания давления, осушительным патроном (с CaCl2) и магнитной мешалкой, к смеси 14,0 г (50 ммоль) 4,4'-ди-трет-бутилдифенилметана, 168 мг (3 ммоль) порошка железа и 50 мл дихлорметана по каплям добавляли раствор 16,8 г (105 ммоль) брома в 25 мл дихлорметана при энергичном перемешивании в течение примерно 1/2 ч. Для получения продукта с хорошим выходом температуру реакционной смеси должны были поддерживать от 10 до 20 С, поэтому при необходимости для охлаждения реакционной смеси использовали холодную водяную баню. Далее реакционную смесь дополнительно перемешивали в течение 1 ч при комнатной температуре. Затем полученную смесь промывали дважды 75 мл насыщенного раствора Na2SO3 для удаления следов брома. Органический слой отделяли и водный слой дополнительно промывали 75 мл дихлорметана. Объединенный органический экстракт сушили над безводным CaCl2 и упаривали досуха. Полученную кристаллическую массу растворяли в приблизительно 50 мл гексанов. Этот раствор в гексанах пропускали через слой силикагеля Silica Gel 60 (диаметр приблизительно 50 мм, длина приблизительно 30 мм) для удаления неорганических солей и полимерных примесей. В дополнение к этому силикагелевый слой промывали 300-400 мл гексанов. Объединенный гексановый элюат упаривали досуха. Остаток подвергали кристаллизации из примерно 200 мл горячего этанола (96%). Кристаллы выпадали в осадок при температуре -30 С, их отделяли, промывали 50 мл холодного этанола и сушили в вакууме (12 мм Hg (133-267 Па. Получили 14,9 г желтоватого кристаллического продукта с выходом 68%. Элементный анализ для C21H26Br2. Вычислено: С 57,55; Н 5,98. Найдено: С 57,65; Н 5,91. 1 Н ЯМР (CDCl3):7.59 (d, J = 2.0 Гц, 2 Н), 7.23 (dd, J = 2.0 Гц, J = 8.1 Гц, 2 Н), 6.92 (d, J = 8.1 Гц, 2 Н),4.13 (s, 2H), 1.30 (s, 18H). 13 С(1 Н) ЯМР (CDCl3):151.4, 136.0, 130.3, 129.7, 124.8, 124.6, 41.0, 34.5, 31.2. Получение лиганда 2,4,4-триметил-N-E,2E)-2-[(E)-1,1,3.3-тетраметилбутил]имино)этилиден)-2 пентанамина (L1). В круглодонной колбе емкостью 2000 мл перемешивали смесь 56,3 г (0,39 ммоль) 40% глиоксаля,100 г (0,78 ммоль) трет-октиламина и 1200 см 3 воды в течение 4 ч при комнатной температуре. Полученный осадок отделяли и промывали приблизительно 200 мл холодной воды. Белый порошок растворяли в 300 мл дихлорметана. Этот раствор сушили над Na2SO4 и затем упаривали досуха. Получили 101 г белого твердого вещества с выходом 93%. Элементный анализ для C18H36N2. Вычислено: С 77,08; Н 12,94. Найдено: С 76,92; Н 13,05. 1 Н ЯМР (CDCl3):7.94 (s, 2H), 1.69 (s, 4H), 1.29 (s, 12H), 0.92 (s, 18H). 13 С(1 Н) ЯМР (CDCl3):157.5, 61.9, 55.9, 32.0, 31.6, 29.31. В трехгорлую круглодонную колбу емкостью 1000 мл помещали 9,83 г (150 ммоль) активированной цинковой пыли, которую предварительно промывали 2% HCl, водой, этанолом, затем эфиром, сушили в вакууме и хранили в атмосфере азота, 43,8 г (100 ммоль) 2,2'-дибром-4,4'-ди-трет-бутилдифенилметана и 5,02 г (20 ммоль) Et4NI. Колбу вакуумировали и затем заполняли сухим азотом. К этой смеси добавляли 600 мл ТГФ и смесь 840 мг (3 ммоль) 2,4,4-триметил-N-Е,2 Е)-2-[(Е)-1,1,3,3-тетраметилбутил]иминоэтилиден)-2-пентанамина и 924 мг (3 ммоль) NiBr2 (ДМЭ), полученную в защитной камере с перчатками. Полученную смесь слегка нагревали при энергичном перемешивании с использованием магнитной мешалки. После начала реакции (через 2-3 мин) внешнее нагревание прерывали, поскольку реакция является экзотермической. Тем не менее, эту смесь кипятили с обратным холодильником в течение по меньшей мере 1 ч. Затем полученную смесь охлаждали до комнатной температуры и упаривали досуха, используя роторный испаритель. К остатку добавляли 300 мл насыщенного водного NH4Cl и 300 мл гексанов. Эту смесь энергично перемешивали до достижения полной экстракции продукта гексанами. Затем органический слой отделяли и водный слой дополнительно экстрагировали 2300 мл гексанов. Объединенный экстракт сушили над Na2SO4 и затем пропускали через короткую колонку с силикагелем Silica Gel 60 (4063 мкм, диаметр приблизительно 50 мм, длина приблизительно 50 мм). Слой силикагеля дополнительно промывали 300 мл гексанов. Объединенный элюат упаривали, используя роторный испаритель. После дополнительной сушки полученного бесцветного масла в вакууме (1-2 мм Hg (1,33-2,67 Па остаток становился твердым. Получили 29,8 г указанного в заголовке продукта в виде белого твердого вещества с выходом 98%. Элементный анализ для C21H28. Вычислено: С 90,59; Н 9,41. Найдено: С 90,45; Н 9,50. 1 Н ЯМР (CDCl3):7.81 (d, J = 1.5 Гц, 2H), 7.45 (d, J = 8.0 Гц, 2H), 7.33 (dd, J = 1.5 Гц, J = 8.0 Гц, 2 Н),4.13 (s, 2H), 1.30 (s, 18H). 13 С(1 Н) ЯМР (CDCl3):149.8, 141.8, 140.8, 124.5, 123.8, 116.3, 36.0, 34.8, 31.7. Пример 2. Другие лиганды L2-L5 получали, используя реакцию конденсации первичного амина с дикарбонильным соединением. Получение N-E,2E)-2-[(E)-1,1-диметилэтил]имино)этилиден)-2-метил-2-пропанамина (L2). В круглодонную колбу емкостью 250 мл к смеси 36,3 г (0,25 ммоль) 40% глиоксаля и 150 мл воды при энергичном перемешивании добавляли 38,5 г (0,50 ммоль) трет-бутиламина. Эта реакция была сильно экзотермической, и образовывался слой легкого масла. Полученную смесь охлаждали до комнатной температуры, и органический слой затвердевал. Образовавшееся твердое вещество отделяли и промывали приблизительно 200 мл холодной воды. Это твердое вещество растворяли в 200 мл дихлорметана. Раствор затем сушили над Na2SO4 и упаривали досуха. Получили 41,2 г кристаллического продукта белого цвета с выходом 98%. Элементный анализ для C10H10N2. Вычислено. С 71,37; Н 11,98. Найдено: С 71,25; Н 12,09. 1 Н ЯМР (CDCl3):7.95 (s, 4H), 1.27 (s, 18H). 13 С(1 Н) ЯМР (CDCl3):157.5, 57.8, 29.1. Получение N-[(Е,2 Е)-2-(1-адамантилимино)этилиден]-1-адамантанамина (L3). В круглодонной колбе емкостью 500 мл перемешивали в течение 5 ч смесь 11,3 г (60 ммоль) 1 адамантиламина гидрохлорида, 205 мл (30 ммоль) 0,292 М NaOH и 8,70 г (30 ммоль) 40% глиоксаля. Продукт экстрагировали 4200 мл эфира. Объединенный экстракт сушили над Na2SO4 и упаривали досу-4 015300 ха. Полученное твердое вещество подвергали перекристаллизации из 200 мл гексанов. Полученный продукт еще раз подвергали перекристаллизации из новой порции 200 мл гексанов. Полученное кристаллическое твердое вещество сушили в вакууме. Получили 7,81 г (80%) белого твердого вещества с выходом 80%. Элементный анализ для C22H32N2. Вычислено: С 81,43; Н 9,94. Найдено: С 81,47; Н 9,97. 1 Н ЯМР (CDCl3):7.93 (s, 2 Н), 2.15 (s, 6H), 1.77-1.63 (m, 24H). 13 С(1 Н) ЯМР (CDCl3):157.8, 58.5, 42.7, 36.4, 29.4. Получение N-[(Е,2 Е)-2-(циклогексилимино)этилиден]циклогексанамина (L4). В круглодонной колбе емкостью 500 мл смесь 12,8 г (129 ммоль) циклогексиламина, 23,4 г (65 ммоль) 40% глиоксаля и 50 мл воды перемешивали в течение 2 суток. Полученный осадок отделяли и промывали 50 мл холодной воды. Твердое вещество растворяли в 50 мл дихлорметана. Этот раствор сушили над Na2SO4 и упаривали досуха. Остаток подвергали перекристаллизации из 200 мл эфира. Получили 10,3 г белого твердого вещества с выходом 72%. Элементный анализ для C14H24N2. Вычислено: С 76,31; Н 10,98. Найдено: С 76,22; Н 11,00. 1 Н ЯМР (CDCl3):7.94 (s, 2H), 3.23-3.10 (m, 2H), 1.87-1.62 (m, 5H), 1.58-1.44 (m, 2H), 1.43-1.15 (m,3 Н). 13 С(1 Н) ЯМР (CDCl3):159.8, 69.1, 33.7, 25.2, 24.3. Получение Смесь 1,82 г (10 ммоль) аценафтохинона, 3,54 г (20 ммоль) 2,6-диизопропиланилина и 40 мл ледяной уксусной кислоты кипятили с обратным холодильником в течение 2 ч и затем перемешивали в течение 12 ч при комнатной температуре. Полученный осадок отделяли, промывали 20 мл холодной воды и сушили под вакуумом. Получили 3,89 г темно-желтого твердого вещества с выходом 78%. Элементный анализ для C36H40N2. Вычислено: С 86,35; Н 8,05. Найдено: С 86,43; Н 8,14. 1 Н ЯМР (CDCl3):7.87 (d, J = 8.1 Гц, 2 Н), 7.36 (dd, J = 8.1 Гц, J = 7.2 Гц, 2 Н), 7.31-7.20 (m, 6 Н), 6.64 известен и коммерчески доступен. Пример 3. Замещенный флуорен получали, следуя тому же общему способу, который описан в примере 1 с лигандами L1-L6, в различных условиях, которые указаны в табл. 1. Во всех процедурах получения в качестве восстановителя использовали цинковый порошок. Выход для каждого синтеза также приведен в табл. 1. Пример 4. Лиганд L6 использовали для оценки влияния изменения природы и количества возможной солиHCl, HBr или HI на стадии III. Для всех примеров использовали 20 мол.% NiCl2 и лиганда L6, температура реакции составляла 70 С, время реакции составляло 15 ч. Природа соли, количество соли и выход указаны в табл. 2. Таблица 2 Из этой таблицы можно сделать вывод, что выход продукта увеличивается с увеличением размеров ионов, составляющих соль. Таким образом, предпочтительно использовать соли, у которых как катион,так и анион являются по возможности более крупными. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения замещенных флуоренов, включающий стадии:I) алкилирования дифенилметана по меньшей мере 2 экв. алкилирующего агента общей формулыRa, Rb и Rc, каждый независимо, выбраны из алкила, арила, гетероарила, циклоалкила или алкенила,имеющего от 1 до 20 атомов углерода, при условии, что атом углерода каждого из Ra, Rb и Rc непосредственно связан с четвертичным атомом углерода,в присутствии кислоты Льюиса с соответствующей операцией выделения;II) бромирования продукта стадии I 2,01-2,05 экв. брома при температуре немного ниже комнатной температуры в присутствии кислоты Льюиса или металла, способного образовывать кислоту Льюиса в реакционной среде, с последующей операцией выделения, включающей удаление избытка брома с использованием подходящего реагента и перекристаллизацию после стандартной обработки;III) взаимодействия продукта, полученного на стадии II, с восстановителем в присутствии катализатора, включающего лиганд общей формулы где Rm, Rn, Rp и Rq, каждый независимо, выбраны из гидрокарбила, имеющего от 1 до 30 атомов углерода, или любого органического заместителя при условии, что галоген не может быть непосредственно связан с N, при этом любые два соседних R могут быть соединены с образованием кольца, и где Е являются одинаковыми или разными и каждый независимо выбран из С, N, Р или В; и соединения любого металла группы 10, предпочтительно безводной соли любого металла группы 10, и возможно, в зависимости от типа и количества катализатора, в присутствии соли HCl, HBr или HI. 2. Способ по п.1, где в комплексе CXRaRbRc на стадии I X представляет собой хлор и Ra, Rb и Rc,каждый независимо, выбраны из алкила, имеющего от 1 до 6 атомов углерода. 3. Способ по п.2, где алкилирующий агент представляет собой t-BuCl. 4. Способ по любому из пп.1-3, где на стадии I кислота Льюиса представляет собой AlCl3. 5. Способ по любому из пп.1-4, где количество кислоты Льюиса составляет от 0,01 до 5 мас.% в расчете на массу всех реагентов на стадии I. 6. Способ по любому из пп.1-5, где на стадии II катализатор представляет собой Fe. 7. Способ по любому из пп.1-6, где в лиганде на стадии III заместители Rm и Rn, непосредственно связанные с азотом, являются стерически затрудненными. 8. Способ по любому из пп.1-7, где количество лиганда составляет от 0,01 до 20 мол.% включительно. 9. Способ по любому из пп.1-8, где восстановитель на стадии III представляет собой цинк в виде порошка. 10. Способ по любому из пп.1-9, где соединение металла группы 10 на стадии III представляет собой безводную соль Ni. 11. Способ по любому из пп.1-10, где возможная соль HCl, HBr или HI на стадии III представляет собой Et4NI, KBr, NaBr, KI, NaI или CsBr. 12. Способ по любому из пп.1-11, где конечный продукт представляет собой 3,6-ди-третбутилфлуорен.
МПК / Метки
МПК: C07C 1/28, C07C 13/567
Метки: флуоренов, синтез, замещенных
Код ссылки
<a href="https://eas.patents.su/8-15300-sintez-zameshhennyh-fluorenov.html" rel="bookmark" title="База патентов Евразийского Союза">Синтез замещенных флуоренов</a>
Стереоспецифический синтез хиральных 1-арил- и 1-гетероарил-2-замещенных этил-2-аминов

Номер патента: 1364
Опубликовано: 26.02.2001
Авторы: Грондар Люк, Пауэрс Мэттью Р., Казимир Жан-Поль, О'брайен Майкл К, Робин Даниэль, Леон Патрик
МПК: A61P 9/12, C07D 203/24, C07C 303/38...
Метки: 1-арил, стереоспецифический, 1-гетероарил-2-замещенных, этил-2-аминов, хиральных, синтез
Формула / Реферат:
1. Способ стереоспецифического синтеза [(1-необязательно замещенный арил)- или (1-необязательно замещенный гетероарил)]-2-замещенного этил-2-амина, имеющего хиральный атом в положении 2, включающий взаимодействие 2-амино-2-замещенного этилового спирта, имеющего хиральный атом в положении 2, с [(необязательно замещенный арил)- или (тригалогенметил)сульфонил]-галогенидом или ангидридом в присутствии основания с образованием [(N-арилсульфонил)- или...
Способ получения замещенных бензотиазиноиндолов

Номер патента: 12750
Опубликовано: 30.12.2009
Авторы: Камбхампати Рама Састри, Дешпанде Амол Динкар, Котхмиркар Прабхакар, Ширсатх Викас Шрикришна, Джасти Венкатесварлу, Рамакришна Венката Сатья Нироги
МПК: C07D 513/06
Метки: получения, замещенных, бензотиазиноиндолов, способ
Формула / Реферат:
1. Способ получения замещенных бензотиазиноиндолов общей формулы (I) где R1, R2 и R4 могут быть одинаковыми или различными и, каждый независимо, представляют водород, хлор, фтор, амино, нитро, циано, CHO, (C1-С3)алкил, пергалоген(C1-С3)алкил, (C1-С3)алкокси, арил, аралкил, аралкокси, (С5-С7)гетероциклил, (С5-С7)гетероциклилалкил, (С5-С7)гетероциклилокси, ацил, ацетил, алкиламино, аминоалкил, амид, гидроксиалкил, группу карбоновой кислоты и ее...
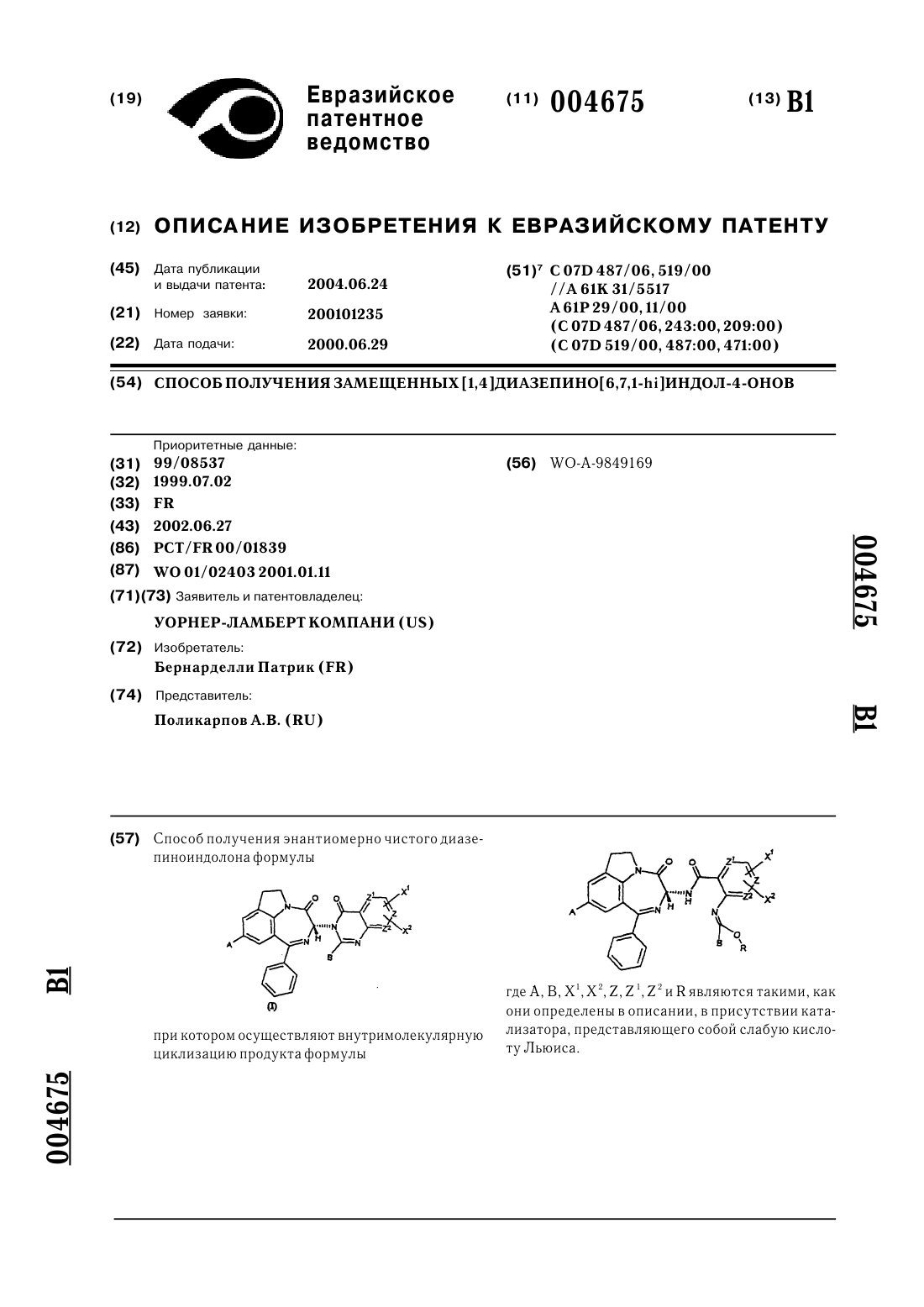
Способ получения замещенных [1,4]диазепино[6,7,1-hi]индол-4-онов
Номер патента: 4675
Опубликовано: 24.06.2004
Автор: Бернарделли Патрик
МПК: C07D 487/06, A61P 29/00, A61K 31/5517...
Метки: 1,4]диазепино[6,7,1-hi]индол-4-онов, способ, получения, замещенных
Формула / Реферат:
1. Способ получения диазепиноиндолона формулы где A представляет собой C1-C4 алкил, C1-C4 алкокси, нитро или NH2; B представляет собой C1-C4 алкил; Z представляет собой CH, тогда Z1 и Z2 оба представляют собой CH или N, или Z представляет собой N, тогда Z1 и Z2 представляют собой CH; X1 и X2 независимо представляют собой водород или -C(=O)OR5, где R5 представляет собой водород или C1-C6 алкил, его солей и сольватов, которые являются...
Способ получения замещенных пиридинкарбоновых кислот

Номер патента: 3054
Опубликовано: 26.12.2002
Авторы: Циммерманн Курт, Штайнвендер Эрих, Штайнбауер Герхард, Вресснеггер Эрнст
МПК: A01N 43/40, C07D 213/807
Метки: пиридинкарбоновых, кислот, получения, способ, замещенных
Формула / Реферат:
1. Способ получения замещенных пиридинкарбоновых кислот путем озонолиза хинолинов, отличающийся тем, что хинолин формулы который в положении 2, и/или 3, и/или 4 замещен остатком R3, а также в положении 6 и/или 7 - остатком R4, причем R1 и R2 означают Н или C1-С3-алкильную группу и R3 и R4 - инертную в условиях реакции группу и, по меньшей мере, один из остатков R1 и R2 не обозначает Н, в кислом водном растворе при температуре от -5 до +40шС...
Региоселективный синтез производных рапамицина

Номер патента: 4331
Опубликовано: 29.04.2004
Авторы: Фортье Женевьев, Нурельдин Раззак, Шо Чиа-Ченг, Селлстедт Джон Гамильтон, Чил Глория Карен
МПК: C07F 7/18, C07D 498/18
Метки: рапамицина, производных, синтез, региоселективный
Формула / Реферат:
1. Способ получения простого 31-силилового эфира рапамицина, включающий (a) взаимодействие рапамицина с силилирующим агентом с образованием простого 31, 42-бис-силилового эфира рапамицина; и (b) гидролиз простого 31, 42-бис-силилового эфира холодной разбавленной кислотой с получением простого 31-силилового эфира рапамицина. 2. Способ получения сложного 42-эфира или простого эфира рапамицина, имеющего структуру в которой R представляет сложный...