Селективные бетта 3-адренергические агонисты
Номер патента: 2778
Опубликовано: 29.08.2002
Авторы: Дрост Кристин А., Нил Дэвид А., Рито Кристофер Дж., Винтер Марк А., Кроуелл Томас А., Белл Майкл Г., Макдональд Джон Х.III, Мэттьюс Дональд П., Джесудейсон Синтия Д., Шукер Энтони Дж.







Формула / Реферат
1. Соединение формулы I

где Х1 представляет -О-, -ОСН2-;
R2 представляет Н, (C1-C4)алкил или фенил;
R3 представляет водород;
R4 является необязательно замещенным гетероциклом или группой, выбранной из группы, состоящей из

X2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода;
R5 представляет Н или (C1-C4)алкил;
R6 представляет Н или (C1-C4)алкил;
или R5 и R6 вместе с атомом углерода, к которому они присоединены, образуют (С3-С6)циклоалкил;
или R6 вместе с Х2 и с атомом углерода, к которому присоединен Х2, образуют (С3-С8)циклоалкил;
или R6 вместе с X2, с атомом углерода, к которому присоединены оба Х2 и R6 и R4 образуют

при условии, что R5 представляет Н;
R7 представляет водород;
R8 представляет независимо Н, галоген или (C1-C4)алкил;
R9 представляет галоген, CN, OR10, (C1-C4)алкил, (C1-C4)галоидалкил, CO2R2, CONR11R12, CONH((C1-C4)алкил), СОNН(С1-С4алкокси), SR2, CSNR2, CSNR11R12, SO2R2, SO2NR11R12, SOR2, NR11R12, фенил, необязательно замещенный группой -CONR11R12, насыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота или (C2-C4)алкенил, замещенный CN, СО2R2 или CONR11R12;
R10 представляет (C1-C4)алкил, (С1-С4)галогеналкил, (СН2)nС3-С8)циклоалкил, (СН2)nфенил, необязательно замещенный CN, CONR11R12 или СОNН(С1-С4алкокси), (СН2)n-насыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота, необязательно замещенный одним или двумя заместителями, выбранными из CN, CONR11R12 или (C1-C4) алкокси;
R11 и R12 независимо представляют Н, (C1-C4)алкил или вместе с атомом азота, к которому они присоединены, образуют морфолинил, пиперидинил, пирролидинил или пиперазинил;
A1 и A2 независимо представляют О, S, NH, CH2, NCH3 или NСН2СН3;
m равен 0 или 1;
n равен 0, 1, 2 или 3;
или его фармацевтически приемлемая соль, при условии, что когда R4 представляет

то ни R5, ни R6 не являются водородом.
2. Соединение по п.1, где R10 представляет фенил или пиридил; причем указанный фенил или пиридил замещены -CONR11R12.
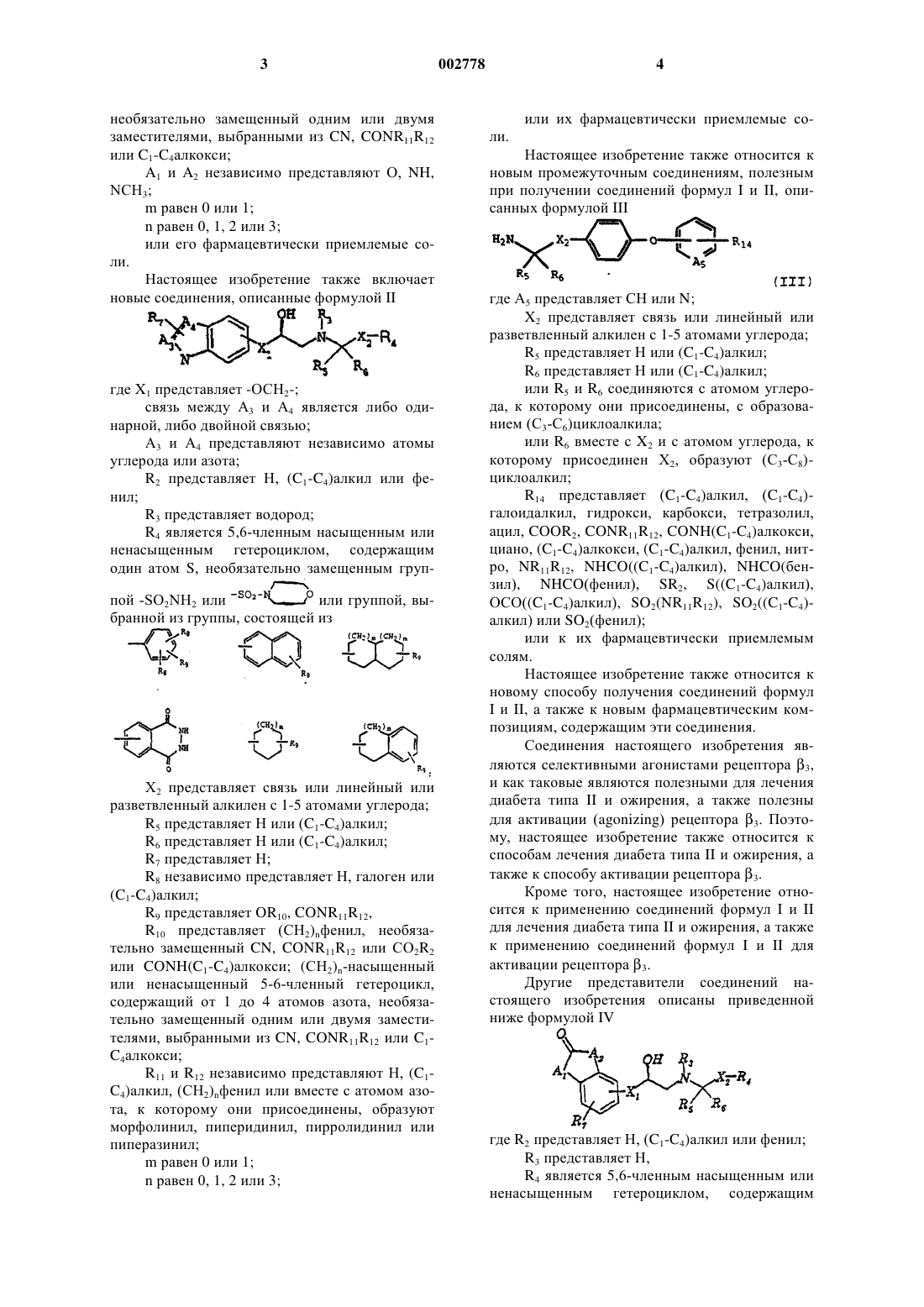
3. Соединение формулы II

где Х1 представляет -О-, -ОСН2-;
связь между А3 и А4 является либо одинарной, либо двойной связью;
А3 и А4 представляют независимо атомы углерода или азота;
R2 представляет Н, (С1-С4)алкил или фенил;
R3 представляет водород;
R4 является необязательно замещенным гетероциклом или группой, выбранной из группы, состоящей из

Х2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода;
R5 представляет С1-C4алкил;
R6 представляет С1-С4алкил;
R7 представляет водород;
R8 представляет независимо Н, галоген или (C1-C4)алкил;
R9 представляет OR10 или CONR11R12;
R10 представляет (СН2)nфенил, необязательно замещенный CN, CONR11R12, CO2R2 или CONH(С1-С4алкокси), (СН2)n-насыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота, необязательно замещенный одним или двумя заместителями, выбранными из CN, CONR11R12 или (C1-C4)алкокси;
R11 и R12 независимо представляют Н, (C1-C4)алкил, (СН2)nфенил или вместе с атомом азота, к которому они присоединены, образуют морфолинил, пиперидинил, пирролидинил или пиперазинил;
m равен 0 или 1;
n равен 0, 1, 2 или 3;
или его фармацевтически приемлемая соль.
4. Соединение по п.3 формулы

где A5 представляет СН или N;
R7 представляет Н;
X1 представляет -ОСН2-; и
Х2 представляет метилен или этилен.
5. Соединение по п.3, выбранное из группы, состоящей из

или их фармацевтически приемлемых солей; или

или

6. Соединение по п.3, где А4 представляет N и А3 представляет СН или N.
7. Соединение по п.6 формулы

где A5 представляет СН или N.
8. Соединение по п.7, где R7 представляет Н, X1 представляет -OCH2-, Х2 представляет метилен или этилен и R10 представляет фенил или пиридил, причем указанные фенил или пиридил замещены CONR11R12, СО2R2, CN.
9. Соединение по п.8, выбранное из группы, состоящей из


или их фармацевтически приемлемых солей.
10. Применение соединения по любому из пп.1-9 для получения фармацевтического средства.
11. Применение соединения по любому из пп.1-9 для лечения диабета типа II.
12. Применение соединения по любому из пп.1-9 для лечения ожирения.
13. Применение соединения по любому из пп.1-9 для активации рецептора b 3.
14. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение по любому из пп.1-9, в сочетании с одним или несколькими фармацевтически приемлемыми носителями, наполнителями или разбавителями.
15. Соединение формулы III

где A5 представляет СН или N;
Х2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода;
R5 представляет Н или (C1-C4)алкил;
R6 представляет Н или (C1-C4)алкил;
или R5 и R6 вместе с атомом углерода, к которому они присоединеэы, образуют (С3-С6)циклоалкил;
или R6 вместе с Х2 и с атомом углерода, к которому присоединен Х2, образуют (С3-С8)циклоалкил;
R14 представляет (C1-C4)алкил, (C1-C4)галоидалкил, гидрокси, карбокси, тетразолил, ацил, COOR2, CONR11R12, CONH((C1-C4)алкокси), циано, (C1-C4)алкокси, (C1-C4)алкил, фенил, нитро, NR11R12, NНСО((C1-C4)алкил), NНСО(бензил), NHCO(фенил), SR2, S((C1-C4)алкил), ОСО((C1-C4)алкил), SO2(NR11R12), SO2((C1-C4)алкил) или SO2(фенил);
или его фармацевтически приемлемые соли.
16. Способ получения соединения по п.1 формулы IA

где А5 представляет СН или N;
включающий на стадии 1 гидролиз соединения формулы IB

и на стадии 2 взаимодействие продукта со стадии 1 с образованием соли присоединения кислоты.
17. Способ получения соединения по п.1, включающий на стадии 1 взаимодействие эпоксида формулы (XI)

с амином формулы (В)

и на стадии 2 взаимодействие продукта со стадии 1 с образованием соли присоединения кислоты.
18. Соединение формулы I

в котором
X1 представляет -ОСН2-, -SCH2- или связь;
R2 и R3 независимо представляют Н, С1-C4алкил или арил;
R4 представляет необязательно замещенный гетероцикл или группу, выбранную из

X2 является связью или представляет прямой или разветвленный алкилен, содержащий от 1 до 5 атомов углерода;
R5 представляет С1-С4алкил;
R6 представляет С1-C4алкил;
R7 представляет Н, галоген, гидрокси, С1-С4алкил, C1-С4галогеналкил, арил, CN, COOR2, CONHR2, NHCOR2, OR2, NHR2, SR2, SO2R2, SO2NHR2 или SOR2;
R8 представляет Н, галоген или С1-С4алкил;
R9, представляет CN, ОR10, CONR11R12;
R10 представляет (СН2)nфенил, необязательно замещенный CN, CONR11R12, CO2R2 или CONH(С1-C4алкокси), (СН2)n-насыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота, необязательно замещенный одним или двумя заместителями, выбранными из CN, CONR11R12 или (C1-С4)алкокси;
R11 и R12 независимо представляют Н, С1-C4алкил, или вместе с атомом азота, к которому они присоединены, образуют морфолинил, пиперидинил, пирролидинил или пиперазинил;
A1 и A2 независимо представляют О, S, NH, СН2, NCH3 или NCH2CH3;
m равно 0 или 1;
n равно 0, 1, 2 или 3;
или его фармацевтически приемлемая соль.
19. Соединение по п.18, где
R5 и R6 представляют метил;
R7 представляет Н, галоген, гидрокси, (С1-С4)алкил, (C1-C4)алкокси, NH2, SR2, SO2R2 или SOR2;
A1 и A2 представляют NH; и R4 представляет

Текст
1 Область техники, к которой относится изобретение Настоящее изобретение относится к области медицины, в частности, к лечению диабета типа II и ожирения. Конкретнее, настоящее изобретение относится к селективным агонистам адренергического рецептора 3, полезным при лечении диабета типа II и ожирения. Предпосылки создания изобретения В настоящее время предпочтительным лечением инсулиннезависимого диабета типа II, а также ожирения является диета и физические упражнения, для того чтобы снизить вес и повысить чувствительность к инсулину. Однако пациенты обычно плохо соблюдают режим. Проблема осложняется тем фактом, что в настоящее время не существует одобренных лекарственных средств для адекватного лечения диабета II или ожирения. Данное изобретение относится к эффективному и своевременному лечению этих серьезных заболеваний. Одной из терапевтических возможностей,которая выяснена недавно, является взаимосвязь между стимуляцией адренергических рецепторов и антигипергликемическим действием. Показано, что соединения, которые действуют как агонисты 3-рецептора, обнаруживают заметное действие на липолиз, термогенез и уровень глюкозы в сыворотке на животных моделях диабета типа II (инсулиннезависимого диабета). Рецептор 3, который обнаружен в различных типах человеческой ткани, в том числе в жировой ткани человека, обладает приблизительно 50% гомологией и субтипами рецепторов 1 и 2 и к тому же значительно меньше распространен. Важность рецептора 3 раскрыта сравнительно недавно, так как аминокислотная последовательность человеческого рецептора была выяснена только в конце 1980-х годов. В последнее время появилось множество публикаций, сообщающих об успехах в поисках агентов,которые стимулируют рецептор. Несмотря на эти последние разработки, остается необходимость в разработке селективного агониста рецептора 3, который обладает минимальной агонистической активностью в отношении рецепторов 1 и 2. В патенте США 5013761,Beedle et al., описаны индолилпропаноламины. Настоящее изобретение относится к соединениям, которые являются селективными агонистами рецептора 3. Как таковые, эти соединения приводят к эффективному увеличению чувствительности к инсулину при лечении диабета типа II и других недомоганий, связанных с рецептором 3, побочных действий, связанных с сердечной деятельностью и тремором. Краткое изложение сущности изобретения Настоящее изобретение включает новые соединения, описанные формулой IR4 является 5,6-членным насыщенным или ненасыщенным гетероциклом, включающим один атом S, необязательно замещенным группой -SO2NH2 или или группой, выбранной из группы, состоящей из Х 2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода;R9 представляет галоген, CN, OR10, (C1-C4)алкил, (C1-C4)галоидалкил, CO2R2, CONR11R12,CONH(C1-C4)алкил или CONH(C1-C4)алкокси,SR2, CSNR2, CSNR11R12, SO2R2, SO2NR11R12,SOR2, NR11R12, фенил, необязательно замещенный группой -CONR11R12, насыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота или (C2-C4)алкенил,замещенныеR10 представляет (C1-C4)алкил, (C1-C4)галоидалкил,(CH2)n(C3-C8)циклоалкил,(СН 2)nфенил, необязательно замещенный CN,CONR11R12 или CONH(C1-C4)алкокси; (СН 2)nнасыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота, 3 необязательно замещенный одним или двумя заместителями, выбранными из CN, CONR11R12 или C1-C4 алкокси;n равен 0, 1, 2 или 3; или его фармацевтически приемлемые соли. Настоящее изобретение также включает новые соединения, описанные формулой II где X1 представляет -ОСН 2-; связь между А 3 и А 4 является либо одинарной, либо двойной связью; А 3 и А 4 представляют независимо атомы углерода или азота;R4 является 5,6-членным насыщенным или ненасыщенным гетероциклом, содержащим один атом S, необязательно замещенным группой -SО 2NН 2 или или группой, выбранной из группы, состоящей из Х 2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода;R9 представляет OR10, CONR11R12,R10 представляет (СН 2)nфенил, необязательно замещенный CN, CONR11R12 или СО 2R2 или CONH(C1-C4)алкокси; (СН 2)n-насыщенный или ненасыщенный 5-6-членный гетероцикл,содержащий от 1 до 4 атомов азота, необязательно замещенный одним или двумя заместителями, выбранными из CN, CONR11R12 или C1C4 алкокси; 4 или их фармацевтически приемлемые соли. Настоящее изобретение также относится к новым промежуточным соединениям, полезным при получении соединений формул I и II, описанных формулой III где А 5 представляет СН или N;X2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода;R14 представляет (C1-C4)алкил, (C1-C4)галоидалкил, гидрокси, карбокси, тетразолил,ацил, COOR2, CONR11R12, CONH(C1-C4)алкокси,циано, (C1-C4)алкокси, (C1-C4)алкил, фенил, нитро, NR11R12, NHCOC1-C4)алкил), NHCO(бензил), NHCO(фенил), SR2, SC1-C4)алкил),ОСОC1-C4)алкил), SO2(NR11R12), SO2C1-C4)алкил) или SO2(фенил); или к их фармацевтически приемлемым солям. Настоящее изобретение также относится к новому способу получения соединений формулI и II, а также к новым фармацевтическим композициям, содержащим эти соединения. Соединения настоящего изобретения являются селективными агонистами рецептора 3,и как таковые являются полезными для лечения диабета типа II и ожирения, а также полезны для активации (agonizing) рецептора 3. Поэтому, настоящее изобретение также относится к способам лечения диабета типа II и ожирения, а также к способу активации рецептора 3. Кроме того, настоящее изобретение относится к применению соединений формул I и II для лечения диабета типа II и ожирения, а также к применению соединений формул I и II для активации рецептора 3. Другие представители соединений настоящего изобретения описаны приведенной ниже формулой IVR3 представляет Н,R4 является 5,6-членным насыщенным или ненасыщенным гетероциклом, содержащим 5 один атом S, необязательно замещенным групили группой, выпой -SO2NH2 или бранной из группы, состоящей изR9 представляет галоген, CN, OR10, (C1-C4)алкил, (C1-C4)галоидалкил, CO2R2, CONR11R12,CONH(C1-C4)алкил или CONH(C1-C4)алкокси,SR2, CSNR2, CSNR11R12, SO2R2, SO2NR11R12,SOR2, NR11R12, фенил, необязательно замещенный группой -CONR11R12, насыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота или (C2-C4)алкенил, необязательно замещенные CN, CO2R2 или CONR11R12;R10 представляет независимо (C1-C4)алкил,(C1-C4)галоидалкил, (СН 2)n(С 3-С 8)циклоалкил,(СН 2)nфенил, необязательно замещенный CN,CONR11R12 или CONH(C1-C4)алкокси; (СН 2)nнасыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота,необязательно замещенный одним или двумя заместителями, выбранными из CN, CONR11R12 или C1-C4 алкокси;R11 и R12 независимо представляют Н, (C1C4)алкил, или соединяются с атомом азота, с которым они связаны с образованием морфолинила, пиперидинила, пирролила или пиперазина;X1 представляет -ОСН 2-,Х 2 отсутствует или представляет линейный или разветвленный алкилен с 1-5 атомами углерода;n равен 0, 1, 2 или 3; или их фармацевтически приемлемыми солями или сольватами. Подробное описание изобретения В настоящем изобретении, как они описываются в описании и формуле изобретения, ис 002778 6 пользуются термины, определение которых дается ниже. В контексте настоящего изобретения приведенные ниже термины нельзя интерпретировать по отдельности или вместе для описания химических структур, которые являются неустойчивыми или образование которых невозможно. Термин "галоген" представляет фтор, хлор,бром или иод. Термин "(C1-C4)алкил" представляет циклическую, линейную или разветвленную алкильную группу с одним-четырьмя атомами углерода, такую как метил, этил, н-пропил, изопропил, циклопропил, н-бутил, изобутил, вторбутил, трет-бутил и подобные группы. "Галоидалкил" представляет один из таких алкилов,замещенный одним или несколькими атомами галогена, предпочтительно одним-тремя атомами галогена. Примером галоидалкила является трифторметил. "Алкокси" представляет алкильную группу, ковалентно связанную посредством-О- связи. Термин "линейный или разветвленный алкилен с 1-5 атомами углерода" представляет линейную или разветвленную алкиленовую группу с одним-пятью атомами углерода. Разветвленный алкилен может иметь одну или несколько точек разветвления. Линейный или разветвленный алкилен с 1-5 атомами углерода может быть, необязательно, ненасыщенным у одного или нескольких атомов углерода. Таким образом, линейный или разветвленный алкилен с 1-5 атомами углерода включает алкиленовые,алкениленовые и алкилиденовые группы с 1-5 атомами углерода. Примерами являются метилен, этилен, пропилен, бутилен, -СН(СН 3)СН 2-,-СH(С 2 Н 5)СН 2-, -СН(СН 3)СН(СН 3)-,-СН 2 С(СН 3)2-, -CH2CH(СН 3)СН 2-, -С(СН 3)2 СН=,-СН=СНСН 2-, -СН=СН- и подобные группы."Ацильная" группа, одна или в сочетании,является производной от алкановой кислоты с одним-семью атомами углерода. Термин "ацил" также включает группы, производные от арилкарбоновых кислот. Обозначение "- - -", когда оно используется в сочетании со связью, показывает, что связь может быть либо двойной, либо одинарной связью. Термин "необязательно замещенный", используемый здесь, обозначает необязательное замещение одной-тремя, предпочтительно одной или двумя группами, выбираемыми независимо из галогена, (C1-C4)галоидалкила, гидрокси, карбокси, тетразолила, ацила, COOR2,CONR11R12, CONHC1-C4)алкокси), циано, (C1C4)алкокси, (C1-C4)алкила, фенила, бензила,нитро,NR11R12,NHCOC1-C4)алкила),NHCO(бензила), NHCO(фенила), SR2, SC1-C4)алкил),ОСОC1-C4)алкил),SO2(NR11R12),SO2C1-C4)алкил) или SO2(фенил); при условии,что такое замещение не нарушает биологиче 7 скую активность, определенную в настоящем описании.R11 и R12 независимо представляют Н, (C1C4)алкил или соединяются с атомом азота, с которым они связаны, с образованием морфолинила, пиперидинила, пирролидинила или пиперазинила. Термин "гетероцикл" представляет устойчивое, необязательно замещенное или незамещенное, насыщенное или ненасыщенное 5- или 6-членное кольцо, причем указанное кольцо содержит от одного до четырех гетероатомов,которые являются одинаковыми или разными и которые выбирают из группы, состоящей из атомов серы, кислорода и азота; и когда гетероцикл содержит два смежных атома углерода,смежные атомы углерода могут образовать структуру в виде группы формулы -СН=СН-; при условии, что (1) когда гетероциклическое кольцо имеет 5 членов, в число гетероатомов входят не более двух атомов серы или атомов кислорода, но не оба; и (2) когда гетероциклическое кольцо имеет 6 членов и является ароматическим, сера и кислород не присутствуют. Гетероцикл может присоединяться к любому атому углерода или азота, который дает устойчивую структуру. Гетероцикл может быть необязательно замещенным. Примерами гетероцикла являются пиразол, пиразолил, имидазол,изоксазол, триазол, тетразол, оксазол, 1,3 диоксолон, тиазол, оксадиазол, тиадиазол, пиридин, пиримидин, пиперазин, морфолин, пиразин, пирролидин, пиперидин, оксазолидон, оксазолидиндион, имидазолидинон. Термин "уходящая группа", который используется в описании, понятен для специалиста в этой области техники. В общем, уходящая группа является любой группой или атомом,который усиливает электрофильность атома, к которому она присоединена для замещения. Предпочтительными уходящими группами являются п-нитробензолсульфонат, трифлат, мезилат, тозилат, имидат, хлор, бром и иод. Выражение "фармацевтически эффективное количество", используемое здесь, представляет количество соединения изобретения, которое способно к активации рецептора 3 у млекопитающих. Конкретная доза соединения, вводимая по настоящему изобретению, будет, конечно, определяться конкретными обстоятельствами, связанными с пациентом, включая вводимое соединение, способ введения, конкретное состояние, которое лечат, и подобные соображения. Термин "единичная дозированная форма" относится к физически отдельным формам, подходящим в качестве единичных доз для людей и других млекопитающих, причем каждая единица содержит предварительно определенное количество активного вещества, вычисленное для получения желаемого терапевтического дейст 002778 8 вия, в сочетании с подходящим фармацевтическим носителем. Термин "лечение", используемый здесь,описывает оказание помощи и уход за пациентом с целью борьбы с заболеванием, состоянием или нарушением, и включает введение соединения настоящего изобретения для предупреждения появления симптомов или осложнений, или для устранения заболевания, состояния или нарушения. Термин "активация", используемый здесь,означает стимуляцию или воздействие на рецептор для выявления фармакологического ответа. Термин "селективный" означает преимущественное действие как агониста на рецептор 3, а не на рецептор 1 или 2. Как правило, соединения настоящего изобретения показывают различие минимум в двадцать раз (предпочтительно различие свыше 50 х) в дозе, необходимой для их действия как агонистов рецептора 3, и дозе, требуемой для равноценного действия как агонистов 1 и 2, при измерении методом анализа функционального агониста. Соединения показывают это различие во всем интервале доз. Таким образом, активные по отношению к 3 соединения ведут себя как агонисты рецептора 3 при значительно меньших концентрациях, с меньшей токсичностью, в силу их минимального "агонизма" к другим рецепторам. Как отмечалось ранее, настоящее изобретение относится к способу лечения диабета типаII и ожирения, включающему введение нуждающемуся в таком лечении млекопитающему соединений формул I и II. Предпочтительные варианты осуществления настоящего изобретения приводятся в нижеследующих разделах. Предпочтительными соединениями являются соединения формул I и II,где R5 и R6 независимо представляют (C1-C4)алкил. Х 2 представляет изопропилен, этилен, метилен или связь. Другими предпочтительными соединениями являются соединения формул I и II,где R5 и R6 представляют метил или этил,Х 2 представляет метилен или этилен,R8 представляет водород,R8 представляет галоген,R9 представляет OR10, 9R9 представляет CONR11R12,R9 представляет CN,R9 представляет необязательно замещенный арил,R10 представляет необязательно замещенный (СН 2)nфенил, (СН 2)n-необязательно замещенный насыщенный или ненасыщенный 5-6 членный гетероцикл, содержащий от 1 до 4 атомов азота. Особенно предпочтительными соединениями являются следующие соединения:(все изомеры) 4-(3-[N-(2-[4-карбамоилфенил)метил]-1-метилпропил)амино]-2-гидроксипропокси)-1,3-дигидро-2 Н-бензимидазол-2-она. Наиболее предпочтительные соединения имеют строение Благодаря своим кислотным группам, соединения формул I и II включают фармацевтически приемлемые соли присоединения оснований. К таким солям относятся соли, производные от неорганических оснований, таких как гидроксиды, карбонаты, бикарбонаты аммония и щелочных и щелочно-земельных металлов и подобных, а также соли, образованные органическими аминами, такими как алифатические и ароматические амины, алифатические диамины,гидроксиалкамины, и подобными аминами. Такими основаниями, полезными при получении солей настоящего изобретения, являются, таким 10 образом, гидроксид аммония, карбонат калия,бикарбонат натрия, гидроксид кальция, метиламин, диэтиламин, этилендиамин, циклогексиламин, этаноламин и т.п. Благодаря своим основным группам, соединения формул I и II также могут существовать в виде фармацевтически приемлемых солей присоединения кислот. Кислоты, обычно используемые для образования таких солей, включают неорганические кислоты, например, хлористо-водородную, бромисто-водородную, иодисто-водородную, серную и фосфорную кислоту, а также органические кислоты, такие как паратолуолсульфоновая,метансульфоновая,щавелевая, парабромфенилсульфоновая, угольная, янтарная, лимонная, бензойная, уксусная кислота, и родственные неорганические и органические кислоты. Таким образом, к таким фармацевтически приемлемым солям относятся сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты,дигидрофосфаты, метафосфаты, пирофосфаты,хлориды, бромиды, иодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты,изобутираты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацаты,фумараты, малеаты, 2-бутин-1,4-диоаты, 3 гексен-2,5-диоаты, бензоаты, хлорбензоаты,гидроксибензоаты, метоксибензоаты, фталаты,ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты,гиппураты, (3-гидроксибутираты, гликоляты,малеаты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты, манделаты и подобные соли. Обнаружено, что могут существовать различные стереоизомерные формы соединений формулы I и II. Соединения можно получить в виде рацематов, и можно их использовать как таковые. Поэтому рацематы, отдельные энантиомеры, диастереомеры или их смеси составляют часть настоящего изобретения. Если нет других указаний, при описании соединения или ссылки на него в настоящем описании, указанная ссылка или описание включает все рацематы, отдельные энантиомеры, диастереомеры или их смеси. Также обнаружено, что могут существовать различные таутомерные формы соединений формул I и II, и все таутомерные формы являются частью настоящего изобретения. Если нет других указаний, при описании соединения или ссылки на него в настоящем описании, указанная ссылка или описание включают все таутомерные формы или их смеси. Соединения формул I и II получают так,как описано в приведенных далее схемах и примерах. Схемы I и II описывают методику получения конечных соединений настоящего изобретения. Схемы III-V описывают методику получения промежуточных соединений, необ 11 ходимых для получения конечных соединений настоящего изобретения. Схема I На схеме I X1, X2, R1, R2, R4, R5 и R6 имеют те же значения, которые указаны ранее. Реакцию по схеме I проводят в условиях, известных из уровня техники для аминирования эпоксидов. Например, эпоксид (А) можно соединить с амином (В) в спирте, предпочтительно в этаноле, при температуре от комнатной до температуры кипения реакционной смеси. Предпочтительно реакцию проводят при условиях, описанных, в общем, в работе Atkins et al., Tetrahedron Lett. 27:2451 (1986). Эти условия включают смешивание реагентов в присутствии триметилсилилацетамида в полярном апротонном растворителе, таком как ацетонитрил, диметилформамид(ДМФА),ацетон,диметилсульфоксид (ДМСО), диоксан, диметиловый эфир диэтиленгликоля (диглим), тетрагидрофуран (ТГФ), или в других полярных апротонных растворителях, в которых реагенты растворяются. Предпочтительно растворителем является ДМСО. Реакцию проводят при температурах в интервале от 0 С до кипения с обратным холодильником. Некоторые из соединений настоящего изобретения получают путем нового комбинаторного/параллельного синтеза. Такой синтез описан на схеме II. Схема II На схеме II X1, X2, R1, R2, R4, и R5 имеют те же значения, которые указаны ранее. R6 представляет Н. Реакцию по схеме II проводят предпочтительно добавлением в стеклянную емкость не вступающего в реакцию растворителя, например метанола, ДМФА, метиленхлорида или ацетонитрила, амина (IV) и кетона (V). Раствор встряхивают, чтобы дать возможность образоваться имину, и обрабатывают борогидридной смолой Amberlite IRA400 (Aldrich). Суспензию затем встряхивают еще в течение 24 ч для прохождения восстановления до вторичного амина. В емкость добавляют метиленхлорид и полистиролбензальдегидную смолу (Frechet, J.M. etal., J. Am. Chem. Soc., 93:492 (1971, чтобы удалить избыток исходного первичного амина. Суспензию встряхивают предпочтительно в течение ночи. Затем суспензию фильтруют через ватную пробку, и оставшееся твердое вещество промывают метанолом. Упаривание в токе воздуха с последующей сушкой в вакуумной печи при комнатной температуре в течение нескольких часов дает желаемый продукт достаточной чистоты. 12 При использовании гидрохлорида амина необходима модификация схемы II. Добавление связанного со смолой основания к исходной реакционной смеси перед восстановлением или удалением примесей позволяет проходить нужной реакции. Образование имина с использованием гидрохлоридных солей амина, альдегида или кетона и связанного со смолой основания и амина можно проводить, используя две различные смолы: поли-4-винилпиридина, коммерчески доступного от Aldrich, и смолы (VIII), синтезированной посредством реакции смолы Merrifield с пиперидином (схема IIа). Схема IIа На схеме IIа PS означает полистирол. Как поли-4-винилпиридин, так и смола (VIII) промотируют образование имина. Реакцию схемы II можно также проводить традиционным способом. Восстановительное аминирование, описанное на схеме II, хорошо известно из уровня техники. Его проводят, как правило, смешиванием исходных амина и кетона в растворителе и добавлением восстановителя. Растворителями обычно являются низшие спирты, ДМФА и подобные растворители. Можно использовать самые разные восстановители, а наиболее часто используют борогидрид натрия и цианоборгидрид натрия. Реакцию осуществляют обычно при температуре от комнатной до температуры кипения растворителя. Продукты реакции выделяют способами, хорошо известными в технике. Исходные вещества на схеме II кетон и амин можно получить методами, известными и принятыми специалистами в этой области техники. Синтез исходных веществ описан, в общем, на схемах III и IV. Схема III На схеме III R1 имеет ранее указанные значения, R13 представляет ОН или SH. Эквимолярные количества ароматического соединения(соединение IX) и (2S)-(+)-глицидил-3-нитробензолсульфоната (соединение X) растворяют в инертном растворителе, например, ацетоне,и обрабатывают 1,1 эквивалентом нереакционноспособного акцептора кислоты, например,К 2 СО 3. Затем суспензию при перемешивании кипятят с обратным холодильником в течение 13 16-20 ч. Растворитель удаляют в вакууме. Остаток разделяют между хлороформом или другим органическим растворителем и водой. Органический слой сушат над Na2SO4 и концентрируют в вакууме с получением эпоксида (XI) с достаточной чистотой (95 %) и выходом (85-100%). Эпоксид (XI) растворяют в спирте, предпочтительно в метаноле, и обрабатывают одним эквивалентом дибензиламина. Раствор предпочтительно перемешивают при кипячении с обратным холодильником в течение трех-четырех часов, и затем охлаждают до температуры окружающей среды. В колбу добавляют приблизительно 10 эвивалентов формиата аммония,затем 10% палладий-на-угле и суспензию энергично перемешивают при кипячении с обратным холодильником в течение 30-45 мин. Реакционную смесь затем фильтруют через целит,концентрируют в вакууме до минимального объема и обрабатывают 1,1 эквивалентом 1,0 М безводного раствора НСl в эфире. Раствор упаривают досуха. Твердый остаток растирают с пентаном, с получением продуктов достаточной чистоты (97%) и с достаточным выходом (60100%). При желании можно провести дополнительную очистку, пропуская через невысокий слой диоксида кремния, элюируя СНСl3, а затем СНСl3/МеОН(25:5:1). Альтернативно, эпоксид (XI) обрабатывают раствором метанола, насыщенным газообразным аммиаком, и перемешивают при комнатной температуре в плотно закрытой пробирке в течение 16 ч. Этот раствор затем упаривают, а остаток подвергают стандартной очистке,например, колоночной хроматографии или перекристаллизации. Затем необязательно получают гидрохлорид посредством добавления газообразного НСl в эфире. Реакция по схеме III описана также в патенте США 5013761, Beedle et al., и в указанных в нем ссылках. Патент США 5013761 включен сюда в качестве ссылки. Кетонные составляющие на схеме II, которые либо неизвестны в технике, либо недоступны коммерчески, получают в соответствии со схемой IV. Схема IV На схеме IV R4 и R5 такие же, как определены ранее. Обозначение - - - показывает необязательное разветвление. Предпочтительно R4 является замещенным фенилом. Реакция, описанная на схеме IV, относится к реакции Хека и описана в работе A.J. Chalk et al., J. Org. Chem.,41: 1206 (1976). Взаимодействие производят обработкой соединения (XIII) арилпалладиевым реагентом. Арилпалладиевый реагент получаютin situ обработкой соединения (XIV) палладийтриарилфосфиновым комплексом. Реакцию обычно проводят при условиях, известных в технике. Дополнительные амины типа, в котором X2 является метиленом, R4 представляет арил, и R10 представляет арил, гетероцикл, необязательно замещенный арил или необязательно замещенный гетероцикл, которые реагируют аналогично схеме I, получают по схеме V. Схема V Соединения формулы (XVII) можно получить взаимодействием арилалкиловых спиртов формулы (XVI) с избытком (5 моль/экв.) соединения формулы XVIA) способами, хорошо известными в технике (см. Ж. прикл. кин., том. 45,1573-77 (1972); рус.). Эту реакцию также можно проводить смешиванием реагентов в апротонном растворителе в диглиме и добавлением трет-бутоксида калия (0,5 моль/экв.). Затем реакционную смесь кипятят с обратным холодильником и удаляют воду. После того как удаление воды завершается, как правило, в течение 2-8 ч, в зависимости от масштаба реакции, полученный в результате раствор обрабатывают водными реагентами, в том числе промывкой кислыми растворами, и продукт реакции выделяют кристаллизацией. Соединения формулы(XVIII) можно получить гидрированием соответствующих соединений формулы (XVII) в присутствии катализатора на основе благородного металла. Гидрирование можно проводить при давлении водорода от 138 до 414 кПа (от 20 до 60 ф/д 2) и с различными растворителями,катализаторами и при температурах, хорошо известных в технике. Предпочтительно реакцию проводят при давлении водорода 345 кПа (50 ф/д 2) над 5% палладием-на-угле, смоченном этанолом 2 В 3. Соединение (XVII) загружают в реактор вместе с одним эквивалентом уксусной кислоты, разбавляют метанолом, нагревают до 50 С и обрабатывают водородом в течение 5-24 ч, в зависимости от масштаба реакции. Продукт выделяют в виде соли уксусной кислоты после обработки методами, хорошо известными в технике. Специалистам понятно, что соединения формулы (XVIII) можно ввести в сочетание с самыми разными ароматическими галогенидами с получением заявленных простых эфиров. Сочетание можно проводить в соответствии с методами, известными в технике, и предпочтительно его проводят смешиванием исходных веществ в N,N-диметилацетамиде и толуоле в присутствии карбоната калия. Затем 15 реакционную смесь кипятят с обратным холодильником в течение 5-24 ч и удаляют воду. Продукт обычно выделяют водной обработкой после выпаривания реакционного растворителя на роторе. Сырой продукт можно очистить способами, хорошо известными в технике. Специалистам понятно, что амины, полученные по схеме V, можно использовать для получения соединений настоящего изобретения. Схема V также описывает получение новых промежуточных соединений формулы III где A5 представляет СН или N; Х 2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода;R14 представляет (C1-C4)алкил, (C1-C4)галоидалкил, гидрокси, карбокси, тетразолил,ацил, COOR2, CONR11R12, CONH C1-C4)алкокси), циано, (C1-C4)алкокси, (C1-C4)алкил,фенил, нитро, NR11R12, NHCOC1-C4)алкил),NНСО(бензил), NHCO(фенил), SR2, SC1-C4)алкил),ОСОC1-C4)алкил),SO2(NR11R12),SО 2C1-C4)алкил или SO2-фенил; или их фармацевтически приемлемых солей. Соединения формулы III полезны для получения соединений формул I и II, и как таковые представляют дополнительный объект настоящего изобретения. Другим объектом настоящего изобретения является способ получения новых соединений формулы IA где A5 представляет СН или N; который включает на стадии 1 гидролиз соединения формулы IB и на стадии 2 взаимодействие продукта со стадии 1 с образованием соли присоединения кислоты. Стадию первую указанного способа можно проводить, используя различные агенты, известные в технике, однако, предпочтительно проводить ее с использованием одного из следующих агентов: полифосфорной кислоты, H2O2 и К 2 СО 3 в диметилсульфоксиде, Н 2O2 и гидро 002778 16 ксида аммония, Н 2 О 2 и гидроксида натрия, гидроксида калия и трет-бутанола, или воды и НСl. Стадия 2 указанного способа включает добавление агента, способного к образованию соли присоединения кислоты с продуктом со стадии 1. Стадию 2 можно проводить многими способами, известными в технике, включая добавление неорганической кислоты или другой кислоты к раствору продукта со стадии 1. Другим объектом настоящего изобретения является способ получения соединений формулI и II, который включает на стадии 1 взаимодействие эпоксида формулы (XI) и на стадии 2 реакцию продукта со стадии 1 с образованием соли присоединения кислоты. Указанный способ можно осуществить,используя различные агенты, известные в технике, однако, предпочтительно осуществлять его взаимодействием амина и эпоксида в растворителе при повышенной температуре. Предпочтительными растворителями являются низшие спирты, диметилформамид, диметилсульфоксид, ацетон и подобные растворители. Реакцию обычно проводят при температуре в интервале от температуры окружающей среды до температуры кипения растворителя. Наиболее предпочтительно проводить ее в этаноле при 4060 С. Стадию 2 можно проводить многими способами, известными в технике, включая добавление неорганической кислоты или другой кислоты к раствору продукта со стадии 1. Исходные вещества для соединений, описанных на схемах I, II, III, IV или V, являются либо коммерчески доступными, известными в технике, либо их можно получить методами,известными в технике или описанными здесь. Препаративные примеры и примеры Следующие далее препаративные примеры и примеры приводятся только для дополнительной иллюстрации изобретения. Объем настоящего изобретения не должен истолковываться как состоящий только из приведенных ниже примеров. В приведенных далее примерах и препаративных примерах для обозначения температуры плавления, спектров ядерного магнитного резонанса, масс-спектров, высокоэффективной жидкостной хроматографии на силикагеле, газовой хроматографии, для N,Nдиметилформамида, палладия-на-угле, тетрагидрофурана, этилацетата, тонкослойной хроматографии и элементного анализа используются аббревиатуры т. пл., ЯМР, МС, ВЭЖХ, ГХ, 17 ДМФА, Pd/C, ТГФ, EtOAc, ТСХ и ЭА соответственно. Термины "ЭА", "ТСХ", "ЯМР" и "МС" в препаративных примерах показывают, что указанные данные соответствуют нужной структуре. Препаративные примеры с 1 по 18 включают методику, требуемую для получения гетероциклических этаноламинов, используемых в реакции по схеме II для получения конечных соединений настоящего изобретения. Препаративный пример 1. (S)-3-(2-Амино 3-нитрофенокси)-1,2-эпоксипропан. Раствор 2-амино-3-нитрофенола (5,95 г,38,6 ммоль) и (2S)-(+)-глицидил-3-нитробензолсульфоната (10,0 г, 38,6 ммоль) в 150 мл ацетона обрабатывают 1,1 эквивалентом К 2 СО 3 (5,86 г,42,4 ммоль) и перемешивают при кипячении с обратным холодильником в течение 18 ч. Суспензию охлаждают до температуры окружающей среды; твердые вещества отфильтровывают; и фильтрат упаривают в вакууме досуха. Полученное твердое вещество обрабатывают хлороформом и водой и водный слой один раз экстрагируют хлороформом. Органические слои объединяют и сушат над Na2SO4 и концентрируют в вакууме, получая 8,0 г (99%) оранжевого твердого вещества. ТСХ ( Rf = 0,5, СНСl3) и ЯМР показывают чистоту 95%, так что вещество используют без дополнительной очистки. ЯМР. Препаративный пример 2. (S)-[3-(N,NДибензиламино)-2-гидроксипропокси]-2-амино 3-нитробензол.(8,05 мл, 42,0 ммоль, d = 1,026). Смесь перемешивают при кипячении с обратным холодильником в течение 10 ч, а затем охлаждают до 0 С. Полученный в результате оранжевый осадок отфильтровывают и промывают холодным метанолом, затем сушат и получают 12,1 г (78%) бледно-оранжевого твердого вещества, которое по данным ЯМР и ТСХ-анализа является чистым. Указанное вещество используют без дополнительной очистки. ЯМР. Препаративный пример 3. (S)-[3-(N,N,Дибензиламино)-2-гидроксипропокси]-2,3 диаминобензол.(S)-[3-(N,N-Дибензиламино)-2-гидроксипропокси]-2-амино-3-нитробензол (10,6 г, 26,0 ммоль) суспендируют в 1 л смеси этанола с водой (2:1) при температуре окружающей среды и обрабатывают избытком бикарбоната натрия(54,34 г, 0,31 моль). Оранжевая реакционная смесь постепенно, в течение 1 ч, становится бесцветной и смесь дополнительно перемешивают при температуре окружающей среды в течение 16 ч. Суспензию фильтруют и фильтрат упаривают в вакууме до получения остатка в виде белого твердого вещества. Этот остаток 18 обрабатывают хлороформом и водой и органический слой дважды промывают рассолом. Объединенные органические экстракты концентрируют в вакууме и получают 8,8 г коричневого масла. Соединение быстро перекристаллизовывают из толуола и получают 7,96 г (81%) бледно-коричневых игольчатых кристаллов. ЯМР. Препаративный пример 4. (S)-4-[2 Гидрокси-3-(N,N-дибензиламина)пропокси]-1,3 дигидро-2 Н-бензимидазол-2-он.(S)-[3-(N,N-Дибензиламино)-2-гидроксипропокси]-2,3-диаминобензол (4,4 г, 11,6 ммоль) суспендируют в смеси толуола (60 мл) и 2N НСl (100 мл) при энергичном перемешивании при температуре окружающей среды. Добавляют избыток трифосгена (17,3 г 58,3 ммоль) и продолжают перемешивание в течение 14 ч. Двухфазную смесь осторожно гасят и нейтрализуют бикарбонатом натрия, что вызывает выпадение не совсем белого осадка, образующегося на поверхности раздела. Осадок отфильтровывают и сушат в вакууме и получают 4,35 г (93%) палевого твердого вещества, которое используют без дополнительной очистки. ТСХ, ЯМР и МС показывают высокую чистоту этого промежуточного соединения. Препаративный пример 5. (S)-4-[2 Гидрокси-3-аминопропокси]-1,3-дигидро-2 Нбензимидазол-2-он.(4,35 г, 10,8 ммоль) растворяют в метаноле (200 мл) и обрабатывают большим избытком формиата аммония (13,0 г, 0,21 моль), а затем добавляют 10% палладий-на-угле (1,5 г). Суспензию перемешивают при кипячении с обратным холодильником в течение 3 ч. После охлаждения суспензии реакционную смесь фильтруют через целит. Фильтрат концентрируют в вакууме до бледно-коричневого масла, которое при стоянии постепенно кристаллизуется. Полученное в результате твердое вещество растирают с хлороформом, содержащим метанол, фильтруют и получают 1,56 г (65%) нужного продукта в виде бледно-серого твердого вещества. ЯМР. МС. Препаративный пример 6. (S)-4-[2 Диметил-трет-бутилсилил)окси-3-(дибензиламино)пропокси]бензимидазол.(S)-[3-(N,N-Дибензиламино)-2-гидроксипропокси]-2,3-диаминобензол (1 г, 2,1 ммоль) растворяют в N,N-диметилформамиде (10 мл) и добавляют имидазол (0,27 г, 4,0 ммоль) и третбутилдиметилсилилхлорид (0,6 г, 4,0 ммоль). Раствор перемешивают при температуре окру 19 жающей среды в течение 18 ч, а затем обрабатывают хлороформом и водой. Объединенные органические экстракты сушат над сульфатом натрия и упаривают в вакууме и получают нужный бензимидазол (1,3 г, 96%). ЯМР. Препаративный пример 7. (S)-4-[2 Диметил-трет-бутилсилил)окси-3-аминопропокси]бензимидазол.(S)-4-[2-Диметил-трет-бутилсилил)окси-3 дибензиламино)пропокси]бензимидазол (1,27 г,2,5 ммоль) растворяют в метаноле (140 мл) и обрабатывают избытком формиата аммония(1,64 г, 25,0 ммоль), а затем 10% палладием-наугле (410 мг). Полученную в результате суспензию перемешивают при кипячении с обратным холодильником в течение 1 ч. После охлаждения реакционную смесь фильтруют через слой целита. Фильтрат концентрируют в вакууме до коричневого масла (780 мг, 97%). ЯМР. Препаративный пример 8. (S)-4-[2 Гидрокси-3-аминопропокси]бензимидазол.(S)-4[2-Диметил-трет-бутилсилил)окси-3 аминопропокси]бензимидазол (10 мг, 31 ммоль) растворяют в ТГФ (1 мл) и смесь охлаждают до 0 С. Добавляют тетрабутиламмонийфторид (1 мл, 1,0 М раствор в ТГФ). Реакционную смесь перемешивают при этой температуре в течение 4 ч. Реакцию гасят, добавляя воду. Упаривание водного слоя дает нужный спирт. ЯМР. МС. Препаративный пример 9. (S)-3-(4-Амино 3-нитрофенокси)-1,2-эпоксипропан. Раствор 4-амино-3-нитрофенола (2,54 г,16,5 ммоль) и (2S)-(+)-глицидил-3-нитробензолсульфоната (4,27 г, 16,5 ммоль) в 50 мл ацетона обрабатывают 1,1 эквивалентом К 2 СО 3 (2,50 г,18,1 ммоль), и перемешивают при кипячении с обратным холодильником в течение 20 ч. Суспензию охлаждают до температуры окружающей среды, и упаривают в вакууме досуха. Полученное в результате твердое вещество обрабатывают хлороформом и водой и водный слой экстрагируют хлороформом. Органические слои объединяют и сушат над МgSO4, упаривают в вакууме и получают 3,0 г (87%) твердого оранжевого вещества. ТСХ и ЯМР показывают чистоту 95%, так что указанное вещество используют без дополнительной очистки. ЯМР. Препаративный пример 10. (S)-[3-(N,NДибензиламино)-2-гидроксипропокси]-4-амино 3-нитробензол. 20 Эпоксид препаративного примера 9 (3,0 г,14,3 ммоль) растворяют в 100 мл метанола и обрабатывают дибензиламином (3,02 мл, 15,7 ммоль). Смесь перемешивают при кипячении с обратным холодильником в течение 6 ч, и после охлаждения удаляют растворитель в вакууме. Полученное в результате твердое оранжевое вещество (5,8 г, 100%) используют без дополнительной очистки. ЯМР. Препаративный пример 11. (S)-[3-(N,NДибензиламино)-2-гидроксипропокси]-3,4 диаминобензол. Нитроанилин препаративного примера 10(4,89 г, 12,0 ммоль) суспендируют в смеси этанола (400 мл) с водой (300 мл) при температуре окружающей среды и обрабатывают бикарбонатом натрия (12,1 г, 144 ммоль, 12 экв.) и гидросульфитом натрия (25,1 г, 144 ммоль, 12 экв.). В течение 3 ч реакционная смесь постепенно становится бесцветной. Реакционную смесь обрабатывают хлороформом и рассолом. Органический слой промывают несколько раз рассолом,сушат над сульфатом магния, и концентрируют в вакууме. Получают коричневое масло. ЯМР. Препаративный пример 12. (S)-5-[2 Гидрокси-3-(N,N-дибензиламино)пропокси]-1,3 дигидро-2 Н-бензимидазол-2-он. Диамин препаративного примера 11 (2,1 г,5,6 ммоль) суспендируют в смеси толуола (40 мл) и 2N НСl (70 мл) при температуре окружающей среды и при энергичном перемешивании. Добавляют избыток трифосгена (17,3 г,58,3 ммоль). Перемешивание продолжают в течение 14 ч. Двухфазную смесь осторожно гасят и нейтрализуют бикарбонатом натрия, что вызывает выпадение не совсем белого осадка, образующегося на поверхности раздела. Осадок отфильтровывают и сушат в вакууме и получают 1,06 г (47%) твердого серого вещества, которое используют без дополнительной очистки. ТСХ, ЯМР и МС указывают на высокую чистоту промежуточного соединения. Препаративный пример 13. (S)-5-[2 Гидрокси-3-амино)пропокси]-1,3-дигидро-2 Нбензимидазол-2-он. Соединение препаративного примера 12(0,75 г, 1,9 ммоль) растворяют в метаноле (100 мл) и обрабатывают избытком формиата аммония (0,7 г, 11,2 ммоль), а затем 10% палладиемна-угле (400 мг). Суспензию перемешивают при кипячении с обратным холодильником в течение 3 ч. После охлаждения суспензии реакционную смесь фильтруют через целит и фильтрат упаривают в вакууме до серовато-черного твердого вещества (0,25 г 60%). ЯМР. МС. 21 Препаративный пример 14. (S)-5-[2(Диметил-трет-бутилсилил)окси-3-(дибензиламино)пропокси]бензимидазол. Это соединение получают способом, аналогичным способу препаративного примера 6. ЯМР. МС. Препаративный пример 15. (S)-5-[2(Диметил-трет-бутилсилил)окси-3-аминопропокси]бензимидазол. Это соединение получают способом, аналогичным способу препаративного примера 7. ЯМР. МС. Препаративный пример 16. (S)-4-[2 Гидрокси-3-аминопропокси]бензимидазол. Нужный аминоспирт получают по способу, аналогичному способу препаративного примера 8. ЯМР. МС. Препаративный пример 17. 4-[(2S)-2,3 Оксопропокси]-2(3 Н)бензоксазолон. Раствор 4-гидрокси-2(3 Н)бензоксазолона(1,00 г, 6,6 ммоль) и (2S)-(+)-глицидил-3 нитробензолсульфата (1,72 г, 6,6 ммоль) в 50 мл ацетона обрабатывают 1,1 эквивалентом К 2 СО 3(1,01 г, 7,3 ммоль) и перемешивают при кипячении с обратным холодильником в течение 4 ч. Суспензию охлаждают до температуры окружающей среды и растворитель выпаривают в вакууме досуха. Полученное в результате твердое вещество обрабатывают хлороформом и водой и водный слой экстрагируют один раз хлороформом. Органические слои объединяют и сушат над Na2SO4 и упаривают в вакууме и получают твердое белое вещество. Флэшхроматография (хлороформ/метанол, 9/1) дает моноалкилированный продукт (0,55 г, 40%). ЯМР. МС. Препаративный пример 18. (S)-4-[2 Гидрокси-3-аминопропокси]-2(3 Н)бензоксазолон.(2 мл) охлаждают до -78 С, используя баню с сухим льдом и ацетоном. В реакционной смеси конденсируют газообразный аммиак (2 мл). Реакционный сосуд закрывают, и дают постепенно, в течение ночи, нагреваться до комнатной температуры. Реакцию прекращают, открывая реакционный сосуд, и дают возможность испариться газообразному аммиаку. ЯМР. МС.(20,3 г, 71,1 ммоль) в абсолютном этаноле (200 мл) нагревают при 55 С в течение 12 ч. Все твердые вещества переходят в раствор при 50 С. После завершения реакции растворитель выпаривают досуха. Остаток снова растворяют в этилацетате и промывают насыщенным раствором бикарбоната натрия. Слои разделяют и водную фазу экстрагируют этилацетатом. Два органических слоя объединяют и промывают рассолом. Фазы разделяют и органический слой сушат над сульфатом натрия и удаляют растворитель. Колоночная хроматография с элюированием 20% МеОН/СНСl3 дает 7,26 г (62%) продукта реакции. ЯМР. МС. Препаративный пример 20. (S)-4-(4-[2-(N[3-(2,3-Диаминофенокси)-2-гидроксипропил]амино)-2-метилпропил]фенокси)бензамид. Нитроанилин препаративного примера 19(0,484 г, 0,98 ммоль) растворяют в абсолютном этаноле (40 мл) и обрабатывают раствором бикарбоната натрия (0,50 г, 5,95 ммоль) в воде (10 мл), а затем раствором гидросульфита натрия(1,41 г, 8,1 ммоль) в воде (15 мл). Как только окраска полностью исчезнет, растворители выпаривают. Остаток промывают водой для удаления избытка солей. Оставшееся аморфное вещество растворяют в метаноле и фильтруют методом гравитационной фильтрации. Растворитель выпаривают и получают 0,454 г (100%) продукта. ЯМР. МС. Препаративный пример 21. 4-третБутилдиметилсилилоксииндол. Раствор 4-гидроксииндола (3,0 г, 22,5 ммоль), трет-бутилдиметилсилилхлорида (5,09 г, 33,8 ммоль) и имидазола (3,83 г, 56,3 ммоль) в диметилформамиде (60 мл) перемешивают при комнатной температуре в течение 24 ч. Добавляют водный раствор хлорида аммония (100 мл) и экстрагируют несколько раз этилацетатом. Органические слои объединяют, сушат над сульфатом магния и выпаривают и получают сырое масло. Флэш-хроматография (10% этилацетат/гексан) дает нужный продукт (5,55 г,100%) в виде белого твердого вещества. ЯМР. Препаративный пример 22. 4-третБутилдиметилсилилокси-2-фенилиндол. 4-трет-Бутилдиметилсилилоксииндол (4,67 г, 18,9 ммоль) растворяют в ТГФ (100 мл) при-78 С в атмосфере азота и раствор обрабатывают бутиллитием (13,0 мл, 1,6 М раствор в гексане, 20,8 ммоль), добавляя его по каплям в течение 10 мин. После перемешивания в течение 30 мин через раствор в течение 20 мин пропускают газообразный диоксид углерода. Прозрачному раствору позволяют нагреваться до комнатной температуры и наблюдают энергичное выделение пузырьков газа. Избыток диоксида углерода удаляют в вакууме на роторном испарителе при комнатной температуре, в то время как растворитель концентрируют до объема приблизительно 50 мл. Добавляют дополнительное количество ТГФ (60 мл) и раствор охлаждают до-78 С. К этой смеси по каплям, в течение 10 мин, добавляют трет-бутиллитий (12,2 мл, 20,8 ммоль) и смесь перемешивают в течение 2 ч при-78 С. Добавляют по каплям хлорид трибутилолова (5,4 мл, 19,8 ммоль). После перемешивания в течение 1,5 ч, холодный раствор выливают в смесь толченого льда с водой (100 г) и добавляют насыщенный раствор хлорида аммония до тех пор, пока раствор не станет кислым. Водный раствор экстрагируют эфиром (3100 мл), сушат над сульфатом магния и концентрируют и получают 12,68 г желтого масла. К раствору полученного таким образом 1 карбокси-2-(трибутилстаннил)индола (12,68 г) в этаноле (100 мл) добавляют иодбензол (2,1 мл,18,9 ммоль) и хлорид бис(трифенилфосфин) палладия(II) (0,60 г, 0,85 ммоль). Смесь выдерживают при кипячении с обратным холодильником в течение 48 ч. Смесь охлаждают до комнатной температуры, фильтруют через слой целита и концентрируют при пониженном давлении. Флэш-хроматография (5% этилацетата/гексан) дает 4-трет-бутилдиметилсилилокси 2-фенилиндол в виде белого твердого вещества. ЯМР. МС. Препаративный пример 23. 4-Гидрокси-2 фенилиндол. 4-трет-Бутилдиметилсилилокси-2-фенилиндол (55 г, 0,17 ммоль) растворяют в ТГФ (10 мл) при 0 С и обрабатывают тетрабутиламмонийфторидом (0,5 мл, 1,0 М раствор ТГФ, избыток). После перемешивания при этой температуре в течение 10 мин реакцию прекращают, добавляя насыщенный раствор хлорида аммония. Смесь экстрагируют несколько раз этилацетатом, сушат над сульфатом магния и упаривают, с получением сырого масла. Флэш-хроматография (5% этилацетата/гексан) дает нужный фенол (30 мг, 84,8%) в виде белого твердого вещества. ЯМР. МС. В препаративных примерах с 24 по 34 описаны синтезы соединений, осуществленные по комбинаторной/параллельной схеме II. Препаративный пример 24. 4-(3 Оксобутил)-1-(2-оксазолидин)бензол. 4-Бром-1-(2-оксазолидин)бензол (3,0 г,13,3 ммоль), 3-бутен-2-ол (1,4 г, 20 ммоль) Рd(ОАс)2 (60 мг, 0,26 ммоль), (о-толил)3 Р (158 мг, 0,52 ммоль), бикарбонат натрия (1,34 г, 15,9 ммоль) в 30 мл N-метилпирролидинона нагревают в атмосфере азота при 130 С в течение 1 ч. Затем реакционную смесь охлаждают и обрабатывают этилацетатом и водой. Объединенные органические слои промывают водой и затем сушат (Na2SO4), фильтруют и концентрируют в вакууме и получают 2,6 г желтоватокоричневого масла. Очистка с помощью флэшхроматографии (силикагель, этилацетат/гексан 1:1) дает 1,9 г бледно-желтого масла, которое после сушки в вакууме кристаллизуется. Перекристаллизацией из гексана получают 1,47 гN,N-диметилацетамиде (50 мл) и нагревают при 150 С в течение 4 ч в атмосфере азота. Затем реакционную смесь выливают в 800 мл ледяной воды. Постепенно кристаллизующееся твердое вещество отфильтровывают и получают 13 г сырого продукта. Это вещество перекристаллизовывают из этанола с водой (3:1) и получают 10,48 г (79%) бледно-коричневых кристаллов; т. пл. 64-66 С. ЭА. ЯМР. МС. Препаративный пример 25.(6,0 г, 22,6 ммоль) и карбонат калия (1,0 г, 7,2 ммоль) суспендируют в ДМСО (50 мл) и суспензию охлаждают до 0 С на ледяной бане. Добавляют постепенно водный 30% пероксид водорода (6 мл) и смесь перемешивают при 0 С в течение 1 ч. Реакцию прекращают, выливая смесь в 500 мл воды, а затем собирают выпавший белый осадок и промывают водой. Это вещество перекристаллизовывают из 300 мл этанола и получают 5,35 г (84%) белых кристаллов; т. пл. 169-172 С. ЯМР. МС.CH2Cl2 (10 мл) обрабатывают трифенилметилхлоридом (2,79 г, 10 ммоль) и диизопропилэтиламином (2,0 мл, 11,5 ммоль) и перемешивают в течение 40 мин при комнатной температуре. Реакционную смесь концентрируют в вакууме и обрабатывают этилацетатом и водой. Органический слой промывают насыщенным раствором NаНСО 3, затем рассолом, сушат(Na2SO4) и упаривают в вакууме с получением 3,48 г не совсем белого твердого вещества. Растирание этого вещества с диэтиловым эфиром дает 3,04 г (84%) белого твердого вещества; т. пл. 162-165 С. ЯМР. МС. ЭА. Препаративный пример 27. 5-[4-(2 Оксобутил)феноксиметил]тетразол.(180 г, 4,5 ммоль, 60%, в минеральном масле) в атмосфере азота. Через 15 мин ледяную баню удаляют и дают раствору возможность нагреваться до комнатной температуры в течение 45 мин. Реакционную смесь охлаждают до 5 С и обрабатывают 2-трифенилметил-5-хлорметилтетразолом (1,08 г, 3 ммоль) и перемешивают при комнатной температуре в течение 3 ч. Реакционную смесь выливают в EtOAc (300 мл) и промывают водой, а затем рассолом. Органический слой сушат (MgSO4) и упаривают в вакууме, получают желтое твердое вещество. Это вещество суспендируют в смеси МеОН (100 мл) и ТГФ (50 мл) и обрабатывают 4N раствором НСl в диоксане (7,5 мл, 30 ммоль). Получающуюся в результате смесь перемешивают в течение 1,5 ч, а затем упаривают в вакууме и получают желтовато-коричневое твердое вещество. Остаток хроматографируют на колонке с силикагелем, элюируя 33-100% этилацетата в гексане. Получают 235 мг (32%) белого твердого вещества; т. пл. 148-150 С. ЯМР. МС. ЭА. Препаративный пример 28. 3-[4-(2 Оксобутил)феноксиметил]пиридин. 4-(4-Гидроксифенил)-2-бутанон (4,11 г, 25 ммоль) и карбонат калия (10,37 г, 75 ммоль) и ацетоне (30 мл) обрабатывают гидрохлоридом 3-пиколилхлорида (4,27 г, 26 ммоль) в атмосфере азота. Смесь перемешивают при кипячении с обратным холодильником в течение 21 ч, причем реакция проходит примерно на 50%. Добавляют иодид калия (2,0 г, 13 ммоль, 0,5 экв.) и 26 через 3 ч на ТСХ не наблюдают присутствия пиколилхлорида. Летучие вещества удаляют в вакууме и полученный в результате твердый остаток обрабатывают EtOAc и водой. Объединенные органические слои промывают водой,насыщенным раствором NaHCO3, 10% раствором Nа 2SO3 и затем рассолом. Органический слой сушат (МgSO4) и концентрируют в вакууме, получают 4,8 г желтого масла. Это вещество очищают на Waters Prep 2000LC, элюируя 1080% этилацетата в гексане в течение 45 мин и получают 2,20 г (34%) масла, которое при стоянии отверждается. Т. пл. 35-37 С. ЯМР. МС. ЭА. Препаративный пример 29. 2,6-Диметокси 4-[4-(2-оксобутил)фенокси]-1,3,5-тиразин.(1,62 г, 30 ммоль) в метаноле (150 мл) в атмосфере азота. После перемешивания в течение 1 ч метанол удаляют в вакууме, а остаток суспендируют в ацетоне (200 мл). Суспензию обрабатывают 4,6-диметокси-2-хлортриазином и кипятят с обратным холодильником в течение 3 ч. Летучие вещества удаляют в вакууме, а остаток обрабатывают этилацетатом и водой. Органические слои сушат (МgSO4) и упаривают в вакууме и получают 10,28 г белого полутвердого вещества. Это вещество очищают на Water Prep 2000LC, элюируя градиентом 20-60% этилацетата в гексане в течение 55 мин и получают 4,43 г (49%) бесцветного масла. ЯМР. МС. ЭА. Препаративный пример 30. 2-[4-(2 Оксобутил)фенокси]-5-карбоксамидопиридин. 4-(4-Гидроксифенил)-2-бутанон (3,28 г, 20 ммоль) в безводном ДМФА (150 мл) обрабатывают NaH (1,2 г, 30 ммоль, 60%, в минеральном масле) в атмосфере азота. Раствор перемешивают в течение 30 мин при температуре окружающей среды и затем обрабатывают 6 хлороникотинамидом (3,13 г, 20 ммоль). Реакционную смесь перемешивают при 60 С в течение 1,5 ч, а затем при 90 С в течение 5 ч. Реакционной смеси дают возможность остыть, выливают в 50% раствор хлорида аммония и экстрагируют EtOAc. Органический слой сушат(MgSO4) и концентрируют в вакууме азеотропной отгонкой с ксилолом и получают 11,4 г коричневого масла. Это вещество очищают наWaters Prep 2000LC посредством элюирования с 75-100 5 EtOAc в течение 60 мин. Полученное в результате вещество растирают с холодным Способом, подобным способу в описанных выше примерах, 3-(4-гидроксифенил)-2-пропанон (2,25 г, 15 ммоль) обрабатывают NaH (0,9 г,22,5 ммоль, 60%, в минеральном масле), а затем вводят во взаимодействие с 6-хлорникотинамидом (2,34 г, 15 ммоль). При последующей обработке вещество очищают на Waters Prep 2000LC, и получают 1,28 г (32%) светло-желтое твердое вещество. Т. пл. 172-174 С. ЯМР. МС. ЭА. Препаративный пример 32. 4-[(2 Оксоциклогексил)метил]фенилметаннитрил.(10,5 г, 76 ммоль) в ТГФ (200 мл) кипятят с обратным холодильником в течение 24 ч. Прохождение реакции наблюдают с помощью ГХ. Реакционную смесь разбавляют водой и удаляют ТГФ при пониженном давлении. Водную часть экстрагируют EtOAc, сушат (МgSO4) и получают 19,3 г белого твердого вещества, которое имеет 74% чистоту по газовой хроматографии. Это твердое вещество перекристаллизуют из гексана с EtOAc и получают 7,75 г белых кристаллов, которые, по данным ГЖХ, имеют 100% чистоту. Вторую порцию в 3,65 г получают при добавлении к фильтрату еще некоторого количества гексана. Всего получают 11,4 г (67%) 1[(4-цианофенил)метил]-1-метоксикарбонил-2 оксоциклогексанкарбоксилата; т. пл. 82-84 С. ЯМР. МС. Под слоем азота смесь 1-[(4 цианофенил)метил]-1-метоксикарбонил-2-оксоциклогексанкарбоксилата (7,6 г, 28 ммоль), цианида натрия (2,1 г, 42 ммоль) и ДМСО (100 мл) нагревают при 115 С в течение 1,5 ч. Прохождение реакции контролируют с помощью ГХ. Реакционную смесь охлаждают и обрабатывают водой, EtOAc и рассолом. Органический слой промывают водой и сушат (MgSO4). После концентрирования получают сырой продукт в виде желтовато-коричневого масла. Очистка с помощью plug-хроматографии (200 г силикагеля,15% EtOAc в гексане) дает 3,3 г (55%) продукта в виде бесцветного масла. ЯМР. МС. Препаративный пример 33. 4-[(2 Оксоциклогексил)метил]бензамид. Раствор соединения препаративного примера 28 (2,5 г, 11,7 ммоль) в ДМСО (20 мл) охлаждают на ледяной бане. Добавляют твердый К 2 СО 3 (500 мг), а затем сразу же 30% Н 2 О 2 (3 мл). Через 20 мин ТСХ (EtOAc/гексан, 3/7) показывает следовое количество исходного вещества. Ледяную баню убирают и реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь разбавляют 500 мл воды и белое твердое вещество собирают и сушат и получают 2,44 г (90%) нужного амида. Продукт перекристаллизовывают из смеси EtOAc с гексаном (1:9) и получают 2,02 г названного в заголовке соединения в виде белых кристаллов; т. пл. 167-170 С. ЯМР. МС. Препаративный пример 34. Этиленкеталь 2-тетралон-6-карбоновой кислоты. 6-Бром-2-тетралон (2,0 г, 8,89 ммоль) растворяют в толуоле (50 мл) и обрабатывают избытком этиленгликоля (4,88 мл, 88,9 ммоль) и катализатором п-толуолсульфоновой кислотой(15 мг). Раствор перемешивают при кипячении с обратным холодильником в течение 16 ч и удаляют воду из реакционной смеси, используя насадку Дина-Старка. После охлаждения до температуры окружающей среды толуольный раствор промывают 2 х 1N NaOH, 1 х водой, 1 х рассолом, сушат над Nа 2SO4 и концентрируют в вакууме и получают 2,23 г (93%) 6-бром-2 тетралонэтиленкеталя в виде коричневого масла, которое используют без дополнительной очистки. 6-Бром-2-тетралонэтиленкеталь (2,2 г, 8,15 ммоль) растворяют в безводном ТГФ (30 мл),охлаждают раствор до -78 С и обрабатывают трет-бутиллитием (12,05 мл, 20,4 ммоль, 1,7 М раствор в пентане) в атмосфере азота. После перемешивания в течение 30 мин через реакционную смесь в течение 20 мин при -78 С пропускают сухой диоксид углерода. Затем суспензии дают возможность нагреться до температуры окружающей среды. Раствор гасят водой и подкисляют 1N HCl и затем экстрагируют 2 хEtOAc. Органические экстракты промывают рассолом, сушат над Na2SO4 и концентрируют в вакууме до бледно-коричневого масла. Маслянистый остаток вносят в колонку с диоксидом кремния для флэш-хроматографии и элюируют 30-50% EtOAc в гексане, с получением 1,06 г(55%) этиленкеталя тетралон-6-карбоновой кислоты в виде медленно кристаллизующегося твердого вещества. ЯМР. МС. Препаративный пример 35. 2-Тетралон-6 карбоксамид.(50 мл) вместе с N-гидроксисукцинимидом (260 мг, 2,76 ммоль) при 0 С и обрабатывают небольшим избытком 1,3-дициклогексилкарбодиимида (502 мг, 2,50 ммоль). Смеси дают возможность нагреться до температуры окружающей среды в течение 30 мин, и в это время образуется тонкодисперсный белый осадок. Добавляют хлорид аммония (333 мг, 6,23 ммоль) и триэтиламин (1,58 мл, 12,5 ммоль, d = 0,797). Раствор перемешивают при температуре окружающей среды в течение 16 ч. Суспендированную мочевину и соли отфильтровывают, а раствор концентрируют в вакууме до бесцветного масла. Это масло вносят в колонку для флэшхроматографии с диоксидом кремния и элюируют 50-100% EtOAc в гексане и получают 250 мг (64%) этиленкеталя 2-тетралон-6-карбоксамида в виде белого твердого вещества, чистого по данным ЯМР, ТСХ. Этиленкеталь 2-тетралон-6-карбоксамида(50 мл) при температуре окружающей среды в течение 48 ч. Летучие вещества удаляют в вакууме, а остаток растирают с этилацетатом. Твердое вещество отфильтровывают, промывают и сушат и получают 77,5 мг (38%) 2 тетралон-6-карбоксамида в виде белого порошка, чистого по ЯМР, ТСХ. МС. Препаративный пример 36. 2-Тетралон-6 морфолинамид.(50 мл) вместе с N-гидроксисукцинимидом (260 мг, 2,76 ммоль) при 0 С и обрабатывают небольшим избытком 1,3-дициклогексилкарбодиимида (502 мг, 2,50 ммоль). Смеси дают возможность нагреваться до температуры окружающей среды в течение 30 мин, и в это время образуется тонкодисперсный белый осадок. Добавляют морфолин (0,91 мл, 10,4 ммоль) и раствор перемешивают при температуре окружающей среды в течение 16 ч. Суспендированную мочевину отфильтровывают, а раствор концентрируют в вакууме до бесцветного масла. Это масло вносят в колонку для флэшхроматографии с диоксидом кремния и элюируют 50-100% EtOAc в гексане и получают 323 мг (51%) этиленкеталя 2-тетралон-6-морфолинамида в виде постепенно кристаллизующе 002778 30 гося твердого вещества, чистого по данным ЯМР, ТСХ. Этиленкеталь 2-тетралон-6-морфолинамида (323 мг, 1,06 ммоль) и катализатор птолуолсульфоновую кислоту перемешивают в ацетоне (50 мл) при температуре окружающей среды в течение 48 ч. ТСХ показывает наличие смеси этиленкеталя 2-тетралон-6-морфолинамида и нужного продукта, и далее раствор кипятят с обратным холодильником в течение 16 ч. Летучие вещества удаляют в вакууме, а остаток вносят в колонку для флэш-хроматографии с диоксидом кремния и элюируют 50-100% EtOAc в гексане и получают 27 мг (10%) 2-тетралон-6 морфолинамида в виде медленно кристаллизующегося твердого вещества, чистого по ЯМР,ТСХ. МС. Препаративный пример 37. (R)-4-[2 Гидрокси-3-(N,N-дибензиламино)пропокси]-1,3 дигидро-2 Н-бензимидазол-2-тион.[3-(N,N-Дибензиламино)-2-гидроксипропокси]-2,3-диаминобензол (400 мг, 1,1 ммоль) растворяют в смеси метиленхлорида (70 мл) и пиридина (35 мл) и охлаждают до 0 С. Добавляют диметиламинопиридин (DMAP; 311 мг, 2,5 ммоль) и вводят по каплям тиофосген (179 мг,0,119 мл, 1,6 ммоль). Через 30 мин добавляют дополнительно эквивалент тиофосгена и смесь перемешивают в течение 5 ч. Осторожно добавляют воду и полученную в результате двухфазную смесь экстрагируют метиленхлоридом. Органический раствор сушат над МgSO4 и фильтруют и упаривают и остается коричневое масло(445 мг, прибл. 100%). МС. ЯМР. Названное выше в заголовке соединение превращают в амин для реакции по схеме I известными методами. Препаративный пример 38. 4-(2-Метил-2 нитропропил)фенол.(100,08 г, 806 ммоль), 2-нитропропана (400 мл,4,45 моль) и диглима (800 мл) нагревают до 38 С. Добавляют трет-бутилат калия (45,29 г,403,6 ммоль) и смесь кипятят с обратным холодильником при 132 С с ловушкой Дина-Старка. Вода начинает собираться в ловушке и собирается с высокой скоростью приблизительно в течение 1,5 ч. Когда накопление воды замедляется (примерно через 2,5 ч), удаляют растворитель частями (30-40 мл каждая) каждые тридцать минут. Во время сбора воды и удаления растворителя температура поднимается от 132 до 149 С. Через 4 ч по данным ВЭЖХ-анализа остается менее 1% 4-гидроксибензилового 31 спирта. Нагревательный кожух удаляют и дают реакционной смеси остыть. Когда температура составляет 100 С, добавляют воду (200 мл) и раствору дают остыть до комнатной температуры. Удаляют растворитель в вакууме на роторном испарителе до тех пор, пока не останется 593 г раствора. Добавляют воду (500 мл) иEtOAc (500 мл) и слои разделяют (слои разделяются плохо, и добавление 20% водного раствора NaCl эффекта не дает). Водный слой экстрагируют EtOAc (200 мл) и объединенные органические слои экстрагируют 1N HCl (500 мл) и водой (300 мл). Органический слой перегоняют в вакууме с получением 261 г масла, к которому добавляют EtOAc (160 мл). Быстро, при энергичном перемешивании, в течение 30 мин, добавляют гептан (3,4 л), и продукт кристаллизуется с образованием твердого бежевого вещества (112,36 г, выход 71%, чистота 98% по данным ВЭЖХ-анализа). Другую порцию кристаллов можно получить из фильтрата посредством его концентрирования и отфильтровывания твердого вещества, или посредством более полного концентрирования раствора и добавления гептана для кристаллизации. ЯМР. ЭА. Препаративный пример 39. Соль уксусной кислоты 4-(2-амино-2-метилпропил)фенола. В реактор высокого давления емкостью 3,8 л (1 галлон) загружают 4-(2-метил-2 нитропропил)фенол (120 г, 614 ммоль), НОАс(35,2 мл, 614 ммоль), 5% палладий-на-угле (24 г), смоченный EtOH 2B3 (60 мл) и МеОН (1230 мл). Смесь при перемешивании (600 об./мин) нагревают до 50 С, продувают реактор N2 и подают H2 до создания давления 345 кПа (50 ф/д 2). Через 15,5 ч реактор продувают N2, и фильтруют охлажденную смесь. Осадок на фильтре промывают МеОН и фильтрат концентрируют с получением 514 г суспензии на роторном испарителе. К этой суспензии при энергичном перемешивании добавляют EtOAc (2 л). После перемешивания в течение 1 ч полученные кристаллы отфильтровывают и промывают небольшим количеством EtOAc. Продукт сушат в течение ночи в вакуумной печи при 45 С и получают 118,83 г (86%) продукта в виде мелких белых игольчатых кристаллов (т. пл. 211-216 С,разл.). Это вещество имеет чистоту 99% по данным анализа ВЭЖХ, в то время как из маточной жидкости получают дополнительные 9,00 г вещества, чистота которого, как обнаружено, составляет только 88%. Препаративный пример 40. 2-(4-(2-Амино 2-метилпропил)фенокси)-5-карбоксамидопиридин. Смесь уксуснокислой соли 4-(2-амино-2 метилпропил)фенола (45,06 г, 200 ммоль), порошкообразного К 2 СО 3 (69,1 г, 500 ммоль), 6 хлороникотинамида (31,32 г, 200 ммоль),DMAC (622 мл) и изооктана (70 мл) медленно нагревают с обратным холодильником при 140 С. Для сбора воды, образующейся в реакции, используют ловушку для воды, наполненную изооктаном, а кипячение с обратным холодильником поддерживают в течение 5,5 ч. Дают смеси остыть до комнатной температуры, и твердое вещество отфильтровывают и промывают EtOAc. Фильтрат концентрируют в вакууме до образования 88,6 г твердого вещества,которое растворяют в EtOAc (500 мл). К этому раствору добавляют воду (800 мл), 1N HCl (200 мл) и МеОН (50 мл). Доводят рН этой смеси до 7,2 с помощью конц. HCl, и водный слой отделяют и промывают метил-трет-бутиловым эфиром (500 мл). Продукт кристаллизуется при добавлении 10N NaOH (20 мл), который увеличивает рН до 11. Эту величину рН поддерживают,добавляя в ходе кристаллизации по необходимости 10N NaOH (90 мин). Продукт отфильтровывают, промывают водой и сушат в вакууме при 45 С, получают 53,11 г (93%) белого твердого вещества, которое по данным ВЭЖХанализа чистое 98%. 1H ЯМР (300 МГц,ДМСО-d6). Данные ЯМР соответствуют нужному продукту. Препаративный пример 41. 4-(4-(2-Амино 2-метилпропил)фенокси)бензонитрил. Смесь уксуснокислой соли 4-(2-амино-2 метилпропил)фенола (45,06 г, 200 ммоль), порошкообразного К 2 СО 3 (69,1 г, 500 ммоль) иDMAC (550 мл) нагревают до 75-100 С. Добавляют толуол и смесь медленно нагревают с обратным холодильником при 134 С. Температуру кипения смеси поднимают путем отгонки толуола и воды в ловушку для воды, пока температура на достигнет 141 С. Затем дают смеси остыть до температуры ниже 100 С, при которой добавляют 4-фторбензонитрил (24,46 г, 202 ммоль) вместе с 50 мл толуола. Смесь снова нагревают с обратным холодильником при 140 С, причем воду собирают в наполненную толуолом и водой ловушку в течение 4 ч. Смеси дают остыть до комнатной температуры и отфильтровывают твердые вещества и промывают толуолом. Фильтрат концентрируют на роторном испарителе до образования 77 г сиропа,который растворяют в EtOAc (400 мл). Этот раствор экстрагируют водой (400 мл) и водный слой снова экстрагируют EtOAc (100 мл). Объе 33 диненные органические слои промывают водой(3400 мл) и концентрируют в вакууме до 53,4 г (100%) масла, которое, по данным ВЭЖХанализа, чистое 98%. 1H ЯМР (300 МГц,ДМСО-d6). Данные ЯМР соответствуют нужному продукту. Препаративный пример 42. 4-(4-(2-Амино 2-метилпропил)фенокси)бензонитрил. Водный 30% H2O2 (62,1 мл, 548 ммоль) добавляют по каплям к смеси 4-(4-(2-амино-2 метилпропил)фенокси)бензонитрила (53,2 г, 200 мл), К 2 СО 3 (15,78 г, 114 ммоль) и ДМСО (270 мл) в течение 20 мин, в то время как температуру поддерживают на уровне 20 С с помощью охлаждающей бани. Смесь перемешивают при этой температуре в течение 1 ч после завершения добавления и затем постепенно добавляют воду (209 мл). Суспензию охлаждают на ледяной бане при перемешивании в течение 1 ч, и продукт реакции затем отфильтровывают, промывают водой и сушат в вакууме при 50 С. Получают 55,0 г (97%) твердого белого вещества. Анализ методом ВЭЖХ показывает чистоту 99%. 1H ЯМР (300 МГц, ДМСО-d6). Данные ЯМР соответствуют нужному продукту. Препаративный пример 43. 2-(4-(2-Амино 2-метилпропил)фенокси)-5-карбоксинитрилпиридин. Смесь уксуснокислой соли 4-(2-амино-2 метилпропил)фенола (22,53 г, 100 ммоль), порошкообразного К 2 СО 3 (34,55 г, 250 ммоль) иDMAC (275 мл) нагревают до 100 С. Добавляют толуол (94 мл) и смесь медленно нагревают с обратным холодильником. Температуру кипения смеси поднимают путем отгонки толуола и воды в ловушку для воды, пока температура не достигнет 140 С. Затем дают смеси остыть до температуры ниже 100 С и добавляют 2 хлороникотинонитрил (13,86 г, 100 ммоль) при орошении толуолом (50 мл). Смесь снова нагревают с обратным холодильником и поднимают температуру кипения смеси до 140 С, как ранее. Затем ловушка для воды наполняется толуолом,и кипячение продолжают с обратным холодильником в течение 40 мин, когда ВЭЖХ показывает, что 2-хлороникотинонитрила не осталось, но реакция не завершена. После охлаждения реакционной смеси до температуры ниже температуры кипения добавляют дополнительно 2 хлороникотинонитрил (0,63 г, 4,5 ммоль) и продолжают кипячение с обратным холодильником в течение 90 мин. Реакционную смесь охлаждают до комнатной температуры, и отфильтровы 002778 34 вают твердые вещества и промывают толуолом. Фильтрат концентрируют на роторном испарителе до образования 41 г сиропа, который растворяют в EtOAc (200 мл). Этот раствор промывают водой (200 мл) и водный слой снова экстрагируют EtOAc (50 мл). Объединенные органические слои промывают водой (3200 мл) и концентрируют в вакууме до 26,93 г твердого вещества, 100% от теории. ВЭЖХ показывает чистоту 94,3%. Данные 1H ЯМР (300 МГц,ДМСО-d6) соответствуют нужному продукту. Перечисленные ниже соединения получают способом, аналогичным показанному на схеме IV, и/или способами, описанными в препаративных примерах 24-33, или известными методами. Пример 1 представляет комбинаторный/параллельный способ получения соединений настоящего изобретения на матрице. Пример 1. Расставляют в сетку 58 4-мл ампулы с завинчивающимися крышками. В каждую из ампул в восьми рядах сетки добавляют 33 мкмоля кетона (из препаративных примеров 24-37, 44-111 или коммерчески доступного) и один кетон на ряд в виде исходного раствора в метаноле (0,5 М, 65 мкл). Если растворимость представляет проблему, используют смесь ацетонитрила с метанолом или ДМФА. В ампулы в каждой колонке сетки добавляют 50 мкмоль гидрохлорида амина - один гидрохлорид амина на колонку (из препаративных примеров 1-16 или доступный коммерчески), в виде исходного раствора в метаноле (0,5 М, 100 мкл). Затем в каждую ампулу добавляют смолу VIII (18-20 мг,1,01 мэк/г, 70-90 мкэк основания). Затем каждую ампулу закрывают футерованной тефлоном крышкой. Затем суспензии встряхивают в течение 24 ч, после чего содержимое каждой ампулы обрабатывают приблизительно 30 мг (2,5 ммоль ВН 4-/г смолы, 75 мкмоль) борогидрированной смолы Amberlite IRA400 (Aldrich Chemical). Крышки снова закрывают и ампулы встряхивают еще в течение 24 ч, а затем в каждую ампулу добавляют 150 мкл метиленхлорида и 40 мг (1 ммоль/г смолы, 0,4 ммоль) полистиролбензальдегидной смолы (Frechet, J.M.; Schuerch,C.J., Am. Chem. Soc., 1971, 93, 492), чтобы удалить избыток исходного первичного амина, и суспензию встряхивают в течение 1 дня. Содержимое каждой ампулы затем фильтруют через ватный вкладыш. Оставшуюся смолу промывают тремя небольшими порциями метанола(приблизительно 200 мкл). Полученные в результате растворы затем обрабатывают 20 мкл конц. НСl (120 мкмоль), чтобы обеспечить образование соли НСl амина, являющегося продуктом реакции, затем содержимое каждой ампулы разбавляют до объема приблизительно 4 мл и по 1 мл каждого раствора переносят в тарированные 4-мл ампулы с завинчивающимися крышками. Растворам дают испаряться досуха в вытяжном шкафу в токе воздуха, а затем ампулы помещают в вакуумную печь на 24 ч при комнатной температуре. Полученные в результате остатки затем взвешивают и передают непосредственно на испытания, не проводя дополнительной очистки. Подобным образом упаривают основную массу вещества (75%). Ниже приводится перечень матриц дополнительных примеров 2-201. Эти соединения 39 получают с использованием комбинаторного/параллельного метода по настоящему изобретению. Все условия реакций, указанных в таблице, одинаковы и соответствуют, по существу, схеме II и примеру 1. Используемые сетки одинаковы, и описаны в верхнем углу матрицы 58. Изменяющиеся функциональные группы 40 показаны в рядах и колонках. Кетоны и амины,указанные в таблице, получают в соответствии со схемами и препаративными примерами, описанными здесь, или методами, известными в технике. 4-(Оксиранилметокси)-1 Н-индол-2-карбоксамид (в диоксане 2:1) (0,304 г, 0,36 ммоль, 1 экв.) и продукт препаративного примера 40(0,312 г, 1,09 ммоль), 3 экв.) суспендируют в абсолютном этаноле (15 мл). Суспензию нагревают до 50 С, в результате чего все реагенты переходят в раствор, и нагревание продолжают в течение 12 ч. После завершения реакции смесь охлаждают до комнатной температуры, и постепенно образуются кристаллы. Твердое белое вещество отфильтровывают и сушат при пониженном давлении и получают 0,190 г (выход метилпропил)фенокси)бензамида (21,0 г, 73,8 ммоль) в метаноле (260 мл) нагревают при перемешивании при 45 С в течение 22 ч. Затем смесь нагревают еще в течение 4 ч при 60 С. Реакционную смесь концентрируют в вакууме до маслянистого остатка. Этот остаток обрабатывают этилацетатом (200 мл), водой (35 мл) и 1N HCl (33 мл). Органический раствор промывают два раза 1N раствором HCl (2 мл) в воде(33 мл). Этилацетатный раствор сушат надNa2SO4, фильтруют и фильтрат концентрируют в вакууме до бледно-желтого пенного остатка. Сырой продукт очищают флэш-хроматографией с 400 г силикагеля 230-400 меш и этилацетатом и этанолом (градиент от 5:1 до 4:1). Упаривание соответствующих фракций дает 14,8 г (84,5%) нужного продукта в виде белого пенистого твердого вещества. Данные 1 Н ЯМР (ДМСО-d6) соответствуют нужному продукту. Пример 204. Гидрохлорид (S)-4-(3-[N-(2[4-(4-карбамоилфенокси)фенил]-1,1-диметилэтил)амино]-2-гидроксипропокси)индола. Раствор продукта примера 203 (11,48 г,24,24 ммоль) в этилацетате (150 мл) при перемешивании обрабатывают, постепенно добавляя, 1 М раствором НСl в этилацетате (24 мл, 24 ммоль) при температуре окружающей среды. К образующемуся в результате белому осадку добавляют еще EtOAc (50 мл) и суспензию перемешивают приблизительно в течение 1 ч при температуре окружающей среды. Продукт в 45 виде суспензии фильтруют под давлением через фильтр из нержавеющей стали в атмосфере азота. Собранный продукт держат под постоянным током азота в течение приблизительно 2 ч. Затем фильтр помещают в вакуумную печь на ночь при 60 С. Продукт сушат до постоянного веса в сушильной печи при 75 С и получают 10,38 г (84,1%) продукта в виде белого твердого вещества. 1 Смесь 4-(2-амино-2-метилпропил)фенокси)-5-карбоксамидпиридина (21,11 г, 74,00 ммоль), (S)-(+)-4-(оксиранилметокси)-1 Н-индола (7,00 г, 37,0 ммоль), НОАс (90,4 мг, 1,48 ммоль) и воды (12 мл) в МеОН (260 мл) перемешивают при 60 С в течение 19,25 ч. Смесь охлаждают и концентрируют в вакууме до масла. Этот остаток растворяют в EtOAc (185 мл) и воде (75 мл), и полученные в результате слои разделяют. Органический слой экстрагируют растворами 1N HCl (34 мл), 1N HCl/MeOH (30 мл/5 мл), 1N HCl/вода/МеОН (15 мл/20 мл/10 мл) и 1N HCl (10 мл). Объединенные кислые водные экстракты (содержащие избыток исходного амина и продукты реакции) промываютEtOAc (40 мл). Органические слои объединяют и отбрасывают. Делают рН водного слоя слегка щелочным (рН 7,0 -7,5), добавляя 5N NaOH (10 мл) и 1N HCl (1 мл). Затем водный слой экстрагируют EtOAc (100 мл, 250 мл). Поднимают слегка рН водного слоя добавлением 5N NaOH(0,25 мл). Водный слой разбавляют водой (5 мл) и МеОН (5 мл) и затем экстрагируют EtOAc (250 мл). Повышают рН водного слоя добавлением 5N NaOH (1 мл) и водный слой еще раз экстрагируют EtOAc (250 мл). Снова повышают рН водного слоя, добавляя 5N NaOH (1 мл), и водный слой снова экстрагируют EtOAc (250 мл). Объединенные органические экстракты щелочного водного слоя концентрируют в вакууме приблизительно до 300 мл. Органический слой промывают водой (50 мл), и затем концентрируют в вакууме до 16,64 г масла. Очистка 16,31 г этого масла флэш-хроматографией на силикагеле 230-400 меш с использованием в качестве элюента смеси хлороформ/метанол/ 28% аммиак (25:4:0,1) дает в качестве продук 002778 46 та 13,35 г (76,02%) свободного основания. Данные 1H ЯМР (ДМСО-d6) соответствуют нужному продукту. Раствор свободного основания (11,86 г 25,00 ммоль) в EtOAc (280 мл) в изопропаноле(20 мл) при перемешивании подкисляют, добавляя по каплям 34 мл (приблизит 25 ммоль HCl) приблизительно 0,725 М раствора HCl (газ) вEtOAc. Полученную в результате суспензию перемешивают в течение 2 ч при температуре окружающей среды. Смесь фильтруют (под давлением азота). Остаток на фильтре дважды промывают EtOAc (220 мл) и сушат в вакууме при 50 С. Получают 12,48 г (97,65%) продукта белого цвета. 1H ЯМР соответствует наружному продукту и показывает небольшие количества Диамин, полученный в препаративном примере 20 (0,304 г, 0,65 ммоль), растворяют в ледяной уксусной кислоте (10 мл) и сразу обрабатывают раствором нитрата натрия (0,047 г,0,68 ммоль) в воде (5 мл). Реакционную смесь перемешивают в течение 5 мин, а затем упаривают досуха. Полученный в результате остаток очищают с использованием колоночной хроматографии при элюировании 20% МеОН в СНСl3,и получают 0,27 г (87%) твердого вещества. МС: m/z (%) = 475,9 (100%), 249,0 (5%),950,7 (5%). 1H ЯМР (300 МГц, d-MeOH):1.35(8H, m); 7.80 (2 Н, d). Примечание. В приведенных выше спектрах ЯМР аббревиатуры означают следующее: Неочищенный 4-(4-(2-(N-2S)-3-(2,3 диаминофенокси)-2-гидроксипропил)амино)-2 метилпропил)фенокси)бензамид (1,49 г, 3,2 ммоль), полученный в препаративном примере 20, растворяют в 1N HCl (100 мл). Добавляют толуол (100 мл) и двухфазную смесь обрабатывают трифосгеном (4,7 г, 16 ммоль) и энергично перемешивают в течение 18 ч. В ходе реакции на стенках реакционного сосуда оседает смола. Жидкость декантируют, а смолу растворяют в МеОН. Раствор выливают на диоксид кремния и хроматографируют на 200 г диоксида кремния с элюированием смесью EtOAc, воды и нпропанола (80 об./15 об./5 об., встряхивают и используют верхний слой) для удаления исходного вещества. Затем колонку элюируют СНСl3/МеОН/NН 4OН (25 об./5 об./1 об.) и получают продукт реакции. После концентрирования и азеотропной отгонки с EtOH получают 715 мг(46%) белой пены. МС. Данные 1H ЯМР (ДМСО-d6) соответствуют нужному продукту. Полученное выше свободное основание(528 мг, 1,08 ммоль) растворяют в EtOH и обрабатывают 4N раствором НСl в диоксане (0,75 мл, 3,0 ммоль). Раствор упаривают в вакууме и получают 595 мг гидрохлорида в виде белой пены. МС. Данные 1H ЯМР (ДМСО-d6) соответствуют нужному продукту. Как отмечалось ранее, соединения настоящего изобретения являются сильными селективными агонистами адренергического рецептора 3. Эту фармакологическую активность определяют с помощью анализа функционального агониста 3. 49 Функциональные агонисты Анализ 3 Клеточные линии. Экспрессируют ДНК h2 из плазмиды 57537, полученной из Американской коллекции типовых культур. Адренергические рецепторыh1 и h3 клонируют из человеческих геномных библиотек, используя метод полимеразной цепной реакции с вырожденными зондами. Первичные рецепторы клонируют, экспрессируют и секвенируют для подтверждения идентичности с опубликованными последовательностями (h1: Т.Frielle et al. (1993) Molecular Pharmacology 44: 264-270). Затем эти рецепторы экспрессируют в варианте DXB-11 СНО-клеток, используя вектор, восстанавливающий тетрагидрофолятредуктазу и устойчивость к гидромицину. Известна экспрессирующая крысиный рецептор 3 клеточная линия СНО (Mol/pharm., Vol. 40, pp. 89599 (1991. Клетки СНО выращивают в 10% диализованной FBS/DMEM с высоким содержанием глюкозы/0,1% пролина. Анализ сАМР (цАМФ). Собирают соскобом клеточные мембраны вышеуказанной клеточной линии с использованием гипотонического 25 мМ Hepes (рН 7,4),1 мМ ЭДТК, 20 мкг/мл лейпептина, 1 мМ ФМСФ-буфера, с последующим дифференциальным центрифугированием. Мембраны инкубируют в 25 мМ трис (рН 7,6), 0,2% БСА, 2,6 мМ Мg, 0,8 мМ АТФ, 0,1 мМ GTP, 5 мМ креатинфосфата, 50 Е/мл креатинкеназы, 0,2 мМIBMX, при 32 С. Добавляют агонисты и продолжают инкубацию в течение 15 мин. Образовавшийся сАМР анализируют с использованием метода иммунного анализа с флуоресцентным индикатором. Анализ интактных клеток выполняют, используя суспендированные клетки, извлеченные из культуральных емкостей обработкой трипсином. Клетки предварительно инкубируют с 0,5 мМ IBMX при 37 С. Добавляют агонисты и продолжают инкубацию в течение 15 мин. Инкубацию прекращают нагреванием суспензии в кипящей воде. При этих soleus инкубациях сАМР и cGMP анализируют с помощью РИА(Amerscham). Соединения настоящего изобретения являются агонистами рецептора 3. В технике в качестве неселективного агониста 3 признан изопротеренол, и его широко используют в качестве вещества для сравнения при оценке активности соединений. См. Trends in Pharm. Sci.,15: 3 (1994). В анализе функциональных агонистов 3 соединения показывают, по меньшей мере, 30%, предпочтительно 50% и наиболее предпочтительно свыше 85% от ответа изопротенерола при однократной дозе 50 мкмоль. Титрование ответа дозы на описанные агонисты дает величины EC50 10 мкМ, предпочтительно 1 ммоль. При функциональном анализе титро 002778 50 вание дозы дает EC50 для изопротенерола 1,1 + 0,5 мкМ. При скрининге в функциональном анализе по отношению к рецепторам 1 и 2 эксперименты с титрованием дозы показывают, что с соединениями изобретения наблюдается весьма сниженное раздражение рецепторов, или вовсе не наблюдается. Это определяют измерением истиной активности (максимального полученного ответа) по сравнению с изопротеренолом. Заявленные соединения формулы I являются селективными агонистами рецептора 3 и имеют истинную активность 3% от ответа на изопротенерол. Таким образом, соединения настоящего изобретения являются селективными агонистами адренергического рецептора 3. В качестве агонистов 3 соединения полезны для лечения состояний у млекопитающего,при которых, как показано, роль играет рецептор 3. Предпочтительным млекопитающим объектом лечения является человек. Соотношение между модуляцией рецептора 3 и лечением заболеваний, таких как диабет типа II и ожирение, точно установлено. Другими такими известными состояниями являются желудочнокишечные расстройства, такие как расстройства двигательной функции желудка и моторики кишечника, астма и депрессия. Таким образом,соединения настоящего изобретения полезны для лечения воспалительного заболевания кишечника (болезни Крона или неспецифического язвенного заболевания), синдрома раздраженной толстой кишки, демпинг-синдрома неспецифической диареи, астмы и депрессии. При лечении млекопитающих, не относящихся к человеку, соединения настоящего изобретения полезны для повышения привеса и/или повышения эффективности усвоения кормов,и/или повышения мясной массы тела, и/или снижения показателя падежа при родах и повышение степени постнатального выживания. Соединения формул I и II предпочтительно включают в композиции перед введением. Следовательно, еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы I или II и один или несколько фармацевтически приемлемых носителей, разбавителей или наполнителей. Фармацевтические композиции настоящего изобретения получают известными методами с использованием хорошо известных и легко доступных ингредиентов. При приготовлении композиций настоящего изобретения обычно активный ингредиент смешивают с носителем,или разбавляют носителем, или включают в носитель, который может находиться в форме капсулы, саше, пакета или другой емкости. Когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким 51 веществом, которое действует как наполнитель,эксципиент или среда для активного ингредиента. Таким образом, композиции могут существовать в форме таблеток, пилюль, порошков,лепешек, саше, капсул, эликсиров, суспензий,эмульсий, растворов, сиропов, аэрозолей (в виде твердого или в жидкой среде), мягких и жестких желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильно упакованных порошков. Конкретными примерами подходящих носителей, эксципиентов и разбавителей являются лактоза, декстроза, сахароза, сорбит, маннит,крахмалы, аравийская камедь, фосфат кальция,альгинаты, тракагант, желатин, силикат кальция, микрокристаллическая целлюлоза, поливинилпирролидон, целлюлоза, водный сироп, метилцеллюлоза, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло. Композиции могут дополнительно содержать смазывающие вещества, смачиватели, эмульгирующие и суспендирующие агенты, консерванты, подслащиватели или корригенты. Композиции изобретения могут быть составлены так,чтобы обеспечить быстрое, длительное или отсроченное выделение активного ингредиента после введения пациенту. Композиции предпочтительно составляют в виде стандартной лекарственной формы, причем каждая доза содержит примерно от 0,1 до примерно 500 мг, предпочтительно и примерно от 5 до примерно 200 мг активного ингредиента. Однако следует иметь в виду, что вводимая доза будет определяться лечащим врачом с учетом сопутствующих обстоятельств, в том числе состояния, от которого лечат, выбора соединения для введения и выбранного способа введения, и,следовательно, вышеуказанные интервалы доз не предназначены для ограничения объема изобретения каким-либо образом. Соединения можно вводить различными путями, включая пероральный, ректальный, трансдермальный,подкожный, местный, внутривенный, внутримышечный или интраназальный способы введения. При всех показаниях типичная суточная доза составляет от 0,05 до 20 мг/кг активного соединения настоящего изобретения. Предпочтительные суточные дозы составляют примерно от 0,1 до примерно 10 мг/кг и наиболее предпочтительно примерно от 0,1 до примерно 5 мг/кг. Однако в случае местного введения стандартная доза составляет примерно от 1 до примерно 500 мкг соединения на см 2 обрабатываемой ткани. Предпочтительно вводимое количество соединения будет находиться в интервале примерно от 30 до примерно 300 мкг/см 2, предпочтительно примерно от 50 до примерно 200 мкг/см 2 и наиболее предпочтительно примерно от 60 до примерно 100 мкг/см 2. Приведенные далее примеры композиции являются только иллюстративными и не пред 002778 52 назначены для какого-либо ограничения объема изобретения. Композиция 1 Получают твердые желатиновые капсулы,используя перечисленные далее ингредиенты. Количество,мг/капсулу Гидрохлорид (S)-4-(3-([N-(2-[4-(4 карбамоилфенокси)фенил]-1,1-диметилэтил)амино]-2-гидроксипропокси) индола Крахмал, сухой Стеарат магния Всего Указанные выше ингредиенты смешивают и заполняют ими твердые желатиновые капсулы в количестве 460 мг. В приведенном выше описании описаны принципы, предпочтительные варианты и способы работы по настоящему изобретению Однако, настоящее изобретение, которое здесь защищается, не должно истолковываться как ограниченное конкретными описанными формами, так как они должны рассматриваться как иллюстративные, а не ограничительные. Специалисты в этой области техники могут осуществить вариации и изменения без отхода от сущности настоящего изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы IR4 является необязательно замещенным гетероциклом или группой, выбранной из группы,состоящей изX2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода; 53 или R6 вместе с Х 2 и с атомом углерода, к которому присоединен Х 2, образуют (С 3-С 8) циклоалкил; или R6 вместе с X2, с атомом углерода, к которому присоединены оба Х 2 и R6 и R4 образуютR9 представляет галоген, CN, OR10, (C1-C4) алкил, (C1-C4)галоидалкил, CO2R2, CONR11R12,СОNН(С 1-С 4 алкокси),CONHC1-C4)алкил),SR2, CSNR2, CSNR11R12, SO2R2, SO2NR11R12,SOR2, NR11R12, фенил, необязательно замещенный группой -CONR11R12, насыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота или (C2C4)алкенил, замещенный CN, СО 2R2 илиR10 представляет (C1-C4)алкил, (С 1 С 4)галогеналкил,(СН 2)nС 3-С 8)циклоалкил,(СН 2)nфенил, необязательно замещенный CN,CONR11R12 или СОNН(С 1-С 4 алкокси), (СН 2)nнасыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота,необязательно замещенный одним или двумя заместителями, выбранными из CN, CONR11R12 или (C1-C4) алкокси;n равен 0, 1, 2 или 3; или его фармацевтически приемлемая соль, при условии, что когда R4 представляет то ни R5, ни R6 не являются водородом. 2. Соединение по п.1, где R10 представляет фенил или пиридил; причем указанный фенил или пиридил замещены -CONR11R12. 3. Соединение формулы II где Х 1 представляет -О-, -ОСН 2-; связь между А 3 и А 4 является либо одинарной, либо двойной связью; А 3 и А 4 представляют независимо атомы углерода или азота;R4 является необязательно замещенным гетероциклом или группой, выбранной из группы,состоящей из Х 2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода;CONH(С 1-С 4 алкокси), (СН 2)n-насыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота, необязательно замещенный одним или двумя заместителями,выбранными из CN, CONR11R12 или (C1-C4)алкокси;n равен 0, 1, 2 или 3; или его фармацевтически приемлемая соль. 4. Соединение по п.3 формулы где A5 представляет СН или N;X1 представляет -ОСН 2-; и Х 2 представляет метилен или этилен. 5. Соединение по п.3, выбранное из группы, состоящей из или их фармацевтически приемлемых солей; илиN и А 3 представляет СН или N. 7. Соединение по п.6 формулы где A5 представляет СН или N. 8. Соединение по п.7, где R7 представляет Н, X1 представляет -OCH2- и Х 2 представляет метилен или этилен, и R10 представляет фенил или пиридил, причем указанные фенил или пиридил замещены CONR11R12, СО 2R2, CN. 9. Соединение по п.8, выбранное из группы, состоящей из 56 или их фармацевтически приемлемых солей. 10. Применение соединения по любому из пп.1-9 для получения фармацевтического средства. 11. Применение соединения по любому из пп.1-9 для лечения диабета типа II. 12. Применение соединения по любому из пп.1-9 для лечения ожирения. 13. Применение соединения по любому из пп.1-9 для активации рецептора 3. 14. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение по любому из пп.1-9, в сочетании с одним или несколькими фармацевтически приемлемыми носителями, наполнителями или разбавителями. 15. Соединение формулы III где A5 представляет СН или N; Х 2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода;R14 представляет (C1-C4)алкил, (C1-C4) галоидалкил, гидрокси, карбокси, тетразолил,ацил, COOR2, CONR11R12, CONHC1-C4) алкокси), циано, (C1-C4)алкокси, (C1-C4)алкил,фенил, нитро, NR11R12, NНСОC1-C4)алкил),NНСО(бензил), NHCO(фенил), SR2, SC1-C4) алкил),ОСОC1-C4)алкил),SO2(NR11R12),SO2C1-C4)алкил) или SO2(фенил); или его фармацевтически приемлемые соли. 16. Способ получения соединения по п.1 формулы IA где А 5 представляет СН или N; включающий на стадии 1 гидролиз соединения формулы IB и на стадии 2 взаимодействие продукта со стадии 1 с образованием соли присоединения кислоты. 17. Способ получения соединения по п.1,включающий на стадии 1 взаимодействие эпоксида формулы (XI) и на стадии 2 взаимодействие продукта со стадии 1 с образованием соли присоединения кислоты. 18. Соединение формулы IR4 представляет необязательно замещенный гетероцикл или группу, выбранную изCONH(С 1-C4 алкокси), (СН 2)n-насыщенный или ненасыщенный 5-6-членный гетероцикл, содержащий от 1 до 4 атомов азота, необязательно замещенный одним или двумя заместителями,выбранными из CN, CONR11R12 или (C1 С 4)алкокси;R11 и R12 независимо представляют Н, С 1C4 алкил или вместе с атомом азота, к которому они присоединены, образуют морфолинил, пиперидинил, пирролидинил или пиперазинил;n равно 0, 1, 2 или 3; или его фармацевтически приемлемая соль. 19. Соединение по п.18, гдеX2 является связью или представляет прямой или разветвленный алкилен, содержащий от 1 до 5 атомов углерода;
МПК / Метки
МПК: C07D 209/04, A61K 31/416
Метки: 3-адренергические, селективные, бетта, агонисты
Код ссылки
<a href="https://eas.patents.su/30-2778-selektivnye-betta-3-adrenergicheskie-agonisty.html" rel="bookmark" title="База патентов Евразийского Союза">Селективные бетта 3-адренергические агонисты</a>
Селективные β3-адренергические агонисты.

Номер патента: 1217
Опубликовано: 25.12.2000
Авторы: Мэттьюз Дональд П., Рито Кристофер Дж., Белл Майкл Г., Винтер Марк А., Кровелл Томас А., Эврард Дебора А., Шакер Энтони Дж., Макдональд Джон Х.
МПК: C07D 257/04, A61K 31/41
Метки: агонисты, селективные, beta;3-адренергические
Формула / Реферат:
1. Соединение формулы где R1 представляет ОН, галоген, SO2NHR2, CO2R2, CONHR2, NHCOR2, -NH (необязательно замещенный арил), СF3 или CF2H, причем термин "арил" обозначает необязательно замещенный фенил или нафтил; R1' представляет Н, галоген, C1-C4 алкил, ОН, SO2NHR2, СO2R2, CONHR2, NHCOR2, СF3 или CF2H; R2 представляет Н, C1-C4 алкил или арил, где арил определен как указано выше; R3 представляет Н или C1-C4 алкил; R4 представляет остаток,...
Бетта-сульфонил гидроксамовые кислоты в качестве ингибиторов матричных металлопротеиназ.

Номер патента: 1460
Опубликовано: 23.04.2001
Авторы: Харпер Дональд Э., Варпехоски Марта А.
МПК: C07C 317/44, A61K 31/164, A61P 1/04...
Метки: ингибиторов, металлопротеиназ, бетта-сульфонил, качестве, матричных, кислоты, гидроксамовые
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемые соли, где R1 представляет собой a) С4-12алкил, b) C4-12алкенил, c) C4-12алкинил, d) -(СН2)h-С3-8циклоалкил, e) -(СН2)h-арил, f) -(СН2)h-арил, замещенный C1-4алкилом, C1-4алкокси, галогеном, -NO2, -CF3, -CN, -N(С1-4алкил)2, g) -(СН2)h-гет или h) -(CH2)h-гет, замещенный C1-4алкилом или галогеном; R2 представляет собой a) C1-12алкил, b) C1-12алкил, замещенный от одного до трех...
Таблетки, содержащие бетта-лактамный антибиотик, и способ их изготовления.

Номер патента: 1089
Опубликовано: 30.10.2000
Автор: Ямагути Хисами
МПК: A61K 31/545
Метки: бетта-лактамный, антибиотик, таблетки, способ, содержащие, изготовления
Формула / Реферат:
1. Таблетка, содержащая b-лактамный антибиотик, которая включает от 60 до 85 вес.% b-лактамного антибиотика, от 1 до 10 вес.% гидроксипропилцеллюлозы с низкой степенью замещения и/или поперечно-сшитого поливинилпирролидона в качестве дезинтегратора и от 0,5 до 2 вес.% связующего в одной таблетке. 2. Таблетка по п.1, в которой связующим является поливинилпирролидон, гидроксипропилцеллюлоза или гидроксипропилметилцеллюлоза. 3. Таблетка по п.1...
Предыдущий патент: Способ получения гидроксамовых кислот
Следующий патент: Искусственный остров, опора искусственного острова и способ сооружения искусственного острова
Случайный патент: Способы и композиции для перорального введения белковых и пептидных терапевтических средств