Мускариновые агонисты
Номер патента: 6853
Опубликовано: 28.04.2006
Авторы: Хитчкок Стефен Эндрю, Джеймисон Джеймс Эндрю, Аллен Дженнифер Ребекка, Тернер Уилльям Уилсон Мл., Лиу Бин
























Формула / Реферат
1. Соединение формулы

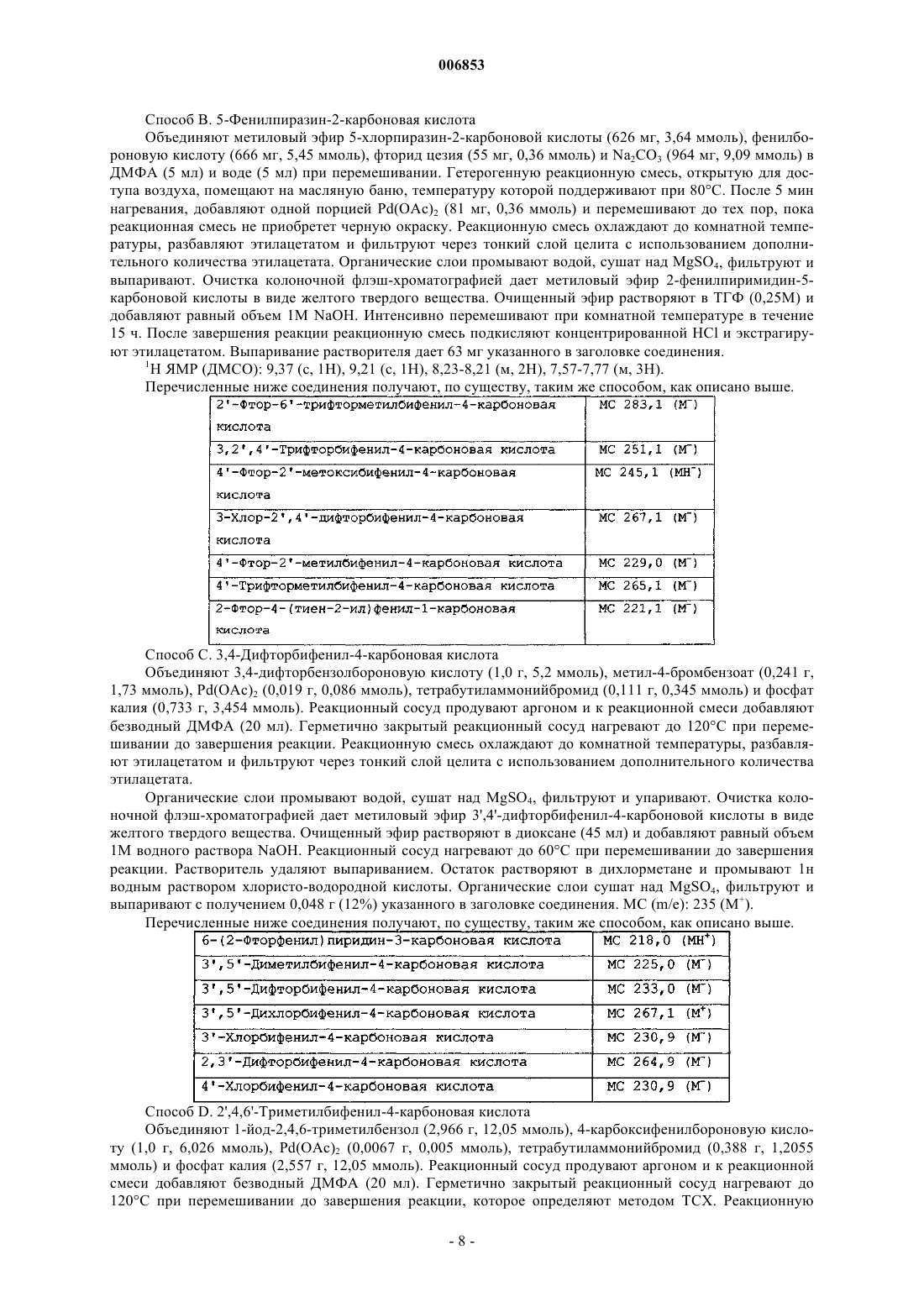
где Q, X, Y и Z независимо выбраны из группы, состоящей из CR1 и N, при условии, что не более чем два из Q, X, Y и Z представляют собой N, и по меньшей мере два из Q, X, Y и Z представляют собой СН; или Y представляет собой СН, Z представляет собой СН и группа Q=X представляет собой S, с образованием тиофенового кольца;
R1, в каждом случае независимо, выбран из группы, состоящей из водорода, галогена, С1-С4-алкокси и C1-C4-алкила;
R2 выбран из группы, состоящей из галогена; С1-С4-алкокси; С1-С4-алкила; С3-С8-циклоалкила; циано; трифторметила; пиридинила, необязательно замещенного одним-двумя заместителями, независимо выбранными из группы, включающей галоген, С1-С4-алкокси и С1-С4-алкил; тиенила, необязательно замещенного одним заместителем, выбранным из группы, включающей галоген, С1-С4-алкокси и С1-С4-алкил; фенила, необязательно замещенного одним-тремя заместителями, независимо выбранными из группы, включающей галоген, С1-С4-алкокси, С1-С4-алкил, трифторметил и циано; и пирролила, необязательно замещенного одним-двумя заместителями, независимо выбранными из группы, включающей галоген, C1-C4-алкокси и С1-С4-алкил;
R3 выбран из группы, состоящей из фенила, необязательно замещенного одним-тремя заместителями, независимо выбранными из группы, включающей галоген, С1-С4-алкокси, С1-С4-алкил, трифторметил, циано и нитро; нафтила, необязательно замещенного одним-тремя заместителями, независимо выбранными из группы, включающей галоген, С1-С4-алкокси, С1-С4-алкил, трифторметил, циано и нитро; гетероарила, представляющего собой стабильное ненасыщенное пяти- или шестичленное кольцо, содержащее от 1 до 2 гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, и необязательно замещенного одним или двумя заместителями, независимо выбранными из группы, включающей галоген, С1-С4-алкокси и С1-С4-алкил; или 1,3-бензодиоксолила, необязательно замещенного одним заместителем, выбранным из группы, включающей галоген, С1-С4-алкокси и С1-С4-алкил;
R4 выбран из группы, состоящей из водорода, гидрокси и фтора;
R5 выбран из группы, состоящей из водорода, галогена, С1-С4-алкокси и С1-С4-алкила;
Ra выбран из группы, состоящей из водорода и метила;
t имеет значение один, два или три и
m имеет значение один или два;
или его фармацевтически приемлемые аддитивные соли.
2. Соединение по п.1, где Ra представляет собой метил, R5 представляет собой водород, R4 представляет собой гидрокси, t имеет значение 1, m имеет значение 1, и которое имеет транс-стереохимию в положении 1 и 2, как показано ниже

3. Соединение по п.1, где каждый из Q, X, Y и Z представляет собой СН.
4. Соединение по любому из пп.1-3, где R2 представляет собой фенил.
5. Соединение по любому из пп.1-4, где R3 представляет собой фенил, монозамещенный галогеном.
6. Соединение по любому из пп.1-4, где R3 представляет собой фенил, монозамещенный фтором.
7. Соединение по любому из пп.1-4, где R3 представляет собой фенил, монозамещенный фтором в пара-положении.
8. Соединение по п.1, которое представляет собой (R)-(6-(l-((4-фтор-бензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1-ил)амид бифенил-4-карбоновой кислоты.
9. Фармацевтическая композиция, включающая соединение по любому из пп.1-8 и один или несколько фармацевтически приемлемых носителей, эксципиентов и разбавителей для этого соединения.
10. Применение соединения по любому из пп.1-8 в качестве фармацевтического средства.
11. Применение соединения по любому из пп.1-8 для изготовления лекарственного средства для применения в лечении расстройств, связанных с мускариновыми рецепторами.
12. Применение соединения по любому из пп.1-8 для изготовления лекарственного средства для применения в лечении расстройств познавательной способности.
13. Применение соединения по любому из пп.1-8 для изготовления лекарственного средства для применения в лечении болезни Альцгеймера.
14. Применение соединения по любому из пп.1-8 для изготовления лекарственного средства для применения в лечении шизофрении.
15. Применение соединения по любому из пп.1-8 для изготовления лекарственного средства для применения в лечении легкого ухудшения познавательной способности.
16. Применение соединения по любому из пп.1-8 для изготовления лекарственного средства для применения в лечении ухудшения познавательной способности, связанного с шизофренией.
Текст
006853 Настоящее изобретение относится к области фармацевтической и органической химии и предлагает соединения, которые являются активными на мускариновых рецепторах. Соединения по настоящему изобретению являются мускариновыми агонистами. Более конкретно,соединения по настоящему изобретению являются селективными агонистами мускаринового рецептора М-1. В качестве таких агонистов они являются полезными для лечения различных расстройств центральной нервной системы и других систем организма. Такие расстройства включают когнитивные расстройства, расстройство дефицита внимания - гиперактивности (ADHD), ожирение, болезнь Альцгеймера,психозы, включая шизофрению, а также соединения являются полезными для снижения внутриглазного давления, например, такого как при глаукоме. Некоторые индано-подобные соединения описываются как полезные для лечения состояний, связанных с нарушенной функцией мускариновой холинергической системы, в публикации РСТWO 97/25983, опубликована 24 июля 1997, и WO 99/04778, опубликована 4 февраля 1999. Настоящее изобретение предлагает соединения формулы I Где Q, X, Y и Z независимо выбраны из группы, состоящей из CR1 и N, при условии, что не более чем два из Q, X, Y и Z представляют собой N, и по меньшей мере два из Q, X, Y и Z представляют собой СН; илиR2 выбран из группы, состоящей из галогена; С 1-С 4-алкокси; С 1-С 4-алкила; С 3-С 8-циклоалкила; циано; трифторметила; пиридинила, необязательно замещенного одним-двумя заместителями, независимо выбранными из группы, включающей галоген, С 1-С 4-алкокси и С 1-С 4-алкил; тиенила, необязательно замещенного одним заместителем, выбранным из группы, включающей галоген, С 1-С 4-алкокси и С 1-С 4 алкил; фенила, необязательно замещенного одним-тремя заместителями, независимо выбранными из группы, включающей галоген, С 1-С 4-алкокси, С 1-С 4-алкил, трифторметил и циано; и пирролила, необязательно замещенного одним-двумя заместителями, независимо выбранными из группы, включающей галоген, С 1-С 4-алкокси и С 1-С 4-алкил;R3 выбран из группы, состоящей из фенила, необязательно замещенного одним-тремя заместителями, независимо выбранными из группы, включающей галоген, С 1-С 4-алкокси, С 1-С 4-алкил, трифторметил, циано и нитро; нафтила, необязательно замещенного одним-тремя заместителями, независимо выбранными из группы, включающей галоген, С 1-С 4-алкокси, С 1-С 4-алкил, трифторметил, циано и нитро; гетероарила, необязательно замещенного одним или двумя заместителями, независимо выбранными из группы, включающей галоген, С 1-С 4-алкокси и С 1-С 4-алкил; или 1,3-бензодиоксолила, необязательно замещенного одним заместителем, выбранным из группы, включающей галоген, С 1-С 4-алкокси и С 1-С 4 алкил;Ra выбран из группы, состоящей из водорода и метила;m имеет значение один или два; или его фармацевтически приемлемые аддитивные соли. Настоящее изобретение также предлагает фармацевтические композиции, включающие соединение формулы I и фармацевтически приемлемый разбавитель. Поскольку соединения формулы I являются агонистами мускаринового рецептора М-1, соединения формулы I являются полезными для лечения различных расстройств, связанных с мускариновыми рецепторами, включая когнитивные расстройства (включая возрастные когнитивные расстройства, легкое ухудшение познавательной функции, ухудшение познавательной функции, связанное с шизофренией, и вызванное химиотерапией ухудшение познавательной функции), ADHD, расстройства настроения(включая болезнь Альцгеймера, вызванную СПИДом деменцию, сосудистую деменцию и деменцию с отсутствием четкой гистологии), болезнь Паркинсона и хорею Гентингтона. Также соединения по настоящему изобретению полезны для лечения хронического колита, включая болезнь Крона. Кроме того,соединения по настоящему изобретению являются полезными для лечения боли (включая острую боль и хроническую боль), ксеростомии (сухость во рту), заболевание телец Леви (включая заболевание диф-1 006853 фузных телец Леви), афазии, т.е. нарушения речи (включая первичную афазию и синдромы первичной афазии), и гипотензивных синдромов. В другом варианте воплощения настоящее изобретение предлагает способы лечения расстройств,связанных с мускариновыми рецепторами, включающие введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы I. То есть в настоящем изобретении предлагается использование соединения формулы I или содержащей его фармацевтической композиции для получения лекарственного средства для лечения расстройств, связанных с мускариновыми рецепторами. В настоящем изобретении также предлагается соединение формулы I для применения в терапии. Перечисленные ниже термины, как они использованы в данном описании, имеют следующие значения. Термин "галоген" относится к атомам хлора, фтора, брома или иода. Термин "С 1-С 4-алкил" относится к линейной или разветвленной алкильной цепи, содержащей от одного до четырех атомов углерода, и включает метил, этил, н-пропил, изопропил, н-бутил, втор-бутил,изобутил и трет-бутил. Термин "С 3-С 6-циклоалкил" относится к циклопропилу, циклобутилу, циклопентилу, циклогексилу,циклогептилу и циклооктилу. Термин "С 1-С 4-алкокси" относится к линейной или разветвленной алкильной цепи, содержащей от одного до четырех атомов углерода, связанной с атомом кислорода, и включает метокси, этокси, нпропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси. Термин "гетероарил" используют для обозначения стабильного ненасыщенного пяти- или шестичленного кольца, содержащего от 1 до 2 гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы. Примеры гетероарила включают пиридинил, пиримидинил, пиразинил, пирролил, оксазолил, изоксазолил, имидазолил, тиазолил, пиридазинил, фурил, тиенил и т.п. Предпочтительными гетероарильными группами являются тиенил, пиридинил и фурил. Соединения по настоящему изобретению образуют фармацевтически приемлемые кислотноаддитивные соли с широким рядом органических и неорганических кислот и включают физиологически приемлемые соли, которые часто используют в фармацевтической химии. Такие соли также являются частью настоящего изобретения. "Фармацевтически приемлемую аддитивную соль" получают из фармацевтически приемлемой кислоты, как хорошо известно из уровня техники. Такие соли включают фармацевтически приемлемые соли, перечисленные в Journal Pharmaceutical Science. 66, 2-19 (1977), которые известны специалистам в данной области. Типичные неорганические кислоты, используемые для получения таких солей, включают хлористо-водородную, бромисто-водородную, йодисто-водородную, азотную, серную, фосфорную, фосфорноватистую, метафосфорную, пирофосфорную и т.п. Соли, полученные из органических кислот, таких как алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые и гидроксиалкандиовые кислоты, ароматические кислоты,алифатические и ароматические сульфоновые кислоты, также могут быть использованы. Подобные фармацевтически приемлемые соли включают, таким образом, хлорид, бромид, йодид, нитрат, ацетат, фенилацетат, трифторацетат, акрилат, аскорбат, бензоат, хлорбензоат, динитробензоат, гидроксибензоат,метоксибензоат, метилбензоат, о-ацетоксибензоат, изобутират, фенилбутират, -гидроксибутират, бутин-1,4-дикарбоксилат, гексин-1,4-дикарбоксилат, капринат, каприлат, циннамат, цитрат, формиат, фумарат, гликолят, гептаноат, гиппурат, лактат, малат, малеат, гидроксималеат, малонат, манделат, мезилат, никотинат, изоникотинат, оксалат, фталат, терефталат, пропиолат, пропионат, фенилпропионат, салицилат, себацат, сукцинат, суберат, бензолсульфонат, п-бромбензолсульфонат, хлорбензолсульфонат,этилсульфонат, 2-гидроксиэтилсульфонат, метилсульфонат, нафталин-1-сульфонат, нафталин-2 сульфонат, нафталин-1,5-сульфонат, п-толуолсульфонат, ксилолсульфонат, тартрат и т.п. Настоящее изобретение включает стереоизомеры и таутомеры соединений формулы I. В данном описании используют обозначения Cahn-Prelog-Ingold, (R)- и (S)-, а также "цис" и "транс" обозначения соответствующей стереохимии для указания конкретных изомеров и соответствующей стереохимии. Как это обычно бывает с любыми группами фармацевтически активных соединений, некоторые группы являются предпочтительными для конечных целей применения. Ниже определяются предпочтительные классы.i) Q, X, Y и Z, каждый, представляют собой CR1, при условии, что по меньшей мере два из Q, X, Y иn) Один из Q, X, Y и Z представляет собой CF, а другие представляют собой СН. о) Q представляет собой CF, а каждый из X, Y и Z представляет собой СН.q) R2 представляет собой фенил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, С 1-С 4-алкокси, С 1-С 4-алкила, трифторметила и циано.s) R3 представляет собой фенил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, С 1-С 4-алкокси, С 1-С 4-алкила, трифторметила, циано и нитро.bb) Ra представляет собой метил, R5 представляет собой водород, R4 представляет собой гидрокси, t имеет значение 1, m имеет значение 1, и R3 представляет собой фенил, монозамещенный фтором в параположении. Указанные выше пункты могут быть объединены для определения дополнительных предпочтительных классов соединений. Соединения формулы I, в которой R4 представляет собой гидрокси, получают с использованием методик, указанных на схеме А. На схеме А все заместители, если не указано иное, имеют значение, определенное выше, и все реагенты хорошо известны и являются общепризнанными в уровне техники. На схеме А, стадия а, соединение формулы (1) разделяют с получением, по существу, чистого соединения формулы (2). Соединение формулы (1) можно легко получить способами, хорошо известными и признанными в уровне техники, например, такими как указаны в публикациях РСТWO 97/25983,опубликована 24 июля 1997; и WO 99/04778, опубликована 4 февраля 1999. Как он использован в данном описании, термин "по существу чистый" относится к энантиомерной чистоте. Желаемую для конечных получаемых соединений формулы I стереохимию можно удобным образом включить в схему А, стадия а,путем разделения соединений формулы I. Дальнейшая обработка разделенных соединений формулы (I),посредством стадий b, с, d и необязательной стадии е, которые описаны ниже, обеспечивает, по существу, чистые соединения формулы I. Могут быть получены, по существу, чистые соединения формулы I,являющиеся энантиомерно чистыми больше чем на 80%, предпочтительно больше чем на 90%, более предпочтительно больше, чем на 95%, наиболее предпочтительно больше чем на 97%. Соединение формулы (I) можно разделить методами хиральной хроматографии или фракционной кристаллизации диастереомерных кислотно-аддитивных солей. Для этой цели, как ожидают, должно подходить большое разнообразие таких солей. При воплощении на практике настоящего изобретения было обнаружено, что изомеры миндальной кислоты являются наиболее подходящими. Например, соединение формулы (I) подвергают контактированию с выбранной кислотой. Как правило, можно использовать от около 0,4 молярных эквивалента до большого избытка выбранной кислоты,при этом предпочтительным является количество от около 0,4 до 1,5 молярных эквивалентов и наиболее предпочтительно от около 0,5 до 1,1 молярных эквивалентов. Разделение в типичном случае осуществляют кристаллизацией кислотно-аддитивной соли из раствора. В частности, полезными являются такие растворители, как низшие спирты, в том числе метанол. Может быть выгодным использовать небольшие количества воды с выбранным(и) растворителем(ями) для осуществления разделения в разумном объеме. Также может быть выгодным использование антирастворителя. Как он используется в данном описании,термин "антирастворитель" относится к растворителю, в котором соль значительно менее растворима в сравнении с выбранным растворителем(ями). Предпочтительно, чтобы антирастворитель, когда его используют, был смешиваемым с другим(и) выбранным(и) растворителем(ями). Подходящие антирастворители включают простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир и т.п.,и низшие алкилацетаты, такие как метилацетат, этилацетат, изопропилацетат, пропилацетат, изобутилацетат, втор-бутилацетат, бутилацетат, амилацетат, изоамилацетат и т.п., и алканы, такие как пентан,гексан, гептан, циклогексан и т.п. Когда используют рацемическую смесь, необходимо использовать антирастворитель осторожно, чтобы избежать кристаллизации соли нежелательной диастереомерной соли. В типичном случае кристаллизацию осуществляют при начальной температуре около 40 С до температуры кипения выбранного растворителя(ей). Затем смесь охлаждают с получением соли. Может быть выгодным использование затравки. Предпочтительно медленное охлаждение кристаллизационного раствора. Наиболее удобно охлаждение кристаллизационного раствора до температур в интервале от температуры окружающей среды до около -20 С. Соль собирают с использованием методов, хорошо известных из уровня техники, включая фильтрацию, декантацию, центрифугирование, выпаривание, сушку и т.п. Соединение формулы (2) можно использовать непосредственно в виде кислотно-аддитивной соли выбранной кислоты. Альтернативно, перед использованием соединение формулы (2) может быть выделено в виде другой кислотно-аддитивной соли после обмена кислот или может быть выделено в виде-4 006853 основания путем экстракции в щелочных условиях, как это хорошо известно и принято в уровне техники. Специалисту в данной области должно быть понятно, что описанное соединение формулы (2) находится в транс-конфигурации в положении 1 и 2 инданового ядра. Цис-соединения можно легко получить из таких транс-соединений путем защиты амина, инверсии гидроксильного центра, с последующим удалением защиты, если это необходимо. Существует множество способов, позволяющих осуществить инверсию гидроксильных центров, например, при помощи реакции Мицунобу (Mitsunobu) с подходящими карбоновыми кислотами, включая уксусную кислоту и бензойную кислоту, с последующим гидролизом. Схема реакций А, стадия b описывает получение соединения формулы (3). Понятно, что соединение формулы (3) может представлять собой соединение, в котором R является такой группой, которая желательна для конечного продукта формулы I, как указано выше. R может также объединяться с карбонилом с образованием защитной группы, такой как трет-ВОС, которую затем можно удалить до введения группы R, желательной для конечного продукта формулы I. Выбор и использование подходящих защитных групп хорошо известен и принят в данной области техники (Protecting Groups in Organic Synthesis, Theodora Greene (Wiley-Interscience. Например, когда R представляет собой такую группу, которая является желательной для конечного продукта, реакцию конденсации, показанную на стадии b, осуществляют с использованием подходящей кислоты или галогенангидрида, полученного из такой кислоты. Подходящие кислоты включают различные замещенные бензойные кислоты и галогенангидриды этих кислот, гетероарильные кислоты и галогенангидриды этих кислот и различные биарилкарбоновые кислоты и галогенангидриды этих кислот. Примеры включают бифенилкарбоновую кислоту и 3-фторбифенил-4-карбоновую кислоту. Например, соединение формулы (2) подвергают контактированию с подходящей кислотой с получением соединения формулы (3). Такие реакции конденсации являются обычными в синтезе пептидов и могут быть использованы методы синтеза, используемые для осуществления таких реакций. Например,для способствования такому ацилированию можно использовать хорошо известные агенты конденсации,такие как связанные на смоле реагенты и карбодиимиды, с использованием или без использования хорошо известных добавок, таких как N-гидроксисукцинимид, 1-гидроксибензотриазол и т.п. Реакцию удобно проводить в инертном апротонном полярном разбавителе, таком как диметилформамид (ДМФА), метиленхлорид (дихлорметан), хлороформ, ацетонитрил, тетрагидрофуран (ТГФ) и т.п. Обычно реакцию осуществляют при температуре от около 0 до около 60 С, и обычно для этого требуется от около 1 до около 24 ч. После завершения реакции продукт формулы (3) выделяют традиционными способами,включая экстракцию, осаждение, хроматографию, фильтрацию, растирание, кристаллизацию и т.п. Альтернативно, например, соединение формулы (2) подвергают контактированию с хлорангидридом подходящей кислоты с получением соединения формулы (3). Такие хлорангидриды кислот являются коммерчески доступными, или их легко можно получить из соответствующих кислот способами, хорошо известными из уровня техники, такими как воздействие трихлоридом фосфора, трибромидом фосфора,оксихлоридом фосфора, пентахлоридом фосфора, тионилхлоридом, тионилбромидом или оксалилхлоридом, с использованием или без использования небольшого количества диметилформамида, в инертном растворителе, таком как толуол, метиленхлорид или хлороформ; при температуре от около 0 до 80 С. Реакцию обычно осуществляют в течение периода времени от 1 до 24 ч. Хлорангидрид кислоты может быть выделен и очищен или часто его используют непосредственно сразу, то есть с выделением и/или очисткой или без них. В реакциях конденсации обычно используют подходящее основание в качестве акцептора кислоты, образуемой в ходе реакции. Подходящие основания включают, например, гидроксид натрия, гидроксид калия, пиридин, триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин и т.п. Реакцию обычно осуществляют в растворителе, таком как метиленхлорид, хлороформ, тетрагидрофуран и т.п., или в условиях реакции Шоттена-Бауманна, в смеси растворителей, таких как метиленхлорид,этилацетат, толуол и вода. Обычно реакцию конденсации осуществляют при температуре от около -20 С до около 80 С, и обычно требуется от около 1 до около 24 ч. После завершения реакции продукт формулы (3) выделяют традиционными способами, включая экстракцию, осаждение, хроматографию, фильтрацию, растирание, кристаллизацию и т.п. Схема реакции А, стадия с описывает восстановление нитрогруппы с получением соединения формулы (4). Такие восстановительные реакции можно осуществлять различными способами, хорошо известными из уровня техники. Например, соединение формулы (3) можно гидрировать над катализатором, таким как палладий-науглероде, с получением соединения формулы (4). Такое гидрирование обычно осуществляют в растворителе, и для этого подходящими являются различные растворители, например метанол, этанол, изопропанол, тетрагидрофуран или этилацетат, или их смеси. Гидрирование можно осуществлять при начальном давлении водорода 20-180 ф./кв.дюйм (137-1241 кПа). Реакцию обычно осуществляют при температуре от около 0 до около 60 С. Для осуществления реакции обычно требуется от 1 ч до 3 дней. Продукт может быть выделен и очищен традиционными способами, хорошо известными из уровня техники, такими как фильтрация, экстракция, выпаривание, растирание, осаждение, хроматография и перекристаллизация и т.п.-5 006853 На схеме А, стадия d соединение формулы (4) подвергают контактированию с подходящим амидинобразующим агентом с получением соединения формулы I. Подходящие амидинобразующие агенты включают 1-метилтио-1-метил-N-(4-фторбензил)-N-метилиммонийтрифлат и 1-метилтио-1-метил-N-(4 фторбензил)-N-метилиммониййодид. Специалисту в данной области должно быть понятно, что подходящие амидинобразующие агенты могут быть получены заранее или in situ, если это необходимо. Например, соединение формулы (4) подвергают контактированию с примерно 1-3 эквивалентами подходящего амидинобразующего агента. Реакцию обычно осуществляют в безводном растворителе,таком как метиленхлорид, толуол или тетрагидрофуран, при температуре от около -20 до 50 С. Реакцию осуществляют с использованием подходящего основания, такого как пиридин, коллидин или триэтиламин. Для осуществления реакции обычно требуется от 1 до 18 ч. Продукт может быть выделен и очищен традиционными способами, хорошо известными из уровня техники, такими как гашение реакции, фильтрация, экстракция, выпаривание, растирание, осаждение, хроматография и перекристаллизация и т.п. Как легко должно быть понятно, когда R представляет собой защитную группу, вводимую на стадии b, такую защитную группу можно удалить после осуществления стадии d, и полученный амин может быть конденсирован с подходящей кислотой или хлорангидридом кислоты, как описано и выше для стадии b, с получением соединения формулы I. Некоторые соединения формулы I являются промежуточными соединениями для других конечных соединений формулы I. Например, когда R2 представляет собой йод, можно использовать другой реагент,например, 2-(трибутилстаннил)тиофен или 2-(трибутилстаннил)пиридин, для вытеснения йода как удаляемой группы и замещения его другой группой R2, которая является желательной для конечного продукта. На схеме А, на необязательной стадии е, которая не показана, получают кислотно-аддитивную соль соединения формулы I с использованием фармацевтически приемлемой кислоты. Получение кислотноаддитивной соли хорошо известно и применяется в уровне техники. Соединения формулы I, в которой R4 представляет собой водород, получают из соединений формулы (3) или из аминзащищенных соединений формулы (2) путем деоксигенирования. Такие реакции деоксигенирования легко осуществимы с использованием методик, хорошо известных из уровня техники,например, Larock, Comprehensive Organic Transformations, pp.44-52 (1999). Альтернативно, соединения формулы I, в которой R4 представляет собой водород, получают способом, показанным на схеме В. На схеме В все заместители, если не указано иное, имеют значение, определенное выше, и все реагенты хорошо известны из уровня техники. Схема В Схема реакций В, стадия а, описывает восстановительное аминирование соединения формулы (5) с получением соединения формулы (6). Такие реакции восстановительного аминирования осуществляют в различных условиях. Реакцию, описанную на схеме В, стадия а, можно осуществить с использованием аммиака или защищенного амина, такого как бензиламин, дибензиламин и т.п., с последующим удалением защиты с получением соединения формулы (6). Например, соединение формулы (5) подвергают взаимодействию с избыточным количеством аммиака и цианоборгидрида натрия с получением соединения формулы (6). Как хорошо известно из уровня техники, в ходе осуществления таких реакций может быть выгодным отслеживание и регулирование-6 006853 уровня рН. Реакцию осуществляют в растворителе, таком как метанол, этанол, изопропанол и вода или смеси таких растворителей. Реакцию обычно осуществляют при температуре от около 0 до около 60 С, и для осуществления реакции обычно требуется от около 1 до около 24 ч. После завершения реакции продукт формулы (6) выделяют традиционными способами, включая экстракцию, осаждение, хроматографию, фильтрацию, растирание, кристаллизацию и т.п. Показанные на схеме реакций В стадии b, с, d и необязательную стадию е осуществляют способами,описанными для схемы А, стадии b, с, d и необязательная стадия е, с получением соединения формулы I. Соединения формулы I, в которых R4 представляет собой фтор, получают из соединения формулы(3) или из аминзащищенных соединений формулы (2) с использованием методов галогенирования, хорошо известных из уровня техники, например, описанных в Larock, Comprehensive Organic Transformations, pp. 689-701 (1999). Настоящее изобретение далее будет проиллюстрировано при помощи примеров и препаративных примеров. Эти примеры и препаративные примеры являются чисто иллюстративными и никоим образом не предназначены для ограничения данного изобретения. Термины, используемые в примерах и препаративных примерах имеют их обычное значение, если не указано иное. Например, "С" относится к градусам Цельсия; "М" означает молярный или молярность;"ммоль" означает миллимоль или миллимоли; "г" означает грамм или граммы; "мл" означает миллилитр или миллилитры; "т.пл." означает температуру плавления; "насыщенный раствор соли" относится к насыщенному водному раствору хлорида натрия; и т.д. В спектрах 1 Н ЯМР все химические сдвиги указаны по шкале , если не указано иное. Методики сочетания Способ А. 2'-Хлорбифенил-4-карбоновая кислота Объединяют метил-4-бромбензоат (1,0 г, 4,65 ммоль), 2-хлорфенилбороновую кислоту (799 мг, 5,1 ммоль), Pd(OAc)2 (51 мг, 0,46 ммоль) и карбонат натрия (1,5 г, 13,9 ммоль) в ДМФА (20 мл) и воде (2,0 мл) при перемешивании. Реакционную смесь продувают аргоном, добавляют трифенилфосфин (61 мг,0,23 ммоль) и снова продувают аргоном. Герметично закрытую реакционную смесь помещают на масляную баню, температуру которой поддерживают при 80 С и оставляют для перемешивания на 1 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и фильтруют через тонкий слой целита с использованием дополнительного количества этилацетата. Органические слои промывают водой, сушат над MgSO4, фильтруют и выпаривают. Очистка колоночной флэшхроматографией дает метиловый эфир 2'-хлорбифенил-4-карбоновой кислоты в виде желтого твердого вещества. Очищенный эфир растворяют в ТГФ (0,25 М) и добавляют равный объем 1 М NaOH. Интенсивно перемешивают при комнатной температуре в течение 15 ч. После завершения реакции реакционную смесь подкисляют концентрированной НСl и экстрагируют этилацетатом. Выпаривание растворителя дает 762 мг (67%) указанного в заголовке соединения. МС (m/е): 231,1 (М-). Перечисленные ниже соединения получают, по существу, таким же способом, как описано выше.-7 006853 Способ В. 5-Фенилпиразин-2-карбоновая кислота Объединяют метиловый эфир 5-хлорпиразин-2-карбоновой кислоты (626 мг, 3,64 ммоль), фенилбороновую кислоту (666 мг, 5,45 ммоль), фторид цезия (55 мг, 0,36 ммоль) и Nа 2 СО 3 (964 мг, 9,09 ммоль) в ДМФА (5 мл) и воде (5 мл) при перемешивании. Гетерогенную реакционную смесь, открытую для доступа воздуха, помещают на масляную баню, температуру которой поддерживают при 80 С. После 5 мин нагревания, добавляют одной порцией Pd(OAc)2 (81 мг, 0,36 ммоль) и перемешивают до тех пор, пока реакционная смесь не приобретет черную окраску. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и фильтруют через тонкий слой целита с использованием дополнительного количества этилацетата. Органические слои промывают водой, сушат над MgSO4, фильтруют и выпаривают. Очистка колоночной флэш-хроматографией дает метиловый эфир 2-фенилпиримидин-5 карбоновой кислоты в виде желтого твердого вещества. Очищенный эфир растворяют в ТГФ (0,25 М) и добавляют равный объем 1 М NaOH. Интенсивно перемешивают при комнатной температуре в течение 15 ч. После завершения реакции реакционную смесь подкисляют концентрированной НСl и экстрагируют этилацетатом. Выпаривание растворителя дает 63 мг указанного в заголовке соединения. 1H ЯМР (ДМСО): 9,37 (с, 1 Н), 9,21 (с, 1 Н), 8,23-8,21 (м, 2 Н), 7,57-7,77 (м, 3 Н). Перечисленные ниже соединения получают, по существу, таким же способом, как описано выше. Способ С. 3,4-Дифторбифенил-4-карбоновая кислота Объединяют 3,4-дифторбензолбороновую кислоту (1,0 г, 5,2 ммоль), метил-4-бромбензоат (0,241 г,1,73 ммоль), Pd(OAc)2 (0,019 г, 0,086 ммоль), тетрабутиламмонийбромид (0,111 г, 0,345 ммоль) и фосфат калия (0,733 г, 3,454 ммоль). Реакционный сосуд продувают аргоном и к реакционной смеси добавляют безводный ДМФА (20 мл). Герметично закрытый реакционный сосуд нагревают до 120 С при перемешивании до завершения реакции. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и фильтруют через тонкий слой целита с использованием дополнительного количества этилацетата. Органические слои промывают водой, сушат над MgSO4, фильтруют и упаривают. Очистка колоночной флэш-хроматографией дает метиловый эфир 3',4'-дифторбифенил-4-карбоновой кислоты в виде желтого твердого вещества. Очищенный эфир растворяют в диоксане (45 мл) и добавляют равный объем 1 М водного раствора NaOH. Реакционный сосуд нагревают до 60 С при перемешивании до завершения реакции. Растворитель удаляют выпариванием. Остаток растворяют в дихлорметане и промывают 1 н водным раствором хлористо-водородной кислоты. Органические слои сушат над MgSO4, фильтруют и выпаривают с получением 0,048 г (12%) указанного в заголовке соединения. МС (m/е): 235 (М+). Перечисленные ниже соединения получают, по существу, таким же способом, как описано выше. Способ D. 2',4,6'-Триметилбифенил-4-карбоновая кислота Объединяют 1-йод-2,4,6-триметилбензол (2,966 г, 12,05 ммоль), 4-карбоксифенилбороновую кислоту (1,0 г, 6,026 ммоль), Pd(OAc)2 (0,0067 г, 0,005 ммоль), тетрабутиламмонийбромид (0,388 г, 1,2055 ммоль) и фосфат калия (2,557 г, 12,05 ммоль). Реакционный сосуд продувают аргоном и к реакционной смеси добавляют безводный ДМФА (20 мл). Герметично закрытый реакционный сосуд нагревают до 120 С при перемешивании до завершения реакции, которое определяют методом ТСХ. Реакционную-8 006853 смесь охлаждают до комнатной температуры. Добавляют к реакционной смеси метилйодид (1,0 мл, 36,63 ммоль) и продолжают перемешивание до завершения реакции. Реакционную смесь разбавляют этилацетатом и фильтруют через тонкий слой целита с использованием дополнительного количества этилацетата. Органические слои промывают водой, сушат над MgSO4, фильтруют и упаривают. Очистка колоночной флэш-хроматографией дает метиловый эфир 2',4',6'-триметилбифенил-4-карбоновой кислоты в виде желтого твердого вещества. Очищенный эфир растворяют в диоксане (45 мл) и воде (5 мл), содержащей 5 экв. LiOH, при перемешивании при 60 С. По завершении растворения растворитель выпаривают, реакционную смесь подкисляют хлористоводородной кислотой и экстрагируют этилацетатом. Органические слои сушат над MgSO4, фильтруют и упаривают с получением 0,023 г (16%) указанного в заголовке соединения. МС (m/е): 239,1 (М-). Перечисленные ниже соединения получают, по существу, таким же способом, как описано выше. Способ Е. 2',4'-Дифторбифенил-4-карбоновая кислота Объединяют 4-карбометоксифенилбороновую кислоту (1,021 г, 5,67 ммоль), 1-бром-2,4 дифторбензол (1,000 г, 5,181 ммоль), Pd(OAc)2 (0,113 г, 0,50 ммоль), трифенилфосфин (0,149 г, 0,505 ммоль) и карбонат натрия (1,664 г, 0,568 ммоль). Реакционный сосуд продувают аргоном. Добавляют ДМФА (20 мл) и воду (2,0 мл) при перемешивании. Герметично закрытую реакционную смесь помещают на масляную баню при температуре 80 С и оставляют для перемешивания на 24 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и фильтруют через тонкий слой целита с использованием дополнительного количества этилацетата. Органические слои промывают водой, сушат над MgSO4, фильтруют и упаривают. Очистка колоночной флэш-хроматографией дает метиловый эфир 2',4'-дифторбифенил-4-карбоновой кислоты в виде желтого твердого вещества. Очищенный эфир растворяют в диоксане (5 мл) и добавляют 5 М NaOH (1 мл). Интенсивно перемешивают при 50 С в течение 15 ч. После завершения перемешивания реакционную смесь подкисляют концентрированной НСl и экстрагируют этилацетатом. Выпаривание растворителя дает 300 мг (24,7%) указанного в заголовке соединения. МС (m/е): 233,0 (M-). Способ F. 6-(2,6-Дифторфенил)пиридин-3-карбоновая кислота Метиловый эфир 6-хлорпиридин-3-карбоновой кислоты (6,86 г, 40 ммоль) растворяют в толуоле(100 мл) и нагревают до 90 С. Добавляют несколькими порциями оксибромид фосфора (25 г, 87 ммоль) и продолжают нагревание в течение 3 ч. Реакционную смесь охлаждают до комнатной температуры и выливают на воду со льдом. Реакционную смесь экстрагируют этилацетатом и органические слои снова промывают водой, затем NаНСО 3. Органические слои объединяют, сушат над MgSO4, фильтруют и упаривают до образования оранжевого твердого вещества (8,1 г, 94%), которое, по определению 1 Н-ЯМР,представляет собой смесь 8:1 метилового эфира 6-бромпиридин-3-карбоновой кислоты:метилового эфира 6-хлорпиридин-3-карбоновой кислоты. Полученную выше смесь (0,225 г, 1,04 ммоль) объединяют с гексаметилдиоловом (0,375 г, 1,15 ммоль), Pd(OAc)2 (21 мг, 0,09 ммоль) и трифенилфосфином (25 мг, 0,09 ммоль) в толуоле (5 мл). Продувают N2 и перемешивают при 80 С в течение 18 ч. Реакционную смесь охлаждают до комнатной температуры. Добавляют раствор 1-бром-2,6-дифторбензола (250 мг, 1,29 ммоль) в толуоле (1 мл) с последующим добавлением Pd(OAc)2 (21 мг, 0,09 ммоль) и трифенилфосфина (25 мг, 0,09 ммоль). ПродуваютN2 и перемешивают при 80 С в течение еще 18 ч. Реакционную смесь охлаждают до комнатной температуры. Растворитель выпаривают и очищают колоночной хроматографией (диоксид кремния, 10% этилацетат в гексане) с получением 50 мг (выход 20%) этилового эфира 6-(2,6-дифторфенил)пиридин-3 карбоновой кислоты. Полученный эфир гидролизуют 1 н раствором гидроксида натрия (0,22 мл, 0,22 ммоль) в метаноле (3 мл) при комнатной температуре в течение 3 дней. Летучие вещества удаляют в вакууме и остаток объединяют с 1 н раствором хлористо-водородной кислоты. Твердое белое вещество собирают фильтрованием, промывают водой и сушат в вакууме с получением 30 мг (выход 63%) указанного в заголовке соединения. МС (m/е) : 235,9 (МН+). Способ G. 3-фторбифенил-4-карбоновая кислота Объединяют метил 2-фтор-4-бромбензоат (1,25 г, 5,36 ммоль), фенилбороновую кислоту (1,30 г,10,72 ммоль) и CsF (2,02 г, 13,40 ммоль) в ДМФА (25 мл) и воде (3,0 мл) при перемешивании. Гетерогенную реакционную смесь, открытую для доступа воздуха, помещают на масляную баню, температуру которой поддерживают при 80 С. Через 5 мин нагревания одной порцией добавляют Pd(OAc)2 (120 мг,0,536 ммоль) и перемешивают до тех пор, пока реакционная смесь не приобретет черную окраску. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и фильтруют через тонкий слой целита с использованием дополнительного количества этилацетата. Органические слои промывают водой, сушат над MgSO4, фильтруют и выпаривают. Очистка колоночной флэшхроматографией дает метиловый эфир 3-фторбифенил-4-карбоновой кислоты в виде твердого вещества.-9 006853 Очищенный эфир растворяют в ТГФ (0,25 М) и добавляют равный объем 1 М раствора NaOH. Интенсивно перемешивают при комнатной температуре в течение 15 ч. После завершения перемешивания реакционную смесь подкисляют концентрированной НСl и экстрагируют этилацетатом. Выпаривание растворителя дает 965 мг (84%) указанного в заголовке соединения. МС (m/е): 214,9 (М-). Перечисленные ниже соединения получают, по существу, таким же способом, как описано выше. Способ Н. 2-Фтор-6-фенилпиридин-3-карбоновая кислота 2,6-Дифторпиридин (5,0 мл, 5,51 ммоль) растворяют в безводном ТГФ (30 мл) и охлаждают до -40 С. Добавляют по каплям в течение 5 мин раствор фениллития (1,8 М раствор в гексане, 30,6 мл). Полученную пурпурную реакционную смесь перемешивают при -40 С в течение 30 мин и доводят до комнатной температуры. Реакцию гасят водой и раствор несколько раз экстрагируют этилацетатом. Органические экстракты объединяют, сушат над MgSO4, фильтруют и упаривают на силикагеле. Очистка колоночной флэшхроматографией дает 2-фтор-6-фенилпиридин 1,0 г (12%) в виде желтого масла. Раствор литийдиизопропиламида (LDA) (3,46 ммоль) в безводном ТГФ (6 мл) охлаждают до -78 С. К охлажденному раствору LDA добавляют через канюлю 2-фтор-6-фенилпиридин в безводном ТГФ (6 мл). Перемешивают при температуре -78 С в течение 30 мин, затем в течение 10 мин через раствор барботируют газообразный диоксид углерода. Реакционной смеси дают нагреться до комнатной температуры и затем смесь продувают аргоном. Реакционную смесь экстрагируют 1 М раствором NaOH и органические слои отбрасывают. Реакционную смесь подкисляют концентрированной НСl и экстрагируют этилацетатом. Органический слой сушат над MgSO4, фильтруют и упаривают с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (405 мг, 65%). МС (m/е): 216,1 (М-). Способ J. 3,5-Дифторбифенил-4-карбоновая кислота Объединяют 1-бром-3,5-дифторбензол (0,863 мл, 7,50 ммоль) и фенилбороновую кислоту (1,22 г,10,00 ммоль) и подвергают условиям, описанным в способе G, с получением 1,3 г 3,5-дифторбифенила. Растворяют неочищенный 3,5-дифторбифенил (1/3 г, 6,83 ммоль) в ТГФ (14 мл) и охлаждают до -78 С. Получают LiTMP добавлением BuLi (1,6M раствор в гексане, 5,33 мл) к тетраметилпиперидину (ТМР) (1,4 мл, 1,25 экв.), при температуре -78 С в ТГФ (14 мл). Охлажденный LiTMP добавляют через канюлю к охлажденному 3,5-дифторбифенилу и реакционную смесь перемешивают при температуре -78 С в течение 1 ч. Через полученный раствор в течение 5 мин барботируют газообразный диоксид углерода, реакционную смесь нагревают до комнатной температуры, выливают в 50 мл 1 М раствора NaOH и экстрагируют 50 мл EtOAc. Органический слой сливают. Оставшийся водный слой подкисляют концентрированной НСl и дважды экстрагируют EtOAc. Органические слои сушат над MgSO4, фильтруют и выпаривают с получением 1,22 г указанного в заголовке соединения в виде белого твердого вещества (77%). МС (m/е): 233,1 (М-). Способ К. 3,2',6'-Трифторбифенил-4-карбоновая кислота Объединяют метил 4-бром-2-фторбензоат (3,66 г, 15,75 ммоль), 4,4,5,5,4',4',5',5'-октаметил-2,2'-би 1,3,2-диоксабороланил (5,0 г, 19,68 ммоль) и ацетат калия (4,63 г, 47,19 ммоль) в ДМСО (40 мл) и полученный раствор продувают аргоном. Добавляют PdCl2 (1,1'-бис(дифенилфосфино)ферроцен)2 (10 мол.%,1,35 г) и раствор снова продувают аргоном. Реакционную смесь нагревают до 80 С в течение 3 ч и охлаждают до комнатной температуры. Реакционную смесь промывают водой, экстрагируют этилацетатом и концентрируют. Полученное черное масло снова растворяют в смеси этилацетат:гексан 1:2, фильтруют через тонкий слой силикагеля и концентрируют. Получают метиловый эфир 2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)бензойной кислоты в виде желтого масла. Полученное желтое масло растворяют в ацетоне (100 мл) и объединяют с NaIO4 (10,1 г, 47,25 ммоль), NH4OAc (3,63 г, 47,25 ммоль) и водой (50 мл) при комнатной температуре. Перемешивают при комнатной температуре в течение 18 ч, переносят в делительную воронку и несколько раз экстрагируют этилацетатом. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют с получением 3,0 г 3-фтор-4-карбометоксибензолбороновой кислоты в виде не совсем белого твердого вещества. Полученную бороновую кислоту (800 мг, 4,04 ммоль) и 2,6-дифторбромбензол (1,17 г, 6,06 ммоль) объединяют с использованием методики, описанной в Способе G, с получением 380 мг указанного в заголовке соединения. МС (m/е): 251,1 (М-). Способ L. 6-Фенилпиридазин-3-карбоновая кислота 6-Фенилпиридазин-3-ол (5,0 г, 29,06 ммоль) растворяют в толуоле (100 мл) и нагревают до 90 С. Добавляют несколькими порциями оксибромид фосфора (25 г, 87,19 ммоль) и реакционную смесь нагревают в течение 30 мин. Полученный желтый раствор охлаждают до комнатной температуры, выливают на воду со льдом и экстрагируют этилацетатом. Органические слои снова промывают водой и 1 М рас- 10006853 твором NaOH, сушат над MgSO4, фильтруют и выпаривают до получения желтого твердого вещества. Перекристаллизация из СНCl3 дает 2,17 г 3-бром-6-фенилпиридазина. 3-Бром-6-фенилпиридазин (1,0 г, 4,25 ммоль) объединяют с ДМФА (5 мл), МеОН (5 мл), триэтиламином (1,18 мл, 8,50 ммоль) и Pd(OAc)2 (76 мг, 0,33 ммоль) и для полученной смеси создают условия вакуума. Добавляют 1,1'-бис(дифенилфосфино)ферроцен (235 мг, 0,42 ммоль) и снова создают вакуум. Через полученный раствор в течение 5 мин барботируют газообразный диоксид углерода и реакционную смесь помещают в атмосферу диоксида углерода при давлении 50 ф./кв.дюйм (345 кПа). Полученный раствор нагревают при 50 С в течение 18 ч. Реакционную смесь охлаждают до комнатной температуры,разбавляют водой и экстрагируют этилацетатом. Органические слои сушат над MgSO4, фильтруют и выпаривают на силикагеле и затем подвергают колоночной флэш-хроматографии. Гидролиз в условиях, описанных в способе А, дает 80 мг указанного в заголовке соединения. 1 Н-ЯМР (CDCl3): 8,24 (д, 1 Н, J=8,8 Гц), 8,18-8,15 (м, 2 Н), 8,0 (д, 1 Н, J=9,2 Гц), 7,56-7,55 (м, 3 Н). Способ М. 6-(4-Фторфенил)пиридин-3-карбоновая кислота Объединяют при перемешивании метиловый эфир 6-бромпиридин-3-карбоновой кислоты (1,03 г,4,78 ммоль), 4-фторфенилбороновую кислоту (1,88 г, 13,41 ммоль) и фторид цезия (2,55 г, 16,78 ммоль) в ДМФА (25 мл) и воде (4 мл). Гетерогенную реакционную смесь, открытую для доступа воздуха, помещают на масляную баню, температуру которой поддерживают при 80 С. После 5 мин нагревания одной порцией добавляют Рd(ОАс)2 (150 мг, 0,67 ммоль). Через 17 ч реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом и фильтруют через тонкий слой целита с использованием дополнительного количества этилацетата. Органические слои промывают водой, сушат над MgSO4,фильтруют и упаривают. Очистка колоночной флэш-хроматографией дает метиловый эфир 6-(4 фторфенил)пиридин-3-карбоновой кислоты в виде желтого твердого вещества. Очищенный эфир растворяют в ТГФ (0,25 М) и добавляют равный объем 1 М раствора NaOH. Интенсивно перемешивают при комнатной температуре в течение 15 ч. После завершения перемешивания реакционную смесь подкисляют концентрированной НСl и белый осадок собирают фильтрованием. Сушка в вакууме дает 385 мг(37%) указанного в заголовке соединения. МС (m/е): 218,1 (МН+). Следующее соединение получают, по существу, таким же способом, как описано выше. Способ N. 6-(4-Фтор-2-метилфенил)пиридин-3-карбоновая кислота Объединяют при перемешивании метиловый эфир 6-бромпиридин-3-карбоновой кислоты (387 мг,1,79 ммоль), 4-фтор-2-метилфенилбороновую кислоту (338 мг, 2,19 ммоль), Рd(ОАс)2 (40 мг, 0,18 ммоль),фторид цезия (27 мг, 0,18 ммоль) и карбонат натрия (570 мг, 5,38 ммоль) в ДМФА (6 мл) и воде (6 мл). Реакционную смесь продувают N2, добавляют трифенилфосфин (47 мг, 0,18 ммоль) и снова продуваютN2. Герметично закрытую реакционную смесь помещают на масляную баню, температуру которой поддерживают при 80 С, и оставляют при перемешивании на 17 ч. Реакционную смесь охлаждают до комнатной температуры и пропускают через тонкий слой силикагеля. Колонку промывают дихлорметаном(100 мл), а затем водным раствором метанола (100 мл, 3 метанол/1 вода). Объединенные фракции концентрируют в вакууме и остаточное твердое вещество суспендируют в воде (10 мл). Фильтруют для удаления черного твердого вещества и подкисляют 1 н хлористо-водородной кислотой до рН 4. Образуется белый осадок, который собирают фильтрованием и сушат с получением 306 мг (74%) указанного в заголовке соединения. МС (m/е): 231,9 (МН+). Перечисленные ниже соединения получают, по существу, таким же способом, как описано выше. К 224 г (5,55 моль, 1,2 экв.) гидрида натрия (60% дисперсия в минеральном масле) в виде суспензии в ТГФ (8,75 л) медленно добавляют раствор 375 г (5,13 моль, 1,12 экв.) N-метилацетамида в ТГФ (1,76 л). Через 30 мин после добавления 25% раствора добавляют 875 г (4,63 моль, 1 экв.) 4-фторбензилбромида и в течение следующих 3 ч одновременно добавляют остальное количество растворов N-метилацетамида и 4-фторбензилбромида. Для поддержания температуры ниже 40 С используют водную баню. Получен- 11006853 ную смесь перемешивают в течение ночи и выливают в смесь 20% NH4Cl (2,5 л), воды (6,5 л) и этилацетата (9 л). Слои разделяют и водный слой экстрагируют обратно в этилацетат (4,5 л, затем 2 л). Органические слои объединяют и промывают водой (4 л), затем насыщенным раствором соли (7 л). Органический слой сушат (Na2SO4) и растворитель удаляют с получением остатка. Остаток растворяют в ацетонитриле (7 л) и гептане (1,75 л). Слои разделяют и ацетонитрильный слой снова промывают гептаном (1,75 л). Гептановые слои объединяют и экстрагируют обратно в ацетонитрил (0,5 л). Ацетонитрильные слои объединяют и упаривают с получением 0,814 кг N-метил-N-(4 фторбензил)ацетамида.N-Метил-N-(4-фторбензил)ацетамид (0,500 кг, 2,76 моль) растворяют в ТГФ (11,5 л). Добавляют пентасульфид фосфора (0,737 кг, 1,65 моль, 0,6 экв.) и смесь нагревают при кипячении с обратным холодильником в течение 1-2 ч. Через 5 ч кипячения с обратным холодильником смеси дают охладиться до комнатной температуры, отфильтровывают твердое вещество и промывают 12,5 л ТГФ. Фильтрат объединяют с аналогичным фильтратом от отдельной реакции и концентрируют с получением 0,978 кг остатка. Остаток растворяют и хроматографируют на 2,7 кг силикагеля, используя СН 2 Сl2, с получением 1,01 кг твердого вещества. Полученное твердое вещество суспендируют в метиленхлориде (1 л) в течение 1530 мин, добавляют гептан (5 л), смесь охлаждают до 0-5 С и перемешивают в течение 2 ч. Твердое вещество собирают фильтрованием и сушат с получением 0,814 кг N-метил-N-(4-фторбензил)тиоацетамида. К 2,30 кг (11,6 моль) N-метил-N-(4-фторбензил)ацетамида добавляют 11,5 л ацетонитрила и 2,52 кг(17,7 моль, 1,5 экв.) метилйодида. Смесь нагревают до 35 С в течение 21 ч. Концентрируют до половины объема на роторном испарителе и добавляют 14 л метил-трет-бутилового эфира (МТВЕ). Объем снова уменьшают наполовину и добавляют еще 14 л МТВЕ. Полученную суспензию охлаждают до 0 С, твердое вещество собирают фильтрованием и сушат с получением 3,92 кг 1-метилтио-1-метил-N-(4 фторбензил)-N-метилиммониййодида в виде белого твердого вещества. К 6,20 кг (35,0 моль) 1,2-эпокси-6-нитроиндана добавляют 85 л концентрированного раствораNH4OH и 28 л воды. Смесь нагревают при 36 С в течение 21 ч и дают охладиться до комнатной температуры. Реакционную смесь фильтруют через слой влажного целита (10 кг) и фильтровальную лепешку промывают водой. К влажной лепешке добавляют 155 л метанола, 1,3 л воды и 5,80 кг (38,1 моль, 1,09 экв.) (S)-(+)-миндальной кислоты. Смесь нагревают в течение 2 ч при 55 С и фильтруют через пропитанный углеродом фильтровальный патрон. Объем фильтрата уменьшают перегонкой в вакууме до около 35 л и добавляют 130 л EtOAc. Уменьшают объем перегонкой в вакууме до около 65 л. Смесь охлаждают до-8 С и перемешивают в течение 8 ч. Суспензию фильтруют и твердый остаток сушат с получением 7,6 кг твердого вещества. Полученное твердое вещество суспендируют в 30 л метанола и 0,3 л воды и смесь нагревают при температуре кипения с обратным холодильником в течение 0,5 ч. Смесь охлаждают до 45 С в течение 0,5 ч и перемешивают в течение 12 ч с последующим охлаждением до 22 С и перемешиванием в течение 10 ч. Твердое вещество собирают фильтрованием и сушат с получением 2,7 кг (S)манделата 1(R)-амино-2(R)-гидрокси-6-нитроиндана. К смеси толуола (9,6 л) и 1 н водного раствора NaOH (4,8 л, 4,8 моль, 2,6 экв.) добавляют (S)манделат 1(R)-амино-2(R)-гидрокси-6-нитроиндана (0,64 кг, 1,85 моль). Через 1 ч добавляют 4 бифенилкарбонилхлорид (0,44 кг, 2,0 моль, 1,1 экв.) порциями в течение 20-30 мин. Спустя 22 ч твердое вещество фильтруют в вакууме и последовательно промывают 0,5 л толуола, 2 л воды и 2 л толуола. Фильтровальную лепешку сушат с получением 0,74 кг (R)-(6-нитро-2-гидроксииндан-1-ил)амида бифенил-4-карбоновой кислоты. К 1,914 кг (R)-(6-нитро-2(R)-гидроксииндан-1-ил)амида бифенил-4 карбоновой кислоты, полученного аналогичным способом, добавляют 38,2 л этилацетата. Суспензию перемешивают в течение 18 ч, твердое вещество собирают фильтрованием, сушат с получением 1,76 кг(R)-(6-нитро-2(R)-гидроксииндан-1-ил)амида бифенил-4-карбоновой кислоты в виде белого твердого вещества. Объединяют суспензию 0,176 кг 10% Pd-C (на 50% смоченный водой) и 1,7 кг (R)-(6-нитро-2(R)гидроксииндан-1-ил)амида бифенил-4-карбоновой кислоты в 17,5 л ДМФА с водородом (50 ф./кв.дюйм,345 кПа). Спустя 19 ч реакционную смесь фильтруют, добавляют порцию раствора в ДМФА в (5 л) к воде (10 л) и перемешивают суспензию в течение 2 ч (повторяют дважды для обработки всего объема реакционной смеси). Суспензии фильтруют вместе и полученную фильтровальную лепешку промывают водой (3x7 л). Фильтровальную лепешку сушат с получением 1,42 кг (R)-(6-амино-2(R)-гидроксииндан 1-ил)амида бифенил-4-карбоновой кислоты.(0,954 кг, 2,81 моль) и 4-диметиламинопиридин (34,5 г, 0,281). Смесь перемешивают в течение 24 ч и растворитель удаляют в вакууме. Полученную пену растворяют в СН 2 Сl2 (12,5 л) и органическую фазу промывают 1,0 н раствором НСl (1x4 л и 1x3 л), 1,0 М раствором NaOH (1x2,4 л) и насыщенным раствором NaCl (1x4 л). Органическую фазу отделяют, сушат (Na2SO4), фильтруют и растворитель удаляют с получением твердого вещества. Полученное твердое вещество растворяют в ацетонитриле (9 л) с одновременным нагреванием до 35-40 С. Примерно через 30 мин добавляют затравочные кристаллы с получением густой белой суспензии. Смесь охлаждают до -15 С и перемешивают при этой температуре в те- 12006853 чение 1-2 ч. Суспензию фильтруют и сушат с получением 1,10 кг указанного в заголовке соединения в виде частичного ацетонитрильного сольвата. 1 Н-ЯМР (CDCl3)7,90 (д, 2, J=8,6), 7,69 (д, 2, J=8,6), 7,63 (д, 2, J=8,2), 7,48 (т, 2, J=8,2, 7,6), 7,41 (д,1, J=7,3), 7,24 (дд, 2, J=8,5, 5,2), 7,14 (д, 1, J=7,9), 7,04 (т, 2, J=8,7), 6,72-6,63 (м, 3), 5,31 (т, 1, J=5,6), 4,84(16,7 г, 0,11 моль, 1,1 экв.) в 173 мл метанола и 3,6 мл воды. Нагревают при температуре кипения с обратным холодильником и затем добавляют 200 мл этилацетата. Вносят затравочные кристаллы и дают охладиться до 23 С. После перемешивания в течение 4 ч при 23 С реакционную смесь охлаждают в течение 3 ч до -3 С, затем фильтруют и промывают холодным раствором 40% метанола и 60% этилацетата с получением твердого вещества. Твердое вещество сушат в вакуумной печи при 45 С в течение 16 ч с получением 59:41 смеси диастереомерной соли, с преимущественным содержанием 1(R)-амино-2(R)гидрокси-6-нитроиндана. Полученную смесь 59:41 диастереомерной соли объединяют с 35 мл метанола и 0,32 г воды. Нагревают до около 64 С, дают охладиться до около 45 С и перемешивают в течение 14 ч, а затем при 23 С в течение 14 ч, с получением твердого вещества. Твердое вещество собирают фильтрованием, промывают метанолом и сушат в вакуумной печи при 45 С в течение 26 ч с получением 2,64 г (S)-манделата 1(R)амино-2(R)-гидрокси-6-нитроиндана высокой энантиомерной чистоты. HRMS (МС высокого разрешения) (m/z): 194,0691. ИК (СНСl3) 1347, 1074 см-1. Объединяют 40,2 г (S)-манделата 1(R)-амино-2(R)-гидрокси-6-нитроиндана (116,1 ммоль), 320 мл воды, 650 мл этилацетата, 26,6 г ди-трет-бутилдикарбоната (121,9 ммоль, 1,05 экв.) и добавляют еще 200 мл этилацетата. Добавляют по каплям 120 мл 1 н водного раствора гидроксида натрия (120 ммоль, 1,03 экв.). Через 15 ч образуется твердое вещество. Твердое вещество собирают и промывают водой (3 раза) и этилацетатом (3 раза). Сушат с получением 19,4 г 1(R)-(трет-бутоксикарбониламино)-2(R)-гидрокси-6 нитроиндана в виде белого твердого вещества (57%):MC (m/z): 295 (M+l). []D=-111 (с=1, МеОН). Объединяют 30,5 г 1(R)-(трет-бутоксикарбониламино)-2(R)-гидрокси-6-нитроиндана (0,11 моль),800 мл ТГФ, 800 мл этилацетата и 6,3 г 5% палладия на угле. Гидрируют при давлении водорода 50 ф./кв.дюйм (345 кПа) в течение 2 ч. Катализатор удаляют фильтрованием и растворитель выпаривают с получением 29,5 г 1(R)-(трет-бутоксикарбониламино)-2(R)-гидрокси-6-аминоиндана: MC (m/z): 265(М+1). ИК (КВr) 1699, 1625, 1535 см-1. []D =-122 (с=1, МеОН). К раствору NaH (6,86 г, 0,172 моль, 60% в минеральном масле, 1,3 экв.) в ТГФ (250 мл) добавляют по каплям раствор N-метилацетамида (11,6 г, 0,159 моль, 1,2 экв.) в ТГФ (90 мл). Примерно через 20 мин добавляют 4-фторбензилбромид (25 г, 0,132 моль). Перемешивают примерно в течение 62 ч, затем выливают на воду со льдом (300 мл) и экстрагируют этилацетатом (400 мл и 200 мл). Органические слои объединяют и промывают водой (300 мл), сушат над Na2SO4, фильтруют и концентрируют с получением масла. Масло растворяют в ацетонитриле и экстрагируют гексаном для удаления минерального масла с получением 22,79 г N-метил-N-(4-фторбензил)ацетамида: т.пл.=48-54 С; Rf=0,45 (4% МеОН/метиленхлорид); 1 Н ЯМР (CDCl3) 7,27-6,93 (м, 4), 4,5 (д, 2), 2,91 (с, 3), 2,14 (с, 3). Объединяют N-метил-N-(4-фторбензил)ацетамид (364,8 г, 2,01 моль) и ТГФ (9 л). Перемешивают с получением раствора, затем добавляют пентасульфид фосфора (537,7 г, 1,21 моль). Через 45 мин нагревают до кипения с обратным холодильником. Через 3 ч смеси дают охладиться и перемешивают в течение ночи с получением твердого вещества. Твердое вещество удаляют фильтрованием и фильтровальную лепешку промывают ТГФ (4 л). Фильтрат упаривают с получением остатка, растворяют в метиленхлориде (500 мл) и пропускают через короткую колонку с силикагелем 60 (1,2 кг) предварительно обработанную гептаном. Элюируют 4 л смеси гептан/метиленхлорид (1:1), а затем 100% метиленхлоридом с получением, после выделения и сушки, 217,98 г N-метил-N-(4-фторбензил)тиоацетамида: т.пл.=99-104 С;(с, 1,2) и 3,15 (с, 1.8), 2,72 (с, 1,2) и 2,69 (с, 1,8). Примечание: Наличие частичных протонов, как полагают, обусловлено ротамерами. В атмосфере азота объединяют N-метил-N-(4-фторбензил)тиоацетамид (14,03 г, 0,0711 моль) и метиленхлорид (140 мл) и охлаждают на водной бане со льдом. Добавляют по каплям метилтрифторметан- 13006853 сульфонат (9,66 мл, 0,085 моль, 1,2 экв.). Через 15 мин ледяную баню удаляют и перемешивают в течение 2 час. Растворитель удаляют в вакууме с получением 25,89 г (100%) 1-метилтио-1-метил-N-(4 фторбензил)-N-метиламмонийтрифлата. В атмосфере азота объединяют 1-метилтио-1-метил-N-(4-фторбензил)-N-метиламмонийтрифлат (5 г, 13,8 ммоль), метиленхлорид (50 мл) и 1(R)-(трет-бутоксикарбониламино)-2(R)-гидрокси-6-аминоиндан(3,65 г, 13,8 ммоль). Добавляют пиридин (0,15 мл). Через 2,5 ч образуется твердое вещество. Твердое вещество собирают фильтрованием, промывают минимальным количеством метиленхлорида и сушат в вакуумной печи с получением 6,29 г (79%) 1(R)-(трет-бутоксикарбониламино)-2(R)-гидрокси-6-(1-метилN'-(4-фторбензил)-N'-метиламидино)индантрифлата в виде белого твердого вещества, т.пл.=123-132 С. Трифторуксусную кислоту (ТФУК, 66 мл) охлаждают на бане со льдом и добавляют порциями 1(R)-(трет-бутоксикарбониламино)-2(R)-гидрокси-6-(1-метил-N'-(4-фторбензил)-N'-метиламидино)индантрифлат (29,65 г, 0,051 моль) вместе с 3 мл метиленхлорида. Перемешивают на ледяной бане в течение 10 мин, затем дают нагреться до комнатной температуры и перемешивают в течение 1,5 ч. Реакционную смесь распределяют между метиленхлоридом (500 мл) и ледяной водой (500 мл). Добавляют 2 М водный раствор гидроксида натрия (450 мл) и слои разделяют. Водный слой экстрагируют метиленхлоридом (250 мл), органические слои объединяют и промывают водой (300 мл). Органический слой охлаждают, добавляют 1 н водный раствор гидроксида натрия (76 мл) и воду (76 мл) и перемешивают с получением N'-(3-амино-2-гидроксииндан-5-ил) -N- (4-фторбензил) -N-метилацетамидина. К N'-(3-амино-2-гидроксииндан-5-ил)-N-(4-фторбензил)-N-метилацетамидину, полученному описанным выше способом, добавляют порциями 4-бифенилкарбонилхлорид (11,05 г, 0,051 моль, 1 экв.). Добавляют еще 50 мл воды. Через 2 ч слои разделяют, органический слой экстрагируют водой (300 мл),сушат над Nа 2SO4, фильтруют и упаривают с получением 27,06 г указанного в заголовке соединения в виде белой пены. Кристаллизуют часть (10 г) указанного в заголовке соединения из 10 мл/г ацетонитрила с получением 5,96 г указанного в заголовке соединения: т.пл. 103-111 С. МС (m/z): 508 (М+1). Указанное в заголовке соединение, полученное путем перекристаллизации, как описано выше, суспендируют в смеси 45 мл этилацетата и 45 мл гексана в течение 16 ч с получением указанного в заголовке соединения, т.пл. 139-142 С. Объединяют указанное в заголовке соединение, имеющее т.пл.=139-142 С (1 г), и абсолютный этанол (12 мл). Нагревают до около 50 С до растворения твердого вещества. Добавляют по каплям деионизированную воду (4,5 мл) с последующим добавлением затравочных кристаллов полиморфа, плавящегося при температуре около 150-152 С. Охлаждают до температуры около 23 С в течение 1,5 ч с получением густой суспензии. Полученную суспензию охлаждают на ледяной бане, фильтруют и сушат в вакуумной печи при 50-60 С с получением 0,76 г указанного в заголовке соединения: т.пл.=150-152 С. Как должно быть понятно специалисту в данной области, альтернативное название указанного в заголовке соединения - 6-(1-4-фторбензил)метиламино)этилиденамино)-2-гидрокси-1-бифениламидоиндан. Пример 2-2. Объединяют 50,0 г S-манделата 1(R)-амино-2(R)-гидрокси-6-нитроиндана (0,144 моль) и 750 мл толуола. Добавляют 375 мл (0,375 моль, 2,6 экв.) 1 н водного раствора гидроксида натрия с последующим добавлением 470 мл воды. Перемешивают примерно в течение 22 мин, затем добавляют порциями 35,5 г(0,159 моль, 1,1 экв.) 4-бифенилкарбонилхлорида в течение примерно 6 мин. Через 3,5 ч полученную суспензию фильтруют, фильтровальную лепешку промывают толуолом, сушат в вакууме при 50 С в течение 18 ч с получением 56,1 г 1(R)-(4-бифенилкарбониламино)-2(R)-гидрокси-6-нитроиндана: ИК (КВr,см-1): 3293, 1640, 1549, 1528, 1345, 1329, 1086, 739; HRMS рассчитано для C22H18N2O4: 374,1267; найдено: 374,1266. Объединяют 55,7 г 1(R)-(4-бифенилкарбониламино)-2(R)-гидрокси-6-нитроиндана, 550 мл ДМФА и 2,8 г катализатора 10% Pd/C. Гидрируют при давлении водорода 50 ф./кв.дюйм (345 кПа) при температуре окружающей среды в течение 4,75 ч. Реакционную смесь фильтруют и фильтрат разбавляют 1200 мл воды с получением суспензии. Суспензию перемешивают в течение 30 мин, фильтруют, промывают водой и сушат в вакууме при 50 С. Высушенный продукт объединяют с примерно 1 л воды и суспендируют в течение 30 мин, фильтруют, промывают водой и сушат в вакууме при 50 С с получением 45,4 г- 14006853 до растворения и затем добавляют пентасульфид фосфора (537,7 г, 1,21 моль). Через 45 мин нагревают при кипячении с обратным холодильником в течение 3 ч, затем охлаждают и перемешивают в течение ночи с получением твердого вещества. Твердое вещество собирают фильтрованием и фильтровальную лепешку промывают ТГФ (4 л), фильтрат упаривают в вакууме с получением остатка, остаток растворяют в метиленхлориде (около 500 мл) и пропускают через слой силикагеля 60 (1,2 кг), предварительно обработанный гептаном. Элюируют 4 л смеси гептан/метиленхлорид (1:1), а затем 100% метиленхлоридом, с получением 217,98 г N-метил-N-(4-фторбензил)тиоацетамида (55%): т.пл.=99-104 С; Rf=0,42 (метиленхлорид); 1 Н ЯМР (CDCl3) 7,35-7,26 (м, 1), 7,14-6,96 (м, 3), 5,28 (с, 1,2) и 4,79 (с, 0,8), 3,42 (с, 1,2) и 3,15 (с, 1.8), 2,72 (с, 1,2) и 2,69 (с, 1,8). Примечание: Наличие частичных протонов, как полагают, обусловлено ротамерами. К суспензии N-метил-N-(4-фторбензил)тиоацетамида (10 г, 50,6 ммоль) в ацетонитриле (50 мл) одной порцией добавляют метилйодид (10,76 г, 75,8 ммоль). Нагревают при 35 С в течение 46 ч, затем охлаждают до около 23 С. Объем реакционной смеси уменьшают до около 25 мл упариванием и затем добавляют метил-трет-бутиловый эфир (50 мл). Объем снова уменьшают до 25 мл упариванием и затем разбавляют дополнительными 50 мл метил-трет-бутилового эфира с получением твердого вещества. Охлаждают до 0 С, фильтруют, промывают 15 мл холодного метил-трет-бутилового эфира и сушат с получением 16,89 г 1-метилтио-1-метил-N-(4-фторбензил)-N-метиламмониййодида в виде желтого твердого вещества, т.пл. 142-150 С. МС (Электрораспыление): теоретически рассчитано для иминиевой частиC11H15FNS: 212; найдено: 212. Объединяют 1(R)-(4-бифенилкарбониламино)-2(R)-гидрокси-6-аминоиндан 10,0 г (0,029 моль), 4 диметиламинопиридин (DMAP) 0,4 г (0,0032 моль, 0,011 экв.), 200 мл ацетона и 10,8 г 1-метилтио-1 метил-N-(4-фторбензил)-N-метиламмониййодида (концентрация 96%, 0,0305 моль, 1,05 экв.). Спустя 6 ч реакционную смесь концентрируют с получением пены. Полученную пену объединяют с 120 мл толуола,перемешивают при температуре окружающей среды в течение 19,5 чс, фильтруют, сушат в вакууме при 50 С с получением остатка, йодистоводородной соли указанного в заголовке соединения, которая характеризуется следующими данными ЯМР: 1 Н ЯМР (CDCl3, 300 МГц):8,02 (д, J=9,0 Гц, 2 Н), 7,82-7,93 (м,1 Н), 6,90-7,69 (м, 14 Н), 4,85-5,25 (м, 2 Н), 4,60-4,80 (м, 2 Н), 3,45 (с, 2 Н), 3,00-3,20 (м, 2 Н), 2,60-2,75 (м,1 Н), 2,10-2,35 (м, 4 Н). Остаток растворяют в 200 мл метиленхлорида и экстрагируют 200 мл 1,0 М водного раствора гидроксида натрия, а затем 200 мл насыщенного раствора соли. Органический слой сушат над MgSO4, фильтруют и упаривают в вакууме с получением 15 г указанного в заголовке соединения. Объединяют 5,0 г соединения, полученного выше, и 200 мл метиленхлорида. Добавляют 1,00 г угляDARCO, перемешивают в течение 1 ч и затем фильтруют через слой Hyflo. Упаривают в вакууме с получением 4,0 г остатка. Остаток растворяют в абсолютном этаноле (около 40 мл) и добавляют деионизированную воду (12 мл) с получением твердого вещества. Суспендированное твердое вещество нагревают до 55 С в течение 30 мин, дают охладиться до комнатной температуры и перемешивают. Спустя 24 ч фильтруют, промывают 10 мл 10:3 смеси ЕtOН:вода, сушат в вакууме с получением 1,5 г указанного в заголовке соединения: т.пл. 108-112 С. Как должно быть понятно специалисту в данной области, альтернативное название указанного в заголовке соединения 6-(1-4-фторбензил)метиламино)этилиденамино)-2-гидрокси-1-бифениламидоиндан. Пример 3-1. Объединяют 4-бром-2-фторбензойную кислоту (10 г, 45,7 ммоль), метанол (100 мл) и концентрированную серную кислоту (5,0 мл). Нагревают при кипячении с обратным холодильником. Через 16 ч охлаждают до комнатной температуры и упаривают в вакууме с получением белого твердого вещества. Твердое вещество растворяют в этилацетате (около 40 мл), экстрагируют 2x80 мл насыщенного водного раствора бикарбоната натрия и 1x80 мл насыщенного раствора соли, сушат над MgSO4, фильтруют и упаривают в вакууме с получением 9,32 г (88%) метил 4-бром-2-фторбензоата в виде белого твердого вещества. 1 Н ЯМР (ДМСО-d6, 300 МГц) 8,80 (т, J=8,4 Гц, 1 Н), 7,72 (дд, J=10,8 Гц, 1,8 Гц, 1 Н), 7,54 (ддд, J=8,4 Гц, 1,8 Гц, 0,6 Гц, 1 Н), 3,83 (с, 3 Н); ИК (см-1, КВr): 3010, 2995, 1723, 1603, 1484, 1438, 1406, 1294, 1277,1095, 885; Анализ. Вычислено для C8H6BrFO2: С, 41,23; Н, 2,60; найдено: С, 40,97; Н, 2,61. Объединяют метил 4-бром-2-фторбензоат (9,3 г, 40,0 ммоль), фенилбороновую кислоту (9,75 г, 80,0 ммоль) и фторид цезия (32,6 г, 100,1 ммоль), ДМФА (190 мл) и деионизированную воду (50 мл). Нагре- 15006853 вают до 80 С и добавляют Pd(OAc)2 (303 г, 4,0 ммоль). Спустя 20 мин охлаждают до комнатной температуры, фильтруют через HyFlo с использованием 100 мл этилацетата, фильтрат экстрагируют 5% водным раствором хлорида лития (2x100 мл), 1,0 М водным раствором гидроксида натрия (2x50 мл) и насыщенным раствором соли (2x100 мл). Органическую фазу отделяют, сушат над MgSO4, фильтруют и упаривают в вакууме с получением 8,85 г (96%) метил 3-фторбифенил-4-карбоксилата в виде белого твердого вещества. 1 Н ЯМР (СDСl3, 300 МГц) 8,01 (т, J=7,8 Гц, 1 Н), 7,58-7,62 (м, 2 Н), 7,41-7,51 (м, 4 Н) , 7,37 (м,1 Н), 3,96 (с, 3 Н); ИК (см-1, КВr): 3033, 2954, 1721, 1622, 1437, 1409, 1298, 1289, 1266, 1097. Анализ. Вычислено для C14H11FO2: С, 73,03, Н, 4,82. Найдено: С, 73,06, Н, 4,86. Объединяют метил 3-фторбифенил-4-карбоксилат (8,18 г, 35,5 ммоль), ТГФ (225 мл) и 1,0 М водный раствор гидроксида натрия (225 мл) . Нагревают при 50 С в течение 8 ч. Охлаждают до комнатной температуры, добавляют 1,0 М водный раствор хлористоводородной кислоты (300 мл) и экстрагируют этилацетатом (2x200 мл). Органическую фазу отделяют, сушат над MgSO4, фильтруют и упаривают в вакууме с получением 7,12 г (93%) 3-фторбифенил-4-карбоновой кислоты в виде белого твердого вещества. 1 Н ЯМР (ДМСО-d6, 300 МГц) 13,22 (шир.с, 1 Н), 7,93 (т, J=8,l Гц, 1 Н), 7,74-7,78 (м, 2 Н), 7,60-7,66 (м,2 Н), 7,40-7,52 (м, 3 Н); Ж (см-1, КВr): 3035, 2666, 2575, 1699, 1621, 1563, 1408, 1298, 1265, 1193, 904; Анализ. Вычислено для C13H9FO2: С, 72,22, н, 4,20. Найдено: С, 72,18, Н, 4,35. Объединяют 10,46 г 3-фторбифенил-4-карбоновой кислоты (0,048 моль), 432 мл метиленхлорида, 7 капель диметилформамида и 5,44 мл (0,062 моль, 1,3 экв.) оксалилхлорида. Через 2 ч растворитель выпаривают с получением 3-фторбифенил-4-карбонилхлорида в виде твердого вещества. Объединяют 20,96 г 1-метилтио-1-метил-N-(4-фторбензил)-N-метиламмонийтрифлата (0,58 моль),53 мл пиридина и 15,33 г 1(R)-(трет-бутоксикарбониламино)-2(R)-гидрокси-6-аминоиндана (0,58 моль, 1 экв.). Через 4,2 ч выпаривают на роторном испарителе для удаления большей части пиридина, добавляют этилацетат и удаляют на роторном испарителе с получением остатка. Остаток хранят в течение ночи при 0 С, добавляют этилацетат (400 мл) и экстрагируют 200 мл 1 н водным раствором гидроксида натрия,затем 200 мл воды. Органический слой сушат над Na2SO4, фильтруют и упаривают с получением остатка. Остаток хроматографируют на силикагеле, элюируя градиентом 3-10% метанола в метиленхлориде, с получением 18,85 г (76%) 1(R)-трет-бутоксикарбониламино)-2(R)-гидрокси-6-(1-метил-N'-(4-фторбензил)-N'-метиламидино)индана: МС m/z=428 (M+1), т.пл.=123-132 С. Объединяют 18,86 г 1(R)-(трет-бутоксикарбониламино)-2(R)-гидрокси-6-(1-метил-N'-(4-фторбензил)-N'-метиламидино)индана (0,044 моль) и 110 мл трифторуксусной кислоты на ледяной бане. После завершения добавления ледяную баню удаляют и перемешивают при комнатной температуре в течение 2,5 ч. Реакционную смесь упаривают на роторном испарителе с получением остатка. Остаток растворяют в 100 мл метиленхлорида и охлаждают на ледяной бане до около 13 С, добавляют 1 н водный раствор гидроксида натрия (272 мл), с последующим добавлением 11,26 г 3-фторбифенил-4 карбонилхлорида (0,048 моль, 1,1 экв.), растворенного в 120 мл метиленхлорида. Добавляют еще 30 мл 1 н водного раствора гидроксида натрия и перемешивают при около 10 С в течение 40 мин. Реакционную смесь распределяют между водой и метиленхлоридом. Слои разделяют и органический слой экстрагируют водой, сушат и концентрируют на роторном испарителе с получением 20,67 г остатка. Остаток хроматографируют на силикагеле, элюируя градиентом 3-10% метанола в метиленхлориде, и затем проводят вторую хроматографию на силикагеле с использованием Prep 2000, элюируя градиентом 3-10% метанола в метиленхлориде, с получением 11,34 г (49%) указанного в заголовке соединения: МСm/z=526 (М+1). Как должно быть понятно специалисту в данной области, альтернативное название указанного в заголовке соединения 6-(1-4-фторбензил)метиламино)этилиденамино)-2-гидрокси-1-(2-фторбифениламидо)индан. Пример 4-1.(0,2 мл). Смесь перемешивают в течение 16 ч, затем добавляют полистирольную трисаминовую смолу 6-циано Растворяют эквимолярные количества N'-(3-амино-2-гидроксииндан-5-ил)-N-(4-фторбензил)-Nметилацетамидина (229 мг, 0,70 ммоль) и 6-циано-3-карбоксипиридина (0,70 ммоль, 103 мг) в безводном ДМФА (4,0 мл). Добавляют триэтиламин (0,4 87 мл, 3,50 ммоль) с последующим добавлением гексафторфосфата бензотриазол-1-илокситрисдиметиламинофосфония (296 мг, 0,70 ммоль). Реакционную смесь оставляют для перемешивания на 0,5 ч при комнатной температуре. Реакционную смесь разбавляют водой (100 мл) и экстрагируют EtOAc (3x50 мл). Объединенные органические слои сушат над MgSO4,фильтруют и концентрируют. Неочищенный реакционный продукт растворяют в ТГФ (5,0 мл). Добавляют гидроксидную смолу (смола BIO-RAD AG 1-X8, 20-50 меш - промытая водой), 1 М раствор NaOH,MeOH, простой эфир и сушат в вакууме, пока раствор не станет щелочным, и перемешивают при температуре окружающей среды в течение 24 ч. Реакционную смесь фильтруют, промывают дополнительным количеством ТГФ, выпаривают на силикагеле и очищают колоночной флэш-хроматографией с использо- 19006853 ванием EtOAc/гексан с получением 253 мг (79%) указанного в заголовке соединения в виде белого твердого вещества. МС (m/е): 458 (М+). Примеры с 5-2 по 5-8 получают, по существу, таким же способом, как пример 1-1. Растворяют N'-(3(R)-амино-2(R)-гидроксииндан-5-ил)-N-(4-фторбензил)-N-метилаацетамидин (42 мг, 0,13 ммоль) и 6-(2-хлорфенил)пиридин-3-карбоновую кислоту (103 мг, 0,70 ммоль) в безводном ДМФА (2,5 мл). Добавляют триэтиламин (0,178 мл) с последующим добавлением гексафторфосфата бензотриазол-1-илокситрисдиметиламинофосфония (54 мг, 0,13 ммоль). Реакционную смесь оставляют для перемешивания при комнатной температуре до завершения реакции. Реакционную смесь разбавляют водой (100 мл) и экстрагируют EtOAc. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют. Проводят очистку колоночной флэш-хроматографией с использованием смеси СНСl3/МеОН с выделением 276 мг (35,7%) указанного в заголовке соединения в виде твердого вещества. МС (m/е): 543,0 (М+). Примеры с 6-2 по 6-4 получают, по существу, таким же способом, как пример 6-1. Объединяют (R)-(6-(1-4-фторбензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1-ил)амид 4-йодфенил-1-карбоновой кислоты (0,131 г, 0,235 ммоль), 2-(трибутилстаннил)пиридин (0,129 г, 0,352 ммоль) и тетракис(трифенилфосфин)палладий (0) (0,406 г, 0,352 ммоль) в диоксане при 80 С. Нагревают при перемешивании до завершения реакции. Растворитель удаляют выпариванием. Полученный остаток разбавляют этилацетатом и перемешивают с равным объемом насыщенного раствора фторида калия в течение 3 ч. Раствор фильтруют через слой целита. Органический слой отделяют от водного слоя и сушат над сульфатом магния. Органический растворитель удаляют выпариванием и проводят очистку флэш-хроматографией с использованием дихлорметана и метанола с получением 0,035 г (30%) указанного в заголовке соединения в виде твердого вещества. МС (m/е): 509,2 (М+). Пример 8-1. Объединяют 2',4',6'-трифторбифенил-4-карбоновую кислоту (66 мг, 0,26 ммоль), EDC (53 мг, 0,28 ммоль) и N-гидроксисукцинимид (0,33 мг, 0,28 ммоль) в дихлорметане и перемешивают до завершения реакции. Полученный раствор промывают 1 н. раствором хлористо-водородной кислоты. Органический слой сушат над сульфатом магния и концентрируют. Остаток объединяют с N'-(3(R)-амино-2(R)гидроксииндан-5-ил)-N-(4-фторбензил)-N-метилацетамидином (42 мг, 0,13 ммоль) в дихлорметане и перемешивают до завершения реакции. Растворитель удаляют выпариванием и остаток очищают флэшхроматографией с использованием дихлорметана и метанола с получением 37 мг указанного в заголовке соединения. МС (m/е): 562,0 (М+). Пример 9-1. Перемешивают смесь 3,4'-дифторбифенил-4-карбоновой кислоты (160 мг, 0,684 ммоль), Nгидроксисукцинимида (79 мг, 0,684 ммоль) и DCC (141 мг, 0,684 ммоль) в 15 мл метиленхлорида при комнатной температуре в течение 2 ч. Объединяют трет-бутиловый эфир (6-(1-4 фторбензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1-ил)карбаминовой кислоты (256 мг, 0,62 ммоль) с ТФУК (2 мл) при 0 С и перемешивают в течение 2 ч. Выпаривают ТФУК при пониженном давлении. Остаток растворяют в метиленхлориде и упаривают досуха (эту процедуру повторяют три раза). Добавляют 5 мл метиленхлорида и 1 мл триэтиламина. Полученный раствор добавляют к указанной выше смеси и перемешивают при комнатной температуре в течение 12 ч. Полученную смесь выливают в метиленхлорид, промывают водой, сушат над Nа 2SO4 и концентрируют. Остаток очищают колоночной хроматографией (силикагель, 3% МеОН в CH2Cl2) с получением 199 мг указанного в заголовке соединения в виде белого твердого вещества (выход 55%). 1 Н-ЯМР (CDCl3)8,21 (1 Н, т, J=8,4 Гц), 7,58 (2 Н, дд, J=8,8 и 4,8 Гц), 7,32 (1 Н, д, J=13,2 Гц), 7,267,23 (2 Н, м), 7,19-7,12 (3 Н, м), 7,04 (2 Н, т, J=8,8 Гц), 6,68 (1 Н, с), 6,66 (1 Н, с), 5,32 (1 Н, т, J=5,2 Гц), 4,824,72 (1 Н, м), 4,64 (2 Н, с), 4,56 (1 Н, кв, J=6,4 Гц), 3,32 (1 Н, дд, J=15,6 и 8,0 Гц), 3,00 (3 Н, с), 2,96 (1 Н, дд,J=15,2 и 8,4 Гц), 1,96 (3 Н, с). МС 544 (МН+). Примеры с 9-2 по 9-6 получают, по существу, таким же способом, как пример 9-1. 4-бром К смеси тиоацетамида (10,04 г, 133,64 ммоль) и К 2 СО 3 (45,80 г, 331,38 ммоль) в 100 мл ТГФ при 0 С добавляют по каплям фталоилхлорид (28,49 г, 140,32 ммоль). Через 2 ч температуру реакции повышают до 25 С и оставляют для перемешивания еще на 2 ч, затем снова охлаждают реакционную смесь до 0 С. Реакцию гасят добавлением по каплям 125 мл ледяной воды. Реакционную смесь экстрагируют EtOAc(2x). Органический слой сушат над MgSO4 и растворитель удаляют в вакууме с получением 3,7 г Nтиоацетилизоиндол-1,3-диона в виде неочищенного красноватого твердого вещества. Растворяют N-метилфурфуриламин (117,2 мг, 1,05 ммоль) в 20 мл Et2O при 25 С. Добавляют Nтиоацетилизоиндол-1,3-дион (294,4 мг, 1,43 ммоль) и оставляют для перемешивания на 24 ч. К реакционной смеси добавляют MeOTf (181,7 мг, 1,11 ммоль) и оставляют для перемешивания еще на 23 ч. Сливают Et2O с масла и масло растирают с Et2O (эту процедуру повторяют три раза). Из маслянистого остатка удаляют в вакууме любое избыточное количество Et2O с получением 320,4 мг неочищенного тиоимидата в виде масла. Полученный неочищенный продукт (161,1 мг, 0,483 ммоль) растворяют в 10 мл пиридина и добавляют N-(6-амино-2-гидроксииндан-1-ил)-4-бромбензамид (107,5 мг, 0,310 ммоль). Реакционную смесь оставляют для перемешивания при 25 С на 22 ч. Растворитель удаляют в вакууме и остаток распределяют между CH2Cl2 и насыщенным водным раствором NаНСО 3. Органический слой сушат надMgSO4. Фильтруют и растворитель удаляют в вакууме с получением 14 6,2 мг неочищенного продукта. Проводят очистку хроматографией Biotage (1% MeOH/EtOAc) с получением 63,6 мг указанного в заголовке соединения в виде не совсем белого твердого вещества (43%). МС (m/е): 483 (М+1). Пример 11-1. Стадия а) Объединяют трет-бутиловый эфир (6-амино-2-гидроксииндан-1-ил)карбаминовой кислоты (3,7 г,14,0 ммоль) с 10 мл ТФУК при 0 С. Смесь перемешивают в течение 1 ч и выпаривают досуха. К остатку добавляют 6,5 мл триэтиламина и 30 мл метиленхлорида. Полученную смесь медленно объединяют с раствором бензотриазол-1-илового эфира 4-бромбензойной кислоты в 15 мл метиленхлорида при 0 С. Полученную смесь перемешивают в течение 12 ч при комнатной температуре. Фильтруют с получением 3,4 г (выход 70%) N-(6-амино-2-гидроксииндан-1-ил)-4-бромбензамида в виде твердого вещества. МС 348 (МН+).- 22006853 Стадия b). Добавляют ацетилхлорид (9,28 г, 118 ммоль) к смеси триэтиламина (16,0 г, 158 ммоль) и 4 фторбензиламина (9,90 г, 78,8 ммоль) в 200 мл этилацетата при 0 С и перемешивают в течение 12 ч. К полученной смеси добавляют 100 мл воды. Водный слой экстрагируют этилацетатом (3x100 мл). Органические слои объединяют и промывают насыщенным раствором соли, затем сушат над безводнымNa2SO4. Растворитель удаляют при пониженном давлении с получением N-(4-фторбензил)ацетамида(13,5 г) с выходом 100% в виде желтого масла. К N-(4-фторбензил)ацетамиду (13,5 г, 80,8 ммоль) в 200 мл тетрагидрофурана при 0 С добавляют NaH (6,5 г, 162 ммоль) и перемешивают в течение 4 ч. Затем к полученой смеси добавляют метилйодид (22,9 г, 161,6 ммоль) и перемешивают в течение 12 ч. Смесь выливают в 200 мл воды. Водный слой экстрагируют метиленхлоридом (3x200 мл). Органические слои объединяют и промывают насыщенным раствором соли, затем сушат над безводным Na2SO4. Растворитель удаляют при пониженном давлении. Остаток очищают колоночной хроматографией (силикагель,5% ацетона в гексане, 50% ацетона в гексане) с получением N-(4-фторбензил)-N-метилацетамида (9,1 г) с выходом 64% в виде желтого масла. К N-(4-фторбензил)-N-метилацетамиду (9,1 г, 50,3 ммоль) в 200 мл толуола добавляют реагент Лавессона (Lawesson) (20,3 г, 50,3 ммоль) и нагревают до 100 С в течение трех часов. Растворитель удаляют при пониженном давлении. Остаток очищают колоночной хроматографией (силикагель, 2% метиленхлорида в гексане, 100% метиленхлорид) с получением N-(4 фторбензил)-N-метилтиоацетамида (5,6 г) 56,5% в виде желтого твердого вещества: МС 198 (МН+). Стадия с). К N-(4-фторбензил)ацетамиду (0,9 г, 5,4 ммоль) в 50 мл толуола добавляют реагент Лавессона (1,8 г, 3,6 ммоль) и нагревают до 80 С в течение 12 ч. Растворитель удаляют при пониженном давлении. Остаток очищают колоночной хроматографией (силикагель, Ацетон:Гексан=20:80) с получением смеси. Промывают эфиром и нерастворимые твердые примеси отделяют фильтрованием. Растворитель удаляют при пониженном давлении с получением N-(4-фторбензил)тиоацетамида (0,7 г) с выходом 71% в виде желтого твердого вещества: МС 184 (МН+). Стадия d). К раствору N-(3-фторбензил)тиоацетамида (0,106 г, 0,580 ммоль) в 10 мл СН 2 Сl2 при комнатной температуре добавляют метилтрифторметансульфонат (0,190 г, 1,16 ммоль). Полученную смесь перемешивают в течение 30 мин и растворитель удаляют при пониженном давлении. Остаток растворяют в 5 мл пиридина. Затем к раствору добавляют соль трифторуксусной кислоты N-(6-амино-2-гидроксииндан-1 ил)-4-бромбензамида. Перемешивают в течение трех часов. Удаляют пиридин при пониженном давлении. Остаток очищают колоночной хроматографией (силикагель/гексан:ацетон=7:3, 1:1) с получением 40 мг указанного в заголовке соединения с выходом 14% в виде белого твердого вещества: МС 496 (МН+). Примеры с 11-2 по 11-17 получают, по существу, таким же способом, как пример 11-1.(R)-(6-(1-4-Фторбензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1-ил)амид бифенил-4 карбоновой кислоты Объединяют трет-бутиловый эфир (6-амино-2-гидроксииндан-1-ил)карбаминовой кислоты (1,5 г,5,68 ммоль) с 5 мл ТФУК при 0 С. Полученную смесь перемешивают в течение 1 ч и затем выпаривают- 24006853 досуха. К остатку добавляют 3,0 мл триэтиламина и 30 мл метиленхлорида. К полученной смеси добавляют раствор 2,5-диоксопирролинид-1-илового эфира бифенил-4-карбоновой кислоты (1,76 г, 5,96 ммоль) в 15 мл метиленхлорида. Полученную смесь перемешивают в течение 12 ч. Растворитель выпаривают и остаток очищают колоночной хроматографией (силикагель/ МеОН:СН 2 Сl2 9:1) с получением 1,88 г (выход 96%) (6-амино-2-гидроксииндан-1-ил)амида бифенил-4-карбоновой кислоты: МС 345(МН+). К раствору N-(4-фторбензил)тиоацетамида (0,2 г, 1,0 ммоль) в 5 мл СН 2 С 12 при комнатной температуре добавляют метилтрифторметансульфонат (0,290 г, 1,76 ммоль). Полученную смесь перемешивают в течение 30 мин и растворитель удаляют при пониженном давлении. Остаток растворяют в 5 мл пиридина. Затем к раствору добавляют (6-амино-2-гидроксииндан-1-ил)амид бифенил-4-карбоновой кислоты. Перемешивают в течение 12 ч. Удаляют пиридин при пониженном давлении. Остаток очищают колоночной хроматографией (силикагель/гексан:ацетон=7:3, 1:1) с получением 78 мг указанного в заголовке соединения с выходом 35% в виде белого твердого вещества: 1 Н ЯМР (ДМСО-d6)8,78 (1 Н, д, J=8,8 Гц),8,04 (2 Н, д, J=8,8 Гц), 7,79 (2 Н, д, J=8,4 Гц), 7,74 (2 Н, дд, J=7,4, 1,6 Гц), 7,50 (2 Н, т, J=8,0 Гц), 7,41 (1 Н, т,J=7,2 Гц), 7,28 (2 Н, дд, J=8,8, 6,6 Гц), 7,14 (2 Н, т, J=8,8 Гц), 7,04 (1 Н, т, J=7,6 Гц), 6,78 (1 Н, д, J=7,2 Гц),6,37 (1 Н, с), 5,33 (1 Н, д, J=5,6 Гц), 5,27 (1 Н, т, J=7,6 Гц), 4,59 (2 Н, с), 4,45 (1 Н, кв, J=6,0 Гц), 3,10 (1 Н, дд,J=14,8, 7,2 Гц), 2,90 (3 Н, с), 2,68 (1 Н, дд, J=14,8, 7,2 Гц), 1,88 (3 Н, с); МС 508 (МН+). Примеры с 12-2 по 12-7 получают, по существу, таким же способом, как пример 12-1.NaOH (4,8 мл, 4,8 ммоль) в смеси 50 мл толуола и 50 мл воды в течение 30 мин. Медленно добавляют бифенил-4-карбонилхлорид (570 мг, 2,64 ммоль) и перемешивают в течение четырех часов при комнатной температуре. Твердое вещество удаляют фильтрованием и промывают толуолом. Растворитель выпаривают при пониженном давлении. (2-Гидрокси-7-нитро-1,2,3,4-тетрагидронафталин-1-ил)амид бифенил-4-карбоновой кислоты получают с выходом 54% в виде белого твердого вещества: 1 Н ЯМР (ДМСОd6)8,87 (1 Н, д, J=8,4 Гц), 8,03 (2 Н, д, J=8,4 Гц), 7,99 (2 Н, д, J=7,6 Гц), 7,79 (2 Н, д, J=8,4 Гц), 7,74 (2 Н, д,J=8,4 Гц), 7,50 (2 Н, т, J=7,2 Гц), 7,43 (2 Н, т, J=6,8 Гц), 5,22 (1 Н, д, J=4,4 Гц), 5,11 (1 Н, т, J=7,6 Гц), 3,954,01 (1 Н, м), 2,87-3,07 (2 Н, м), 2,08-2,15 (1 Н, м), 1,81-1,94 (1 Н, м); МС 389 (МН+). Указанное выше соединение (0,25 г, 0,64 ммоль) восстанавливают в 10 мл ДМФА с использованием 48 мг 10% Pd-C при комнатной температуре и давлении 60 ф./кв.дюйм (414 кПа) в течение ночи с получением (7-амино-2 гидрокси-1,2,3,4-тетрагидронафталин-1-ил)амида бифенил-4-карбоновой кислоты, который получают с выходом 96% в виде желтого масла. МС 359 (МН+).- 25006853 К раствору N-4-фторбензил-N-метилтиоацетамида (0,145 г, 0,736 ммоль) в 10 мл СН 2 Сl2 при комнатной температуре добавляют метилтрифторметансульфонат (0,150 г, 0,915 ммоль). Полученную смесь перемешивают в течение 30 мин и растворитель удаляют при пониженном давлении. Затем добавляют(7-амино-2-гидрокси-1,2,3,4-тетрагидронафталин-1-ил)амид бифенил-4-карбоновой кислоты (0,22 г, 0,61 ммоль) с последующим добавлением 5 мл пиридина. Полученную смесь перемешивают в течение 12 ч. После выпаривания пиридина остаток очищают колоночной хроматографией (силикагель, 5% МеОН в СН 2 Сl2) с получением 10 мг указанного в заголовке соединения с выходом 2,6% в виде светло-желтого твердого вещества: 1 Н-ЯМР (CDCl3)7,89 (2 Н, д, J=8,0 Гц), 8,66 (2 Н, д, J=8,4 Гц), 7,61 (2 Н, д, J=7,2 Гц),7,47 (2 Н, т, J=7,6 Гц), 7,39 (1 Н, т, J=7,6 Гц), 7,21 (1 Н, дд, J=6,0, 8,8 Гц), 6,98-7,05 (3 Н, м) , 6,72 (1 Н, с),6,62 (1 Н, дд, J=2,0, 8,4 Гц), 6,56 (1 Н, д, J=7,2 Гц), 5,22 (1 Н, т, J=7,6 Гц), 4,60 (2 Н, с), 3,98-4,04 (2 Н, м), 3,41 Исходя из 3,4-дифторбензиламина, N-(3,4-дифторбензил)-N-метилтиоацетамида получают, по существу, так же, как на стадии b) примера 11-1, и получают N-(3,4-дифторбензил)-N-метилтиоацетамид с выходом 66% в виде желтого твердого вещества: МС 216 (МН+). К раствору N-(3,4-дифторбензил)-N-метилтиоацетамида (0,145 г, 0,736 ммоль) в 10 мл СН 2 Сl2 при комнатной температуре добавляют метилтрифторметансульфонат (0,750 г, 3,49 ммоль). Полученную смесь перемешивают в течение 30 мин и растворитель удаляют при пониженном давлении. Затем добавляют трет-бутиловый эфир (6-амино-2-гидроксииндан-1-ил)карбаминовой кислоты (0,78 г, 2,97 ммоль) с последующим добавлением 5 мл пиридина. Полученную смесь перемешивают в течение 12 ч. После выпаривания пиридина остаток очищают колоночной хроматографией (силикагель, 3% МеОН в CH2Cl2) с получением 0,98 г трет-бутилового эфира (6-(1-3,4-дифторбензил)метиламино)этилиденамино)-2 гидроксииндан-1-ил)карбаминовой кислоты в виде белого твердого вещества: МС 4 46 (МН+). К трет-бутиловому эфиру(R)-(6-(1-3,4-дифторбензил)метиламино)этилиденамино)-2(R)гидроксииндан-1-ил)карбаминовой кислоты (180 мг, 4,04 ммоль) добавляют 5 мл трифторуксусной кислоты и перемешивают в течение 30 мин. Растворитель удаляют при пониженном давлении. Остаток растворяют в 15 мл СН 2 Сl2. Затем к полученному раствору добавляют 2,5-диоксипирролидин-3-иловый эфир 3-фторбифенил-4-карбоновой кислоты (150 мг, 4,7 9 ммоль) и триэтиламин (0,6 мл, 4,0 ммоль) и перемешивают в течение 12 ч. Смесь выливают в метиленхлорид, промывают водой, сушат над Na2SO4 и затем концентрируют. Остаток очищают колоночной хроматографией (силикагель, 3% МеОН в СН 2 Сl2) с получением 35 мг указанного в заголовке соединения в виде белого твердого вещества с выходом 16%: 1 Н-ЯМР (CD3OD)7,88 (2 Н, т, J=8,0 Гц), 7,7 (2 Н, д, J=8,4 Гц), 7,60 (2 Н, дд, J=8,0, 1,2 Гц), 7,47-7,55 (3 Н,м) , 7,43-7,47 (1 Н, м) , 7,21-7,32 (2 Н, м), 7,18 (1 Н, д, J=8,0 Гц), 7,11-7,14 (1 Н, м), 6,71 (2 Н, дд, J=10,0, 1,2 Гц), 5,47 (1 Н, д, J=6,8 Гц), 4,69 (2 Н, с), 4,54 (1 Н, кв, J=7,2 Гц), 4,54 (1 Н, кв, J=6,4 Гц), 3,29 (1 Н, кв, J=7,2 Гц), 3,07 (3 Н, с), 2,88 (1 Н, кв, J=7,8 Гц), 1,99 (3 Н, с); МС 544 (МН+). Пример 15-1. Объединяют 1-аминоиндан (1,0 г, 8,1 ммоль) с 6,6 мл дымящей НNO3 при -10 С. Полученную смесь перемешивают в течение 30 мин, выливают на лед и затем перемешивают в течение 30 мин. Твердое вещество собирают, промывают водой и сушат с получением 0,614 г (42%) продукта. Смешивают с дитрет-бутилдикарбонатом (0,929 г, 4,26 ммоль) в ТГФ. Полученную смесь объединяют с насыщенным водным раствором К 2 СО 3 для доведения уровня рН до 11. Перемешивают в течение 8 ч, выливают вCH2Cl2, промывают водой, сушат и концентрируют. Остаток растворяют в 1:1 смеси эфира и гексана,затем выдерживают при -10 С в течение ночи. Твердое вещество собирают и сушат с получением 0,57 гCuCl (1,25 г, 12,6 ммоль) в 50 мл метанола при 0 С в течение 15 мин медленно добавляют КВН 4 (1,59 г,29,4 ммоль). Полученную смесь перемешивают в течение 1 ч и затем пропускают через слой Florisil. Выпаривают ТГФ и твердое вещество растворяют в EtOAc. Смесь промывают водой, сушат и концентрируют с получением 0,76 г (73%) трет-бутилового эфира (6-аминоиндан-1-ил)карбаминовой кислоты. 1HЯМР (CDCl3) d 6,99 (1 Н, д, J=8,0 Гц), 6,65 (1 Н, с), 6,56 (1 Н, дд, J=8,0 и 2,0 Гц), 5,09 (1 Н, кв, J=8,0 Гц),4,70 (1 Н, шир.д, J=7,2 Гц), 3,60 (2 Н, с), 2,86-2,79 (1 Н, м) , 2,75-2,67 (1 Н, м), 2,58-2,47 (1 Н, м), 1,78-1,69(1 Н, м), 1,48 (3 н, с), 2,86-2,79 (1 Н, м), 2,75-2,67 (1 Н, м), 2,58-2,47 (1 Н, м), 1,78-1,69 (1 Н, м), 1,48 (3 Н, с). К раствору N-4-фторбензил-N-метилтиоацетамида (0,662 г, 3,4 ммоль) в 10 мл эфира при комнатной температуре добавляют MeI (0,623 г, 4,4 ммоль). Полученную смесь перемешивают в течение 3 ч и эфир выпаривают в вакууме. Добавляют трет-бутил эфир (6-аминоиндан-1-ил)карбаминовой кислоты (0,7 г,2,28 ммоль) с последующим добавлением 5 мл пиридина. Полученную смесь перемешивают в течение 12 час. Выпаривают пиридин и остаток очищают колоночной хроматографией (силикагель, 3% МеОН в(1 Н, т, J=6,8 Гц), 4,69 (2 Н, с), 3,05 (3 Н, с), 2,97-2,91 (1 Н, м), 2,84-2,77 (1 Н, м), 2,55-2,48 (1 Н, м), 1,97 (3 Н,с), 1,52 (9 Н, с). Перемешивают смесь 2'-трифторметилбифенил-4-карбоновой кислоты (100 мг, 0,376 ммоль), Nгидроксисукцинимида (43 мг, 0,376 ммоль) и DCC (77 мг, 0,376 ммоль) в 15 мл метиленхлорида при комнатной температуре в течение 2 ч. Объединяют трет-бутиловый эфир (6-(1-(4 фторбензил)метиламино)этилиденамино)индан-1-ил)карбаминовой кислоты (129 мг, 0,313 ммоль) с ТФУК (2 мл) при 0 С и перемешивают в течение 2 ч. Выпаривают ТФУК при пониженном давлении. Остаток растворяют в метиленхлориде и упаривают досуха; эту процедуру повторяют три раза. Добавляют 5 мл метиленхлорида и 1 мл триэтиламина. Полученный раствор добавляют к полученной выше смеси и перемешивают при комнатной температуре в течение 12 ч. Смесь выливают в метиленхлорид,промывают водой, сушат над Nа 2SО 4 и концентрируют. Остаток очищают колоночной хроматографией(силикагель, 3% МеОН в СН 2 Сl2) с получением 82 мг указанного в заголовке соединения в виде белого твердого вещества (выход 47%). МС 560 (МН+); 1 Н-ЯМР (CDCl3) d 7,83 (2 Н, д, J=8,0 Гц), 7,75 (1 Н, д,J=7,6 Гц), 7,56 (1 Н, т, J=7,2 Гц), 7,49 (1 Н, т, J=8,0 Гц), 7,40 (2 Н, д, J=8,0 Гц), 7,31 (1 Н, д, J=8,0 Гц), 7,267,21 (3 Н, м), 7,15 (1 Н, д, J=7,6 Гц), 7,02 (2 Н, т, J=8,8 Гц), 6,75 (1 Н, с), 6,64 (1 Н, Д, J=7,2 Гц), 5,67 (1 Н, кв,J=7,6 Гц), 4,61 (2 Н, кв, J=10,0 Гц), 2,97 (3 Н, с), 2,94-2,83 (1 Н, м), 2,75-2,68 (1 Н, м), 2,00-1,70 (4 Н, шир.с),1, 69-1,58 (1 Н, м). Примеры с 15-2 по 15-7 получают, по существу, таким же способом, как пример 15-1.- 27006853 Пример Р-1. Полугидрат (R)-(6-(l-4-фторбензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1-ил)амида бифенил-4-карбоновой кислоты К 2,86 кг сольвата (R)-(6-(1-4-фторбензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1 ил)амида бифенил-4-карбоновой кислоты добавляют 21,8 л метанола. Полученный раствор пропускают через углеродный фильтр и фильтрат промывают 24 л метанола. К раствору в течение 35 мин добавляют 5,7 кг воды с последующим добавлением 15 г затравочных кристаллов полугидрата (R)-(6-(l-4 фторбензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1-ил)амида бифенил-4-карбоновой кислоты. Через 20 мин добавляют 1,15 кг воды с последующим добавлением 15 г затравочных кристаллов. Через 1 ч добавляют еще 1,15 кг воды в течение 30 мин с последующим добавлением 15 г затравочных кристаллов. Через 10 мин добавляют 3,4 кг воды в течение 1 ч и суспензию перемешивают при комнатной температуре в течение 1 ч и при 0 С в течение 45 мин. Твердое вещество собирают фильтрованием,промывают холодным раствором 11,4 л метанола и 2,9 л воды и сушат с получением 2,19 кг указанного в заголовке соединения в виде белого твердого вещества. Пример Р-2. Полугидрат (R)-(6-(l-4-фторбензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1-ил)амида бифенил-4-карбоновой кислоты Сольват (R)-(6-(1-4-фторбензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1-ил)амида бифенил-4-карбоновой кислоты (2,0 г) растворяют в метаноле (24 мл) при 20-23 С. К полученному раствору добавляют воду (5 мл) с последующим добавлением затравочных кристаллов (20 мг). Смесь перемешивают в течение 2 ч при 20-23 С, затем охлаждают до 0-5 С. Смесь фильтруют, промывают раствором метанола (8 мл) и воды (2 мл) и сушат при 50-60 С в вакууме в течение 16 ч с получением 1,66 г указанного в заголовке соединения. Пример Р-3. Полугидрат (R)-(6-(l-4-фторбензил)метиламино)этилиденамино)-2(R)-гидроксииндан-1-ил)амида бифенил-4-карбоновой кислоты Объединяют раствор сольвата 6-(1-4-фторбензил)метиламино)этилиденамино)-2-гидрокси-1 бифениламиноинданацетонитрила (101 г) и метанола (1,2 л) с Darco G-60 (5 г). После перемешивания в течение 15-30 мин при 15-25 С смесь фильтруют и отфильтрованное твердое вещество промывают метанолом (0,4 л). К объединенному фильтату добавляют воду (0,4 л), промывают и добавляют затравочные кристаллы полугидрата (1,5 г). Смесь перемешивают 2-3 ч при 15-25 С, затем охлаждают до 0-5 С и перемешивают еще 90 мин. Смесь фильтруют, промывают 0-5 С раствором метанола (0,8 л) и воды (0,2 л) и сушат при 47-53 С в вакууме в течение 20 ч с получением 88,7 г указанного в заголовке соединения. Соединения по настоящему изобретению можно вводить отдельно или в форме фармацевтической композиции, т.е. в сочетании с фармацевтически приемлемыми носителями или эксципиентами, содержание и природа которых определяются растворимостью и химическими свойствами выбранного соединения, выбранным путем введения и стандартной фармацевтической практикой. Соединения по настоящему изобретению, хотя они эффективны сами по себе, можно готовить и вводить в форме их фармацевтически приемлемых солей, в целях стабильности, удобства, растворимости и т.п. На практике соединения формулы I обычно вводят в форме фармацевтических композиций, т.е. в смеси с фармацевтически приемлемыми носителями или разбавителями. Таким образом, настоящее изобретение предлагает фармацевтические композиции, включающие соединение формулы I и фармацевтически приемлемый разбавитель. Настоящее изобретение также предлагает подходящую упаковку, включая этикетку, содержащую фармацевтические композиции,включающие соединение формулы I. Соединения формулы I можно вводить различными путями. При осуществлении лечения пациента,страдающего заболеванием, указанным в данном описании, соединение формулы I можно вводить в любой форме или любым путем, которые делают такое соединение биодоступным в эффективном количестве, включая пероральный и парентеральный пути. Например, соединения формулы I можно вводить перорально, путем ингаляции, подкожно, внутримышечно, внутривенно, чрезкожно, интраназальным,ректальным, внутриглазным путем, путем местого применения, сублингвальным, буккальным путем и т.п. Для лечения заболеваний, указанных в данном описании, пероральное введение обычно является предпочтительным. Специалист в области составления фармацевтических препаратов легко выберет подходящую форму и путь введения в зависимости от конкретных характеристик выбранного соединения, подлежащего лечению состояния или расстройства, стадии расстройства или состояния и других соответствующих обстоятельств. (Remington's Pharmaceutical Sciences, 18th Edition, Mack Publishing Co. (1990. Фармацевтические композиции получают способом, хорошо известным в области фармацевтики. Носитель или эксципиент могут быть твердыми, полутвердыми или жидкими веществами, которые могут служить в качестве носителя или среды для активного ингредиента. Подходящие носители или эксципиенты хорошо известны из уровня техники. Фармацевтические композиции могут быть приспособлены для перорального применения, применения путем ингаляции, парентерального или местного при- 28006853 менения, и их можно вводить пациенту в форме таблеток, капсул, аэрозолей, препаратов для ингаляции,суппозиториев, растворов, суспензий и т.п. Соединения по настоящему изобретению можно вводить перорально, например, с инертным разбавителем, или в виде капсул или спрессованными в таблетки. Для перорального терапевтического применения соединения могут быть включены вместе с эксципиентами в форму таблеток, драже, капсул, эликсиров, суспензий, сиропов, облаток, жевательных резинок и т.п. Такие препараты должны содержать по меньшей мере 4% соединения по настоящему изобретению в качестве активного ингредиента, но это количество может меняться в зависимости от конкретной формы, и удобно, когда оно составляет от 4% до около 70% от массы единицы лекарственной формы. Количество соединения, присутствующего в композиции, должно быть таким, чтобы можно было получить подходящую дозу. Предпочтительные композиции и препараты по настоящему изобретению могут быть определены специалистом в данной области. Таблетки, пилюли, капсулы, драже и т.п. могут также содержать один или несколько из следующих адъювантов: связующие, такие как микрокристаллическая целлюлоза, смола трагаканта или желатин; эксципиенты, такие как лактоза, разрыхлители, такие как альгиновая кислота, Primogel, кукурузный крахмал и т.п.; смазывающие вещества, такие как стеарат магния или Sterotex; агенты скольжения, такие как коллоидный диоксид кремния; и могут быть добавлены подсластители, такие как сахароза или сахарин, или отдушки, такие как перечная мята, метилсалицилат или апельсиновая отдушка. Когда лекарственная форма представляет собой капсулу, она может содержать, в дополнение к указанным выше веществам, жидкий носитель, такой как полиэтиленгликоль или жирное масло. Другие лекарственные формы могут содержать другие различные вещества, модифицирующие физическую форму лекарственной формы, например покрытия. Так, таблетки или пилюли могут быть покрыты сахаром, шеллаком или другими веществами для покрытия. Сироп может содержать, в дополнение к соединениям по настоящему изобретению, сахарозу, в качестве подсластителя, и некоторые консерванты, красители и отдушки. Вещества,используемые для получения таких различных композиций, должны быть фармацевтически чистыми и нетоксичными в используемых количествах. Для перорального и парентерального терапевтического применения соединения по настоящему изобретению могут быть включены в растворы или суспензии. Такие препараты обычно содержат по меньшей мере 0,1% соединения по настоящему изобретению, но этот количество может варьировать от 0,1 до 90% в расчете на массу препарата. Количество присутствующего в таких композициях соединения формулы I такое, чтобы можно было получить подходящую дозу. Растворы или суспензии также могут включать один или несколько из следующих адъювантов: стерильные разбавители, такие как вода для инъекций, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатообразующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты регулирования тоничности, такие как хлорид натрия или декстроза. Препараты для парнетерального введения могут быть заключены в ампулу из стекла или пластика. Предпочтительные композиции и препараты могут быть определены специалистом в данной области. Композиции по настоящему изобретению можно также вводить местным путем, при этом носитель в подходящем случае включает раствор, мазь или гелевую основу. Основа может, например, включать одно или несколько из следующих веществ: вазелин, ланолин, полиэтиленгликоли, пчелиный воск, минеральное масло, разбавители, такие как вода и спирты, и эмульгаторы и стабилизаторы. Препараты для местного применения могут содержать соединение формулы I или его фармацевтически приемлемую соль в концентрации от около 0,1 до около 10% мас./об. (масса на единицу объема). Соединения формулы I являются агонистами мускариновых рецепторов M-1. Более того, соединения формулы I являются селективными агонистами такого конкретного мускаринового рецептора. Соединения по настоящему изобретению обладают особенно полезными свойствами, связанными с их биодоступностью, фармакокинетикой, безопасностью и эффективностью. Мускариновые агонисты, включая их профиль связывания с подтипами, могут быть идентифицированы методами, известными из уровня техники. В одном своем воплощении настоящее изобретение обеспечивает способы лечения расстройств,связанных с мускариновыми рецепторами, которые включают введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы I. Так настоящее изобретение рассматривает различные расстройства, описанные как подлежащие лечению в данной заявке, и другие, которые можно лечить такими агонистами, как признано специалистами в данной области. Различные расстройства, которые можно лечить мускариновыми агонистами, являются известными в соответствии с установленной и принятой классификацией, тогда как другие неизвестны. Например,познавательная способность представляет собой сложный и иногда трудно определяемый феномен. Однако признано, что познавательная способность включает различные "области". Эти области включают краткосрочную память, долгосрочную память, рабочую память, исполнительную функцию и внимание. Понятно, что соединения по настоящему изобретению являются полезными для лечения рас- 29006853 стройств, характеризующихся дефицитом в любой из познавательных (когнитивных) областей, перечисленных выше, или в других аспектах познавательной способности. Таким образом, термин "когнитивные расстройства" включает любое расстройство, характеризующееся дефицитом в одной или нескольких когнитивных областях, включая, но не ограничиваясь этим, краткосрочную память, долгосрочную память, рабочую память, исполнительную функцию и внимание. Одно когнитивное расстройство, подлежащее лечению в соответствии с настоящим изобретением,представляет собой возрастное ухудшение познавательной способности. Это расстройство недостаточно охарактеризовано в существующем уровне техники, но оно включает ухудшение в когнитивных областях, особенно в областях памяти и внимания, что сопутствует старению. Другим когнитивным расстройством является незначительное ухудшение познавательной способности. Это расстройство также недостаточно охарактеризовано в существующем уровне техники, но оно включает ухудшение в когнитивных областях, и полагают, что оно представляет группу пациентов, большинство из которых имеют начальную стадию болезни Альцгеймера. Другим когнитивным расстройством является ухудшение познавательной способности, связанное с шизофренией. Взаимосвязь между когнитивными нарушениями и другими симптомами шизофрении еще не совсем изучена. Согласно наблюдениям, у некоторых людей имеются проблемы познавательной способности задолго до проявления определенных симптомов заболевания, тогда как у других ухудшение познавательной способности проявляется после первого эпизода и с последующими приступами. Еще одно когнитивное расстройство представляет собой ухудшение познавательной способности, индуцированное химиотерапией. Люди, прошедшие курс химиотерапии от рака могут испытывать ухудшение познавательной функции, и такое ухудшение может быть длительным. Также на ухудшение познавательной способности могут влиять различные инсульты, включая удар,ишемию, гипоксию, воспаление, инфекционные процессы, и недостаточная познавательная способность может быть результатом хирургических операций на сердце и по введению трансплантата, удара, церебральной ишемии, травмы спинного мозга, травмы головы, перинатальной гипоксии, фетального алкогольного синдрома, остановки сердца и гипогликемического повреждения нервных клеток, сосудистой деменции, мульти-инфарктой деменции, бокового амилотрофического склероза, химиотерапии и рассеянного склероза, что приводит к таким последствиям как дефицит познавательной способности, который можно лечить в соответствии с настоящим изобретением. Когда расстройства, которые можно лечить мускариновыми агонистами, являются известными в соответствии с установленной и принятой классификацией, такие классификации можно найти в различных источниках. Например, в настоящее время четвертое издание Diagnostic and Statistical Manual ofMental Disorders (DSM-IV) (1994, American Psychiatric Association, Washington, D.C.) обеспечивает диагностический инструмент для идентификации многих расстройств, описанных в этом справочнике. Также International Classification of Diseases, tenth Revision (ICD-10) обеспечивает классификацию для многих расстройств, описанных в этом справочнике. Специалисту в данной области должно быть понятно,что существуют альтернативные номенклатуры, нозологии и системы классификации для описанных в данной заявке расстройств, включая те, которые описаны в DSM-IV и ICD-10, и что терминология и системы классификации меняются с развитием медицинской науки. В особенно предпочтительных воплощениях настоящее изобретение обеспечивает способы лечения расстройств, выбранных из группы, включающей когнитивные расстройства (включая возрастные когнитивные расстройства, легкое ухудшение познавательной функции, ухудшение познавательной функции,связанное с шизофренией, и ухудшение познавательной функции, вызванное химиотерапией), ADHD,расстройства настроения (включая депрессию, мании, биполярные расстройства), психоз (в частности,шизофрению и расстройства типа шизофрении), деменцию (включая болезнь Альцгеймера, деменцию,вызванную СПИДом, сосудистую деменцию и деменцию с отсутствием четкой гистологии), болезнь Паркинсона, хорею Гентингтона, боль (включая острую боль и хроническую боль), ксеростомию (сухость во рту), заболевание телец Леви (включая заболевание диффузных телец Леви), афазию, т.е. нарушение речи (включая первичную афазию и синдром первичной афазии), гипотензивные синдромы и хронический колит (включая болезнь Крона), которые включают введение нуждающемуся в этом пациенту эффективного количества соединения формулы I. То есть настоящее изобретение обеспечивает для применения соединение формулы I или фармацевтическую композицию, содержащую такое соединение, для лечения расстройств, связанных с мускариновыми рецепторами. Должно быть понятно, что термины "лечение" и "лечить" включают улучшение симптоматологии,связанной с каждым из расстройств, связанных с мускариновыми рецепторами, которые описаны в данной заявке. Также должно быть понятно, что специалист может воздействовать на расстройства посредством лечения пациента, у которого наблюдается такое расстройство, или посредством профилактического лечения пациента, который, как считают, подвержен такому расстройству, эффективным количеством соединения формулы I. Таким образом, термины "лечение" и "лечить" относятся ко всем процессам,где возможно замедление, прерывание, приостановка, регулирование или остановка развития расстройств, описанных в данной заявке, но необязательно означают полное устранение всех симптомов, и,как подразумевается, также включают в себя профилактическое лечение таких расстройств. Должно быть понятно, что настоящее изобретение включает сопутствующее лечение расстройств,- 30
МПК / Метки
МПК: C07C 251/00
Метки: агонисты, мускариновые
Код ссылки
<a href="https://eas.patents.su/30-6853-muskarinovye-agonisty.html" rel="bookmark" title="База патентов Евразийского Союза">Мускариновые агонисты</a>
Селективные β3-адренергические агонисты.

Номер патента: 1217
Опубликовано: 25.12.2000
Авторы: Рито Кристофер Дж., Макдональд Джон Х., Винтер Марк А., Белл Майкл Г., Эврард Дебора А., Кровелл Томас А., Мэттьюз Дональд П., Шакер Энтони Дж.
МПК: A61K 31/41, C07D 257/04
Метки: агонисты, beta;3-адренергические, селективные
Формула / Реферат:
1. Соединение формулы где R1 представляет ОН, галоген, SO2NHR2, CO2R2, CONHR2, NHCOR2, -NH (необязательно замещенный арил), СF3 или CF2H, причем термин "арил" обозначает необязательно замещенный фенил или нафтил; R1' представляет Н, галоген, C1-C4 алкил, ОН, SO2NHR2, СO2R2, CONHR2, NHCOR2, СF3 или CF2H; R2 представляет Н, C1-C4 алкил или арил, где арил определен как указано выше; R3 представляет Н или C1-C4 алкил; R4 представляет остаток,...
Селективные бетта 3-адренергические агонисты

Номер патента: 2778
Опубликовано: 29.08.2002
Авторы: Кроуелл Томас А., Шукер Энтони Дж., Нил Дэвид А., Мэттьюс Дональд П., Макдональд Джон Х.III, Винтер Марк А., Белл Майкл Г., Рито Кристофер Дж., Джесудейсон Синтия Д., Дрост Кристин А.
МПК: C07D 209/04, A61K 31/416
Метки: бетта, 3-адренергические, селективные, агонисты
Формула / Реферат:
1. Соединение формулы I где Х1 представляет -О-, -ОСН2-; R2 представляет Н, (C1-C4)алкил или фенил; R3 представляет водород; R4 является необязательно замещенным гетероциклом или группой, выбранной из группы, состоящей из X2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода; R5 представляет Н или (C1-C4)алкил; R6 представляет Н или (C1-C4)алкил; или R5 и R6 вместе с атомом углерода, к которому они присоединены,...
Агонисты и антагонисты рецепторов 5-ht2c

Номер патента: 5820
Опубликовано: 30.06.2005
Авторы: Нильссон Бьерн, Рингберг Эрик, Пелькман Беньямин, Йенссон Маттиас, Тор Маркус, Тейбрант Ян, Нильссон Йонас
МПК: C07D 241/18, A61P 25/00, A61K 31/497...
Метки: рецепторов, антагонисты, агонисты, 5-ht2c
Формула / Реферат:
1. Соединение общей формулы (Ib) где X1 представляет -O-, -S- или -N(R27)-; Y1 представляет -O-, -S-, -N(R27)- или -CH2-; Ra представляет C3-8-циклоалкил, арил или гетероарил, каждый из которых может быть, необязательно, замещен в одном или в нескольких положениях независимо друг от друга C1-6-алкилом, C1-6-алкокси, фтор-C1-6-алкокси, 2,2,2-трифторэтокси, C3-5-алкинилокси, C3-5-алкенилокси, диметиламино-C1-6-алкокси, метиламино-C1-6-алкокси,...
Агонисты в2-адренергического рецептора

Номер патента: 3453
Опубликовано: 26.06.2003
Авторы: Чои Сеок-Ки, Моран Эдмунд Дж.
МПК: A61K 39/395, A61K 39/00, A61K 38/00...
Метки: в2-адренергического, агонисты, рецептора
Формула / Реферат:
1. Соединение формулы (II) где Ar1 и Ar3 независимо выбирают из группы, включающей арил, гетероарил и гетероциклил; Ar2 выбирают из группы, включающей фенилен, где группы W и X присоединены в 1,2-, 1,3 и 1,4 положениях фениленового кольца; циклогексилен, необязательно замещенный метилом, где группы W и X присоединены в 1,3 и 1,4 положениях циклогексиленового кольца, и пиперазинилен, где группы W и X присоединены в 1,4 положениях...
Агонисты β2-адренергического рецептора

Номер патента: 4263
Опубликовано: 26.02.2004
Авторы: Моран Эдмунд Дж., Чои Сеок-Ки
МПК: A61P 11/00, A61K 31/165, A61K 31/135...
Метки: рецептора, агонисты, beta;2-адренергического
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль. 2. Соединение формулы где стереохимия при *C и **C представляет (RS) и (RS); (R) и (R); (R) и (S); (S) и (R) или (S) и (S); или его фармацевтически приемлемая соль. 3. Соединение по п.2, где стереохимия при *C представляет (R) и стереохимия при **C представляет (R). 4. Соединение по п.2, где стереохимия при *C представляет (R) и стереохимия при **C представляет (S). 5....
Предыдущий патент: Бис(n,n’-бис(2-галоэтил)амино)фосфороамидаты в качестве противоопухолевых агентов
Следующий патент: Производные аминохинолина и его применение в качестве лигандов аденозина а3
Случайный патент: Способы для интрадермальной, трансдермальной или трансмукозальной доставки биологически активных веществ