Соединение и фармацевтическая композиция для ингибирования экспрессии vcam-1
Номер патента: 9370
Опубликовано: 28.12.2007
Авторы: Хоонг Ли К., Самерс Патриция К., Менг Чарльз К.




















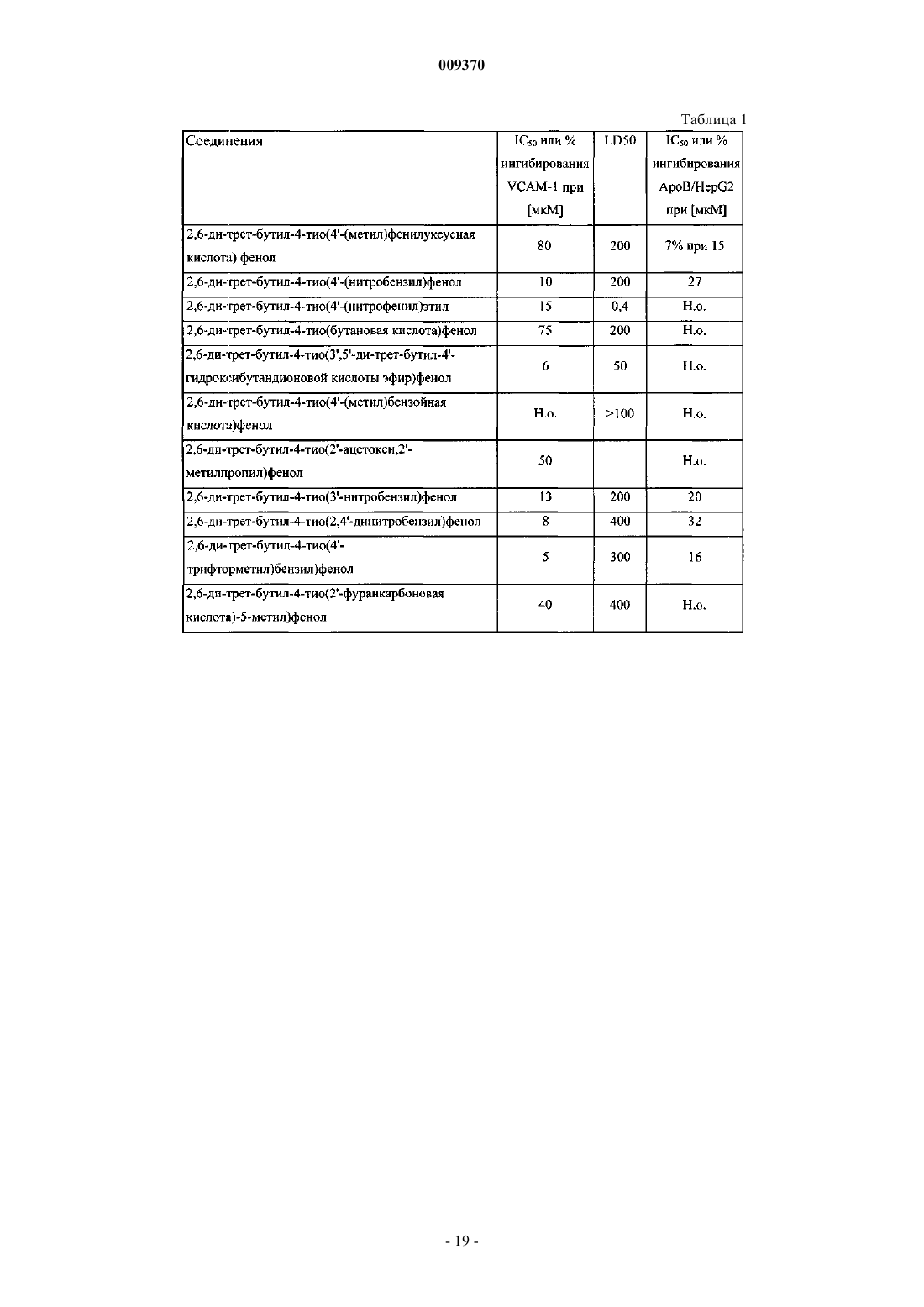
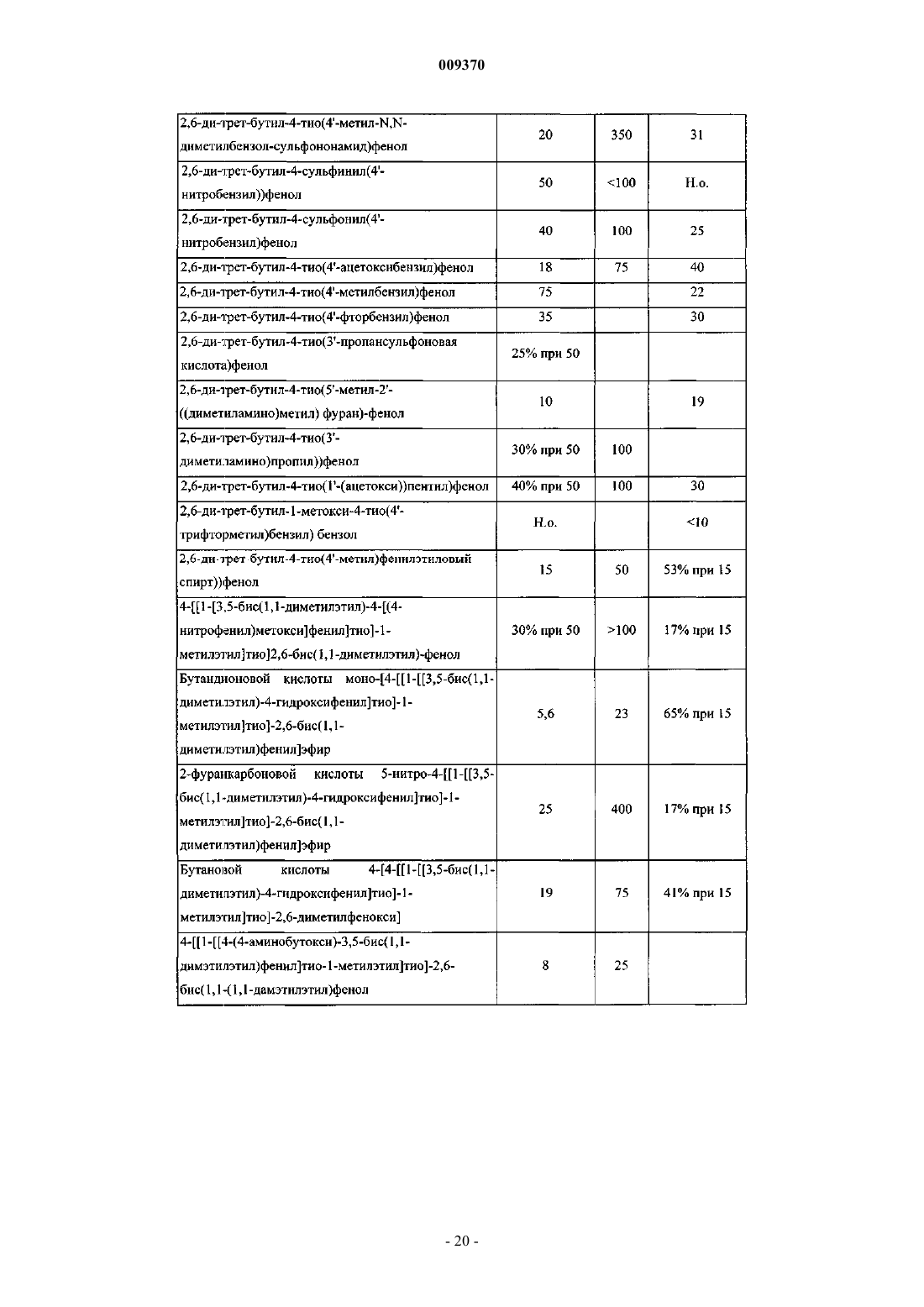
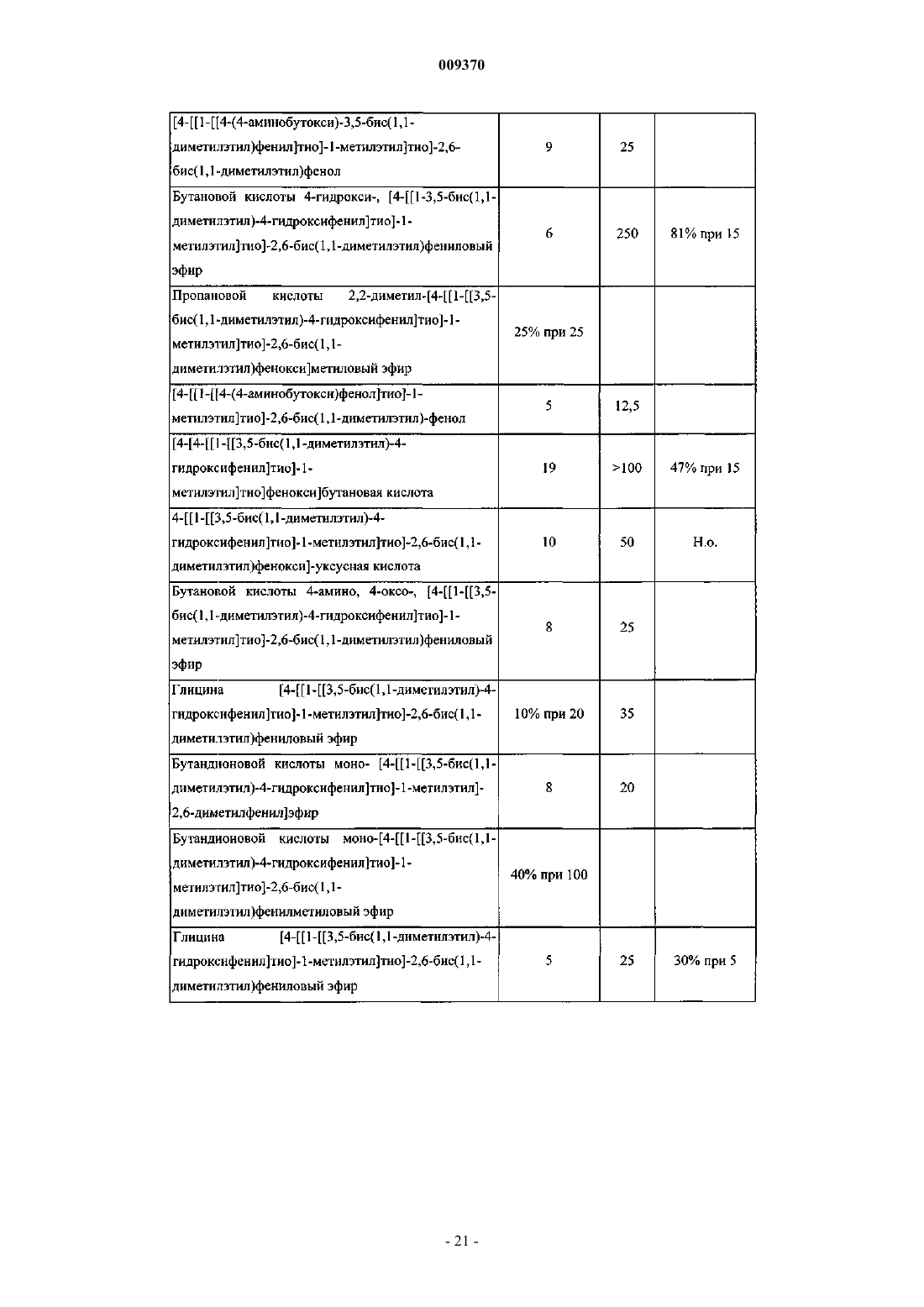
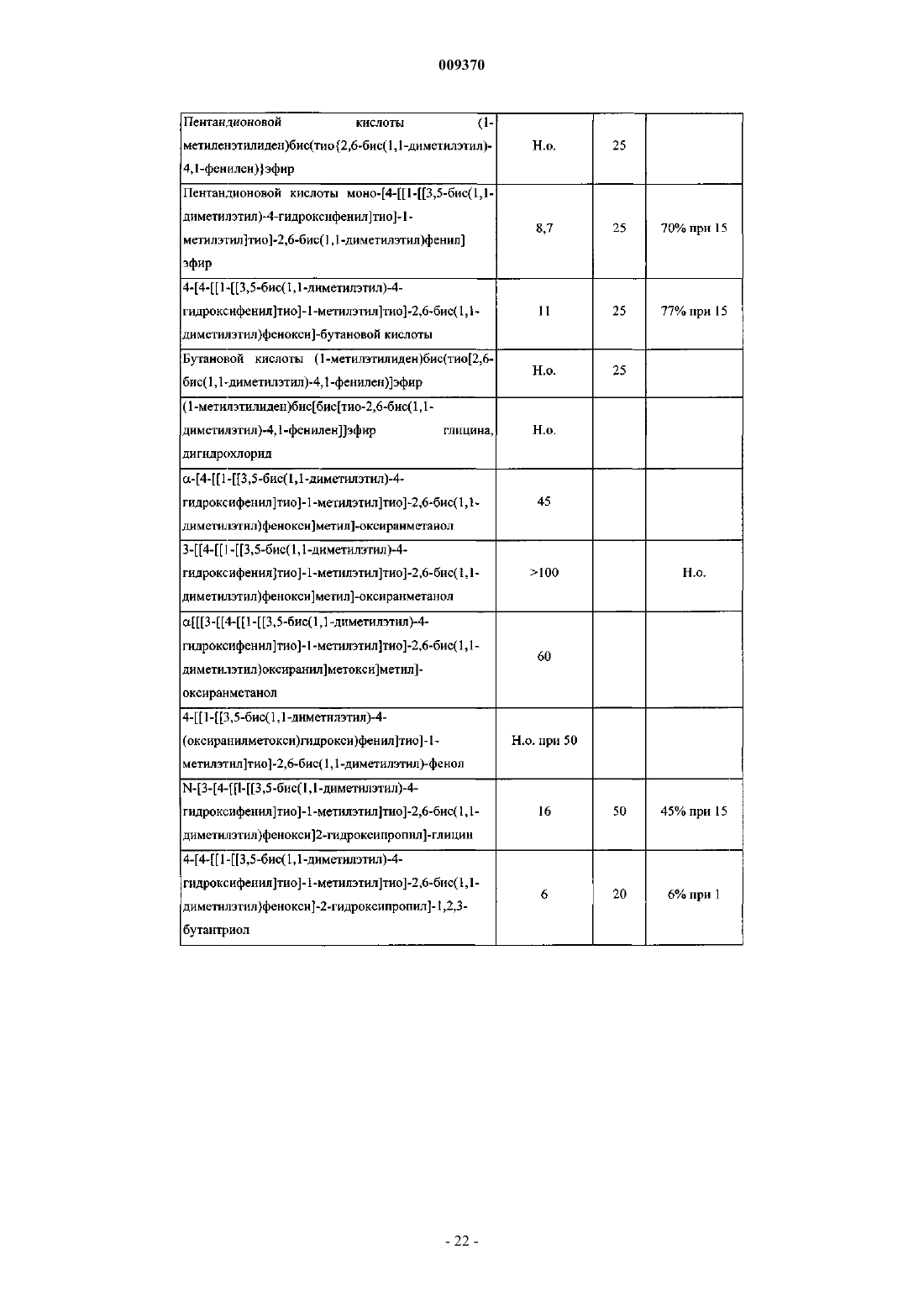




Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль.
2. Фармацевтическая композиция для лечения заболевания, опосредованного экспрессией VCAM-1, содержащая эффективное количество соединения формулы

или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
3. Фармацевтическая композиция по п.2, где заболевание является сердечно-сосудистым заболеванием.
4. Фармацевтическая композиция по п.3, где сердечно-сосудистое заболевание представляет собой выбранное из группы, состоящей из атеросклероза, рестеноза после ангиопластики, заболевания, поражающего коронарную артерию, стенокардии и заболевания, поражающего капилляры.
5. Фармацевтическая композиция по п.2, где заболевание является воспалительным заболеванием.
6. Фармацевтическая композиция по п.5, где воспалительное заболевание представляет собой выбранное из группы, состоящей из ревматоидного артрита, остеоартрита, бронхиальной астмы, дерматита, рассеянного склероза и псориаза.
7. Фармацевтическая композиция по п.3, содержащая указанное соединение в комбинации с другим сердечно-сосудистым препаратом, выбранным из группы, состоящей из средств, снижающих уровень липидов, ингибиторов агрегации тромбоцитов, антитромботических средств, блокаторов кальциевых каналов, ингибиторов ангиотензин-превращающего фермента (АСЕ) и b-блокаторов.
8. Фармацевтическая композиция по п.5, содержащая указанное соединение в комбинации с другим противовоспалительным препаратом.
9. Фармацевтическая композиция по п.2, являющаяся лекарственной формой, которая представляет собой таблетку, драже, пастилку или капсулу.
10. Фармацевтическая композиция по п.9, где лекарственная форма содержит от приблизительно 5 до приблизительно 1500 мг указанного соединения.
11. Фармацевтическая композиция по п.9, где лекарственная форма содержит от приблизительно 25 до приблизительно 750 мг указанного соединения.
12. Фармацевтическая композиция по п.2, где фармацевтически приемлемый носитель является подходящим для орального введения.
13. Фармацевтическая композиция по п.2, где фармацевтически приемлемый носитель является подходящим для введения посредством ингаляции.
14. Фармацевтическая композиция по п.2, где фармацевтически приемлемый носитель является подходящим для внутривенного введения.
15. Фармацевтическая композиция по п.2, где фармацевтически приемлемый носитель является подходящим для парентерального введения.
16. Фармацевтическая композиция по п.2, где фармацевтически приемлемый носитель является подходящим для внутрибрюшинного введения.
17. Фармацевтическая композиция по п.2, где фармацевтически приемлемый носитель является подходящим для подкожного введения.
18. Фармацевтическая композиция по п.2, где фармацевтически приемлемый носитель является подходящим для местного введения.
19. Фармацевтическая композиция по п.2, где фармацевтически приемлемый носитель является подходящим для внутримышечного введения.
Текст
009370 Настоящее изобретение относится к соединению и к композициям для ингибирования экспрессииVCAM-1, и, в частности, для лечения заболеваний, опосредуемых VCAM-1, включая сердечнососудистые и воспалительные заболевания. Предпосылки создания изобретения Ишемическая болезнь сердца (ИБС) остается ведущей причиной смертности в промышленно развитых странах. Основной причиной ИБС является атеросклероз, заболевание, характеризующееся отложением липидов на стенке артерии, приводящим к сужению просвета сосуда, и, в конечном счете, к ригидности сосудистой системы. Атеросклероз, манифестирующий своим главным клиническим осложнением, ишемической болезнью сердца, продолжает оставаться главной причиной смертности в промышленно развитых странах. В настоящее время хорошо известно, что развитие атеросклероза может начинаться с локального повреждения эндотелия артерии с последующей пролиферацией гладкомышечных клеток артерии, от среднего слоя и до внутреннего слоя с одновременным отложением липидов и накопления пенистых клеток в очаге повреждения. По мере развития атеросклеротическая бляшка постепенно все больше и больше закупоривает пораженный кровеносный сосуд, что может, в конечном счете, привести к ишемии или к инфаркту. Поэтому, было бы желательно разработать способы подавления развития атеросклероза у нуждающихся в этом пациентов. Сердечно-сосудистые заболевания связаны с некоторыми этиологическими факторами, которыми являются гиперхолестеринемия, гиперлипидемия и экспрессия VCAM-1 в эндотелиальных клетках сосудов. Экспрессия VCAM-1 Адгезия лейкоцитов к эндотелию представляет собой фундаментальный ранний процесс при целом ряде воспалительных состояний, включая атеросклероз, аутоиммунные заболевания и бактериальные и вирусные инфекции. Рекрутинг лейкоцитов в эндотелий начинается тогда, когда индуцибельные молекулы-рецепторы адгезии на поверхности эндотелиальных клеток взаимодействуют с противорецепторами на иммуннокомпетентных клетках. Эндотелиальные клетки сосудов определяют, какой рекрутируется тип лейкоцитов (моноциты, лимфоциты или нейтрофилы) путем селективной экспрессии специфических молекул адгезии, таких как молекула адгезии клеток сосудов-1 (VCAM-1), молекула межклеточной адгезии-1 (ICAM-1) и Е-селектин. На самой ранней стадии формирования атеросклеротического очага происходит локальная эндотелиальная экспрессия VCAM-1 и селективный рекрутинг мононуклеарных лейкоцитов, которые экспрессируют интегриновый противорецептор VLA-4. Поскольку VLA-4 селективно экспрессируется на моноцитах и лимфоцитах, а не на нейтрофилах, VCAM-1 играет важную роль в опосредовании селективной адгезии мононуклеарных лейкоцитов. Последующее превращение лейкоцитов в пенистные макрофаги приводит к синтезу широкого ряда воспалительных цитокинов, факторов роста и хемоаттрактантов, которые способствуют росту рекрутинга лейкоцитов и тромбоцитов, пролиферации гладкомышечных клеток, активации эндотелиальных клеток и синтезу внутриклеточного матрикса, характеризующего созревание атеросклеротической бляшки.VCAM-1 является медиатором хронических воспалительных заболеваний, таких как бронхиальная астма, ревматоидный артрит и аутоиммунный сахарный диабет. Так, например, известно, что у астматиков наблюдается повышенная экспрессия VCAM-1 и ICAM-1. Pilewski, J.M., Am.J.Respir.Cell Mol. Biol 12 1-3 (1995); Ohkawara, Y., et al., Am. J.Respir.Cell Mol. Biol. 12: 4-12 (1995). Кроме того, блокирование интегриновых рецепторов для VCAM-1 и ICAM-1 (VLA-4 и LFA-1 соответственно) приводит к подавлению реакций ранней и поздней фазы у сенсибилизированных овальбумином экспериментальных крыс в модели аллергической реакции дыхательных путей. Rabb, 11. A., et al., Am.J.Respir. Care Med. 149, 11861191 (1994). Наблюдалась также повышенная экспрессия эндотелиальных молекул адгезии, включаяVCAM-1, в капиллярной сети синовиальной оболочки при ревматоидном артрите. Koch, A.E. et al., Lab.invest. 64, 313-322 (1191); Morales-Ducret, J. et al., Immunol., 149, 1421-1431 (1992). Нейтрализующие антитела, направленные против VCAM-1 или его противорецептора VLA-4, могут задерживать начало развития сахарного диабета у мышиной модели (у мышей NOD), у которых спонтанно развивается это заболевание. Yang X.D. et al., Proc.Natl. Acad. Sci., USA, 90, 10494-10498 (1993); Burkly, L.C. et al., Diabetes 43, 523-534 (1994); Baron, J.I. et al., J. Clin. Invest., 93, 1700-1708 (1994). Моноклональные антитела против VCAM-1 могут также оказывать благоприятный эффект в животных моделях, отторжения аллотрансплантата, что дает основание предположить, что ингибиторы экспрессии VCAM-1 могут быть использованы для профилактики отторжения трансплантата. Oroez, C.G. et al., Immunol. Lett. 32, 7-12VCAM-1 экспрессируется клетками как в мембраносвязанной форме, так и в растворимой форме. Было показано, что растворимая форма VCAM-1 индуцирует хемотаксис эндотелиальных клеток сосудовin vitro и стимулирует ангиогенный ответ в роговице крыс. Koch, A.F. et al., Nature 376, 517-519 (1995). Ингибиторы экспрессии растворимого VCAM-1, возможно, обладают терапевтической ценностью для лечения заболеваний с сильным ангиогенным компонентом, включая рост опухолей и метастазирование.VCAM-1 экспрессируется в культуре сосудистых эндотелиальных клеток человека после активации(TNF-a). Эти факторы не являются селективными для активации экспрессии молекул клеточной адгезии. В патенте США 5380747 (Medford et al.) описано использование дитиокарбаматов, таких как дитиокарбамат пирролидина, для лечения сердечно-сосудистых и других воспалительных заболеваний. В патенте США 5750351 (Medford et al.) и в WO 95/30415 (Emory University) описан факт индукции полиненасыщенными жирными кислотами ("PUFA") и их гидропероксидами ("ox-PUFA"), которые являются важными компонентами окислительно-модифицированного липопротеина низкой плотности(ЛНП), экспрессии VCAM-1, но не молекулы межклеточной адгезии (ICAM-1) и не Е-селектин; в эндотелиальных клетках аорты человека посредством механизма, который не опосредуется цитокинами или другими нецитокиновыми сигналами. Этот факт является фундаментальным открытием важных и ранее неизвестных каскадов биологических реакций в ходе VCAM-1-опосредованного иммунного ответа. В качестве неограничивающих примеров можно указать, что линолевая кислота, линоленовая кислота, арахидоновая кислота, линолеилгидропероксид (13-HPODE) и гидропероксид арахидоновой кислоты (15-НРЕТЕ) индуцируют экспрессию гена VCAM-1 на клеточной поверхности, но не гена ICAM-1 или Е-селектина. Насыщенные жирные кислоты (такие как стеариновая кислота) и мононенасыщенные жирные кислоты (такие как олеиновая кислота) не индуцируют экспрессию VCAM-1, ICAM-1 или Еселектина. Индуцирование VCAM-1 под действием PUFA и их гидропероксидов подавляется дитиокарбаматами, включая дитиокарбамат пирролидина (PDTC). Это свидетельствует о том, что указанное индуцирование опосредуется окисленной сигнальной молекулой, и что это индуцирование предотвращается тогда,когда окисление молекулы блокируется (то есть окисления не происходит), и наоборот (то есть сигнальная молекула восстанавливается), или когда каким-либо другим образом блокируется взаимодействие окислительно-восстановительного модифицированного сигнала с его регуляторной мишенью. Клетки, которые постоянно подвергаются воздействию превышающих нормальные уровни концентраций полиненасыщенных жирных кислот или их окисленных аналогов, могут инициировать иммунный ответ, который не является нормальным и является аномально высоким относительно реальной угрозы, и который приводит к патологическому состоянию. Сверхчувствительность сосудистых эндотелиальных клеток к PUFA и ox-PUFA может способствовать ускорению образования, например, атеросклеротических бляшек. В результате данных находок в WO 95/30415 был описан метод лечения атеросклероза; постангиопастического рестенозирования; заболеваний коронарных артерий; стенокардии; заболевания мелких артерий; и других сердечно-сосудистых заболеваний, а также воспалительных заболеваний, которые не относятся к сердечно-сосудистым заболеваниям, но которые опосредуются VCAM-1, причем указанный метод предусматривает удаление, снижение концентрации или предотвращение образования окисленных полиненасыщенных жирных кислот, включая, но не ограничиваясь ими, окисленную линолевую(C189,12), линоленовую (C186,9,12), арахидоновую (С 205,8,11,14) и эйкозатриеновую (С 208,11,14) кислоты. Неограничивающими примерами воспалительных заболеваний, не относящихся к сердечнососудистым заболеваниям, опосредуемых VCAM-1, являются ревматоидный артрит, остеоартрит, бронхиальная астма, дерматит и рассеянный склероз. Гиперхолестеринемия и гиперлипидемия Гиперхолестеринемия является важным фактором риска, ассоциированным с сердечно-сосудистым заболеванием. Липопротеины сыворотки являются носителями липидов в кровотоке. Липопротеины классифицируются по их плотности: хиломикроны, липопротеины очень низкой плотности (ЛПОНП), липопротеины низкой плотности (ЛПНП) и липопротеины высокой плотности (ЛПВП). Хиломикроны участвуют, главным образом, в транспорте пищевых триглицеридов и холестерина из кишечника в жировую ткань и печень. ЛПОНП обеспечивают доставку эндогенно синтезированных триглицеридов из печени в жировую ткань и в другие ткани. ЛПНП транспортируют холестерин в периферические ткани и регулируют содержание эндогенного холестерина в этих тканях. ЛПВП транспортируют холестерин из периферических тканей в печень. Холестерин, откладывающийся на стенках артерий, происходит почти исключительно из ЛПНП. BrownGoldstein, Ann. Rev. Biochem., 52, 223 (1983); Miller, Ann.Rev.Med. 31, 97 (1980). У пациентов с низкими уровнями ЛПНП атеросклероз развивается редко.Steinberg, et al., (N.Eng. J.Mecl. 1989; 320:915-924) высказали предположение, что модификация липопротеина низкой плотности (ЛПНП) в окисленно-модифицированный ЛПНП (ок-ЛПНП) под действием активных молекул кислорода является основным процессом, который инициирует и стимулирует развитие атеросклероза. Окисленный ЛПНП представляет собой сложную структуру, состоящую, по крайней мере, из нескольких химически отличных окисленных соединений, каждое из которых, по отдельности или в сочетании друг с другом, могут модулировать активированную цитокинами экспрессию генов молекул адгезии. Гидропероксиды жирных кислот, такие как линолеилгидропероксид (13-HPODE), продуцируются из свободных жирных кислот под действием липоксигеназ и являются важным компонентом окисленного ЛПНП. Было высказано предположение, что наработка окисленных липидов инициируется под действием-2 009370 системы клеточных липоксигеназ, и что эти окисленные липиды затем превращаются в ЛПНП. Это происходит после цепной реакции образования ЛПНП в среде, катализируемой переходными металлами и/или сульфгидрильными соединениями. Предварительные исследования показали, что модификация жирных кислот в культуре эндотелиальных клеток может приводить к изменению их восприимчивости к окислительному повреждению, а добавление в среду полиненасыщенных жирных кислот (PUFA) способствует усилению восприимчивости к окислительному повреждению. Добавление насыщенных или мононенасыщенных жирных кислот в культуру эндотелиальных клеток снижает их восприимчивость к окислительному повреждению, тогда как добавление полиненасыщенных жирных кислот (PUFA) усиливает восприимчивость этих клеток к окислительному повреждению. Анализ нативных и омыленных жидких экстрактов ЛПНП с использованием обращенно-фазовой ВЭЖХ продемонстрировал, что 13-HPODE представляет собой преобладающую окисленную жирную кислоту в ЛПНП, окисленном активированными моноцитами человека. Хроническое воздействие окисленных ЛПНП приводит к продуцированию окислительного сигнала, направленного на сосудистые эндотелиальные клетки, вероятно, посредством соответствующего гидропероксида жирной кислоты, что способствует селективному усилению цитокин-индуцированной экспрессии гена VCAM-1. Посредством механизма, который пока еще точно не выяснен, участки стенок сосудов, предрасположенных к атеросклерозу, предпочтительно, захватывают ЛПНП из кровотока. Посредством пока еще не совсем ясного механизма, ЛПНП в клетках эндотелия, гладкой мышцы и/или воспалительной ткани затем превращается в окисленный ЛПНП. В противоположность ЛПНП, который поглощается посредством рецептора ЛПНП, моноциты мгновенно поглощают окисленный ЛПНП посредством рецептораскэвенджера, экспрессия которого, в отличие от рецептора ЛПНП, не ингибируется по мере возрастания внутриклеточного содержания липида. Поэтому моноциты продолжают поглощать окисленный ЛПНП и становятся перегруженными липидом магрофагальными, пенистыми клетками, которые образуют жирное пятно. В настоящее время имеется большое число данных, свидетельствующих о том, что гиперхолестеринемия является важным фактором риска, ассоциированным с заболеваниями сердца. Так, например, в декабре 1984 г., на конференции Национального института здоровья, совещательной группой специалистов по выработке экспертной оценки был сделан вывод, что снижение повышенных уровней холестерина в крови (особенно уровней холестерина липопротеина низкой плотности в крови) будет способствовать снижению риска сердечных приступов, обусловленных ишемической болезнью сердца. Обычно, холестерин переносится в кровь теплокровных животных в определенных липидпротеиновых комплексах, таких как хиломикроны, липопротеины очень низкой плотности (ЛПОНП),липопротеины низкой плотности (ЛПНП) и липопротеины высокой плотности (ЛПВП). Широко известно, что ЛПНП функционирует таким образом, что это непосредственно приводит к отложению холестерина ЛПНП на стенки кровеносных сосудов, а ЛПВП функционирует таким образом, что это приводит к захвату этим липопротеином высокой плотности холестерина со стенок сосудов и транспортировки его в печень, где он подвергается метаболизму [BrownGoldstein, Ann.Rev. Biochem., 52, 223 (1983); Miller,Ann. Rev.Med. 31, 97 (1980)]. Так, например, при различных эпидемиологических исследованиях было обнаружено, что уровни холестерина ЛПНП четко коррелируют с риском возникновения ишемической болезни сердца, тогда как уровни холестерина ЛПВП связаны обратной зависимостью с возникновением ишемической болезни сердца [Patton et al., Clin. Chem., 29, 1980 (1983)]. Обычно в данной области считается, что снижение аномально высоких уровней холестерина ЛПНП является эффективной терапией не только при лечении гиперхолестеринемии, но также и при лечении атеросклероза. Кроме того, данные, полученные на основе экспериментов на животных и лабораторных исследований, указывают, что перекисное окисление липидов ЛПНП, таких как ненасыщенные жирные кислоты,являющихся компонентами сложных эфиров холестерина ЛПНП и фосфолипидов, способствует накоплению холестерина в моноцитах/макрофагах, которые, в конечном счете, трансформируются в пенистые клетки и начинают накапливаться в субэндотелиальном пространстве сосудистой стенки. Накопление пенистых клеток на стенках сосудов считается ранним процессом в образовании атеросклеротической бляшки. Таким образом, очевидно, что перекисное окисление липида ЛПНП является важным фактором,способствующим накоплению холестерина на стенках сосудов и последующего образования атеросклеротической бляшки. Так, например, было показано, что моноциты/макрофаги поглощают и разлагают нативный ЛПНП с относительно низкой скоростью без какого-либо заметного накопления холестерина. Напротив, окисленный ЛПНП поглощается указанными моноцитами/макрофагами со значительно более высокой скоростью и с заметным накоплением холестерина [Parthasarathy el al., J Clin.invest., 77, 641(1986)]. Поэтому было бы желательно разработать способы ингибирования перекисного окисления ЛПНП-липида у нуждающегося в этом пациента. Повышенные уровни холестерина ассоциируются с рядом патологических состояний, включая рестенозирование, стенокардию, церебральный атеросклероз и ксантому. Поэтому было бы желательно разработать способ снижения уровня холестерина в плазме у пациентов с развитием или с риском развития рестенозирования, стенокардии, церебрального артериосклероза, ксантомы и других патологических состояний, ассоциируемых с повышенными уровнями холестерина в крови.-3 009370 Поскольку было определено, что гиперхолестеринемия обусловлена повышенными уровнями ЛПНП (гиперлипидемия), то были сделаны попытки снижения уровней ЛПНП посредством терапии с применением диеты. Существует несколько классов лекарственных средств, которые обычно используются для снижения уровней ЛПНП, включая секвестранты желчных кислот, никотиновую кислоту (ниацин) и ингибиторы 3-гидрокси-3-метилглутарил-кофермент А (HMG-CoA)-редуктазы. Пробукол и производные фибрата иногда используются в качестве дополнительной терапии, обычно в комбинации с другими лекарственными средствами. Было установлено, что ингибиторами HMG-CoA-редуктазы являются статины и вастатины. Статины, среди прочих, относятся к наиболее эффективным средствам, предлагаемым в настоящее время на фармацевтическом рынке для лечения гиперхолестеринемии, и включают правастатин (Pravchol, Bristol Myers Squibb), аторвастатин (Warner Lambert/Pfizer), симвастатин (Zocor, Merck), ловастатин (Mevacor, Merck) и флувастатин (Lescoi). Имеющиеся данные позволяют предположить, что атерогенное действие липопротеина низкой плотности (ЛПНП) может быть частично опосредовано их окислительной модификацией. Было показано, что пробукол обладает сильнодействующими противоокислительными свойствами и блокирует окислительную модификацию ЛПНП. В соответствии с этим фактом было показано, что пробукол фактически замедляет прогрессирование атеросклероза у кроликов с дефицитом рецептора ЛПНП, как обсуждается в работе Carew et al., Proc.Natl. Acad. ScL, USA, 84:7725-7729 (1987). По всей вероятности, пробукол является эффективным благодаря своей высокой растворимости в липидах, и транспортируется липопротеинами, защищая их, тем самым, от окислительного повреждения. По своей химической структуре пробукол относится к широко используемым пищевым добавкам 2,[3]-трет-бутил-4-гидроксианизолу (ВНА) и 2,6-ди-трет-бутил-4-метилфенолу (ВНТ). Пробукол имеет полное химическое название: 4,4'-(изопропилидендитио)-бис-(2,6-ди-трет-бутилфенол). Пробукол используют, главным образом, для снижения уровней холестерина в сыворотке у пациентов с гиперхолестеринемией. Пробукол обычно вводят в форме таблеток, выпускаемых под торговым знаком Lorelco. К сожалению, пробукол почти не растворяется в воде, а поэтому, он не может быть инъецирован внутривенно. Действительно, пробукол очень плохо абсорбируется клетками in vitro из-за его плохой смешиваемости с буферами и со средами для культивирования клеток. Твердый пробукол плохо всасывается в кровь и выводится из организма в почти неизмененном виде. Кроме того, таблетированная форма пробукола абсорбируется при существенно различных скоростях и в различных количествах у различных пациентов. В одном исследовании (Heeg et al., Plasma Levels of Probucol in Man AfterSingle and Repeated Oral Doses, La Nouvelle Presse Medicale, 9:2990-2994 (1980 было обнаружено, что максимальные уровни пробукола в сыворотке у различных пациентов отличаются почти в 20 раз. В другом исследовании, Kazuya et al., J.Lipid Res. 32:197-204 (1991), наблюдалось включение приблизительно менее 1 мкг пробукола на 106 клеток при инкубировании эндотелиальных клеток в течение 24 ч с 50 мкМ пробукола. В патенте США 5262439 (Parthasarathy) описаны аналоги пробукола с повышенной водорастворимостью, в которых одна или обе гидроксильные группы заменены сложноэфирными группами, что приводило к увеличению водорастворимости этого соединения. В одном варианте это производное выбирают из группы, состоящей из сложного моно- или диэфира янтарной кислоты, глутаровой кислоты,адипиновой кислоты, себериновой кислоты, себациновой кислоты, азелаиновой кислоты или малеиновой кислоты и пробукола. В другом варианте, производным пробукола является сложный моно- или диэфир,где этот сложный эфир содержит алкильную или алкенильную группу, которая включает функциональную группу, выбранную из группы, состоящей из карбокислотной группы, аминогруппы, соли аминогруппы, амидных групп и альдегидных групп. В ряде патентов Франции указывается, что некоторые производные пробукола представляют собой гипохолестеринемические и гиполипидемические средства: Fr 2168137 (эфиры бис-4-гидроксифенилтиоалкановой кислоты); Fr 2140771 (эфиры тетралинилфеноксиалкановой кислоты и пробукола);Fr 2140769 (бензофурилоксиалканово-кислотные производные пробукола), Fr 2134810 (бис-(3-алкил-5 трет-алкил-4-тиазол-5-карбокси)фенилтио)алканы; Fr 2133024 (бис-(4-никотиноилоксифенилтио)пропаны) и Fr 2130975 (бис-(4-(феноксиалканоилокси)фенилтио)алканы). В патенте США 5155250 (Parker et al.) указывается, что 2,6-диалкил-4-силилфенолы являются противоатеросклеротическими средствами. В публикации РСТWO 95/15760, опубликованной 15 июня 1995 г., эти же самые соединения были описаны как средства, снижающие уровни холестерина в сыворотке. В патенте США 5608095 (Parker et al.) указывается, что алкилированные 4-силилфенолы ингибируют перекисное окисление ЛПНП, понижают уровни холестерина в плазме и ингибируют экспрессию VCAM-1, а поэтому, они могут быть использованы для лечения атеросклероза. В ряде Европейских патентных заявок и в Shionogi Seiyaku Kabushiki Kaisha описаны тиоэфиры фенола для использования в лечении артериосклероза. В Европейской патентной заявке 348203 описаны тиоэфиры фенола, которые ингибируют денатурацию ЛПНП и включение ЛПНП макрофагами. Эти соединения могут быть использованы в качестве противоартериосклеротических средств. Гидроксаминовокислотные производные этих соединений описаны в Европейской патентной заявке 405788 и используются для лечения артериосклероза, язвы, воспаления и аллергии. В патенте США 4954514 (Kita etal.) описаны карбамоил- и цианопроизводные тиоэфиров фенола. В патенте США 4752616 (Hall et al.) описаны арилтиоалкилфенилкарбоновые кислоты для лечения тромботических заболеваний. Описанные соединения могут быть использованы в качестве ингибиторов агрегации тромбоцитов для лечения коронарных и церебральных тромбозов и для ингибирования,среди прочих, бронхостеноза. В ряде патентов Adir et Compagnie описаны замещенные феноксиизомасляные кислоты и сложные эфиры, которые могут быть использованы в качестве антиоксидантов и гиполипидемических средств. Этот ряд патентов включает патенты США 5206247 и 5627205 (Regnier et al., который соответствует Европейской патентной заявке 621255) и Европейскую патентную заявку 763527. В WO 97/15546 (Nippon Shinyaku Co. Ltd) описаны карбоново-кислотные производные для лечения артериального склероза, ишемической болезни сердца, церебрального инфаркта и рестенозирования после РТСА (чрескожной катетерной коронаропластики). Компания Dow Chemicai Company является правоприемником патентов на гиполипидемические 2(3,5-ди-трет-бутил-4-гидроксифенил)тиокарбаксамиды. Так, например, в патентах 4029812, 4076841 и 4078084 (Wagner et al.) описаны эти соединения для снижения уровней липидов в сыворотке крови, а особенно уровней холестерина и триглицеридов. Поскольку в настоящее время сердечно-сосудистые заболевания являются ведущей причиной смертности в Соединенных Штатах, и 90% из всех сердечно-сосудистых заболеваний диагностируется как атеросклероз, то существует крайняя необходимость в разработке новых способов и фармацевтических средств для его лечения. Важным этапом для достижения этой цели является идентификация и модификация специфических окисленных биологических соединений, которые действуют как селективные регуляторы экспрессии медиаторов воспалительного процесса, а в частности, VCAM-1. Более общей целью является разработка селективных способов подавления экспрессии генов восприимчивости к окислительно-восстановительным процессам или подавления активации генов восприимчивости к окислительно-восстановительным процессам. Следовательно, целью настоящего изобретения является получение новых соединений, композиций и способов для лечения сердечно-сосудистых и воспалительных заболеваний. Другой целью настоящего изобретения является получение новых соединений и композиций, которые могут быть использованы в качестве ингибиторов перекисного окисления липидов ЛПНП. Другой целью настоящего изобретения является получение новых соединений и композиций, которые могут быть использованы в качестве противоатеросклеротических средств. Другой целью настоящего изобретения является получение новых соединений и композиций, которые могут быть использованы в качестве средств, снижающих уровни липидов ЛПНП. Другой целью настоящего изобретения является получение новых соединений, композиций и способов селективного ингибирования экспрессии VCAM-1. Другой целью настоящего изобретения является разработка способа лечения заболевания, опосредуемого экспрессией или супрессией гена восприимчивости к окислительно-восстановительным процессам, например, гена МСР-1, IL-6 и рецептора тромбина. Сущность изобретения Настоящее изобретение относится к соединению, фармацевтической композиции и лекарственной форме, которые могут быть использованы для лечения заболеваний, опосредуемых VCAM-1, где фармацевтическая композиция содержит соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Соединением формулы (I) является Настоящее изобретение, в основном, относится к фармацевтической композиции и лекарственной форме для лечения сердечно-сосудистых и воспалительных заболеваний у пациента, нуждающегося в таком лечении, где указанному пациенту вводится фармацевтическая композиция, содержащая эффективное количество соединения формулы (I). Подробное описание настоящего изобретения Термин "защищенный", используемый в настоящем описании, если это не оговорено особо, относится к группе, которая присоединена к атому кислорода, азота или фосфора для предотвращения его последующей реакции или для других целей. Специалистам в области органического синтеза известен широкий ряд кислород- и азотзащитных групп. Термин "фармацевтически приемлемые соли или комплексы" означает соли или комплексы, которые сохраняют желаемую биологическую активность соединений настоящего изобретения и обладают минимальным нежелательным токсическим действием. Неограничивающими примерами таких солей-5 009370 являются: (а) кислотно-аддитивные соли, образованные неорганическими кислотами (например, хлористо-водородной кислотой, бромисто-водородной кислотой, серной кислотой, фосфорной кислотой, азотной кислотой и т.п.), и соли, образованные органическими кислотами, такими как уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памовая кислота, альгиновая кислота, полиглутаминовая кислота, нафталинсульфоновая кислота, нафталиндисульфоновая кислота и полигалактуроновая кислота; (b) основноаддитивные соли, образованные катионами металлов, таких как цинк, кальций, висмут, барий, магний,алюминий, медь, кобальт, никель, кадмий, натрий, калий и т.п., или образованные катионами аммиака,N,N-дибензилэтилендиамина, D-глюкозамина, тетраэтиламмония или этилендиамина; или (с) комбинации соединений (а) и (b), например таннатная соль цинка или т.п. В объем этого определения входят также фармацевтически приемлемые четвертичные соли, известные специалистам, которыми конкретно является четвертичная соль аммония формулы -NR+A-, где R определен выше, a A представляет противоион, включая хлорид, бромид, иодид, -О-алкил, толуолсульфонат, метилсульфонат, сульфонат, фосфат или карбоксилат (такой как бензоат, сукцинат, ацетат, гликолят, малеат, малат, цитрат, тартрат, аскорбат,бензоат, циннамоат, манделоат, бензилоат и дифенилацетат). Заболеваниями, опосредованными VCAM-1, являются, но не ограничиваются, атеросклероз, постангиопластическое рестенозирование, заболевание коронарных артерий, стенокардия, заболевание мелких артерий и другие сердечно-сосудистые заболевания, а также воспалительные заболевания, не относящиеся к сердечно-сосудистым заболеваниям, такие как ревматоидный артрит, остеоартрит, бронхиальная астма, дерматит, рассеянный склероз и псориаз. Соединением настоящего изобретения является соединение формулы (I) Соединение формулы (I) может быть получено с использованием известной методики и техники или их рутинных модификаций. Общая схема для получения соединения формулы (I) представлена в схеме А. Схема А. Определенное количество пробукола (поставляемого фирмой Sigma Chemicals) в 0,1 М растворе тетрагидрофурана обрабатывали 2 экв. гидрида натрия и перемешивали в течение 30 мин при комнатной температуре. К реакционной смеси добавляли 3 экв. хлорангидрида или ангидрида кислоты, и реакционную смесь перемешивали в течение 16 ч при комнатной температуре. Реакцию гасили 1 н. водной HCl и разбавляли этилацетатом. Водный слой удаляли и этилацетатный слой промывали водой, а затем насыщенным водным раствором хлорида натрия. Этилацетатный раствор сушили сульфатом магния, фильтровали под давлением или в вакууме, а затем концентрировали. Продукт очищали с помощью хроматографии на силикагеле. Исходные соединения для использования в общих методах синтеза, представленных в вышеуказанных реакционных схемах, являются легко доступными или могут быть легко получены стандартными методами. Пробукол является легко доступным соединением, поставляемым фирмой Sigma Chemicals. В нижеследующих примерах проиллюстрирован стандартный синтез, описанный в схеме А. Следует отметить, что эти примеры представлены лишь в иллюстративных целях и не должны рассматриваться как ограничение объема настоящего изобретения. Пример 1. Пентандионовой кислоты 4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1 метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенилметиловый эфир. Описание реакции. Пробукол (2,8 г, 5,5 ммоль) растворяли в ТГФ (25 мл), добавляли 60% гидрид натрия в минеральном масле (528 мг, 13,2 ммоль), а затем добавляли метилхлорформилбутират (0,751 мл, 6,6 ммоль). Через 2 ч реакцию гасили метанолом (3 мл), а затем водой (10 мл). Реакционную смесь экстрагировали эфиром(50 мл), концентрировали и хроматографировали на силикагеле, элюируя градиентом концентраций смеси эфира/гексана (0:100)эфира/гексана (20:80). В результате реакции было получено 500 мг продукта. 1H-ЯМР (400 МГц, CDCl3)7,63 (с, 2 Н), 7,45 (с, 2 Н), 5,82 (с, 1 Н), 3,71 (с, 3H), 2,73 (т, J=7,6 Гц, 2 Н),2,50 (т, J=7,2 Гц, 2 Н), 2,07 (пент, J=7,6 Гц, 2 Н), 1,47 (с, 6 Н), 1,44 (с, 18 Н), 1,34 (с, 18 Н). Пример 2. 4-1-3,5-бис-(1,1-Диметилэтил)-4-[(4-нитрофенил)метокси]фенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенол. Описание реакции. Раствор пробукола (0,19 ммоль; 100 мг) в сухом ДМФ (1 мл) перемешивали и обрабатывали гидридом натрия (0,28 ммоль; 11 мг, 60% дисперсия в минеральном масле), а затем 4-нитробензилиодидом(0,24 ммоль; 63 мг). Смесь перемешивали в течение 18 ч, за которые она приобретала желто-зеленую-6 009370 окраску. Эту смесь гасили солевым раствором и экстрагировали 32 мл Et2O. Объединенные органические слои сушили MgSO4, фильтровали и концентрировали на роторном испарителе с получением коричневого масла. После очистки с помощью радиальной хроматографии (2-мм пластина; элюент: смесь гексана-СН 2 СН 2 (1:1 получали продукт в виде желтого твердого вещества (53 мг, выход 43%). 1H-ЯМР (400 МГц, CDCl3)8,06 (д, J=7,6 Гц, 2 Н), 7,35 (с, 2 Н), 7,14 (д, J=7,2 Гц, 2 Н), 6,79 (с, 2 Н),5,41 (с, 1 Н), 3,13 (с, 2 Н), 1,45-1,43 (перекрывающийся с, 21 Н), 1,14 (с, 21 Н). Пример 3. Бутандионовой кислоты моно[4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1 метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенил]эфир. Описание реакции. В 50-миллилитровую колбу-сборник добавляли пробукол (1,0 г, 1,93 ммоль) и тетрагидрофуран (16 мл). К раствору добавляли 60% гидрид натрия в минеральном масле (0,23 г, 5,75 ммоль). К мутной белой смеси добавляли ангидрид янтарной кислоты (0,58 г, 5,8 ммоль) в ТГФ (12 мл). Реакционная смесь приобретала темно-пурпурную окраску и была перемешана в течение 3 ч при комнатной температуре. Эту темно-пурпурную реакционную смесь подкисляли 1 н. HCl (25 мл) и дважды экстрагировали этилацетатом (50 мл). Органические экстракты сушили MgSO4, фильтровали и концентрировали с получением оранжевого твердого вещества. Это оранжевое твердое вещество растворяли в эфире и хроматографировали на силикагеле с градиентом концентрации смеси гексана/эфира (70:30)гексана/эфира (0:100). Соответствующие фракции объединяли и концентрировали с получением белого твердого вещества (170 мгм, 0,276 ммоль, 14%). ТСХ (силикагель, смесь эфира/гексана (60:40)+10 капель HOAc, Rf=0,35); 1 Н-ЯМР (400 МГц, CDCl3)7,61 (с, 2 Н), 7,43 (с, 2 Н), 5,38 (с, 1 Н), 2,97 (т, J=6,8 Гц, 2 Н), 2,76 (т,J=6,8 Гц, 2 Н), 1,45 (с, 8 Н), 1,42 (с, 16 Н), 1,32 (с, 18 Н). Пример 4. 2-Фуранкарбоновой кислоты 5-нитро-4-1-3,5-бис-(1,1-диметилэтил)-4 гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фениловый эфир. Описание реакции. Раствор 0,39 ммоль (200 мг) пробукола в сухом ТГФ (3,9 мл) обрабатывали гидридом натрия (0,58 ммоль; 23 мг; 60% дисперсия в минеральном масле) и перемешивали в течение 10 мин при комнатной температуре. После добавления 4-нитрофуроилхлорида (0,77 ммоль; 136 мг) прозрачная смесь становилась пурпурной. Смесь перемешивали в течение 47 ч, после чего она становилась коричневой и наблюдалось выпадение осадка. Эту реакционную смесь разбавляли Et2O (40 мл), промывали Н 2 О (15 мл), а затем сушили Na2SO4 и концентрировали на роторном испарителе с получением неочищенного оранжево-желтого твердого вещества. После очистки с помощью радиальной хроматографии (2 мм SiO2 пластина, элюент: смесь гексана-CH2Cl2 (1:1 получали 4,4'-(изопропилиденди-тио)[О-(5"-нитро-2"фуроил)-2',6'-ди-трет-бутилфенол]-[2,6-ди-трет-бутилфенол] (83 мг, выход 33%). 1 Н-ЯМР (400 МГц, CDCl3)7,70 (с, 2 Н), 7,50 (д, J=4,0 Гц, 1 Н), 7,45 (д, J=3,6 Гц, 1 Н), 7,45 (с, 2 Н),5,39 (с, 1 Н), 1,50 (с, 6 Н), 1,45 (с, 14 Н), 1,35 (с, 22 Н). Пример 5. 4-[4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6 диметилфенокси]бутановая кислота. Описание реакции. 4,4'-(Изопропилидендитио)[2',6'-ди-метилфенол][2,6-ди-трет-бутилфенол] (0,55 ммоль; 0,24 г) растворяли в сухом ДМФ (5,5 мл). К смеси добавляли гидрид натрия (1,38 ммоль; 33 мг), а затем метил-4 иодбутират (0,83 ммоль; 188 мг). Полученную смесь перемешивали при комнатной температуре в течение 4,5 ч, за которые она приобретала зеленую окраску. Реакцию гасили 0,3 н. HCl (прибл. 6 мл), после чего смесь становилась желтой. Затем ее разбавляли Et2O (25 мл), после чего промывали H2O (10 мл) и солевым раствором (10 мл). Раствор сушили MgSO4, a затем концентрировали на роторном испарителе. После очистки с помощью ЖХСД (SiO2; градиент растворителя: 100% гексан, затем смесь гексанаEt2O=95:5, затем смесь гексана-Et2O=90:10, затем смесь гексана-Et2O=80:20) получали желаемое промежуточное соединение в виде желтого масла (197 мг; выход 67%). Это масло (0,35 ммоль; 187 мг) растворяли в смеси МеОН-ТГФ-Н 2O (4:1:1) (3,5 мл). Затем добавляли моногидрат LiOH (1,05 ммоль; 44 мг) и смесь перемешивали в течение 1,75 ч при комнатной температуре. Затем реакционную смесь подкисляли путем добавления 0,1 н. HCl до pH 4. После экстракции 315 мл EtOAc, сушки объединенных экстрактовMgSO4 и концентрирования на роторном испарителе получали неочищенный продукт. В результате очистки с помощью ЖХСД (SiO2; градиент растворителя: 100% гексансмесь 60:40 Et2 О-гексана (подкисление следовым количеством уксусной кислоты получали 4,4'-(изопропилидендитио) [О-(-масляная кислота)-2',6'-диметилфенол] [2,6-ди-трет-бутилфенол] в виде желтой пены (100 мг; выход 55%). 1 Н-ЯМР (400 МГц, CDCl3)7,44 (с, 2 Н), 7,25 (с, 2 Н), 5,38 (с, 1 Н), 3,83 (приб.т, J=6,0 Гц, 2 Н), 2,68(приб.т, J=8,0 Гц, 2 Н), 2,25 (с, 6 Н), 2,14 (м, 2 Н), 1,47 (с, 6 Н), 1,45 (с, 18 Н). Пример 6. 4-1-4-(4-Аминобутокси)-3,5-бис-(1,1-диметилэтил)фенил]тио]-1-метилэтил]тио]-2,6 бис-(1,1-диметилэтил)фенол. Описание реакции. 4,4'-(Изопропилидендитио)[2',6'-диметилфенол][2,6-ди-трет-бутилфенол] (1,44 ммоль, 622 мг) растворяли в сухом ДМФ (14,4 мл) и обрабатывали гидридом натрия (3,6 ммоль; 144 мг). После этого до-7 009370 бавляли иодид тетрабутиламмония (0,72 ммоль; 266 мг), а затем (N-бромбутил)фталимид (2,2 ммоль; 608 мг). Смесь перемешивали при комнатной температуре в течение 17 ч, за которые она приобретала темно-зеленую окраску. Эту смесь гасили 0,3 н. HCl (6 мл), а затем разбавляли Et2O (100 мл). Затем ее промывали Н 2O (50 мл) и солевым раствором (50 мл). Водный слой обрабатывали NaCl, а затем подвергали обратной экстракции Et2O. Объединенные органические слои сушили MgSO4, после чего концентрировали на роторном испарителе. После очистки с помощью ЖХСД (SiO2; градиент растворителя: 100% гексансмесь гексана-Et2O (75:25 получали желаемое промежуточное соединение в виде желто-коричневого масла (750 мг; выход 82%). Это промежуточное соединение (0,89 ммоль; 563 мг) растворяли в сухом ДМФ (8,9 мл) и обрабатывали гидратом гидразина (27 ммоль; 0,83 мл). Смесь перемешивали в течение 42 ч при комнатной температуре. Реакционную смесь обрабатывали 1 н. HCl (8,9 мл) и перемешивали в течение 1,5 ч; после чего добавляли NaHCO3 для доведения pH до 7, а затем экстрагировали EtOAc (215 мл) и сушили MgSO4. После удаления растворителя на роторном испарителе с последующей очисткой посредством ЖХСД (SiO2; градиент растворителей: смесь МеОН-CH2Cl2 (50:50)(приб. т, J=6,8 Гц, 2 Н), 2,24 (с, 6 Н), 1,83 (м, 2 Н), 1,66 (м, 2 Н), 1,45 (с, 6 Н), 1,42 (с, 18 Н). Пример 7. 4-1-4-(4-Аминобутокси)-3,5-бис-(1,1-диметилэтил)фенил]тио]-1-метилэтил]тио]-2,6 бис-(1,1-диметилэтил)фенол. Описание реакции. Пробукол (9,7 ммоль; 5 г) растворяли в сухом ДМФ (14,5 мл) и обрабатывали (Nбромбутил)фталимидом (13,6 ммоль; 3,82 г) и KF на окиси алюминия (48,4 ммоль; 7,03 г). Реакционную смесь перемешивали в течение 18 ч при комнатной температуре, а затем в течение 4 ч при 80 С. Смесь фильтровали через воронку из спеченного стекла и остаток промывали H2O (10 мл) и Et2O (10 мл). Фильтрат разбавляли Et2O (200 мл), после чего промывали H2O (50 мл) и солевым раствором (50 мл). Объединенные органические слом сушили MgSO4 и концентрировали на роторном испарителе. В результате очистки посредством ЖХСД (SiO2; градиент растворителя: 100% гексансмесь гексана-Et2O,80:20) получали желаемое промежуточное соединение в виде коричневой пены (346 мг; выход 5%). Это промежуточное соединение (0,44 ммоль; 314 мг) растворяли в ДМФ (4,4 мл) и обрабатывали гидратом гидразина (13 ммоль; 0,41 мл), в результате чего появлялась зеленая окраска. Полученную смесь перемешивали при комнатной температуре в течение 16 ч, обрабатывали 1 н. HCl (4,7 мл) и перемешивали еще 1,5 ч. Затем добавляли NaHCO3 для доведения смеси до pH 7. После экстракции 230 мл EtOAc,промывки органических экстрактов солевым раствором (20 мл), сушки MgSO4 и удаления растворителя на роторном испарителе получали зеленую жидкость. После очистки посредством ЖХСД (SiO2; градиент растворителя: 100% CH2Cl2 смесь CH2Cl2-MeOH, 90:10) получали 4,4'-(изопропилидендитио)[О(аминобутил)-2',6'-ди-трет-бутилфенол][2,6-ди-трет-бутилфенол] в виде желтого твердого вещества (182 мг; выход 71%). 1H-ЯМР (400 МГц, CDCl3)7,52 (с, 2 Н), 7,44 (с, 2 Н), 5,28 (с, 1 Н), 5,25 (шир.с, 2 Н), 3,72-3,69 (м, 2 Н),2,92-2,88 (м, 2 Н), 1,94-1,90 (м, 2 Н), 1,73-1,69 (м, 2 Н), 1,43 (с, 22 Н), 1,40 (с, 20 Н). Пример 8. Бутановой кислоты 4-гидрокси-,4-1-3,5-бис-(1,1-диметилэтил)-4 гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фениловый эфир. Описание реакции. К раствору соединения примера 22 (6,17 г, 10 ммоль) в ТГФ (200 мл), охлажденному до -78 С, медленно добавляли боран-метилсульфид (10 мл, 2 М раствор в ТГФ). Полученную смесь перемешивали в течение ночи в атмосфере азота с нагреванием охлаждающей бани до комнатной температуры. Затем смесь охлаждали до 0 С, добавляли хлористый водород (37%, 4 мл) и смесь перемешивали в течение ночи при комнатной температуре. Смесь выпаривали с получением остатка и распределяли между этилацетатом (100 мл) и солевым раствором (100 мл). Органическую фазу промывали 1 н. раствором гидроксида натрия (100 мл), а затем солевым раствором (100 мл), после чего сушили сульфатом магния и упаривали. После проведения хроматографии на силикагеле (дихлорметан) получали целевое соединение в виде вязкого остатка. После кристаллизации из гексана/дихлорметана были получены белые кристаллы (5,5 г). Т.пл. 138-139 С. 1 Н-ЯМР (400 МГц, CDCl3)7,63 (с, 2 Н), 7,45 (с, 2 Н), 5,38 (с, 1 Н), 3,76 (т, 2 Н), 2,79 (т, 2 Н), 2,01 (м,2 Н), 1,47 (с, 6 Н), 1,44 (с, 18 Н), 1,34 (с, 18 Н). Пример 9. Пропановой кислоты 2,2-диметил-[4-1-3,5-бис-(1,1-диметилэтил)-4 гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенокси]метиловый эфир. К суспензии пробукола (5,17 г, 10 ммоль) в ацетонитриле (30 мл) добавляли хлорметилпивалат(6,0 г, 40 ммоль) и фторид калия (8,0 г, 40% на окиси алюминия). Полученную смесь перемешивали в атмосфере азота при нагревании с обратным холодильником в течение 18 ч. После охлаждения до комнатной температуры ее фильтровали и промывали дихлорметаном (100 мл). Фильтрат промывали солевым раствором (100 мл), сушили сульфатом магния и упаривали. После хроматографии на силикагеле(смесь гексана/дихлорметана=4:1) получали целевое соединение в виде желтого масла (0,39 г). 1 Н-ЯМР (400 МГц, CDCl3)7,59 (с, 2 Н), 7,45 (с, 2 Н), 5,49 (с, 2 Н), 5,38 (с, 1 Н), 1,464 (с, 6 Н), 1,457 (с,18 Н), 1,445 (с, 18 Н), 1,28 (с, 9 Н). Пример 10. 4-1-4-(4-Аминобутокси)фенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенол. Описание реакции. Раствор 4,4'-(изопропилидендитио)[фенол][2,6-ди-трет-бутилфенола] (0,18 ммоль; 75 мг) в 2 мл сухого ДМФ перемешивали и обрабатывали 0,23 ммоль (9 мг) гидрида натрия (60% дисперсия в минеральном масле) с получением желтой смеси. После этого добавляли N-(4-бромбутил)фталимид (0,22 ммоль; 63 мг), а затем 0,22 ммоль (33 мг) Nal. Смесь нагревали до 120 С, в результате чего появлялась темнозеленая окраска. Через 24 ч с помощью ТСХ (SiO2; элюент: CH2Cl2; визуализация: УФ, ПМА/уголь) обнаруживали лишь следовые количества исходного продукта. Реакционную смесь охлаждали до комнатной температуры, а затем гасили 3 мл (каждого) Et2O и насыщенного NaCl. Водный слой подвергали обратной экстракции 3 мл Et2O; объединенные органические слои сушили MgSO4, фильтровали и концентрировали на роторном испарителе с получением темно-коричневого масла. В результате очистки с помощью колоночной хроматографии (SiO2, 20170-мм колонки; элюент: CH2Cl2) получали желаемое промежуточное соединение с выходом 69% (69 мг). Это промежуточное соединение (0,11 ммоль; 69 мг) растворяли в 1 мл ДМФ и перемешивали. Затем добавляли гидрат гидразина (0,16 ммоль; 8 мкл), в результате чего цвет менялся с желтого на глубокий сине-зеленый. Реакционную смесь перемешивали при комнатной температуре в течение 1 недели, за которые эта смесь становилась прозрачно-желтой. С помощью ТСХ обнаруживали присутствие исходного продукта. Затем добавляли дополнительное количество гидрата гидразина (10,3 ммоль; 0,5 мл); смесь перемешивали еще 24 ч, после чего исходный продукт был полностью израсходован. Смесь гасили путем добавления 12 н. HCl для доведения pH до 3. После перемешивания в течение 5 мин добавляли NaHCO3 для нейтрализации кислоты (конечное значение pH 7). После добавления EtOAc водный слой подвергали обратной экстракции 22 мл EtOAc. Объединенные органические слои сушили MgSO4, фильтровали и концентрировали на роторном испарителе. Неочищенный продукт очищали с помощью колоночной хроматографии (SiO2, колонка 15110 мм; элюент:EtOH) с получением 4,4'-(изопропилидендитио)[О-(аминобутил)фенол][2,6-ди-трет-бутилфенола] (25 мг; выход 48%) в виде желтого твердого вещества. 1H-ЯМР (400 МГц, CDCl3)7,50 (д, J=8,4 Гц, 2 Н), 7,42 (с, 2 Н), 6,83 (д, J=8,4 Гц, 2 Н), 5,36 (шир.с,1 Н), 3,97 (шир.м., 2 Н), 3,10 (шир.м., 2 Н), 2,01-1,86 (перекрывающийся м, 4 Н), 1,43 (с, 24 Н). Пример 11. 4-[4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенилтио]тио]-1-метилэтил]тио]фенокси]бутановая кислота. Описание реакции. Раствор 4,4'-(изопропилидендитио)[фенол][2,6-ди-трет-бутилфенола] (0,6 ммоль; 242 мг) в 6 мл сухого ДМФ перемешивали и обрабатывали 1,3 ммоль (53 мг) гидрида натрия (60% дисперсия в минеральном масле) с получением коричневого раствора. Реакционную смесь обрабатывали 0,9 ммоль (131 мкл) метил-4-иодбутирата. Ход реакции прослеживали с помощью ТСХ с использованием CH2Cl2 в качестве элюента (визуализация с помощью УФ, ПМА/уголь). Через 24 ч ТСХ-анализ темно-зеленой смеси обнаруживал присутствие преобладающего продукта. Смесь гасили насыщенным NaCl и Et2O. Водный слой подвергали обратной экстракции 26 мл Et2O; объединенные органические слои сушили MgSO4, фильтровали и концентрировали на роторном испарителе с получением неочищенного масла. В результате проведения колоночной хроматографии (SiO2; колонка 20185 мм) с использованием CH2Cl2 получали 232 мг (выход 77%) желаемого промежуточного соединения, которое растворяли и перемешивали в 3 мл смеси МеОН-ТГФ-H2O (4:1:1). Бледно-желтый раствор обрабатывали 0,92 ммоль (39 мг) моногидратаLiOH с получением зеленовато-желтого раствора. Смесь перемешивали при комнатной температуре до тех пор, пока весь исходный продукт не был израсходован (приб. 18 ч), а затем обрабатывали 12 н. HCl до pH 2 (желтая смесь). После этого добавляли Et2O (5 мл); водный слой подвергали обратной экстракции 25 мл Et2O. Объединенные органические слои сушили MgSO4, фильтровали, а затем концентрировали на роторном испарителе с образованием неочищенного твердого вещества. После очистки с помощью колоночной хроматографии (SiO2; колонка 15180 мм; элюент: смесь гексана-EtOH, 3:1) получали 4,4'-(изопропилидендитио)[O-(-масляная кислота)фенол][2,6-ди-трет-бутилфенол] в виде масла (62 мг; выход 40%). 1H-ЯМР (400 МГц, CDCl3)7,47 (д, J=8,0 Гц, 2 Н), 7,40 (с, 2 Н), 6,80 (д, J=7,6 Гц, 2 Н), 5,35 (с, 1 Н),3,93 (шир.м, 2 Н), 2,51 (шир.м., 2 Н), 2,07 (шир.м, 2 Н), 1,42 (с, 18 Н), 1,25 (с, 6 Н). Пример 12. [4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1 диметилэтил)фенокси]уксусная кислота. Описание реакции. К диметилформамиду (1,5 мл) добавляли пробукол (0,5 г, 0,967 ммоль) и этил-2-иодацетат (0,31 г,1,45 ммоль) и 40% фторид калия на окиси алюминия (0,7 г) и реакционную смесь перемешивали в течение 24 ч. Реакционную смесь разбавляли эфиром (25 мл), фильтровали и промывали водой (25 мл).-9 009370 Эфирный слой сушили MgSO4, фильтровали и концентрировали. Полученное масло очищали с помощью радиальной хроматографии на силикагеле при элюировании смесью эфира/гексана (5:95) с получением 160 мг сложного этилового эфира данного продукта. Этот сложный этиловый эфир растворяли в смеси ТГФ:Н 2O:МеОН (4:1:1) (4 мл) и добавляли смесь LiOH-H2O (50 мг), а затем реакционную смесь перемешивали в течение 1 ч. Реакционную смесь нейтрализовали 1 н. HCl, экстрагировали эфиром (210 мл),сушили MgSO4, фильтровали и концентрировали. После хроматографии на силикагеле и элюирования смесью эфира/гексана (50:50) получали 90 мг продукта. 1H-ЯМР (400 МГц, CDCl3)7,55 (с, 2 Н), 7,40 (с, 2 Н), 5,35 (с, 1 Н), 4,40 (с, 2 Н), 1,43 (с, 6 Н), 1,41 (с,9 Н), 1,39 (с, 9 Н). Пример 13. 4-1-3,5-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-(1,1 диметилэтил)фениловый эфир глицина. Описание реакции. К раствору пробукола (3,0 г, 5,8 ммоль) в ТГФ (58 мл) добавляли 60% гидрид натрия (1,16 г, 29,0 ммоль) и реакционную смесь перемешивали в течение 0,5 ч при комнатной температуре. Затем добавляли хлорангидрид фталоилглицина и реакционную смесь перемешивали еще 0,5 ч. Реакционную смесь разбавляли этилацетатом (150 мл), гасили водой (5 мл), а затем промывали водой (250 мл) и солевым раствором (150 мл). Органический слой сушили MgSO4, фильтровали и концентрировали. Полученное масло хроматографировали на силикагеле, элюируя смесью 10% этилацетата/гексана, а затем 20% этилацетата/гексана, с получением 610 мг сложного эфира фталоилглицина. Сложный эфир фталоилглицина растворяли в ДМФ (8,6 мл) и добавляли гидрат гидразина (0,136 мл, 2,34 ммоль), после чего реакционную смесь перемешивали в течение ночи. Затем добавляли 1 н. HCl (5 мл) и реакционную смесь перемешивали еще 1 ч. Реакционную смесь разбавляли этилацетатом (25 мл) и промывали NaHCO3 (вод.) (110 мл). Этил-ацетатный слой сушили MgSO4, фильтровали, концентрировали и хроматографировали на силикагеле, элюируя смесью 1% метанола/метиленхлорида, а затем смесью 1,5% метанола/метиленхлорида с получением 334 мг продукта. 1(с, 18 Н), 1,33 (с, 18 Н). Пример 14. Пентандионовой кислоты моно[4-1-3,5-бис-(1,1-диметил)-4-гидроксифенил]тио]-1 метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенил]эфир. Описание реакции. В 50-миллилитровую колбу-сборник добавляли пробукол (1,0 г, 1,93 ммоль) и тетрагидрофуран (20 мл). К этому раствору добавляли 60% гидрид натрия в минеральном масле (0,16 г, 4 ммоль). К мутной белой смеси добавляли ангидрид глутаровой кислоты (0,170 г, 3 ммоль) в ТГФ (12 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь подкисляли 1 н. HCl(25 мл) и дважды экстрагировали этилацетатом (50 мл). Органические экстракты сушили MgSO4, фильтровали и концентрировали с получением желтого масла. Желтое масло растворяли в эфире и хроматографировали на силикагеле с градиентом концентрации смеси гексана/эфира=70:30 гексана/эфира=0:100. Соответствующие фракции объединяли и концентрировали с получением белого твердого вещества. 1(15 мл). К реакционной смеси добавляли 40% фторид калия на окиси алюминия (7 г, 48 моль) и перемешивание продолжали при комнатной температуре в течение ночи. Зеленую реакционную смесь фильтровали в делительной воронке, разбавляли этилацетатом (50 мл), а затем промывали водой (220 мл) и насыщенным водным раствором хлорида натрия (120 мл). Этилацетатный слой сушили MgSO4, фильтровали, концентрировали и хроматографировали на силикагеле при элюировании градиентом концентрации смеси метиленхлорида/гексана=10:90 метиленхлорида/гексана=60:40. Соответствующие фракции собирали и концентрировали с получением 442 мг белого твердого вещества. Сложный метиловый эфир растворяли в смеси ТГФ:МеОН:Н 2O (4:1:1) (5 мл) и добавляли гидроксид лития (63 мг, 1,5 ммоль). Через 2,5 ч реакция была полностью завершена, после чего ее гасили 1 н. HCl (3 мл) и экстрагировали этилацетатом (15 мл). Этилацетатный раствор промывали насыщенным водным раствором хлорида натрия (3 мл), сушили MgSO4, фильтровали и концентрировали. Затем проводили хроматографию на силикагеле с использованием в качестве элюента градиент смеси растворителей: эфира/гексана=10:90 эфира/гексана=50:50, в результате чего получали 308 мг продукта. 1(1,6 мл, 20 ммоль) и фторид калия (2,9 г, 20 ммоль, 40% на окиси алюминия). Полученную смесь перемешивали в атмосфере азота с обратным холодильником в течение 18 ч. После охлаждения до комнатной температуры ее выливали в дихлорметан (150 мл), промывали водой (2100 мл), сушили магнием и упаривали. После хроматографии на силикагеле (гексан/дихлорметан, 2:1, 1:1, 1:2, а затем дихлорметан) получали(0,66 мл, 10 ммоль), трифенилфосфин (2,62 г, 10 ммоль) и диэтилазодикарбоксилат (1,57 мл, 10 ммоль). Полученную смесь перемешивали в атмосфере азота при нагревании с обратным холодильником в течение 48 ч, а затем упаривали. После хроматографии на силикагеле (смесь гексана/дихлорметана=4:1, 2:1) получали целевое соединение в виде вязкого остатка (1,01 г). 1 Н-ЯМР (400 МГц, CDCl3)7,56 (с, 2 Н), 7,44 (с, 2 Н), 5,39 (с, 1 Н), 4,40 (м, 1 Н), 3,75 (м, 1 Н), 3,39 (м,1 Н), 2,91 (м, 1 Н), 2,77 (м, 1 Н), 1,40-1,49 (м, 42 Н). Пример 18. N-[3-[4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6 бис-(1,1-диметилэтил)фенокси]-2-гидроксипропил]глицин. К суспензии 4-1-3,5-бис-(1,1-диметилэтил)-4-(оксиранилметокси)фенил]тио]-1-метилэтил]тио]2,6-бис-(1,1-диметилэтил)фенола (0,17 г, 0,28 ммоль) в этаноле (10 мл) добавляли глицин (43 мг, 0,57 ммоль) и триэтиламин (1 мл). Полученную смесь перемешивали в атмосфере азота при нагревании с обратным холодильником в течение ночи. После нагревания смесь превращалась в раствор. Затем эту смесь упаривали. После хроматографии на силикагеле (смесь дихлорметана/метанола, 10:11:1), получали целевое соединение (99 мг). 1 Н-ЯМР (400 МГц, CDCl3)7,52 (с, 2 Н), 7,43 (с, 2 Н), 5,37 (с, 1 Н), 4,58 (шир.с, 1 Н), 3,79 (шир.м.,2 Н), 3,67 (м, 1 Н), 3,30 (м, 1 Н), 3,21 (м, 1 Н), 3,13 (м, 1 Н), 1,43 (с, 18 Н), 1,41 (с, 6 Н), 1,38 (с, 18 Н). Пример 19. 4-4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенокси]-2-гидроксипропил]-1,2,3-бутантриол. К раствору -4-4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенокси]метил]оксиранметанола (0,15 г) в ацетонитриле (5 мл) добавляли воду (1 мл) и концентрированную серную кислоту (10 капель). Полученную смесь перемешивали в течение 48 ч при комнатной температуре, после чего выливали в солевой раствор (50 мл), экстрагировали дихлорметаном(350 мл), сушили сульфатом магния и упаривали. После хроматографии на силикагеле (дихлорметан/этилацетат 3:1) получали целевое соединение в виде бесцветного вязкого остатка (26 мг). 1 Н-ЯМР (400 МГц, CDCl3)7,54 (с, 2 Н), 7,43 (с, 2 Н), 5,36 (с, 1 Н), 4,27 (м, 2 Н), 3,98 (м, 2 Н), 3,75 (м,2 Н), 1,36-1,44 (м, 42 Н). Пример 20.(10 мл) добавляли 1 н. раствор гидроксида натрия (1,5 мл). Полученную смесь перемешивали с обратным холодильников в течение 3 дней, а затем упаривали. Остаток распределяли между этилацетатом (50 мл) и солевым раствором (50 мл). Органическую фазу промывали солевым раствором (50 мл), сушили сульфатом магния и упаривали. После хроматографии на силикагеле (гексан/дихлорметан 1:1, дихлорметан, а затем дихлорметан/этилацетат 4:1) получали 3-[41-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенокси]-1,2-пропандиол (58 мг) и смесь, которая содержала 4-1-3,5-бис-(1,1-диметилэтил)-4-(3 этокси-2-гидроксипропокси)фенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенол и которую снова пропускали через колонку (гексан/этилацетат 5:1), и получали 4-1-3,5-бис-(1,1-диметилэтил)-4(3-этокси-2-гидроксипропокси)фенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенол в чистой форме (52 мг). 4-1-3,5-бис-(1,1-Диметилэтил)-4-(3-этокси-2-гидроксипропокси)фенил]тио]-1-метилэтил]тио]2,6-бис-(1,1-диметилэтил)фенол. 1 Н-ЯМР (400 МГц, CDCl3)7,55 (с, 2 Н), 7,45 (с, 2 Н), 5,38 (с, 1 Н), 4,35 (м, 1 Н), 4,11 (м, 1 Н), 3,83(м,2 Н), 3,62 (м, 1 Н), 3,57 (м, 2 Н), 1,43-1,46 (м, 42 Н), 1,22 (т, 3H). 3-[4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1 диметилэтил)фенокси]-1,2-пропандиол; 1 Н-ЯМР (400 МГц, CDCl3)7,56 (с, 2 Н), 7,45 (с, 2 Н), 5,38 (с, 1 Н), 4,32 (м, 1 Н), 3,94 (дд, 1 Н), 3,85 (м,1 Н), 3,77 (м, 1 Н), 3,66 (м, 1 Н), 1,40-1,44 (м, 42 Н). Пример 21. 4-1-3,5-бис-(1,1-Диметилэтил)-4-этоксифенил]тио]-1-метоксиэтил]тио]-2,6-бис-(1,1 диметилэтил)фенол; 3-[4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1 диметилэтил)фенокси]-2-пропеновой кислоты этиловый эфир, [Е]-; 3-[4-1-3,5-бис-(1,1-диметилэтил)-4-этоксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1 диметилэтил)фенокси]-2-пропеновой кислоты этиловый эфир, (Е)-. К раствору пробукола (5,16 г, 10 ммоль) в ТГФ (50 мл) добавляли этилпропиолат (1,2 мл, 12 ммоль) и триэтиламин (7 мл, 50 ммоль). Полученную смесь перемешивали в атмосфере азота при нагревании с обратным холодильником в течение выходных дней. После охлаждения до комнатной температуры смесь выливали в солевой раствор (100 мл), экстрагировали дихлорметаном (2100 мл); сушили сульфатом магния и упаривали. После хроматографии на силикагеле (смесь гексана/дихлорметана (9:1)дихлорметан) получали 4-[1-[3,5-бис-(1,1-диметилэтил)-4-этоксифенил]тио-1-метилэтил]тио-2,6-бис-(1,1 диметилэтил)фенол;(квад., 2 Н), 1,25-1,48 (м, 48 Н). Пример 22. Бутандионовой кислоты моно[4-1-3,5-бис-(1,1-диметилэтил)-4-метоксифенил]тио]-1 метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенил]эфир. Описание реакции. Соединение примера 22 (1,13 г, 1,83 ммоль) растворяли в ДМФ (3,6 мл) и добавляли 60% гидрид натрия (183 мг, 4,6 ммоль), а затем через 0,25 ч добавляли метилиодид (0,342 мл, 5,5 ммоль). Реакционную смесь перемешивали в течение ночи. Реакционную смесь гасили водой (2 мл) и разбавляли эфиром(50 мл). Эфирный слой промывали водой (210 мл) и насыщенным водным раствором хлорида натрия(110 мл), а затем сушили MgSO4, фильтровали и концентрировали. После колоночной хроматографии на силикагеле и элюирования градиентом концентрации смеси 0:100 эфира/гексана 40:60 эфи- 12009370 ра/гексана получали 556 мг диметилированного продукта. Этот продукт растворяли в смеси ТГФ:МеОН:Н 2O (4:1:1) (5 мл) и добавляли гидроксид лития (63 мг, 1,5 ммоль). Через 2,5 ч реакцию завершали и гасили 1 н. HCl (3 мл) и экстрагировали этилацетатом (15 мл). Этилацетатный слой промывали насыщенным водным раствором хлорида натрия (3 мл), сушили MgSO4, фильтровали и концентрировали. После хроматографии на силикагеле и элюирования градиентом растворителя эфира/гексана=10:90 эфира/гексана=50:50 получали 400 мг продукта. 1 Н-ЯМР (400 МГц, CDCl3)7,62 (с, 2 Н), 7,45 (с, 2 Н), 5,37 (с, 1 Н), 3,71 (с, 3H), 2,75 (т, J=7,2 Гц, 2 Н),2,55 (т, J=7,2 Гц, 2 Н), 2,09 (м, 2 Н), 1,46 (с, 6 Н), 1,44 (с, 18 Н), 1,42 (с, 18 Н). Пример 23. 4-1-4-[2-[4-(Диметиламино)фенил]этокси]-3,5-бис-(1,1-диметилэтил)фенил]тио]-1 метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенол. Описание реакции. Пробукол (1,16 ммоль; 600 мг) растворяли в ТГФ (11,6 мл) и обрабатывали трифенилфосфином (2,3 ммоль; 608 мг), диэтилазодикарбоксилатом (2,3 ммоль; 0,37 мл) и 4-(диметиламино)фенэтиловым спиртом (2,3 ммоль; 383 мг). Коричневую смесь перемешивали при нагревании с обратным холодильником в течение 41,5 ч. Растворитель удаляли на роторном испарителе с получением коричневого масла. В результате хроматографической очистки получали 4,4'-(изопропилидендитио)[О-(4"(диметиламино)фенэтил)-2',6'-ди-трет-бутилфенол][2,6-ди-трет-бутилфенол] в виде масла (256 мг; выход 33%). 1(перекрывающийся с, 42 Н). Пример 24. 4,4'-[(1-Метилэтилиден)бис-[тио[2,6-бис-(1,1-диметилэтил)-4,1-фенилен]окси-2,1 этандиилбис-[N,N-диметилбензоламин]. Описание реакции. Пробукол (1,16 ммоль; 600 мг) растворяли в ТГФ (11,6 мл) и обрабатывали трифенилфосфином (2,3 ммоль; 608 мг), диэтилазодикарбоксилатом (2,3 ммоль, 0,37 мл) и 4-(диметиламино)фенэтиловым спиртом (2,3 ммоль, 383 мг). Коричневую смесь перемешивали при нагревании с обратным холодильником в течение 41,5 ч. Растворитель удаляли на роторном испарителе с получением коричневого масла. После хроматографической очистки получали 4,4'-(изопропилидендитио)бис-[(4"-(диметиламино)фенэтил)-2,6 ди-трет-бутилфенол] в виде светло-розового твердого вещества (155 мг, выход 16%). 1(приб.т, J=7,6, 8,8 Гц, 4 Н), 3,09 (приб.т, J=8,0, 8,8 Гц, 4 Н), 2,93 (с, 12 Н), 1,43-1,42 (перекрывающийся с,42 Н). Пример 25. Моно[4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6 бис-(1,1-диметилэтил)фенилбутандиоат] L-аргинина. Описание реакции. К раствору соединения примера 22 (1,67 г, 2,7 ммоль) в метаноле (30 мл) добавляли L-аргинин (0,47 г, 2,7 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 ч, а затем фильтровали. Фильтрат упаривали и остаток растворяли в минимальном количестве эфира. Затем добавляли гексан, в результате чего осаждалось целевое соединение. Это соединение фильтровали и сушили в вакууме с получением беловатого твердого вещества (1,75 г). Т.пл. 185-190 С. 1H-ЯМР (400 МГц, CDCl3)7,60 (с, 2 Н), 7,42 (с, 2 Н), 5,37 (с, 1 Н), 3,64 (шир.с, 1 Н), 3,11 (шир.с, 2 Н),2,96 (шир.с, 2 Н), 2,58 (шир.с, 2 Н), 1,41-1,44 (м, 26 Н), 1,23-1,31 (м, 20 Н). Пример 26. 3-[4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенокси]-2-пропеновая кислота, (Е)-. К раствору 3-[4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенокси]-2-пропеновой кислоты этилового эфира, (Е)-, (0,16 г, 0,26 ммоль) в ТГФ (5 мл) добавляли воду (2 мл) и моногидрат гидроксида лития (42 мг, 1 ммоль). Полученную смесь перемешивали в течение ночи при нагревании с обратным холодильником. После охлаждения до комнатной температуры эту смесь выливали в дихлорметан (50 мл), промывали солевым раствором, сушили сульфатом магния и упаривали. После хроматографии на силикагеле (смесь гексана/этилацетата=4:1) получали целевое соединение в виде вязкого остатка (22 мг). 1 Н-ЯМР (400 МГц, CDCl3)7,63 (с, 2 Н), 7,44 (с, 2 Н), 6,52 (д, 1 Н), 5,39 (с, 1 Н), 5,08 (д, 1 Н), 1,47 (с,6 Н), 1,44 (с, 18 Н), 1,42 (с, 18 Н). Пример 27. 6-O-[4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенил]-1,2,3,4-бис-О-(1-метилэтилиден)D-галактопираноза. К раствору пробукола (2,58 г, 5 ммоль) и 1,2,3,4-ди-O-изопропилиден-D-галактопиранозы (1,8 мл,10 ммоль) в ТГФ (100 мл) добавляли трифенилфосфин (2,62 г, 10 ммоль) и диэтилазодикарбоксилат (1,57 мл, 10 ммоль). Полученную смесь перемешивали в атмосфере азота при нагревании с обратным холодильником в течение 72 ч. Затем эту смесь упаривали. После хроматографии на силикагеле (смесь циклогексана/этилацетата, 30:1) получали целевое соединение (0,16 г).H-ЯМР (400 МГц, CDCl3)7,53 (с, 2 Н), 7,45 (с, 2 Н), 5,60 (с, 1 Н), 4,65 (м, 2 Н), 4,35 (м, 4 Н), 1,59 (с,6 Н), 1,44 (с, 18 Н), 1,43 (с, 18 Н), 1,37 (с, 6 Н), 1,33 (с, 6 Н). Пример 28. 4-1-4-[3-(Диметиламино)пропокси]-3,5-бис-(1,1-диметилэтил)фенил]тио]-1 метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенол. Описание реакции. Пробукол (0,5 г, 0,97 ммоль) растворяли в ТГФ, охлаждали до 0 С, и добавляли 3 гидроксипропилдиэтиламин (0,287 мл, 1,94 ммоль), а затем добавляли трифенилфосфин (0,508 г, 1,94 ммоль) и диэтилазодикарбоксилат (0,31 мл, 1,94 ммоль). Реакционную смесь нагревали до температуры перегонки и это нагревание продолжали в течение 30 ч. Реакционную смесь концентрировали и очищали с помощью хроматографии на силикагеле при элюировании смесью метанола/эфира (20:80) с получением продукта. 1H-ЯМР (400 МГц, CDCl3)7,52 (с, 2 Н), 7,27 (с, 2 Н), 3,74 (т, J=1,6 Гц, 2 Н), 2,56 (кв., J=7,2 Гц, 2 Н),2,03 (пент., J=7,6 Гц, 2 Н), 1,44 (кв., J=3,2 Гц, 4 Н), 1,42 (с, 24 Н), 1,25 (т, J=3,3 Гц, 6 Н). Пример 29. N-4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенокси]ацетил]глицин. Описание реакции. К 4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтилфенокси)уксусной кислоте (50 мг, 0,087 ммоль) в метиленхлориде (0,87 мл) добавляли гидрохлорид этилового эфира глицина (15,8 мг, 0,11 ммоль), гидрохлорид 1-(3-диметиламинопропил-3 этилкарбодиимида) (22 мг, 0,11 ммоль) и диметиламинопиридин (28 мг, 0,23 ммоль). Реакционную смесь перемешивали в течение ночи, а метиленхлорид выпаривали. Реакционную смесь разбавляли эфиром (10 мл), промывали водой (23 мл), сушили MgSO4, фильтровали и концентрировали. Неочищенную смесь очищали с помощью хроматографии на силикагеле и элюировали смесью эфира/гексана (50:50) с получением 50 мг сложного этилового эфира желаемого продукта. Этот сложный этиловый эфир растворяли в смеси ТГФ:Н 2O:МеОН (2:1:1) (1 мл) и добавляли LiOH-H2O (15 мг), после чего реакционную смесь перемешивали в течение 1 ч. Эту реакционную смесь нейтрализовали 1 н. HCl, экстрагировали эфиром(210 мл), сушили MgSO4, фильтровали и концентрировали с получением 25 мг продукта. 1 Н-ЯМР (400 МГц, CDCl3)7,56 (с, 2 Н), 7,42 (с, 2 Н), 5,39 (шир.с, 1 Н), 4,31 (с, 2 Н), 4,22 (д, J=5,2 Гц,2 Н), 1,44 (с, 6 Н), 1,42 (с, 9 Н), 1,39 (с, 9 Н). Пример 30. N-4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенокси]ацетил]глутаминовая кислота. Описание реакции. К [4-1-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтилфенокси)уксусной кислоте (100 мг, 0,174 ммоль) в метиленхлориде (1,8 мл) добавляли гидрохлорид диэтилового эфира глутаминовой кислоты (54 мг, 0,22 ммоль), гидрохлорид 1-(3-диметиламинопропил 3-этилкарбодиимида) (44 мг, 0,22 ммоль) и диметиламинопиридин (55 мг, 0,45 ммоль). Реакционную смесь перемешивали в течение ночи, а метиленхлорид выпаривали. Реакционную смесь разбавляли эфиром (10 мл), промывали водой (23 мл), сушили MgSO4, фильтровали и концентрировали. Неочищенную смесь очищали с помощью хроматографии на силикагеле и элюировали смесью эфира/гексана (50:50) с получением 130 мг диэтилового эфира желаемого продукта. Этот диэтиловый сложный эфир растворяли в смеси ТГФ: Н 2O: МеОН (2:1:1) (3 мл), добавляли LiOH-H2O (100 мг) и реакционную смесь перемешивали в течение 1 ч. Реакционную смесь нейтрализовали 1 н. HCl, экстрагировали эфиром (210 мл), сушили MgSO4, фильтровали и концентрировали с получением 45 мг продукта. 1 Н-ЯМР (400 МГц, CDCl3)7,57 (с, 2 Н), 7,42 (с, 2 Н), 5,37 (с, 1 Н), 4,83 (м, 1 Н), 4,28 (с, 2 Н), 2,56 (м,2 Н), 1,44 (с, 6 Н), 1,43 (с, 9 Н), 1,41 (с, 9 Н). Пример 31. N-[3-[4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6 бис-(1,1-диметилэтил)фенокси]-2-гидроксипропил]-ди-L-глутаминовой кислоты диэтиловый эфир. Описание реакции. К суспензии 4-1-3,5-бис-(1,1-диметилэтил)-4-(оксиранилметокси)фенил]тио]-1-метилэтил]тио]2,6-бис-(1,1-диметилэтил)фенола (0,12 г, 0,20 ммоль) и гидрохлорида диэтилового эфира L-глутаминовой кислоты (0,24 г, 1 ммоль) в этаноле (15 мл) добавляли триэтиламин (2 мл). Полученную смесь перемешивали в атмосфере азота при нагревании с обратным холодильником в течение 18 ч. Затем ее упаривали. После хроматографии на силикагеле (смесь дихлорметана/метанола, 5:1) получали желтое масло, которое снова подвергали колоночной хроматографии (смесь дихлорметана/метанола, 10:1) с получением целевого соединения в виде белого вязкого остатка (16 мг). 1 Н-ЯМР (400 МГц, CDCl3)7,53 (с, 2 Н), 7,42 (с, 2 Н), 5,36 (с, 1 Н), 4,90 (м, 1 Н), 3,85 (м, 2 Н), 3,553,75 (м, 7 Н), 2,01 (м, 2 Н), 1,39-1,42 (м, 48 Н), 1,23 (м, 2 Н). Пример 32. 2-Пропеновой кислоты 4-[4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1 метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенокси]бутиловый эфир. К раствору пробукола (2,58 г, 5 ммоль) в ТГФ (50 мл) добавляли 4-гидроксибутилакрилат (1,0 мл,10 ммоль), трифенилфосфин (2,62 г, 10 ммоль) и диэтилазодикарбоксилат (1,57 мл, 10 ммоль). Получен- 14009370 ную смесь перемешивали в атмосфере азота при нагревании с обратным холодильником в течение выходных дней. Затем ее упаривали. После хроматографии на силикагеле (смесь гексана/дихлорметана=4:1) получали целевое соединение в виде коричневого масла (0,92 г). 1 Н-ЯМР (400 МГц, CDCl3)7,54 (с, 2 Н), 7,46 (с, 2 Н), 6,42 (дд, 1 Н), 6,14 (дд, 1 Н), 5,84 (дд, 1 Н), 5,38(с, 1 Н), 4,23 (т, 2 Н), 3,75 (т, 2 Н), 1,97 (м, 2 Н), 1,82 (м, 2 Н), 1,46 (с, 6 Н), 1,45 (с, 18 Н), 1,42 (с, 18 Н). Пример 33. 4-1-3,5-бис-(1,1-Диметилэтил)-4-(4-гидроксибутокси)фенил]тио]-1-метилэтил]тио]2,6-бис-(1,1-диметилэтил)фенол. К суспензии 4-[4-[1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио-2,6-бис(1,1-диметилэтил)фенокси]бутилового эфира 2-пропеновой кислоты (0,82 г) в метаноле (20 мл) добавляли карбонат калия (0,5 г). Полученную смесь перемешивали в атмосфере азота при комнатной температуре в течение ночи. Затем эту смесь выливали в воду (50 мл), экстрагировали дихлорметаном (250 мл),сушили сульфатом магния и упаривали. После хроматографии на силикагеле (смесь гексана/этилацетата=4:1) получали целевое соединение в виде бесцветного масла (0,52 г). 1H-ЯМР (400 МГц, CDCl3)7,54 (с, 2 Н), 7,46 (с, 2 Н), 3,71-3,77 (м, 4 Н), 1,96 (м, 2 Н), 1,72 (м, 2 Н),1,46 (с, 6 Н), 1,45 (с, 18 Н), 1,43 (с, 18 Н). Пример 34. 6-О-[4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенил]D-глюкопираноза. К раствору пробукола (1,8 г, 3,5 ммоль) в ТГФ (20 мл) добавляли 1,2,3,4-тетра-O-актилDглюкопиранозу (1,0 г, 2,9 ммоль), трифенилфосфин (0,92 г, 3,5 ммоль) и диэтилазодикарбоксилат (0,55 мл, 3,5 ммоль). Полученную смесь перемешивали в атмосфере азота при нагревании с обратным холодильником в течение 2 ч. Затем ее упаривали. После хроматографии на силикагеле (смесь гексана/этилацетата, 4:1) получали целевое соединение в виде беловатого твердого вещества (0,92 г). 1 Н-ЯМР (400 МГц, CDCl3)7,53 (с, 2 Н), 7,45 (с, 2 Н), 5,80 (д, 1 Н), 5,38 (с, 1 Н), 5,33 (дд, 1 Н), 5,16(с, 3H), 1,45 (с, 18+6 Н), 1,38 (с, 18 Н). Пример 35. 1-Н-тетразол-1-бутановой кислоты 4-1-3,5-бис-(1,1-диметилэтил)-4 гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фениловый эфир. К раствору 4-гидрокси-4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]2,6-бис-(1,1-диметилэтил)фенилового эфира бутановой кислоты (60 мг, 0,1 ммоль) в ТГФ (10 мл) добавляли 1 Н-тетразол (14 мг, 0,2 ммоль), трифенилфосфин (52 мг, 0,2 ммоль) и диэтилазодикарбоксилат (0,03 мл, 0,2 ммоль). Полученную смесь перемешивали в атмосфере азота при нагревании с обратным холодильником в течение 2 ч. Затем ее упаривали. После хроматографии на силикагеле (смесь гексана/этилацетата, 4:1) получали целевое соединение в виде масла (57 мг). 1H-ЯМР (400 МГц, CDCl3)8,56 (с, 1 Н), 7,64 (с, 2 Н), 7,45 (с, 2 Н), 5,39 (с, 1 Н), 4,84 (т, 2 Н), 2,74 (т,2 Н), 2,47 (м, 2 Н), 1,47 (с, 6 Н), 1,45 (с, 18 Н), 1,33 (с, 18H). Пример 36. 4-1-3,5-бис-(1,1-Диметилэтил)-4-[(3-гидрокси-1-пропенил)окси]фенил]тио]-1 метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенол. К раствору 3-[4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенокси]-2-пропеновой кислоты, этилового эфира, (Е)- (65 мг, 0,1 ммоль) в ТГФ (15 мл) добавляли алюмогидрид лития (1 мл, 1 М раствор в ТГФ). Полученную смесь перемешивали в атмосфере азота при комнатной температуре в течение ночи. Затем добавляли насыщенный раствор хлорида аммония (20 м.п.) и смесь перемешивали в течение 0,5 ч. После экстракции дихлорметаном (350 мл) органическую фазу сушили сульфатом магния и выпаривали. После проведения хроматографии на силикагеле (смесь гексана/этилацетата, 4:1) получали целевое соединение в виде масла (46 мг). 1H-ЯМР (400 МГц, CDCl3)7,61 (с, 2 Н), 7,45 (с, 2 Н), 5,99 (д, 1 Н), 5,39 (с, 1 Н), 4,84 (м, 1 Н), 4,46 (м,2 Н), 1,47 (с, 6 Н), 1,45 (с, 18 Н), 1,42. Пример 37. N6-4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенокси]ацетил]-L-лизин. Описание соединения. К [4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенокси]уксусной кислоте (150 мг, 0,26 ммоль) в метиленхлориде (1,8 мл) добавляли гидрохлорид метилового эфира лизина (79 мг, 0,34 ммоль), гидрохлорид 1-(3-диметиламинопропил-3 этилкарбодиимида) (130 мг, 0,67 ммоль) и диметиламинопиридин (82 мг, 0,67 ммоль). Реакционную смесь перемешивали в течение ночи, а метиленхлорид выпаривали. Реакционную смесь разбавляли эфиром (10 мл), промывали водой (23 мл), сушили MgSO4, фильтровали и концентрировали. Неочищенную смесь очищали с помощью хроматографии на силикагеле и элюировали смесью эфира/гексана=50:50, а затем смесью эфира/гексана=70:30 с получением сложного метилового эфира желаемого продукта. Этот сложный метиловый эфир растворяли в смеси ТГФ:H2O:МеОН (2:1:1) (3 мл), добавляли LiOH-H2O (50 мг) и реакционную смесь перемешивали в течение 1 ч. Реакционную смесь концентрировали и очищали на силикагеле, элюируя смесью метанола/гексана (20:80) с получением 67 мг продукта. 1(1 г) и смесь перемешивали в атмосфере азота в течение ночи при комнатной температуре. Затем эту смесь выливали в воду (200 мл), экстрагировали этилацетатом (3150 мл), промывали солевым раствором (100 мл), сушили магнием и упаривали. После хроматографии на силикагеле (смесь дихлорметана/метанола, 10:15:1) получали целевое соединение в виде беловатого твердого вещества (0,26 г). 1 Н-ЯМР (400 МГц, CDCl3)7,52 (с, 2 Н), 7,44 (с, 2 Н), 5,36 (с, 1 Н), 5,31 (с) и 4,78 (шир.с, 1 Н), 3,304,38 (шир.м, 6 Н), 1,38-1,43 (м, 42 Н). Пример 39. 6-О-[4-1-3,5-бис-(1,1-Диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенил]-D-глюцит. К раствору 6-O-[4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис(1,1-диметилэтил)фенил]-D-глюкопиранозы (70 мг) в ТГФ (5 мл) добавляли борогидрид натрия и смесь перемешивали в атмосфере азота при комнатной температуре в течение 2 ч. Затем добавляли насыщенный хлорид аммония (2 мл) и смесь перемешивали еще 1 ч. Затем смесь выливали в воду (50 мл) и экстрагировали дихлорметаном (350 мл). Органическую фазу сушили сульфатом магния и упаривали. После хроматографии на силикагеле (смесь дихлорметана/метанола=100:12) получали целевое соединение в виде белого твердого вещества (19 мг). 1 Н-ЯМР (400 МГц, CDCl3)7,54 (с, 2 Н), 7,44 (с, 2 Н), 5,36 (с, 1 Н), 4,35 (м, 1 Н), 3,30-4,10 (м, 7 Н),1,40-1,44 (м, 42 Н). Пример 40. Бутановой кислоты [4-гидрокси(2-гидроксифенокси)фосфинил]-окси-4-[1-[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фениловый эфир. К раствору 4-гидрокси-, 4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1-метилэтил]тио]2,6-бис-(1,1-диметилэтил)фенилового эфира бутановой кислоты (60 мг, 0,1 ммоль) в пиридине (1 мл) добавляли 1,2-фениленфосфохлоридат (21 мг, 0,11 ммоль) и смесь перемешивали в течение 1 ч в атмосфере азота при комнатной температуре. Затем смесь упаривали и остаток растворяли в дихлорметане(10 мл). После этого добавляли воду (1 мл) и уксусную кислоту (0,5 мл), и смесь перемешивали в течение 0,5 ч. Смесь выливали в воду (50 мл) и экстрагировали дихлорметаном (250 мл). Органическую фазу сушили сульфатом магния и упаривали. После хроматографии на силикагеле (смесь дихлорметана/метанола=5:1) получали целевое соединение в виде белого твердого вещества (21 мг). 1 Н-ЯМР (400 МГц, CDCl3)7,58 (с, 2 Н), 7,44 (с, 2 Н), 7,15 (шир.с, 1 Н), 6,87 (шир.с, 2 Н), 6,71 (шир.с,1 Н), 5,37 (с, 1 Н), 3,97 (шир.с, 2 Н), 2,48 (шир.с, 2 Н), 1,83 (шир.с, 2 Н), 1,45 (с, 6 Н), 1,43 (с, 18 Н), 1,24 (с,18 Н). Пример 41. Бутановой кислоты 4-гидрокси-3,3-диметил-, [4-[1-3,5-бис-(1,1-диметилэтил)-4 гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фениловый эфир. Описание реакции. В колбу добавляли пробукол (2,3 г, 4,46 ммоль) и тетрагидрофуран (23 мл). К раствору добавляли 60% гидрид натрия в минеральном масле (0,23 г, 5,75 ммоль). К непрозрачной белой смеси добавляли ангидрид 2,2-диметилянтарной кислоты (1 г, 7,6 ммоль). Реакционную смесь перемешивали в течение 3 ч при комнатной температуре. Темно-пурпурную реакционную смесь подкисляли 1 н. HCl (25 мл) и два раза экстрагировали этилацетатом (50 мл). Органические экстракты сушили MgSO4, фильтровали и концентрировали. Неочищенную продуктовую смесь растворяли в простом эфире и хроматографировали на силикагеле с градиентом концентраций смеси гексана/эфира 70:300:100 гексана/эфира. Соответствующие фракции объединяли и концентрировали, в результате чего получали 700 мг белого твердого продукта. Этот белый твердый продукт (214 мг, 0,332 ммоль) растворяли в ТГФ (6 мл), а затем добавляли боран-диметилсульфид (2 М в ТГФ, 0,665 ммоль) и реакционную смесь перемешивали в течение 6 ч. Реакцию гасили путем добавления концентрированной HCl (0,100 мл) и реакционную смесь перемешивали в течение ночи. Реакционную смесь разбавляли простым эфиром (25 мл), промывали водой (15 мл),NaHCO3 (15 мл) и солевым раствором (15 мл). Эфирный слой сушили MgSO4, фильтровали и концентрировали. После радиальной хроматографии на силикагеле и элюирования градиентом концентраций от 100:0 смеси гексана/эфира до 50:50 смеси гексана/эфира получали 85 мг продукта. 1 Н-ЯМР (400 МГц, CDCl3)7,64 (с, 2 Н), 7,46 (с, 2 Н), 5,39 (с, 1 Н), 3,48 (д, J=6,8 Гц, 2 Н), 2,73 (с, 2 Н),1,47 (с, 6 Н), 1,45 (с, 9 Н), 1,35 (с, 9 Н), 1,11 (с, 6 Н). Пример 42. Бутановой кислоты 4-(сульфокси)-,1-4-3,5-бис-(1,1-диметилэтил)-4 гидроксифенил]тио]-1-метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фенил]эфир. Описание реакции. Бутановой кислоты 4-гидрокси-, 4-1-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]тио]-1 метилэтил]тио]-2,6-бис-(1,1-диметилэтил)фениловый эфир (12,5 г, 20,75 ммоль) растворяли в ДМФ (150 мл) и добавляли комплекс "триоксид серы триметиламин" (12,5 г, 87,5 ммоль). Смесь перемешивали в- 16009370 течение ночи при комнатной температуре. Затем смесь упаривали и остаток растворяли в дихлорметане(100 мл), промывали водой (250 мл). Водную фазу экстрагировали дихлорметаном (75 мл). Объединенную органическую фазу сушили сульфатом магния и упаривали. После хроматографии на силикагеле(дихлорметан/метанол 10:1, 5:1) получали остаток, который использовали в следующей стадии реакции. Вышеуказанный продукт растворяли в ТГФ (200 мл). Затем добавляли NaOH (0,8 г, 20 ммоль) в воде (5 мл). Смесь перемешивали в течение 2 ч при комнатной температуре, а затем упаривали. После фильтрации собирали желтоватое твердое вещество, которое сушили до постоянной массы (9,23 г). Настоящее изобретение также относится к использованию соединений формул (I) и (II) для ингибирования перекисного окисления липида ЛПНП и ингибирования прогрессирования атеросклероза у пациентов, нуждающихся в этом. Используемый в настоящем описании термин "пациент" относится к теплокровным животным или млекопитающим, а в частности, к человеку, которые нуждаются в терапии, описанной в настоящем изобретении. Нижеследующие примеры иллюстрируют использование соединений формулы (I) настоящего изобретения, эти примеры приводятся лишь в целях иллюстрации, и не должны рассматриваться как некое ограничение объема изобретения. Пример 43. Протокол скрининга липидов и определения IC50. Получение HEPG2. Клетки HEPG2 засевали в 10 мл MEM, 10% FBS, 1 мМ пирувата натрия. Затем клетки инкубировали в инкубаторе для тканевой культуры. Эти клетки распределяли по 496-луночному планшету с MEM,10% FBS, 1 мМ пирувата натрия и оставляли для роста приблизительно до 50%-ной конфлюэнтности, а затем собирали. 1-й день: Обработка Клетки обрабатывали желаемой концентрацией соединений в 100 мкл DMEM, 1% RSA в течение 24 ч. Эти соединения растворяли в ДМСО. Для IC50 диапазон концентраций составлял 10-40 мкМ, причем каждую концентрацию дублировали три раза. В тот же самый день 496-луночный планшет NunclmmunoSorb покрывали 100 мкл мышиных моноклональных антител 1D1 против АроВ человека (разведение 1:1000 в 1PBS, pH 7,4). Покрытые планшеты оставляли на ночь. 2-й день: ApoB-ELISA Покрытый планшет 3 раза промывали 1PBS, pH 7,4, 0,05% Твином 20. В выбранные лунки добавляли 100 мкл стандартов. Стандарты АроВ получали при концентрациях 6,25; 3,12, 1,56; 0,78; 0,39 нг,и каждую концентрацию использовали в тройных дубликатах. Для образцов: в каждую лунку, соответствующую данному образцу, добавляли 90 мкл 1PBS, pH 7,4, 0,05% Твин 20. 10 мкл среды переносили из обработанного планшета с HEPG2 в ELISA-планшет с АроВ. Этот планшет инкубировали в течение 2 ч при комнатной температуре, слегка помешивая. Покрытый планшет промывали 31XPBS, pH 7,4, 0,05% Твином 20. Затем добавляли 100 мкл поликлонального овечьего антитела против АроВ человека (разведение 1:2000 в 1PBS, pH 7,4, 0,05% Твине 20) от Boehringer Mannheim. Затем проводили инкубирование при комнатной температуре 1 ч слегка помешивая. Покрытый планшет промывали 3 раза 1PBS, pH 7,4, 0,05% Твином 20. Затем добавляли 100 мкл кроличьего антиовечьего IgG (разведение 1:2000 в 1PBS, pH 7,4, 0,05% Твине 20). Затем проводили инкубирование в течение 1 ч при комнатной температуре, слегка помешивая. Покрытый планшет промывали 3 раза 1PBS, pH 7,4, 0,05% Твином 20. Затем добавляли 100 мкл субстрата (10 мл дистиллированной воды, 100 мкл ТМВ (10 мг/мл) и 1 мкл перекиси водорода). Реакционную смесь оставляли для проявления окраски и реакцию прекращали путем добавления 25 мкл 8 н. серной кислоты. Лунки считывали на микропланшет-ридере MicroPlate Reader при 450 нм. Строили график накопления АроВ в средах, выраженной как процент от контроля для каждого образца, в зависимости от его концентрации. Исходя из этого графика определяли IC50. Пример 44. Анализ на VCAM-1. Распределение клеток. Два-четыре конфлюентных планшета Р 150 трипсинизировали и клетки переносили в коническую центрифужную 50-мл пробирку. Эти клетки осаждали, ресуспендировали и подсчитывали с использованием метода исключения трипанового синего. Клетки ресуспендировали при концентрации 36000 клеток/мл и брали 1-мл аликвоту от каждой лунки. Клетки распределяли по 24-луночным планшетам для тканевых культур. На следующий день клетки в каждой лунке должны достигать приблизительно 90-95% конфлюентности. Клетки не должны быть старше 8 пересевов. Получение соединения. Водорастворимые соединения. Соединения сначала скринировали при 50 мкМ и 10 мкМ. Исходный 50 мМ раствор для каждого- 17009370 соединения приготавливали в культуральной среде. Этот исходный раствор разводили до 5 и 1 мМ. При добавлении 10 мкл 5 мМ раствора в лунку (1 мл среды/лунку) конечная концентрация достигала 50 мкМ. Добавление 10 мкл 1 мМ раствора в лунку давало конечную концентрацию 10 мкМ. Не растворимые в воде соединения. Соединения, которые не растворяются в культуральной среде, ресуспендировали в ДМСО при концентрации 25 мМ. Затем исходный раствор разводили до конечной концентрации в культуральной среде. Старую среду отсасывали и добавляли 1 мл новой среды с соединением. Так, например, если конечная концентрация составляла 50 мкМ, то добавляли 2 мкл 25 мМ исходного раствора на 1 мл культуральной среды. 50 мМ раствор разводили до более низких концентраций. Добавление соединений. Соединения добавляли в планшет (каждое соединение добавляли в дубликате). Один планшет использовали для экспрессии VCAM, а один планшет - для экспрессии ICAM. Сразу после добавления соединений, в каждую лунку добавляли TNF. Обычно в каждую лунку добавляли 100 ед./мл TNF. Поскольку каждый лот TNF варьируется в зависимости от числа единиц, то для определения оптимальной концентрации каждый новый лот титровали. Поэтому эта концентрация менялась. Если использовали партию 100 ед./шт., то TNF разводили до 10 ед./мкл, и в каждую лунку добавляли 10 мкл. Планшеты инкубировали в течение ночи (приблизительно 16 ч) при 37 С в 5% СО 2. На следующий день планшеты исследовали под микроскопом для того, чтобы определить, имеются ли какие-нибудь признаки токсичности. Были зарегистрированы все погибшие клетки, дебрис или морфологические изменения, а также нерастворимые соединения (частицы или помутнение). Пример 45. ELISA-анализ. Для оценки МСР-1, среду (500 мкл) хранили и замораживали при -70 С. Клетки промывали один раз приблизительно 1 мл/лунку сбалансированным солевым раствором Хэнкса (HBSS) или PBS. Промывочный раствор осторожно удаляли и планшет обертывали бумажными полотенцами. Затем добавляли либо 250 мкл/лунку HBSS + 5% FCCS в контрольные лунки (лунки, не содержащие "первого" антитела),либо 250 мкл/лунку "первого" антитела, разведенного в HBSS + 5% PCS. Лунки инкубировали 30 мин при 37 С. Лунки промывали два раза 5 мл HBSS или PBS на лунку, и после последней промывки планшеты осторожно заворачивали в бумажные полотенца. К каждой лунке, включая контрольные лунки (без"первого" антитела), добавляли 250 мкл/лунку конъюгированного с пероксидазой хрена (HRP) "второго" антитела, разведенного в HBSS + 5% FCS. Затем инкубировали 30 мин при 37 С. Лунки промывали четыре раза 5 мл HBSS или PBS на лунку, и после последней промывки планшеты осторожно заворачивали в бумажные полотенца. Затем добавляли 250 мкл/лунку раствора субстрата. После этого инкубировали в темноте при комнатной температуре до тех пор, пока не стала развиваться адекватная окраска (синяя). Отмечали период времени, в течение которого осуществляли инкубирование (обычно 15-30 мин). Затем добавляли 75 мкл/лунку стоп-раствор (8 н. серной кислоты) и осуществляли считывание оптической плотности при 450 нм (А 450 нм). Антитела и растворы. 1. Раствор субстрата приготавливают непосредственно перед использованием и этот раствор содержит: Вода 10 мл 30% перекись водорода 1 мкл ТМВ (3,3',5,5'-тетраметилбензидин) 100 мкл исходный раствор ТМВ: к 10 мг ТМВ добавляют 1 мл ацетона. Раствор хранят при 4 С в месте, защищенном от света. 2. Ab против УСДУ-1: исходный раствор 1 мкг/мкл - конечная концентрация 0,25 мкг/мл смешивают 25 мкл исходного раствора VCAM-1 (Southern Biotechnology) и 10 мл HBSS + 5% FCS. 3. Ab против ICAM-1: исходный раствор 1 мкг/мкл - конечная концентрация 0,25 мкг/мл, смешивают 25 мкл исходного раствора ICAM-1 (Southern Biotechnology) и 10 мл HBSS + 5% FCS. 4. "Второе" антитело: HRP-конъюгированный козий антимышиный IgG, разведенный в соотношении 1:500. Смешивают 20 мкл исходного раствора (Southern Biotechnology) и 10 мл HBSS + 5% FCS. Степень ингибирования соединений формулы (I) определяли с помощью анализов, описанных в примерах 43-45. Результаты представлены в табл. 1. Фармацевтические композиции. Млекопитающие, а в частности человек, страдающие любыми из вышеперечисленных состояний,могут быть подвергнуты лечению путем местного, системного или чрескожного введения композиции,содержащей эффективное количество соединения формулы (I), или формулы (II), или его фармацевтически приемлемой соли, необязательно, в фармацевтически приемлемом носителе или разбавителе. Данную композицию вводят подкожно, внутривенно, внутрибрюшинно, внутримышечно, парентерально, перорально, через слизистую оболочку, путем ингаляции, чрескожно с использованием пластыря пролонгированного высвобождения, или местно, в эффективных интервалах доз, подходящих для лечения данного состояния. Эффективная доза может быть легко определена стандартными методами и путем наблюдения результатов, полученных в аналогичных случаях. При определении эффективной дозы необходимо учитывать ряд факторов, включая, но не ограничиваясь ими, тип пациента, его вес, возраст и общее состояние здоровья; конкретное заболевание; степень прогрессирования или тяжести этого заболевания; восприимчивость данного пациента; конкретное вводимое соединение; способ введения; биологическую усвояемость вводимого препарата; выбранный режим введения доз; и использование дополнительных лекарственных средств. Типичные дозы для системного введения при всех состояниях, описанных в данном изобретении, варьируются от 0,1 до 500 мг/кг веса тела в день в форме разовой суточной дозы или дробных суточных доз. Предпочтительный интервал доз для лечения данных состояний составляет 5-1500 мг в день. Более предпочтительный интервал доз для лечения данных состояний составляет 25-750 мг в день. Типичные дозы для местного применения составляют в пределах от 0,001 до 100 мас.% активного соединения. Данное соединение вводят в течение периода времени, достаточного для ослабления нежелательных симптомов и клинических признаков, ассоциируемых с заболеванием, подвергаемым лечению. Активное соединение включают в фармацевтически приемлемый носитель или разбавитель в количестве, достаточном для обеспечения доставки в организм пациента терапевтического количества соединения in vivo при отсутствии серьезных токсических эффектов. Концентрация активного соединения в лекарственной композиции зависит от скорости абсорбции,инактивации и выведения лекарственного средства, а также от других факторов, известных специалистам. При этом следует отметить, что значения доз могут также варьироваться в зависимости от тяжести состояния, подвергаемого лечению. Кроме того, следует отметить, что для любого конкретного индивидуума, конкретный режим введения доз должен быть скорректирован в соответствии с периодом времени, индивидуальной потребностью и профессиональной оценкой специалиста, который осуществляет назначение по введению композиции и наблюдение за введением данной композиции, и что интервал- 25009370 доз, указанный в данном описании, приводится лишь в качестве примера и не должен рассматриваться как ограничение объема или применения заявленной композиции. Активный ингредиент может быть введен в виде разовой дозы, либо он может быть разделен на несколько дробных доз, которые могут быть введены через различные интервалы времени. Предпочтительным способом введения активного соединения для системной доставки является пероральное введение. Пероральные композиции, в основном, включают инертный разбавитель или пищевой носитель. Они могут быть включены в желатиновые капсулы или спрессованы в таблетки. Для перорального введения в терапевтических целях, активное соединение может быть введено вместе с носителями и использовано в форме таблеток, пастилок или капсул. В состав данной композиции могут быть включены фармацевтически приемлемые связующие средства и/или добавки. Таблетки, пилюли, капсулы, пастилки и т.п. могут содержать любой из нижеследующих ингредиентов или соединения аналогичной природы, а именно связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; носитель, такой как крахмал или лактоза; дезинтегрирующее средство, такое как альгиновая кислота, примогель или кукурузный крахмал; смазывающее вещество, такое как стеарат магния или sterotes; вещество, увеличивающее скольжение, такое как коллоидальная двуокись кремния; подслащивающее средство, такое как сахароза или сахарин; или корригент, такой как перечная мята, метилсалицилат или апельсиновый корригент. Если стандартной лекарственной формой является капсула, то, помимо вышеуказанных ингредиентов, она может содержать жидкий носитель, такой как нелетучее масло. Кроме того, стандартные лекарственные формы могут содержать различные другие вещества, модифицирующие физическую форму стандартной лекарственной формы, например, оболочку из сахара, шеллака или других энтеросолюбильных средств. Соединение или его соль могут быть введены в качестве компонента эликсира, суспензии, сиропа,облатки, жевательной резинки или т.п. Помимо активных соединений сироп может содержать сахарозу в качестве подслащивающего средства, а также он может содержать консерванты, красители и подкрашивающие вещества и отдушки. Данное соединение может быть также смешано с другими активными соединениями, не обладающими нежелательным действием, или с другими веществами, усиливающими желательный эффект. Активные соединения могут быть введены в комбинации с другими лекарственными препаратами, используемыми для лечения сердечно-сосудистых заболеваний, включая средства, снижающие уровень липидов, такие как пробукол и никотиновая кислота; ингибиторы агрегации тромбоцитов, такие как аспирин; антитромботические средства, такие как кумадин; блокаторы кальциевых каналов, такие как варапамил,дилтиазем и нифедипин; ингибиторы ангиотензин превращающего фермента (АСЕ), такие как каптоприл и эналоприл; и -блокаторы, такие как пропаналол, тербуталол и лабеталол. Соединения могут быть также введены в комбинации с нестероидными противовоспалительными средствами, такими как ибупрофен, индометацин, фенопрофен, мефенаминовая кислота, флуфенаминовая кислота, сулиндак. Данное соединение может быть также введено вместе с кортикостроидами. Растворы или суспензии, используемые для парентерального, чрескожного, подкожного или местного введения, могут включать в себя следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия, хелатообразующие средства, такие как этилендиаминотетрауксусная кислота; буферы, такие как ацетатный, цитратный или фосфатный и средства для коррекции изотоничности раствора, такие как хлорид натрия или декстроза. pH может быть скорректирован путем добавления кислот или оснований, таких как хлористоводородная кислота или гидроксид натрия. Препарат для парентерального введения может быть включен в ампулы, в одноразовые шприцы или в стеклянные или пластиковые флаконы для многократных доз. Предпочтительными носителями для внутривенного введения являются физиологический раствор,бактериостатическая вода, Cremophor Et (BASF, Parsippany, NJ) или забуференный фосфатом физиологический раствор (PBS). В предпочтительном варианте осуществления изобретения активные соединения приготавливают в виде готовой препаративной формы в сочетании с носителями, которые предохраняют данное соединение от быстрого выведения из организма, например, такой как готовая препаративная форма с регулируемым высвобождением активного соединения, включая имплантаты и системы доставки в виде микрокапсул. Могут быть использованы биологически разлагаемые и биологически совместимые полимеры,такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Методы получения таких готовых форм известны специалистам. Указанные материалы могут быть также закуплены у фирм Alza Corporation и Nova Pharmaceuticals, inc. Предпочтительными фармацевтически приемлемыми носителями также являются липосомные суспензии (включая липосомы, направляемые на инфицированные клетки моноклональными антителами против вирусных антигенов).- 26009370 Они могут быть получены методами, известными специалистам, например, описанными в патенте США 4522811 (который вводится в настоящее описание посредством ссылки). Так, например, липосомные препараты могут быть получены путем растворения соответствующего липида(ов) (таких как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин, арахадоилфосфатидилхолин и холестерин) в неорганическом растворителе, который затем выпаривают, в результате чего остается тонкая пленка осушенного липида на поверхности контейнера. После этого, в этот контейнер вводят водный раствор соединения. Затем этот контейнер взбалтывают вручную для отделения липидного материала от стенок контейнера и для диспергирования липидных скоплений, в результате чего образуется липосомная суспензия. Подходящие наполнители или носители для местного применения могут быть приготовлены стандартными методами в виде таких форм, как лосьоны, суспензии, мази, кремы, гели, тинктуры, спреи,порошки, пасты, пластыри для чрескожного введения с пролонгированным действием, суппозитории для ректального, вагинального, интраназального введения или для перорального введения через слизистую оболочку. Помимо других материалов, перечисленных выше для системного введения, для изготовлении композиций для местного применения могут быть использованы загущающие средства, смягчающие вещества и стабилизаторы. В качестве примеров могут служить загущающие средства, которыми являются вазелин, пчелиный воск, ксантановая камедь или полиэтилен; увлажнители, такие как сорбит; мягчители,такие как минеральное масло, ланолин и их производные, или сквален. Исходя из вышеуказанного подробного описания изобретения, для каждого специалиста очевидно,что в данное изобретение могут быть внесены модификации и изменения, относящиеся к соединениям настоящего изобретения, ингибирующим супрессию VCAM-1, и к способам лечения заболеваний, опосредованных экспрессией VCAM-1. Такие модификации и изменения не должны выходить за рамки объема нижеследующей формулы изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы или его фармацевтически приемлемая соль. 2. Фармацевтическая композиция для лечения заболевания, опосредованного экспрессией VCAM-1,содержащая эффективное количество соединения формулы или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. 3. Фармацевтическая композиция по п.2, где заболевание является сердечно-сосудистым заболеванием. 4. Фармацевтическая композиция по п.3, где сердечно-сосудистое заболевание представляет собой выбранное из группы, состоящей из атеросклероза, рестеноза после ангиопластики, заболевания, поражающего коронарную артерию, стенокардии и заболевания, поражающего капилляры. 5. Фармацевтическая композиция по п.2, где заболевание является воспалительным заболеванием. 6. Фармацевтическая композиция по п.5, где воспалительное заболевание представляет собой выбранное из группы, состоящей из ревматоидного артрита, остеоартрита, бронхиальной астмы, дерматита,рассеянного склероза и псориаза. 7. Фармацевтическая композиция по п.3, содержащая указанное соединение в комбинации с другим сердечно-сосудистым препаратом, выбранным из группы, состоящей из средств, снижающих уровень липидов, ингибиторов агрегации тромбоцитов, антитромботических средств, блокаторов кальциевых каналов, ингибиторов ангиотензин-превращающего фермента (АСЕ) и -блокаторов. 8. Фармацевтическая композиция по п.5, содержащая указанное соединение в комбинации с другим противовоспалительным препаратом. 9. Фармацевтическая композиция по п.2, являющаяся лекарственной формой, которая представляет собой таблетку, драже, пастилку или капсулу. 10. Фармацевтическая композиция по п.9, где лекарственная форма содержит от приблизительно 5 до приблизительно 1500 мг указанного соединения. 11. Фармацевтическая композиция по п.9, где лекарственная форма содержит от приблизительно 25- 27009370 до приблизительно 750 мг указанного соединения. 12. Фармацевтическая композиция по п.2, где подходящим для орального введения. 13. Фармацевтическая композиция по п.2, где подходящим для введения посредством ингаляции. 14. Фармацевтическая композиция по п.2, где подходящим для внутривенного введения. 15. Фармацевтическая композиция по п.2, где подходящим для парентерального введения. 16. Фармацевтическая композиция по п.2, где подходящим для внутрибрюшинного введения. 17. Фармацевтическая композиция по п.2, где подходящим для подкожного введения. 18. Фармацевтическая композиция по п.2, где подходящим для местного введения. 19. Фармацевтическая композиция по п.2, где подходящим для внутримышечного введения. фармацевтически приемлемый носитель является фармацевтически приемлемый носитель является фармацевтически приемлемый носитель является фармацевтически приемлемый носитель является фармацевтически приемлемый носитель является фармацевтически приемлемый носитель является фармацевтически приемлемый носитель является фармацевтически приемлемый носитель является
МПК / Метки
МПК: C07C 323/00
Метки: экспрессии, vcam-1, соединение, композиция, фармацевтическая, ингибирования
Код ссылки
<a href="https://eas.patents.su/30-9370-soedinenie-i-farmacevticheskaya-kompoziciya-dlya-ingibirovaniya-ekspressii-vcam-1.html" rel="bookmark" title="База патентов Евразийского Союза">Соединение и фармацевтическая композиция для ингибирования экспрессии vcam-1</a>
Циклическое пептидное соединение и его применение, бициклопептидное соединение, пептидное соединение и его применение, фармацевтическая композиция

Номер патента: 2819
Опубликовано: 31.10.2002
Авторы: Кондон Стефен М., Мориз Изабелль
МПК: A61P 19/00, C07K 7/64, A61K 38/12...
Метки: циклическое, пептидное, применение, соединение, композиция, фармацевтическая, бициклопептидное
Формула / Реферат:
1. Циклическое пептидное соединение формулы X-A10-A11-A12-A13-A14-A15-A16-A17-A18-A19-A20-A21-A22-A23-A24-A25-A26-A27-Y, или его фармацевтически приемлемая соль, или пролекарственный предшественник, где Х выбирают из группы, состоящей из (a) R1a-Ao-A1-A2-A3-A4-A5-A6-A7-A8-A9-, (b) R1a-А2-А3-А4-А5-А6-А7-А8-А9-, (c) R1b-А3-А4-А5-А6-А7-А8-А9-, (d) R1a-A4-A5-A6-A7-A8-A9-, (e) R1a-A5-A6-A7-A8-A9-, (f) R1a-А6-А7-А8-А9-, (g) R1a-A7-A8-A9-, (h)...
Производные никотинамида, их применение, фармацевтическая композиция, способ лечения и способ ингибирования изоферментов фдэ4 d

Номер патента: 3528
Опубликовано: 26.06.2003
Авторы: Уотсон Джон Уэсли, Марфэт Энтони, Ченг Джон Бин, Клайнмен Эдуард Фокс, Даплэнтьер Аллен Джейкоб, Чеймберс Роберт Джеймс
МПК: C07D 213/82, A61K 31/455, A61P 1/04...
Метки: лечения, производные, способ, фармацевтическая, никотинамида, применение, ингибирования, композиция, изоферментов, фдэ4
Формула / Реферат:
1. Производные никотинамида формулы (I) или их фармацевтически приемлемые соли, где t равно 0 или 1; A представляет собой кислород или NH; R1 представляет собой (C3-C7)циклоалкил, (C6-C10)арил или насыщенную или ненасыщенную циклическую или бициклическую (C3-C7)гетероциклическую группу, содержащую в качестве гетероатома от одного до четырех гетероатомов, независимо выбранных из группы, состоящей из кислорода, серы, азота и NR9, где R9 является...
5-замещенные-3-(1,2,3,6-тетрагидропиридин-4-ил)- и 3-(пиперидин-4-ил)-1н-индолы и их фармацевтически приемлемые соли и сольваты, фармацевтическая композиция на их основе, способ активации рецепторов 5-нт1 и способ ингибирования нейронной белковой транссудации.

Номер патента: 1113
Опубликовано: 30.10.2000
Авторы: Шос Джон Мехнет, Дрессман Брюс Энтони, Фриц Джеймс Эрвин, Кэлдор Стефен Уоррен, Рокко Винсент Патрик, Томпсон Деннис Чарльз, Ниссен Джеффри Скотт, Одиа Джеймс Эдмунд, Кох Дэниел Джеймс, Дрост Джеймс Джозеф
МПК: A61K 31/4439, C07D 401/04
Метки: способ, активации, белковой, рецепторов, приемлемые, транссудации, фармацевтическая, основе, фармацевтически, композиция, 5-нт1, ингибирования, 5-замещенные-3-(1,2,3,6-тетрагидропиридин-4-ил, соли, нейронной, сольваты, 3-(пиперидин-4-ил)-1н-индолы
Формула / Реферат:
1. 5-Замещенные -3-(1,2,3,6-тетрагидропиридин-4-ил)-3-(пиперидин-4-ил)индолы общей формулы I в которой А-В представляет -CH-CH2- или -С=СН-; R представляет Н или C1-C6-алкил; R1 представляет Н или С1-С4-алкил; Х представляет -C(О)NR4R15, -NR5R6, -NR7SO2R8, -NHC(Q)NR10R11, -NHC(O)OR12 или NR13C(O)R14; где Q представляет О или S; R4 представляет гетероарил, замещенный гетероарил, гетероарил(C1-C4 алкил) или замещенный гетероарил(C1-C4...
Фармацевтическая композиция, способ лечения, способ ингибирования иммунного ответа

Номер патента: 1200
Опубликовано: 25.12.2000
Авторы: Хохман Паула С., Браунинг Джеффри Л., Бенджамин Кристофер Д.
МПК: G01N 33/577, A61K 38/17, A61P 1/00...
Метки: ответа, ингибирования, композиция, иммунного, фармацевтическая, лечения, способ
Формула / Реферат:
1. Фармацевтическая композиция, содержащая терапевтически эффективное количество блокатора рецептора лимфотоксина-b (LT-b-R) и фармацевтически приемлемый носитель. 2. Композиция по п.1, отличающаяся тем, что блокатор LT-b-R выбран из группы, включающей растворимый рецептор лимфотоксина-b с аминокислотной последовательностью SEQ ID ь1, антитело к рецептору лимфотоксина-b (LT-R) и антитело к поверхностному лиганду лимфотоксина (LT-лиганду). 3....
Производные аминогуанидина и алкоксигуанидина (варианты) и способ их получения (варианты), фармацевтическая композиция и способ ингибирования протеолиза у млекопитающего, способ лечения различных заболеваний млекопитающего, способы ингибирования индуцированной тромбином агрегации тромбоцитов и свертывания фибриногена в плазме, тромбина, агрегации тромбоцитов и образования тромбов в крови, устройство для сбора крови, искусственного кровообращения и хранения крови

Номер патента: 3461
Опубликовано: 26.06.2003
Авторы: Солл Ричард М., Маркоутэн Томас П., Томкзук Брюс Э., Лу Тианбао, Сайедем Коллин
МПК: A61K 31/445, A61P 7/02, C07C 279/00...
Метки: получения, лечения, агрегации, алкоксигуанидина, млекопитающего, крови, свертывания, аминогуанидина, способ, фибриногена, тромбов, композиция, протеолиза, способы, индуцированной, заболеваний, варианты, искусственного, ингибирования, тромбина, хранения, фармацевтическая, устройство, различных, производные, сбора, кровообращения, плазме, тромбоцитов, тромбином, образования
Формула / Реферат:
1. Соединение формулы I или его сольват, гидрат или фармацевтически приемлемая соль, где L - означает -C(O)- или -SO2-; R1 означает группу R2 означает группу или R1 и R2 могут быть объединены с атомом азота, к которому они присоединены, с образованием цикла, содержащего от трех до семи атомов, который необязательно содержит дополнительный атом азота или кислорода, и который по выбору является бензо- или пиридоконденсированным циклом, причем...
Предыдущий патент: Производные гидроксизамещенного 1,3,8-триазинспиро[4,5]декан-4-она, полезные для лечения расстройств, опосредованных orl-рецептором
Следующий патент: Способ управляемой гиперстимуляции яичников и фармацевтический набор для использования в таком способе
Случайный патент: Схема для портативных осветительных устройств и портативных перезаряжаемых электронных устройств