Вич-пролекарства, способные расщепляться под действием cd26
Номер патента: 9727
Опубликовано: 28.02.2008
Авторы: Вигеринк Пит Том Берт Поль, Де Кок Херман Аугустинус, Бальзарини Ян



























Формула / Реферат
1. Пролекарство, имеющее формулу

его стереоизомерная форма или его соль,
где n равно 1, 2, 3, 4 или 5;
Y обозначает пролин, аланин, гидроксипролин, дигидроксипролин, тиазолидинкарбоновую кислоту (тиопролин), дегидропролин, пипеколиновую кислоту (L-гомопролин), азетидинкарбоновую кислоту, азиридинкарбоновую кислоту, глицин, серин, валин, лейцин, изолейцин и треонин;
X выбирают из любой аминокислоты с D- или L-конфигурацией;
X и Y в каждом повторе фрагмента [Y-X] выбирают независимо друг от друга и независимо от других повторов;
Z обозначает прямую связь или бивалентную насыщенную углеводородную группу с прямой или разветвленной цепью, содержащую от 1 до 4 атомов углерода;
R1 обозначает арильную, гетероарильную группу, арилоксигруппу, гетероарилоксигруппу, арилоксиС1-4алкильную группу, гетероциклоалкилоксигруппу, гетероциклоакилС1-4алкилоксигруппу, гетероарилоксиС1-4алкильную группу, гетероарилС1-4алкилоксигруппу;
R2 обозначает арилС1-4алкильную группу;
R3 обозначает C1-10алкильную, С2-6алкенильную или С3-7циклоалкилС1-4алкильную группу;
R4 обозначает водород или С1-4алкильную группу;
арильная группа, когда она используется самостоятельно или в комбинации с другой группой,
означает фенильную группу, необязательно замещенную одним или несколькими заместителями,
каждый из которых индивидуально выбирают из группы, включающей С1-4алкильную, гидроксильную группу, С1-4алкилоксигруппу, нитрогруппу, цианогруппу, галоген, аминогруппу, моно- или
ди(С1-4алкил)аминогруппу и амидную группу;
гетероарильная группа, когда она используется самостоятельно или в комбинации с другой группой, означает моноциклический или бициклический ароматический гетероцикл, содержащий один или несколько гетероатомов кислорода, серы или азота, при этом ароматический гетероцикл необязательно может быть замещен по одному или нескольким атомам углерода заместителем, выбранным из группы, включающей С1-4алкильную группу, С1-4алкилоксигруппу, аминогруппу, гидроксильную, арильную, амидную группу, моно- или ди(С1-4алкил)аминогруппу, галоген, нитрогруппу, гетероциклоалкильную и С1-4алкилоксикарбонильную группу и при этом ароматический гетероцикл необязательно может быть замещен по вторичному атому азота С1-4алкильной или арилС1-4алкильной группой;
гетероциклоалкильная группа, когда она используется самостоятельно или в комбинации с другой группой, означает насыщенный или частично ненасыщенный моноциклический или бициклический гетероцикл, содержащий один или несколько гетероатомов кислорода, серы или азота, при этом гетероцикл необязательно может быть замещен по одному или нескольким атомам углерода заместителем, выбранным из группы, включающей С1-4алкильную группу, С1-4алкилоксигруппу, гидроксильную группу, галоген и оксогруппу, и при этом указанный гетероцикл необязательно может быть замещен по вторичному атому азота С1-4алкильной или арилС1-4алкильной группой.
2. Пролекарство по п.1, где каждый X независимо выбирают из природной аминокислоты.
3. Пролекарство по п.1 или 2, где n равно 1, 2 или 3.
4. Пролекарство по любому из пп.1-3, где n равно 2 или 3 и где по крайней мере один X представляет собой гидрофобную или ароматическую аминокислоту.
5. Пролекарство по любому из пп.1-4, где n равно 2 или 3 и где по крайней мере один X представляет собой нейтральную или кислую аминокислоту.
6. Пролекарство по любому из пп.1-5, где n равно 2 или 3 и где по крайней мере один X представляет собой основную аминокислоту.
7. Пролекарство по любому из пп.1-6, где -(Y-X)n содержит аминоконцевые X-Pro, X-Ala, X-Gly,
X-Ser, X-Val или X-Leu.
8. Пролекарство по любому из пп.1-7, где -(Y-X)n содержит аминоконцевые X-пролин или
X-аланин.
9. Пролекарство по любому из пп.1-8, где каждый Y независимо обозначает пролин, аланин,
глицин, серин, валин или лейцин.
10. Пролекарство по любому из пп.1-9, где каждый Y независимо обозначает пролин, или гидроксипролин, или дигидроксипролин, или аланин.
11. Пролекарство по любому из пп.1-10, где каждый Y независимо обозначает пролин или аланин.
12. Пролекарство по любому из пп.1-11, где -(Y-X)n обозначает -(Y-X)1 или 2-Y-Val.
13. Пролекарство по любому из пп.1-12, где -(Y-X)n обозначает -(Y-X)1 или 2-Pro-Val.
14. Пролекарство по любому из пп.1-13, где олигопептид (Y-X)nвыстроен из повторов фрагментов (Y-X), выбранных из группы, включающей Pro-Val, Pro-Asp, Pro-Ser, Pro-Lys, Pro-Arg, Pro-His, Pro-Phe, Pro-Ile, Pro-Leu, Ala-Val, Ala-Asp, Ala-Ser, Ala-Lys, Ala-Arg, Ala-His, Ala-Phe, Ala-Ile и Ala-Leu.
15. Пролекарство по любому из пп.1-14, где R1 обозначает гетероциклоалкилоксигруппу,
гетероарильную группу, гетероарилС1-4алкилоксигруппу, арильную группу или арилоксиС1-4алкильную группу.
16. Пролекарство по любому из пп.1-15, где R1 обозначает гексагидрофуро[2,3-b]фуран-3-илоксигруппу, тетрагидрофуран-3-илоксигруппу, хинолин-2-ильную группу,
тиазол-5-илметилоксигруппу, 3-гидрокси-2-метил-1-фенильную, 2,6-диметилфеноксиметильную группу.
17. Пролекарство по любому из пп.1-16, где R1 обозначает (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илоксигруппу.
18. Пролекарство по любому из пп.1-17, где R2 обозначает фенилметильную группу, R3 обозначает изобутильную группу и R4 обозначает водород.
19. Пролекарство по любому из пп.1-18, где Z обозначает метиленовую группу.
20. Пролекарство по п.1, где пролекарством является

или его соль.
21. Пролекарство по п.1, где пролекарством является

или его соль.
22. Пролекарство по п.1, где пролекарством является

или его соль.
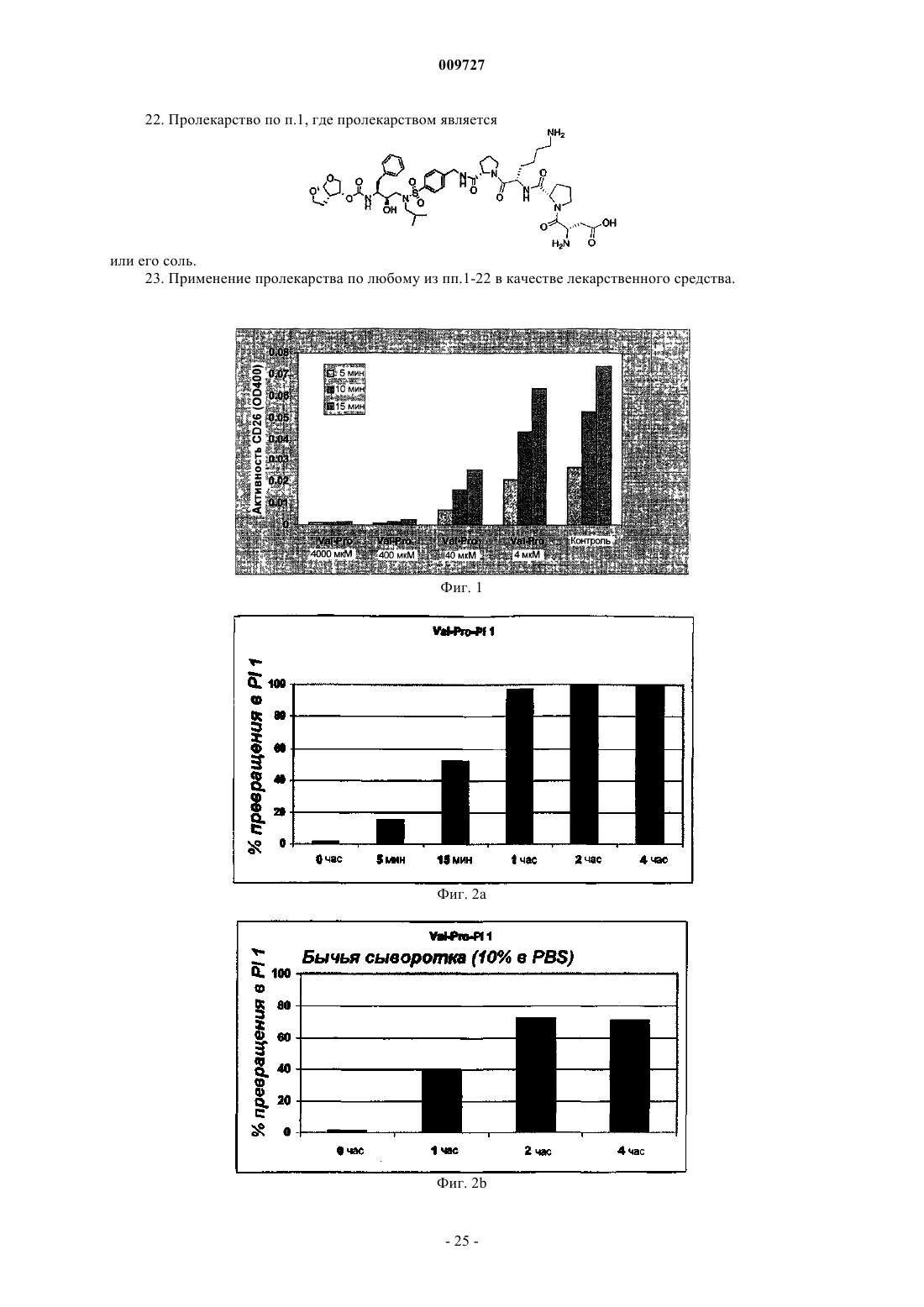
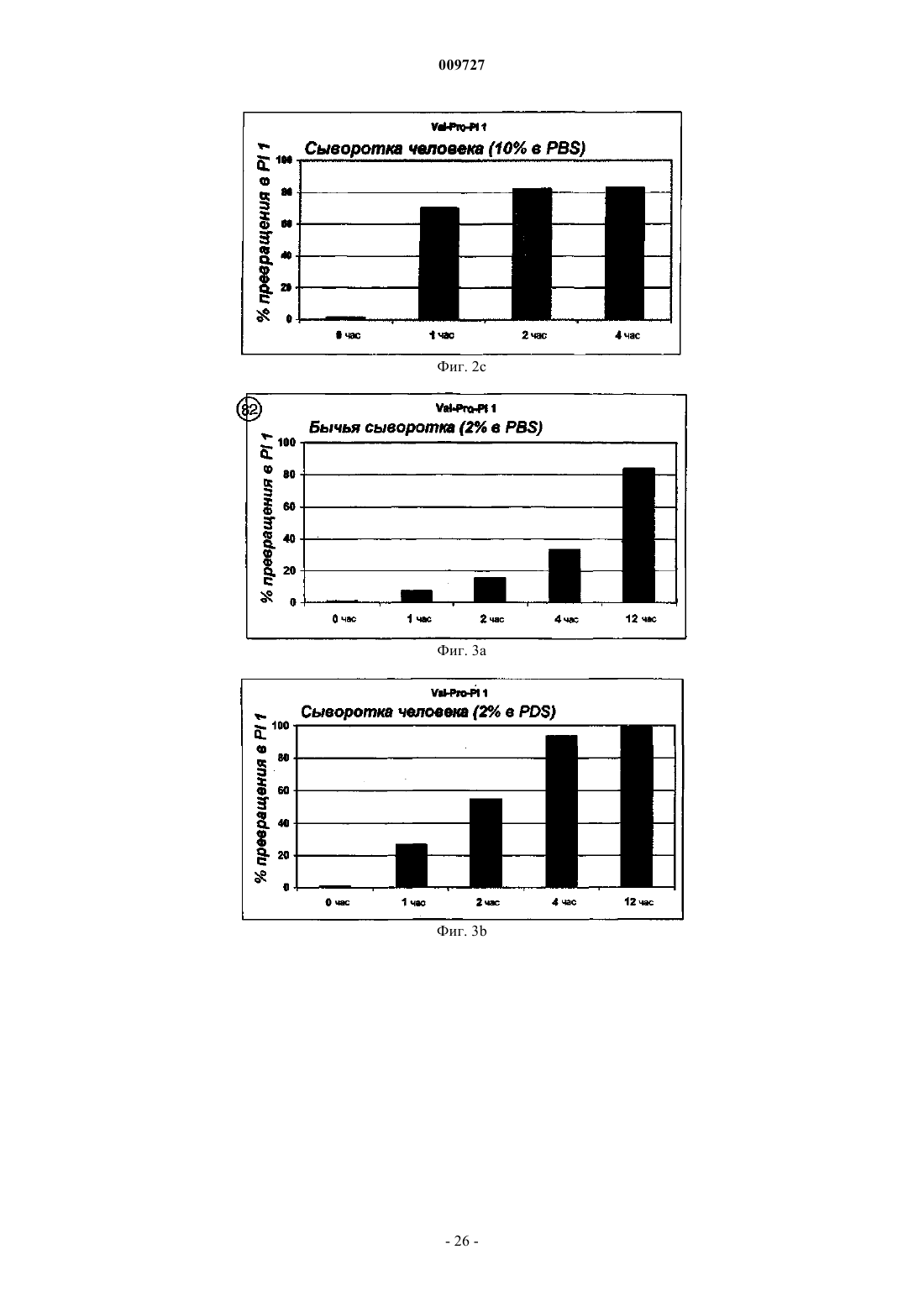
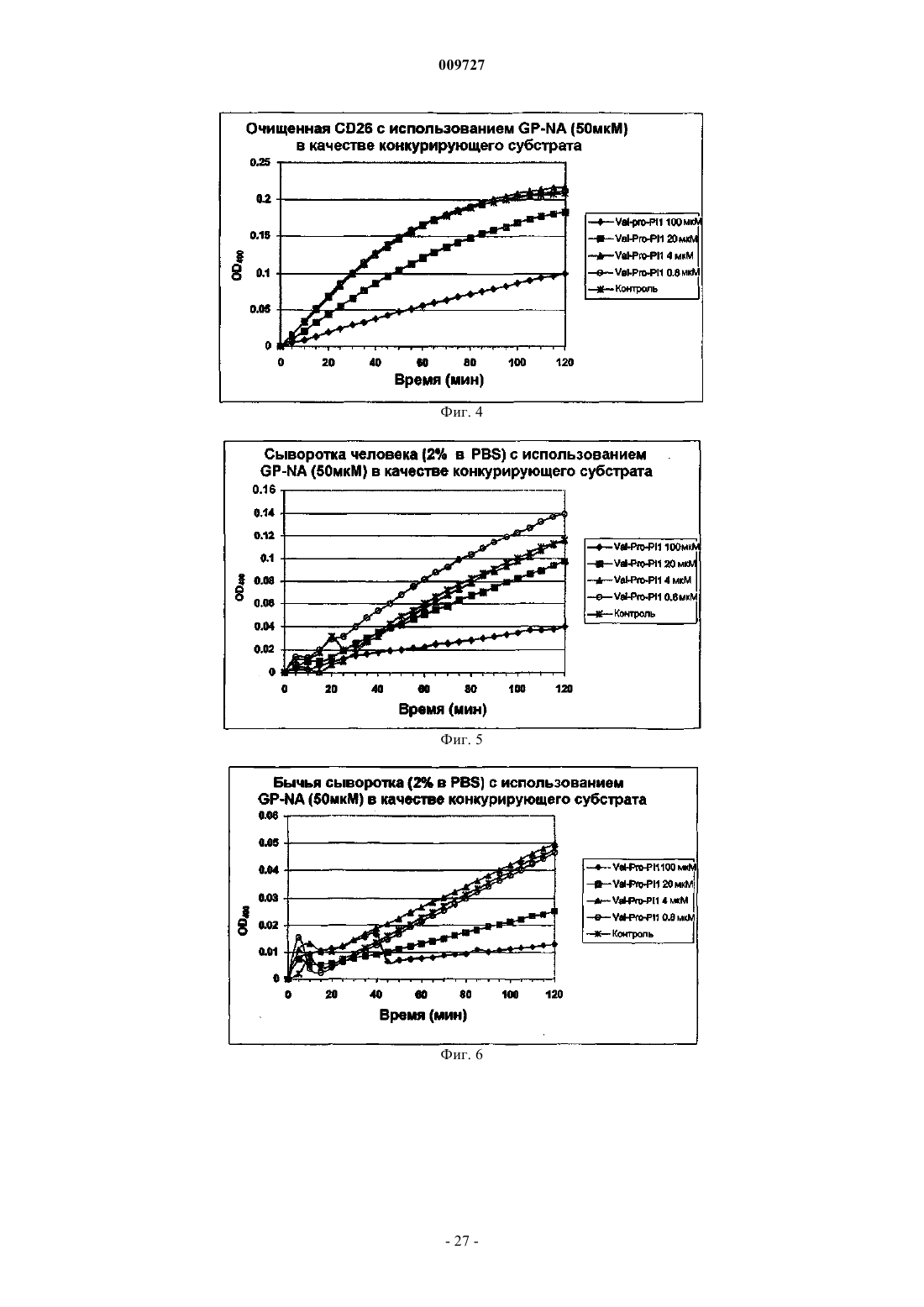
23. Применение пролекарства по любому из пп.1-22 в качестве лекарственного средства.
Текст
009727 Настоящее изобретение относится к пролекарству ВИЧ-ингибирующих соединений, при этом ВИЧингибирующее соединение высвобождается или активируется посредством протеолиза пептидной части молекулы. Настоящее изобретение относится также к способам улучшения перорального усвоения, модифицирования связывания с белком сыворотки, улучшения проникновения через гематоэнцефалический барьер или растворимости и биосовместимости ВИЧ-ингибирующих соединений. Современные методы разработки лекарств (в частности, комбинаторная химия, высокопроизводительный фармакологический скрининг, структурно-направленный дизайн лекарств) поставляют весьма специфичные и действенные молекулы лекарств. Тем не менее, довольно часто эти новые химические структуры обладают неблагоприятными физико-химическими и биофармацевтическими свойствами. Кроме того, в процессе разработки новых терапевтических агентов исследователи фокусируют свое внимание на фармакологических и/или биологических свойствах, придавая меньше значения физикохимическим свойствам веществ. Однако физико-химические свойства (константа диссоциации, растворимость, коэффициент перераспределения, стабильность) лекарственной молекулы оказывают значительное влияние на ее фармацевтическое и биофармацевтическое поведение. Таким образом, в процессе разработки лекарства следует устанавливать и, если необходимо, модифицировать физико-химические свойства веществ. Более того, физико-химические свойства многих лекарственных молекул, уже имеющихся на рынке, не являются оптимальными. В настоящее время соединения, испытываемые в качестве лекарства, часто не принимаются в расчет вследствие их плохой растворимости в воде или неадекватной абсорбции, в результате чего бесчисленное количество усовершенствованных медицинских препаратов остается нереализованным. Другие же продукты успевают попасть на рынок, но не раскрывают свой полный коммерческий потенциал по причинам, связанным с их безопасностью или эффективностью. Пролекарства потенциально способны преодолеть обе эти проблемы. С целью селективного биопревращения пролекарства в активную форму лекарства в технологии используют эндогенные ферменты. Указанная технология способна продлить жизнь новым перспективным кандидатам в лекарства в процессе их разработки и улучшить безопасность и эффективность существующих лекарственных препаратов. Пролекарства в основном представляют собой неактивные производные лекарственных молекул,для которых, для того чтобы они высвободили в организме активное родительское лекарство, требуется осуществить химическую или ферментативную биотрансформацию. Пролекарства разрабатывают для того, чтобы преодолеть нежелательное свойство лекарства. В таком случае указанная технология может быть использована для улучшения физико-химических, биофармацевтических и/или фармакокинетических свойств различных лекарств. Как правило, пролекарство само по себе биологически неактивно. По этой причине, чтобы добиться заметной эффективности после того, как лекарства достигают своей мишени, пролекарства необходимо эффективно превратить в родительские соединения. В общем случае пролекарства разрабатывают с целью улучшить проникновение лекарства через биологические мембраны и добиться лучшей абсорбции лекарства, увеличить продолжительность действия лекарства (обеспечить медленное высвобождение родительского соединения из пролекарства, уменьшить первичную биотрансформацию лекарства), нацелить действие лекарства (в частности, при нацеливании в мозг или опухоль), улучшить его растворимость в воде и повысить стабильность лекарства(внутривенных препаратов, глазных капель и т.п.), улучшить условия для местного нанесения лекарства(в частности, при нанесении лекарства на кожу и в глаза), улучшить химическую/ферментативную стабильность лекарства (в частности, пептидов) или уменьшить побочное действие лекарства. В зависимости от типа лекарства, которое необходимо подвергнуть превращению, уже разработаны многочисленные технологии получения пролекарств. Указанные технологии получения пролекарств включают химические процессы получения циклических пролекарств для пептидов и пептидоподобных веществ, химические процессы фосфонооксиметилирования (РОМ) для солюбилизации третичных аминов, фенолов и пространственно затрудненных спиртов и химические процессы этерификации в целом. Кроме того, стратегии нацеливания лекарства осуществляют путем присоединения групп, способных расщепляться специфическими ферментами, такими как пептиддеформилаза бактерий, расщепляющаяN-концевые формильные группы пептидов, или PSA (специфичный антиген простаты), который используют для нацеливания на рак предстательной железы. По ряду причин связывание пептидов или аминокислот с терапевтическим агентом уже применяли в прошлом. В антисмысловой-антигенной области олигонуклеотиды или интеркаляторы связывают с пептидами с целью повысить усвоение терапевтических агентов клетками. Однако указанные олигонуклеотиды или интеркаляторы необязательно должны высвобождаться после проникновения в клетку, и они не могут считаться пролекарствами. Примером связывания аминокислоты с терапевтическим соединением является валганцикловир, пролекарство на основе сложного L-валилового эфира ганцикловира,который применяют для предупреждения или лечения инфекций цитомегаловируса. После перорального введения пролекарство быстро превращается в ганцикловир под действие эстераз кишечника и печени. Недавно были изучены аланиновые и лизиновые пролекарства новых противоопухолевых бензотриазолов. Уже продемонстрирован опосредованный пептидным переносчиком транспорт через мембрану пролекарственных аналогов нуклеозидов на основе сложных эфиров аминокислот (Han et al., Pharm. Res.-1 009727 1998, 15: 1154-1159; Han et al., Pharm. Res. 1998, 15: 1382-1386). Действительно, было показано, что пероральная биодоступность лекарств может меняться в зависимости от производных пролекарств на основе аминокислот, содержащих аминокислоту, преимущественно с L-конфигурацией. Для абсорбции в кишечнике, оптимальной комбинацией длины цепи и степени разветвления у -углерода аминокислоты предположительно обладает L-валин. Было показано, что hPEPT-1 принимает участие в качестве первичного пути абсорбции при усиленной системной доставке пролекарств на основе сложных эфировL-валина. Недавно была показана необходимость того, чтобы транспортер hPEPT-1 оптимально взаимодействовал со свободной NH2, карбонильной группой и липофильным фрагментом и мог образовывать несколько дополнительных Н-мостиков со своей молекулой-мишенью. Связанные L-валиновыми мостиками аналоги нуклеозидов на основе сложных эфиров могут удовлетворять указанным требованиям к эффективной активности субстрата hPEPT-1 (Friedrichsen et al., Eur. J. Pharm. Sci. 2002, 16: 1-13). Тем не менее, из области техники для улучшения растворимости и биодоступности известно лишь использования аминокислотных пролекарств (связана только одна аминокислота) небольших органических молекул, при этом аминокислота в основном связывается посредством сложноэфирных связей, поскольку под действием эстераз они легко превращаются обратно в свободный терапевтический агент. Известно получение пролекарств с помощью ряда протеаз, таких как аминопептидазы (РСТ заявкаWO 01/68145) и аминотрипептидаза (РСТ заявка WO 02/00263). В РСТ заявке WO 99/67278 описываются ингибиторы CD26/DPPIV, которые становятся активными при процессинге под действием CD26/DPPIV и затем инактивируют протеазу. Тем не менее, сохраняется потребность в новых, альтернативных и лучших методах разработки пролекарств, и ожидается, что эта потребность будет расти, поскольку комбинаторная химия и высокопроизводительный скрининг продолжают поставлять значительное количество новых соединений с большой молекулярной массой, большим значением P (коэффициент перераспределения) или плохой растворимостью в воде. Сущность изобретения Настоящее изобретение относится к новой технологии получения пролекарств, которую можно применять для улучшения растворимости и/или биодоступности терапевтического соединения. Настоящее изобретение включает функционализацию терапевтических соединений с целью улучшить их растворимость и/или биодоступность. В настоящем изобретении предлагаются конъюгаты терапевтических соединений с пептидным фрагментом, в котором указанный конъюгат способен расщепляться дипептидилпептидазой, такой как CD26. Указанная технология может быть далее использована для модулирования связывания белком терапевтического соединения D и нацеливания на специфические места у млекопитающего. Специфическими терапевтическими соединениями являются ВИЧ-ингибирующие соединения, в частности соединения-ингибиторы ВИЧ-протеазы, а именно соединения формулы (Ia). Настоящее изобретение относится к новой технологии получения пролекарств и новых пролекарств для модулирования растворимости, связывания белком и/или биодоступности лекарства. В настоящем изобретении пролекарства представляют собой конъюгаты терапевтического соединения D и пептида,при этом конъюгат способен расщепляться дипептидилпептидазой, более предпочтительно дипептидилпептидазой IV. Кроме того, настоящее изобретение относится к способу получения указанных пролекарств. Настоящее изобретение относится также к технологии получения пролекарств для более селективного нацеливания лекарств, для модифицирования, в частности, усиления доставки лекарств в мозг и лимфатическую систему, и/или увеличения периода полураспада лекарства в плазме. В соответствии с одним аспектом настоящее изобретение относится к фармацевтической композиции, включающей пролекарство терапевтического соединения D. Терапевтическое соединение D не является пептидом или белком и включает концевую первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты. В качестве альтернативы терапевтическое соединение D связано с линкером Am, содержащим первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты. Пролекарство отличается тем, что указанное пролекарство включает указанное терапевтическое соединение D, присоединенное к олигопептиду, при этом указанный олигопептид имеет общую структуру H-[X-Y]n, где n составляет от 1 до 5; X или Y в каждом повторе фрагмента [X-Y] выбирают независимо один от другого и независимо от других повторов; X представляет собой аминокислоту, a Y обозначает L-аминокислоту, выбранную из группы, включающей пролин, аланин, гидроксипролин, дигидроксипролин, тиазолидинкарбоновую кислоту (тиопролин), дегидропролин, пипеколиновую кислоту (L-гомопролин), азетидинкарбоновую кислоту, азиридинкарбоновую кислоту, глицин, серин, валин, лейцин, изолейцин и треонин, и где связывание между карбоксильным концом H-[X-Y]n и аминогруппой в D или линкере Am осуществляется посредством амида. ПептидH-[X-Y]n содержит свободные аминоконцевые группы, т.е. немодифицированные группы NH2. В одном из вариантов осуществления настоящего изобретения терапевтическое соединение является антивирусным лекарством, в частности ингибитором ВИЧ-протеазы, а именно представляет собой соединение формулы (Ia), а пролекарство имеет формулу (I). В одном из вариантов осуществления настоящего изобретения пептид содержит от двух до пяти повторов, способных расщепляться под действием CD26. В другом варианте осуществления настоящего-2 009727 изобретения олигопептид H-[X-Y]n, способный расщепляться под действием CD26, представляет собой тетрапептид или гексапептид, в котором по крайней мере один X представляет собой гидрофобную или ароматическую аминокислоту, или в котором по крайней мере один X представляет собой нейтральную или кислую аминокислоту, или в котором по крайней мере один X представляет собой основную аминокислоту. В конкретном варианте осуществления настоящего изобретения олигопептид H-[X-Y]n представляет собой тетрапептид или гексапептид, выбранный из группы Val-Y-[X-Y]1-2, более конкретноVal-Pro-[X-Y]1-2, с целью обеспечения, в зависимости от выбора X или Y, хорошей абсорбции в кишечнике с последующим медленным или быстрым высвобождением терапевтического соединения, а также с целью модификации растворимости. В одном из вариантов осуществления настоящего изобретения тетрапептид или гексапептид имеет общую структуру Val-Y-[X-Y] или Val-Y-[X-Y]2. В соответствии с одним из вариантов осуществления настоящего изобретения Y представляет собой пролин, или гидроксипролин, или дигидроксипролин, или аланин. В соответствии с другим вариантом осуществления настоящего изобретения X выбирают из валина, аспарагиновой кислоты, серина, лизина,аргинина, гистидина, фенилаланина, изолейцина или лейцина. В соответствии с другим вариантом осуществления настоящего изобретения X выбирают из кислых аминокислот - аспарагиновой кислоты или глутаминовой кислоты с тем, чтобы получить медленное расщепление, из положительно заряженных аминокислот - аргинина, гистидина или лейцина с тем, чтобы добиться быстрого высвобождения терапевтического соединения D. Олигопептид [X-Y]n посредством амидной связующей группы может быть соединен с аминогруппой, имеющейся в органической молекуле/атоме, такой как ароматическая группа терапевтического соединения, в углеводороде, в нуклеозиде или в гетероциклической группе, или в алкильной, алкенильной или алкинильной группах, или в неорганической молекуле/атоме. В одном из вариантов осуществления настоящего изобретения олигопептид H-[X-Y]n посредством амидной связующей группы присоединен к аминогруппе, имеющейся в ароматической группе терапевтического соединения, имеющейся в углеводороде или в нуклеозиде. В альтернативном случае олигопептид Н-[X-Y]n может быть связан с терапевтическим соединением D не прямо, а посредством линкера,содержащего аминогруппу. Подобный линкер может иметь общую структуру олигопептида Am, где значение m находится от 1 до 15, в частности от 1 до 3 или же m=1. A в структуре Am может быть любой аминокислотой. В соответствии с одним из вариантов осуществления настоящего изобретения m=1,A обозначает валин. Пролекарство с подобным линкером имеет общую структуру Н-[X-Y]n-Am-D. Олигопептид Am или аминокислота A присоединены по концевой аминогруппе посредством амидной связи к олигопептиду H-[X-Y]n. Олигопептид Am или аминокислота A присоединены по своему карбоксильному концу посредством амидной или сложноэфирной связи к терапевтическому соединению D. Фармацевтические композиции могут включать пролекарства или лекарства для предупреждения или лечения вирусной инфекции. В одном из вариантов осуществления настоящего изобретения терапевтическое соединение представляет собой антивирусное лекарство, в частности, ингибитор ВИЧ-протеазы, а именно представляет собой соединение формулы (Ia), а пролекарство имеет формулу (I). В соответствии с другим аспектом настоящее изобретение относится к структурному компоненту пролекарства терапевтического соединения D, где указанное терапевтическое соединение D не является пептидом или белком и где терапевтическое соединение D включает концевую первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты, или где терапевтическое соединение D соединено с линкером, содержащим первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты, при этом указанное пролекарство включает указанное терапевтическое соединение D, которое связано мостиковой связью с олигопептидом общей структуры H-[X-Y]n, где n=1-5; X и Y в каждом повторе фрагмента [X-Y] выбирают независимо один от другого и независимо от других повторов и X представляет собой аминокислоту, a Y представляет собой(L-гомопролин), азетидинкарбоновую кислоту, азиридинкарбоновую кислоту, глицин, серин, валин, лейцин, изолейцин и треонин, и где связывание между карбоксильным концом в H-[X-Y]n, и аминогруппой вD осуществляется посредством амида. В одном из вариантов осуществления настоящего изобретения терапевтическое соединение представляет собой антивирусное лекарство, в частности ингибитор ВИЧпротеазы, а именно представляет собой соединение формулы (Ia), а пролекарство имеет формулу (I). В соответствии с одним из вариантов осуществления настоящего изобретения указанное пролекарство при его активировании не оказывает ингибирующего воздействия на фермент CD26/DPPIV. В одном из вариантов осуществления настоящего изобретения олигопептид H-[X-Y]n представляет собой тетрапептид или гексапептид, в котором по крайней мере один X представляет собой гидрофобную или ароматическую аминокислоту, или в котором по крайней мере один X представляет собой нейтральную или кислую аминокислоту, или в котором по крайней мере один X представляет собой основную аминокислоту. В конкретном варианте осуществления настоящего изобретения олигопептид H-[X-Y]n построен из повторов фрагментов [X-Y], выбранных из группы Val-Pro, Asp-Pro, Ser-Pro, Lys-Pro, Arg-Pro, His-Pro,Phe-Pro, Ile-Pro, Leu-Pro, Val-Ala, Asp-Ala, Ser-Ala, Lys-Ala, Arg-Ala, His-Ala, Phe-Ala, Ile-Ala и Leu-Ala.-3 009727 В соответствии с одним из вариантов осуществления настоящего изобретения Y обозначает пролин, или гидроксипролин, или дигидроксипролин, или аланин. В соответствии с одним из вариантов осуществления настоящего изобретения олигопептид [X-Y]n посредством амидной связующей группы соединен с аминогруппой, имеющейся в ароматической группе терапевтического соединения, имеющейся в углеводороде или имеющейся в нуклеозиде. В альтернативном случае олигопептид [X-Y]n связан с терапевтическим соединением D не прямо, а посредством линкера, содержащего аминогруппу. Указанный линкер представляет собой органическую молекулу (т.е. алкиламиногруппу, пептид или комбинацию обоих). В одном из вариантов осуществления настоящего изобретения количество аминокислот m в линкере между олигопептидом, способным расщепляться под действием CD26, и терапевтическим соединением D, составляет от 1 до 15. В конкретном варианте осуществления настоящего изобретения подобный линкер может иметь общую структуру олигопептида Am,где значение m составляет 1-15, в частности от 1 до 3 или m=1. A в структуре Am может быть любой аминокислотой. В соответствии с одним из вариантов осуществления настоящего изобретения m=1,A обозначает валин. Пролекарство с подобным линкером имеет общую структуру Н-[X-Y]n-Am-D. В соответствии с одним из вариантов осуществления настоящего изобретения пролекарство представляет собой пролекарство терапевтического соединения для предупреждения или лечения вирусной инфекции. В соответствии с другим вариантом осуществления настоящего изобретения пролекарство представляет собой пролекарство ингибитора ВИЧ-протеазы с общей структурной формулой (I). В соответствии с другим аспектом настоящее изобретение относится к способу модулирования(увеличения или уменьшения) растворимости в воде, и/или связывания белком плазмы, и/или биодоступности терапевтического соединения D путем связывания пептида с указанным терапевтическим соединением, а полученный при этом конъюгат способен расщепляться дипептидилпептидазой. В соответствии с одним из вариантов осуществления настоящего изобретения дипептидилпептидаза представляет собой CD26, а терапевтическое соединение D не является пептидом или белком и терапевтическое соединение D включает концевую первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты, или же терапевтическое соединение D присоединено к линкеру, содержащему первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты, и где олигопептид имеет общую структуру H-[X-Y]n, где n составляет от 1 до 5; X и Y в каждом повторе фрагмента [X-Y] выбирают независимо один от другого и независимо от других повторов; X представляет собой аминокислоту, a Y представляет собой L-аминокислоту, выбранную из группы, включающей пролин, аланин, гидроксипролин, дигидроксипролин, тиазолидинкарбоновую кислоту(тиопролин), дегидропролин, пипеколиновую кислоту (L-гомопролин), азетидинкарбоновую кислоту,азиридинкарбоновую кислоту, глицин, серин, валин, лейцин, изолейцин и треонин, и где связывание между карбоксильным концом в H-[X-Y]n и аминогруппой в D осуществляется посредством амида. В соответствии с одним из вариантов осуществления настоящего изобретения олигопептид Н-[X-Y]n представляет собой тетрапептид или гексапептид, в котором по крайней мере один X представляет собой гидрофобную или ароматическую аминокислоту, или в котором по крайней мере одинX представляет собой нейтральную или кислую аминокислоту, или в котором по крайней мере одинX представляет собой основную аминокислоту. В одном из вариантов осуществления настоящего изобретения терапевтическое соединение представляет собой антивирусное лекарство, в частности ингибитор ВИЧ-протеазы, а именно представляет собой соединение формулы (Ia), а пролекарство имеет формулу (I). Другой аспект настоящего соединения относится к способу получения пролекарства, в котором пролекарство способно расщепляться под действием дипептидилпептидазы, причем способ включает стадию связывания терапевтически активного лекарства D и пептида со структурой H-[X-Y]n, при этом полученный в результате конъюгат способен расщепляться под действием CD26. В соответствии с одним из вариантов осуществления настоящего изобретения дипептидилпептидаза представляет собой CD26, а терапевтическое соединение D не является пептидом или белком и терапевтическое соединение D включает концевую первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты, или же терапевтическое соединение D присоединено к линкеру, содержащему первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты,где олигопептид состоит из общей структуры Н-[X-Y]n, где значение n составляет от 1 до 5; X и Y в каждом повторе фрагмента [X-Y] выбирают независимо один от другого и независимо от других повторов;X представляет собой аминокислоту, a Y представляет собой L-аминокислоту, выбранную из группы,включающей пролин, аланин, гидроксипролин, дигидроксипролин, тиазолидинкарбоновую кислоту (тиопролин), дегидропролин, пипеколиновую кислоту (L-гомопролин), азетидинкарбоновую кислоту, азиридинкарбоновую кислоту, глицин, серин, валин, лейцин, изолейцин и треонин, и где связывание между карбоксильным концом в H-[X-Y]n и аминогруппой в D осуществляется посредством амида. В соответствии с одним из вариантов осуществления настоящего изобретения, олигопептид Н-[X-Y]n представляет собой тетрапептид или гексапептид, в котором по крайней мере один X представляет собой гидрофобную или ароматическую аминокислоту, или в котором по крайней мере один X представляет собой нейтральную или кислую аминокислоту, или в котором по крайней мере один X представляет собой основ-4 009727 ную аминокислоту. В одном из вариантов осуществления настоящего изобретения терапевтическое соединение представляет собой антивирусное лекарство, в частности ингибитор ВИЧ-протеазы, а именно представляет собой соединение формулы (Ia), а пролекарство имеет формулу (I). В соответствии с другим аспектом, настоящее изобретение относится к применению пролекарства терапевтического соединения D при изготовлении лекарственного средства для лечения или предупреждения вирусной инфекции. Терапевтическое соединение D не является пептидом или белком, и терапевтическое соединение D включает концевую первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты, или же терапевтическое соединение D присоединено к линкеру, содержащему первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты, причем в указанном пролекарстве указанное терапевтическое соединение D связано с олигопептидом, при этом указанный олигопептид состоит из общей структуры H-[X-Y]n, где значение n составляет от 1 до 5; X и Y в каждом повторе фрагмента [X-Y] выбирают независимо один от другого и независимо от других повторов, X представляет собой аминокислоту, a Y представляет собой(L-гомопролин), азетидинкарбоновую кислоту, азиридинкарбоновую кислоту, глицин, серин, валин, лейцин, изолейцин и треонин, и где связывание между карбоксильным концом в H-[X-Y]n и аминогруппой вD осуществляется посредством амида. В одном из вариантов осуществления настоящего изобретения терапевтическое соединение представляет собой антивирусное лекарство, в частности ингибитор ВИЧпротеазы, а именно представляет собой соединение формулы (Ia), а пролекарство имеет формулу (I). Наконец, в соответствии с еще одним аспектом, настоящее изобретение относится к способу получения пролекарства с использованием пептида с общей структурой H-[X-Y]n для получения пролекарства терапевтического соединения D, способного расщепляться под действием CD26. Терапевтическое соединение D не является пептидом или белком, и терапевтическое соединение D включает концевую первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты, или же терапевтическое соединение D присоединено к линкеру, содержащему первичную или вторичную аминогруппу, способную связываться с карбоксильной группой аминокислоты. Пролекарство характеризуется тем, что в указанном пролекарстве указанное терапевтическое соединение D связано с олигопептидом, при этом указанный олигопептид имеет общую структуру H-[X-Y]n, где значение n составляет от 1 до 5; X и Y в каждом повторе фрагмента [X-Y] выбирают независимо один от другого и независимо от других повторов; X представляет собой аминокислоту, a Y представляет собой L-аминокислоту, выбранную из группы, включающей пролин, аланин, гидроксипролин, дигидроксипролин, тиазолидинкарбоновую кислоту (тиопролин), дегидропролин, пипеколиновую кислоту (L-гомопролин), азетидинкарбоновую кислоту, азиридинкарбоновую кислоту, глицин, серин, валин, лейцин, изолейцин и треонин, и где связывание между карбоксильным концом в H-[X-Y]n и аминогруппой в D осуществляется посредством амида. В одном из вариантов осуществления настоящего изобретения терапевтическое соединение представляет собой антивирусное лекарство, в частности ингибитор ВИЧ-протеазы, а именно представляет собой соединение формулы (Ia), а пролекарство имеет формулу (I). В одном из вариантов осуществления настоящего изобретения пептид имеет от двух до пяти повторов, способных расщепляться под действием CD26. В другом варианте осуществления настоящего изобретения количество m аминокислот в линкере Am между расщепляющимся под действием CD26 олигопептидом и терапевтическим соединением составляет 1, а A обозначает валин. В другом варианте осуществления настоящего изобретения олигопептид H-[X-Y]n, способный расщепляться под действием CD26,представляет собой тетрапептид или гексапептид, в котором по крайней мере один X представляет собой гидрофобную или ароматическую аминокислоту, или в котором по крайней мере один X представляет собой нейтральную или кислую аминокислоту, или в котором по крайней мере один X представляет собой основную аминокислоту. В конкретном варианте осуществления настоящего изобретения олигопептид H-[X-Y]n представляет собой тетрапептид или гексапептид, выбранный из группы Val-Pro-[X-Y]1-2 таким образом, чтобы он обладал хорошей абсорбцией в кишечнике с последующим медленным или быстрым, в зависимости от выбора X, высвобождением терапевтического соединения. В конструкцииH-[X-Y]n-D терапевтическое соединение D имеет первичную (NH2) или вторичную (NH) аминогруппу,которая связана с группой СООН аминокислоты на карбоксильном конце пептида H-[X-Y]n. В том случае, если терапевтическое соединение D не имеет групп NH2 или NH или же группы NH или NH2 не могут вступать в реакцию (вследствие, в частности, стерических затруднений), то терапевтическое соединение D можно ввести во взаимодействия с линкером, который после указанного взаимодействия имеет группы NH2 или NH, способные реагировать с группой СООН аминокислоты на карбоксильном конце пептида Н-[X-Y]n. В соответствии с одним из вариантов осуществления настоящего изобретения каждый Y независимо обозначает пролин, или гидроксипролин, или дигидроксипролин, или аланин. В одном из вариантов осуществления настоящего изобретения олигопептид H-[X-Y]n посредством амидной связующей группы присоединен к аминогруппе, имеющейся в ароматической группе терапевтического соединения, в углеводороде или в нуклеозиде. В альтернативном случае олигопептид H-[X-Y]n связан с терапевтическим-5 009727 соединением D не прямо, а посредством линкера, содержащего аминогруппу. Подобный линкер может иметь общую структуру олигопептида Am, где значение m составляет от 1 до 15, в частности от 1 до 3 или m=1. A в структуре Am может быть любой аминокислотой. В соответствии с одним из вариантов осуществления настоящего изобретения m=1, a A обозначает валин. Пролекарство с подобным линкером имеет общую структуру Н-[X-Y]n-Am-D. Олигопептид Am или аминокислота A соединены своим аминовым концом с олигопептидом H-[X-Y]n посредством амидной группы. Олигопептид Am или аминокислота A соединены своим карбоксильным концом с терапевтическим соединением D посредством амидной или сложноэфирной группы. Фармацевтические композиции могут включать пролекарства лекарств для предупреждения или лечения вирусной инфекции. В одном из вариантов осуществления настоящего изобретения терапевтическое соединение представляет собой антивирусное лекарство, в частности ингибитор ВИЧ-протеазы, а именно представляет собой соединение формулы (Ia), а пролекарство имеет формулу (I). Краткое описание фигур Фиг. 1 показывает ингибирующее действие различных концентраций дипептида Val-Pro наCD26-катализируемое превращение хромофорного субстрата GP-pNA (25 мкМ) в GP+pNA по прошествии 5 (левый прямоугольник), 10 (средний прямоугольник) или 15 мин (правый прямоугольник) реакции. Каталитическую реакцию CD26 измеряют путем регистрации усиления поглощения при 400 нм, вызываемого высвобождением pNA. Фиг. 2 показывает превращение пролекарства Val-Pro-PI 1 в PI 1 (ингибитор ВИЧ-протеазы) в зависимости от времени. A: CD26; В: бычья сыворотка; С: сыворотка человека (в обоих случаях концентрация 10% в забуференном фосфатом физиологическом растворе, PBS). Фиг. 3 показывает превращение пролекарства Val-Pro-PI 1 в PI 1 (ингибитор ВИЧ-протеазы) в зависимости от времени. а: бычья сыворотка (2% в PBS); b: сыворотка человека (2% в PBS). Фиг. 4 показывает ингибирующее (конкурентное) действие Val-Pro-PI 1 на CD26-катализируемое превращение GP-pNA в GP+pNA (желтый). Фиг. 5 показывает ингибирующее (конкурентное) действие Val-Pro-PI 1 на CD26-катализируемое превращение GP-pNA в GP+pNA (желтый) в 2% сыворотке человека (в PBS). Фиг. 6 показывает ингибирующее (конкурентное) действие Val-Pro-PI 1 на CD26-катализируемое превращение GP-pNA в GP+pNA (желтый) в 2% бычьей сыворотке (в PBS). Подробное описание изобретения Термин "пролекарство или пролекарства" в контексте настоящего описания относится в основном к неактивным производным (или производным со значительно сниженной активностью, т.е. менее чем 20%, менее чем 10%, менее чем 5% или даже менее чем 1% остаточной активностью нефункционализованной лекарственной молекулы) терапевтического соединения, которому для того, чтобы высвободить активное родительское соединение, требуется химическая или ферментативная трансформация. Пролекарство по настоящему изобретению имеет общую структуру H-[X-Y]n-D. Химические свойства этого пролекарства детально поясняются ниже. Пролекарства разрабатывают с целью преодолеть нежелательное свойство лекарства. В таком случае эта технология может быть применена для улучшения физикохимических, биофармацевтических и/или фармакокинетических свойств различных лекарств. Обычно пролекарство само по себе биологически неактивно. Поэтому для того, чтобы добиться значительной эффективности, после того как лекарство достигает своей мишени, пролекарства необходимо эффективно превращать в родительские соединения. Эту активацию можно осуществить с помощью ферментов,которые находятся в организме, или же ферменты вводят совместно с пролекарством. В общем случае пролекарства разрабатывают с целью улучшения проникновения лекарства через биологические мембраны с тем, чтобы добиться лучшей абсорбции лекарства, удлинения продолжительности действия лекарства (с целью более медленного высвобождения родительского лекарства из пролекарства, уменьшения первичной биотрансформации лекарства), нацеливания действия лекарства (в частности, нацеливания в мозг или на опухоль), улучшения растворимости в воде и повышения стабильности лекарства (внутривенных препаратов, глазных капель и т.п.), улучшения местного нанесения лекарства(в частности, доставки лекарства на кожу или в глаза), улучшения химической/ферментативной устойчивости лекарства (в частности, пептидов) или снижения побочных эффектов лекарства. Термин "терапевтическое соединение D" в контексте настоящего описания относится к любому агенту, оказывающему благотворное воздействие на заболевание, к любому агенту, который используется или будет в будущем использоваться в качестве терапевтического средства для определенных заболеваний или расстройств. Этот термин относится также ко всем молекулам, которые все еще находятся на стадии открытия или разработки и которые еще не подтвердили свою эффективность и безопасность. Он включает небольшие органические молекулы, белки, пептиды, олигонуклеотиды, углеводороды, алифатические цепочки углерода, ароматические соединения и их аналоги и производные. Терапевтическое соединение D с (концевой) аминогруппой, в частности с первичной или вторичной аминогруппой, относится к терапевтическим соединениям со свободной аминогруппой (первичной или вторичной), а именно группой NHR, где R может быть водородом (первичная группа) или любой другой химической группой, известной из области техники. Аминогруппа может быть связана с терапевтиче-6 009727 ским соединением D посредством насыщенного или ненасыщенного атома углерода, посредством карбонильной группы или же может быть частью более широкого круга функциональных групп (амидных,карбаматных и т.п.), которые включают аминогруппу, однако аминогруппа при всех обстоятельствах должна быть способной реагировать с аминокислотой с тем, чтобы образовать стабильные амидные связи. В конкретном варианте осуществления настоящего изобретения аминогруппа NHR терапевтического соединения относится к функциональной группе аминогрупп и не относится к более широкому кругу общих функциональных групп, таких как амидные или карбаматные группы. Терапевтическое соединение также может быть соединено с олигопептидом посредством линкера. Указанный линкер может иметь любую органическую структуру, при этом он включает аминокислоты и содержит группу NHR, как указано выше."CD26" в контексте настоящего описания относится к дипептидилпептидазе IV (ЕС 3.4.14.5) в том виде, когда она связана с мембраной, и в свободной форме. Синонимами для CD26 являются DPPIV,DPP4, CD26/DPPIV или ADCP2 (белок 2, комплексующий аденозиндеаминазу). В контексте настоящего описания термин "дипептидилпептидаза(ы)" обозначает ферменты, обладающие активностью дипептидиламинопептидазы, т.е. способностью удалять дипептид из аминоконцевой стороны субстрата путем расщепления второй CO-NH амидной связи в субстрате. Другие ферменты,помимо CD26, обладающие сравнимой с CD26 активностью и протеолитической специфичностью (в частности, пролилолигопептидазы) обозначают как "дипептидилпептидаза(ы)". "Дипептидилпептидаза IV" означает CD26. В контексте настоящего описания термин "пептид" или "олигопептид" относится к двум или нескольким аминокислотам, которые соединены амидными связями. При упоминании в связи с терапевтическим соединением D, термин пептид или олигопептид относится к двум или нескольким аминокислотам, которые соединены амидными связями, источником которых являются группы СООН пептида иNH2 или NH группы терапевтического соединения D или линкера, присоединенного к терапевтическому соединению. Длина пептида обозначается греческими буквами, предшествующими слову -пептид (дипептид, трипептид, тетрапептид, пентапептид, гексапептид, гептапептид, октапептид, нонапептид, декапептид и т.д.). В настоящем изобретении предлагается новая технология получения пролекарств, которая основана на связывании пептида с терапевтическим агентом, при этом амидная связь конъюгата способна расщепляться под действием дипептидилпептидазы, такой как CD26. В этом случае растворимость, биодоступность и эффективность терапевтического соединения D в целом может модулироваться более значительно. Гликопротеин CD26 на поверхности лимфоцитов относится к уникальному классу ассоциированных с мембранами пептидаз. Он отличается спектром разнообразных функциональных свойств и идентичен дипептидилпептидазе IV (DPP IV, ЕС 3.4.14.5). DPP IV является членом семейства пролилолигопептидаз(POP; EC 3.4.21.26), группы атипичных серинпротеиназ, способных гидролизовать пролильную связь. Имеющая 766 аминокислот в длину CD26 заякоривается к бислойной липидной мембране клетки с помощью одиночного гидрофобного сегмента и имеет короткий цитоплазматический хвост из шести аминокислот (Abbott et al., Immunogenetics. 1994, 40: 331-338). Мембранный якорь присоединен к большой внеклеточной гликозилированной области, обогащенной цистеином области и С-концевому каталитическому домену (Abott et al., см. выше). CD26 сверхэкспрессируется в эпителиальных клетках (в частности,проксимальных почечных канальцах, кишечнике) и в нескольких типах эндотелиальных клеток и фибробластах, а также в субпопуляциях лейкоцитов (Hegen, M. в: Leukocyte Typing VI. Kishimoto, Т., ed. Garland Publishing, 1997, p. 478-481). CD26 в виде растворимой формы содержится в семенных жидкостях,плазме и спинно-мозговой жидкости. У него отсутствует внутриклеточный хвост и трансмембранная область (De Meester et al., Rev. Immunol. Today. 1999, 20: 367-375). Помимо своей экзопептидазной активности CD26 специфично связывается с несколькими белками вне своего субстрат-связывающего сайта, в частности с аденозиндеаминазой (Trugnan et al. In: Cell-Surface Peptidases in Health and Disease.CD26 наделена интересной (дипептидил)пептидазной каталитической активностью и обладает высокой селективность по отношению к пептидам с пролином или аланином в предпоследнем положении Nконца разнообразных природных пептидов. Несколько цитокинов, гемапоэтических факторов роста, нейропептидов и гормонов имеют общий фрагмент X-Pro или X-Ala на своем N-конце, и было показано, что они являются эффективными субстратами для фермента (обзор в De Meester et al., Rev. Immunol. Today. 1999, 20: 367-375 и Mentlein, Regul.Pept. 1999, 85: 8-24). Вещество Р является примером природного пептида с 11 аминокислотными остатками, содержащего последовательность Arg-Pro-Lys-Pro [SEQ ID NO:1, последовательность с номером идентификации: 1] на его Н-конце и способного расщепляться под действием CD26 в активный гептапептид путем постадийного высвобождения Arg-Pro и Lys-Pro (Ahmad et al., Pharmacol. Exp. Ther. 1992,260: 1257-1261). CD26 может отрезать дипептиды от очень маленьких природных пептидов, в частности от пентапептида энтеростатина (Val-Pro-Asp-Pro-Arg) [SEQ ID NO:2] (Bouras et al., Peptides. 1995, 16: 399-405), до больших пептидов [включая хемокины RANTES, и SDF-1, и IP-10 (68-77 аминокислот)],-7 009727 содержащих, соответственно, последовательности Ser-Pro, Lys-Pro и Val-Pro на их аминовом концеet al., FEBS Lett. 1998, 431: 236-240; Proost et. al., FEBS Lett. 1998, 432: 73-76). Хотя для CD26 наблюдали сравнительно ограниченную специфичность по отношению к субстратам(предпоследний Pro или Ala), более низкие скорости иногда наблюдали в тех случаях, когда предпоследними аминокислотами вместо Pro или Ala являлись Gly, Ser, Val или Leu (De Meester et al., см. ссылку,приведенную выше). Кроме того, тип концевой аминокислоты имеет значение для окончательной каталитической эффективности CD26. Наблюдается уменьшение предпочтения при переходе от гидрофобных (в частности, алифатических: Val, Ile, Leu, Met и ароматических: Phe, Tyr, Trp) к основным (в частности, Lys, Arg, His), к нейтральным (в частности, Gly, Ala, Thr, Cys, Pro, Ser, Gln, 7Asn) и кислым (в частности, Asp, Glu) аминокислотам в качестве предпочтительной первой аминокислоты у аминового конца для эффективного разрезания пептида под действием CD26 (De Meester et al., см. ссылку, приведенную выше). Кроме того, распознаются также неприродные аминокислоты. Наблюдаемое под действиемCD26 двойное усечение выделяемого макрофагом хемокина (MDC), при котором последовательно теряется Gly1-Pro2, а затем Tyr3-Gly4, заставляет предположить, что активность CD26 по отношению к субстрату может быть менее ограничена, как обычно полагают, лишь предпоследними Pro или Ala (Proost, P.et al., J. Biol. Chem. 1999, 274: 3988-3993). Были также идентифицированы многие другие гидролазы (ЕС 3), в частности пептидазы (ЕС 3.4),более конкретно аминопептидазы (ЕС 3.4.11), такие как пролиламинопептидаза (ЕС 3.4.11.5) и X-Pro аминопептидаза (ЕС 3.4.11.9). Кроме того, помимо CD26, существуют другие дипептидазы (ЕС 3.4.13),пептидилдипептидазы (ЕС 3.4.15) и дипептидилпептидазы (ЕС 3.4.14, эта ЕС-группа включает также трипептидилпептидазы). Дипептидилпептидаза I (ЕС 3.4.14.1) встречается в лизосомах и отщепляет дипептид от пептида с консенсусной последовательностью X1-X2-X3, за исключением случая, когда X1 обозначает Arg или Lys, а X2 или X3 обозначает Pro. Дипептидилпептидаза II (ЕС 3.4.14.2) представляет собой лизосомальную пептидазу, которая обладает максимальной активностью при кислом pH и высвобождает дипептиды из олигопептидов (предпочтительно трипептидов) с последовательностью X1-X2-X3,где X2 предпочтительно обозначает Ala или Pro. DPP III (EC 3.4.14.4) представляет собой цитозольную пептидазу и отщепляет дипептиды от пептида, содержащего четыре или более остатков дипептидилдипептидазы (ЕС 3.4.14.6). X-Pro дипептидилпептидаза (ЕС 3.4.14.11) представляет собой микробную пептидазу, обладающую схожей с CD26 активностью. Некоторые из них обнаруживаются у людей и других млекопитающих, в то время как другие продуцируются микроорганизмами, такими как дрожжи и грибы. Они в первую очередь различаются аминокислотной последовательностью, а также различаются своей специфичностью при распознании аминокислотных последовательностей. Кроме того, поиск в базе данных DPP IV выявляет новые пролин-специфичные дипептидазы (DPP8, DPP9, DPP10) (Qi et al., Biochem.J. (2003) 373: 179-189). Большинство этих пролин-специфичных дипептидаз встречаются внутри клеток лизосом и активны при кислом значении pH. Лишь DPP IV встречается как белок, прикрепленный к мембране вне клетки, или как секретируемый белок. Таким образом, в соответствии с одним из вариантов осуществления настоящего изобретения соединения по настоящему изобретению способны расщепляться внеклеточной или прикрепленной к мембране дипептидилпептидазой при нейтральном значенииpH. Настоящее изобретение показывает, что пептидильные пролекарственные производные эффективно превращаются в родительское соединение под действием экзодипептидилпептидазной активности CD26. Настоящее изобретение также показывает, что пептидильные пролекарственные производные внеклеточно превращаются в родительское терапевтическое соединение. Поскольку L-валиновый фрагмент может вовлекаться в дипептидильный подход при разработке пролекарств, то указанная технология может оказаться мощным инструментом для того, чтобы сделать липофильные соединения не только заметно более растворимыми в воде и обладающими меньшим связыванием к белку, но и увеличить их пероральную биодоступность и доставку в плазму родительского соединения, а также обеспечить более селективную доставку родительского лекарства в CD26 экспрессирующие клетки. На основании вышеуказанных наблюдений в настоящем изобретении предлагается новая технология получения пролекарств с целью модулирования растворимости, связывания белком плазмы и/или увеличения биодоступности лекарства. В других вариантах осуществления настоящего изобретения разрабатываются пролекарства для более селективного нацеливания лекарства, для усиления доставки лекарств в мозг и лимфатическую систему и/или увеличения периода полураспада лекарства в плазме. В настоящем изобретении предлагаются новые пролекарства, отличающиеся тем, что указанные пролекарства способны расщепляться дипептидилпептидазой CD26 или другими ферментами, обладающими той же активностью и протеолитической специфичностью, что и CD26. В предпочтительном варианте осуществления настоящего изобретения пролекарства по настоящему изобретению представляют собой конъюгаты пептид-лекарство и их производные, которые включают аминокислотную последовательность, содержащую сайты расщепления для дипептидилпептидаз, таких как CD26. Таким образом, в настоящем изобретении предлагается также терапевтическая композиция пролекарства, включающая тера-8 009727 певтическое соединение D, связанное с пептидом посредством амидной связи, которая специфично расщепляется дипептидилпептидазами, такими как CD26. Терапевтическое соединение D может быть связано с карбоксильной группой аминокислоты как непосредственно, так и посредством связующей группы. В предпочтительном варианте осуществления настоящего изобретения терапевтическое соединениеD и пептид непосредственно связаны с помощью амидной связи. Терапевтическое соединение D может иметь свободную аминогруппу (первичную или вторичную), которая может связываться с карбоксильной группой аминокислоты, более предпочтительно, с -карбоксильной группой. В другом предпочтительном варианте осуществления настоящего изобретения терапевтическое соединение D и пептид связаны посредством линкера, при этом линкер может быть непептидного или пептидного типа. Если связь между терапевтическим соединением D и пептидом осуществляется посредством линкера, то соединение линкера с первой аминокислотой преимущественно осуществляется с помощью амидной связи. Линкер может быть присоединен к терапевтическому соединению D посредством связи любого типа и с помощью химических групп, известных специалистам, более предпочтительно - с помощью ковалентной связи. Линкер может сохраняться у терапевтического соединения D неограниченное количество времени после расщепления или же может быть впоследствии удален либо с помощью дальнейших реакций с агентами, имеющимися у млекопитающего, либо на стадии саморасщепления. Внешние агенты, которые могут оказывать воздействие на расщепление линкера, включают ферменты, белки, органические и неорганические реагенты, протоны и другие агенты. В тех вариантах осуществления настоящего изобретения,когда линкер остается присоединенным к лекарству, линкер может быть любой группой, которая не ингибирует в заметной степени активность лекарства после расщепления пептида. В других вариантах осуществления настоящего изобретения линкер является саморасщепляющимся. Саморасщепляющиеся линкеры являются такими линкерами, которые склонны отделяться от лекарства после расщепления пептида под действием дипептидилпептидазы, такой как CD26. Механизмы, которые лежат в основе саморасщепления линкеров, являются, например, внутримолекулярной циклизацией или спонтанным SN1 сольволизом и высвобождением лекарства в процессе расщепления пептида. Некоторые примеры линкеров представлены авторами Atwell et al. (Atwell et al., J. Med. Chem. 1994, 37: 371-380). Линкеры в общем случае включают первичные амины, которые образуют амидные связи с карбоксильным концом пептида. Линкеры могут также содержать карбоновую кислоту, которая образует амидную связь с первичным амином лекарства. Линкеры могут связываться с лекарством с использованием одной или нескольких реакций, выбранных из реакций, доступных специалисту в данной области техники. В одном из вариантов осуществления настоящего изобретения протеазой, которая может применяться для протеолиза пролекарства, является CD26. Полученные экспериментальные данные свидетельствуют о том, что в процессе расщепления CD26 принимает во внимание лишь дипептидную структуру. Активности фермента не мешает присутствие терапевтического соединения D непосредственно вслед за амидной связью между пролином и лекарственной молекулой. Аналогично, необязательно наличие дополнительного пептида или других молекул линкеров между дипептидом и лекарством. Более того, благодаря экспрессии в тканях (как в раковых, так и нормальных тканях) различных органов (от высоких уровней к низким уровням: почка, легкое, надпочечник, тощая кишка, печень, околоушная слюнная железа, селезенка, яичко, а также кожа, сердце, поджелудочная железа, мозг, спинной мозг, сыворотка) и клетках различных типов (таких как тимоциты, клетки эндотелия, лимфоциты, клетки микроглии) может быть рассмотрен ряд применений, в том числе ряд терапевтических применений. Скорость протеолиза дипептида можно модулировать путем модифицирования аминоконцевой аминокислоты и/или второй аминокислоты. Одновременно или независимо от модулирования гидролиза могут быть модифицированы физико-химические свойства дипептидного пролекарства. Терапевтические соединения, которые могут использоваться в пролекарствах по настоящему изобретению, включают любые лекарства (за исключением белковых или пептидных лекарств, таких как пептидные гормоны), которые могут прямо и косвенно связываться с пептидом, и при этом конъюгат способен расщепляться под действием дипептидилпептидазы, такой как CD26. В дополнение к известным терапевтическим соединениям настоящее изобретение может быть также применено для новых лекарственных молекул, которые в настоящее время находятся в стадии разработки, или к лекарственным молекулам, которые уже используются в клиниках. В еще одном предпочтительном варианте осуществления настоящего изобретения терапевтическое соединение D представляет собой небольшую органическую молекулу и не является пептидом, белком, интеркалирующим агентом или олигонуклеотидом или их аналогами (такими как HNA, PNA и т.д.). Терапевтические молекулы могут быть активны при сердечно-сосудистых, неврологических болезнях, заболеваниях дыхательных путей, онкологических заболеваниях, расстройствах обмена веществ, могут быть активны в области иммунологии, урологии, при борьбе с инфекциями и воспалениями, а также во всех других областях терапии. Наконец, в еще одном предпочтительном варианте осуществления настоящего изобретения терапевтическое соединение D обладает антивирусной активностью. В наиболее предпочтительном варианте осуществления настоящего изобретения терапевтическое соединение D обладает активностью против ВИЧ. Предпочтительными являются лекарства, содержащие первичные аминогруппы. Присутствие первичной аминогруппы позволяет сформировать амидную связь между лекарством и пептидом. Первичные-9 009727 амины могут сразу же входить в структуру лекарств или они могут быть введены в лекарства с помощью химического синтеза. В соответствии с биофармацевтической системой классификации (BCS) Федерального агентства по лекарственным препаратам (FDA) лекарственные вещества классифицируют следующим образом: Класс I - высокая проницаемость, высокая растворимость; Класс II - высокая проницаемость, низкая растворимость; Класс III - низкая проницаемость, высокая растворимость; Класс IV - низкая проницаемость, низкая растворимость. Каким образом лекарства классифицируют в соответствии с этой системой классификации, описывается в руководстве к BCS. В предпочтительном варианте осуществления настоящего изобретения терапевтические соединения D, которые могут использоваться по настоящему изобретению, выбирают из классов II, III и IV. В настоящем изобретении предлагаются пролекарства, которые способны расщепляться под действием дипептидилпептидаз. Дипептидилпептидазы могут быть выбраны из группы пептидаз (ЕС 3.4), а более конкретно - из группы аминопептидаз (ЕС 4.4.11), таких как пролиламинопептидаза (ЕС 3.4.11.5) иX-Pro аминопептидаза (ЕС 3.4.11.9), из группы дипептидаз (ЕС 3.4.13), пептидилдипептидаз(ЕС 3.4.15) и дипептидилпептидаз (ЕС 3.4.14, эта ЕС-группа включает также трипептидилпептидазы),таких как дипептидилпептидаза I (ЕС 3.4.14.1), II (ЕС 3.4.14.2), III (ЕС 3.4.14.4), IV (ЕС 3.4.14.5), дипептидилдипептидаза (ЕС 3.4.14.6) и X-Pro дипептидилпептидаза (ЕС 3.4.14.11). В предпочтительном варианте осуществления настоящего изобретения пролекарство способно расщепляться под действием дипептидилпептидаз, которые имеются у млекопитающих или, более предпочтительно, у людей. В еще более предпочтительном варианте осуществления настоящего изобретения пролекарство способно расщепляться под действием дипептидилпептидазы IV (CD26), как находящейся на поверхности клетки, так и существующей в растворимой форме, которая содержится в семенных жидкостях, плазме и спинномозговой жидкости. Существование двух различных типов CD26 позволяет использовать пролекарства для активации как на клеточной мембране, так и в имеющихся в организме жидкостях. В соответствии с конкретным вариантом его осуществления настоящее изобретение относится к соединениям-пролекарствам формулы (I) их стереоизомерным формам и их солям,где n обозначает 1, 2, 3, 4 или 5;X выбирают из любой аминокислоты с D- или L-конфигурацией;X и Y в каждом повторе фрагмента [Y-X] выбирают независимо друг от друга и независимо от других повторов;Z обозначает прямую связь или бивалентную насыщенную углеводородную группу с прямой или разветвленной цепью, содержащую от 1 до 4 атомов углерода;R4 обозначает водород или С 1-4 алкильную группу; арильная группа, когда она используется самостоятельно или в комбинации с другой группой, означает фенильную группу, необязательно замещенную одним или несколькими заместителями, каждый из которых индивидуально выбирают из группы, включающей С 1-4 алкильную, гидроксильную группу,С 1-4 алкилоксигруппу,нитрогруппу,цианогруппу,галоген,аминогруппу,моноили ди(С 1-4 алкил)аминогруппу и амидную группу; гетероарильная группа, когда она используется самостоятельно или в комбинации с другой группой,означает моноциклический или бициклический ароматический гетероцикл, содержащий один или несколько гетероатомов кислорода, серы или азота, при этом ароматический гетероцикл необязательно может быть замещен по одному или нескольким атомам углерода заместителем, выбранным из группы,включающей С 1-4 алкильную группу, С 1-4 алкилоксигруппу, аминогруппу, гидроксильную, арильную,амидную группу, моно- или ди(С 1-4 алкил)аминогруппу, галоген, нитрогруппу, гетероциклоалкильную и С 1-4 алкилоксикарбонильную группу и при этом указанный ароматический гетероцикл необязательно мо- 10009727 жет быть замещен по вторичному атому азота С 1-4 алкильной или арилС 1-4 алкильной группой; гетероциклоалкильная группа, когда она используется самостоятельно или в комбинации с другой группой, означает насыщенный или частично ненасыщенный моноциклический или бициклический гетероцикл, содержащий один или несколько гетероатомов кислорода, серы или азота, при этом гетероцикл необязательно может быть замещен по одному или несколькими атомам углерода заместителем,выбранным из группы, включающей С 1-4 алкильную группу, С 1-4 алкилоксигруппу, гидроксильную группу, галоген и оксогруппу и при этом указанный гетероцикл необязательно может быть замещен по вторичному атому азота С 1-4 алкильной или арилС 1-4 алкильной группой. Термин С 1-4 алкил в качестве группы или части группы обозначает моновалентные насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 4 атомов углерода. Примеры подобных С 1-4 алкильных радикалов включают метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, втор-бутильную, трет-бутильную группу и т.п. Термин C1-6 алкил в качестве группы или части группы обозначает моновалентные насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода. Примеры подобных C1-6 алкильных радикалов включают метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, втор-бутильную, трет-бутильную, 2-метилбутильную, пентильную, изоамильную, гексильную, 3-метилпентильную группу и т.п. Термин C1-10 алкил в качестве группы или части группы обозначает моновалентные насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 10 атомов углерода. Примеры подобных C1-10 алкильных радикалов включают примеры C1-6 алкильных радикалов и гептильную, октильную, нонильную, децильную, 3-этилгептильную группу и т.п. Термин С 2-6 алкенил в качестве группы или части группы обозначает моновалентные насыщенные углеводородные радикалы с прямой или разветвленной цепью, имеющие по крайней мере одну двойную связь и содержащие от 2 до 6 атомов углерода. Примеры подобных С 2-6 алкенильных радикалов включают этенильную, пропенильную, 1-бутенильную, 2-бутенильную, изобутенильную, 2-метил-1 бутенильную, 1-пентенильную, 2-пентенильную, 1-гексенильную, 2-гексенильную, 3-гексенильную,3-метил-2-пентенильную группу и т.п. Термин "галоген", когда он используется самостоятельно или в комбинации с другой группой, является общим термином для обозначения фтора, хлора, брома и йода. Термин С 3-7 циклоалкил, когда он используется самостоятельно или в комбинации с другой группой,является общим термином для обозначения циклопропильной, циклобутильной, циклопентильной, циклогексильной и циклогептильной группы. Для терапевтического применения солями соединений-пролекарств по настоящему изобретению являются такие соли, в которых противоион является фармацевтически или физиологически приемлемым. Тем не менее, соли, содержащие фармацевтически неприемлемый противоион, также могут найти применение, например, при приготовлении или очистке фармацевтически приемлемого соединения по настоящему изобретению. Все соли, как фармацевтически приемлемые, так и фармацевтически неприемлемые, входят в объем настоящего изобретения. Фармацевтически приемлемые или физиологически переносимые формы кислотно-аддитивных солей, которые могут образовывать соединения-пролекарства по настоящему изобретению, могут быть легко приготовлены с использованием соответствующих кислот, таких как, например, неорганические кислоты, такие как галогеноводородные кислоты, в частности хлористо-водородная или бромистоводородная кислота, серная, азотная, фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропановая, гидроксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты. И, наоборот, указанные формы кислотно-аддитивных солей могут быть превращены в форму свободного основания путем обработки подходящим основанием. Соединения-пролекарства по настоящему изобретению, содержащие кислый протон, могут быть также превращены в их нетоксичные основно-аддитивные соли с металлами или аминами путем обработки соответствующими органическими и неорганическими основаниями. Подходящие формы солей с основаниями включают, например, аммониевые соли, четвертичные аммониевые соли, соли щелочных и щелочно-земельных металлов, в частности соли лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, например бензатиновые, N-метильные, D-глюкаминовые, гидрабаминовые соли, и соли с аминокислотами, такими как, например, аргинин, лизин и т.п. И, наоборот, указанные формы основно-аддитивных солей могут быть превращены в свободную кислотную форму путем обработки подходящей кислотой. Термин "соли" включает также гидраты и формы присоединения растворителя, которые могут образовывать соединения-пролекарства по настоящему изобретению. Примерами подобных солей являются, например, гидраты, алкоголяты и т.п. Термин "соли" включает также кватернизацию атомов азота в соединениях по настоящему изобретению. Основной азот может быть кватернизован с помощью любого- 11009727 агента, известного специалистам в данной области техники, который включает, например, низшие алкилгалогениды, диалкилсульфаты, галогенпроизводные с длинной цепью и арилалкилгалогениды. Соединения-пролекарства по настоящему изобретению могут также существовать в их таутомерной форме. Предполагается, что подобные формы, хотя они в явном виде не указаны в вышеприведенной формуле, входят в объем настоящего изобретения. В одном из вариантов осуществления настоящего изобретения концевые аминогруппы концевой аминокислоты пептидной связи, образованной фрагментами -(Y-X)n, необязательно могут содержать одну или две кэппинг-группы, выбранные из ацетильной, сукцинильной, бензилоксикарбонильной, глутарильной, морфолинокарбонильной и С 1-4 алкильной группы. В одном из вариантов осуществления настоящего изобретения каждый X независимо выбирают из природных аминокислот. В одном из вариантов осуществления настоящего изобретения каждый X независимо обозначаетL-аминокислоту, выбранную из группы, включающей аланин, аргинин, аспарагин, аспарагиновую кислоту, цистеин, глутамин, глутаминовую кислоту, глицин, гистидин, изолейцин, лейцин, лизин, метионин,фенилаланин, пролин, серин, треонин, триптофан, тирозин или валин. В одном из вариантов осуществления настоящего изобретения каждый Y независимо обозначает пролин, аланин, глицин, серин, валин или лейцин; предпочтительно каждый Y независимо обозначает пролин или аланин. В одном из вариантов осуществления настоящего изобретения n обозначает 1, 2 или 3. В одном из вариантов осуществления настоящего изобретения -(Y-X)n обозначает-(Y-X)1 или 2-Y-Val, более предпочтительно -(Y-X)n обозначает -(Y-X)1 или 2-Pro-Val. В одном из вариантов осуществления настоящего изобретения олигопептид (Y-X)n выстроен из повторов фрагментов (Y-X), выбранных из группы, включающей Pro-Val, Pro-Asp, Pro-Ser, Pro-Lys,Pro-Arg, Pro-His, Pro-Phe, Pro-Ile, Pro-Leu, Ala-Val, Ala-Asp, Ala-Ser, Ala-Lys, Ala-Arg, Ala-His, Ala-Phe,Ala-Ile и Ala-Leu. В одном из вариантов осуществления настоящего изобретения R1 обозначает гетероциклоалкилоксигруппу, гетероарильную группу, гетероарилС 1-4 алкилоксигруппу, арильную группу или арилоксиС 1-4 алкильную группу. В одном из вариантов осуществления настоящего изобретенияR1 обозначает гексагидрофуро[2,3-b]фуранилоксигруппу, тетрагидрофуранилоксигруппу, хинолинильную группу, тиазолилметилоксигруппу, арильную, арилоксиметильную группу. В одном из вариантов осуществления настоящего изобретенияR1 обозначает гексагидрофуро[2,3-b]фуран-3-илоксигруппу, тетрагидрофуран-3-илоксигруппу, хинолин-2-ильную группу, тиазол-5-илметилоксигруппу, 3-гидрокси-2-метил-1-фенильную, 2,6-диметилфеноксиметильную группу. В одном из вариантов осуществления настоящего изобретения(3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-илоксигруппу,(3S)-тетрагидрофуран-3-илоксигруппу,хинолин-2-ильную группу,тиазол-5-илметилоксигруппу,3-гидрокси-2-метил-1-фенильную,2,6-диметилфеноксиметильную группу. Представляющими интерес группами соединений являются такие группы соединений формулы (I),для которых применимы одно или несколько следующих ограничений:Y обозначает пролин; каждый X независимо выбирают из валина, аспарагиновой кислоты, лизина или пролина;R4 обозначает водород. Представляющими интерес соединениями являются такие соединения формулы (I) или любые из указанных подгрупп соединений формулы (I), в которых R2 обозначает фенилметильную группу. Представляющими интерес соединениями являются такие соединения формулы (I) или любые из указанных подгрупп соединений формулы (I), в которых R3 обозначает С 1-4 алкильную группу, в частности R3 обозначает изобутильную группу. Представляющими интерес соединениями являются такие соединения формулы (I) или любые из указанных подгрупп соединений формулы (I), в которых R4 обозначает водород. Представляющими интерес соединениями являются такие соединения формулы (I) или любые из указанных подгрупп соединений формулы (I), в которых R2 обозначает фенилметильную группу, R3 обозначает изобутильную группу и R4 обозначает водород. Представляющими интерес соединениями являются такие соединения формулы (I) или любые из указанных подгрупп соединений формулы (I), в которых Z обозначает метилен.- 12009727 Представляющими интерес соединениями являются такие соединения формулы (I) или любые из обозначает указанных подгрупп соединений формулыR1 гексагидрофуро[2,3-b]фуранилоксигруппу, тетрагидрофуранилоксигруппу, хинолинильную группу, тиазолилметилоксигруппу, арильную, арилоксиметильную группу; R2 обозначает фенилметильную группу,R3 обозначает изобутильную группу и R4 обозначает водород. Конкретную группу соединений составляют такие соединения формулы (I) или любые из указанных подгрупп соединений формулы (I), в которыхY обозначает пролин или аланин; каждый X независимо выбирают из встречающейся природной аминокислоты;Z обозначает прямую связь или метилен;R1 обозначает гетероциклоалкилоксигруппу, гетероарильную группу, гетероарилС 1-4 алкилоксигруппу, арильную или арилоксиС 1-4 алкильную группу;R4 обозначает водород. Кроме того, конкретную группу соединений составляют такие соединения формулы (I) или любые из указанных подгрупп соединений формулы (I), в которыхY обозначает пролин; каждый X независимо выбирают из встречающейся природной аминокислоты;R4 обозначает водород. Предпочтительными пролекарствами являются такие пролекарства, в которых терапевтическим соединением является и их солевые формы. В частности, аминоконцевую часть пептида в пролекарстве составляют X-Pro, X-Ala, X-Gly, X-Ser,X-Val или X-Leu, где X обозначает любую аминокислоту или ее изомеры (т.е. имеющие L- илиD-конфигурацию). Другие дипептиды, в которых вторую позицию занимают гидроксипролин, дигидроксипролин, тиазолидинкарбоновая кислота (тиопролин), дегидропролин, пипеколиновая кислота (Lгомопролин), азетидинкарбоновая кислота и азиридинкарбоновая кислота, также способны расщепляться под действием CD26. В предпочтительном варианте осуществления настоящего изобретения аминоконцевая часть пептида содержит X-пролин или X-аланин. В таком случае аминокислоты могут быть выбра- 13009727 ны из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глутамина, глутаминовой кислоты, глицина, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина,треонина, триптофана, тирозина, валина и их производных. Могут быть также включены модифицированные (в частности, гидроксипролин) аминокислоты и аминокислоты неприродного происхождения. В другом предпочтительном варианте осуществления настоящего изобретения длина пептида составляет от 2 до 10 аминокислот, а потому может содержать 2, 3, 4, 5, 6, 7, 8, 9 или 10 аминокислот. В другом предпочтительном варианте осуществления настоящего изобретения пептид содержит повторяющиеся фрагменты [X-Y]n, в которых X обозначает любую аминокислоту, Y выбирают из Pro, Ala,Gly, Ser, Val или Leu, a значение n выбирают из 1, 2, 3, 4 или 5. В другом более предпочтительном варианте осуществления настоящего изобретения указанный пептид представляет собой дипептид. В еще более предпочтительном варианте осуществления настоящего изобретения дипептид представляет собой Lys-Pro. В другом еще более предпочтительном варианте осуществления настоящего изобретения аминокислоты имеют L-конфигурацию. Аминовый конец пептида может также содержать обычные кэппинггруппы. Подобные кэппинг-группы включают ацетильную, сукцинильную, бензилоксикарбонильную,глутарильную, морфолинокарбонильную, метильную и многие другие группы, известные из данной области техники. Специалисты могут провести замены с тем, чтобы получить пептиды, обладающие лучшим профилем, касающимся растворимости, биодоступности и способности к нацеливанию конъюгата. Таким образом, изобретение включает вышеописанные пептидные последовательности, а также их аналоги и производные, при условии, что конъюгаты сохраняют способность расщепляться под действием дипептидилпептидазы, такой как CD26. В другом варианте осуществления настоящего изобретения пептиды в линкере по настоящему изобретению состоят из одного или нескольких повторяющихся дипептидов X-Y со структурой [X-Y]n,способных расщепляться под действием CD26, включают одну или несколько аминокислот между пептидом, способным отщепляться под действием CD26, и пролекарством и имеют общую структуру[X-Y]n-Am. Здесь A обозначает любую аминокислоту. Связь между олигопептидом [X-Y]n и последующей аминокислотой A является амидной связью, которая позволяет осуществить протеолиз под действием CD26. Связь между двумя аминокислотами A и между аминокислотой A и пролекарством может быть как амидной связью, так и сложноэфирной связью. Величина m может составлять от 1 до 15. В одном из вариантов осуществления настоящего изобретения m равно 1 и m может быть отделено от пролекарства гидролизом с помощью эстеразы или аминопептидазы. В другом варианте осуществления настоящего изобретения олигопептид [X-Y]n, способный расщепляться под действием CD26, представляет собой пептид, в котором по крайней мере один X обозначает гидрофобную или ароматическую аминокислоту, или же в котором по крайней мере один X обозначает нейтральную или кислую аминокислоту, или же в котором по крайней мере один X обозначает основную аминокислоту. Для того чтобы добиться желаемого эффекта, с целью модулировать гидрофобность и/или скорость протеолиза более длинных пептидов (n равно 3, 4, 5), более чем один X должен иметь специфический тип боковых цепей. Кроме того, на скорость протеолиза, гидрофобность, растворимость, биодоступность и эффективность пролекарства может оказать влияние выбор Y. Наконец, в еще одном из вариантов осуществления настоящего изобретения пептиды с общей структурой [X-Y]n представляют собой тетрапептиды или гексапептиды со структурой, выбранной из группы XF-Y-XF-Y, XF-Y-XS-Y, XS-Y-XF-Y, XS-Y-XS-Y, XB-Y-X-Y, X-Y-XB-Y и XB-Y-XB-Y,или гексапептид со структурой, выбранной из группы XF-Y-XF-Y-XF-Y, XS-Y-XF-Y-XF-Y,XF-Y-XS-Y-XF-Y, XF-Y-XF-Y-XS-Y, XF-Y-XS-Y-XS-Y, XS-Y-XF-Y-XS-Y, XS-Y-XS-Y-XF-Y иXB-Y-XB-Y-XB. Здесь F означает быстрый, a XF обозначает аминокислоту, которая получается при быстром высвобождении дипептида под действием CD26 (например, кислые и нейтральные аминокислоты,такие как аспарагиновая кислота и глутаминовая кислота); S означает медленный, a XS обозначает аминокислоту, которая вызывает медленное высвобождение дипептида под действием CD26 (например, заряженные и нейтральные аминокислоты); В означает основный, а XВ обозначает основную аминокислоту (Lys, Arg и His), которая приводит к умеренному высвобождению содержащего заряд или гидрофильного дипептида. Подобные комбинации позволяют осуществить специально приготовленные комбинации пептидов,которые придают пролекарству вполне определенную скорость разложения вместе с вполне определенной гидрофобностью. Например, разложение гидрофобного пролекарства с дипептидом Tyr/Phe-Pro может быть отсрочено за счет присутствия дополнительного аминоконцевого дипептида Gly-Pro, что приводит к образованию тетрапептидного пролекарства Gly-Pro-Tyr/Phe-Pro [SEQ ID NO: 3]. Гидрофобность может быть еще более усилена добавлением дополнительного дипептида Tyr/Phe-Pro, что приводит к получению гексапептидного пролекарства Gly-Pro-Tyr/Phe-Pro-Tyr/Phe-Pro [SEQ ID NO: 4]. Если необходимо получить содержащее заряд пептидное пролекарство, характеризующееся медленной скоростью- 14009727 высвобождения, то Asp-Pro-Lys-Pro [SEQ ID NO: 5] может оказаться более предпочтительным по сравнению с Gly-Pro. Специалист может подобрать другие комбинации, в которых тетрапептид или гексапептид позволяет модулировать растворимость или скорость разложения пептидного пролекарства под действием CD26. Для других целей пролин может быть заменен аланином. Физико-химические свойства и скорость разложения нерасщепленного, частично расщепленного и полностью расщепленного пролекарства можно оценить путем определения времени его удерживания в методе хроматографии с обращенной фазой. Настоящее изобретение показывает, что пептидильные производные пролекарства формулы (I) эффективно превращаются в родительское терапевтическое соединение формулы (Ia) где значения R1, R2, R3, R4 и Z определены для соединений формулы (I) под действием экзодипептидилпептидазной активности CD26. Известно, что указанные терапевтические соединения формулы (Ia) обладают ингибирующей активностью по отношению к ВИЧ-протеазе и описаны в ЕР 656887, ЕР 715618, ЕР 810209, US 5744481,US 5786483, US 5830897, US 5843946, US 5968942, US 6046190, US 6060476, US 6248775, WO 99/67417,и все они включены в настоящее описание посредством ссылки. Благодаря тому, что терапевтические соединения формулы (Ia) являются ингибиторами репликации ВИЧ, соединения-пролекарства формулы (I) применимы при лечении ВИЧ-инфицированных теплокровных животных, в частности людей. Связанные с ВИЧ состояния, которые предупреждают или лечат с помощью соединений по настоящему изобретению, включают СПИД (AIDS), СПИД-ассоциированный комплекс (ARC), прогрессирующую генерализованную лимфаденопатию (PGL), а также хронические заболевания центральной нервной системы, вызываемые ретровирусами, такими как, например, ВИЧопосредованная деменция и рассеянный склероз. Таким образом, соединения-пролекарства по настоящему изобретению могут применяться в качестве лекарственных средств против вышеуказанных состояний или применяться в способе лечения вышеуказанных состояний. Указанное применение в качестве лекарственного средства или применение в способе лечения включает системное введение эффективного терапевтического количества соединения формулы (I) ВИЧ-инфицированным теплокровным животным, в частности, ВИЧ-инфицированным людям. Следовательно, соединения-пролекарства по настоящему изобретению могут быть использованы при изготовлении лекарственных средств, применимых для лечения состояний, связанных с ВИЧ-инфекцией. Термин "стереохимически изомерные формы соединений" по настоящему изобретению, который ранее использован в настоящем описании, обозначает все возможные соединения, образованные теми же атомами, соединенными той же самой последовательностью связей, однако имеющие различные трехмерные структуры, не являющиеся взаимозаменяемыми, которые могут иметь соединения по настоящему изобретению. Если иное не упоминается или не оговаривается, химическое обозначение соединения охватывает смесь всех возможных стереохимически изомерных форм, которые может иметь указанное соединение. Указанная смесь может включать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Предполагается, что в объем настоящего изобретения входят все стереохимически изомерные формы соединений по настоящему изобретению как в чистой форме, так и в смеси друг с другом. Чистые стереоизомерные формы соединений и промежуточных соединений, которые рассматриваются в настоящем описании, определяются как изомеры, в значительной степени свободные от других энантиомерных или диастереомерных форм той же самой основной молекулярной структуры указанных соединений или промежуточных соединений. В частности, термин "стереоизомерно чистый" относится к соединениям или промежуточным соединениям, обладающим стереоизомерным избытком, составляющим по крайней мере 80% (т.е. минимум 80% одного изомера и максимум 20% других возможных изомеров) вплоть до стереоизомерного избытка 100% (т.е. 100% одного изомера и отсутствие другого), в частности, относится к соединениям или промежуточным соединениям, обладающим стереоизомерным избытком от 90 до 100%, более предпочтительно обладающим стереоизомерным избытком от 94 до 100% и наиболее предпочтительно обладающим стереоизомерным избытком от 97 до 100%. Термины"энантиомерно чистый" и "диастереомерно чистый" следует понимать аналогично, однако в этом случае они относятся к энантиомерному и диастереомерному избыткам соответственно рассматриваемой смеси. Чистые стереоизомерные формы соединений и промежуточных соединений по настоящему изобретению могут быть получены с помощью известных из области техники методик. Например, энантиомеры могут быть отделены друг от друга с помощью селективной кристаллизации их диастереомерных солей с оптически активными кислотами. В альтернативном случае энантиомеры могут быть разделены хроматографическими методами с использованием хиральных неподвижных фаз. Указанные чистые стереохи- 15009727 мически изомерные формы могут быть также получены из соответствующих чистых стереохимически изомерных форм подходящих исходных соединений, при условии, что реакция протекает стереоспецифично. Если необходим конкретный изомер, то указанное соединение предпочтительно синтезируют с использованием стереоспецифических методов синтеза. В указанных методах синтеза преимущественно используют энантиомерно чистые исходные соединения. Диастереомерные рацематы соединений по настоящему изобретению могут быть отдельно получены известными способами. Подходящими способами физического разделения, которые могут быть преимущественно использованы, являются, например, селективная кристаллизация и хроматография, в частности колоночная хроматография. Соединения могут содержать один или несколько асимметрических центров и, таким образом, могут существовать в виде различных стереоизомерных форм. Абсолютная конфигурация каждого асимметрического центра, который может присутствовать в соединениях, может быть указана с помощью стереохимических идентификаторов R и S, при этом указанные обозначения R и S соответствуют правилам, приведенным в Pure Appl. Chem. 1976, 45, 11-30. В одном из вариантов осуществления настоящего изобретения атом углерода, несущий заместительR2, имеет конфигурацию "S", а соседний атом углерода, несущий гидроксильный заместитель, имеет конфигурацию "R". Подразумевается также, что настоящее изобретение включает все изотопы атомов, содержащихся в соединениях по настоящему изобретению. Изотопы включают такие атомы, которые имеют тот же атомный номер, но другие массовые числа. В качестве общего примера, не ограничивающего настоящее изобретение, изотопы водорода включают тритий и дейтерий. Изотопы углерода включают С-13 и С-14. В настоящем изобретении далее предлагается способ получения пролекарства формулы (I), где пролекарство способно расщепляться дипептидилпептидазой, такой как CD26. Указанный способ получения пролекарства формулы (I) включает стадию связывания терапевтически активного лекарства формулы (Ia) и пептида. В более предпочтительном варианте осуществления настоящего изобретения терапевтически активное лекарство или пептид на первой стадии функционализуют с тем, чтобы можно было позднее связывать терапевтическое соединение формулы (Ia) и пептид посредством амидной связи. Специалистам известны многие приемлемые способы связывания карбоксильных групп и аминогрупп с образованием амидных связей. В общем случае терапевтические соединения формулы (Ia) могут быть получены, как описано в ЕР 656887, ЕР 715618, ЕР 810209, US 5744481, US 5786483, US 5830897, US 5843946, US 5968942,US 6046190, US 6060476, US 6248775, WO 99/67417. Общая химия пептидов не представляет сложности для специалиста в данной области техники и включает связывание природных аминокислот, с целью получения пептида. Известно несколько подходов к проведению химического процесса, из которых наиболее часто применяются химические способы с использованием флуоренилметилоксикарбонила (Fmoc) или трет-бутилоксикарбонила (Boc). FieldsG.B. приводит подробное описание химических методов для пептидов, которые могут быть применены для связывания аминокислот друг с другом или с терапевтическим соединением D (Fieldsin Methods inMolecular Biology, vol. 35: Peptide Synthesis Protocols Humana Press Inc.: Totawa. 1994, p. 17-27). Могут быть использованы твердофазные химические методы, а также химические методы с использованием растворов (AthertonSheppard Solid Phase Peptide Synthesis IRL Press: Oxford-New York-Tokyo, 1989). Может потребоваться использование методов защиты, при этом функциональные группы терапевтического соединения, которые не могут взаимодействовать в процессе получения пролекарства, блокируются путем связывания их защитной группой. В частности, соединения-пролекарства формулы (I) могут быть получены из исходных соединений формулы (Ia) с помощью хорошо известных методов химии пептидов. Схема 1- 16009727 Например, аминокислоты могут быть связаны с терапевтическим соединением с образованием пептидных связей, как показано на схеме 1. Указанную реакцию связывания можно проводить в подходящем инертном для данной реакции растворителе, таком как N,N-диметилформамид, ацетонитрил, дихлорметан, тетрагидрофуран, или в любом другом растворителе, который растворяет реагенты, используя защищенную по аминогруппе аминокислоту формулы PG-Y-OH, где PG (защитной группой) может быть, например, Boc (трет-бутилоксикарбонил), Cbz (бензилоксикарбонил) или Fmoc, в присутствии связывающего агента, такого как DCC (дициклогексилкарбодиимид) или EDCI (гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодимида) и HOAt (1-гидрокси-7-азабензотриазол) или функционально эквивалентное им соединение. У полученного указанным образом пептида защиту можно удалить с помощью обычных методов удаления защиты, таких как, например, удаление защитной группы с помощью трифторуксусной кислоты в дихлорметане. Указанные стадии связывания и последующего удаления защитной группы можно повторить, используя PG-Y-OH в качестве реагента для получения требуемой пептидной связи. Некоторые из аминокислот, такие как, например, лизин и аспарагиновая кислота, могут потребовать второй защитной группы и их можно представить формулой PG-(XPG)-ОН или PG-(YPG)-ОН. Кроме того, в указанных выше реакциях могут применяться реагенты, имеющие формулуPG-X-Y-OH, или PG-(X)n-OH, или PG-(X-Y)n-OH. В представленных схемах получения продукты реакции могут быть выделены из реакционной среды и, если необходимо, подвергнуты дальнейшей очистке с помощью общеизвестных из области техники методов, таких как, например, экстракция, кристаллизация, дистилляция, растирание в растворителе и хроматография. Соединения формулы (I), процесс получения которых описан выше, могут быть синтезированы в виде смеси стереоизомерных форм, в частности в виде рацемических смесей энантиомеров, которые могут быть отделены друг от друга с помощью следующих известных из области техники способов разделения. Рацемические соединения формулы (I) могут быть превращены в соответствующие диастереомерные солевые формы по реакции с подходящей хиральной кислотой. Указанные диастереомерные солевые формы затем разделяют, например, селективной или дробной кристаллизацией, а энантиомеры выделяют из них с помощью щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает жидкостную хроматографию с применением хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы могут быть также получены из соответствующих чистых стереохимически изомерных форм подходящих исходных соединений при условии, что реакция протекает стереоспецифично. Если требуется специфический изомер, то указанное соединение предпочтительно будет синтезироваться с применением стереоспецифических способов получения. В указанных методах преимущественно используют энантиомерно чистые исходные соединения. Существуют способы прогнозирования растворимости соединения. Например, в J. Chem. Inf. Comput. Sci 1998 May-June, 38(3): 450-6 описывают прогнозирование растворимости лекарств в воде на основе молекулярной топологии и с помощью моделирования с использованием нейронных сетей. В самом деле, могут быть определены все параметры, касающиеся растворимости и биодоступности (pKa, коэффициент перераспределения и т.д.). В монографии "Drug Bioavailability: Estimation of Solubility, Permeability, Absorption and Bioavailability" приводится исчерпывающий обзор указанных параметров и их определение или прогнозирование (ISBN 352730438X). Коэффициенты перераспределения являются мерой липофильности. Выраженные в численном виде как значения "log Р", они представляют собой отношение между концентрациями веществ в двух несмешивающихся друг с другом фазах, таких как вода/октанол или вода/липосомы, и могут быть легко рассчитаны. Вещества с большими значениями log P лучше растворяются в жирах и маслах, чем в воде. Это усиливает их способность проникать через липидные (на основе жиров) мембраны в теле за счет пассивной диффузии и, тем самым, увеличивает их потенциал для абсорбции. Многие лекарства имеют значения log P между единицей и четырьмя, что делает их пригодными для использования в пероральных способах доставки. Лекарства с большими значениями log P обычно плохо растворимы в воде. Они могут растворяться в липидах, однако они не могут растворяться в желудочно-кишечном тракте, а потому они не могут диффундировать через стенки кишечника. Если они попадают в мембрану, то они могут захватываться там и в результате вызывать токсическое действие. Коэффициент перераспределения также может быть рассчитан. Способ прогнозирования для log P,разработанный компанией Molinspiration (miLog P1.2), основывается на влиянии групп. Величину влияния групп получают путем сопоставления рассчитанных величин log P с экспериментально полученными величинами log P с использованием обучающего набора, который включает несколько тысяч молекул,напоминающих лекарственные молекулы. Описание способа можно найти на сайтеwww.molinspiration.com/services/logp.html (QSAR 15, 403, 1996). Имеется множество других программ для определения величин log P. В конкретном варианте осуществления настоящего изобретения пролекарства формулы (I) могут применяться в качестве лекарственного средства. В другом варианте осуществления настоящего изобретения пролекарства формулы (I) могут применяться при изготовлении лекарства для предупреждения- 17009727 или лечения ВИЧ-инфекции. В настоящем изобретении предлагается также способ предупреждения или лечения ВИЧ-инфекции путем введения пролекарства формулы (I) по настоящему изобретению. Пролекарства формулы (I) можно вводить любому хозяину, в том числе человеку, отличному от человека животному и млекопитающим в количестве, эффективном для предупреждения или лечения ВИЧ-инфекции. С целью дальнейшей оптимизации фармакокинетического профиля пролекарств формулы (I) они могут вводиться в сочетании с подходящими средствами доставки (в частности, в виде микрокапсул,микросфер, биоразлагаемых полимерных пленок, систем доставки на основе липидов, таких как липосомы и липидные пены, в виде вязких жидких лекарственных средств и способных поглощаться механических барьеров), пригодными для поддержания необходимых концентраций пролекарств или терапевтического соединения D по месту распространения болезни. Пролекарство или "лекарственное средство" может вводиться с помощью подходящего способа, известного специалисту. Способы введения терапевтических агентов, известные из области техники, включают парентеральное, например внутривенное (в частности, для ингибиторов антител), внутрибрюшинное, внутримышечное, внутрикожное введение, и эпидермальное, включая подкожное и внутрикожное,пероральное введение или нанесение на поверхность слизистых оболочек, в частности интраназальное введение с использованием ингаляций аэрозольных суспензий, имплантацию в мышцы или другие ткани субъекта. Предусматривается также использование суппозиториев и местных, локально наносимых препаратов. В зависимости от пути и места введения можно рассматривать пролекарства с более гидрофобными или более гидрофильными пептидными фрагментами. В соответствии с настоящим изобретением пролекарства формулы (I) вводят в количестве, эффективном для того, чтобы предупредить, ослабить или вылечить ВИЧ-инфекцию. Наиболее эффективные способы введения и схемы дозировки для лекарств или "лекарственных средств" в способах по настоящему изобретению зависят от тяжести ВИЧ-инфекции, здоровья пациента,предыстории предшествующего лечения, возраста, веса, роста, пола, восприимчивости к лечению и от заключения лечащего врача. Таким образом, назначаемое количество пролекарства, а также количество и время проведения последующих введений пролекарства определяются опытным специалистом в области медицины, который проводит терапию в зависимости от ответной реакции на лечение индивидуального субъекта. Вначале опытные практики могут легко определить указанные параметры, проводя испытания на безопасность и эффективность с помощью тестов на животных моделях, а также на людях при проведении клинических испытаний составов пролекарств. После введения эффективность терапии с применением пролекарств оценивают различными способами, включая анализ клинической картины. Таким образом, соединения по настоящему изобретению могут применяться для животных, преимущественно для млекопитающих, в частности для людей, как самостоятельные фармацевтические средства, в смеси один с другим или в форме фармацевтических препаратов. Далее, настоящее изобретение относится к фармацевтическим препаратам, которые содержат эффективную дозу по крайней мере одного пролекарства формулы (I) вместе с обычными фармацевтически безвредными наполнителями и вспомогательными веществами. Фармацевтические препараты обычно содержат от 0,1 до 90 мас.% пролекарства. Фармацевтические препараты могут быть получены способами, которые знакомы специалисту. С этой целью по меньшей мере одно пролекарство по настоящему изобретению вместе с одним или несколькими твердыми или жидкими фармацевтическими наполнителями и/или вспомогательными средствами и, если необходимо, в комбинации с другими фармацевтически активными соединениями, превращают в подходящую для введения форму или дозировочную форму, которую затем можно использовать в качестве фармацевтического средства при лечении людей или в ветеринарии. Подходящие фармацевтические носители для использования в указанных фармацевтических композициях и составах хорошо известны специалистам, и в объеме настоящего изобретения не существует каких-либо специальных ограничений для их выбора. Подходящими носителями или наполнителями,известными специалисту, являются физиологический раствор, раствор Рингера, раствор декстрозы, раствор Хенка, нелетучие масла, этилолеат, 5% декстрозы в физиологическом растворе, вещества, которые повышают изотоничность (такие как сахара или хлорид натрия) и химическую стабильность, буферы и консерванты. Другие подходящие носители включают любые носители, которые сами не индуцируют продукцию антител, вредных для индивида, получающего композицию, такие как белки, полисахариды,полимолочные кислоты, полигликолевые кислоты, полимерные аминокислоты и сополимеры аминокислот. Они могут также включать добавки, такие как смачиватели, диспергаторы, связующие вещества,адгезивы, эмульгаторы, растворители, покрытия, антибактериальные и противогрибковые агенты (например, фенол, сорбиновую кислоту, хлорбутанол) и т.п., при условии, что они совместимы с практикой применения фармацевтических препаратов, в частности, представлять собой носители и добавки, которые не приносят продолжительного вреда млекопитающим. Фармацевтические композиции по настоящему изобретению могут быть приготовлены любыми известными способами, например путем смешивания до однородного состояния, путем формирования в виде покрытия и/или путем измельчения активных ингредиентов, в одну или в несколько стадий, вместе с выбранным материалом носителя, а если это- 18009727 возможно, то другие добавки, такие как поверхностно-активные агенты, могут быть приготовлены путем микронизации, например, с целью получить их в форме микросфер, которые обычно имеют диаметр приблизительно 1-10 мкм, в частности, для изготовления микрокапсул для контролируемого или замедленного высвобождения активных ингредиентов. Подходящими поверхностно-активными агентами, которые следует использовать в фармацевтических композициях по настоящему изобретению, являются неионные, катионогенные и/или анионогенные вещества, обладающие хорошими эмульгирующими, диспергирующими и/или смачивающими способностями. Подходящие анионогенные поверхностно-активные вещества включают как водорастворимые мыла, так и водорастворимые синтетические поверхностно-активные агенты. Подходящими мылами являются соли щелочных или щелочно-земельных металлов, аммониевые соли незамещенных или замещенных высших жирных кислот (С 10-C22), в частности натриевые или калиевые соли олеиновой или стеариновой кислоты, или смесей природных жирных кислот, получаемых из кокосового и талового масла. Синтетические поверхностно-активные вещества включают натриевые или кальциевые соли полиакриловой кислоты; жирные сульфонаты или сульфаты; сульфонированные производные бензимидазола и алкиларилсульфонаты. Жирные сульфонаты или сульфаты обычно существуют в виде солей щелочных или щелочно-земельных металлов, незамещенных аммониевых солей или аммониевых солей, замещенных алкильными или ацильными радикалами, которые содержат от 8 до 22 атомов углерода, в частности,существуют в виде натриевой или кальциевой соли лигносульфоновой кислоты или додецилсульфоновой кислоты, или смеси сульфатов жирных спиртов, полученных из природных жирных кислот, в виде солей щелочных или щелочно-земельных металлов эфиров серной или сульфоновой кислоты (таких как лаурилсульфат натрия) и сульфоновых кислот аддуктов жирный спирт/оксид этилена. Подходящие сульфонированные производные бензимидазола преимущественно содержат от 8 до 22 атомов углерода. Примерами алкиларилсульфонатов являются натриевые, кальциевые или алканоламиновые соли додецилбензолсульфоновой кислоты или дибутилнафталинсульфоновой кислоты или продукта связывания нафталинсульфоновой кислоты с формальдегидом. Пригодны также соответствующие фосфаты, в частности,соли эфиров фосфорной кислоты и аддукта п-нонилфенола с оксидом этилена и/или оксидом пропилена,или фосфолипиды. Подходящими для указанной цели фосфолипидами являются природные (получаемые из животных или растительных клеток) или синтетические фосфолипиды цефалинового или лецитинового типа, такие как, например, фосфатидилэтаноламин, фосфатидилсерин, фосфатидилглицерин, лизолецитин, кардиолипин, диоктанилфосфатидилхолин, дипальмитоилфосфатидилхолин и их смеси. Подходящие неионные поверхностно-активные вещества включают полиэтоксилированные и полипропоксилированные производные алкилфенолов, жирных спиртов, жирных кислот, алифатических аминов или амидов, содержащих в молекуле по меньшей мере 12 атомов углерода, алкиларенсульфонаты и диалкилсульфосукцинаты, такие как полигликолевые эфирные производные алифатических и циклоалифатических спиртов, насыщенных и ненасыщенных жирных кислот и алкилфенолов, при этом указанные производные преимущественно содержат от 3 до 10 гликолевых эфирных групп, от 8 до 20 атомов углерода в (алифатическом) углеводородном фрагменте и от 6 до 18 атомов углерода в алкильном фрагменте алкилфенола. Другими подходящими неионными поверхностно-активными веществами являются водорастворимые аддукты полиэтиленоксида с полипропиленгликолем, этилендиаминполипропиленгликолем, содержащим от 1 до 10 атомов углерода в алкильной цепи, при этом указанные аддукты содержат от 20 до 250 этиленгликолевых эфирных групп и/или от 10 до 100 пропиленгликолевых эфирных групп. Подобные соединения обычно содержат от 1 до 5 этиленгликолевых единиц на одну пропиленгликолевую единицу. Типичными примерами неионных поверхностно-активных веществ являются нонилфенолполиэтоксиэтанол, полигликолевые эфиры касторового масла, аддукты полипропилен/полиэтиленоксид,трибутилфеноксиполиэтоксиэтанол, полиэтиленгликоль и октилфеноксиполиэтоксиэтанол. Сложные эфиры жирных кислот и полиэтиленсорбитана (такие как триолеат полиоксиэтиленсорбитана), глицерина, сорбитана, сахарозы и пентаэритрита также являются подходящими неионными поверхностноактивными веществами. Подходящие катионогенные поверхностно-активные вещества включают четвертичные аммониевые соли, преимущественно галогениды, содержащие 4 углеводородных радикала, необязательно замещенные галогеном, фенильной группой, замещенной фенильной группой или гидроксильной группой; например, четвертичные аммониевые соли, содержащие в качестве N-заместителя по меньшей мере один С 8-С 22 алкильный радикал (в частности, цетил, лаурил, пальмитил, миристил, олеил и т.п.) и в качестве других заместителей - незамещенные или галогензамещенные низший алкильный, бензильный и/или гидроксинизший алкильный радикалы. Более детальное описание поверхностно-активных агентов, подходящих для целей настоящего изобретения, может быть найдено, например, в "McCutcheon's Detergents and Emulsifiers Annual" (MC Publishing Crop., Ridgewood, New Jersey, 1981), "Tensid-Taschenbuch", 2nd ed. (Hanser Verlag, Vienna, 1981) и"Encyclopaedia of Surfactants" (Chemical Publishing Co., New York, 1981). С целью контролирования продолжительности действия активного ингредиента в композиции могут быть добавлены дополнительные ингредиенты. Таким образом, композиции с контролируемым высвобождением лекарства могут быть получены за счет выбора соответствующих полимерных носителей,- 19009727 таких как, например, полиэфиры, полиаминокислоты, поливинилпирролидоны, сополимеры этилена и винилацетата, метилцеллюлоза, карбоксиметилцеллюлоза, сульфат протамина и т.п. Скорость высвобождения лекарства и продолжительность его действия можно также контролировать путем заключения активного ингредиента внутрь частиц, в частности микрокапсул, полимерного вещества, такого как гидрогели, полимолочная кислота, гидроксиметилцеллюлоза, полиметилметакрилат и другие вышеприведенные полимеры. Подобные способы включают коллоидные системы доставки лекарства, такие как липосомы, микросферы, микроэмульсии, наночастицы, нанокапсулы и т.п. В зависимости от пути введения фармацевтическая композиция может потребовать применения защитных покрытий. Фармацевтические формы,подходящие для использования в качестве инъекций, включают стерильные водные растворы или неводные растворы или дисперсии (суспензии, эмульсии) и стерильные порошки для приготовления из них импровизированных препаратов. Обычные носители для этой цели, таким образом, включают биосовместимые водные буферные растворы, этанол, глицерин, пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, органические эфиры для инъекций, такие как этилолеат и т.п.,и их смеси. Парентеральные носители включают раствор хлорида натрия, декстрозу Рингера, декстрозу и хлорид натрия, лактаты Рингера или нелетучие масла. Внутривенные носители включают жидкие и питательные подкрепляющие добавки, подкрепляющие электролиты (такие как подкрепляющие электролиты на основе декстрозы Рингера) и т.п. Могут также присутствовать консерванты и другие добавки, такие как, например, антимикробные средства, антиоксиданты, хелатообразующие агенты и инертные газы и т.д. Изобретение далее подробно описывается в следующих примерах, которые не ограничивают объем изобретения, изложенный в формуле изобретения. Примеры Экспериментальная часть: приготовление соединений формулы (I). Примеры, описывающие приготовление соединений-пролекарств формулы (I), основываются на ингибиторе ВИЧ-протеазы, имеющего формулу который далее обозначают как PI 1. Пример 1. Val-Pro-PI 1.N,N-диметилформамида. Добавляют EDCI (0,36 г; 1,9 ммоль) и HOAt (0,023 г; 0,17 ммоль) и перемешивают при комнатной температуре в течение 20 ч. Реакционную смесь выливают в Н 2 О и дважды экстрагируют этилацетатом. Органические слои объединяют, промывают насыщенным раствором соли и сушат над Na2SO4. Растворитель удаляют, и полученный сырой продукт очищают колоночной хроматографией(элюент: этилацетат). Соединение 1.2 получают в виде твердого вещества белого цвета (выход 55%, чистота 95% по данным ЖХ-МС). К раствору соединения 1.2 (0,77 г; 0,9 ммоль) в 10 мл CH2Cl2 добавляют 10 мл трифторуксусной кислоты. После перемешивания реакционной смеси при комнатной температуре в течение 3 ч растворитель удаляют. Сырую смесь очищают колоночной хроматографией и получают 0,42 г соединения 1.3 (выход 61%; чистота 95% по данным ЖХ-МС).N,N-диметилформамида. Добавляют EDCI (1,18 г; 6,18 ммоль) и HOAt (0,077 г; 0,5 ммоль) и перемешивают в течение 36 ч. Добавляют этилацетат и 0,1 н. HCl и полученную реакционную смесь 3 раза экстрагируют этилацетатом. Органические слои объединяют, промывают 0,1 н. HCl, H2O, насыщенным раствором NaHCO3, водой и насыщенным раствором соли. После сушки над Na2SO4 и упаривания растворителя получают пенообразный продукт белого цвета (4,39 г, 103%). После его растирания в диизопропиловом эфире получают 3,9 г соединения 2.2 (выход 93%, чистота 97% по данным ЖХ-МС). Смесь соединения 2.2 (3,7 г; 4,8 ммоль) и 15 мл трифторуксусной кислоты в 40 мл CH2Cl2 перемешивают при комнатной температуре в течение 2 ч. После упаривания растворителя сырую смесь перераспределяют между этилацетатом и насыщенным раствором NaHCO3. Органический слой отделяют,промывают насыщенным раствором соли и сушат над Na2SO4. Сырой продукт вновь суспендируют в диизопропиловом эфире и после фильтрации получают 2,73 г соединения 2.3 (выход 85%, чистота 90% по данным ЯМР).N,N-диметилформамида добавляют EDCI (0,32 г; 1,7 ммоль) и HOAt (0,02 г; 0,15 ммоль). Оставляют перемешиваться на ночь при комнатной температуре, а затем реакционную смесь перераспределяют между этилацетатом и 0,1 н. HCl. Водный слой экстрагируют 3 раза, органические слои объединяют, промывают с помощью 0,1 н. HCl, водой, насыщенным раствором NaHCO3 и водой. После сушки над Na2SO4 растворитель удаляют, и сырой продукт растирают в диизопропиловом эфире. Получают 1,12 г соединения 2.4 Удаление защиты у соединения 2.4 с получением соединения 2.5 проводят аналогично методике удаления защиты у соединения 2.2 с получением соединения 2.3. Используя аналогичные процедуры, приведенные в примерах 1 и 2, Boc-Lys(Fmoc)-ОН конденсируют с соединением 3.1 (получают по примеру 2) и получают соединение 3.2. После удаления защитной группы Boc получают соединение 3.3. Затем Boc-Pro-OH конденсируют с соединением 3.3 и получают соединение 3.4, у которого затем удаляют защитную группу Boc и получают соединение 3.5. Соединение 3.5 конденсируют с Boc-Asp(OtBu)-ОН, получая соединение 3.6, у которого вначале удаляют защитную группу Boc, а затем с помощью диметиламина в тетрагидрофуране удаляют защитную группу Fmoc и получают соединение 3.8, соответствующее Asp-Pro-Lys-Pro-PI 1. Пример 4. Превращение Val-Pro-PI 1 в PI 1 под действием очищенной CD26, сыворотки человека и бычьей сыворотки. Пептидное (Val-Pro) производное PI I (Val-Pro-PI 1) вводят во взаимодействие с очищенной CD26(фиг. 2 а-с) в 10- или 2% сыворотке человека или бычьей сыворотке, разбавленной забуференным фосфатом физиологическим раствором (PBS) (фиг. 2 а-с и 3 а, 3b). Val-Pro-PI 1 эффективно превращается в PI 1 при всех условиях проведения испытаний. В течение 60 мин Val-Pro-PI 1 полностью превращается в PI 1 под действием очищенной CD26. 10% бычья сыворотка или сыворотка человека превращает от 40 до 70% Val-Pro-PI 1 в PI 1 в течение 1 ч (фиг. 2 а-с). 2% бычья сыворотка или сыворотка человека, соответственно, превращает 8 и 25% Val-Pro-PI 1 в PI 1. Через 4 ч 35 и 95% соединения гидролизуется бычьей сывороткой или сывороткой человека, соответственно (фиг. 3 а, 3b). В присутствии 50 мкМ GP-pNA (глицилпролил-пара-нитроанилид), 100 мкМ Val-Pro-PI 1 эффективно конкурирует с субстратом за CD26 (фиг. 4). Кроме того, 20 мкМ Val-Pro-PI 1 может ингибировать высвобождение pNA из GP-pNA главным образом за счет конкурентного ингибированияCD26-катализируемой реакции. Val-Pro-PI 1 даже более эффективно, чем очищенная CD26, ингибирует превращение GP-pNA в pNA под действием 2%-ной бычьей сыворотки в PBS (фиг. 5). Кроме того,Val-Pro-PI 1 полностью ингибирует катализируемое сывороткой человека (2% в PBS) превращение GPpNA в pNA (фиг. 6). Пример 5. Разделением соединений Val-Pro-PI 1 и PI 1. Соединения разделяют на обращенной фазе RP-8 (Merck) в градиенте буферного раствора A (50 мМNaH2PO4+5 мМ раствора сульфоновой кислоты в гептане с pH 3,2) и буферного раствора В (ацетонитрил). 02 мин: 2% буфера В; 28 мин: 20% буфера В; 810 мин: 25% буфера В; 1012 мин: 35% буфера В; 1230 мин: 50% буфера В; 3035 мин: 50% буфера В; 3540 мин: 2% буфера В; 4045 мин: 2% буфера В. Значения Rt для Val-Pro-PI 1 и PI 1, соответственно, составляют 18,7 и 17,7 мин.- 22009727 Пример 6. Фармацевтические композиции. Капсулы. Соединение формулы (I) растворяют в органическом растворителе, таком как этанол, метанол или метиленхлорид, преимущественно, в смеси этанола и метиленхлорида. Полимеры, такие как сополимер поливинилпирролидона с винилацетатом (PVP-VA) или гидроксипропилметилцеллюлозу (НРМС),обычно с вязкостью 5 мПас, растворяют в органических растворителях, таких как этанол, метанол, метиленхлорид. Преимущественно полимер растворяют в этаноле. Растворы полимера и соединения смешивают, а затем сушат разбрызгиванием. Отношение соединение/полимер выбирают от 1/1 до 1/6. Промежуточные значения составляют 1/1,5 и 1/3. Предпочтительным является соотношение 1/6. После осушки разбрызгиванием твердую дисперсию помещают в предназначенные для введения капсулы. Загрузка лекарства на одну капсулу, в зависимости от размера используемой капсулы, составляет от 50 до 100 мг. Таблетки с покрытием. Приготовление ядра таблетки. Смесь 100 г соединения формулы (I), 570 г лактозы и 200 г крахмала тщательно смешивают, а затем смачивают раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона приблизительно в 200 мл воды. Влажный порошок просеивают, сушат и вновь просеивают. Затем добавляют 100 г микрокристаллической целлюлозы и 15 г гидрированного растительного масла. Все тщательно смешивают и спрессовывают в таблетки, получая 10000 таблеток, каждая из которых содержит 10 мг пролекарства формулы (I). Покрытие. К раствору 10 г метилцеллюлозы в 75 мл денатурированного этанола добавляют раствор 5 г этилцеллюлозы в 150 мл дихлорметана. Затем добавляют 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. Расплавляют 10 г полиэтиленгликоля и растворяют в 75 мл дихлорметана. Последний раствор добавляют к первому раствору, а затем добавляют 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной суспензии красителя, после чего смесь гомогенизуют. Ядра таблеток покрывают полученной смесью в аппарате для нанесения покрытия. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Пролекарство, имеющее формулу его стереоизомерная форма или его соль,где n равно 1, 2, 3, 4 или 5;X выбирают из любой аминокислоты с D- или L-конфигурацией;X и Y в каждом повторе фрагмента [Y-X] выбирают независимо друг от друга и независимо от других повторов;Z обозначает прямую связь или бивалентную насыщенную углеводородную группу с прямой или разветвленной цепью, содержащую от 1 до 4 атомов углерода;R4 обозначает водород или С 1-4 алкильную группу; арильная группа, когда она используется самостоятельно или в комбинации с другой группой,означает фенильную группу, необязательно замещенную одним или несколькими заместителями,каждый из которых индивидуально выбирают из группы, включающей С 1-4 алкильную, гидроксильную группу, С 1-4 алкилоксигруппу, нитрогруппу, цианогруппу, галоген, аминогруппу, моно- или ди(С 1-4 алкил)аминогруппу и амидную группу; гетероарильная группа, когда она используется самостоятельно или в комбинации с другой группой, означает моноциклический или бициклический ароматический гетероцикл, содержащий один или несколько гетероатомов кислорода, серы или азота, при этом ароматический гетероцикл необязательно может быть замещен по одному или нескольким атомам углерода заместителем, выбранным из группы,включающей С 1-4 алкильную группу, С 1-4 алкилоксигруппу, аминогруппу, гидроксильную, арильную,амидную группу, моно- или ди(С 1-4 алкил)аминогруппу, галоген, нитрогруппу, гетероциклоалкильную и- 23009727 С 1-4 алкилоксикарбонильную группу и при этом ароматический гетероцикл необязательно может быть замещен по вторичному атому азота С 1-4 алкильной или арилС 1-4 алкильной группой; гетероциклоалкильная группа, когда она используется самостоятельно или в комбинации с другой группой, означает насыщенный или частично ненасыщенный моноциклический или бициклический гетероцикл, содержащий один или несколько гетероатомов кислорода, серы или азота, при этом гетероцикл необязательно может быть замещен по одному или нескольким атомам углерода заместителем, выбранным из группы, включающей С 1-4 алкильную группу, С 1-4 алкилоксигруппу, гидроксильную группу,галоген и оксогруппу, и при этом указанный гетероцикл необязательно может быть замещен по вторичному атому азота С 1-4 алкильной или арилС 1-4 алкильной группой. 2. Пролекарство по п.1, где каждый X независимо выбирают из природной аминокислоты. 3. Пролекарство по п.1 или 2, где n равно 1, 2 или 3. 4. Пролекарство по любому из пп.1-3, где n равно 2 или 3 и где по крайней мере один X представляет собой гидрофобную или ароматическую аминокислоту. 5. Пролекарство по любому из пп.1-4, где n равно 2 или 3 и где по крайней мере один X представляет собой нейтральную или кислую аминокислоту. 6. Пролекарство по любому из пп.1-5, где n равно 2 или 3 и где по крайней мере один X представляет собой основную аминокислоту. 7. Пролекарство по любому из пп.1-6, где -(Y-X)n содержит аминоконцевые X-Pro, X-Ala, X-Gly,X-Ser, X-Val или X-Leu. 8. Пролекарство по любому из пп.1-7, где -(Y-X)n содержит аминоконцевые X-пролин илиX-аланин. 9. Пролекарство по любому из пп.1-8, где каждый Y независимо обозначает пролин, аланин,глицин, серин, валин или лейцин. 10. Пролекарство по любому из пп.1-9, где каждый Y независимо обозначает пролин, или гидроксипролин, или дигидроксипролин, или аланин. 11. Пролекарство по любому из пп.1-10, где каждый Y независимо обозначает пролин или аланин. 12. Пролекарство по любому из пп.1-11, где -(Y-X)n обозначает -(Y-X)1 или 2-Y-Val. 13. Пролекарство по любому из пп.1-12, где -(Y-X)n обозначает -(Y-X)1 или 2-Pro-Val. 14. Пролекарство по любому из пп.1-13, где олигопептид (Y-X)n выстроен из повторов фрагментов(Y-X), выбранных из группы, включающей Pro-Val, Pro-Asp, Pro-Ser, Pro-Lys, Pro-Arg, Pro-His, Pro-Phe,Pro-Ile, Pro-Leu, Ala-Val, Ala-Asp, Ala-Ser, Ala-Lys, Ala-Arg, Ala-His, Ala-Phe, Ala-Ile и Ala-Leu. 15. Пролекарство по любому из пп.1-14, где R1 обозначает гетероциклоалкилоксигруппу,гетероарильную группу, гетероарилС 1-4 алкилоксигруппу, арильную группу или арилоксиС 1-4 алкильную группу. 16. Пролекарство по любому из пп.1-15, где R1 обозначает гексагидрофуро[2,3-b]фуран-3 илоксигруппу,тетрагидрофуран-3-илоксигруппу,хинолин-2-ильную группу,тиазол-5-илметилоксигруппу, 3-гидрокси-2-метил-1-фенильную, 2,6-диметилфеноксиметильную группу. 17. Пролекарство по любому из пп.1-16, где R1 обозначает (3R,3aS,6aR)-гексагидрофуро[2,3b]фуран-3-илоксигруппу. 18. Пролекарство по любому из пп.1-17, где R2 обозначает фенилметильную группу, R3 обозначает изобутильную группу и R4 обозначает водород. 19. Пролекарство по любому из пп.1-18, где Z обозначает метиленовую группу. 20. Пролекарство по п.1, где пролекарством является или его соль. 23. Применение пролекарства по любому из пп.1-22 в качестве лекарственного средства.
МПК / Метки
МПК: C07K 5/065, C07D 493/04, C07K 5/103, C07K 5/068, C07H 15/252, C07K 5/062, C07K 5/072, C07K 5/083, C07K 5/11, A61K 47/48, C07K 5/06
Метки: действием, способные, расщепляться, вич-пролекарства
Код ссылки
<a href="https://eas.patents.su/30-9727-vich-prolekarstva-sposobnye-rasshheplyatsya-pod-dejjstviem-cd26.html" rel="bookmark" title="База патентов Евразийского Союза">Вич-пролекарства, способные расщепляться под действием cd26</a>
Трансформированные грибы, в частности рода chrysosporium, способные к синтезу гетерологичных полипептидов

Номер патента: 5682
Опубликовано: 28.04.2005
Авторы: Синицин Аркадий Пантелеймонович, Пант Питер Ян, Берлингейм Ричард Пол, Буссон Жан Кристоф, Олсон Филип Терри, Пиннонен Кристин Мари, Ван Зейл Корнелия Мария Йоханна, Парриш Мартин, Эмалфарб Марк Аарон
МПК: C12N 15/80
Метки: chrysosporium, полипептидов, частности, рода, синтезу, грибы, способные, гетерологичных, трансформированные
Формула / Реферат:
1. Трансформированный микроорганизм рода Chrysosporium, содержащий последовательность нуклеиновой кислоты, кодирующую заданный полипептид, функционально связанную с гомологичным или гетерологичным участком, регулирующим экспрессию, распознаваемым указанным микроорганизмом Chrysosporium, и, необязательно, с секреторной сигнальной последовательностью, полученный путем трансформации исходного микроорганизма Chrysosporium генетической конструкцией,...
Высокоэффективные способные к слиянию везикулы, способ их получения и фармацевтическая композиция, их содержащая

Номер патента: 8497
Опубликовано: 29.06.2007
Авторы: Вадруччи Соня, Бруннер Йозеф, Цурбригген Ринальдо
МПК: A61K 9/127
Метки: способные, высокоэффективные, содержащая, композиция, получения, фармацевтическая, слиянию, способ, везикулы
Формула / Реферат:
1. Способные к слиянию везикулы, составленные из виросомных и липосомных липидов, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, причем способные к слиянию везикулы содержат слитые белки с различными параметрами слияния или фрагменты слитых белков, при этом фрагменты имеют активность слияния и параметры слияния полного слитого белка, по меньшей мере, в той степени, которая необходима для...
Микроорганизмы rhodococcus sp., способные превращать ацетонитрил в амид, полученый из этих микроорганизмов фермент с нитрилгидратазной активностью и способ их применения

Номер патента: 8600
Опубликовано: 29.06.2007
Авторы: Робинс Карен Трейси, Нагасава Тору
МПК: C12N 9/78, C12P 13/02, C12N 1/20...
Метки: этих, микроорганизмов, амид, микроорганизмы, нитрилгидратазной, способ, превращать, фермент, ацетонитрил, активностью, полученый, rhodococcus, способные, применения
Формула / Реферат:
1. Микроорганизм, представляющий собой штамм Rhodococcus sp. FZ4, депонированный под регистрационным номером DSM 13597, и его функционально эквивалентные варианты и мутанты, способные превращать ацетонитрил в амид. 2. Нитрилгидратаза, полученная из микроорганизма по п.1 и характеризующаяся: а) Km 2,84+1,00 мМ при использовании в качестве субстрата ацетонитрила и Km 80,5+15,0 мМ при использовании в качестве субстрата 3-цианпиридина; б)...
Штаммы микроорганизма rhodococcus, способные превращать нитрил в амид, и способ получения амидов

Номер патента: 9075
Опубликовано: 26.10.2007
Авторы: Робинс Карен Трейси, Нагасава Тору
МПК: C12P 13/02, C12N 1/20, C12N 9/88...
Метки: амид, rhodococcus, микроорганизма, способ, превращать, нитрил, штаммы, способные, получения, амидов
Формула / Реферат:
1. Штамм микроорганизма Rhodococcus sp. GF270 DSM 12211, а также его функционально эквивалентные варианты и мутанты, способные превращать нитрил в амид. 2. Штамм микроорганизма Rhodococcus sp. GF376 DSM 12175, а также его функционально эквивалентные варианты и мутанты, способные превращать нитрил в амид. 3. Способ получения амидов, отличающийся тем, что нитрил, применяемый в качестве субстрата, превращают в соответствующий амид с помощью штаммов...
Микроорганизмы amycolatopsis, способные превращать нитрил в амид, фермент с нитрилгидратазной активностью, полученный из указанных микроорганизмов, и способы их использования

Номер патента: 6444
Опубликовано: 29.12.2005
Авторы: Робинс Карен Трейси, Нагасава Тору
МПК: C12P 13/02, C12N 1/20
Метки: amycolatopsis, способы, микроорганизмов, микроорганизмы, превращать, указанных, способные, нитрилгидратазной, полученный, нитрил, амид, использования, активностью, фермент
Формула / Реферат:
1. Микроорганизм Amycolatopsis sp.NA31 (DSM 11616), а также его варианты и мутанты, способные превращать нитрил в амид. 2. Микроорганизм Amycolatopsis sp.NA40 (DSM 11617), а также его варианты и мутанты, способные превращать нитрил в амид. 3. Фермент с нитрилгидратазной активностью, полученный из микроорганизмов рода Amycolatopsis, характеризующийся а) оптимумом активности при pH 6,5+0,1, б) оптимумом активности при температуре между 35 и 40шC...
Предыдущий патент: Способ извлечения золота
Следующий патент: Триазиновые соединения и их применение в медицине