Производные гексагидро-7-1н-азепин-2-илгексановой (или гексеновой) кислоты в качестве ингибиторов индуцибельной синтазы оксида азота
Номер патента: 5817
Опубликовано: 30.06.2005
Авторы: Франчик Таддеус С., Триведи Махима, Мурманн Алан Э., Хансен Дональд У., Ауасти Алок К., Уэббер Роналд Кит, Снайдер Джеффри С.


























Формула / Реферат
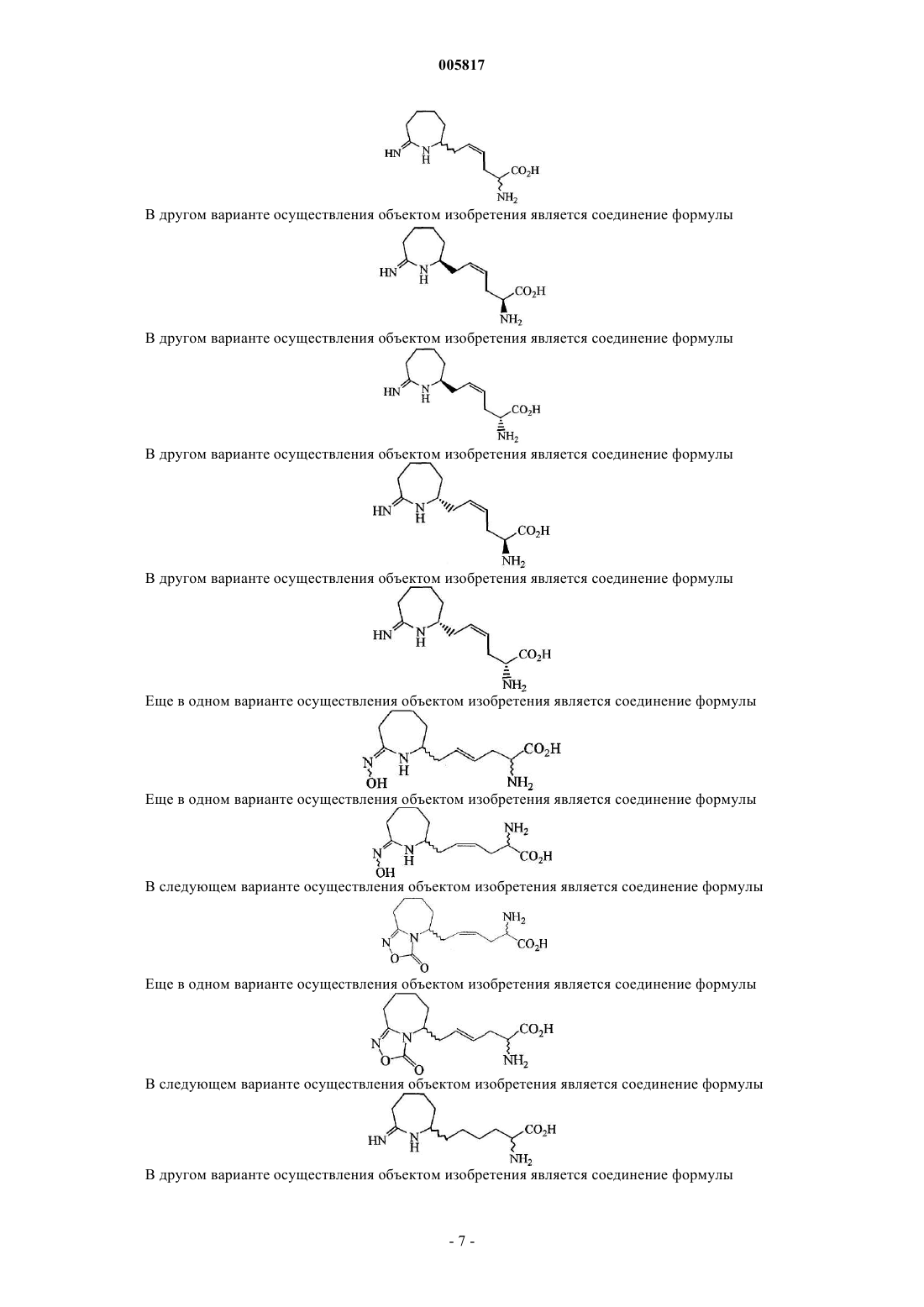
1. Соединение формулы

или его фармацевтически приемлемая соль.
2. Соединение формулы

или его фармацевтически приемлемая соль.
3. Соединение по п.2, где соединение представлено формулой

4. Соединение формулы

5. Соединение формулы

или его фармацевтически приемлемая соль.
6. Соединение по п.5, где соединение представлено формулой

или его фармацевтически приемлемая соль.
7. Соединение по п.5, где соединение представлено формулой

или его фармацевтически приемлемая соль.
8. Соединение по п.5, где соединение представлено формулой

или его фармацевтически приемлемая соль.
9. Соединение по п.5, где соединение представлено формулой

или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция, содержащая соединение по п.5.
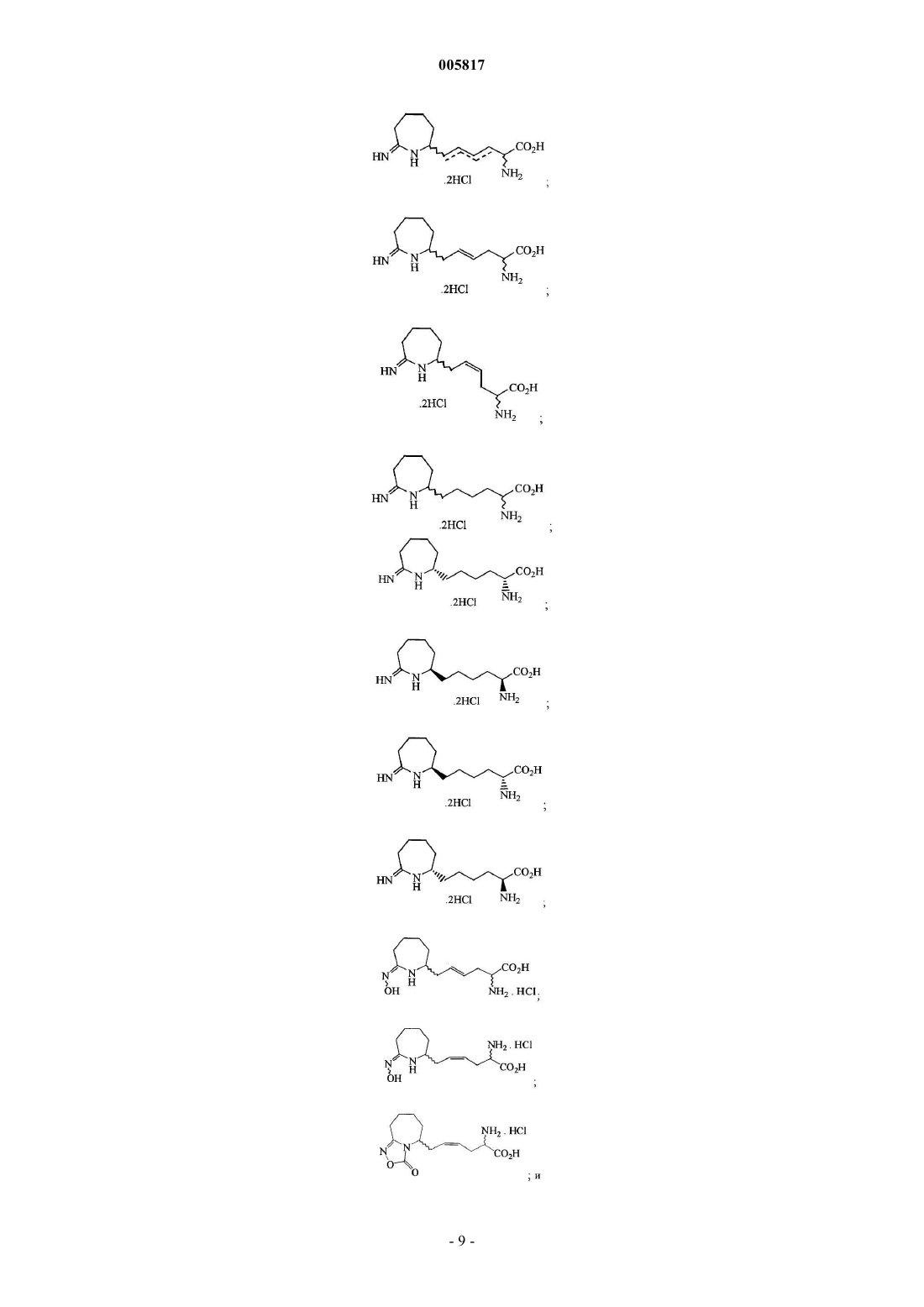
11. Соединение, выбранное из группы, включающей

12. Соединение по п.11, представляющее собой

13. Соединение по п.11, представляющее собой

14. Соединение по п.11, представляющее собой

15. Соединение по п.11, представляющее собой

16. Соединение по п.11, представляющее собой

17. Соединение по п.11, представляющее собой

18. Соединение формулы

или его фармацевтически приемлемая соль.
19. Соединение формулы

или его фармацевтически приемлемая соль.
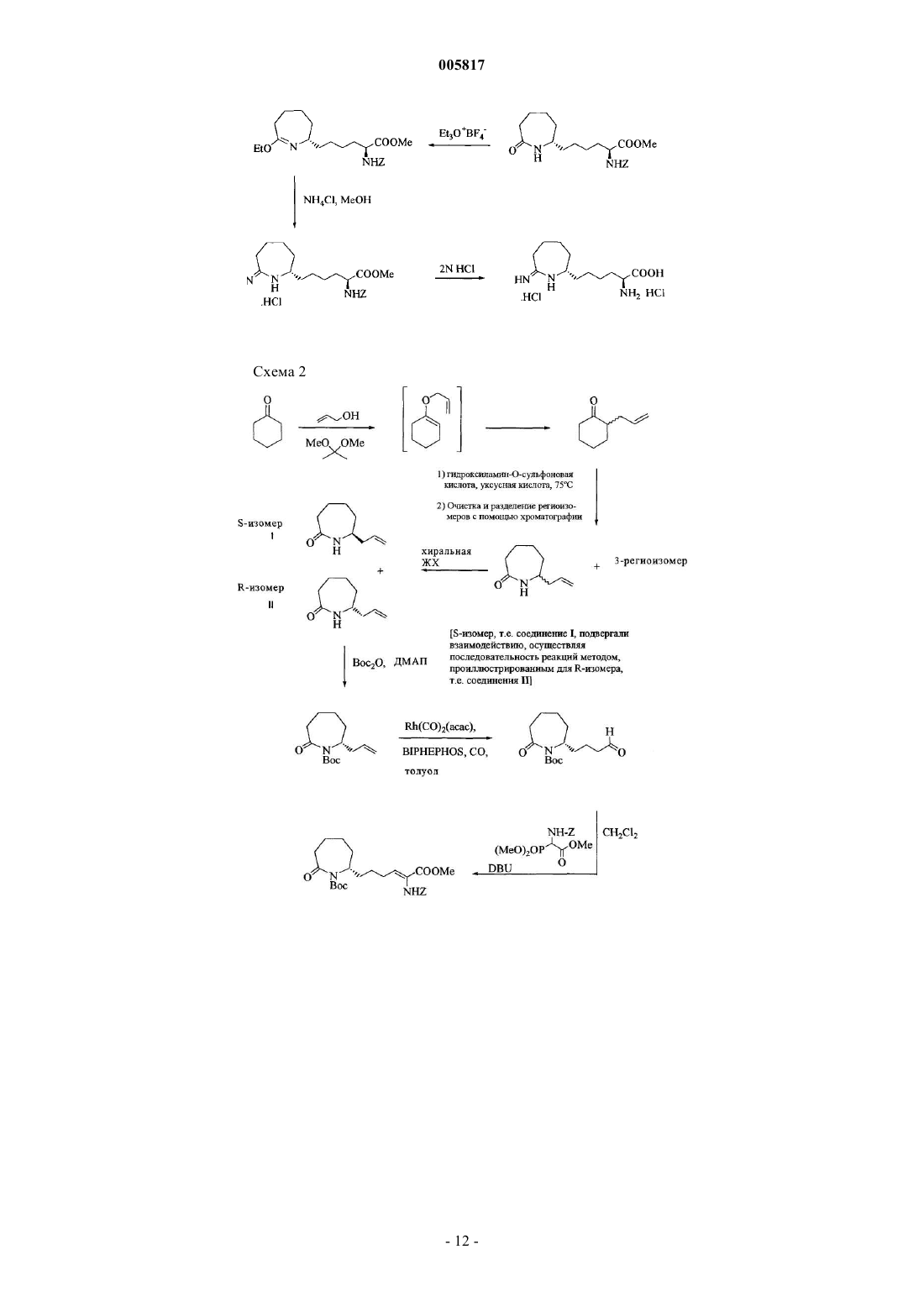
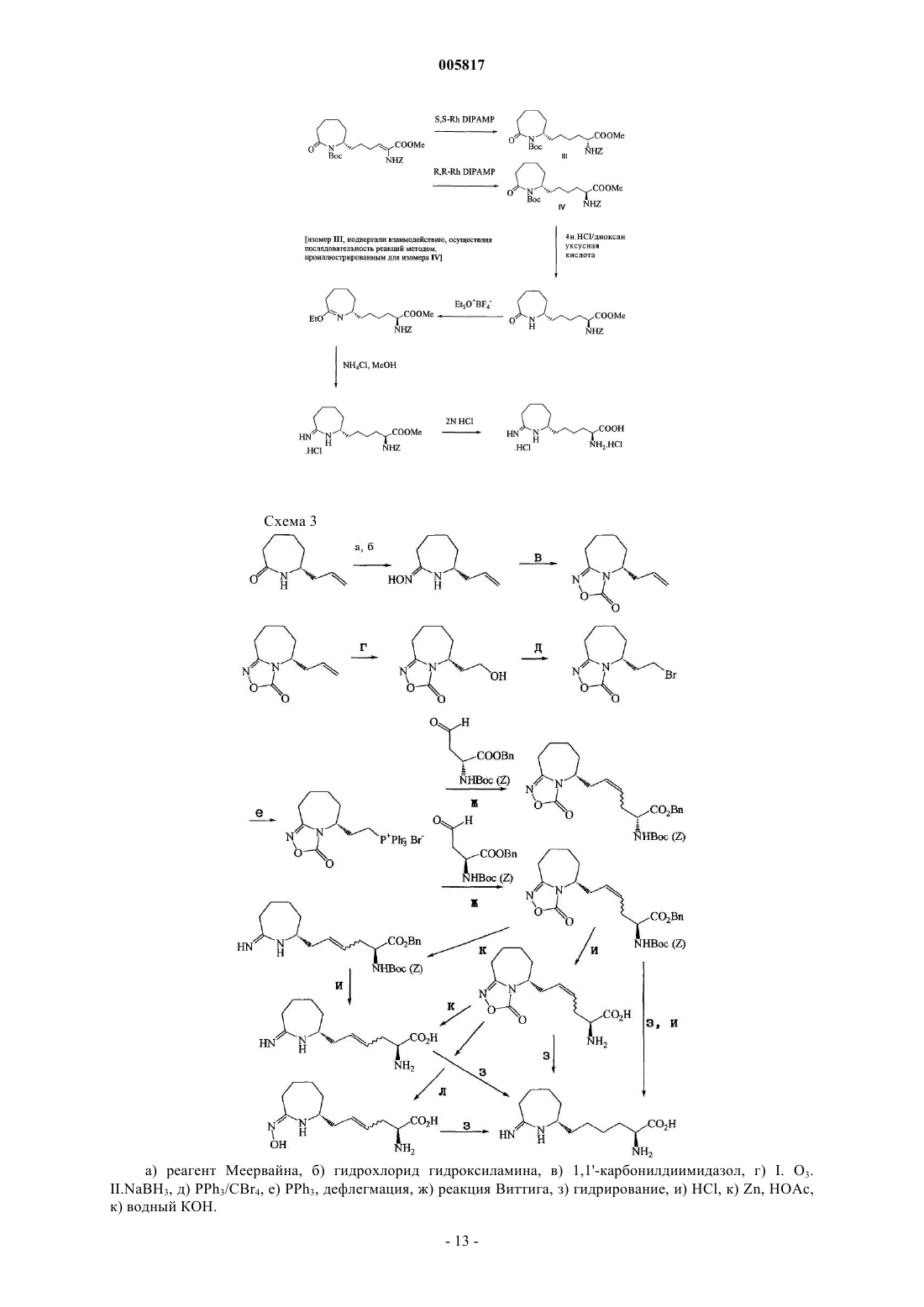
Текст
005817 Область техники, к которой относится изобретение Настоящее изобретение относится к производным гомоиминопиперидинилгексановой кислоты и их применению в терапии, прежде всего к их применению в качестве ингибиторов синтазы оксида азота. Предпосылки создания изобретения С начала 1980-х годов известно, что вызываемая ацетилхолином релаксация сосудов зависит от эндотелия сосудов. В настоящее время установлено, что эндотелиальный фактор релаксации (EDRF) представляет собой оксид азота (NO), который продуцируется в эндотелии сосудов синтазой оксида азота(NOS). Вазодилататорная активность NO известна уже более 100 лет. Кроме того, NO представляет собой обладающую активностью субстанцию, образующуюся из амилнитрила, глицерилтринитрата и других азотистых вазодилататоров. Тот факт, что NO представляет собой EDRF, подтверждается открытием биохимического пути синтеза NO из аминокислоты L-аргинина с помощью фермента NO-синтазы. Оксид азота является эндогенным стимулятором растворимой гуанилатциклазы. Помимо того, что он вызывает зависящую от эндотелия релаксацию, NO участвует во многих биологических реакциях, в том числе в цитотоксичности фагоцитов и взаимодействиях типа клетка-клетка в центральной нервной системе. Существуют по меньшей мере три перечисленные ниже типа NO-синтазы:(I) конститутивный Са/кальмодулинзависимый фермент, локализованный в эндотелии, приводящий к высвобождению NO в ответ на стимуляцию рецептора или физический стимул,(II) конститутивный Са/кальмодулинзависимый фермент, локализованный в головном мозге, приводящий к высвобождению NO в ответ на стимуляцию рецептора или физический стимул, и(III) независящий от Са, который индуцируется после активации клеток гладкой мускулатуры сосуда, макрофагов, эндотелиальных клеток и многочисленных других клеток эндотоксином и цитокинами. После экспрессии эта индуцибельная синтаза оксида азота (далее в настоящем описании обозначенная как iNOS) непрерывно обеспечивает синтез больших количеств NO в течение длительных периодов времени. Оксид азота, продуцируемый каждым из двух конститутивных ферментов, принимает участие в механизме трансдукции, лежащем в основе нескольких физиологических реакций. Продуцируемый индуцибельным ферментом NO представляет собой молекулу, обладающую цитотоксическим действием в отношении опухолевых клеток и инвазивных микроорганизмов. По-видимому, побочные действия, вызываемые избыточным производством NO, в частности патологическая вазодилатация и повреждение ткани, могут быть в основном обусловлены NO, синтезируемым с помощью iNOS. В настоящее время все возрастает количество доказательств того, что NO может участвовать в дегенерации хрящевой ткани, которое происходит при определенных состояниях, таких как артрит, известно также, что синтез NO увеличивается при ревматоидном артрите и остеоартрите. Некоторые из предложенных для терапевтического применения ингибиторов NO-синтазы не обладают селективным действием (неселективные NO-синтазы); они ингибируют как конститутивные, так и индуцибельные NO-синтазы. При применении таких не обладающих селективным действием ингибиторов NO-синтазы необходимо принимать особые меры предосторожности для того, чтобы избежать возможных серьезных последствий чрезмерного ингибирования конститутивной NO-синтазы, такие последствия включают гипертензию и возможный тромбоз и повреждение ткани. В частности, было рекомендовано в случае терапевтического применения L-NMMA (неселективный ингибитор NO-синтазы) для лечения токсического шока осуществлять в процессе лечения непрерывное наблюдение за величиной кровяного давления. Таким образом, хотя неселективные ингибиторы NO-синтазы являются ценными с терапевтической точки зрения при условии принятия соответствующих мер предосторожности, ингибиторы NO-синтазы, обладающие селективным действием в том смысле, что они ингибируют индуцибельную NO-синтазу в существенно большей степени, чем конститутивные изоформы NO-синтазы, могут быть даже более предпочтительными с точки зрения терапии и их применение может быть более простым (S. Moncada и Е. Higgs, FASEB J., 9, 1319-1330, 1995). В патенте US5854234, содержание которого полностью включено в настоящее описание в качестве ссылки, описаны соединения, которые обладают способностью ингибировать синтез оксида азота и предпочтительно ингибировать индуцибельную изоформу синтазы оксида азота. Краткое изложение сущности изобретения При создании изобретения были выявлены соединения, которые являются обладающими высокой селективностью ингибиторами индуцибельной синтазы оксида азота (iNOS). В широком смысле настоящее изобретение относится к новым соединениям, фармацевтическим композициям, способу получения новых соединений, способу приготовления фармацевтических композиций и способам применения указанных соединений и композиций для ингибирования или модуляции синтеза оксида азота в организме пациента, нуждающегося в таком ингибировании или модуляции, путем введения соединения, которое обладает способностью ингибировать или модулировать индуцибельную изоформу синтазы оксида азота в большей степени, чем конститутивные изоформы синтазы оксида азота. Другим объектом настоящего изобретения является способ уменьшения уровней оксида азота в организме пациента, нуждающегося в таком уменьшении. Соединения по настоящему изобретению об-1 005817 ладают ценной ингибирующей синтазу оксида азота активностью и следует ожидать, что их можно применять для лечения или профилактики заболевания или состояния, для которого существенную роль играет синтез или избыточный синтез оксида азота. Соединения по настоящему изобретению можно применять среди прочего для лечения воспаления у пациента или для лечения других опосредуемых синтазой оксида азота нарушений, например, в качестве аналгетика при лечении боли и головной боли. Соединения по настоящему изобретению можно применять для лечения боли, включая соматогенную (как ноцицептивную, так и невропатическую) боль, как острую, так и хроническую, и их можно применять в ситуации, в частности при невропатической боли, в которой, как правило, применяют обычные НСПВЛ (нестероидные противовоспалительные лекарственные средства), опиоидные аналгетики или определенные антиконвульсивные средства. Состояния, которые можно облегчать с помощью соединений по настоящему изобретению путем ингибирования производства NO из L-аргинина, включают состояния, связанные с артритом. Например,соединения по настоящему изобретению можно применять для лечения артрита, включая (но не ограничиваясь ими) ревматоидный артрит, спондилоартропатии, подагрический артрит, остеоартрит, системную красную волчанку, ювенильный артрит, острый ревматоидный артрит, энтеропатический артрит,невропатический артрит, псориатический артрит и пиогенный артрит. Кроме того, соединения по изобретению можно применять для лечения астмы, бронхита, менструальных спазмов (например, дисменореи), преждевременных родов, тендинита, бурсита, состояний, связанных с кожными заболеваниями, таких как псориаз, экзема, ожоги, солнечный ожог, дерматит, панкреатит, гепатит и послеоперационное воспаление, включая воспаление после офтальмической хирургической операции, такой как хирургическая операция по поводу катаракты и хирургическая операция на хрусталике. Соединения по изобретению можно применять также для лечения желудочно-кишечных состояний, таких как воспалительное заболевание кишечника, болезнь Крона, гастрит, синдром раздраженной толстой кишки и неспецифический язвенный колит. Соединения по изобретению можно применять для лечения воспаления и повреждения тканей при таких заболеваниях, как заболевания сосудов, мигрень, нодозный периартериит, тироидит, апластическая анемия, болезнь Ходжкина, склеродома, ревматическая лихорадка, диабет типа I, нейромышечное заболевание суставов, в том числе тяжелая псевдопаралитическая миастения, заболевание белого вещества, в том числе рассеянный склероз, саркоидоз, нефротический синдром, синдром Бехчета, полимиозит, гингивит, нефрит, гиперчувствительность, отек после повреждения, ишемия миокарда и т.п. Соединения можно применять также для лечения глазных заболеваний, таких как глаукома, ретинит, ретинопатии,увеит, глазная фотофобия и воспаление и боль, связанные с острым повреждением глазной ткани. Среди областей применения соединений по настоящему изобретению наибольший интерес представляет собой лечение глаукомы, особенно в том случае, когда симптомы глаукомы обусловлены производством оксида азота, например в случае опосредуемого оксидом азота повреждения нерва. Соединения можно применять также для лечения легочных воспалений, таких как воспаления, связанные с вирусными инфекциями и кистозным фиброзом. Соединения можно применять также для лечения определенных нарушений центральной нервной системы, таких как деменция коры головного мозга, включая болезнь Альцгеймера, и повреждение центральной нервной системы в результате удара, ишемии и травмы. Эти соединения можно применять также для лечения аллергического ринита, респираторного дистресссиндрома, синдрома эндотоксинового бактериально-токсического шока и атеросклероза. Соединения можно применять также для лечения боли, включая, но не ограничиваясь ими, послеоперационную боль,зубную боль, мышечную боль, боль, сопровождающую височно-нижнечелюстной синдром, и боль, обусловленную раком. Соединения можно применять также для предупреждения деменций, таких как болезнь Альцгеймера. Помимо того, что соединения можно применять для лечения людей, указанные соединения можно применять также в ветеринарии для лечения домашних животных, экзотических животных и сельскохозяйственных животных, включая млекопитающих и других позвоночных. Наиболее предпочтительными животными являются лошади, собаки и кошки. Соединения по настоящему изобретению можно применять в качестве дополнительных терапевтических средств, заменяя полностью или частично другие обычные противовоспалительные лекарственные средства, например применять в сочетании со стероидами, НСПВС, селективными ингибиторами СОХ-2, ингибиторами матриксных металлопротеаз, ингибиторами 5-липоксигеназы, антагонистамиLTB4 (лейкотриен В 4) и ингибиторами LTA4-гидролазы. Другие состояния, при которых соединения по настоящему изобретению могут обладать преимуществами в отношении ингибирования NO, включают сердечно-сосудистую ишемию, диабет (типа I или типа II), застойную сердечную недостаточность, миокардит, атеросклероз, мигрень, глаукому, аневризму аорты, рефлюкс-эзофагит, диарею, синдром раздраженной толстой кишки, кистозный фиброз, эмфизему,астму, бронхоэктаз, гипералгезию (аллодинию), ишемию головного мозга (включая фокальную ишемию,связанный с тромбозом удар, и глобальную ишемию (например, возникающую после остановки сердца, рассеянный склероз и другие заболевания центральной нервной системы, опосредуемые NO, например болезнь Паркинсона. Другие нейродегенеративные заболевания, которые можно облегчать путем-2 005817 ингибирования производства NO, включают дегенерацию нерва или некроз нерва, обусловленные такими заболеваниями, как гипоксия, гипогликемия, эпилепсия, а также в случаях повреждений центральной нервной системы (ЦНС) (таких как травма спинного мозга и головного мозга) - конвульсии и токсичность, обусловленные избыточным давлением кислорода, деменции, такие, например, как пресенильная деменция и деменция, связанная со СПИДом, кахексия, хорея Сиденгама, болезнь Гентингтона, амиотрофический боковой склероз, болезнь Корсакова, имбецильность, связанная с нарушением сосудов головного мозга, нарушения сна, шизофрения, депрессия, депрессия или другие симптомы, связанные с предменструальным синдромом (ПМС), состояние страха и септический шок. Соединения по настоящему изобретению предпочтительно следует применять для лечения также и других заболеваний или состояний, в том числе для лечения или предупреждения толерантности к опиатам у пациентов, нуждающихся в продолжительном применении аналгетиков из класса опиатов, и толерантности к бензодиазепинам у пациентов, принимающих бензодиазепины, и другого привыкания, например привыкания к никотину, алкоголизма, и нарушений питания. Соединения и способы по настоящему изобретению можно применять также для лечения или предупреждения симптомов отмены, например для лечения или предупреждения симптомов отмены опиата, алкоголя или табака. Соединения по настоящему изобретению можно применять также для предупреждения повреждения ткани при терапии совместно с использованием антибактериальных или антивирусных агентов. Соединения по настоящему изобретению можно применять также для ингибирования производстваNO из L-аргинина в случае системной гипотензии, которая связана с септическим и/или токсическим гемморагическим шоком, который может вызываться широким разнообразием агентов; при терапии с использованием цитокинов, таких как TNF, IL-1 и IL-2; и в качестве адъюванта для кратковременной иммуносупрессии при лечении с использованием трансплантатов. Соединения по изобретению можно применять для предупреждения или лечения рака, такого как колоректальный рак и рак молочной железы, легкого, предстательной железы, мочевого пузыря, шейки матки и кожи. Кроме того, настоящее изобретение относится к применению соединений по настоящему изобретению для лечения и предупреждения неоплазий. Неоплазии, которые можно лечить или предупреждать с помощью соединений и способов по настоящему изобретению, включают рак головного мозга, рак кости, лейкоз, такой, например, как лимфоцитарный лейкоз, лимфому, эпителиальную неоплазию(эпителиальную карциному), такую как базально-клеточная карцинома, аденокарциному, рак желудочнокишечного тракта, такой как рак губы, рак рта, рак пищевода, рак тонкой кишки и рак желудка, рак ободочной кишки, рак печени, рак мочевого пузыря, рак поджелудочной железы, различные виды раков мочеполовых органов, такие как рак яичника, рак шейки матки, рак вульвы, и рак легкого, рак молочной железы и рак кожи, такой как плоскоклеточная карцинома, меланома, и базально-клеточная карцинома,рак предстательной железы, карцинома почечных клеток и другие известные виды рака, которые поражают эпителиальные клетки в организме. Соединения по настоящему изобретению можно применять также для лечения неоплазий, возникающих в мезенхимальных клетках. Предпочтительно подлежащую лечению неоплазию выбирают из группы, включающей рак желудочно-кишечного тракта, рак печени,рак мочевого пузыря, рак поджелудочной железы, рак яичника, рак предстательной железы, рак шейки матки, рак вульвы, рак легкого, рак молочной железы и рак кожи, такой как плоскоклеточная карцинома и базально-клеточная карцинома. Соединения и способы по настоящему изобретению можно применять также для лечения фиброза, возникающего при лучевой терапии. Соединения и способы по настоящему изобретению можно применять для лечения пациентов, имеющих аденоматозные полипы, включая семейный аденоматозный полипоз (САП). Кроме того, соединения и способы по настоящему изобретению можно применять для предупреждения образования полипов у пациентов, имеющих риск заболевания САП. Лечение, при котором соединение по настоящему изобретению применяют совместно с другим обладающим антинеопластической активностью агентом, позволяет получать синергетическое действие или в альтернативном варианте снижать побочные токсические действия, связанные с химиотерапией, в результате уменьшения терапевтической дозы вызывающего побочные действия агента, необходимой для оказания терапевтического действия, или в результате непосредственного ослабления симптомов токсических побочных действий, вызванных обладающим побочными действиями агентом. Кроме того,соединение по настоящему изобретению можно применять в качестве вспомогательного средства при лучевой терапии для уменьшения побочных действий или повышения эффективности. Согласно настоящему изобретению второй агент, который можно в терапевтических целях применять совместно с соединением по настоящему изобретению, представляет собой любой терапевтический агент, обладающий способностью ингибировать фермент циклооксигеназу-2 ("СОХ-2"). Предпочтительно такие ингибирующие СОХ-2 агенты обладают селективностью в отношении ингибирования СОХ-2 по сравнению с ингибированием фермента циклооксигеназы-1 ("СОХ-1"). Такой ингибитор СОХ-2 называют "селективным ингибитором СОХ-2". Более предпочтительно, соединение по настоящему изобретению можно в терапевтических целях применять совместно с селективным ингибитором СОХ-2, для которого соотношение уровня селективного ингибирования СОХ-2 и уровня ингибирования СОХ-1 селективным ингибитором СОХ-2 в опыте in vitro составляет по меньшей мере 10:1, более предпочтительно по меньшей-3 005817 мере 30:1 и наиболее предпочтительно по меньшей мере 50:1. Селективные ингибиторы СОХ-2, которые можно применять в терапевтических целях в сочетании с соединениями по настоящему изобретению,включают целекоксиб, валдекоксиб, деракоксиб, эторикоксиб, рофекоксиб, АВТ-963 (2-(3,4 дифторфенил)-4-(3-гидрокси-3-метил-1-бутокси)-5-[4-(метилсульфонил)фенил-3(2 Н)]пиридазинон, описанный в заявке РСТ WO 00/24719) или мелоксикам. Соединение по настоящему изобретению можно предпочтительно применять в терапевтических целях в сочетании с пролекарством селективного ингибитора СОХ-2, например парекоксибом. Другой химиотерапевтический агент, который можно применять в сочетании с соединением по настоящему изобретению, можно выбирать, например, из списка, который не является исчерпывающим и не ограничивает объем изобретения, включающего следующие соединения: альфа-дифторметилорнитин (DFMO), 5-FU-фибриноген, акантифолиевую кислоту, аминотиадиазол,натрийбреквинар, кармофур, Ciba-Geigy CGP-30694, циклопентилцитозин, стеарат цитарабинфосфата,конъюгаты цитарабина, Lilly DATHF, Merrel Dow DDFC, дезагуанин, дидезоксицитидин, дидезоксигуанозин, дидокс, Yoshitomi DMDC, доксифлуридин, Wellcome EHNA, MerckCo. ЕХ-015, фазарабин,флоксуридин, фосфат флударабина, 5-флуороурацил, N-(2'-фуранидинил)-5-флуороурацил, Daiichi Seiyaku FO-152, изопропилпирролизин, Lilly LY-188011, Lilly LY-264618, метобензаприм, метотрексат,Wellcome MZPES, норспермидин, NCI NSC-127716, NCI NSC-264880, NCI NSC-39661, NCI NSC-612567,Warner-Lambert PALA, пентостатин, пиритрексим, пликамицин, Asahi Chemical PL-AC, Takeda TAC-788,тиогуанин, тиазофурин, Erbamont TIF, триметрексат, ингибиторы тирозинкиназы, ингибиторы тирозинпротеинкиназы, Taiho UFT, урицитин, Shionogi 254-S, альдо-фосфамидные аналоги, алтретамин, анаксирон, Boehringer Mannheim BBR-2207, бестрабуцил, будотитан, Wakunaga СА-102, карбоплатин, кармустин, Chinoin-139, Chinoin-153, хлорамбуцил, цисплатин, циклофосфамид, American Cyanamid CL286558, Sanofi CY-233, циплатат, Degussa D-19-384, Sumimoto DACHP(Myr)2, дифенилспиромустин, диплатинацитостатик, производные дистамицина Erba, Chugai DWA-2114R, ITI E09, элмустин, ErbamontSKF-104864, Sumitomo SM-108, Kuraray SMANCS, SeaPharm SP-10094, спатол, производные спироциклопропана, спирогерманий, юнимед, SS Pharmaceutical SS-554, стриполдинон, стиполдион, Suntory SUN 0237, Suntory SUN 2071, супероксиддисмутаза, Toyama T-506, Toyama T-680, taxol, Teijin TEI-0303, тенипосид, талибластин, Eastman Kodak TJB-29, токотриенол, топостин, Teijin TT-82, Kyowa Hakko UCN01, Kyowa Hakko UCN-1028, украин, Eastman Kodak USB-006, сульфат винбластина, винкристин, виндесин, винестрамид, винорелбин, винтриптол, винзолидин, витанолиды, Yamanouchi YM-534, урогуанилин,комбретастатин, доластатин, идарубицин, эпирубицин, эстрамустин, циклофосфамид, 9-амино-2-(S)камптотецин, топотекан, иринотекан (камптосар), экземестан, декапептил (трипторелин) или омега-3 жирная кислота. Примерами радиозащитных агентов, которые можно применять в комбинированной терапии в сочетании с соединениями по настоящему изобретению, являются AD-5, адхнон, аналоги амифостина, детокс, димесна, 1-102, ММ-159, N-ацилированные дегидроаланины, TGF-Genentech, типротимод, амифостин, WR-151327, FUT-187, трансдермальный кетопрофен, набуметон, супероксиддисмутаза (Chiron) и супероксиддисмутаза (Enzon). Соединения по настоящему изобретению можно применять также для лечения или предупреждения связанных с ангиогенезом нарушений или состояний, например роста опухоли, метастазов, дегенерации желтого пятна и атеросклероза. Кроме того, настоящее изобретение относится также к терапевтическим комбинациям, предназначенным для лечения или предупреждения офтальмических нарушений или состояний, таких как глаукома. Например, соединения по настоящему изобретению можно применять в составе терапевтических композиций в сочетании с лекарственным средством, которое обладает способностью понижать внутриглазное давление у пациентов, страдающих глаукомой. Такие обладающие способностью понижать внутриглазное давление лекарственные средства включают (не ограничиваясь ими) латанопрост, травопрост, биматопрост или унопростол. Можно применять терапевтическую композицию, содержащую соединение по настоящему изобретению и лекарственное средство, обладающее способностью понижать внутриглазное давление, поскольку предполагается, что действие каждого из них обусловлено различными механизмами. В другом варианте композиции по настоящему изобретению соединения по настоящему изобретению можно применять в составе терапевтических композиций в сочетании с антигиперлипедимическим или снижающим уровень холестерина лекарственным средством, таким как антигиперлипедемическое лекарственное средство на основе бензотиепина или бензотиазепина. Примеры антигиперлипедемических лекарственных средств на основе бензотиепина, которые можно применять в терапевтических композициях по настоящему изобретению, приведены в патенте US5994391, включенном в настоящее описание в качестве ссылки. Некоторые антигиперлипедемические лекарственные средства на основе бензотиазепина описаны в WO 93/16055. В альтернативном варианте антигиперлипедимическое или снижающее уровень холестерина лекарственное средство, которое можно применять в сочетании с соединением по настоящему изобретению, может представлять собой ингибитор HMG СоА редуктазы (3 гидрокси-3-метилглутарил-СоА редуктаза). Примерами ингибиторов HMG СоА редуктазы, которые можно применять в терапевтических композициях по настоящему изобретению, являются, в частности,бенфлуорекс, флувастатин, ловастатин, провастатин, симвастатин, аторвастатин, церивастатин, бервастатин, ZD-9720 (описан в заявке РСТWO 97/06802), ZD-4522 (кальциевая соль CAS147098-20-2; натриевая соль CAS No. 147098-18-8 описаны в европейском патентеЕР 521471), BMS 180431 (CAS No. 129829-03-4) или NK-104 (CAS No. 141750-63-2). Терапевтическую композицию, включающую соединение по настоящему изобретению и обладающее антигиперлипедимическим или снижающим уровень холестерина действием лекарственное средство, можно применять, например, для снижения риска образования атеросклеротических повреждений в кровеносных сосудах. Например, атеросклеротические повреждения часто возникают в областях воспаления в кровеносных сосудах. Установлено, что лекарственное средство, обладающее антигиперлипедимическим или снижающим уровень холестерина действием, в результате снижения уровней липидов в крови уменьшает риск образования атеросклеротических повреждений. Не ограничивая объем изобретения одним механизмом действия, можно предполагать, что одной из возможностей является то, что соединения, входящие в композицию по настоящему изобретению, действуют совместно, что обеспечивает более высокий уровень контроля атеросклеротических по-5 005817 вреждений, например уменьшение воспаления кровеносных сосудов в сочетании с уменьшением уровней липидов в крови. Согласно другому варианту осуществления изобретения соединения по настоящему изобретению можно применять в сочетании с другими соединениями или в рамках других терапевтических подходов для лечения таких состояний или нарушений центральной нервной системы, как мигрень. Например,соединения по настоящему изобретению можно применять в составе терапевтической композиции в сочетании с кофеином, агонистом 5-HT-1B/1D (например, триптаном, таким как суматриптан, наратриптан,золмитриптан, ризатриптан, алмотриптан или фроватриптан), антагонистом допамина D4 (например,сонепипразолом), аспирином, ацетаминофеном, ибупрофеном, индометацином, напроксеном натрия,изометептеном, дихлоралфеназоном, буталбиталом, эргоалкалоидом (например, эрготамином, дигидроэрготамином, бромкриптином, эргоновином или метилэргоновином), трициклическим антидепрессантом(например, амитриптилином или нортриптилином), серотонергическим антагонистом (например, метисергидом или ципрогептадином), бета-адренергическим антагонистом (например, пропранололом, тимололом, атенололом, надололом или метпрололом) или ингибитором моноаминооксидазы (например, фенелзином или изокарбоксазидом). Следующий объект изобретения относится к терапевтической композиции, включающей соединение по настоящему изобретению и опиоид. Опиоиды, которые можно применять в композициях по настоящему изобретению, включают (но не ограничиваясь ими) морфин, метадон, гидроморфон, оксиморфон, леворфанол, леваллорфан, кодеин, дигидрокодеин, дигидрогидроксикодеинон, пентазоцин, гидрокодон, оксикодон, налмефен, эторфин, леворфанол, фентанил, суфентанил, DAMGO, буторфанол, бупренорфин, налоксон, налтрексон, СТОР, дипренорфин, бета-фуналтрексамин, налоксоназин, налорфин,пентазоцин, налбуфин, налоксонбензоилгидразон, бремазоцин, этилкетоциклазоцин, U50488, U69593,спирадолин, норбиналторфимин, налтриндол, DPDPE, [D-la2, glu4 ]делторфин, DSLET, метэнкефалин,леуэнкафалин, бета-эндорфин, динорфин А, динорфин В и альфа-неоэндорфин. Преимущество композиции по настоящему изобретению, включающей опиоид, заключается в том, что применение соединения по настоящему изобретению позволяет уменьшать дозу опиоида, снижая тем самым риск возникновения или серьезность побочных опиоидных действий, таких как зависимость от опиоидов. Подробное описание изобретения В наиболее широком смысле изобретение относится к соединениям формулы В другом варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы Еще в одном варианте осуществления объектом изобретения является соединение формулы Еще в одном варианте осуществления объектом изобретения является соединение формулы В следующем варианте осуществления объектом изобретения является соединение формулы Еще в одном варианте осуществления объектом изобретения является соединение формулы В следующем варианте осуществления объектом изобретения является соединение формулы В другом варианте осуществления объектом изобретения является соединение формулы Еще в одном варианте осуществления объектом изобретения является соединение формулы И еще в одном варианте осуществления объектом изобретения является соединение формулы Следующим объектом изобретения является соединение, представленное формулой Под объем семейства указанных выше соединений подпадают также их фармацевтически приемлемые соли. Понятие фармацевтически приемлемые соли включает обычно применяемые соли, образованные с щелочными металлами, соли, образованные с щелочно-земельными металлами, и кислотноаддитивные соли, образованные со свободными кислотами или соли присоединения свободных оснований. Природа соли не имеет решающего значения при условии того, что соль является фармацевтически приемлемой. Пригодные фармацевтически приемлемые кислотно-аддитивные соли соединений формулыI можно получать из неорганической кислоты или из органической кислоты. Примерами неорганических кислот являются соляная, бромисто-водородная, йодисто-водородная, азотная, карбоновая, серная и фосфорная кислоты. Пригодными органическими кислотами являются кислоты, выбранные из классов алифатических, циклоалифатических, ароматических, арилифатических, гетероциклических, карбоциклических и сульфоновых органических кислот, примерами которых являются муравьиная, уксусная,пропионовая, янтарная, гликолевая, глюконовая, молочная, яблочная, винная, лимонная, аскорбиновая,глюкуроновая, малеиновая, фумаровая, пировиноградная, аспартамовая, глутаминовая, бензойная, антраниловая, мезиловая, салициловая, парагидроксибензойная, фенилуксусная, миндальная, эмбоновая(памовая), метансульфоновая, этансульфоновая, бензолсульфоновая, сульфанильная, стеариновая, циклогексиламиносульфоновая, альгиновая, галактуроновая кислоты. Соответствующими фармацевтически приемлемыми солями соединений присоединения оснований являются соли, образованные с металлами,такими как алюминий, кальций, литий, магний, калий, натрий и цинк, или органические соли, образованные с N,N-дибензилэтилендиамином, холином, хлорпрокаином, диэтаноламином, этилендиамином, меглумином (N-метилглюкамином) и прокаином. Все эти соли можно получать обычными методами из соответствующих соединений, подвергая взаимодействию, например, соответствующую кислоту или основание с рассматриваемым соединением. Примерами солей по изобретению, которые не ограничивают объем изобретения, являются Хотя соединения можно применять в виде чистых химических веществ, предпочтительно включать их в состав фармацевтической композиции. Еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение или его фармацевтически приемлемую соль или сольват наряду с одним или несколькими фармацевтически приемлемыми носителями и необязательно в сочетании с одним или несколькими дополнительными терапевтическими действующими веществами. Носитель(ли) должен(ны) быть приемлемым(и) в том смысле, что он(и) должен(ны) быть совместимым(и) с другими ингредиентами, входящими в состав композиции, и не должен(ны) наносить вред реципиенту. Указанные композиции включают композиции, предназначенные для перорального, парентерального (в том числе подкожного, внутрикожного, внутримышечного, внутривенного и внутрисуставного),ректального и местного (в том числе кожного, трансбуккального, подъязычного и внутриглазного) введения, хотя выбор наиболее предпочтительного пути введения может зависеть, например, от состояния и заболевания реципиента. Композиции предпочтительно могут находиться в виде стандартной дозируемой формы и их можно приготавливать любыми методами, известными в области фармацевтики. Во всех методах используется стадия объединения соединения или его фармацевтически приемлемой соли или сольвата с носителем, который состоит из одного или нескольких вспомогательных веществ. Как правило, композиции приготавливают путем однородного и тщательного смешения действующего вещества с жидкими носителями, или тонкоизмельченными твердыми носителями, или ими обоими и затем при необходимости придают продукту требуемую форму. Композиции по настоящему изобретению, предназначенные для перорального введения, могут находиться в виде отдельных стандартных форм, таких как капсулы, саше, или таблетки, каждая из которых содержит заранее определенное количество действующего вещества; в виде порошка или гранул; в виде раствора или суспензии в водной или неводной среде; или в виде жидкой эмульсии типа масло в воде или жидкой эмульсии типа вода в масле. Действующее вещество может находиться также в виде болюса, электуария или пасты. Таблетку можно изготавливать путем прессования или литья необязательно с использованием одного или нескольких вспомогательных действующих веществ. Спрессованные таблетки можно изготавливать путем прессования с помощью пригодной машины действующего вещества, находящегося в свободно текучей форме, такой как порошок или гранулы, необязательно в смеси со связующим веществом,замасливателем, инертным разбавителем, поверхностно-активным агентом или разрыхлителем. Таблетки, получаемые путем литья, можно изготавливать с использованием соответствующей машины с помощью литья под давлением смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. На таблетки необязательно можно наносить покрытие или насечки и их можно изготавливать таким образом, чтобы обеспечивать замедленное или контролируемое высвобождение содержащегося в них действующего вещества. Композиции для парентерального введения включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические агенты и растворенные вещества, которые обеспечивают изотоничность композиции с кровью конкретного реципиента; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители. Композиции могут находиться в контейнерах, содержащих одну дозу или несколько доз, например,в запечатанных ампулах и пузырьках, и их можно хранить в полученном путем сушки вымораживанием(лиофилизацией) состоянии, которое непосредственно перед применением требует лишь добавления стерильного жидкого носителя, например физиологического раствора, воды для инъекций. При необходимости растворы и суспензии для инъекций можно приготавливать из описанных выше стерильных порошков, гранул и таблеток. Композиции для ректального введения могут представлять собой суппозитории, изготовленные с применением обычных носителей, таких как кокосовое масло или полиэтиленгликоль. Композиции для местного введения в полость рта, например, трансбуккально или сублингвально,включают лепешки, содержащие действующее вещество в придающей приятный вкус основе, такой как сахароза и аравийская камедь или трагакант, и пастилки, содержащие действующее вещество в такой основе, как желатин и глицерин или сахароза и аравийская камедь. Предпочтительно стандартные дозируемые формы содержат эффективную дозу действующего вещества, указанную ниже в настоящем описании, или определенную ее часть. Следует понимать, что в дополнение к ингредиентам, конкретно указанным выше, композиции по изобретению могут включать другие агенты, обычно применяемые в данной области для рассматриваемого типа композиции, например, композиции, предназначенные для перорального введения, могут включать корригенты.- 10005817 Соединения по изобретению можно вводить перорально или путем инъекции в дозе от 0,001 до 2500 мг/кг в день. Дозы для взрослых людей, как правило, составляют от 0,005 мг до 10 г/день. Таблетки или другие предназначенные для введения дискретные формы, как правило, должны содержать количество соединения по изобретению, которое обладает эффективностью при применении такой дозы или при применении нескольких таких доз, например, они представляют собой стандартные дозируемые формы, содержащие от 0,5 до 200 мг, как правило, от приблизительно 0,5 до 100 мг. Соединения предпочтительно вводят перорально или путем инъекции (внутривенной или подкожной). Точное количество соединения, вводимого пациенту, находится в компетенции штатного врача больницы. Следует иметь ввиду, что вводимая доза должна зависеть от многих факторов, таких как возраст и пол пациента, точно установленное заболевание, подлежащее лечению, и степень его серьезности. Путь введения может варьироваться также в зависимости от состояния и степени серьезности заболевания. Соединения по настоящему изобретению могут находиться в таутомерных или стереоизомерных формах. Предполагается, что все такие соединения, включая R- и S-энантиомеры, диастереоизомеры, dизомеры, l-изомеры, их рацемические смеси и иные их смеси, подпадают под объем настоящего изобретения. Под объем изобретения подпадают также фармацевтически приемлемые соли таких таутомерных,геометрических или стереоизомерных форм. Некоторые из описанных соединений содержат один или несколько стереоцентров и следует иметь ввиду, что под объем изобретения подпадают R-, S- и смеси R- и S-форм, которые имеют место для каждого присутствующего стереоцентра. Для осуществления на практике настоящего изобретения можно применять следующие реакционные схемы. Схема 1- 13005817 Приведенные ниже примеры служат для иллюстрации изобретения и не направлены на ограничение его объема. Специалистам в данной области должно быть очевидно, что указанные соединения могут быть получены в рамках общепринятых вариаций условий и процессов, применяемых в описанных ниже методах получения. Пример 1. Тригидрат гидрохлорида (R,2S)аминогексагидро-7-имино-1 Н-азепин-2-гексановой кислоты Трехгорлую колбу объемом 3 л продували азотом, после чего в нее вносили циклогексанон (1,27 моля, 132 мл) и 500 мл толуола. Эту смесь охлаждали при перемешивании до 0 С и добавляли 157,2 г(1,1 экв.) трет-бутоксида калия. После перемешивания указанной смеси в течение 1 ч обнаруживали изменение окраски и текстуры, после чего к перемешиваемой с помощью механической мешалки смеси добавляли по каплям в течение 1 ч раствор 5-пентенилбромида (1,27 моля, 136 мл) в 100 мл толуола. Реакционной смеси давали нагреться до 25 С и ее перемешивали в течение ночи. Затем ее разбавляли с помощью 800 мл 1 н. раствора KHSO4 и органическую фазу сушили (MgSO4), фильтровали и упаривали досуха, получая 208,5 г неочищенного продукта. После этого продукт очищали с помощью вакуумной дистилляции (с использованием водяного отсасывающего насоса), получая указанный в заголовке продукт с выходом 47%. 1 Н-ЯМР (CDCl3,част./млн): 1,0-2,4 (m, 13 Н), 4,9-5,1 (m, 2 Н), 5,7-5,9 (m, 1 Н). Пример 1 б) Продукт, полученный согласно методу, описанному в примере 1 а) (93,67 г, 0,563 моля), вносили вместе с EtOH (600 мл), водой (300 мл), NaOAc (101,67 г, 1,24 моля) и NH2OHHCl (78,31 г, 1,13 моля) в трехгорлую колбу объемом 3 л. Эту реакционную смесь при перемешивании кипятили с обратным холодильником в течение 16 ч и затем перемешивали при 25 С еще в течение 24 ч. Растворитель полностью удаляли при пониженном давлении и остаток распределяли между диэтиловым эфиром (Et2O, 500 мл) и водой (200 мл). Водный слой экстрагировали простым эфиром (3 х 200 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме, получая указанный в заголовке оксим (121,3 г, выход 100% в виде неочищенного продукта). 1 Трехгорлую колбу объемом 3 л продували азотом и затем в нее вносили гексаметилдисилоксан(471,7 мл, 2,2 моля), толуол (500 мл) и пентоксид фосфора (203,88 г, 1,4 моля). Эту гетерогенную смесь кипятили с обратным холодильником до получения прозрачного раствора (в течение приблизительно 1,5 ч). После охлаждения смеси до комнатной температуры к указанной выше реакционной смеси добавляли в течение 1 ч при 25 С оксим, полученный согласно методу, описанному в примере 1 б (102,1 г, 0,563 моля), в 200 мл толуола. Реакционную смесь перемешивали еще в течение 4-6 ч (проверку осуществляли с помощью ТСХ: 50% ЭА (этилацетат) в Hex, I2), после чего при тщательном перемешивании сливали на ледяную воду. К этой охлажденной на льду суспензии добавляли 250 г NaCl и значение рН образовавшейся смеси доводили до 5 путем добавления твердого карбоната калия. Указанную суспензию экстрагировали диэтиловым эфиром (Et2O) (3 х 500 мл) и объединенные органические фракции сушили надMgSO4, фильтровали и концентрировали в вакууме, получая неочищенную смесь региостереоизомеров лактамов (84,6 г). Пример 1 г) После этого продукт, полученный согласно методу, описанному в примере 1 в), подвергали хроматографии (силикагель:ацетонитрил) с целью очистки и разделения региостереоизомеров. Из неочищен- 14005817 ного образца выделяли 7-пентенильный региостереоизомер с выходом 50% и с помощью хиральной хроматографии выделяли отдельные энантиомеры, каждый из которых имел выход 43%.R-изомер продукта, полученного согласно методу, описанному в примере 1 г (102,1 г, 0,56 моля),безводный ТГФ (800 мл), ДМАП (68,9 г, 0,56 моля), ди-трет-бутилдикарбонат (Вос 2 О, 99 г, 0,45 моля) вносили в трехгорлую колбу объемом 3 л, которую перед этим продували аргоном. Реакционную смесь нагревали в течение 30 мин до 70 С, после чего добавляли еще 52,8 г Вос 2 О и 200 мл безводного ТГФ. Спустя 30 мин добавляли еще 32 г Вос 2 О и смесь перемешивали в течение 1 ч при 70 С. Добавляли еще 36 г Вос 2 О и смесь перемешивали в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и отгоняли ТГФ при 18-20 С при пониженном давлении. Осадок фильтровали и промывали с помощью 100 мл этилацетата (ЭА) и отбрасывали ( 45 г). ЕА-фильтрат разбавляли дополнительной порцией ЭА объемом 500 мл, после чего промывали 500 мл 1 н. раствора KHSO4, 500 мл насыщенного водного раствора NaHCO3 и 500 мл соляного раствора, затем сушили над безводным Na2SO4 в течение 12 ч. После этого полученный ЭА-экстракт обрабатывали 20 г DARCO, фильтровали через целит, наверху которого находился MgSO4, и концентрировали в вакууме, получая 150 г указанного в заголовке продукта в виде темно-коричневого масла. 1 Трехгорлую колбу объемом 3 л, содержащую продукт, полученный согласно методу, описанному в примере 1 е, (150 г, 0,533 моля), растворенный в 3 л СН 2 Сl2, охлаждали до -78 С. В течение 2,5 ч через раствор пропускали струю О 3 до тех пор, пока окраска реакционной смеси не становилась голубой. Затем раствор барботировали аргоном при температуре от -60 до -70 С до тех пор, пока раствор не становился прозрачным и бесцветным (30 мин). После этого добавляли диметилсульфид (ДМС 500 мл), после чего температуру реакционной смеси доводили до температуры дефлегмации и продолжали кипятить с обратным холодильником в течение 24 ч. Добавляли еще 100 мл ДМС и продолжали кипятить с обратным холодильником в течение 12 ч. Добавляли еще 100 мл ДМС и продолжали кипятить с обратным холодильником еще в течение 12 ч. Затем растворитель и избыток ДМС отгоняли с помощью роторного испарителя при 20 С. Полученный остаток в виде масла желтого цвета разбавляли дважды дистиллированной водой и экстрагировали 3 х 300 мл ЭА. ЭА-слой сушили над безводным MgSO4, обрабатывали 20 гDARCO, фильтровали через тонкий слой целита, наверху которого находился безводный MgSO4, и полностью отгоняли растворитель при пониженном давлении, получая 156 г неочищенного указанного в заголовке продукта в виде масла желто-оранжевого цвета. 1 К образцу триметилового эфира N-(бензилоксикарбонил)альфафосфоноглицина (160 г, 0,48 моля),растворенного в 1 л дихлорметана (CH2Cl2) и охлажденного до 0 С, добавляли раствор ДБУ (110,29 г,0,72 моля) в 100 мл CH2Cl2. Полученную прозрачную бесцветную реакционную смесь перемешивали в течение 1 ч при 0 - 6 С, после чего при температуре от -5 до -1 С по каплям добавляли альдегид, несущий защитную группу Воc, который получали согласно методу, описанному в примере 1 е) (150 г, 0,53 моля) в 600 мл CH2Cl2. Реакционную смесь перемешивали в течение 30 мин при указанной температуре,после чего медленно нагревали до 10 С в течение приблизительно 1 ч. Реакционную смесь промывали 1 н. раствором KHSO4 (500 мл), насыщенным водным раствором NаНСО 3 (200 мл) и 50% водным раствором NaCl (200 мл). Затем органический слой сушили над безводным MgSO4, обрабатывали 40 г DARCO,фильтровали через тонкий слой целита, наверху которого находился безводный MgSO4, и концентрировали, получая 258 г неочищенного указанного в заголовке продукта в виде масла желтого цвета. После хроматографической очистки этого продукта получали 130 г (55%) чистого указанного в заголовке соединения. Элементарный анализ: Рассчитано для C26H36N2O7: С 63,96; Н 7,42; N 5,77. Обнаружено: С 63,42; Н 8,16; N 5,31. 1 К раствору продукта, полученного согласно методу, описанному в примере 1 ж) (91,3 г, 0,19 моля), в МеОН (1 л) добавляли 2,5 г катализатора S,S-Rh-DIPAMP, а затем осуществляли гидрирование. Гидрирование осуществляли в течение 1,5 ч при 25 С в аппарате Парра. Реакционную смесь фильтровали через целит, после чего концентрировали, получая неочищенный указанный в заголовке продукт (90 г, 98%) в виде масла коричневого цвета. 1 К раствору продукта, полученного согласно методу, описанному в примере 1 з) (90 г), в 200 мл ледяной уксусной кислоты добавляли 200 мл 4 н. НСl в диоксане. Реакционную смесь перемешивали в течение 20 мин при 25 С, после чего растворитель полностью отгоняли при пониженном давлении при 40 С, получая масло красно-коричневого цвета. Этот маслянистый продукт обрабатывали 500 мл воды и экстрагировали дихлорметаном (2 х 300 мл). Объединенные органические слои промывали насыщенным раствором бикарбоната натрия (100 мл), сушили над сульфатом магния, фильтровали и полностью отгоняли растворитель, получая неочищенный указанный в заголовке продукт. Этот продукт хроматографировали, получая 45 г (62%) чистого указанного в заголовке соединения. Элементарный анализ: Расcчитано для C21H30N2O5: С 64,02; Н 7,68; N 7,17. Обнаружено: С 63,10; Н 7,88; N 6,60. 1- 16005817 К 45,0 г (0,115 моля) образца продукта, полученного согласно методу, описанному в примере 1 и), в 300 мл дихлорметана после продувки аргоном добавляли 23,0 г (0,121 моля) тетрафторбората триэтилоксония. Эту смесь перемешивали в течение 1 ч при 25 С, после чего добавляли 150 мл насыщенного водного раствора бикарбоната натрия. Дихлорметановый слой отделяли, промывали 150 мл 50% водного раствора NaCl, сушили над сульфатом натрия, фильтровали через целит и концентрировали при 25 С,получая 47,0 г (97%) указанного в заголовке продукта в виде прозрачного масла желтого цвета. Элементарный анализ: Рассчитано для C23H34N2O5: С 60,01; Н 8,19; N 6,69. Обнаружено: С 65,13; Н 8,45; N 6,64. 1 Н-ЯМР (CDCl3 част./млн): 1,2 (t, 3H), 1,25-1,74 (m, 12H), 1,75-1,95 (m, 2 Н), 2,2-2,3 (m, 1 Н), 2,4-2,5 К 7,0 г (0,130 моля) хлорида аммония в 500 мл метанола добавляли 31,2 г указанного в заголовке продукта, полученного согласно методу, описанному в примере 1 к) (45,0 г, 0,107 моля). Реакционную смесь кипятили с обратным холодильником в течение 5 ч при 65 С, после чего растворитель полностью удаляли при пониженном давлении, получая 40 г (87%) неочищенного продукта в виде пенообразной вязкой массы. Этот продукт очищали с помощью хроматографии на колонках, получая 37 г (81%) указанного в заголовке соединения. Элементарный анализ: Рассчитано для C21H31N3O4: С 59,22; Н 7,57; N 9,86; Сl 8,32. Обнаружено для[] =+31,5 при 365 нм. Пример 1) Продукт, указанный в заголовке примера 1 л) (36,0 г, 0,084 моля), в 1 л 2,3 н. НСl кипятили с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры раствор промывали 2x150 мл CH2Cl2 и затем растворитель полностью отгоняли в вакууме, получая 25,6 г (96%) указанного в заголовке аминокислотного производного в виде пены светло-желтого цвета. Элементарный анализ: Рассчитано для C12H23N3O22 НСl: С 46,02; Н 8,01; N 13,39; Сl 22,45. Обнаружено для C12H23N3O2 + 2,2 НСl + 0,1 Н 2 О: С 42,76; Н 8,02; N 12,41; Сl 22,79. ИК (чистый,макс, см-1): 2930, 2861, 1738,1665. 1S-изомер продукта, полученного согласно методу, описанному в примере 1 г) (5,45 г, 0,030 моля),превращали в его Вос-защищенное производное согласно методу, описанному в примере 1 д). После хроматографирования получали 6,3 г (75%) указанного в заголовке продукта. 1 Продукт, полученный согласно методу, описанному в примере 2 а) (6,3 г, 0,025 моля), озонировали согласно методу, описанному в примере 1 е), получая 8,03 г неочищенного указанного в заголовке альдегида, который использовали без дальнейшей очистки. 1 Продукт, полученный согласно методу, описанному в примере 2 б) (8,03 г, 0,024 моля), конденсировали с триметиловым эфиром N-(бензилоксикарбонил)альфафосфоноглицина (7,9 г, 0,024 моля), следуя методу, описанному в примере 1 ж), в результате чего после хроматографирования получали 4,9 г (44%) требуемого указанного в заголовке продукта. 1 Н-ЯМР (CDCl3,част./млн): 1,25 (m, 2H), 1,5 (s, 9H), 1,51-1,9 (bm, 8H), 2,25 (m, 2 Н), 2,5 (m, 1 Н),2,65 (m, 1 Н), 3,75 (s, 3 Н), 4,15-4,25 (m, 1 Н), 5,15 (s, 2H), 6,3-6,4 (bs, 1H), 6,45-6,55 (t, 1H), 7,3-7,4 (m,5 Н). Пример 2 г) Продукт, полученный согласно методу, описанному в примере 2 в) (4,8 г, 0,010 моля), восстанавливали в присутствии катализатора R,R-Rh-DIPAMP с помощью метода, описанного в примере 1 з), получая после хроматографирования 2,9 г (60%) требуемого указанного в заголовке соединения. Пример 2 д) У продукта, полученного согласно методу, описанному в примере 2 г) (2,9 г, 0,006 моля), удаляли защитную группу путем обработки НСl согласно методу, описанному в примере 1 и), получая 2,3 г Продукт, полученный согласно методу, описанному в примере 2 д) (0,56 г, 0,0015 моля), алкилировали с помощью тетрафторбората триэтилоксония согласно методу, описанному в примере 1 к), получая 0,62 г (98%) требуемого указанного в заголовке продукта. Пример 2 ж) Продукт, полученный согласно методу, описанному в примере 2 е) (0,62 г, 0,0015 моля), обрабатывали раствором хлорида аммония в метаноле согласно методу, описанному в примере 1 л), получая после хроматографирования 0,50 г (88%) требуемого указанного в заголовке соединения. Продукт, полученный согласно методу, описанному в примере 2 ж) (0,37 г, 0,0009 моля), растворенный в МеОН, добавляли в предназначенный для осуществления гидрирования аппарат Парра. В этот сосуд добавляли каталитическое количество 5%Pd/C. Впускали водород и в течение 7 ч осуществляли реакцию при комнатной температуре и давлении 5 фунтов/кв. дюйм. Катализатор удаляли фильтрацией, а растворитель полностью удаляли из фильтрата при пониженном давлении, получая 0,26 г (количественный выход) требуемого указанного в заголовке продукта. Пример 2. Раствор продукта, полученного согласно методу, описанному в примере 2 з), в 2 н. НСl (30 мл) выдерживали при температуре дефлегмации в течение 2 ч, после чего охлаждали до комнатной температуры. Растворитель полностью удаляли при пониженном давлении и остаток растворяли в 50 мл воды. Из указанного раствора снова полностью удаляли растворитель при пониженном давлении, после чего его вновь растворяли в 12 мл воды и затем лиофилизировали, получая 0,245 г (71%) указанного в заголовке соединения. Элементарный анализ: Рассчитано для C12H23N3O22,3 НСl1,9 Н 2 О: С 40,10; Н 8,16; N 11,69; Сl 22,69. Обнаружено для C12H23N3O2 + 2,1 НСl + 0,7 Н 2 О: С 40,27; Н 8,28; N 11,62; Сl 22,70. 1 Продукт, полученный согласно методу, описанному в примере 2 в), восстанавливали в присутствии катализатора S,S-Rh-DIPAMP согласно методу, описанному в примере 1 з), получая после хроматографирования требуемый указанный в заголовке продукт. Пример 3 б) У продукта, полученного согласно методу, описанному в примере 3 а), удаляли защитную группу с помощью НСl согласно методу, описанному в примере 1 и), получая требуемое указанное в заголовке соединение. Пример 3 в) Продукт, полученный согласно методу, описанному в примере 3 б), алкилировали с помощью тетрафторбората триэтилоксония согласно методу, описанному в примере 1 к), получая требуемый указанный в заголовке продукт. Пример 3 г)- 19005817 Продукт, полученный согласно методу, описанному в примере 3 в), обрабатывают раствором хлорида аммония в метаноле согласно методу, описанному в примере 1 л), получая после хроматографической очистки требуемое указанное в заголовке соединение. Пример 3). Раствор продукта, полученного согласно методу, описанному в примере 3 г), в 2 н. НСl обрабатывали согласно методу, описанному в примере 1, получая указанное в заголовке соединение. Пример 4. Тригидрат гидрохлорида (S,2S)аминогексагидро-7-имино-1 Н-азепин-2-гексановой кислоты. В круглодонную колбу объемом 22 л, снабженную верхней мешалкой с лопастями, имеющими форму полумесяца, нагревательным кожухом, термопарой и серебряной дистилляционной колонкой с вакуумной рубашкой (5 тарелок), загружали циклогексанон (4500,0 г, 45,85 моля), ацетондиметилацеталь(ПТСК) (0,256 г, 0,001 моля). После начала перемешивания (137 об/мин) сосуд подвергали медленному нагреву, начиная с исходной температуры 70 С. Нагрев постепенно увеличивали до тех пор, пока температура в сосуде не достигала 150 С. Решение о увеличении регулировочных параметров реактора принимали на основе данных о скорости дистилляции. Если скорость дистилляции замедлялась или дистилляция прекращалась, то увеличивали приток тепла. При дополнительном нагреве до 150 С создавались условия для осуществления перегруппировки Кляйзена. После того, как температура в сосуде достигала 150 С и не наблюдалось образования дистиллята, температуру нагревательного кожуха уменьшали и реакционной смеси давали охладиться до 130 С. После этого ПТСК нейтрализовали с помощью 3 капель 2,5 н. NaOH. Затем после снятия с колбы нагревательного кожуха начинали вакуумную отгонку. Для понижения температуры в сосуде применяли испарительное охлаждение и давление постепенно понижали до 40 мм рт.ст. Когда температура в сосуде понижалась до 100 С, нагревательный кожух возвращали в положение, при котором осуществлялся нагрев. Отгоняли непрореагировавший циклогексанон и примеси, имеющие низкую температуру кипения. Температуру в сосуде медленно повышали (максимальная разница между температурой в сосуде и температурой пара составляла -12 С). Продукт выделяли при 109-112 С и давлении 40 мм рт.ст. Выходы, как правило, составляли 40-45%. Фракции, для которых по данным газовой хроматографии (ГХ) площадь пиков составляла 95%, объединяли и подвергали повторной дистилляции, в результате чего получали указанный в заголовке продукт с выходом 55%. 1 Гидроксиламин-О-сульфоновую кислоту (91,8 г), растворенную в уксусной кислоте (470 г), добавляли в колбу Байера объемом 1 л, снабженную механической мешалкой, термопарой, конденсатором,охлажденным до 0 С, и дополнительной воронкой, и нагревали до 70 С. К указанному выше раствору добавляли по каплям в течение приблизительно 40 мин аллилциклогексон (100 г), поддерживая температуру на уровне 70-78 С. В процессе добавления внешний вид реакционной смеси изменялся от суспензии белого цвета до прозрачного раствора оранжевого цвета. После завершения добавления реакционную смесь нагревали и перемешивали в течение еще 5 ч при 75 С. Через каждый час отбирали IPC (управление технологическим процессом) образцы. После завершения реакции уксусную кислоту отгоняли при 50 С при пониженном давлении с помощью роторного испарителя. Затем к остатку добавляли воду (200 мл) и раствор экстрагировали толуолом (2 х 300 мл). Органические слои объединяли, обрабатывали водой (150 мл) и перемешивали в течение 10 мин. Добавляли раствор гидроксида натрия (79,4 г 50%-ного раствора) до тех пор, пока водный слой не становился щелочным (рН 12). Нейтрализацию осуществляли в реакторе, поддерживая температуру ниже 40 С. Затем слои разделяли и толуоловый слой пропускали через фильтр для удаления любых твердых или смолообразных продуктов. После этого раствор упарива- 20005817 ли при 50 С при пониженном давлении с помощью роторного испарителя. Остаток растворяли в смеси толуола (510 мл) и гептана (2040 мл) и нагревали до 60 С в реакторе объемом 3 л. Получали прозрачный раствор желто-оранжевого цвета. Указанный в заголовке продукт начинал кристаллизоваться при 53 С в процессе того, как его медленно охлаждали при перемешивании до 5 С. Твердый продукт фильтровали,промывали гептаном (50 мл) и сушили в течение ночи при 40 С в неглубоком вакууме, получая 66,3 г(60%) указанного в заголовке продукта в виде кристаллов беловатого цвета. Часть этого продукта перекристаллизовывали из толуола и гептана, получая указанное в заголовке соединение в виде твердого кристаллического вещества белого цвета. 1 Рацемическую смесь продуктов, полученных согласно методу, описанному в примере 4 б), подвергали хиральному хроматографическому разделению на колонке типа Chiralpac AS 20 мкм, осуществляя элюирование с помощью 100% ацетонитрила. Обнаружение проводили при длине волны 220 нм. Загружали образец, содержащий 0,08 г/мл ацетонитрила и получали 90%-ный выход отдельных изомеров, каждый из которых имел чистоту 95% ее. Часть продукта, представляющего собой R-изомер, перекристаллизовывали из толуола и гептана, получая указанный в заголовке R-изомер в виде кристаллического твердого вещества белого цвета. В пятигорлой плоскодонной колбе, снабженной капельной воронкой, термометром и механической верхней мешалкой, создавали вакуум и трижды продували азотом. R-изомер лактама, полученный согласно методу, описанному в примере 4 в) (100,0 г, 0,653 моля), ДМАП (7,98 г, 65 ммолей) и Nдиизопропилэтиламин (основание Хюнига, 113,3 г, 0,876 моля) растворяли в толуоле (350 мл) и добавляли ди-трет-бутилдикарбонат (170,2 г, 0,78 моля), растворенный в толуоле (100 мл). (Примечание: реакция протекает лучше, если применяют 2,0 экв. основания Хюнига). Смесь нагревали до 65 С (Примечание: в процессе реакции наблюдалось постоянное выделение газа). Через 1,5 ч добавляли еще одну порцию дитрет-бутилдикарбоната (86,25 г, 0,395 моля), растворенного в толуоле (50 мл). Нагрев продолжали в течение 17 ч, после чего по данным IPC, осуществляемого с использованием ЖХВР, превращение исходного продукта составляло 75%. Добавляли еще 42,78 г ди-трет-бутилдикарбоната (0,196 моля) в толуоле(30 мл) и смесь коричневого цвета нагревали в течение 5,5 ч. После охлаждения до температуры окружающей среды смесь обрабатывали 4 М НСl (215 мл) и водный слой экстрагировали с помощью толуола(2x80 мл). Объединенные органические слои промывали NаНСО 3 (170 мл) и 250 мл воды (примечание: внутреннюю температуру в процессе резкого охлаждения регулировали с помощью внешнего охлаждения смесью лед/вода). Происходило выделение газа. Органический слой упаривали, получая 257,4 г жидкости коричневого цвета. Этот неочищенный продукт очищали путем фильтрации через пробку из SiO2(950 г), используя в качестве элюента смесь толуол /EtOAc, 9/1 (6 л) и смесь толуол /AcOEt, 1/1 (0,5 л), в результате чего получали 139,5 г (51%) указанного в заголовке продукта в виде жидкости желтого цвета. Пример 4 д) Пример 1 е. В автоклав из нержавеющей стали объемом 2 л, снабженный дефлекторами и шестилопастной осевой крыльчаткой для диспергирования газа, загружали Rh(CO)2(acac) (0,248 г, 0,959 ммоля),- 21005817BIPHEPHOS (структура соединения приведена ниже, соединение получали согласно методу, описанному в примере 13 патента US4769498, 2,265 г, 2,879 ммоля), продукт, полученный согласно методу, описанному в примере 4 г) (N-(трет-бутоксикарбонил)-S-7-аллилкапролактам(242,9 г, 0,959 моля) и толуол (965 г). Реактор герметически закрывали и продували 100%-ным монооксидом углерода (8 х 515 кПа). В реакторе повышали давление до 308 кПа (30 фунтов/кв. дюйм) с помощью 100%-ного монооксида углерода и затем добавляли газовую смесь СО/Н 2 (1:1), в результате чего общее давление достигало 515 кПа (60 фунтов/кв. дюйм). При интенсивном механическом перемешивании смесь нагревали до 50 С, при этом добавляли газовую смесь СО/Нг (1:1), поддерживая общее давление на уровне приблизительно 515 кПа (60 фунтов/кв. дюйм). Через 22 ч смесь охлаждали до приблизительно 25 С и осторожно уменьшали давление. После вакуумной фильтрации полученной смеси и упаривания фильтрата при пониженном давлении получали 267,7 г продукта в виде масла светло-желтого цвета. Результаты Н-ЯМР-анализа свидетельствовали о практически количественном превращении исходного продукта с избирательным выходом соответствующего альдегида, описанного в примере 1 е),составляющим приблизительно 96%. Этот продукт в виде масла использовали в следующем примере без дальнейшей очистки. 1 Пример 1 ж). К образцу триметилового эфира N-(бензилоксикарбонил)альфа-фосфоноглицина(901,8 г, 2,7 моля), растворенного в СН 2 Сl2 и охлажденного до 0 С, добавляли раствор ДБУ (597,7 г, 3,9 моля) в CH2Cl2. Эту прозрачную бесцветную смесь перемешивали в течение 1 ч при температуре 0-6 С,после чего по каплям при температуре от -5 до -1 С добавляли образец Вос-защищенного альдегида, полученного согласно методу, описанному в примере 1 е) (812,0 г, 2,9 моля) в CH2Cl2. Реакцию, обработку и очистку проводили согласно методу, описанному в примере 1 ж), получая 1550 г указанного в заголовке продукта, описанного в примере 1 ж), который содержал небольшое количество CH2Cl2. Пример 4 д). К раствору продукта, полученного согласно методу, описанному в примере 1 ж) (100 г,0,20 моля), в МеОН (1 л) добавляли 3 г катализатора RR-Rh-DIPAMP. Гидрирование осуществляли при 25 С в течение 1,5 ч в аппарате Парра. Реакционную смесь фильтровали через целит, после чего концентрировали, получая неочищенный указанный в заголовке примера 4 д) продукт в виде масла коричневого цвета (100 г). 1 К раствору продукта, полученного согласно методу, описанному в примере 1 з) (100 г), в 200 мл ледяной уксусной кислоты добавляли 25 мл 4 н. НСl в диоксане. Реакционную смесь перемешивали в течение 20 мин при 25 С, после чего растворитель полностью отгоняли при пониженном давлении, получая 105 г масла красно-коричневого цвета. Этот маслянистый продукт обрабатывали 500 мл воды и экстрагировали 2 х 300 мл дихлорметана. Объединенные органические слои промывали насыщенным раствором бикарбоната натрия (100 мл), сушили над сульфатом магния и полностью отгоняли растворитель,получая 99,9 г указанного в заголовке продукта в виде масла красно-коричневого цвета. 1(s, 2 Н), 5,5 (d, 1 Н), 6,45 (bs, 1 Н), 7,35 (m, 5 Н). ее = 95% по данным анализа с помощью хиральной ЖХВР. Пример 4 ж)- 22005817 К 30,0 г (0,077 моля) образца продукта, полученного согласно методу, описанному в примере 4 е), в 600 мл дихлорметана, продутого аргоном, добавляли 15,7 г (0,082 моля) тетрафторбората триэтилоксония. Эту смесь перемешивали в течение 1 ч при 25 С, после чего добавляли 300 мл насыщенного водного раствора бикарбоната натрия. Дихлорметановый слой отделяли, промывали 300 мл 50%-ного водного раствора NaCl, сушили над сульфатом натрия, фильтровали через целит и концентрировали при 25 С,получая 31,2 г (97%) указанного в заголовке продукта в виде прозрачного масла желтого цвета. Элементарный анализ: Рассчитано для C23H34N2O5: С 60,01; Н 8,19; N 6,69. Обнаружено для К 4,2 г (0,078 моля) хлорида аммония в 500 мл метанола добавляли 31,2 г продукта, указанного в заголовке примера 4 ж). Реакционную смесь кипятили с обратным холодильником при 65 С в течение 5 ч при пониженном давлении до полного удаления растворителя, в результате чего получали 29 г (92%) неочищенного продукта в виде пенообразной вязкой массы. Этот продукт очищали с помощью хроматографии на колонках, получая 23 г (70%) указанного в заголовке продукта. Элементарный анализ: Рассчитано для C21H31N3O41HCl: С 59,28; Н 7,57; N 9,89; Сl 8,39. Обнаружено для C21H31N3O4 + 1HCl + 1 Н 2 О: С 56,73; Н 7,74; N, 9,40; Сl 8,06. ИК (чистый,маc. см-1): 3136, 30348, 2935, 1716, 1669. 1[]25 = -42,8 (МеОН) при 365 нм. ее = 96% поданным оценки с помощью ЖХВР. Пример 4. Продукт, указанный в заголовке примера 4 з) (23 г), в 500 мл 2 н. НСl кипятили с обратным холодильником в течение 5 ч. Затем растворитель полностью удаляли в вакууме и остаток, повторно растворенный в воде, промывали 2x300 мл СН 2 Сl2 После этого водный слой концентрировали в вакууме, получая 17 г (100%) указанного в заголовке твердого гигроскопического продукта светло-коричневого цвета. Элементарный анализ: Рассчитано для C12H23N3O22HCl: С 45,86; Н 8,02; N 13,37; Сl 22,56. Обнаружено для С 12 Н 23N3 О 2 + 2,1 НСl + 0,7 Н 2 О: С 43,94; Н 8,65; N 12,52; Сl 22,23. ИК (чистый,макс, см-1): 2936, 1742,1669. 1[]25 = +60,0 (МеОН) при 365 нм. ее = 92% по данным измерений с помощью СЕ при= 210 нм. Пример 5. Тригидрат гидрохлорида (R,2S)аминогексагидро-7-имино-1 Н-азепин-2-гексановой кислоты Раствор, содержащий соединение, полученное согласно методу, описанному в примере 4 в) (3,0 г,0,015 моля) в метиленхлориде и метаноле (75/45 мл), охлаждали до -78 С в бане с сухим льдом. Реакционную смесь перемешивали, осуществляя при этом барботирование раствора озоном со скоростью потока 3 мл/мин. После того, как раствор приобретал стабильную темно-синюю окраску, барботирование озоном прекращали и реакционную смесь продували азотом. К холодному раствору очень медленно добавляли борогидрид натрия (2,14 г, 0,061 моля) для того чтобы минимизировать выделение газа в каждый момент времени. К реакционной смеси медленно добавляли ледяную уксусную кислоту, доводя значение рН до 3. Затем реакционную смесь нейтрализовали насыщенным раствором бикарбоната натрия. После этого органическую фазу промывали 3 х 50 мл соляного раствора, сушили над безводным сульфатом магния, удаляли растворитель при пониженном давлении. Продукт в виде слабоокрашенного масла пропускали через пробку из диоксида кремния (15 г), получая 5,15 г, 0,026 моля (64%) спирта. К раствору, полученному согласно методу, описанному в примере 5 а) (5,15 г, 0,026 моля), в метиленхлориде (100 мл), при температуре 0 С, поддерживаемой с помощью ледяной бани, добавляли четырехбромистый углерод (10,78 г, 0,033 моля). Раствор охлаждали с помощью ледяной бани до 0 С. Затем порциями добавляли трифенилфосфин (10,23 г, 0,39 моля) таким образом, чтобы температура не поднималась выше 3 С. Реакционную смесь перемешивали в течение 2 ч и растворитель удаляли в вакууме. Неочищенный продукт очищали с помощью экспресс-хроматографии, получая бромид (5,9 г, 0,023 моля) с выходом 87%. Элементарный анализ: Рассчитано для: C10H16N2O3: С 41,40; Н 5,02; N 10,73; Вr 30,60. Обнаружено: С 41,59; Н 5,07; N 10,60, Вг 30,86. 1(25 мл) добавляли трифенилфосфин (7,17 г, 0,027 моля). Реакционную смесь кипятили с обратным холодильником в масляной бане в течение 16 ч. После охлаждения толуол декантировали со стекловидного твердого продукта. Твердый продукт растирали в течение ночи с диэтиловым эфиром, получая бромид фосфония (10,21 г, 0,020 моля) с выходом 90%. 1 В круглодонную колбу объемом 1 л добавляли лактон N-бензилоксикарбонил-D-гомосерина (97 г,0,442 моля) в этаноле (500 мл). К реакционной смеси добавляли раствор гидроксида натрия (1 М, 50 мл). За ходом реакции наблюдали с помощью тонкослойной хроматографии в течение 12 ч до тех пор, пока- 24005817 не происходило поглощение исходного продукта. Добавляли толуол (60 мл) и растворитель удаляли в вакууме. Остаток использовали на следующих стадиях без дополнительной очистки. Пример 5 д) Остаток, полученный согласно методу, описанному в примере 5 г), суспендировали в ДМФ в круглодонной колбе объемом 1 л. К суспензии добавляли бензилбромид (76,9 г, 0,45 моля, 53,5 мл) и смесь перемешивали в течение 1 ч. Образец быстро охлаждали и анализировали с помощью массспектроскопии для оценки поглощения исходного продукта, при этом не происходила конформация лактона. К реакционной смеси добавляли 1 л этилацетата и 500 мл соляного раствора. Водный слой дополнительно промывали два раза 500 мл этилацетата. Органические слои объединяли, сушили над MgSO4 и концентрировали. После хроматографии на силикагеле получали бензиловый эфир Nбензилоксикарбонил-S-гомосерина в виде твердого вещества белого цвета (80 г). Пример 5 е) В круглодонную колбу объемом 2 л добавляли хлорхромат пиридиния (187 г, 0,867 моля) и силикагель (197 г), суспендированный в CH2Cl2 (600 мл). К суспензии добавляли раствор продукта, полученного согласно методу, описанному в примере 5 д) (80 г, 0,233 моля), в СН 2 Сl2 (600 мл). Смесь перемешивали в течение 4 ч. Результаты анализа с помощью тонкослойной хроматографии позволяли установить, что произошло поглощение исходного продукта. К реакционной смеси добавляли 1 л диэтилового эфира. После этого раствор фильтровали через пробку из целита, а затем через пробку из силикагеля. Растворитель удаляли в вакууме и образовавшееся масло очищали с помощью хроматографии на силикагеле, получая альдегид (58,8 г) с общим выходом 38%. МН+ 342,5, MH+NH4+ 359,5. 1 Н-ЯМР (CDCl3,част./млн) 3,15 (q, 2H), 4,12 (m, 1H), 5,15 (s, 2H), 5,20 (s, 2 Н), 7,31 (m, 10 Н), 972 В трехгорлую колбу объемом 3 л вносили соль фосфония, полученную согласно методу, описанному в примере 5 в (56,86 г, 0,11 моля), которая была высушена над Р 2 О 5 в вакууме, в ТГФ (1 л). Суспензию охлаждали до -78 С в бане с сухим льдом. К холодной суспензии добавляли по каплям KHMDS (220 мл,0,22 моля) таким образом, чтобы температура не поднималась выше -72 С. Реакционную смесь перемешивали при -78 С в течение 20 мин, а затем при -45 С в течение 2 ч. После этого температуру опять понижали до -78 С и по каплям в течение 45 мин добавляли альдегид, полученный согласно методу, описанному в примере 5 е (15,9 г, 0,047 моля), в ТГФ (50 мл). Реакционную смесь перемешивали при -77 С в течение 30 мин, затем нагревали в течение 1 ч до -50 С, после чего нагревали до комнатной температуры в течение 4 ч. К реакционной смеси добавляли этилацетат (200 мл) и насыщенный хлорид аммония. Органические слои собирали, сушили над MgSO4 и концентрировали в вакууме. Неочищенное масло очищали с помощью хроматографии на силикагеле, получая олефин (45,1 г) с выходом 81% в виде вязкого масла светло-желтого цвета. 1H-ЯМР (CDCl3,част./млн) 1,4-2,6 (m, 10 Н), 2,92 (d, 1H), 4,17(m, 1H), 4,38 (m, 1 Н), 5,05 (q, 2 Н),5,40 (m, 2 Н), 7,3 (m, 10 Н). 13 С-ЯМР (CDCl3,част./млн) 29,49, 29,64, 31,32, 39,60, 49,56, 53,98, 61,01, 65,25, 124,14, 127,81,128,20, 128,55, 128,79, 129,30, 130,96, 135,68, 137,31, 152,59, 157,57, 171,61. Пример 5. В пузырек объемом 20 мл добавляли продукт, полученный согласно методу, описанному в примере 5 ж) (19,77 г, 0,039 моля), в диоксане (50 мл) и 4 н. водном растворе НСl (250 мл). К этому раствору добавляли каталитическое количество 10%-ного Pd на угле и вносили в сосуд для гидрирования. В колбе создавали повышенное давление Н 2 (50 фунтов/кв. дюйм) на период времени, составляющий 5 ч. За ходом реакции наблюдали с помощью масс-спектроскопии и устанавливали, когда происходило погложение исходного продукта. Раствор фильтровали через пробку из целита и промывали водой. Растворитель удаляли с помощью лиофилизации, получая указанное в заголовке соединение (7,52 г) с выходом 81%. В трехгорлую колбу объемом 1 л вносили соль фосфония, полученную согласно методу, описанному в примере 5 в) (21,21 г, 0,041 моля), в ТГФ (200 мл). Суспензию охлаждали до -78 С в бане с сухим льдом. К холодной суспензии по каплям добавляли KHMDS (88 мл, 0,044 моля) таким образом, чтобы внутренняя температура не поднималась выше -72 С. Реакционную смесь перемешивали при -78 С в течение 20 мин, а затем при -45 С в течение 1 ч. После этого температуру понижали снова до -78 С и по каплям в течение 45 мин добавляли альдегид (15,9 г, 0,047 моля) (полученный согласно методу, описанному в примере 5 (г-е) с использованием лактона N-бензилоксикарбонил-L-гомосерина) в ТГФ (50 мл). Реакционную смесь перемешивали при -77 С в течение 30 мин, затем нагревали до -50 С в течение 30 мин, после чего нагревали до комнатной температуры в течение 4 ч. К реакционной смеси добавляли этилацетат (100 мл) и насыщенный раствор хлорида аммония. Органические фазы собирали, сушили надMgSO4 и концентрировали в вакууме. Неочищенное масло очищали с помощью хроматографии на силикагеле, получая олефин (9,0 г) с выходом 45% в виде вязкого масла светло-желтого цвета. 1H-ЯМР (CDCl3,част./млн) 1,4-2,6 (m, 10 Н), 2,92 (d, 1H), 4,17 (m, 1H), 4,38 (m, 1 Н), 5,05 (q, 2 Н),5,40 (m, 2 Н), 7,3 (m, 10 Н). 13 С-ЯМР (CDCl3,част./млн) 29,49, 29,64, 31,32, 39,60, 49,56, 53,98, 61,01, 65,25, 124,14, 127,81,128,20, 128,55, 128,79, 129,30, 130,96, 135,68, 137,31, 152,59, 157,57, 171,71. Пример 6). В пузырек объемом 20 мл добавляли продукт, полученный согласно методу, описанному в примере 6 а), в диоксане (5 мл) и 4 н. водном растворе НСl (16 мл). К этому раствору добавляли каталитическое количество 10%-ного Pd на угле и вносили в сосуд для гидрирования. В колбе создавали повышенное давление Н 2 (50 фунтов/кв. дюйм) на период времени, составляющий 5 ч. За ходом реакции наблюдали с помощью масс-спектроскопии и устанавливали, когда происходило поглощение исходного продукта. Раствор фильтровали через пробку из целита и промывали водой. Растворитель удаляли с помощью лиофилизации, получая указанное в заголовке соединение (98,7 г) с выходом 79,4%. МН+ 242,2, MH+NH4+259,2. 1 Н-ЯМР (CD3OD,част./млн) 1,2-2,0 (m, 15H), 2,42 (d, 1H), 2,6 (dd, 1H), 3,49 (m, 1 Н), 3,98 (t, 1H). 13 С-ЯМР (CDCl3,част./млн) 24,43, 25,58, 26,00, 26,10, 32,75, 33,45, 35,31, 53,76, 54,55, 157,27,175,13. Пример 7. (2S,4Z)-2-амино-6-[(2R)-гексагидро-7-имино-1H-азепин-2-ил]-4-гексеновая кислота(1,5 г, 2,97 ммоля), в метаноле (25 мл). Затем к реакционной смеси добавляли 60%-ный раствор ледяной уксусной кислоты (16 мл). Происходило образование осадка. Для растворения твердого продукта добавляли дополнительную порцию метанола (1 мл). Затем к реакционной смеси добавляли порошок цинка(0,200 г). Реакционную смесь облучали ультразвуком в течение 4 ч, при этом в течение указанного пе- 26005817 риода времени температуру поддерживали на уровне 37 С. За ходом реакции наблюдали с помощью ТСХ и МС до тех пор, пока не происходило поглощение исходного продукта и образование определенного количества требуемого продукта. Раствор декантировали с цинка и к фильтрату добавляли 30%-ный раствор ацетонитрила в воде (100 мл). Продукт реакции очищали с использованием 52% ацетонитрила/воды в два погона с помощью препаративной хроматографии на колонке фирмы Waters [градиент от 20 до 70% ацетонитрила в течение 30 мин]. После лиофилизации образовавшегося продукта получали соединение, указанное в заголовке примера 7 а) (1,01 г) с выходом 73% в виде твердого вещества белого цвета. МН+ 464,4, MH+Na+ 486,4. 1H-ЯМР (CD3OD,част./млн): 1,2-2,0 (m, 8 Н), 2,42 (m, 2 Н), 2,6 (m, 5 Н), 3,49 (q, 1H), 4,31 (t, 1H),5,15 (s, 2H), 5,22 (s, 2H), 5,43 (q, 1H), 5,59(q, 1H), 7,25 (bs, 10H). 13 С-ЯМР (CDCl3,част./млн): 24,37, 29,61, 30,76, 32,45, 33,73, 34,42, 55,40, 57,09, 68,06, 68,07,122,3, 124,9, 128,76, 129,09, 129,28, 129,39, 129,51, 129,61, 155,71, 158,35, 173,90. Пример 7). В колбу объемом 250 мл вносили продукт, полученный согласно методу, описанному в примере 7 а) (1,0 г, 2,2 ммоля), в 4 М НСl (100 мл). Реакционную смесь кипятили с обратным холодильником в течение ночи, за ходом реакции наблюдали с помощью МС до тех пор, пока не происходило поглощение исходного продукта и образование определенного количества требуемого продукта. Продукт реакции без дальнейшей обработки очищали с использованием 18% ацетонитрила/воды в два погона с помощью препаративной хроматографии с обращенной фазой на колонке фирмы Waters [градиент от 0 до 30% ацетонитрила в течение 30 мин]. После лиофилизации образовавшегося продукта получали указанный в заголовке продукт (0,34 г) с выходом 64% в виде пены кремового цвета. МН+ 240,3, MH+Na+ 486,4. 1 В колбу объемом 250 мл вносили соединение, полученное согласно методу, описанному в примере 6 а) (2,0 г, 3,9 ммоля), и фенилдисульфид (0,860 г, 3,9 ммоля) в растворе циклогексан (70 мл)/бензол (40 мл). Для удаления из системы кислорода раствор барботировали азотом. Реакционную смесь в течение уикэнда облучали с помощью лампы, испускающей коротковолновое УФ-излучение. За ходом реакции наблюдали с помощью ЖХВР с обычной фазой (этилацетат/гексан). Установлено, что продукт содержал 71% транс-изомера и 29% цис-изомера. Реакционную смесь подвергали УФ-облучению еще в течение трех дней, в результате чего 84% исходного продукта превращалось в транс-изомер, а 16% исходного продукта превращалось в цис-изомер. После очистки с помощью хроматографии получали продукт, указанный в заголовке примере 8 а (0,956 г) с выходом 48%. МН+ 506,1, MH+NH4+ 523,2. 1H-ЯМР (CD3OD,част./млн): 1,2-2,0 (m, 8 Н), 2,42-2,6 (m, 6 Н), 2,91 (dd, 1 Н), 4,19 (m, 1 Н), 4,31 (dt,1H), 5,09 (s, 2H), 5,11 (s, 2H), 5,18 (dt, 1H), 5,27(m, 1 Н), 7,25 (bs, 10H). Пример 8 б). Моногидрохлорид фенилметилового эфира (2S,4E)-6-[(2R)-гексагидро-7-имино-1Hазепин-2-ил]-2-(фенилметокси)карбонил]амино]-4-гексановой кислоты У продукта, полученного согласно методу, описанному в примере 8 а) (0,956 г, 1,9 ммоля), растворенного в МеОН (80 мл), удаляли защитную группу согласно методу, описанному в примере 7 а), с помощью порошкообразного цинка (1,5 г) и смеси 60% НОАс/Н 2 О (40 мл). Образовавшийся продукт очищали с помощью хроматографии с обращенной фазой, получая указанный в заголовке продукт (0,248 г) с выходом 28%.- 27005817 Пример 8. Продукт, полученный согласно методу, описанному в примере 8 б) (0,248 г, 0,53 ммоля),превращали в указанный в заголовке продукт методом, описанным в примере 7, с ипользованием НСl (2 мл), Н 2 О (2 мл), CH3CN (4 мл). Неочищенный продукт очищали с помощью хроматографии с обращенной фазой, получая продукт, указанный в заголовке примера 8 (0,073 г), с выходом 57%. МН+ 240,3, MH+Na+ 486,4. 1 Образец продукта, полученного согласно методу, описанному в примере 6 а), превращали в указанное в заголовке соединение путем взаимодействия с НСl согласно методу, описанному в примере 8. Пример 10. Моногидрохлорид (2S,4E)-2-амино-6-[(5R)-6,7,8,9-тетрагидро-3-оксо-3H,5H-[1,2,4]оксадиазоло[4,3-]азепин-5-ил]-4-гексеновой кислоты Образец продукта, полученного согласно методу, описанному в примере 8 а), превращали в указанное в заголовке соединение путем взаимодействия с НСl согласно методу, описанному в примере 8. Пример 11. Моногидрохлорид (2S,4Z)-2-амино-6-[(2R,7Z)-гексагидро-7-(гидроксиимино)-1H-азепин-2-ил]-4-гексеновой кислоты Образец продукта, полученного согласно методу, описанному в примере 9, превращали в указанное в заголовке соединение путем взаимодействия с водным раствором гидроксида калия или натрия. Пример 12. Моногидрохлорид (2S,4E)-2-амино-6-[(2R,7Z)-гексагидро-7-(гидроксиимино)-1H-азепин-2-ил]- 4-гексеновой кислоты Образец продукта, полученного согласно методу, описанному в примере 10, превращали в указанное в заголовке соединение путем взаимодействия с водным раствором гидроксида калия или натрия. Конкретные новые промежуточные соединения, используемые в синтезе соединений по настоящему изобретению, представляют собой любые энантиомеры, стереоизомеры и геометрические изомеры следующих соединений:- 28005817 Биологическая активность Активность перечисленных выше соединений можно определять с помощью описанных ниже методов анализа. Анализ уровня синтазы оксида азота на основе оценки образования цитруллина Активность синтазы оксида азота (NOS) измеряли, наблюдая за превращением [3 Н]-аргинина в [3 Н]цитруллин (Bredt и Snyder, Proc. Natl. Acad. Sci. U.S.A., 87, 682-685, 1990, Misko и др., Eur. J. Pharm., 233,119-125, 1993). Осуществляли клонирование человеческой индуцибельной NOS (hiNOS), человеческой эндотелиальной конститутивной NOS (hecNOS) и человеческой нейронной конститутивной NOS(hncNOS), каждую из которых получали из РНК, выделенной из человеческой ткани. кДНК человеческой индуцибельной NOS (hiNOS) выделяли из библиотеки лямбда-кДНК, полученной из РНК, экстрагированной из образца ободочной кишки пациента, страдающего неспецифическим язвенным колитом. кДНК человеческой эндотелиальной конститутивной NOS (hecNOS) выделяли из библиотеки лямбда-кДНК,полученной из РНК, экстрагированной из эндотелиальных клеток пупочной вены человека (HUVEC), а кДНК человеческой нейронной конститутивной NOS (hncNOS) выделяют из библиотеки лямбда-кДНК,полученной из РНК, выделенной из мозжечка мертвого человека. Осуществляют экспрессию рекомбинатных ферментов в клетки насекомых линии Sf9 с помощью бакуловирусного вектора (Rodi и др., TheR., Higgs E., ред-ры; Portland Press Ltd., London, стр. 447-450 (1995. Обладающий активностью фермент выделяют из растворимых клеточных экстрактов и частично очищают с помощью хроматографии с использованием ДЭАЭ-сефарозы. Для измерения активности NOS 10 мкл фермента добавляют к 40 мкл 50 мМ Трис (рН 7,6) в присутствии тестируемых соединений или без них и инициируют реакцию путем добавления 50 мкл реакционной смеси, содержащей 50 мМ трис (рН 7,6), 2,0 мг/мл бычьего сывороточного альбумина, 2,0 мМ ДТТ, 4,0 мМ СаСl2, 20 мкМ ФАД (флавинадениндинуклеотид), 100 мкМ тетрагидробиоптерин, 0,4-2,0 мМ НАДФН и 60 мкМ L-аргинин, включающий L-[2,3-3 Н]-аргинин с радиоактивностью 0,9 мкКи. Конечная концентрация L-аргинина в смеси для анализа составляет 30 мкМ. В случае использования hecNOS или hncNOS применяют калмодулин в конечной концентрации 40-100 нМ. После инкубации в течение 15 мин при 37 С реакцию прекращают путем добавления 300 мкл холодного стопбуфера, содержащего 10 мМ ЭГТК (этиленгликольтетрауксусная кислота),100 мМ HEPES, рН 5,5 и 1 мМ цитруллин. [3 Н]-цитруллин отделяют путем хроматографии с использованием катионообменной смолыDowex 50W Х-8 и оценивают радиоактивность с помощью жидкостного сцинтилляционного счетчика. Результаты представлены в табл. 1 в виде значений IC50 тестируемых соединений в присутствии hiNOS,hecNOS и hncNOS. Таблица 1 Анализ in vivo Крысам вводили путем внутрибрюшинной инъекции 10-12,5 мг/кг эндотоксина (ЛПС (липополисахарид для того, чтобы индуцировать системную экспрессию синтазы оксида азота, которая приводит к выраженному повышению уровней нитритов/нитратов в плазме. Тестируемые соединения вводили перорально за 0,5-1 ч до введения ЛПС и уровни нитритов/нитратов в плазме оценивали через 5 ч после введения ЛПС. Соединение из примера 1 (дигидрохлорид (2S,5E)-2-амино-6-фтор-7-[(1-иминоэтил)амино]5-гептеновой кислоты) ингибировало индуцируемое ЛПС увеличение уровней нитритов/нитратов в плазме в зависимости от дозы, что свидетельствует о способности ингибировать активность индуцибельной синтазы оксида азота in vivo, при этом измеренное значение ED50 составляло 3 мг/кг (табл. 2). Таблица 2. Значения ED50, для соединений из перечисленных примеров, определенные в опытах с использованием обработанных эндотоксином крыс Все соединения вводили перорально, если не указано иное. Анализ содержания нитрита с использованием клеток линии RAW Клетки линии RAW 264.7 высевали до достижения конфлуэнтности в 96-луночные планшеты для культур тканей и выращивали в течение ночи (17 ч) в присутствии ЛПС для индуцирования NOS. Ряд,состоящий из 3-6 лунок, не подвергали обработке и он служил в качестве контроля для учета неспецифического базового уровня. Среду удаляли из каждой лунки и клетки промывали раствором КребсаРингера-HEPES (25 мМ, рН 7,4), дополненным 2 мг/мл глюкозы. Затем клетки помещали на лед и инкубировали в течение 1 ч в 50 мл буфера, включающего L-аргинин (30 мМ) и содержащего ингибиторы или- 29005817 без них. Анализ начинали путем нагрева планшета до 37 С в водяной бане в течение 1 ч. Производство нитрита внутриклеточными iNOS характеризуется линейной зависимостью от времени. Для завершения анализа клеточной активности содержащие клетки планшеты помещали на лед, отделяли содержащий нитрит буфер и анализировали содержание нитрита с помощью опубликованного ранее флуоресцентного метода определения содержания нитрита (см. у Т. P. Misko и др., Analytical Biochemistry, 214, 11-16(1993. Анализ эксплантатов человеческого хряща Кусочки костной ткани промывали дважды забуференным фосфатом физиологическим раствором Дульбекко (фирма GibcoBRL) и один раз модифицированной по методу Дульбекко средой Игла (фирмаGibcoBRL) и помещали в чашку Петри в несодержащую фенолового красного минимальную незаменимую среду (MEM) (фирма GibcoBRL). Хрящ разрезали на небольшие эксплантаты массой приблизительно 25-45 мг и одному или два эксплантата помещали в каждую лунку 48-луночного планшета для культур тканей, содержащую 500 мкл культуральной среды. Культуральная среда представляла собой либо обычную модификацию минимальной необходимой среды (среда Игла) с добавлением солей Ирла (фирма GibcoBRL), приготовленную без добавления L-аргинина, L-глутамата и фенолового красного, либо обычную модификацию бессывороточной среды Ньюмена и Тителла (фирма GibcoBRL), приготовленную без добавления L-аргинина, инсулина, аскорбиновой кислоты, L-глутамина и без фенолового красного. Перед применением обе среды дополняли 100 мкМL-аргинина (фирма Sigma), 2 мМ L-глутамина,добавкой 1X HL-1 (фирма BioWhittaker), 50 мг/мл аскорбиновой кислоты (фирма Sigma) и 150 пг/мл рекомбинантного человеческого IL-1 (фирма RD Systems) для индуцирования синтазы оксида азота. После этого добавляли соединения в виде аликвот объемом 10 мкл и эксплантаты инкубировали в течение 18-24 ч при 37 С в атмосфере, содержащей 5% СО 2. Затем образовавшийся в течение суток супернатант отбрасывали и заменяли свежей культуральной средой, содержащей рекомбинантный человеческий IL1 и соединение, и инкубировали еще в течение 20-24 ч. Содержание нитрита в этом супернатанте анализировали флуорометрическим методом (Misko и др., Anal. Biochem., 214, 11-16, 1993). Исследование всех образцов проводили в четырех повторностях. Эксплантаты взвешивали и уровни нитрита стандартизовали по отношению к массе эксплантата. Контрольные культуры, не подвергнутые стимуляции, выращивали в среде, не содержащей рекомбинантный человеческий IL-1. Значения IC50 (табл. 3) определяли на основе графика зависимости процента ингибирования производства нитрита от концентрации ингибитора, полученного для шести различных концентраций ингибитора. Таблица 3 Анализ зависимости ингибирования от времени Оценивали активность соединений в отношении ингибирования изоформ человеческой NOS в зависимости от времени с помощью предварительной инкубации в течение периода времени 0-60 мин соединения вместе с ферментом при 37 С в присутствии компонентов, применяемых в ферментативном анализе цитруллина, за исключением L-аргинина. В моменты времени, равные 0, 10, 21 и 60 мин отбирали аликвоты (10 мкл) и их сразу добавляли к реакционной смеси для ферментативного анализа цитруллина,содержащей L-[2,3-3 Н]аргинин и имеющей конечную концентрацию L-аргинина 30 мкМ в конечном объеме 100 мкл. Реакции давали протекать в течение 15 мин при 37 С и ее прекращали путем добавления стоп-буфера и продукт подвергали хроматографии на колонке с использованием катионообменной смолы Dowex 50W Х-8 согласно методу, описанному для анализа NOS с использованием цитруллина. В качестве % ингибирования активности NOS соединением-ингибитором принимали процент ингибирования активности по отношению к активности контрольного фермента, предварительно инкубированного в течение такого же промежутка времени без ингибитора. Данные, приведенные в табл. 4, представляют собой % ингибирования после 21 и 60 мин предварительной инкубации смеси, содержащей ингибитор и фермент. Таблица 4 На основе приведенного выше описания специалист в данной области может легко понять основные характеристики изобретения и без отклонения от его сущности и объема может реализовать различные изменения и модификации изобретения для приспособления к различным случаям и условиям применения.
МПК / Метки
МПК: C07D 233/12, A61P 29/00, A61K 31/55
Метки: производные, оксида, гексагидро-7-1н-азепин-2-илгексановой, качестве, синтазы, индуцибельной, или, кислоты, азота, ингибиторов, гексеновой
Код ссылки
<a href="https://eas.patents.su/30-5817-proizvodnye-geksagidro-7-1n-azepin-2-ilgeksanovojj-ili-geksenovojj-kisloty-v-kachestve-ingibitorov-inducibelnojj-sintazy-oksida-azota.html" rel="bookmark" title="База патентов Евразийского Союза">Производные гексагидро-7-1н-азепин-2-илгексановой (или гексеновой) кислоты в качестве ингибиторов индуцибельной синтазы оксида азота</a>
Производные 2-аминопиридинов, способ ингибирования синтазы оксида азота, фармацевтические композиции и способы лечения

Номер патента: 3991
Опубликовано: 25.12.2003
Автор: Лоуи Джон Адамс III
МПК: C07D 213/73, A61P 11/00, A61K 31/4439...
Метки: производные, композиции, ингибирования, фармацевтические, 2-аминопиридинов, лечения, способы, оксида, азота, способ, синтазы
Формула / Реферат:
1. Производные 2-аминопиридинов формулы где n и m в мостиковых кольцах независимо равны 1, 2 или 3 и атом углерода в одном из указанных мостиковых колец может быть заменен гетероатомом, выбранным из O, S и N, при условии, что узловой атом углерода может быть заменен только атомом азота, и R1 и R2 независимо выбраны из C1-C6алкила, который может быть линейным, разветвленным или циклическим или может содержать линейную, циклическую либо...
Ингибиторы синтазы оксида азота

Номер патента: 2033
Опубликовано: 24.12.2001
Авторы: Ходсон Хэролд Фрэнсис, Френд Энтони Джозеф, Бимз Ричард Менсфилд, Риз Дэрил Дейвид, Ноулс Ричард Грэм, Френзмен Карл Витолд, Сойер Дейвид Алан, Дрисдейл Мартин Джеймс
МПК: A61P 29/00, C07C 323/58, A61K 31/197...
Метки: оксида, азота, синтазы, ингибиторы
Формула / Реферат:
1. Соединение формулы (I) или его соль, сольват или физиологически функциональное производное. 2. Соединение формулы (I), выбранное из (R,S)-[2-(1-иминоэтиламино)этил]-DL-гомоцистеина, (S)-[2-(1-иминоэтиламино)этил]-L-гомоцистеина и (R)-[2-(1-иминоэтиламино)этил]-D-гомоцистеина или его соль, сольват или физиологически функциональное производное. 3. Соединение формулы (I), которое представляет собой...
Ингибиторы синтазы оксида азота

Номер патента: 3026
Опубликовано: 26.12.2002
Авторы: Янг Роберт Джон, Бесуик Пол Джон, Клинтхаус Сэввас
МПК: C07C 323/58, A61K 31/198, A61P 1/04...
Метки: азота, оксида, синтазы, ингибиторы
Формула / Реферат:
1. Соединение формулы (I) или его соль, сольват или физиологически функциональное производное, где R1 выбран из С1-4алкила, С3-4циклоалкила, С1-4гидроксиалкила и С1-4галогеноалкила. 2. Соединение формулы (I), выбранное из S-[(R)-2-(1-иминоэтиламино)пропил]-L-цистеина, S-[(S)-2-(1-иминоэтиламино)пропил]-L-цистеина, S-[(R/S)-2-(1-иминоэтиламино)пропил]-L-цистеина, S-[(R)-2-(1-иминоэтиламино)пропил]-D-цистеина,...
2-амино-6-(2-замещенные-4-фенокси)-замещенные-пиридины в качестве ингибиторов синтазы окиси азота

Номер патента: 3834
Опубликовано: 30.10.2003
Авторы: Новаковски Йоланта, Волкманн Роберт Альфред, Лауэ Джон Адамс III
МПК: A61K 31/4418, C07D 213/73, A61P 25/00...
Метки: окиси, качестве, азота, синтазы, ингибиторов, 2-амино-6-(2-замещенные-4-фенокси)-замещенные-пиридины
Формула / Реферат:
1. Соединение формулы где R1 и R2 выбраны независимо из группы, включающей водород, галоид, гидрокси, (C1-C6)алкокси, (C1-C7)алкил, (C2-C6)алкенил, (C2-C10)алкоксиалкил; G выбирают из группы, включающей водород, (C1-C6)алкил, (C1-C6)алкокси(C1-C3)алкил, аминокарбонил-(C1-C3)алкил-, (C1-C3)алкиламинокарбонил-(C1-C3)алкил-, ди-[(C1-C3)алкил]аминокарбонил-(C1-C3)алкил и N(R3)(R4)(C0-C4)алкил-, где R3 и R4 независимо выбирают из группы, включающей...
2-аминопиридины, содержащие в качестве заместителей конденсированные кольца, применяемые в качестве ингибиторов nos (no синтазы)

Номер патента: 3839
Опубликовано: 30.10.2003
Автор: Лоуэ Джон Адамс III
МПК: A61K 31/495, C07D 401/10
Метки: заместителей, ингибиторов, применяемые, кольца, синтазы, содержащие, конденсированные, 2-аминопиридины, качестве
Формула / Реферат:
1. Соединение формулы где кольцо A является 5-7-членным насыщенным или ненасыщенным конденсированным кольцом, X является кислородом или связью; n представляет собой целое число от 2 до 6 и R1 и R2 независимо выбирают из водорода (C1-C6)алкила, арила, тетрагидронафталина и аралкила, где указанный арил и арильная часть указанного аралкила представляет собой фенил или нафтил, а алкильная часть является линейной или разветвленной и содержит от 1 до...
Предыдущий патент: Применяемые в качестве медикаментов антихолинергические средства, а также способ их получения
Следующий патент: Сложные эфиры фениламинобензгидроксамовых кислот
Случайный патент: Способ транспортирования, передачи и выгрузки порошкового химического состава