Высокоэффективные способные к слиянию везикулы, способ их получения и фармацевтическая композиция, их содержащая
Номер патента: 8497
Опубликовано: 29.06.2007
Авторы: Вадруччи Соня, Бруннер Йозеф, Цурбригген Ринальдо







Формула / Реферат
1. Способные к слиянию везикулы, составленные из виросомных и липосомных липидов, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, причем способные к слиянию везикулы содержат слитые белки с различными параметрами слияния или фрагменты слитых белков, при этом фрагменты имеют активность слияния и параметры слияния полного слитого белка, по меньшей мере, в той степени, которая необходима для слияния с биологической мембраной клетки-мишени.
2. Способные к слиянию везикулы по п.1, отличающиеся тем, что везикулы являются однослойными.
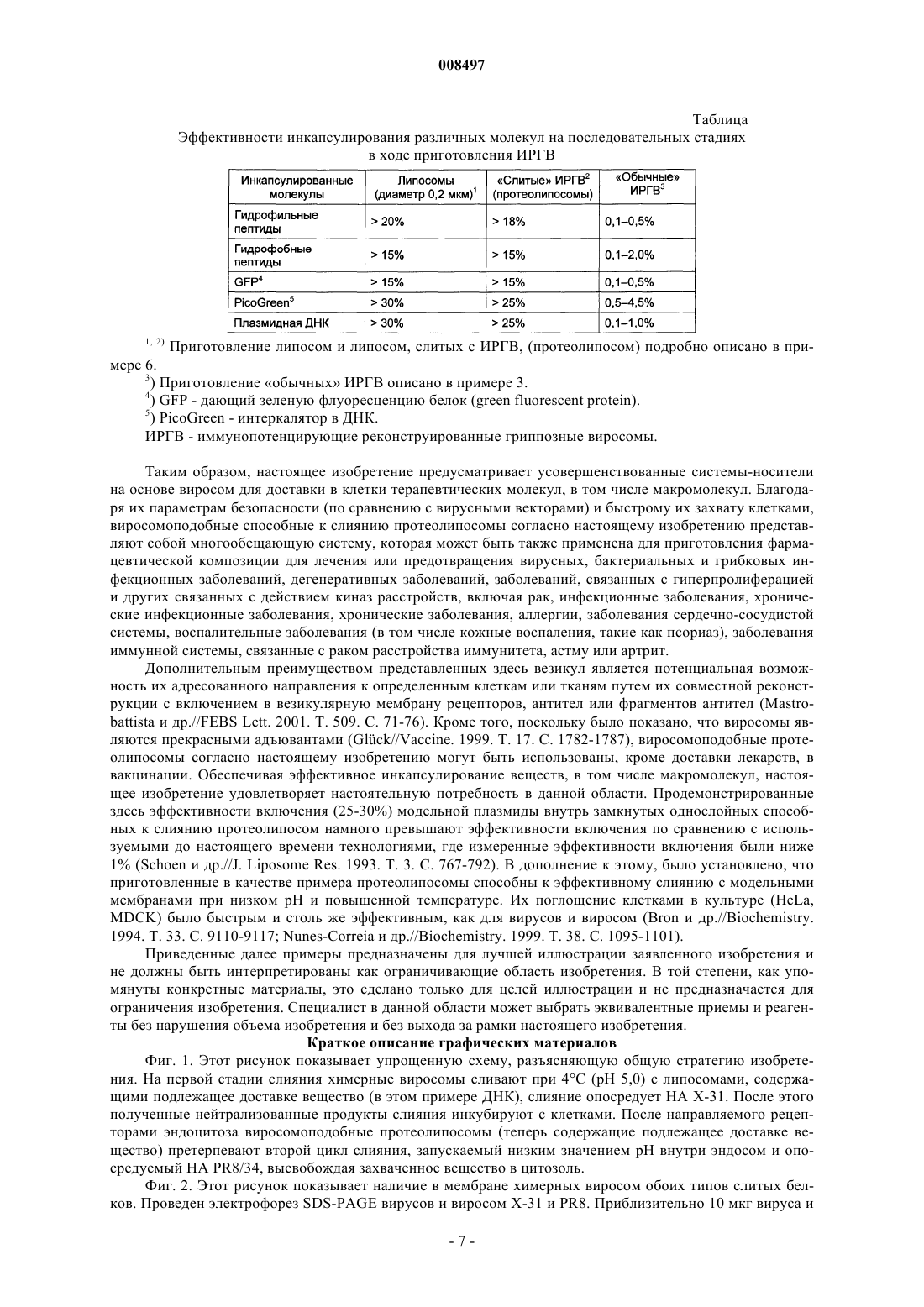
3. Способные к слиянию везикулы по п.1 или 2, отличающиеся тем, что инкапсулированное терапевтическое или иммунологически активное вещество выбрано из группы, содержащей ДНК, РНК, миРНК (малую интерферирующую РНК), белки, пептиды, аминокислоты и фармацевтически активные вещества.
4. Способные к слиянию везикулы по п.3, отличающиеся тем, что инкапсулированное терапевтическое или иммунологически активное вещество выбрано из косметического средства, фармацевтического лекарства, антигена или их смесей.
5. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем, что различные параметры слияния выбраны из температуры, концентрации ионов, кислотности, специфичности к типу клеток и к типу ткани.
6. Способные к слиянию везикулы по п.5, отличающиеся тем, что различные параметры слияния слитых белков зависят от температуры.
7. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем, что слитые белки или их фрагменты извлечены из вирусов.
8. Способные к слиянию везикулы по п.7, отличающиеся тем, что слитые белки или их фрагменты извлечены из вирусов, выбранных из группы, содержащей вирус гриппа, вирус везикулярного стоматита (ВВС), вирус леса Семлики (ВЛС), вирус Сендай и вирус иммунодефицита человека (ВИЧ).
9. Способные к слиянию везикулы по п.8, отличающиеся тем, что слитые белки или их фрагменты извлечены из вируса гриппа.
10. Способные к слиянию везикулы по п.9, отличающиеся тем, что слитые белки или их фрагменты представляют собой гемагглютинины штаммов вируса гриппа Х-31, PR8/34 или A/Singapore или же их фрагменты.
11. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем, что липосомные липиды происходят из группы, включающей гликолипиды, фосфолипиды, катионные липиды, синтетические липиды и холестерин.
12. Способные к слиянию везикулы по п.11, отличающиеся тем, что липосомные липиды включают РОРС и DDAB.
13. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем, что виросомные липиды извлечены из вирусов, выбранных из группы, содержащей вирус гриппа, вирус везикулярного стоматита (ВВС), вирус леса Семлики (ВЛС), вирус Сендай и вирус иммунодефицита человека (ВИЧ).
14. Способные к слиянию везикулы по п.13, отличающиеся тем, что виросомные липиды извлечены из вируса гриппа.
15. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем, что везикулы имеют диаметр в диапазоне от 100 до 300 нм.
16. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем, что везикулы дополнительно содержат рецептор клеточной поверхности, цитокин, фактор роста, антитело или фрагмент антитела, внедренные в мембрану или присоединенные к указанной мембране.
17. Способ приготовления способных к слиянию везикул, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, включающий слияние липосом, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, с виросомами, имеющими слитые белки с различными параметрами слияния или фрагменты слитых белков, причем фрагменты имеют активность слияния и параметры слияния полного слитого белка, по меньшей мере, в той степени, которая необходима для слияния с биологической мембраной клетки-мишени.
18. Способ инкапсулирования по меньшей мере одного терапевтического или иммунологически активного вещества в способные к слиянию везикулы, включающий слияние липосом, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, с виросомами, имеющими слитые белки с различными параметрами слияния или фрагменты слитых белков, причем фрагменты имеют активность слияния и параметры полного слитого белка, по меньшей мере, в той степени, которая необходима для слияния с биологической мембраной клетки-мишени.
19. Способ по п.17 или 18, отличающийся тем, что везикулы являются однослойными.
20. Способ по любому из пп.17-19, отличающийся тем, что инкапсулированное по меньшей мере одно терапевтическое или иммунологически активное вещество выбрано из группы, содержащей ДНК, РНК, миРНК, белки, пептиды, аминокислоты и фармацевтически активные вещества.
21. Способ по п.20, отличающийся тем, что инкапсулированное по меньшей мере одно терапевтическое или иммунологически активное вещество выбрано из косметического средства, фармацевтического лекарства, антигена или их смесей.
22. Способ по любому из пп.17-21, отличающийся тем, что различные параметры слияния слитых белков выбраны из температуры, концентрации ионов, кислотности, специфичности к типу клеток и к типу ткани.
23. Способ по п.22, отличающийся тем, что различные параметры слияния слитых белков или их фрагментов зависят от температуры.
24. Способ по любому из пп.17-23, отличающийся тем, что используемые слитые белки или их фрагменты извлечены из вирусов.
25. Способ по п.24, отличающийся тем, что используемые слитые белки или их фрагменты, извлеченные из вирусов, выбранных из группы, содержащей вирус гриппа, вирус везикулярного стоматита (ВВС), вирус леса Семлики (ВЛС), вирус Сендай и вирус иммунодефицита человека (ВИЧ).
26. Способ по п.25, отличающийся тем, что используемые слитые белки или их фрагменты, извлечены из вируса гриппа.
27. Способ по п.26, отличающийся тем, что слитые белки или их фрагменты представляют собой гемагглютинины штаммов вируса гриппа Х-31, PR8/34 или A/Singapore или же их фрагменты.
28. Способ по любому из пп.17-27, отличающийся тем, что липосомы состоят из липидов, выбранных из группы, включающей гликолипиды, фосфолипиды, катионные липиды, синтетические липиды и холестерин.
29. Способ по п.28, отличающийся тем, что липосомы состоят из РОРС и DDAB.
30. Способ по любому из пп.17-29, отличающийся тем, что виросомы получены из вирусов, выбранных из группы, содержащей вирус гриппа, вирус везикулярного стоматита (ВВС), вирус леса Семлики (ВЛС), вирус Сендай и вирус иммунодефицита человека (ВИЧ).
31. Способ по п.30, отличающийся тем, что виросомы получены из вируса гриппа.
32. Способ по любому из пп.17-31, отличающийся тем, что он дополнительно включает изменение размера способных к слиянию везикул, полученных после слияния липосом, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, с виросомами, имеющими слитые белки с различными параметрами слияния или фрагменты слитых белков, при этом фрагменты имеют активность слияния и параметры слияния полного слитого белка слияния, по меньшей мере, в той степени, которая необходима для слияния с биологической мембраной клетки-мишени.
33. Способ по п.32, отличающийся тем, что изменение размера осуществляется путем экструдирования везикул.
34. Способ по любому из пп.17-33, отличающийся тем, что изменение размера способных к слиянию везикул приводит к тому, что их диаметр находится в диапазоне от 100 до 300 нм.
35. Способ по любому из пп.17-34, отличающийся тем, что к мембране способных к слиянию везикул прикреплен или в мембрану способных к слиянию везикул внедрен по меньшей мере один рецептор клеточной поверхности, цитокин, фактор роста, одно антитело или один фрагмент антитела.
36. Применение способных к слиянию везикул, как они заявлены в пп.1-16, для приготовления фармацевтической композиции, предназначенной для лечения или предотвращения вирусных, бактериальных и грибкотых инфекционных заболеваний, дегенеративных заболеваний, заболеваний, связанных с гиперпролиферацией, и других связанных с действием киназ расстройств, включая рак, инфекционные заболевания, хронические инфекционные заболевания, хронические заболевания, аллергии, заболевания сердечно-сосудистой системы, воспалительные заболевания (в том числе кожные воспаления, такие как псориаз), заболевания иммунной системы, связанные с раком расстройства иммунитета, астму или артрит.
37. Фармацевтическая композиция, содержащая способные к слиянию везикулы, как они определены в любом из пп.1-16.

Текст
008497 Область техники Настоящее изобретение относится к новым способным к слиянию (фузогенным) везикулам как высоко эффективным и универсальным системам упаковки (инкапсулирования) для доставки в клетки и ткани выбранных веществ, таких как нуклеиновые кислоты, белки, пептиды, антигены, фармацевтические препараты и косметические средства. Предшествующий уровень техники Одной из первостепенных задач медицинской терапии является эффективная доставка терапевтических веществ к месту развития заболевания. В то время как некоторые терапевтические вещества могут доставляться в свободной форме, для других необходим носитель или вектор, чтобы они могли достигнуть конечного места назначения и попасть в него. Такая ситуация бывает в случае либо их быстрого исчезновения из места введения, либо их неспособности к прохождению через биологические барьеры,либо их общей токсичности. Для доставки веществ в клетки и ткани требуются векторы - эффективные,маневренные, легкие в приготовлении и безопасные. Существующие в настоящее время способы доставки веществ в эукариотические клетки включают применение либо вирусных, либо невирусных векторов. Вирусные векторы - это вирусы с нарушенной репликацией, у которых часть их кодирующих последовательностей заменена последовательностями терапевтического гена. Хотя рекомбинантные вирусы являются высоко эффективными векторами для доставки и экспрессии генов, их применение в настоящее время ограничено доставкой нуклеиновых кислот, а их параметры безопасности пока еще недостаточно установлены для медицинского применения у людей. Действие большинства невирусных систем доставки слагается из следующих этапов: загрузка вектора доставки представляющим интерес веществом (например, белками, пептидами, нуклеиновыми кислотами, фармацевтическими или иными лекарственными препаратами), эндоцитоз и, в случае доставки гена, адресованное нацеливание на ядро и вхождение в него. Главным недостатком невирусных систем,таких как липосомы, является их низкая эффективность доставки в клетки (Chu и др.; Legendre andSzoka//Pharmaceut. Res. 1992. Т. 9. С. 1235), предположительно из-за отсутствия опосредующих слияние молекул на поверхности липосом. Гибридный тип систем доставки, виросома, объединяет эффективный механизм доставки вирусов с универсальностью и безопасностью невирусных систем доставки. Виросомы представляют собой реконструированную оболочку без инфекционных нуклеокапсидов и генетический материал, который может быть получен из многих вирусов. Эти виросомы функциональны в том смысле, что их способность сливаться с мембраной очень близка к хорошо известной, зависящей от низкого рН активностью слияния с мембраной интактного вируса, которая опосредуется только вирусным белком слияния. Подобно вирусам, виросомы быстро захватываются путем опосредованного рецепторами эндоцитоза. В отличие от вирусных систем, виросомы безопасны, поскольку инфекционный нуклеокапсид вируса удален. Таким образом, виросомы являются многообещающими транспортными системами для доставки широкого спектра различных веществ - либо инкапсулированных в их водную внутреннюю часть, либо совместно реконструированных в их мембранах. Более того, совместная реконструкция различных рецепторов в виросомной мембране позволяет адресованно направлять виросомы к разным клеткам или тканям. Поэтому виросомы, в основном, используют как вакцины, нанося антиген на поверхность виросом. Главным ограничением применяемой в настоящее время методики приготовления виросом является то, что она не обеспечивает высокой эффективности инкапсулирования. При концентрации липида, при которой получают виросомы (1 мМ липид) и при том, что их средний диаметр равен приблизительно 200 нм, внутрь липосом захватывается менее 1% водной фазы (Schoen и др.//J. Liposome Res. 1993. Т. 3. С. 767-792). Такие низкие эффективности включения снижают опосредованную виросомами доставку лекарств или генов в клетки. Поэтому в области доставки терапевтических лекарств, белков и генов существует весьма актуальная задача - разработка новых, не существовавших ранее везикул с повышенной эффективностью загрузки, которые сохраняют полезную способность к слиянию виросом, а также способов их приготовления, загрузки и доставки. Сущность изобретения Одной из проблем в существующей виросомной технологии является отсутствие способов эффективного включения растворенного вещества, например белка, нуклеиновой кислоты или терапевтического лекарства. Настоящее изобретение предусматривает новый подход, который преодолевает проблемы низких эффективностей загрузки виросом. Поэтому настоящее изобретение предусматривает способную к слиянию (фузогенную) везикулу,состоящую из виросомных и липосомных липидов, в которую инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, причем указанная способная к слиянию везикула содержит слитые белки, предпочтительно по меньшей мере два различных слитых белка или их фрагменты с различными характеристиками (параметрами) слияния, при этом указанные фрагменты имеют активность слияния и характеристики полного слитого белка, по меньшей мере, до той степени,которая необходима для слияния с биологической мембраной клетки-мишени. Предпочтительно, везикула однослойная. Везикула имеет диаметр, как правило, в интервале от 100 до 600 нм, предпочтительно от 100 до 300 нм или от 200 до 400 нм, в зависимости от конкретного типа везикулы.-1 008497 Изобретение имеет преимущество в том, что липосомы, которые можно готовить при очень высоких концентрациях липида, имеют высокую эффективность инкапсулирования. Поэтому настоящее изобретение предусматривает методологию, объединяющую высокую способность липосом к загрузке с эффективностью виросом к слиянию с клетками и доставке содержимого, в результате чего существенно возрастает захват растворенных веществ, таких как белки, нуклеиновые кислоты и фармацевтические лекарства, внутрь функциональных химерных липосом контролируемого размера, которые способны эффективно доставлять к клеткам терапевтически или иммунологически активные вещества. В соответствии с этим, в предпочтительном варианте осуществления изобретения целевое вещество вначале загружается или инкапсулируется в липосомы. Липосомные липиды, которые используются в настоящем изобретении, предпочтительно выбраны из группы, состоящей из катионных липидов, синтетических липидов, гликолипидов, фосфолипидов, холестерина или их производных. Фосфолипиды включают преимущественно фосфатидилхолин, сфингомиелин, фосфатидилэтаноламин, фосфатидилсерин,фосфатидилглицерин, фосфатидную кислоту, кардиолипин и фосфатидилинозитол с различным составом жирной ацильной части. Катионные липиды предпочтительно выбраны из группы, состоящей из(N-[1-(2,3 диолеоилокси)пропил]-N,N,N-триметиламмонийхлорид), DODAC (N,N-диолеил-N,N-диметиламмонийхлорид), DDAB (дидодецилдиметиламмонийбромид) и стеариламина или других алифатических аминов и им подобных. Как правило, их рецептура определяется как малые однослойные липосомы в смеси сDOPE (диолеилфосфатидил-этаноламин), который широко используется как липид-помощник для облегчения разрушения эндосомной мембраны. Наиболее предпочтительно липосомные липиды липосом включают POPC/DDAB. На втором этапе липосомы, содержащие целевое вещество, подлежат слиянию с химерными виросомами, имеющими в своих мембранах различные типы слитых белков. Термин слитый белок или его фрагменты относится к белкам или их фрагментам, способным индуцировать реакцию слияния и/или способствовать реакции слияния между способной к слиянию мембраной везикулы и биологической мембраной клетки-мишени. После изменения размера продукты слияния липосомы и виросомы становятся виросомоподобными частицами, или протеолипосомами, которые содержат в своей внутренней полости целевое вещество и еще способны претерпеть второй этап слияния, при других условиях, с биологическими мембранами,чтобы доставить терапевтическое или иммунологически активное вещество. Способная к слиянию везикула настоящего изобретения инкапсулирует по меньшей мере одно вещество, предпочтительно терапевтическое или иммунологически активное вещество, которое предпочтительно выбрано из группы, состоящей из ДНК, РНК, миРНК (малая интерферирующая РНК (siRNA,белков, пептидов, аминокислот и фармацевтически активных веществ или их производных. Предпочтительно, по меньшей мере одно вещество является косметическим средством, фармацевтическим лекарством, антигеном или их смесью. Примеры для косметического вещества выбраны из противопсориазных и противогрибковых веществ для дерматологического применения. Примеры терапевтического вещества или фармацевтических лекарств выбраны из группы, содержащей анестетики, ингибиторы ангиогенеза, противоугревые препараты, антиаллергики, антибиотики и химиотерапевтические препараты для местного применения, антигистамины, противовоспалительные/противоинфекционные агенты, противоопухолевые агенты, антигены, препараты против простейших, антиревматики, антивирусные вакцины, антивирусные препараты,контролирующие апоптоз препараты, антибактериальные вакцины, химиотерапевтические средства, цитостатики, иммуносупрессивные агенты, слабительные средства, психолептики. Предпочтительными примерами для косметического средства являются дегтевые мази и нистатин, а для фармацевтических лекарств - иммунологически активное вещество доксорубицин, винбластин, цисплатин, метотрексат, циклоспорин, ибупрофен, антигены Т-клеток на основе вируса гепатита С (HCV) и связанные с опухолями антигены. Инкапсулирование в липосомы по меньшей мере одного из терапевтических веществ, таких как белки, пептиды, нуклеиновые кислоты, фармацевтические, химиотерапевтические или косметические лекарства, может быть осуществлено любым известным в данной области способом, включая, помимо многих других хорошо известных способов, процедуры, описанные в работах: Monnard и др.//Biochim.Oberholzer и др.//Biochim. Biophys. Acta. 1999. Т. 1416. С. 57-68. В предпочтительном примере осуществления изобретения высокие эффективности инкапсулирования в липосомы достигаются техникой замораживания/оттаивания, примененной для приготовления чистых липидных везикул. С помощью этого метода приблизительно 50% от исходного количества линеаризованной плазмиды длиной свыше 3 тыс. пар нуклеотидов (п.н.) может быть заключено внутрь крупных однослойных везикул (КOB), состоящих предпочтительно из смеси 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина (РОРС) с дидодецилдиметиламмонийбромидом (DDAB) (Monnard и др.//Biochim. Biophys. Acta. 1997. Т. 1329. С. 39-50). В одном из предпочтительных примеров осуществления настоящее изобретение предусматривает способ приготовления виросом, содержащих бинарную смесь вирусных слитых молекул, имеющих раз-2 008497 личающиеся характеристики (параметры) слияния. В другом предпочтительном примере осуществления изобретения для конструирования химерных виросом используют различные вирусные слитые белки гемагглютинины (НА). Как было показано ранее (Tsurudome и др.//J. Biol. Chem. 1992. Т. 267. С. 2022520232), слитые белки НА из различных штаммов вирусов могут иметь существенно различные температурные параметры слияния и инактивации. Например, при рН 5,0 НА из Х-31 эффективно инициирует слияние при низкой температуре, в то время как при том же самом рН для НА из вирусов гриппа PR8/34 или А/Сингапур (A/Singapore) требуется повышенная температура (25 С). Поэтому в предпочтительном примере осуществления изобретения химерные виросомы содержат в своей мембране белки, которые вызывают слияние при двух различных температурах. Различная температурная чувствительность является особенно полезной характеристикой слитых белков, так как это обеспечивает удобный и простой контроль реакций слияния. Как пример, настоящее изобретение демонстрирует, что виросомы, содержащие молекулы НА из вирионов и Х-31, и PR8/34, способны катализировать при рН 5 две различных реакции слияния: первую при низкой температуре (от 0 до 4-10 С), а вторую при повышенной температуре(25 С). Однако в данной области хорошо известны и могут быть использованы для целей настоящего изобретения другие слитые белки с отличающимися параметрами слияния, включая чувствительность к температуре, концентрации ионов, кислотности, специфичность к типу клеток и типу ткани и т.д. Слитые белки с различными параметрами слияния могут быть получены из различных штаммов вируса гриппа,таких как хотя бы MRC-11, Х-97, NIB24, NIB26, Х-47, A/Johannesburg/33 и A/Singapore. Кроме того, для конструирования химерных виросом, способных подвергаться последовательным и раздельным актам слияния, могут быть использованы другие известные слитые белки вирусов, такие как G-белок вируса везикулярного стоматита (ВВС), белок Е 1 вируса леса Семлики (ВЛС) или белок F вируса Сендай. Более того, химерные виросомы и получаемые виросомоподобные протеолипосомы, загруженные целевым терапевтическим веществом, могут содержать слитые белки, нацеливающие виросомоподобные протеолипосомы на специфические клетки или ткани. Так, например, для адресованной доставки терапевтических веществ к специфическим типам клеток или тканей могут быть использованы такие слитые белки,специфичные для определенных рецепторов клеточной поверхности, как белок gp41/gp120 вируса иммунодефицита человека (ВИЧ) (gp120 связывается с рецептором CD4 на CD4+ Т-лимфоцитах и клетках,происходящих от моноцитов/макрофагов). В данной области хорошо известны также другие клеточноили тканеспецифичные слитые белки, которые могут быть легко включены в везикулы согласно настоящему изобретению. Кроме слитых белков, способные к слиянию везикулы согласно настоящему изобретению могут дополнительно содержать (внедренный в поверхность или присоединенный к ней) рецептор клеточной поверхности, цитокин, фактор роста, антитело или фрагмент антитела для улучшения адресованной направленности способной к слиянию везикулы к различным клеткам или тканям. Как показано в соответствии с настоящим изобретением, как только такие химерные виросомы сконструированы, они могут быть слиты при первых выбранных условиях, чтобы запустить слияние первого вирусного слитого белка или пептида с липосомами, содержащими инкапсулированное целевое вещество или целевые вещества, такие как белки, пептиды, нуклеиновые кислоты или фармацевтические лекарства. Изобретение впервые демонстрирует, что слияние химерных виросом с нагруженными веществом липосомами создает виросомоподобные протеолипосомы, в которые не только инкапсулировано целевое вещество, но и которые еще способны направить дополнительное слияние при других условиях,например при других температуре, концентрации ионов и т.д. После надлежащего манипулирования для установления их размера, эти содержащие вещество протеолипосомы могут быть доставлены к клеткам,где они эффективно захватываются. В окружении эндосомы молекулы вторичного слияния могут запустить вторую реакцию слияния между виросомной и эндосомной мембранами. Как показано на фиг. 1,этот многостадийный процесс приводит к переносу инкапсулированных в липосомы веществ внутрь цитозольных отсеков клеток. Процедуры приготовления виросом (именуемых в этом изобретении также иммунопотенцирующими реконструированными гриппозными виросомами, ИРГВ) хорошо известны специалистам в данной области (Вrоn и др.//Methods Enzymol. 1993. Т. 220. С. 313-331). Гриппозные виросомы, например, могут быть реконструированы из исходных липидов вирусной мембраны и гликопротеинов шипов после солюбилизации интактного вируса гриппа октаэтиленгликоль-монодециловым эфиром, седиментации нуклеокапсида (вирусные гликопротеины и липиды остаются в надосадочной жидкости) и удаления детергента из надосадочной жидкости с помощью гидрофобной смолы (Bio-Beads SM2) (Stegmann и др.//Traffic. 1987. Т. 1. С. 598-604). В качестве одного из примеров для приготовления химерных виросом, содержащих гемагглютинины из штаммов вирусов Х-31 и PR8/34, равные количества белков обоих вирусов солюбилизировали с помощью неионного детергента С 12 Е 8. Выделение других вирусных слитых белков является обычной процедурой для специалистов в данной области. После удаления детергента на смоле Bio-Beads SM2 образовались новые оболочки, содержащие оба типа слитых белков. Электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия (SDS-PAGE) описанных в данном примере химерных виросом, содержащих два различных типа слитых белков НА, показал (фиг. 2), что в химерных виросомах реконструированы очень близкие количества слитых белков - здесь НА PR8 и Х-31.-3 008497 Отношение НА/фосфолипид составляет приблизительно 1,4 мг/мкмоль. Для определения морфологии сконструированных химерных виросом их можно охарактеризовать с помощью криоэлектронной микроскопии пропускания (cryo-ТЕМ). Реконструированные частицы оказались однослойными везикулами с диаметром в интервале 150-250 нм; были различимы также в некотором количестве везикулы меньшего размера (фиг. 3). Плотность гемагглютининовых шипов была явно ниже, чем обычно видна в интактных вирусных частицах. Это согласуется с измеренным соотношением НА/фосфолипид для химерных виросом, равным 1,4 мг/мкмоль, что несколько ниже, чем соотношение в интактных вирионах (приблизительно 2 мг/мкмоль). Кроме того, шипы НА находятся на обеих сторонах мембраны с приблизительно равной плотностью. Свойства слияния химерных виросом с липосомами, такие как зависимость активности слияния от определенных значений температуры, рН или другого параметра, можно определить in vitro с помощью анализа смешивания липидов (Struck и др.//Biochemistry. 1981. Т. 20. С. 4093-4099). Этот метод анализа основан на измерении переноса энергии резонанса флуоресценции (fluorescence resonance energy transfer,FRET), в нем используются два флуорофора: N-NBD-PE (донор энергии) и N-Rh-PE (акцептор энергии),находящиеся в одной из двух мембран (например, в липосомах). Может быть также применен вариант этого метода анализа, в котором используется тот же донор N-NBD-PE, но другой акцептор - холестеринантрацен-9-карбоксилат (XAК). Может быть применен также и иной вариант этого метода анализа с использованием того же акцептора N-Rh-PE, но другого донора - Bodipy 530/550-GHPE. При слиянии два флуорофора раздвигаются, и испускаемая донором флуоресценция возрастает (или снижается флуоресценция акцептора). В качестве примера, активность слияния химерных виросом X-31/PR8/34 сравнивали с активностью слияния вирусов Х-31 и PR8, а также соответствующих виросом (фиг. 4). Использовали в качестве мембран-мишеней однослойные липосомы, приготовленные из POPC/POPG (4:1), и два флуорофора (0,6% каждого). Как показано на фиг. 4.1 а, и вирус Х-31, и виросомы X-31 эффективно сливались с липосомами при 4 С и рН 5,0. Поскольку для обоих слитых белков - НА Х-31 и НА PR8/34 необходим кислотный рН, при нейтральном рН слияния не происходило. Предварительная инкубация НА Х-31 при низкой температуре и низком рН почти полностью уничтожала его активность слияния (фиг. 4.1b). Напротив, при 4 С и рН 5,0 вирус и виросомы PR8/34 не только не были способны к слиянию, но и,что важно, не претерпевали инактивации (фиг. 4.2 а). Однако они были способны запустить слияние при 37 С (фиг. 4.2b). Согласно этим данным, могут быть сконструированы химерные виросомы для осуществления двух различных реакций слияния, зависящих от параметров, для которых специфичен каждый из слитых белков. Например, химерные виросомы PR8/34/X-31 могут осуществлять одну реакцию слияния при низкой температуре (4 С, фиг. 4.3 а), а вторую реакцию слияния - при более высокой температуре(37 С, фиг. 4.3b). Неожиданное и важное открытие настоящего изобретения состоит в том, что предварительная инкубация химерных виросом с мембранами-мишенями при низкой температуре и низком рН не исключает активность слияния при повышенной температуре (фиг. 4.3b). Таким образом, настоящее изобретение предусматривает контролируемую реакцию слияния, которая приводит к образованию нагруженных виросом, способных к повторному слиянию при других условиях. Тот факт, что приготовленные липосомы являются однослойными, имеет очень большое значение для последующего этапа слияния с химерными виросомами, поскольку загруженное вещество должно быть высвобождено во внутреннюю полость вновь образующейся везикулы, а не остаться удержанным между мембранными слоями. Чтобы оценить возможность взаимодействия загруженного вещества с использованными липидами и его влияние на морфологию липосом, содержащие плазмиду везикулы были подвергнуты cryo-ТЕМ. Электронные микрофотографии (фиг. 6) показывают, что катионные везикулы были однослойными, размером 100-150 нм. В противоположность другим способам, где содержащие катионные липиды и ДНК препараты представляли собой многослойные, трубчатые структуры (Gerson и др.//Biochemistry. 1993. Т. 32. С. 7143-7151), предлагаемый здесь способ имеет преимущества, так как позволяет получить гомогенные однослойные везикулы. Чтобы продемонстрировать осуществимость этого подхода, была определена эффективность инкапсулирования выбранного терапевтического или иммунологически активного вещества - в данном случае модельной плазмиды размером 6,5 тыс. п.н. После расщепления свободной плазмидной ДНК смесью ДНКазы I с экзонуклеазой III и разделения продуктов гель-хроматографией было определено, что достигнутая эффективность включения составляла приблизительно 40% кольцевой модельной плазмиды длиной 6,5 тыс. п.н. Количественное определение оставшейся интактной ДНК может быть основано на введении в ДНК радиоизотопной метки или на визуальном сопоставлении со стандартными количествами в агарозном геле после окрашивания бромистым этидием, как представлено на фиг. 5. Чтобы удостовериться, что доступная ДНК была разрушена количественно, были проведены контрольные опыты, в которых вся ДНК была доступна (т.е. липосомы были солюбилизированы с помощью детергента (фиг. 5,полоски 9 и 10) или ДНК была на внешней поверхности липосом (фиг. 5, полоски 11 и 12. Таким образом, эти результаты показывают, что устойчивая к действию смеси ДНКазы I и экзонуклеазы III фракция эффективно включена во внутреннюю полость липосом и совершенно недоступна действию ДНКазы I и экзонуклеазы III вследствие (защитных) взаимодействий с катионным липидом. Достигнутое включение(35-40%) весьма значительно и делает данный подход пригодным для терапевтических применений.-4 008497 Вслед за успешным приготовлением химерных виросом и однослойных липосом, загруженных целевым веществом, настоящее изобретение предусматривает слияние этих двух везикулярных систем. Однако имеется несколько главных препятствий, которые необходимо преодолеть до получения требуемых способных к слиянию и загруженных виросом. Поскольку виросомы способны сливаться и с липосомами, и с другими виросомами, а также с первичными продуктами слияния, то было обнаружено, что в смеси нагруженных липосом с химерными липосомами происходит несколько циклов слияния. Эти неконтролируемые реакции слияния приводят к образованию большого процента крупных, неопределенных структур. Такие крупные структуры не подходят для доставки терапевтических веществ к клеткам, и следует ожидать, что их эндоцитоз клетками будет самым серьезным образом поставлен под сомнение. Более того, множественные циклы слияния приводят к получению популяций виросомоподобных конечных продуктов (протеолипосом) с варьированием размеров в широком интервале. Такие гетерогенные по размерам популяции нежелательны для терапевтического применения, так как количество и дозировку инкапсулированного терапевтического вещества невозможно контролировать. Кроме того, инкапсулирование заключенного в исходные липосомы целевого терапевтического или иммунологически активного вещества во внутреннюю полость получаемых химерных виросомоподобных протеолипосом ранее никем не было продемонстрировано. Вызывало сомнения, может ли быть достигнуто успешное инкапсулирование, или слияние приведет к частичному или полному высвобождению целевого терапевтического вещества. Наконец, было сомнительно, сохранят ли слитые везикулы способность к слиянию при втором наборе условий или параметров - как, например, при повышенной температуре. Чтобы исследовать продукты слияния между липосомами и виросомами, содержащими различные слитые белки, образцы слитых везикул были охарактеризованы криоэлектронной микроскопией пропускания (cryo-ТЕМ). Продукты слияния липосом с виросомами представляли собой везикулы с распределением по размерам от 450 до 900 нм (фиг. 7, верхние кадры). Было обнаружено некоторое количество везикул меньшего размера - возможно, исходных виросом (фиг. 7, слева вверху). С другой стороны, оказалось, что все липосомы претерпели по меньшей мере один цикл слияния. Наличие очень больших везикул (900 нм) является доказательством того, что на этапе слияния виросом с липосомами имеют место множественные циклы слияния. Чтобы разрешить проблему неконтролируемого слияния и получать везикулы гомогенного размера, которые могут обеспечить контроль над дозированной доставкой лекарства или вещества, необходимо так воздействовать на везикулы, чтобы получить везикулы однородного размера. В идеале, везикулы большого размера, неподходящего для эндоцитоза, следует уменьшить без потери инкапсулированного содержимого или способности к слиянию. Поэтому настоящее изобретение дополнительно включает преобразование, изменение размера (re-sizing) способных к слиянию везикул,полученных после слияния липосом, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, с виросомами, содержащими слитые белки с различными характеристиками слияния. В соответствии с изобретением, указанное преобразование размера может быть осуществлено путем экструзии везикул. В соответствии с этим настоящее изобретение предусматривает этап снижения размера виросомоподобных протеосом, полученных путем слияния химерных виросом с нагруженными липосомами, который не вызывает ни заметной потери из везикул инкапсулированного вещества, ни заметной инактивации оставшегося функционального слитого белка с сопутствующей утратой способности к слиянию. Поэтому в предпочтительном примере осуществления изобретения продукты реакции слияния виросом с липосомами подвергают экструдированию через мембраны с ядерными порами (nucleopores) с целью уменьшения их размера. Как видно на фиг. 7 (нижние кадры), криоэлектронная микроскопия показывает,что экструзия через поры диаметром 200 нм дает везикулы примерно половинного в сравнении с исходным размера. Большинство частиц имеют диаметр от 100 до 300 нм, видно также очень небольшое количество везикул большего размера (500-600 нм). Тот факт, что этап экструзии приводит к преобразованию размера способных к слиянию виросомоподобных везикул без существенной потери инкапсулированного вещества, является в достаточной степени непредвиденным и полезным результатом, поскольку можно было бы ожидать, что приложенные в процессе экструзии напряжения сдвига приведут к разрушению большей части мембран, вызывая потерю захваченного материала и инактивацию слитых белков. То, что этап экструзии не приводит ни к существенной потере включенной плазмидной ДНК, ни к инактивации белка для вторичного слияния (гемагглютинин PR8), было подтверждено тестированием у экструдированных протеолипосом способности к слиянию и содержания в них ДНК. Анализ методом FRET показал,что способность к слиянию у нагруженных протеолипосом до и после экструзии была одной и той же(фиг. 8). Кроме того, определили содержание плазмиды в экструдированных протеолипосомах. Для этого были приготовлены липосомы, содержащие радиоактивную плазмидную ДНК (оставшуюся свободную ДНК расщепляли ДНКазой и нуклеотиды удаляли с помощью гель-хроматографии, количество включенной ДНК принимали за 100%). Затем эти липосомы подвергали влиянию с виросомами. Одну аликвоту продуктов слияния сразу подвергали действию ДНКазы I, другую - после экструзии сквозь мембрану с ядерными порами. После удаления свободных нуклеотидов определяли радиоактивность, захваченную внутрь протеолипосом. Результаты позволяют сделать заключение, что в ходе этапа первоначального слияния утрачивается не более 15% (стандартное отклонение 5%; n=2), а в ходе этапа слияния и экстру-5 008497 зии - не более 25% (стандартное отклонение 3%; n=2) первоначально захваченной ДНК. Таким образом,радиоактивность, обнаруживаемая в протеолипосомах после экструзии, лишь ненамного ниже, чем до экструзии. То, что слияние липосом с виросомами сохраняет большую часть включенного вещества, даже более удивительно, если принять во внимание сообщение, что опосредованное гемагглютинином слияние мембран является процессом, не обеспечивающим целостности (Shannguan и др.//Biochemistry. 1996. Т. 35. С. 4956-4965). Таким образом, настоящее изобретение предусматривает способ приготовления способных к слиянию протеолипосомных везикул, которые эффективно инкапсулируют целевое вещество для доставки к клеткам и тканям. Захват клетками этих новых протеолипосомных везикул может быть прослежен с помощью флуоресцирующего липида родамин-фосфатидилэтаноламина (Rh-PE), стабильно включенного в двойной мембранный слой. Меченые протеолипосомы добавляли к клеткам MDCK или HeLa (приблизительно 1105 клеток), культивируемым на покровных стеклах в планшетах с 24 ячейками. После инкубации при 4 С в течение 1 ч для обеспечения связывания с клетками, несвязанные частицы удаляли, а клетки инкубировали еще 15 мин при 37 С. После фиксирования формальдегидом клетки анализировали с помощью флуоресцентной микроскопии. После этой (короткой) инкубации при 37 С ясно видно точечное окрашивание по поверхности ядер(фиг. 9 В), указывающее на быстрое поглощение меченых родамином протеолипосом. Этот процесс зависит от температуры, поскольку при 4 С все частицы оставались на поверхности клеток (фиг. 9 А). Более того, он зависит также от наличия в мембране везикул слитого белка, так как лишенные белка Rhлипосомы не поглощались до заметной степени в течение 2 ч. Эти результаты показывают, что химерные протеолипосомы быстро поглощаются; этот процесс во всем подобен направляемому рецепторами эндоцитозу, как в случае вируса гриппа. Подобно этому, к липосомам можно добавить декстран в высокой концентрации. Предполагается, что благодаря меньшему размеру молекул декстрана, в одну протеолипосому могут быть включены несколько меченых молекул. Действительно, теоретически при использованной концентрации декстрана (37,5 мг/мл) в одну липосому может быть заключено приблизительно 170 молекул декстрана. При степени замещения 6 флуорофоров на молекулу декстрана в липосоме могут находиться тысяча молекул Texas-red. Ожидалось, что если инкапсулированный материал высвобождается в цитозоль посредством диффузии, флуоресцентная микроскопия обнаружит окрашивание цитозоля,в отличие от точечного окрашивания поверхности ядер в случае, когда вещество остается удержанным. Как описано в примерах, декстран был захвачен внутрь липосом POPC/DDAB. Липосомы были слиты с химерными виросомами, и продукты слияния были добавлены к клеткам, культивируемым на покровных стеклах. Клетки инкубировали в течение 1 ч при 4 С, затем несвязанный материал отмывали, и клетки инкубировали в течение различных промежутков времени при 37 С. После фиксации 3,7% формальдегидом (объемные проценты) клетки анализировали с помощью флуоресцентной микроскопии. Включение содержащих декстран протеолипосом можно было отметить через 15 мин инкубации при 37 С, что было уже видно для меченых родамином протеолипосом. На этой стадии существенное количество красителя находилось на поверхности клеток (фиг. 10 В). Позднее (фиг. 10 С и 10D) наблюдалось более интенсивное окрашивание поверхности ядер. Для дальнейшей демонстрации осуществимости выбранного подхода были определены эффективности инкапсулирования других выбранных терапевтических веществ. Были использованы гидрофобные и гидрофильные пептиды, белки (например, рекомбинантный дающий зеленую флуоресценцию белок green fluorescent protein, GFP) и малые молекулы (например, PicoGreen). В табл. 1 суммированы эти результаты. При использовании общепринятого способа приготовления (Schoen и др.//J. Liposome Res. 1993. Т. 3. С. 767-792) полученные эффективности инкапсулирования составляли менее 5%. Однако новый подход обеспечил в каждом случае эффективность инкапсулирования по меньшей мере выше 15%-6 008497 Таблица Эффективности инкапсулирования различных молекул на последовательных стадиях в ходе приготовления ИРГВ) Приготовление обычных ИРГВ описано в примере 3. 4) GFP - дающий зеленую флуоресценцию белок (green fluorescent protein). 5) PicoGreen - интеркалятор в ДНК. ИРГВ - иммунопотенцирующие реконструированные гриппозные виросомы. Таким образом, настоящее изобретение предусматривает усовершенствованные системы-носители на основе виросом для доставки в клетки терапевтических молекул, в том числе макромолекул. Благодаря их параметрам безопасности (по сравнению с вирусными векторами) и быстрому их захвату клетками,виросомоподобные способные к слиянию протеолипосомы согласно настоящему изобретению представляют собой многообещающую систему, которая может быть также применена для приготовления фармацевтической композиции для лечения или предотвращения вирусных, бактериальных и грибковых инфекционных заболеваний, дегенеративных заболеваний, заболеваний, связанных с гиперпролиферацией и других связанных с действием киназ расстройств, включая рак, инфекционные заболевания, хронические инфекционные заболевания, хронические заболевания, аллергии, заболевания сердечно-сосудистой системы, воспалительные заболевания (в том числе кожные воспаления, такие как псориаз), заболевания иммунной системы, связанные с раком расстройства иммунитета, астму или артрит. Дополнительным преимуществом представленных здесь везикул является потенциальная возможность их адресованного направления к определенным клеткам или тканям путем их совместной реконструкции с включением в везикулярную мембрану рецепторов, антител или фрагментов антител (Mastrobattista и др.//FEBS Lett. 2001. Т. 509. С. 71-76). Кроме того, поскольку было показано, что виросомы являются прекрасными адъювантами (Glck//Vaccine. 1999. Т. 17. С. 1782-1787), виросомоподобные протеолипосомы согласно настоящему изобретению могут быть использованы, кроме доставки лекарств, в вакцинации. Обеспечивая эффективное инкапсулирование веществ, в том числе макромолекул, настоящее изобретение удовлетворяет настоятельную потребность в данной области. Продемонстрированные здесь эффективности включения (25-30%) модельной плазмиды внутрь замкнутых однослойных способных к слиянию протеолипосом намного превышают эффективности включения по сравнению с используемыми до настоящего времени технологиями, где измеренные эффективности включения были ниже 1% (Schoen и др.//J. Liposome Res. 1993. Т. 3. С. 767-792). В дополнение к этому, было установлено, что приготовленные в качестве примера протеолипосомы способны к эффективному слиянию с модельными мембранами при низком рН и повышенной температуре. Их поглощение клетками в культуре (HeLa,MDCK) было быстрым и столь же эффективным, как для вирусов и виросом (Bron и др.//Biochemistry. 1994. Т. 33. С. 9110-9117; Nunes-Correia и др.//Biochemistry. 1999. Т. 38. С. 1095-1101). Приведенные далее примеры предназначены для лучшей иллюстрации заявленного изобретения и не должны быть интерпретированы как ограничивающие область изобретения. В той степени, как упомянуты конкретные материалы, это сделано только для целей иллюстрации и не предназначается для ограничения изобретения. Специалист в данной области может выбрать эквивалентные приемы и реагенты без нарушения объема изобретения и без выхода за рамки настоящего изобретения. Краткое описание графических материалов Фиг. 1. Этот рисунок показывает упрощенную схему, разъясняющую общую стратегию изобретения. На первой стадии слияния химерные виросомы сливают при 4 С (рН 5,0) с липосомами, содержащими подлежащее доставке вещество (в этом примере ДНК), слияние опосредует НА Х-31. После этого полученные нейтрализованные продукты слияния инкубируют с клетками. После направляемого рецепторами эндоцитоза виросомоподобные протеолипосомы (теперь содержащие подлежащее доставке вещество) претерпевают второй цикл слияния, запускаемый низким значением рН внутри эндосом и опосредуемый НА PR8/34, высвобождая захваченное вещество в цитозоль. Фиг. 2. Этот рисунок показывает наличие в мембране химерных виросом обоих типов слитых белков. Проведен электрофорез SDS-PAGE вирусов и виросом Х-31 и PR8. Приблизительно 10 мкг вируса и-7 008497 3 мкг виросом наносили при невосстанавливающих условиях на полиакриламидный гель в буфере SDSтрис/трицин 10% (объемные проценты) (Schagger и др.//Anal. Biochem. 1987. Т. 166. С. 368-379). Полиакриламидный гель окрашивали Кумасси голубым (Coomassie blue). Полоса 1: вирус Х-31. Полоса 2: виросомы Х-31. Полоса 3: химерные виросомы. Полоса 4: виросомы PR8. Полоса 5: вирус PR8. Фиг. 3. Этот рисунок показывает морфологию химерных виросом, определенную с помощью cryoТЕМ. Это однослойные везикулы с диаметрами в интервале от 150 до 250 нм. Линия масштаба соответствует 100 нм. Фиг. 4. Этот рисунок (фиг. 4.1-4.3) показывает активность слияния вирусов и виросом Х-31 и PR8 и химерных виросом X-31/PR8 с липосомами. Флуоресценцию измеряли непрерывно при длинах волн возбуждения и эмиссии - соответственно 465 и 530 нм. Измерения проводили с помощью флуориметраSPEX-Fluorolog-2 (SPEX Inductries, Inc., Edison, NJ, USA), оборудованного держателем для термостатирования кювет и магнитным перемешивающим устройством. Для калибровки шкалы флуоресценции начальную остаточную флуоресценцию липосом принимали за 0, а флуоресценцию при бесконечном разбавлении пробы - за 100%. Последнюю величину определяли, добавляя тритон Х-100 (0,5 об.%). 1) Вирус и виросомы Х-31, слитые с КOB POPC/POPG. 2) Вирус и виросомы PR8/34, инкубированные с КOB POPC/POPG. 3) Химерные виросомы, инкубированные с КOB POPC/POPG:a) измерения слияния при 4 С;b) вирус и виросомы перед измерениями слияния при 37 С инкубировали при 4 С и рН 5,0 в течение 1 ч с КOB POPC/DDAB. Фиг. 5. Этот рисунок показывает эффективность инкапсулирования ДНК в липосомы POPC/DDAB. Содержащие ДНК липосомы были приготовлены методом замораживания-оттаивания, как описано в примерах, и обрабатывали в течение 3 ч при 37 С смесью ДНКазы I и экзонуклеазы III. После этого инкапсулированную ДНК экстрагировали фенолом и хлороформом и подвергали гель-электрофорезу в 1% агарозном геле. По линии М был пропущен маркер 1 тыс. п.н. плюс набор зон (лестница). В качестве контроля 100 нг (линия 1) и 200 нг (линия 2) плазмидной ДНК наносили прямо на гель. В качестве других контролей были экстрагированы фенолом и хлороформом необработанные (не расщепленные ДНКазой I) липосомы с ДНК в количестве, соответствующем 20 нг (полоса 3), 50 нг (полоса 4), 100 нг (полоса 5) и 200 нг (полоса 6) плазмиды. Водную фазу наносили на гель. Обработанные ДНКазой I липосомы в количествах, соответствующих начальному количеству ДНК 50 нг (полоса 7) и 100 нг (полоса 8), обрабатывали так же, как и предыдущие пробы. Полосы 9 и 10 соответствовали такому же количеству липосом,за исключением того, что липосомы перед обработкой ДНКазой I были растворены в тритоне Х-100(1 об.%). На полосах 11 и 12 - ДНК, которая не была инкапсулирована, но была добавлена к пустым липосомам. Эти липосомы были так же обработаны, как описано выше. Фиг. 6. Этот рисунок показывает на основании cryo-ТЕМ морфологию загруженных липосом. Они однослойны и имеют диаметр от 100 до 150 нм. Линия шкалы соответствует 100 нм. Фиг. 7. Этот рисунок показывает результаты исследования с помощью cryo-ТЕМ продуктов слияния химерных виросом и нагруженных липосом до (верхний ряд) и после (нижний ряд) экструдирования. Перед экструдированием везикулы имеют широкий разброс диаметров, и диаметр некоторых везикул превышает 900 нм. Этот результат указывает на множественность циклов слияния. Шкала в верхнем ряду: 500 нм; в нижнем ряду: слева - 100 нм, справа - 200 нм. Фиг. 8. Этот рисунок демонстрирует способность к слиянию экструдированных везикул, полученных слиянием химерных виросом с липосомами. Важно отметить, что везикулы сохраняют свою способность к слиянию. Протеолипосомы были экструдированы сквозь мембраны с ядерными порами. Флуоресценцию измеряли непрерывно при длинах волн возбуждения и эмиссии соответственно 465 и 530 нм. Измерения проводили с помощью флуориметра SPEX-Fluorolog-2, оборудованного держателем для термостатирования кювет и магнитным перемешивающим устройством. Флуоресценция 100% соответствовала бесконечному разбавлению пробы и определялась после добавления тритона Х-100 (0,5 об.%). Измерения слияния проводили при 37 С и рН 5,0 с КOB POPC/POPG (4:1), содержащими 0,6% N-NBD-PE иN-Rh-РЕ. Фиг. 9. Этот рисунок показывает, что протеолипосомы быстро поглощаются клетками. Меченые родамином протеолипосомы обнаруживаются в клетках MDCK и HeLa. Слитые виросомы связывались с клетками HeLa и MDCK при 4 С в течение 1 ч. После этого несвязанный материал отмывали. Клетки немедленно фиксировали (А) или инкубировали еще 15 мин при 37 С, фиксировали формальдегидом(3,7 об.%) и анализировали с помощью флуоресцентной микроскопии (В). Фиг. 10. Этот рисунок демонстрирует быстрый захват клетками HeLa протеолипосом, загруженных декстраном с флуорофором Texas Red. Клетки HeLa инкубировали с содержащими декстран с флуорофором Texas Red протеолипосомами в течение 1 ч при 4 С. После этого несвязанный материал отмывали-8 008497 и клетки фиксировали формальдегидом (3,7 об.%) (А). В других случаях клетки дополнительно инкубировали при 37 С в течение 15 мин (В), 2 ч (С) или 5 ч (D). После фиксации клетки анализировали с помощью флуоресцентной микроскопии. Увеличения объектива: для (А) - 63, для (В), (С) и (D) - 100. Описание примеров осуществления изобретения Пример 1. Материалы и методы Химические реагенты. Октаэтиленгликоль-моно-(n-додецил)-эфир (OEG, С 12 Е 8) и дидодецилдиметиламмонийбромид(DDAB) были предоставлены фирмой Fluka, поли-L-лизина гидробромид (мол. масса 29300), протамина сульфат, -целлюлоза, микрокристаллическая целлюлоза и 1,2-дипальмитоил-sn-глицеро-3-фосфо-rac-(1 глицерин) (PG) были предоставлены фирмой Sigma (Buchs, Switzerland). Яичный фосфатидил-холин (PC) был получен от фирмы Lipoid (Cham. Switzreland). Фосфатидилэтаноламин (РЕ) был предоставлен R. Berchtold (Biochemical Laboratory, University ofBern, Switzerland). Биогранулы (Bio-Beads) SM2 и биогель A-15m были предоставлены фирмой Bio-RadLaboratories (Glattbrugg, Switzerland). POPC, N-NBD-PE и N-Rh-PE были предоставлены фирмой AvantiPolar Lipids (Birmingham, AL, USA). Панкреатическая ДНКаза I (из поджелудочной железы крупного рогатого скота) с удельной активностью 2000 ед. Куница на мг и экзонуклеаза III (Е. coli, удельная активность 100 ед./мкл) были предоставлены фирмами Roche Diagnostics (Basel, Switzerland) и New EnglandBioLabs (BioConcept, Alschwill, Switzerland). ДНК-полимераза Taq была предоставлена фирмой Promega(Catalys, Wallisellen, Switzerland). N-(4,4-дифтор-5,7-дифенил-4-бор-3 а,4 а-диаза-s-индацен-3-пропионил)1,2-дигексадеканоил-sn-глицеро-3-фосфоэтаноламин (Bodipy 530/550-DHPE), бромистый моноазид этидия (ethidium monoazide bromide, EMA), меченый Texas Red декстран (мол. масса 70000) и реагент PicoGreen были предоставлены фирмой Molecular Probes Europe (Leiden, The Netherlands). Олигонуклеотиды, используемые для полимеразной цепной реакции и для опытов с введением антисмысловых последовательностей, были предоставлены фирмой Microsynth (Balgach, Switzerland). Плазмиды pT7-EGFP,VR1012, EGFP-VR1012 и меченая 35S плазмида были любезно предоставлены Karin Moelling (Institute ofMedical Virology, University of Zurich, Switzerland). Вирусы. Вирусы гриппа: рекомбинантный штамм Х-31 и штамм A/PR/8/34 (PR8), размноженные в аллантоидной полости оплодотворенных яиц (Gerhard//J. Exp. Med. 1976. Т. 144. С. 985-995), были получены от фирмы Berna Biotech (Bern, Switzerland). Рекомбинантный вирус осповакцины vTF7-3 был любезно предоставлен Karin Moelling (Institute of Medical Virology, University of Zurich, Switzerland). Культуры клеток. Клетки MDCK и HeLa поддерживали в минимальной среде Игла MEM (Life Technologies, Basel,Switzerland), дополненной 10% сывороткой коровьего плода (FCS), пенициллином (100 МЕ/мл) и стрептомицином (100 мкг/мл). Пример 2. Приготовление иммунопотенцирующих реконструированных гриппозных виросом (ИРГВ) Виросомы были приготовлены способом, описанным Bron и соавторами (Bron и др.//Methods Enzymol. 1993. Т. 220. С. 313-331). Вкратце, вирус (1,5 мкмоль фосфолипида или 5 мг вирусного белка) осаждали ультрацентрифугированием и солюбилизировали в 0,7 мл 100 мМ OEG в забуференном фосфатом солевом растворе (PBS: 150 мМ NaCl, 5 мМ Na-фосфат, рН 7,4). Нуклеокапсид удаляли ультрацентрифугированием (100000 g, 30 мин). Надосадочную жидкость (содержащую вирусные липиды и белки оболочки) встряхивали (1200 об./мин) с 180 мг влажных биогранул Bio-Beads SM2 в течение 1 ч при комнатной температуре и затем дважды в течение 10 мин с 90 мг Bio-Beads при 1400 об./мин для удаления детергента. После этого препарат виросом центрифугировали через ступенчатый градиент плотности сахарозы(10-40%, масса к объему) в PBS при 130000 g в течение 90 мин. Виросомы собирали с поверхности раздела между слоями сахарозы. Подобным же образом готовили химерные виросомы, содержащие НА из X-31 и PR8. Перед солюбилизацией смешивали равные количества вирусных белков из двух штаммов вируса. Соотношение гемагглютинин/фосфолипид устанавливали, определяя фосфорипид по Bttcher (Bttcher и др.//Anal. Chim. Acta. 1961. Т. 24. С. 202-203) и количественно определяя НА после SDS-PAGE методом экстракции с Кумасси по Ball (Ball//Anal. Biochem. 1986. Т. 155. С. 23-27). Пример 3. Приготовление обычных ИРГВ с инкапсулированными молекулами ИРГВ в основном готовили, как описано Zurbriggen и др. (//Progr. Lipid Res. 2000. Т. 39. С. 3-18). Вкратце, 32 мг яичного PC и 8 мг РЕ растворяли в 2 мл PBS, содержащего 100 мМ OEG (PBS/OEG). Вирус гриппа A/Singapore был очищен, как описано (Skehel and Schild//Virology. 1971. Т. 44. С. 396-408). Количество вируса гриппа, соответствующее 4 мг гемагглютинина (НА) центрифугировали при 100000 g в течение 30 мин, осадок растворяли в 1 мл PBS/OEG. Чтобы приготовить ИРГВ с пептидом, 4 мг пепти-9 008497 да растворяли в 1 мл PBS/OEG. Фосфолипиды, раствор солюбилизированного вируса и раствор пептида смешивали и обрабатывали ультразвуком в течение 1 мин. Эту смесь центрифугировали при 100000 g в течение 1 ч, надосадочную жидкость стерилизовали фильтрованием через поры 0,22 мкм. Затем получали виросомы путем удаления детергента с использованием 180 мг влажных Bio-Beads SM2 (встряхивание в течение 1 ч при комнатной температуре) и затем 3-кратного встряхивания с 90 мг Bio-Beads SM2 в течение 30 мин. Размер ИРГВ определяли по светорассеянию с помощью прибора Zetasizer 1000HS (Malvern Instruments, UK). Для количественного определения инкапсулированного пептида фракцию гомогенных ИРГВ наносили на гель-хроматографическую колонку с сефадексом G50 грубым (Amersham Biosciences, Switzerland) и отделяли от неинкапсулированного пептида. Количественное определение пептида проводили с помощью прибора kta Explorer 10 (Amersham Biosciences, Switzerland), используя обратно-фазную колонку СС 125/4.6 Nucleosil 100-5 С 8 (Macherey-Nagel, Switzerland). Чтобы приготовить ИРГВ с белком (например, GFP-ИРГВ), 10 мкг белка растворяли в 1 млPBS/OEG и проводили дальнейшие процедуры, как описано выше. Чтобы приготовить PicoGreen-ИРГВ,20 мкл базового раствора добавляли к 1 мл PBS/OEG и проводили дальнейшие процедуры, как описано выше. Количественное определение GFP и PicoGreen осуществляли на люминесцентном спектрометреLS 55 (Perkin Elmer Instruments, USA). Были приготовлены химерные виросомы, содержащие НА из вирусов гриппа Х-31 и A/Singapore, но раствор пептида заменили 1 мл PBS/OEG. Перед солюбилизацией смешали равные количества белков НА из двух штаммов вируса. Пример 4. Приготовление крупных однослойных везикул (КOB)POPC/DDAB (12 мг, молярное соотношение 98:2) растворяли в смеси хлороформ/метанол. Затем растворитель удаляли выпариванием, после чего производили сушку в течение ночи при 10-2 мм рт.ст. Высушенную смесь липидов диспергировали в 50 мкл трис-буфера (50 мМ трис, рН 8,0) и обрабатывали ультразвуком в течение 15 мин в камерном излучателе. После добавления подлежащего инкапсулированию вещества концентрацию липидов доводили до 120-160 мМ. Дисперсную систему замораживали в жидком азоте и оттаивали при комнатной температуре 10 раз. Перед экструдированием дисперсную систему липосом разбавляли трис-буфером до концентрации липидов 40 мМ и экструдировали 10 раз - каждый раз через поликарбонатные мембраны с порами 0,2 и 0,1 мкм (Nucleopore Track-Etch Membrane,Sterico, Switzerland), используя самодельный экструдер (аналогичный экструдеру LiposoFast фирмыAvestin Inc.). Плазмидную ДНК инкапсулировали, в основном, как описано Monnard с соавторами (Monnard и др.//Biochim. Biophys. Acta. 1997. Т. 1329. С. 39-50). К обработанным ультразвуком липосомам добавили 10 мкг ДНК (в некоторых опытах содержавшую также меченую 35S плазмидную ДНК) и обрабатывали,как описано выше. Не захваченную в липосомы ДНК расщепляли добавлением к содержащим ДНК липосомам 700-800 ед. панкреатической ДНКазы I, 200 ед. экзонуклеазы III, 5 мМ MgCl2 и 0,1 мМ дитиотреитола (DTT). Для опытов с большими количествами ДНК количество ферментов пропорционально увеличивали. После инкубации в течение 3 ч при 37 С реакцию останавливали добавлением этилендиаминтетраацетата (EDTA) до конечной концентрации 7 мМ. Внешний объем среды отделяли от липосом на центрифужных колонках для гель-хроматографии (Bio-Gel A-15m, предварительно уравновешен трисHCl буфером, рН 8,0). Обычно колонки центрифугировали при 165 g в течение 2 мин. Собирали от 10 до 15 фракций, объем каждой - около 50 мкл. Обычно фракции 2-7 были мутными, остальные не были заметно мутными, что указывало на то, что они не содержали заметного количества липосом. Количественное определение инкапсулированной в КOB ДНК Чтобы определить долю включенной плазмидной ДНК, проводили совместное включение с меченой 35S ДНК. По радиоактивности, изначально добавленной, и радиоактивности, оставшейся связанной с липосомами после экстенсивного расщепления смесью ДНКазы I и экзонуклеазы III и гельхроматографии, можно было определить долю захваченной липосомами ДНК (по измерениям на жидкостно-сцинтилляционном счетчике). Количественное определение проводили также путем визуализации плазмидной ДНК в 1% агарозных гелях до и после расщепления. Для этих опытов плазмидную ДНК экстрагировали фенолом и хлороформом из необработанных липосом и из липосом, обработанных смесью ДНКазы I и экзонуклеазы III и очищенных гель-хроматографией. Содержащий ДНК водный раствор подвергали электрофорезу в геле. Гель окрашивали бромистым этидием, количество плазмидной ДНК в соответствующих зонах определяли путем сравнения с известными количествами плазмиды. Пример 5. Приготовление крупных однослойных везикул (КOB) Растворяли в смеси метанол/хлороформ (2:1) 36,4 мкмоль (27,95 мг) PC и 15,6 мкмоль (10,75 мг) PG(молярное соотношение 70:30). Растворитель удаляли на ротационном испарителе (Rotavapor R-205, Buchi Labortechnik, Switzerland) при 40 С при постепенном усилении вакуума от 30 до 10 кПа. Для получения пептидо-липосом, содержащих гидрофобные пептиды, 2-3,5 мг пептида растворяли в метаноле и добавляли к фосфолипидной смеси перед удалением растворителя.- 10008497 Высушенную липидную пленку увлажняли в 500 мкл PBS. Для получения пептидо-липосом, содержащих гидрофильные пептиды, высушенную липидную пленку увлажняли в 350 мкл PBS с добавлением 2-3,5 мг подлежащего инкапсулированию пептида. Для получения GFP-липосом высушенную липидную пленку увлажняли в 350 мкл PBS с добавлением 10 мкг GFP. Для получения PicoGreen-липосом высушенную липидную пленку увлажняли в 350 мкл PBS с добавлением 1,75 мкл базового раствора PicoGreen. Перед экструдированием объем доводили до 500 мкл добавлением PBS. Дисперсную систему липосом экструдировали 10 раз сквозь поликарбонатные мембраны (Nucleopore Track-Etch membrane,поры 0,1 мкм или 0,2 мкм, Whatman, UK) с помощью экструдера Lipex Extruder (Northern Lipids, Canada). Размер экструдированных липосом определяли по светорассеянию с помощью прибора Zetasizer 1000HS(Malvern Instruments, UK). Для количественного определения инкапсулированного пептида фракцию гомогенных липосом наносили на колонку для гель-хроматографии с сефадексом G50 грубым и отделяли от неинкапсулированного пептида. Количественное определение пептидов производили на приборе ktaExplorer 10 (Amersham Biosciences, Switzerland), используя обратно-фазную колонку СС 125/4.6 Nucleosil 100-5 С 8 (Macherey-Nagel, Switzerland). Количественное определение GFP и PicoGreen выполняли на люминесцентном спектрометре LS 55 (Perkin Elmer Instruments, USA). Пример 6. Приготовление химерных протеолипосом Химерные виросомы (50 мкл) инкубировали с 2 мкл КOB POPC/DDAB при 4 С в PBS (конечный объем 200 мкл), рН доводили до 5,0 добавлением 5 мкл лимонной кислоты (150 мМ, рН доведен до 3,5). После нейтрализации добавлением 1,4 мкл 1 М NaOH продукты слияния подвергали экструдированию 10 раз сквозь поликарбонатные мембраны с диаметром пор 0,2 мкм. Свежие слитые протеолипосомы для каждого опыта готовили заново, так как было замечено, что они склонны к агрегации. Чтобы проанализировать, не приводит ли экструзия к потерям ДНК, содержащие меченую 35S ДНК сливали с химерными виросомами и подвергали экструзии, как описано выше. Затем протеолипосомы обрабатывали ДНКазой I и экзонуклеазой III, как уже было описано выше. На жидкостном сцинтилляционном счетчике определяли долю радиоактивности, оставшуюся связанной с протеолипосомами после гель-хроматографии. Контрольные пробы обрабатывали аналогично, за исключением того, что их либо не подвергали экструдированию, либо подвергали экструдированию, но не обрабатывали нуклеазами. Измерения слияния с помощью FRET Для опытов по слиянию in vitro каждый из N-NBD-PE и N-Rh-PE вводили в КOB, состоящие изPOPC/DDAB (98:2) (1 мг/мл). Измерения флуоресценции проводили при 4 и 37 С в конечном объеме 2 мл PBS (5 мМ фосфат Na, pH 7,4 и 150 мМ NaCl). В кювете присутствовали липосомы (19 мкл) и виросомы (30-50 мкл). После добавления 48 мкл лимонной кислоты (150 мМ, рН доведен до 3,5 добавлениемNaOH) непрерывно регистрировали флуоресценцию при длинах волн возбуждения и эмиссии соответственно 465 и 530 нм. Измерения проводили на флуориметре SPEX Fluorolog-2 (SPEX Industries, Edison,NJ, USA), оборудованном термостатированным держателем кюветы и магнитной мешалкой. Для калибровки шкалы значений флуоресценции начальную остаточную флуоресценцию принимали за 0, а флуоресценцию пробы с бесконечным разбавлением, определенную после добавления тритона Х-100 (конечная концентрация 0,5 об.%) - за 100%. Криоэлектронная микроскопия пропускания (Cryo-TEM) Небольшие капли содержащей виросомы или липосомы дисперсной системы наносили на покрытые углем медные сеточки и немедленно замораживали в жидком этане в системе контролируемого стеклования (Engelhaaf и др.//J. Microsc. 2000. Т. 200. С. 128-139). Стеклованные образцы переносили в модифицированном охлаждаемом держателе (Gatan, Pleasanton, CA, USA) в электронный микроскоп пропускания (Zeiss EM 912 OMEGA), работающий в условиях нулевых потерь при 120 кэВ. Через 5 мин,необходимых для уравновешивания, образцы просматривали при -170 С. Электронные микрофотографии получали в цифровой форме без поправки на фон с помощью охлаждаемой камеры 10241024 CCD(Proscan, Germering, Germany) под контролем системы анализа изображений SYS версия 2.1 (SYS, Mnster, Germany). Пример 7. Приготовление химерных протеолипосом Химерные виросомы (600 мкл в PBS) инкубировали с 200 мкл КOB PC/PG при 15 С в PBS при непрерывном перемешивании. Чтобы инициировать процесс слияния, доводили рН до приблизительно 4,5 добавлением 15 мкл 1 М HCl. После инкубации в течение 30 мин смесь нейтрализовали добавлением 15 мкл 1 М NaOH, и продукты слияния подвергали экструдированию 10 раз сквозь поликарбонатные мембраны с порами 0,2 мкм, как описано в примере 4. Для измерений слияния in vitro методом FRET был разработан альтернативный метод анализа: в КOB, состоящие из PC/PG (70:30), вводили 0,75 мол.%Bodipy530/550-DHPE и 0,25 мол.% Rh-DHPE. Bodipy530/550-DHPE обладает преимуществом перед NNBD-РЕ, так как его коэффициент экстинкции втрое выше и меньше зависит от растворителя. Измерения флуоресценции проводили при различных температурах в интервале от 4 до 42 С в 5 мМ натрийфосфатном буфере рН 7,5 с 100 мМ NaCl в конечном объеме 0,8 мл в полиметилметакрилатных микрокюветах объемом 2,5 мл (VWR, Switzerland) при непрерывном перемешивании. Обычно 1 мкл меченых- 11008497 липосом (0,3 мкмоль фосфолипида) смешивали с 5-50 мкл виросом, слияние инициировали добавлением 3,75-7 мкл 1 М HCl, что давало рН около 4,5. Увеличение флуоресценции регистрировали непрерывно при длинах волн возбуждения и эмиссии соответственно 530 и 550 нм, при щели возбуждения 2,5 нм и щели эмиссии 15,0 нм. Измерения проводили на люминесцентном спектрометре LS 55 (Perkin Elmer Instruments, USA), снабженном термостатированным держателем кювет и магнитной мешалкой. Максимальная флуоресценция при бесконечном разбавлении пробы достигалась после добавления тритона Х 100 (конечная концентрация 0,5 об.%). Пример 8. Опыты по трансфекции Клетки MDCK или HeLa (1105) высевали на планшеты с 24 ячейками в день накануне трансфекции и трансфицировали 1 мкг на ячейку плазмидной ДНК (EGFP-VR1012), используя реагент SuperFect в соответствии с прописью поставщика (Qiagen, Basel, Switzerland). Приготовление меченых родамином химерных протеолипосом 1 мг липидов (POPC/DDAB/N-Rh-PE 98:2:0,5) растворяли в смеси хлороформ/метанол. Затем растворитель удаляли выпариванием с последующей сушкой в течение ночи при 10-2 мм рт.ст. Высушенные липиды диспергировали в 200 мкл PBS. Дисперсную систему 10 раз замораживали в жидком азоте и оттаивали при комнатной температуре. После этого липосомы подвергали экструзии 10 раз - каждый раз через поликарбонатные мембраны с порами 0,2 и 0,1 мкм. Меченые родамином протеолипосомы готовили из 50 мкл КOB с родамином и 100 мкл химерных виросом в общем объеме 200 мкл, как описано выше. Вкратце, дисперсную систему подкисляли и инкубировали в течение 10 мин при 4 С. После нейтрализации 1 М NaOH протеолипосомы подвергали экструдированию 10 раз сквозь поликарбонатную мембрану с порами 0,2 мкм. Для трансфекции содержащими ДНК протеолипосомами высевали такое же количество клеток. Химерные протеолипосомы (10-20 мкл), содержащие плазмиду EGFP-VR1012, инкубировали с клетками в 200 мкл среды (MEM или MEM с добавкой 10% FCS, или среда RPMI с добавкой 0,2% BSA) при 4 С в течение 1 ч или при 37 С в течение 2,5-4 ч, после чего среду удаляли. Затем клетки инкубировали в полной ростовой среде в атмосфере 5% СO2 при 37 С в течение 48-70 ч. Экспрессию EGFP в трансфецированных клетках оценивали с помощью широкопольного микроскопа Axiovert фирмы Zeiss. Пример 9. Исследование включения в клетки меченых родамином протеолипосом Клетки MDCK или HeLa (1105) выращивали до 60-80% заполнения слоя на 12 мм покровных стеклах, покрытых Alcian-blue, и инкубировали с 10 мкл меченых родамином протеолипосом в 100 мкл среды RPMI с добавкой 20 мМ HEPES (гидроксиэтил-пиперазин-этансульфонат) и 0,2% BSA в течение 1 ч при 4 С с защитой от света. Затем клетки тщательно промывали ледяным PBS и инкубировали в полной ростовой среде (MEM с 10% FCS) в атмосфере 5% СO2 при 37 С. В указанные моменты времени клетки фиксировали 3,7 об.% формальдегидом. Покровные стекла монтировали в Moviol и анализировали с помощью широкопольного микроскопа Axiovert фирмы Zeiss. Изображения получали с помощью камерыCCD (Hamamatsu) и стандартных светофильтров. Исследование клеточной локализации ДНК, меченой бромистым моноазидом этидия (ЕМА) Меченую ЕМА плазмидную ДНК готовили, в основном, как было описано (Zabner и др.//J. Biol.Chem. 1995. Т. 270. С. 18997-19007; Serikawa и др.//Biochim. Biophys. Acta. 2000. Т. 1467. С. 419-430). К 200 мкг EGFP-VR1012 в 100 мкл Н 2O добавляли 5 мкг ЕМА (раствор 5 мг/мл в EtOH). После инкубации в течение 15 мин во льду раствор фотоактивировали в течение 10 мин (источник света SUSS LH 1000lamphouse (KARL SUSS, Waterbury Center, VT, USA), снабженный ртутной лампой высокого давленияOSRAM HBO мощностью 350 Вт). Для удаления свободного реагента ДНК трижды осаждали 3-кратным объемом этанола и 1/10 объема 3 М ацетата натрия (рН 5,2). Меченую ДНК ресуспендировали в 70-100 мл Н 2O. Затем меченую ЕМА ДНК либо инкапсулировали в КOB и использовали для опытов по трансфекции с протеолипосомами, либо готовили с SuperFect, как описано выше. Для исследования внутриклеточной локализации меченой ЕМА ДНК использовали широкопольный микроскоп Axiovert фирмы Zeiss. Исследование захвата клетками протеолипосом, содержащих меченый Texas red декстран К 6 мг высушенных липидов (POPC/DDAB 98:2) в 50 мкл буфера трис-HCl (50 мМ трис-HCl, рН 8,0) добавляли 3 мг декстрана, меченого Texas red (мол. масса 70000) в 30 мкл трис-буфера. Липосомы готовили, как описано выше. Свободный декстран отделяли от липосом центрифугированием в градиенте плотности сахарозы. Вкратце, 100 мкл экструдированных липосом смешивали с 900 мкл 60% сахарозы в трис-буфере, затем поверх этого наслаивали по 1 мл 30, 20 и 10% сахарозы. Конечный слой был 500 мкл буфера трис-HCl. Градиенты центрифугировали в течение 3 ч при 35000 об./мин и 20 С в ротореSW55 (Beckman). Содержащие декстран липосомы собирались в тонкий слой в верхних областях градиента и могли быть собраны. Собранную фракцию разбавляли трис-буфером и концентрировали приблизительно до 200 мкл с помощью ультрафильтра в центрифужном фильтровальном устройстве (Millipore,- 12008497Volketswil, Switzerland). Содержащие декстран с Texas red липосомы сливали с химерными виросомами на холоде, как описано выше. Продукты слияния инкубировали с клетками, как уже было описано. Захват декстрана сTexas red в цитозоль и высвобождение анализировали после фиксации клеток с помощью флуоресцентной микроскопии на широкопольном микроскопе Axiovert фирмы Zeiss. Пример 10. Опыты по трансфекции с использованием системы цитоплазматической экспрессии Т 7 Клетки HeLa (3105), выращенные на покровных стеклах, заражали vTF7-3 (множественность инфицирования 5). Через 1 ч инкубационную среду удаляли и клетки трансфицировали протеолипосомами,содержащими pT7-EGFP, или липофектамином, согласно прописи поставщика (Invitrogen AG, Basel,Switzerland). Среду заменяли через 4 ч после трансфекции и клетки инкубировали еще в течение 40-60 ч,после чего экспрессию GFP анализировали с помощью флуоресцентной микроскопии. Выделение плазмиды из трансфицированных клеток и амплификация с помощью полимеразной цепной реакции Клетки MDCK и HeLa выращивали до 60-80% заполнения слоя на чашках диаметром 60 мм. К клеткам добавляли, как описано выше, EGFP-VR1012, инкапсулированную в протеолипосомы, или комплексы с SuperFect. После инкубации в течение различных промежутков времени при 37 С клетки лизировали и плазмиду выделяли экстракцией по Hirt (Hirt//J. Mol. Biol. 1967. Т. 26. С. 365-369). Вкратце, в каждую чашку добавляли 0,4 мл раствора, содержащего 0,6% (масса к объему) SDS и 10 мМ EDTA и инкубировали в течение 20 мин при комнатной температуре. Клетки соскребали резиновой палочкой. Вязкую смесь переносили пипеткой в пробирку Eppendorf, добавляли 100 мкл 5 М раствора NaCl и смесь выдерживали во льду в течение не менее 5 ч. После центрифугирования в течение 4 мин при 14000 об./мин при 4 С надосадочную жидкость аккуратно удаляли и экстрагировали дважды фенолом и один раз хлороформом. Водную фазу осаждали в течение ночи при -20 С 3-кратным объемом этанола с добавкой 0,1 объема 3 М ацетата натрия (рН 5,2). Осадок промывали 70% этанолом и ресуспендировали в 20 мкл Н 2O. Для полимеразной цепной реакции (ПЦР) готовили следующую смесь реагентов в общем объеме 20 мкл: 1-2 мкл выделенной плазмидной ДНК,1 буфер для ДНК-полимеразы Taq,1,5-2 мМ MgCl2,20 мкМ дезоксинуклеозид-трифосфатов (dNTP, каждый по 5 мкМ),1 мкМ каждого праймера,1 ед. ДНК-полимеразы Taq. Использовали следующие праймеры:EGFP-rev (обратный): 5'-GGCTGTTGTAGTTGTACTCC-3' Параметры цикличности: 1 цикл 94 С, 45 с 19-24 циклов 94 С, 45 с 52 С, 45 с 72 С, 45 с 1 цикл 72 С, 10 мин Затем продукты анализировали в 8 об.% полиакриламидном геле толщиной 1 мм. Пропись геля: 1 мл 40 об.% акриламида 1 мл 5 ТВЕ (буфер трис-борат-EDTA) 2,625 мл Н 2O 375 мкл глицерина 30 мкл 10% (масса к объему) APS (персульфат аммония) 6 мкл TEMED (тетраметилэтилендиамин) Конденсация ДНК с поли-L-лизином или протаминсульфатом Комплексы ДНК-полилизин/ДНК-протамин готовили медленным добавлением различных количеств поликатиона (12,5-100 мкг) к 50 мкг плазмидной ДНК и 2,5 мкмоль липидов (POPC/DDAB, 98:2) в общем объеме 0,5 мл Н 2O или PBS. Пример 11. Опыты по введению антисмысловых последовательностей Приготовление протеолипосом, содержащих олигодезоксинуклеотиды с-myc. Из 15 мМ базового раствора олигодезоксинуклеотидов с-myc отбирали 100 мкл (1,5 мкмоль) и добавляли к обработанной ультразвуком липидной дисперсной системы. Все это обрабатывали, как описано для приготовления КOB. Не инкапсулированные олигодезоксинуклеотиды удаляли гель- 13008497 хроматографией, как описано для инкапсулирования плазмидной ДНК (без расщепления нуклеазами). Первые 4 порции элюата, содержавшие липосомы, объединяли и концентрировали до исходного объема(50 мкл) с помощью ультрафильтра в центрифужном фильтровальном устройстве (Millipore, Volketswil,Switzerland). Были использованы олигодезоксинуклеотиды (фосфотиоилированные) со следующими нуклеотидными последовательностями: антисмысловой: 5'-AACGTTGAGGGGCAT-3' беспорядочный: 5'-GAACGGAGACGGTTT-3' КOB, содержащие олигодезоксинуклеотиды, сливали с химерными виросомами и после этого подвергали экструзии. Инкапсулирование олигодезоксинуклеотидов c-myc в виросомы PR8 Чтобы инкапсулировать олигодезоксинуклеотиды с-myc непосредственно в виросомы PR8, 1,5 мкмоль олигодезоксинуклеотидов в 100 мкл Н 2O добавляли к фракции растворенного вируса. Затем виросомы PR8 готовили, как описано выше. Доставка олигонуклеотидов в клетки Клетки HeLa (4-6103 клеток на ячейку) высевали накануне трансфекции в планшеты с 96 ячейками. Для трансфекции олигофектамином (Invitrogen AG, Basej, Switzerland) клетки поддерживали в ростовой среде без пенициллина и стрептомицина. Клетки обрабатывали с-myc (антисмысловым и беспорядочным, "scrambled") фосфотиоилированными олигодезоксинуклеотидами (ОДН), объединенными (комплексированными) с олигофектамином согласно прописи поставщика. Для трансфекции содержащими ОДН протеолипосомами 2-3 мкл слитых виросом инкубировали в течение 3 дней с клетками в общем объеме 100 мкл среды (RPMI с 0,2% BSA или MEM, или MEM с 10%FCS) при 37 С в атмосфере 5% СO2. В некоторых опытах протеолипосомы вначале инкубировали в течение 1 ч при 4 С. Несвязанные частицы отмывали, добавляли питательную среду и проводили инкубацию при 37 С, как описано выше. Пролиферацию клеток тестировали через 70 ч с помощью реагента(Promega, Catalys, Switzerland). После добавления 20 мкл реагента в каждую ячейку и инкубации в течение 1-3 ч при 37 С в атмосфере 5% CO2 регистрировали оптическое поглощение при 450 нм с помощью считывающего устройства для микропланшетов Benchmark microplate reader (Bio-Rad Laboratories,Glattbrugg, Switzerland). Исследование захвата клетками меченых FITC олигодезоксинуклеотидов Клетки HeLa (1105), высеянные на 12 мм покровные стекла, обрабатывали олигофектамином и олигодезоксинуклеотидами, мечеными FITC (флуоресцеин-изотиоцианат), как описано выше. Для обработки химерными протеолипосомами или виросомами PR8 высевали такое же количество клеток. 20 мкл протеолипосом или 10 мкл виросом PR8, содержащих меченые FITC олигодезоксинуклеотиды, инкубировали с клетками в 200 мкл среды (RPMI с 0,2% BSA) при 4 С в течение 1 ч. Затем среду удаляли и клетки инкубировали в полной ростовой среде в атмосфере 5% CO2 при 37 С. В указанные моменты времени клетки фиксировали 3,7 об.% формальдегидом. Покровные стекла монтировали в Moviol и просматривали в широкопольном микроскопе Axiovert фирмы Zeiss. Изображения фиксировали камерой CCD (Hamamatsu) со стандартным светофильтром для FITC. Пример 12. Опыты по слиянию с эритроцитами Свежепромытые куриные эритроциты (0,8%) получали от фирмы Веrnа Biotech (Bern, Switzerland) вPBS+ (7 мМ Na2HPO4, 5 мМ KH2PO4, 138 мМ NaCl, 3 мМ KCl и 1 мМ MgCl2, рН 7,2). Перед каждым опытом эритроциты центрифугировали в течение 5 мин при 830 g и ресуспендировали в PBS (150 мМ NaCl, 5 мМ фосфат Na, рН 7,4) или в дважды дистиллированной воде до одинакового объема (500 мкл). Добавляли различные количества вируса (1-20 мкг белка) и соответствующие количества виросом (PR8, Х-31), химерных виросом и протеолипосом, инкубировали в течение 15 мин при 37 С с легким встряхиванием соответственно при рН 5,0, рН 5,5, рН 6,0 и рН 7,3. После этого пробы нейтрализовали добавлением 1 М NaOH. Эритроциты седиментировали в течение 5 мин при 830 g. Для определения гемолитической активности измеряли поглощение надосадочной жидкости при 541 нм. Процент гемолиза определяли как 100(А-А 0)/(А 100-А 0), где А 100 - поглощение для 100% гемолиза, измеренное для эритроцитов в дистиллированной воде, а А 0 - фоновое поглощение в отсутствие вируса/виросом. Осадок клеток ресуспендировали в 500 мкл PBS. Капли суспензии наносили на покровные стекла и готовили для световой микроскопии. Чтобы инактивировать химерные виросомы, 50 мкл виросом инкубировали в конечном объеме 200 мкл при 37 С. После подкисления добавлением 5 мкл лимонной кислоты (150 мМ, рН 3,5) суспензию инкубировали еще в течение 30 мин при 37 С и снова нейтрализовали добавлением 1,4 мкл 1 М NaOH. Для реакции гемолиза использовали 20 мкл этой смеси. Опыты по введению фотометки Содержащие биотинил-TID-РЕ/16 виросомы PR8 готовили, как описано выше, за тем исключением,что после этапа солюбилизации с помощью C12E8 добавляли 300 нмоль (10% вирусных фосфолипидов)- 14008497 высушенного биотинил-ТID-РЕ/16. Для приготовления содержащих биотинил-TID-РЕ/16 протеолипосом КOB, приготовленные из РОРС/DDAB/биотинил-TID-РЕ/16 (98:2:20) (5 мг/мл), были слиты с химерными виросомами. БиотинилTID-РЕ/16-КOB (20 мкл) инкубировали с 50 мкл химерных виросом в конечном объеме 200 мкл при рН 5,0 и 4 С в течение 10 мин. После этого смесь нейтрализовали и протеолипосомы экструдировали 10 раз через поры 200 нм. Приготовление теней (пустых оболочек) человеческих эритроцитов Свежую человеческую кровь получали из Stiftung Zurcher Blutspendedienst (SRK), BlutspendezentrumZurich (Switzerland). Чтобы выделить красные кровяные клетки, 20 мл крови фильтровали через подушкуNa, рН 7,3), как описано ранее Beutler (Beutler и др.//J. Lab. Clin. Med. 1976. Т. 88. С. 328-333). Фильтрат промывали 4 раза путем седиментации клеток в роторе SS-34 (Sorvall) при 2500 об./мин и 4 С в течение 7 мин и ресуспендирования клеток в PBS. После последней промывки клетки ресуспендировали в 20 мл PBS и готовили (как описано поставщиком) для подсчета в счетчике Sysmex Microcellcounter F-800 (TOA Medical Electronics, Inc., Hamburg,Germany). Тени готовили, в основном, как описано Steck (Steck and Kant//Methods Enzymol. 1974. T. 31. С 172180). Осадок эритроцитов (1 мл) смешивали с 40 мл буфера для гемолиза (5 мМ фосфат, рН 7,4, 1 мМEDTA, 15 мкг/мл PMSF - фенилметилсульфонил фторида). Тени собирали в осадок центрифугированием при 22000 g в течение 10 мин. Аккуратно отсасывали надосадочную жидкость и маленький твердый сгусток, содержащий примесные протеазы. Мембраны ресуспендировали в буфере для гемолиза и промывали еще три раза, пока они не становились кремово-белыми. После этого тени ресуспендировали в 1 мл буфера для гемолиза. Опыты по введению фотометки Содержащие биотинил-TID-РЕ/16 виросомы PR8 готовили, как описано выше, за тем исключением,что после этапа солюбилизации с помощью С 12 Е 8 добавляли 300 нмоль (10% вирусных фосфолипидов) высушенного биотинил-TID-РЕ/16. Для приготовления содержащих биотинил-TID-РЕ/16 протеолипосом приготовленные из РОРС/DDAB/биотинил-TID-РЕ/16 (98:2:20) (5 мг/мл) КOB были слиты с химерными виросомами. Биотинил-TID-РЕ/16-КOB (20 мкл) инкубировали с 50 мкл химерных виросом в конечном объеме 200 мкл при рН 5,0 и 4 С в течение 10 мин. После этого смесь нейтрализовали и протеолипосомы экструдировали 10 раз через поры 200 нм. Биотинил-TID-РЕ/16 протеолипосомы (20 мкл) или 5 мкл биотинил-TID-РЕ/16- PR8 протеолипосомы инкубировали с 20 мкл теней в общем объеме 50 мкл при 37 С в течение 20 мин. Образцы с низким рН инкубировали в указанных выше условиях после подкисления цитратным буфером до рН 5,0. После обратной нейтрализации добавлением 1 М NaOH соответствующие пробы подвергали фотолизу в течение 90 с, помещая их на расстоянии около 10 см от осветителя SUSSLH 1000 lamphouse (KARL SUSS, Waterbury Center, VT), снабженного короткодуговой ртутной лампой высокого давления OSRAM НВО мощностью 350 Вт. Белок для удаления несвязанной метки осаждали 3 кратным объемом смеси хлороформ/метанол 1:2. После этого пробы подготавливали для разделения электрофорезом в присутствии SDS в 8 об.% полиакриламидном геле. Обнаружение биотинилированных белков осуществлялось путем вестерн-блотирования: белки,разделенные SDS-PAGE, переносили электрофорезом на мембрану Immobilon-P PVDF (Millipore,Volketsvil, Switzerland). Блотирование геля осуществляли в течение 2 ч при токе 110 мА в буфере для переноса, содержащем 20% метанол, 20 мМ трис-HCl, 200 мМ глицин. Эффективность блотирования проверяли, используя предварительно окрашенный стандарт белка Kaleidoscope (Bio-Rad Laboratories,Glattbrugg, Switzerland) и окрашивая блотированный гель Кумасси голубым. Фиксацию блота (отпечатка) осуществляли в течение не менее 1 ч при комнатной температуре в буфере TBS-T (137 мМ NaCl, 20 мМ трис-HCl, 0,1 об.% твин-20, рН 7,6) с добавкой 5% BSA. Добавляли первичные антитела (-биотин, Bethyl laboratories, Montgomery, TX, USA), разбавленные 1:1000 в TBS-T,содержащем 5% BSA, и оставляли для связывания на 1 ч. Мембрану промывали в течение 15 мин TBS-T,затем делали 2 коротких промывки (5 мин) TBS-T и одну промывку TBS (137 мМ NaCl, 20 мМ трис-HCl,рН 7,6). Добавляли вторичные антитела (козьи антитела к кроличьеиу иммуноглобулину, конъюгированные с пероксидазой хрена) фирмы Pierce (Socochim, Lausanne, Switzerland), разбавленные 1:20000 в TBST, содержащем 5% BSA. Связывание вели в течение 1 ч при комнатной температуре. Мембрану вновь тщательно промывали, как описано выше. Влажный блот проявляли с помощью проявляющего раствора ECL +Plus (Amersham Biosciences,Dubendorf, Switzerland), как описано поставщиком. Гемолиз и межклеточное слияние с человеческими эритроцитами Опыты по гемолизу и межклеточному слиянию с человеческими эритроцитами проводили, как описано выше для куриных эритроцитов. На каждую пробу брали 6107 красных кровяных клеток.- 15008497 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способные к слиянию везикулы, составленные из виросомных и липосомных липидов, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество,причем способные к слиянию везикулы содержат слитые белки с различными параметрами слияния или фрагменты слитых белков, при этом фрагменты имеют активность слияния и параметры слияния полного слитого белка, по меньшей мере, в той степени, которая необходима для слияния с биологической мембраной клетки-мишени. 2. Способные к слиянию везикулы по п.1, отличающиеся тем, что везикулы являются однослойными. 3. Способные к слиянию везикулы по п.1 или 2, отличающиеся тем, что инкапсулированное терапевтическое или иммунологически активное вещество выбрано из группы, содержащей ДНК, РНК,миРНК (малую интерферирующую РНК), белки, пептиды, аминокислоты и фармацевтически активные вещества. 4. Способные к слиянию везикулы по п.3, отличающиеся тем, что инкапсулированное терапевтическое или иммунологически активное вещество выбрано из косметического средства, фармацевтического лекарства, антигена или их смесей. 5. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем, что различные параметры слияния выбраны из температуры, концентрации ионов, кислотности, специфичности к типу клеток и к типу ткани. 6. Способные к слиянию везикулы по п.5, отличающиеся тем, что различные параметры слияния слитых белков зависят от температуры. 7. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем, что слитые белки или их фрагменты извлечены из вирусов. 8. Способные к слиянию везикулы по п.7, отличающиеся тем, что слитые белки или их фрагменты извлечены из вирусов, выбранных из группы, содержащей вирус гриппа, вирус везикулярного стоматита(ВВС), вирус леса Семлики (ВЛС), вирус Сендай и вирус иммунодефицита человека (ВИЧ). 9. Способные к слиянию везикулы по п.8, отличающиеся тем, что слитые белки или их фрагменты извлечены из вируса гриппа. 10. Способные к слиянию везикулы по п.9, отличающиеся тем, что слитые белки или их фрагменты представляют собой гемагглютинины штаммов вируса гриппа Х-31, PR8/34 или A/Singapore или же их фрагменты. 11. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем,что липосомные липиды происходят из группы, включающей гликолипиды, фосфолипиды, катионные липиды, синтетические липиды и холестерин. 12. Способные к слиянию везикулы по п.11, отличающиеся тем, что липосомные липиды включают РОРС и DDAB. 13. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем,что виросомные липиды извлечены из вирусов, выбранных из группы, содержащей вирус гриппа, вирус везикулярного стоматита (ВВС), вирус леса Семлики (ВЛС), вирус Сендай и вирус иммунодефицита человека (ВИЧ). 14. Способные к слиянию везикулы по п.13, отличающиеся тем, что виросомные липиды извлечены из вируса гриппа. 15. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем,что везикулы имеют диаметр в диапазоне от 100 до 300 нм. 16. Способные к слиянию везикулы по любому из предшествующих пунктов, отличающиеся тем,что везикулы дополнительно содержат рецептор клеточной поверхности, цитокин, фактор роста, антитело или фрагмент антитела, внедренные в мембрану или присоединенные к указанной мембране. 17. Способ приготовления способных к слиянию везикул, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, включающий слияние липосом, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, с виросомами, имеющими слитые белки с различными параметрами слияния или фрагменты слитых белков, причем фрагменты имеют активность слияния и параметры слияния полного слитого белка,по меньшей мере, в той степени, которая необходима для слияния с биологической мембраной клеткимишени. 18. Способ инкапсулирования по меньшей мере одного терапевтического или иммунологически активного вещества в способные к слиянию везикулы, включающий слияние липосом, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, с виросомами, имеющими слитые белки с различными параметрами слияния или фрагменты слитых белков, причем фрагменты имеют активность слияния и параметры полного слитого белка, по меньшей мере, в той степени, которая необходима для слияния с биологической мембраной клетки-мишени. 19. Способ по п.17 или 18, отличающийся тем, что везикулы являются однослойными. 20. Способ по любому из пп.17-19, отличающийся тем, что инкапсулированное по меньшей мере- 16008497 одно терапевтическое или иммунологически активное вещество выбрано из группы, содержащей ДНК,РНК, миРНК, белки, пептиды, аминокислоты и фармацевтически активные вещества. 21. Способ по п.20, отличающийся тем, что инкапсулированное по меньшей мере одно терапевтическое или иммунологически активное вещество выбрано из косметического средства, фармацевтического лекарства, антигена или их смесей. 22. Способ по любому из пп.17-21, отличающийся тем, что различные параметры слияния слитых белков выбраны из температуры, концентрации ионов, кислотности, специфичности к типу клеток и к типу ткани. 23. Способ по п.22, отличающийся тем, что различные параметры слияния слитых белков или их фрагментов зависят от температуры. 24. Способ по любому из пп.17-23, отличающийся тем, что используемые слитые белки или их фрагменты извлечены из вирусов. 25. Способ по п.24, отличающийся тем, что используемые слитые белки или их фрагменты, извлеченные из вирусов, выбранных из группы, содержащей вирус гриппа, вирус везикулярного стоматита(ВВС), вирус леса Семлики (ВЛС), вирус Сендай и вирус иммунодефицита человека (ВИЧ). 26. Способ по п.25, отличающийся тем, что используемые слитые белки или их фрагменты, извлечены из вируса гриппа. 27. Способ по п.26, отличающийся тем, что слитые белки или их фрагменты представляют собой гемагглютинины штаммов вируса гриппа Х-31, PR8/34 или A/Singapore или же их фрагменты. 28. Способ по любому из пп.17-27, отличающийся тем, что липосомы состоят из липидов, выбранных из группы, включающей гликолипиды, фосфолипиды, катионные липиды, синтетические липиды и холестерин. 29. Способ по п.28, отличающийся тем, что липосомы состоят из РОРС и DDAB. 30. Способ по любому из пп.17-29, отличающийся тем, что виросомы получены из вирусов, выбранных из группы, содержащей вирус гриппа, вирус везикулярного стоматита (ВВС), вирус леса Семлики (ВЛС), вирус Сендай и вирус иммунодефицита человека (ВИЧ). 31. Способ по п.30, отличающийся тем, что виросомы получены из вируса гриппа. 32. Способ по любому из пп.17-31, отличающийся тем, что он дополнительно включает изменение размера способных к слиянию везикул, полученных после слияния липосом, в которые инкапсулировано по меньшей мере одно терапевтическое или иммунологически активное вещество, с виросомами, имеющими слитые белки с различными параметрами слияния или фрагменты слитых белков, при этом фрагменты имеют активность слияния и параметры слияния полного слитого белка слияния, по меньшей мере, в той степени, которая необходима для слияния с биологической мембраной клетки-мишени. 33. Способ по п.32, отличающийся тем, что изменение размера осуществляется путем экструдирования везикул. 34. Способ по любому из пп.17-33, отличающийся тем, что изменение размера способных к слиянию везикул приводит к тому, что их диаметр находится в диапазоне от 100 до 300 нм. 35. Способ по любому из пп.17-34, отличающийся тем, что к мембране способных к слиянию везикул прикреплен или в мембрану способных к слиянию везикул внедрен по меньшей мере один рецептор клеточной поверхности, цитокин, фактор роста, одно антитело или один фрагмент антитела. 36. Применение способных к слиянию везикул, как они заявлены в пп.1-16, для приготовления фармацевтической композиции, предназначенной для лечения или предотвращения вирусных, бактериальных и грибковых инфекционных заболеваний, дегенеративных заболеваний, заболеваний, связанных с гиперпролиферацией, и других связанных с действием киназ расстройств, включая рак, инфекционные заболевания, хронические инфекционные заболевания, хронические заболевания, аллергии, заболевания сердечно-сосудистой системы, воспалительные заболевания (в том числе кожные воспаления, такие как псориаз), заболевания иммунной системы, связанные с раком расстройства иммунитета, астму или артрит. 37. Фармацевтическая композиция, содержащая способные к слиянию везикулы, как они определены в любом из пп.1-16.
МПК / Метки
МПК: A61K 9/127
Метки: везикулы, фармацевтическая, содержащая, слиянию, получения, высокоэффективные, способ, композиция, способные
Код ссылки
<a href="https://eas.patents.su/23-8497-vysokoeffektivnye-sposobnye-k-sliyaniyu-vezikuly-sposob-ih-polucheniya-i-farmacevticheskaya-kompoziciya-ih-soderzhashhaya.html" rel="bookmark" title="База патентов Евразийского Союза">Высокоэффективные способные к слиянию везикулы, способ их получения и фармацевтическая композиция, их содержащая</a>
Фармацевтическая композиция, содержащая фенофибрат, и способ ее получения

Номер патента: 4294
Опубликовано: 26.02.2004
Авторы: Шеневьер Филипп, Сюпли Паскаль, Криер Брюно
МПК: A61K 31/216
Метки: способ, фармацевтическая, содержащая, получения, фенофибрат, композиция
Формула / Реферат:
1. Фармацевтическая композиция, содержащая микронизированный фенофибрат, поверхностно-активное вещество и связующее производное целлюлозы в качестве адъюванта солюбилизации, отличающаяся тем, что она содержит количество фенофибрата выше или равное 60 мас.%. 2. Композиция по п.1, отличающаяся тем, что связующим производным целлюлозы, являющимся адъювантом солюбилизации, является гидроксипропилметилцеллюлоза. 3. Композиция по п.2, отличающаяся...
Спироимидазолиновые соединения, способ их получения и фармацевтическая композиция, содержащая их

Номер патента: 2979
Опубликовано: 26.12.2002
Авторы: Ньюмен-Танкреди Адриан, Миллан Марк, Брокко Морисетт, Корди Алекс
МПК: A61P 25/24, C07D 235/02, A61K 31/4184...
Метки: способ, спироимидазолиновые, фармацевтическая, соединения, получения, содержащая, композиция
Формула / Реферат:
1. Спироимидазолиновые соединения формулы (I) в которой А представляет бензольное кольцо, незамещенное или замещенное 1-4 одинаковыми или различными группами, выбранными из линейного или разветвленного (С1-С6)алкила, линейного или разветвленного (С1-С6)алкокси, гидрокси, полигалоген-(С1-С6)алкила, в котором алкильный фрагмент является линейным или разветвленным, и галогена, В представляет имидазолиновое кольцо, как представлено в формулах (Iа)...
Бета- карболиновые соединения, способ их получения и фармацевтическая композиция, их содержащая

Номер патента: 4140
Опубликовано: 26.02.2004
Авторы: Декейн Анн, Пуассонне Гийом, Миллан Марк, Брион Жан-Даниель, Парментье Жан-Жилль, Бутэн Жан, Гольдстейн Соло
МПК: A61K 31/437, C07D 471/04, A61P 25/00...
Метки: beta, способ, фармацевтическая, карболиновые, получения, содержащая, соединения, композиция
Формула / Реферат:
1. Соединения формулы (I) в которой --- представляет простую или двойную связь, способную необязательно сообщать ароматический характер кольцу, несущему ее, R1 представляет группу, выбранную из водорода, прямого или разветвленного (C1-C6)алкила, -R6-арила, -R6-циклоалкила, -R6-гетероцикла, где группы R6 представляют прямую или разветвленную (C1-C6)алкиленовую группу, -CO2R7, где R7 представляет прямую или разветвленную (C1-C6)алкильную группу,...
Новые замещенные димерные соединения, способ их получения и содержащая их фармацевтическая композиция

Номер патента: 3300
Опубликовано: 24.04.2003
Авторы: Иус Саид, Декамп-Франсуа Кароль, Ренар Пьер, Вио Мари-Клод, Гийоме Жераль, Делягранж Филипп, Лефулон Франсуа, Беннежан Каролин, Лезьер Даниель
МПК: A61K 31/353, C07D 403/12, C07C 233/72...
Метки: получения, композиция, димерные, новые, способ, соединения, замещенные, содержащая, фармацевтическая
Формула / Реферат:
1. Соединения формулы (I) A-G1-Cy-G2-Cy-G3-B (I), где A представляет собой группы формулы -NHC(Q)R1 или -NHC(Q)NHR2, где Q обозначает атом кислорода или серы, R1 обозначает линейную или разветвленную (C1-C6)алкильную группу, замещенную или незамещенную атомом галогена, линейную или разветвленную (C2-C6)алкенильную группу, (C3-C8)циклоалькильную группу, арильную или гетероарильную группу, R2 обозначает линейную или разветвленную...
Новые производные арилглицинамида, способ их получения и фармацевтическая композиция , содержащая эти соединения

Номер патента: 2201
Опубликовано: 28.02.2002
Авторы: Эссер Франц, Шнорренберг Герд, Юнг Биргит, Шпек Георг, Доллингер Хорст, Шромм Курт
МПК: A61P 9/00, A61K 31/395, C07D 295/14...
Метки: соединения, фармацевтическая, содержащая, композиция, производные, способ, получения, эти, новые, арилглицинамида
Формула / Реферат:
1. Производные арилглицинамида общей формулы (I) или их фармацевтически приемлемые соли, где Ar означает незамещенный или 1-5-кратнозамещенный фенил или незамещенный или 1-2-кратнозамещенный нафтил, причем заместители фенила и нафтила независимо друг от друга являются галогеном (фтором, хлором, бромом, йодом), алкилом с 1-4 атомами углерода, O-(С1-С4)алкилом, трифторметилом, трифторметокси или NR12R13, где R12 и R13 независимо друг от друга...
Предыдущий патент: Офтальмологический препарат для профилактики и лечения болезненных состояний глаз
Следующий патент: Замещённые соединения [1,4]бенздиоксино[2,3-e]изоиндола, способ их получения и фармацевтические композиции, которые их содержат
Случайный патент: Композиция полимерного предшественника кислоты для обработки подземных пластов