Пиперидинодигидротиенопиримидинсульфоксиды и их применение для лечения хозл и астмы
Номер патента: 24033
Опубликовано: 31.08.2016
Авторы: Ким Суджин, Вэй Сюйдон, Пузе Паскаль А.Ж., Тэмпоун Томас Г., Вертманн Ульрике, Малдер Джейсон Алан, Сенанаяке Крис Х., Ян Биншиоу, Фрутос Рохелио П., Пейтел Нитинчандра, Николаус Петер

























Формула / Реферат
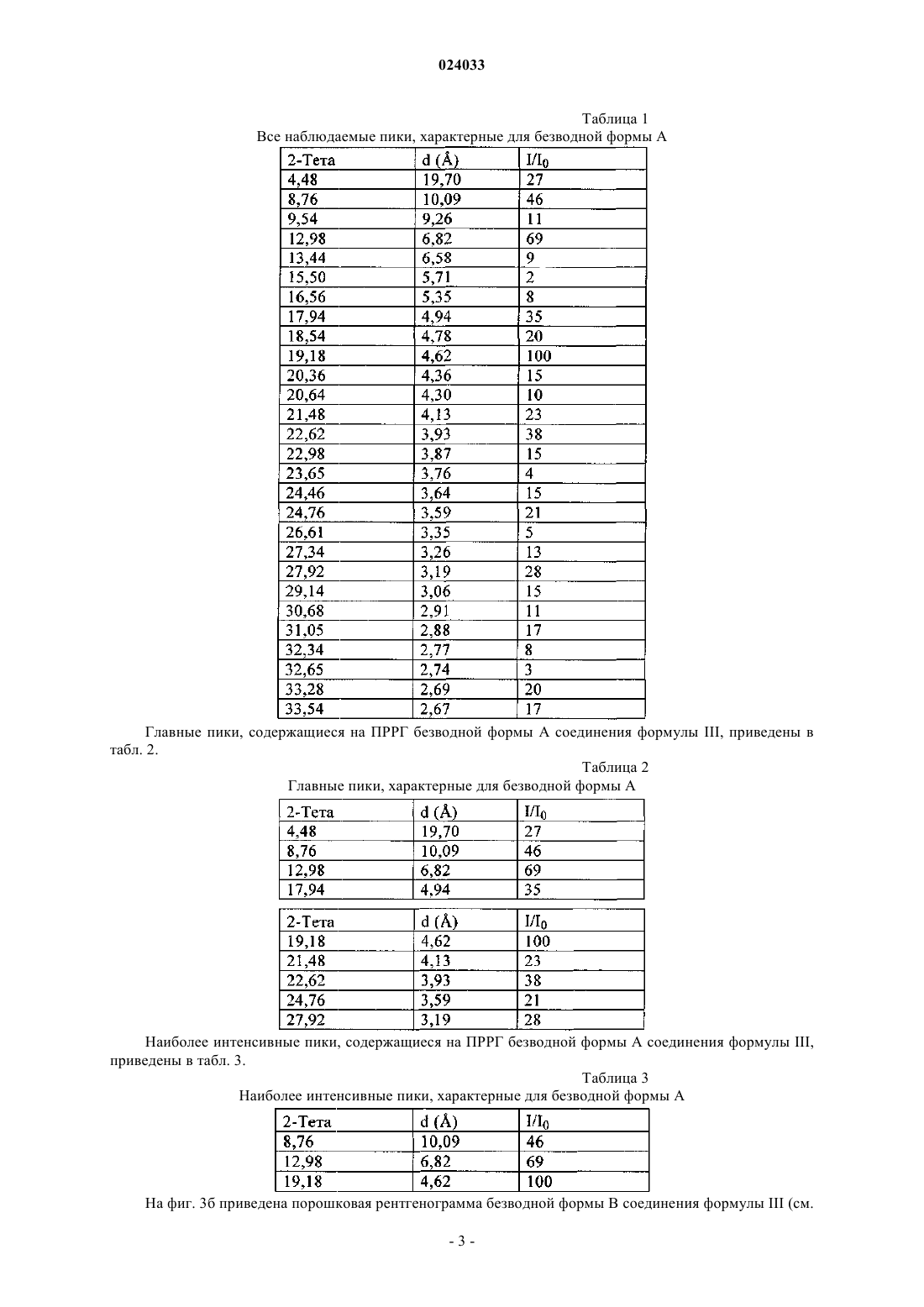
1. Соединение формулы I

в которой кольцо А представляет собой 6-членное ароматическое кольцо, которое необязательно может содержать 1 или 2 атома азота;
R обозначает Cl и
R может быть расположен в пара-, мета- или орто-положении кольца А;
S* обозначает атом серы, который является хиральным центром,
и все его фармацевтически приемлемые соли, энантиомеры и рацематы.
2. Соединение формулы I по п.1, в которой R обозначает Cl и находится в пара-положении кольца А, и все его фармацевтически приемлемые соли, энантиомеры и рацематы.
3. Соединение формулы I по одному из пп.1 или 2, в которой кольцо А выбрано из группы, включающей фенил, пиридинил и пиримидинил, и все его фармацевтически приемлемые соли, энантиомеры и рацематы.
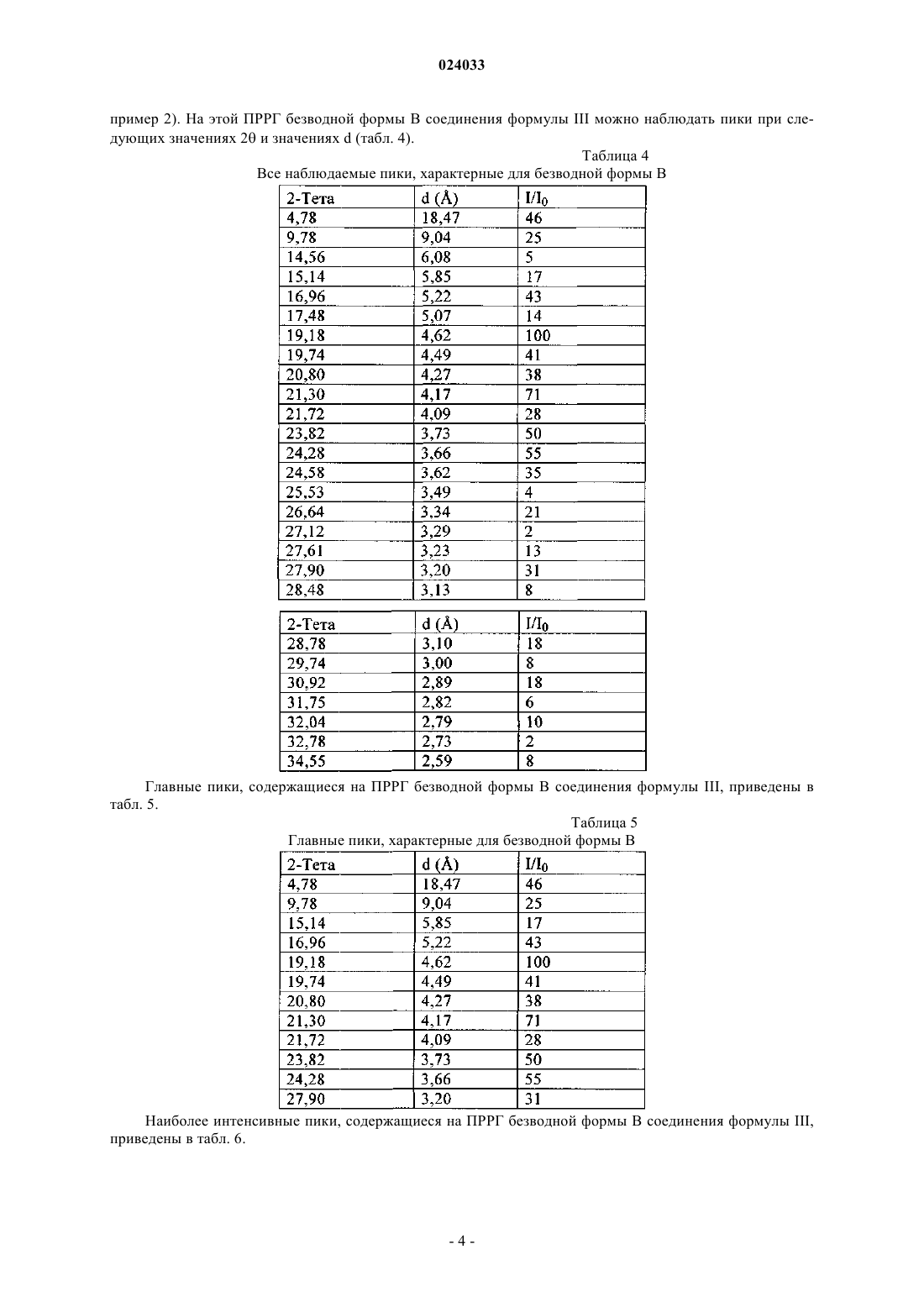
4. Соединение по одному из пп.1-3, которое является соединением формулы II

и все его фармацевтически приемлемые соли, энантиомеры и рацематы.
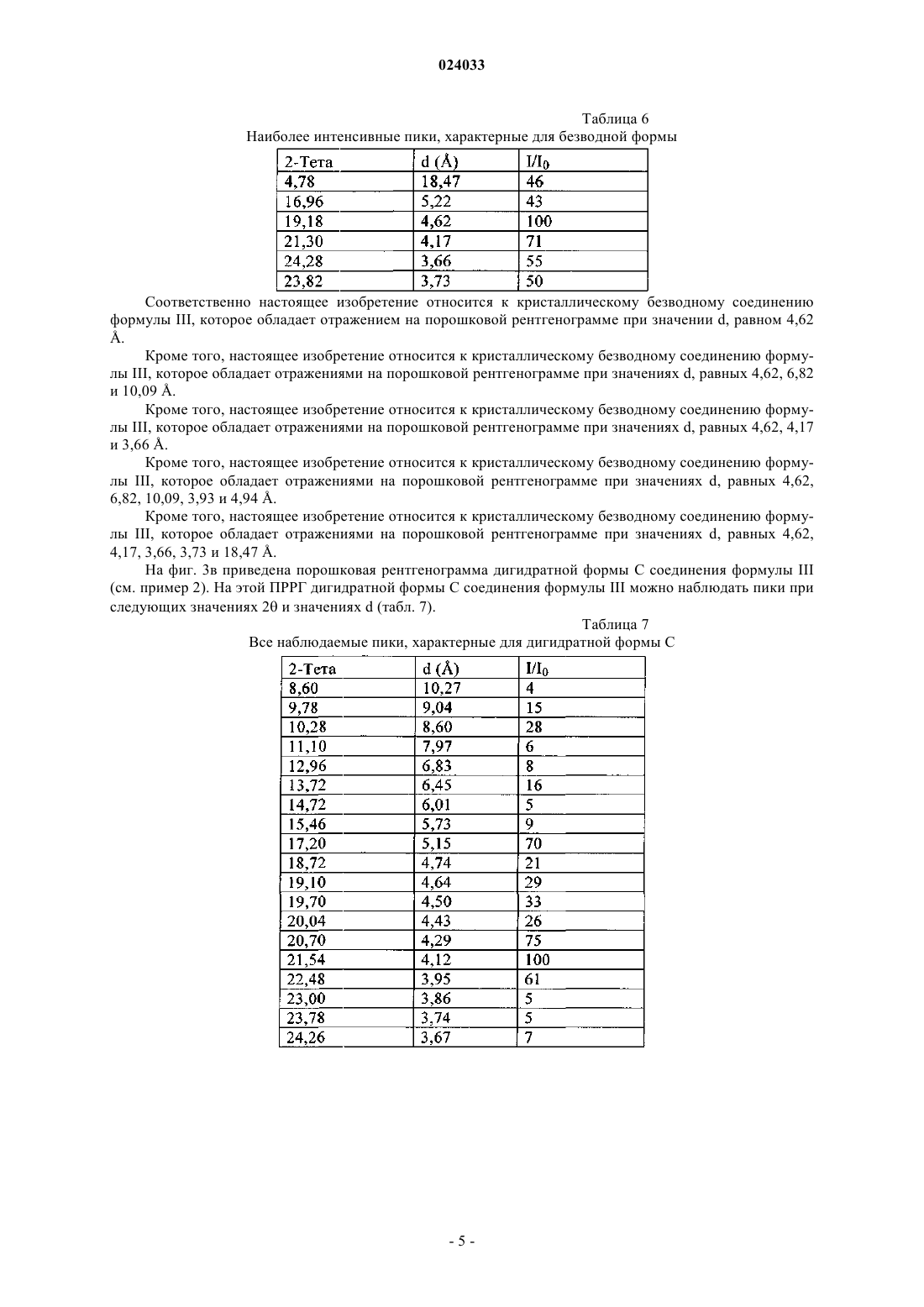
5. Соединение по одному из пп.1-3, которое является соединением формулы III

и все его фармацевтически приемлемые соли, энантиомеры и рацематы.
6. Соединение по одному из пп.1-5, в котором S* обозначает атом серы, который является хиральным центром, обладающим R-конфигурацией.
7. Соединение по одному из пп.1-5, в котором S* обозначает атом серы, который является хиральным центром, обладающим S-конфигурацией.
8. Кристаллическое безводное соединение формулы III по п.5, которое обладает отражением на порошковой рентгенограмме при значении межплоскостного расстояния (d), равном 4,62 Å.
9. Кристаллическое безводное соединение формулы III по п.5, которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,62, 6,82 и 10,09 Å.
10. Кристаллическое безводное соединение формулы III по п.5, которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,62, 4,17 и 3,66 Å.
11. Кристаллическое безводное соединение формулы III по п.5, которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,62, 6,82, 10,09, 3,93 и 4,94 Å.
12. Кристаллическое безводное соединение формулы III по п.5, которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,62, 4,17, 3,66, 3,73 и 18,47 Å.
13. Кристаллический дигидрат соединения формулы III по п.5, который обладает отражением на порошковой рентгенограмме при значении d, равном 4,12 Å.
14. Кристаллический дигидрат соединения формулы III по п.5, который обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,12, 4,29 и 5,15 Å.
15. Кристаллический дигидрат соединения формулы III по п.5, который обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,12, 4,29, 5,15, 3,95 и 3,36 Å.
16. Применение соединения по одному из пп.1-15 для приготовления лекарственного средства, предназначенного для лечения заболевания, которое можно лечить путем ингибирования фермента PDE4.
17. Применение по п.16, где заболевание, которое можно лечить путем ингибирования фермента PDE4, выбрано из группы, включающей респираторное заболевание, желудочно-кишечное заболевание, воспалительное заболевание суставов, кожи или глаз, рак и заболевание периферической или центральной нервной системы.
18. Применение по п.17, где заболевание, которое можно лечить путем ингибирования фермента PDE4, выбрано из группы, включающей респираторное заболевание или заболевание легких, которое сопровождается усилением образования слизи, воспаления, и/или обструктивные заболевания дыхательных путей.
19. Применение по п.18, где заболевание, которое можно лечить путем ингибирования фермента PDE4, выбрано из группы, включающей ХОЗЛ (хроническое обструктивное заболевание легких), идиопатический фиброз легких, альфа-1-антитрипсиновую недостаточность, хронический синусит, астму и хронический бронхит.
20. Применение по п.16, где заболеванием, которое можно лечить путем ингибирования фермента PDE4, является воспалительное заболевание суставов, кожи или глаз, выбранное из группы, включающей ревматоидный артрит, саркоидоз, сухой кератит и глаукому.
21. Применение по п.16, где заболевание, которое можно лечить путем ингибирования фермента PDE4, выбрано из группы, включающей болезнь Крона или язвенный колит.
22. Применение по п.16, где заболевание, которое можно лечить путем ингибирования фермента PDE4, выбрано из группы, включающей депрессию, биполярную или маниакальную депрессию, острые и хронические состояния тревоги, шизофрению, болезнь Альцгеймера, болезнь Паркинсона, острый и хронический рассеянный склероз или острую и хроническую боль и повреждение мозга, вызванное ударом, гипоксией или черепно-мозговой травмой, сухой кератит и глаукому.
23. Фармацевтическая композиция, содержащая соединение по одному из пп.1-15.
24. Фармацевтическая композиция, отличающаяся тем, что она содержит соединение формул I, II или III по одному из пп.1-15 в комбинации с одним или большим количеством активных соединений, выбранных из группы, включающей бета-миметики, кортикостероиды, антихолинергетики, другие ингибиторы PDE4, НСПВС, ингибиторы СОХ2, антагонисты рецептора ЕР4, ингибиторы EGFR, антагонисты LTD4, ингибиторы CCR3, ингибиторы iNOS, ингибиторы MRP4 и ингибиторы SYK.
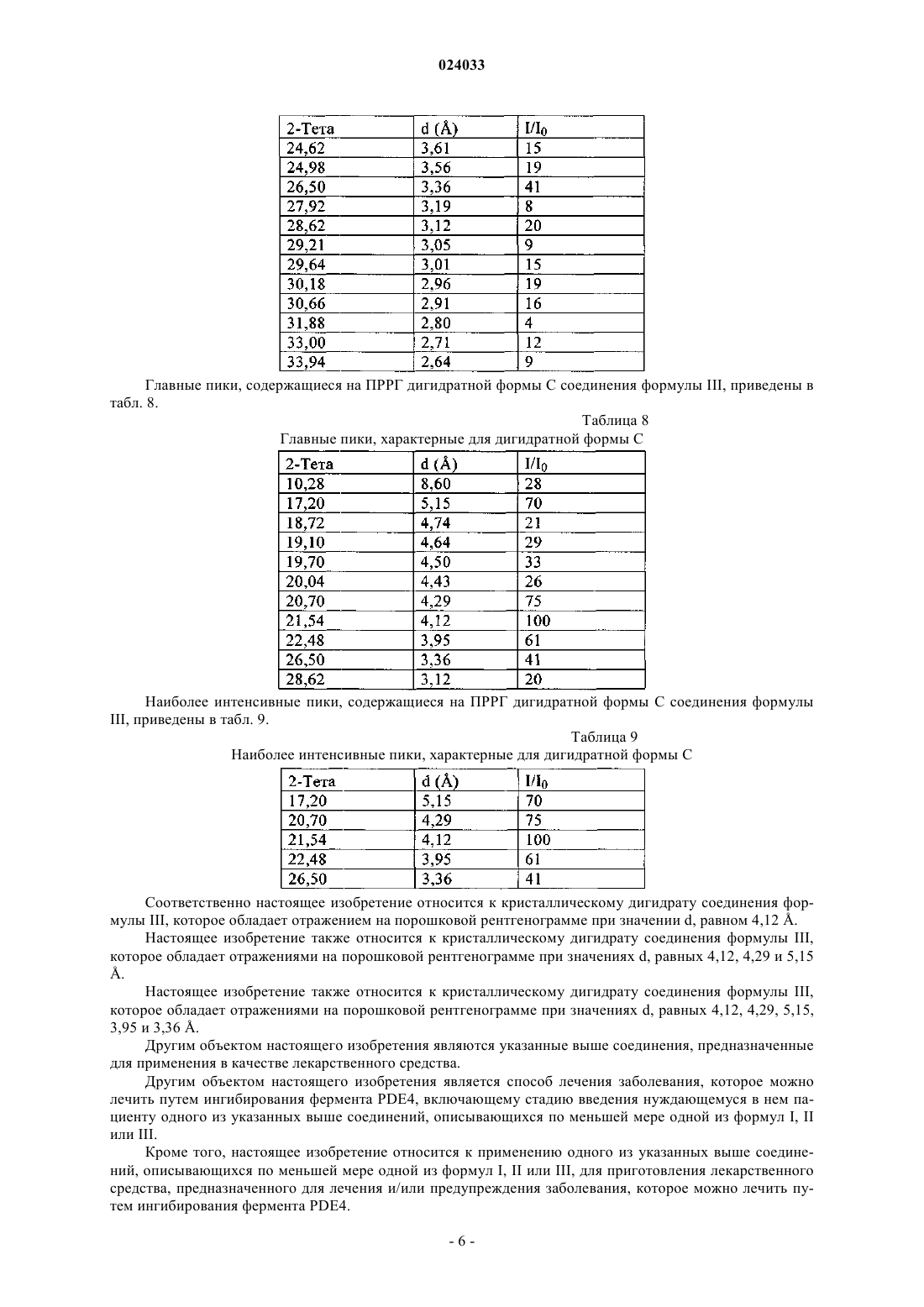
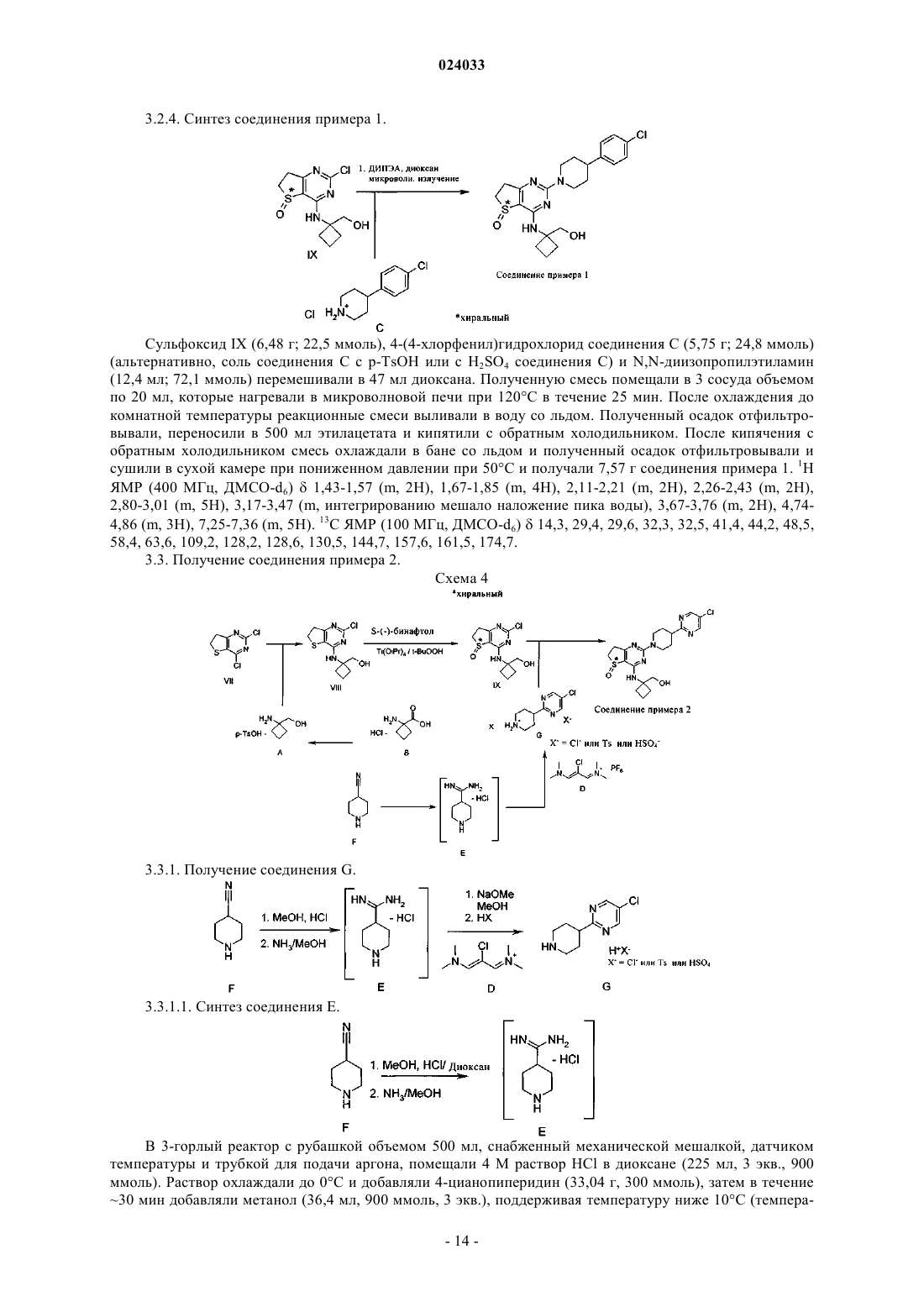
25. Промежуточный продукт формулы VIII

и его соли.
26. Промежуточный продукт формулы IX

в которой S* обозначает атом серы, который является хиральным центром, и его соли.
Текст
ПИПЕРИДИНОДИГИДРОТИЕНОПИРИМИДИНСУЛЬФОКСИДЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ХОЗЛ И АСТМЫ Рохелио П., Ян Биншиоу, Ким Суджин,Малдер Джейсон Алан, Пейтел Нитинчандра, Сенанаяке Крис Х.,Тэмпоун Томас Г., Вэй Сюйдон (US) В изобретении описаны новые пиперидинодигидротиенопиримидинсульфоксиды формулы I в которой кольцо А представляет собой 6-членное ароматическое кольцо, которое необязательно может содержать 1 или 2 атома азота, и в которой R обозначает Cl и может быть расположен в пара-, мета- или орто-положении кольца А, в которой S обозначает атом серы, который является хиральным центром, и все его фармацевтически приемлемые соли, энантиомеры и рацематы, его гидраты и сольваты, и применение этих соединений для лечения воспалительных или аллергических заболеваний дыхательных путей, таких как ХОЗЛ (хроническое обструктивное заболевание легких) или астма.(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) Настоящее изобретение относится к новым пиперидинодигидротиенопиримидинсульфоксидам формулы I в которой кольцо A представляет собой 6-членное ароматическое кольцо, которое необязательно может содержать 1 или 2 атома азота; иR может быть расположен в пара-, мета- или орто-положении кольца A;S обозначает атом серы, который является хиральным центром,и ко всем их фармацевтически приемлемым солям, энантиомерам и рацематам, их гидратам и сольватам, и к применению этих соединений для лечения воспалительных или аллергических заболеваний дыхательных путей, таких как ХОЗЛ (хроническое обструктивное заболевание легких) или астма. Уровень техники В WO 2006/111549 и WO 2007/118793 раскрыты дигидротиенопиримидинсульфоксиды, которые замещены пиперазином вместо пиперидина. В WO 2009/050248 раскрыты пиперидинодигидротиенопиримидины, которые обладают другой схемой замещения по сравнению с соединениями, предлагаемыми в настоящем изобретении. Благодаря их особенной схеме замещения соединения, предлагаемые в настоящем изобретении, являются более эффективными ингибиторами PDE4, чем соединения, раскрытые вWO 2009/050248, и в то же время обеспечивают сведение к минимуму возможности проявления нежелательных побочных эффектов, связанных с желудочно-кишечным трактом. Описание изобретения Согласно изобретению неожиданно установлено, что соединения, предлагаемые в настоящем изобретении, благодаря их особенной схеме замещения являются особенно подходящими для лечения воспалительного заболевания. Кроме того, соединения, предлагаемые в настоящем изобретении, превосходят соответствующие пиперазинодигидротиенопиримидинсульфоксиды, описанные в документе предшествующего уровня техники, WO 2009/050248. Поэтому настоящее изобретение относится к соединениям формулы I в которой кольцо А представляет собой 6-членное ароматическое кольцо, которое необязательно может содержать 1 или 2 атома азота; иR может быть расположен в пара-, мета- или орто-положении кольца A;S обозначает атом серы, который является хиральным центром; и ко всем их фармацевтически приемлемым солям, энантиомерам и рацематам, их гидратам, сольватам и полиморфным формам. Настоящее изобретение также относится к указанным выше соединениям формулы I, в которой R обозначает Cl и предпочтительно находится в пара-положении кольца A, и ко всем их фармацевтически приемлемым солям, энантиомерам и рацематам, их гидратам, сольватам и полиморфным формам. Настоящее изобретение также относится к указанным выше соединениям формулы I, в которой кольцо А выбрано из группы, включающей фенил, пиридинил и пиримидинил, и ко всем их фармацевтически приемлемым солям, энантиомерам и рацематам, их гидратам, сольватам и полиморфным формам. Настоящее изобретение предпочтительно относится к указанным выше соединениям формулы I, в которой кольцо A выбрано из группы, включающей фенил, пиридинил и пиримидинил, и в которой R обозначает заместитель Cl, находящийся в пара-положении, и ко всем их фармацевтически приемлемым солям, энантиомерам и рацематам, их гидратам, сольватам и полиморфным формам. В частности, настоящее изобретение относится к соединению формулы II и ко всем его фармацевтически приемлемым солям, энантиомерам и рацематам, его гидратам, сольватам и полиморфным формам. В частности, настоящее изобретение относится к соединению формулы III и ко всем его фармацевтически приемлемым солям, энантиомерам и рацематам, его гидратам, сольватам и полиморфным формам. Настоящее изобретение также относится к указанным выше соединениям, описывающимся одной из формул I, II или III, в которой S обозначает атом серы, который является хиральным центром, обладающим R-конфигурацией. Настоящее изобретение также относится к указанным выше соединениям, описывающимся одной из формул I, II или III, в которой S обозначает атом серы, который является хиральным центром, обладающим S-конфигурацией. Для соединения формулы III с помощью порошковой рентгенографии (ПРРГ), с помощью термогравиметрического анализа (ТГА) и с помощью дифференциальной сканирующей калориметрии (ДСК) идентифицированы и охарактеризованы три разные полиморфные формы, две разные безводные формы и одна гидратная форма. На фиг. 3 а приведена порошковая рентгенограмма безводной формы А соединения формулы III (см. пример 2). На этой ПРРГ безводной формы А соединения формулы III можно наблюдать пики при следующих значениях 2 и значениях межплоскостного расстояния d (табл. 1). Таблица 1 Все наблюдаемые пики, характерные для безводной формы A Главные пики, содержащиеся на ПРРГ безводной формы А соединения формулы III, приведены в табл. 2. Таблица 2 Главные пики, характерные для безводной формы А Наиболее интенсивные пики, содержащиеся на ПРРГ безводной формы А соединения формулы III,приведены в табл. 3. Таблица 3 Наиболее интенсивные пики, характерные для безводной формы А На фиг. 3 б приведена порошковая рентгенограмма безводной формы В соединения формулы III (см. пример 2). На этой ПРРГ безводной формы В соединения формулы III можно наблюдать пики при следующих значениях 2 и значениях d (табл. 4). Таблица 4 Все наблюдаемые пики, характерные для безводной формы В Главные пики, содержащиеся на ПРРГ безводной формы В соединения формулы III, приведены в табл. 5. Таблица 5 Главные пики, характерные для безводной формы В Наиболее интенсивные пики, содержащиеся на ПРРГ безводной формы В соединения формулы III,приведены в табл. 6. Таблица 6 Наиболее интенсивные пики, характерные для безводной формы Соответственно настоящее изобретение относится к кристаллическому безводному соединению формулы III, которое обладает отражением на порошковой рентгенограмме при значении d, равном 4,62. Кроме того, настоящее изобретение относится к кристаллическому безводному соединению формулы III, которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,62, 6,82 и 10,09 . Кроме того, настоящее изобретение относится к кристаллическому безводному соединению формулы III, которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,62, 4,17 и 3,66 . Кроме того, настоящее изобретение относится к кристаллическому безводному соединению формулы III, которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,62,6,82, 10,09, 3,93 и 4,94 . Кроме того, настоящее изобретение относится к кристаллическому безводному соединению формулы III, которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,62,4,17, 3,66, 3,73 и 18,47 . На фиг. 3 в приведена порошковая рентгенограмма дигидратной формы С соединения формулы III(см. пример 2). На этой ПРРГ дигидратной формы С соединения формулы III можно наблюдать пики при следующих значениях 2 и значениях d (табл. 7). Таблица 7 Все наблюдаемые пики, характерные для дигидратной формы C Главные пики, содержащиеся на ПРРГ дигидратной формы C соединения формулы III, приведены в табл. 8. Таблица 8 Главные пики, характерные для дигидратной формы C Наиболее интенсивные пики, содержащиеся на ПРРГ дигидратной формы C соединения формулыIII, приведены в табл. 9. Таблица 9 Наиболее интенсивные пики, характерные для дигидратной формы C Соответственно настоящее изобретение относится к кристаллическому дигидрату соединения формулы III, которое обладает отражением на порошковой рентгенограмме при значении d, равном 4,12 . Настоящее изобретение также относится к кристаллическому дигидрату соединения формулы III,которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,12, 4,29 и 5,15. Настоящее изобретение также относится к кристаллическому дигидрату соединения формулы III,которое обладает отражениями на порошковой рентгенограмме при значениях d, равных 4,12, 4,29, 5,15,3,95 и 3,36 . Другим объектом настоящего изобретения являются указанные выше соединения, предназначенные для применения в качестве лекарственного средства. Другим объектом настоящего изобретения является способ лечения заболевания, которое можно лечить путем ингибирования фермента PDE4, включающему стадию введения нуждающемуся в нем пациенту одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III. Кроме того, настоящее изобретение относится к применению одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, для приготовления лекарственного средства, предназначенного для лечения и/или предупреждения заболевания, которое можно лечить путем ингибирования фермента PDE4. Кроме того, настоящее изобретение относится к одному из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, предназначенному для лечения и/или предупреждения заболевания, которое можно лечить путем ингибирования фермента PDE4. Настоящее изобретение также относится к указанному выше способу лечения заболевания, которое можно лечить путем ингибирования фермента PDE4, включающему стадию введения нуждающемуся в нем пациенту одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, отличающемуся тем, что подвергающееся лечению заболевание выбрано из группы,включающей респираторное заболевание, желудочно-кишечное заболевание, воспалительное заболевание суставов, кожи или глаз, рак и заболевание периферической или центральной нервной системы. Кроме того, настоящее изобретение относится к применению одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, для приготовления лекарственного средства, предназначенного для лечения и/или предупреждения заболевания, которое можно лечить путем ингибирования фермента PDE4, где подвергающееся лечению заболевание выбрано из группы,включающей респираторное заболевание, желудочно-кишечное заболевание, воспалительное заболевание суставов, кожи или глаз, рак и заболевание периферической или центральной нервной системы. Кроме того, настоящее изобретение относится к одному из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, предназначенному для лечения и/или предупреждения заболевания, которое можно лечить путем ингибирования фермента PDE4, где подвергающееся лечению заболевание выбрано из группы, включающей респираторное заболевание, желудочнокишечное заболевание, воспалительное заболевание суставов, кожи или глаз, рак и заболевание периферической или центральной нервной системы. Настоящее изобретение также относится к указанному выше способу лечения заболевания, которое выбрано из группы, включающей респираторное заболевание или заболевание легких, которое сопровождается усилением образования слизи, воспаления, и/или обструктивные заболевания дыхательных путей, включающему стадию введения нуждающемуся в нем пациенту одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III. Кроме того, настоящее изобретение относится к применению одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, для приготовления лекарственного средства, предназначенного для лечения и/или предупреждения заболевания, выбранного из группы,включающей респираторное заболевание или заболевание легких, которое сопровождается усилением образования слизи, воспаления, и/или обструктивные заболевания дыхательных путей, включающего стадию введения нуждающемуся в нем пациенту одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III. Кроме того, настоящее изобретение относится к одному из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, предназначенному для лечения и/или предупреждения заболевания, выбранного из группы, включающей респираторное заболевание или заболевание легких, которое сопровождается усилением образования слизи, воспаления, и/или обструктивные заболевания дыхательных путей, включающего стадию введения нуждающемуся в нем пациенту одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III. Настоящее изобретение такжеотносится к указанному выше способу лечения заболевания, которое выбрано из группы, включающей ХОЗЛ, хронический синусит, идиопатический фиброз легких, альфа-1 антитрипсиновую недостаточность, астму и хронический бронхит, включающему стадию введения нуждающемуся в нем пациенту одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III. Кроме того, настоящее изобретение к применению одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, для приготовления лекарственного средства,предназначенного для лечения и/или предупреждения заболевания, выбранного из группы, включающей ХОЗЛ, хронический синусит, идиопатический фиброз легких, альфа-1-антитрипсиновую недостаточность, астму и хронический бронхит. Кроме того, настоящее изобретение относится к одному из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, предназначенному для лечения и/или предупреждения заболевания, выбранного из группы, включающей ХОЗЛ, хронический синусит, идиопатический фиброз легких, альфа-1-антитрипсиновую недостаточность, астму и хронический бронхит. Кроме того, настоящее изобретение к применению одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, для приготовления лекарственного средства,предназначенного для лечения и/или предупреждения заболевания, выбранного из группы, включающей ревматоидный артрит, саркоидоз, глаукому и сухой кератит. Кроме того, настоящее изобретение относится к одному из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, предназначенному для лечения и/или предупреждения заболевания, выбранного из группы, включающей ревматоидный артрит, саркоидоз, глаукому и сухой кератит. Настоящее изобретение также относится к указанному выше способу лечения заболевания, которое выбрано из группы, включающей болезнь Крона и язвенный колит, включающему стадию введения нуждающемуся в нем пациенту одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III. Кроме того, настоящее изобретение к применению одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, для приготовления лекарственного средства,предназначенного для лечения и/или предупреждения заболевания, выбранного из группы, включающей болезнь Крона и язвенный колит. Кроме того, настоящее изобретение относится к одному из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, предназначенному для лечения и/или предупреждения заболевания, выбранного из группы, включающей болезнь Крона и язвенный колит. Настоящее изобретение также относится к указанному выше способу лечения заболевания, которое выбрано из группы, включающей депрессию, биполярную или маниакальную депрессию, острые и хронические состояния тревоги, шизофрению, болезнь Альцгеймера, болезнь Паркинсона, острый и хронический рассеянный склероз или острую и хроническую боль, и повреждение мозга, вызванное ударом,гипоксией или черепно-мозговой травмой, включающему стадию введения нуждающемуся в нем пациенту одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II илиIII. Кроме того, настоящее изобретение к применению одного из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, для приготовления лекарственного средства,предназначенного для лечения и/или предупреждения заболевания, выбранного из группы, включающей депрессию, биполярную или маниакальную депрессию, острые и хронические состояния тревоги, шизофрению, болезнь Альцгеймера, болезнь Паркинсона, острый и хронический рассеянный склероз или острую и хроническую боль, и повреждение мозга, вызванное ударом, гипоксией или черепно-мозговой травмой. Кроме того, настоящее изобретение относится к одному из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, предназначенному для лечения и/или предупреждения заболевания, выбранного из группы, включающей депрессию, биполярную или маниакальную депрессию, острые и хронические состояния тревоги, шизофрению, болезнь Альцгеймера, болезнь Паркинсона, острый и хронический рассеянный склероз или острую и хроническую боль, и повреждение мозга, вызванное ударом, гипоксией или черепно-мозговой травмой. В другом объекте настоящее изобретение относится к фармацевтической композиции, содержащей по меньшей мере одно из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III. Другим объектом настоящего изобретения является фармацевтическая композиция, отличающаяся тем, что она содержит по меньшей мере одно из указанных выше соединений, описывающихся по меньшей мере одной из формул I, II или III, в комбинации с одним или большим количеством активных соединений, выбранных из группы, включающей бета-миметики, кортикостероиды, антихолинергетики,другие ингибиторы PDE4, НСПВС (нестероидные противовоспалительные лекарственные средства), ингибиторы СОХ 2, антагонисты рецептора ЕР 4, ингибиторы EGFR, антагонисты LTD4, ингибиторы CCR3,ингибиторы iNOS, ингибиторы MRP4 и ингибиторы SYK. Другим объектом настоящего изобретения является способ получения соединения А в котором НХ обозначает фармацевтически приемлемую кислоту,включающий стадии а) и b), в котором на стадии а) соединение В в котором HY обозначает фармацевтически приемлемую кислоту,восстанавливают бораном, и в котором на стадии b) добавляют фармацевтически приемлемую кислоту НХ и получают соединение А. В одном варианте осуществления указанного выше способа получения соединения А боран, использующийся для восстановления на стадии а), добавляют непосредственно. В другом варианте осуществления указанного выше способа получения соединения А боран, использующийся для восстановления на стадии а), образуется in situ. В предпочтительном варианте осуществления указанного выше способа получения соединения А боран, использующийся для восстановления на стадии а), образуется in situ из комбинации NaBH4 и I2 или из комбинации NaBH4 и BF3-OEt2. В другом предпочтительном варианте осуществления указанного выше способа получения соединения А кислота НХ выбрана из группы, включающей п-толуолсульфоновую кислоту или хлористоводородную кислоту. В другом варианте осуществления указанного выше способа получения соединения А фармацевтически приемлемой кислотой HY, содержащейся в соединении В, является HCl. Другим объектом настоящего изобретения является способ получения соединения C в котором НХ обозначает п-толуолсульфоновую кислоту, хлористо-водородную кислоту или серную кислоту,включающий стадии i)-iii),где на стадии i) 4-цианопиперидин сначала вводят во взаимодействие с кислотой, затем вводят в реакцию с аммиаком и получают промежуточный продукт Е и где на стадии ii) промежуточный продукт Е вводят в реакцию с соединением D в присутствии основания и где на стадии iii) добавляют кислоту Н. В предпочтительном варианте осуществления казанного выше способа получения соединения C на стадии i) 4-цианопиперидин вводят во взаимодействие с хлористо-водородной кислотой и затем вводят в реакцию с аммиаком и получают промежуточный продукт Е. В предпочтительном варианте осуществления указанного выше способа получения соединения C на стадии ii) промежуточный продукт Е вводят в реакцию с соединением D в присутствии метанолята натрия. Другим объектом настоящего изобретения являются промежуточные продукты формулы VIII и их соли. Другим объектом настоящего изобретения являются промежуточные продукты формулы IX и их соли, в которой S обозначает атом серы, который является хиральным центром. Соединения общих формул I-III содержат основные группы. Поэтому соединения общих формул IIII могут образовывать соли с фармацевтически приемлемыми неорганическими кислотами, такими как хлористо-водородная кислота, серная кислота, фосфорная кислота, сульфоновая кислота, или с органическими кислотами, такими (такими как, например, малеиновая кислота, фумаровая кислота, лимонная кислота, винная кислота и уксусная кислота). Как указано выше, для использования в качестве лекарственных средств соединения формул I-III можно превратить в их фармакологически приемлемые соли. Например, эти соединения могут образовывать физиологически и фармакологически приемлемые соли присоединения с неорганическими или с органическими кислотами. Для получения таких солей присоединения с кислотами соединений формулI-III можно использовать, например, хлористо-водородную кислоту, бромисто-водородную кислоту, серную кислоту, фосфорную кислоту, метилсульфоновую кислоту, уксусную кислоту, фумаровую кислоту,янтарную кислоту, молочную кислоту, лимонную кислоту, винную кислоту и малеиновую кислоту. Также можно использовать смеси указанных выше кислот. Соединения формул I-III также могут существовать в виде их отдельных оптических изомеров или энантиомеров, смесей отдельных энантиомеров или в виде их рацематов, и в виде свободных оснований или в виде их солей присоединения с фармакологически приемлемыми кислотами (например, солей присоединения с галогенводородными кислотами, такими как хлористо-водородная кислота или бромистоводородная кислота, или с органическими кислотами, такими как щавелевая кислота, фумаровая кислота,дигликолевая кислота или метилсульфоновая кислота. Соединения, предлагаемые в настоящем изобретении, также могут находиться в виде рацематов, но они также можно находиться в виде одного энантиомера, т.е. в их (R)- или (S)-форме. Как указано выше, фармакологически приемлемые соли соединений формул I-III также являются предпочтительным объектом настоящего изобретения. Эти фармацевтически приемлемые соли соединений формул I-III также могут существовать в виде их гидратов (например, моно- или дигидратов) и/или в виде их сольватов. Сольват соединения формул I, II или III определен в настоящем изобретении как кристаллическая соль соответствующего соединения формул I, II или III, которые содержат в кристаллической решетке молекулы растворителя (например, этанола, метанола и т. п.). Гидрат соединения формул I, II или III определен в настоящем изобретении как кристаллическая соль соответствующего соединения формул I, II или III, которые содержат в кристаллической решетке кристаллизационную воду. Методика синтеза. Получение соединений примеров 1 и 2. Схема 1 В реактор с рубашкой с инертной атмосферой, снабженный капельной воронкой, механической мешалкой, трубкой для подачи N2 и термометром с термопарой, помещали метилтиогликолят (292 г, 2,61 моль) и пиперидин (4,43 г, 0,052 моль). Затем в течение 30 мин медленно добавляли метилакрилат (250 г,2,87 моль), поддерживая температуру, равной примерно 45 С. После завершения добавления смесь перемешивали при 45 С в течение 30 мин. Добавляли пиперидин (17,9 г, 210 ммоль) и перемешивание продолжали при 45 С в течение 30 мин (для поглощения избытка акрилата). Добавляли третбутилметиловый эфир (ТБМЭ) (251 мл), смесь охлаждали до 15 С и добавляли 1 М раствор HCl (251 мл). Смесь перемешивали в течение 5 мин и органический слой собирали и промывали водой (251 мл). Смесь концентрировали до минимального объема путем перегонки при пониженном давлении при 50 С. Добавляли дихлорметан (251 мл) и смесь повторно концентрировали при пониженном давлении путем перегонки при 40-45 С. Неочищенный продукт III (480 г) использовали на следующей стадии без дополнительной очистки. 3.1.2. Синтез метил-3-оксотетрагидротиофен-2-карбоксилата (соединение IV) В сухой реактор с рубашкой с инертной атмосферой, снабженный датчиком температуры, механической мешалкой и капельной воронкой, помещали TiCl4 (1,0 М раствор в CH2Cl2, 1,16 л; 1,16 моль). Содержимое реактора охлаждали до -10 С и при температуре, равной -10 С или ниже, добавляли изопропанол (89,6 мл, 1,16 моль). Смесь перемешивали при -10 С в течение 30 мин и в течение 1 ч медленно добавляли диметил-3-тиаадипат (200 г, 1,01 моль), поддерживая внутреннюю температуру равной -10 С или ниже. Реакционную смесь перемешивали при -10 С в течение еще 30 мин и в течение 1,5 ч медленно добавляли Et3N (489 мл, 3,49 моль), поддерживая внутреннюю температуру равной -10 С или ниже. Смесь перемешивали при температуре, равной -10 С или ниже, в течение 1,5 ч. Медленно добавляли 3 н. раствор HCl (1,01 л; 3,03 моль), поддерживая внутреннюю температуру ниже 10 С. Температуру повышали до 30 С и смесь перемешивали в течение 1 ч. Смеси давали отстояться, органический слой собирали и водный слой дважды экстрагировали дихлорметаном (по 1,5 л для каждой экстракции). Объединенные органические порции дважды промывали водой (по 1,5 л для каждой промывки) и сушили надMgSO4 (40 г). Полученный раствор концентрировали до минимального объема при пониженном давлении при 25-35 С и получали неочищенное соединение IV (148,6 г). Спектральные данные, полученные для соединения IV, соответствовали приведенным в литературе (Liu, H.-J.; Teng, K.N. Can. J. Chem. 1982,60, 437). 3.1.3. Синтез метилового эфира 3-уреидо-4,5-дигидротиофен-2-карбоновой кислоты (соединение V) В реактор с рубашкой с инертной атмосферой, снабженный мешалкой, трубкой для подачи N2 и термометром с термопарой, помещали мочевину (2,16 кг, 35,9 моль). Добавляли метиловый эфир 3 оксотетрагидротиофен-2-карбоновой кислоты (соединение IV, 3,0 кг), затем метанол (4,5 л). При 20-25 С добавляли концентрированную HCl (297 мл, 3,59 моль) и смесь перемешивали при кипячении с обратным холодильником в течение 4-6 ч. Реакционную смесь охлаждали до 0 С и полученное твердое вещество собирали фильтрованием. Осадок на фильтре дважды промывали водой (по 2 л воды для каждой промывки) и сушили в вакуумном сушильном шкафу при 50 С и получали 4,17 кг (83% мас./мас.) соединения V (выход 95%), 1 Н ЯМР (500 МГц, (CD3)2SO)3,10 (dd, 2H, J=8,5, 8,5 Гц), 3,50 (dd, 2H, J=8,5, 8,5 Гц), 3,73 (s, 3 Н), 6,50-7,20 (bs, 2H), 9,47 (s, 1 Н); 13 С ЯМР (125 МГц, (CD3)2SO)28,7, 37,8, 52,4, 100,0,151,6, 154,7, 165,7; ЖХМС (жидкостная хроматография-масс-спектрометрия) (ЭУ (ионизация электронным ударом для C7H11N2O3S, (М+Н)+ рассчитано: 203,0, найдено: 203,0. 3.1.4. Синтез 6,7-дигидротиено[3,2-d]пиримидин-2,4-диола (соединение VI) При комнатной температуре к раствору NaOH (379 г, 9,47 моль) в воде (6,0 л) добавляли соединение V (2,0 кг, 9,47 моль). Полученную выше смесь перемешивали при 85 С в течение 3 ч. После охлаждения до 0 С медленно добавляли концентрированную HCl (861 мл, 10,4 моль) до установления значения рН раствора, равного 0-1. Смесь охлаждали до 0 С, перемешивали в течение 5-10 мин и полученное твердое вещество собирали фильтрованием. Осадок на фильтре дважды тщательно промывали водой (по 1 л для каждой промывки), сушили на воздухе в течение 2-3 ч (с отсасыванием) и затем дополнительно сушили в вакуумном сушильном шкафу при 50 С в течение 12-16 ч и получали 1,67 кг соединения VI. 1H ЯМР (500 МГц, (CD3)2SO)3,11 (dd, 2H, J=8,5, 8,5 Гц), 3,31 (dd, 2 Н, J=8,5, 8,5 Гц), 11,14 (s, 1H), 11,38 (s,1 Н); 13 С ЯМР (125 МГц, (CD3)2SO)29,3, 35,4, 108,5, 150,5, 152,4, 160,4; ЖХМС (ЭУ) для C6H7N2O2S,(M+H)+ рассчитано: 171,0, найдено: 171,0. 3.1.5. Синтез 2,4-дихлортиено[3,2-d]пиримидина (соединение VII) В сухой реактор (реактор 1) с рубашкой с инертной атмосферой, снабженный датчиком температуры, механической мешалкой и капельной воронкой, помещали 800 г твердого соединения VI (4,66 моль). В течение 30 мин-1 ч добавляли 1,5 л (9,31 моль) диэтиланилина, поддерживая температуру равной 25 С или ниже. Внутреннюю температуру повышали до 105-110 С и в реактор (реактор 1) в течение 5-10 мин добавляли 0,68 экв. (868 мл, 34% от полного количества) оксихлорида фосфора. Когда внутренняя температура начинала понижаться, внутреннюю температуру поддерживали равной 110 С и в течение 30-40 мин добавляли оставшийся POCl3 (1,32 экв. или 66% от полного количества). Внутреннюю температуру доводили до равной 105-110 С и смесь перемешивали в течение 18-24 ч или до завершения реакции (по данным анализа с помощью ВЭЖХ (высокоэффективная жидкостная хроматография). Смесь охлаждали до 45 С и при 45 С добавляли ТГФ (тетрагидрофуран) (400 мл). Полученную выше неочищенную смесь помещали во второй сосуд или реактор (сосуд или реактор 2). В реактор 1 помещали 4,8 л воды и охлаждали до 5 С. Затем в реактор 1, содержащий воду, медленно добавляли неочищенную реакционную смесь (содержащуюся в сосуде или реакторе 2), поддерживая температуру равной 5-10 С. Смесь перемешивали при 5 С в течение 30 мин-1 ч и полученное твердое вещество собирали фильтрованием. Осадок на фильтре дважды промывали водой (по 1,6 л для каждой промывки) и осадок на фильтре сушили на воронке на воздухе в течение 6-8 ч и получали 964 г (92% (мас./мас); выход 88%) неочищенного соединения VII. В реактор объемом 10 л помещали дихлорметан (4,6 л). В реактор помещали неочищенное соединение VII и активированный уголь (46,2 г), смесь нагревали при 40 С и перемешивали в течение 20 мин. Полученный раствор фильтровали через фильтрующий материал для удаления древесного угля. Осадок на фильтре дважды промывали дихлорметаном (по 175 мл для каждой промывки). Раствор концентрировали при пониженном давлении до минимального объема, допускающего перемешивание, и оставшийся дихлорметан удаляли путем перегонки с минимальным количеством петролейного эфира. В реактор дополнительно добавляли петролейный эфир (1,3 л), смесь охлаждали до 10 С и перемешивали в течение 1 ч. Полученное твердое вещество собирали фильтрованием и осадок на фильтре дважды промывали петролейным эфиром (по 150 мл для каждой промывки). Осадок на фильтре сушили на воронке(с отсасыванием) на воздухе до высыхания. Полученное твердое соединение VII переносили в подходящий тарированный сосуд и сушили в сушильном шкафу при 50 С в течение 6 ч и получали конечный продукт: 1 Н ЯМР (400 МГц, ДМСО-d6 (ДМСО = диметилсульфоксид 3,45-3,56 (m, 4 Н); 13 С ЯМР (400 МГц, ДМСО-d6)29,3, 36,5, 134,8, 151,0, 154,1, 175,9. 3.2. Получение соединения примера 1. Схема 3 В реактор объемом 2 л в атмосфере азота помещали NaBH4 (28,6 г, 757 ммоль, 2,87 экв.) и ТГФ (500 мл) и смесь охлаждали до -5 С. Готовили раствор I2 (63,6 г, 251 ммоль, 0,95 экв.) в 125 мл ТГФ и его в течение 45 мин медленно добавляли в реактор, поддерживая внутреннюю температуру равной от -5 до 5 С. Затем капельную воронку промывали с помощью 42 мл ТГФ. Затем при -6 С добавляли соединение В (50 г, 264 ммоль, 1 экв.), затем температуру повышали примерно до 5 С. Затем реакционную смесь нагревали при 65 С в течение 23 ч (примечание: за протеканием реакции следили с помощью ГХ (газовая хроматография)/ПИД (пламенный ионизационный детектор), для этого реакцию останавливали с помощью 0,1 мл МеОН и затем получали производное с использованием 0,5 мл смеси ТГФ/уксусный ангидрид/ТЭА (триэтиламин) состава 5/2/2). Затем к реакционной смеси в течение 20 мин медленно добавляли 83 мл МеОН, поддерживая температуру равной 20-27 С. Реакционную смесь концентрировали до минимального объема, допускающего перемешивание, и добавляли 500 мл 2-метилтетрагидрофурана(МеТГФ). Затем добавляли 485 г 25 мас.% водного раствора NaOH (11,5 экв.), твердые вещества растворялись. Слои разделяли и водную фазу дважды экстрагировали с помощью 500 мл 2 метилтетрагидрофурана (МеТГФ). Затем органические вещества фильтровали через слой целита иMgSO4 и промывали с помощью 50 мл 2-метилтетрагидрофурана (МеТГФ). Готовили раствор моногидрата п-толуолсульфоновой кислоты (51 г, 264 ммоль, 1 экв.) в МеТГФ (100 мл) и его добавляли к органическим веществам (альтернативно, можно использовать HCl и получить соль соединения А с HCl). Получали гомогенный светло-желтый раствор. Раствор концентрировали до объема, равного 275-300 мл, и определяли содержание воды. Добавляли дополнительное количество МеТГФ и смесь концентрировали до объема, равного исходному, до тех пор, пока содержание воды не составляло 0,1%. Полученное твердое вещество отфильтровывали и промывали с помощью 50 мл МеТГФ, сушили на воронке в течение ночи и затем дополнительно сушили в вакуумном сушильном шкафу при 50 сС. Собирали 61,71 г соединения А: 1H ЯМР (ДМСО-d6, 400 МГц)1,70-1,92 (m, 2H), 1,94-2,03 (m, 2H), 2,04-2,18 (m, 2H), 2,29 В многогорлый сосуд, снабженный холодильником, термометром с термопарой и трубкой для подачи азота, последовательно помещали промежуточный продукт VII (180 г, 852 ммоль) и соединение А(129 г, 937 молей). Затем при 22 С добавляли ацетонитрил (900 мл) и триэтиламин (594 мл, 4,26 моль) и смесь перемешивали при 75-77 С в течение 12 ч. В течение 20 мин медленно добавляли воду (1,2 л), в смесь при 40 С вносили затравочные кристаллы соединения VIII (0,3 г) и затем смесь в течение 2 ч охлаждали до 25 С. Смесь перемешивали при комнатной температуре в течение еще 12 ч и полученное твердое вещество собирали фильтрованием. Осадок на фильтре промывали смесью вода/MeCN состава 2:1 (400 мл), затем водой (200 мл). Полученное твердое вещество сушили в вакууме при 50 С в течение 12 ч и получали 132 г (выход 57%) соединения VIII. 1 Н ЯМР (400 МГц, CDCl3)1,85-2,05 (m, 2H), 2,102,21 (m, 2H), 2,32-2,41 (m, 2H), 3,27 (dd, J=8,0, 8,4 Гц, 2H), 3,43 (dd, J=8,0, 8,4 Гц, 2H), 3,91 (s, 2 Н), 4,67 (s,1 Н);13 С ЯМР (CDCl3, 100 МГц)14,8, 30,7, 31,2, 36,7, 59,7, 67,6, 114,7, 156,1, 156,2, 168,0. 3.2.3. Синтез соединения IX. В многогорлую колбу объемом 2 л в атмосфере азота при 20 С помещали соединение VIII (122 г,429 ммоль), S-(-)-1,1'-би-2-нафтол (S-(-)-BINOL) (12,4 г, 42,9 ммоль), дихлорметан (608 мл), Ti(OiPr)4(6,54 мл, 21,4 ммоль) и воду (7,72 мл, 429 ммоль) и перемешивали в течение 1 ч. При 21 С одной порцией добавляли трет-бутилгидропероксид (70% раствор в воде, 62,3 мл, 472 ммоль); смесь становилась полностью гомогенной и температура повышалась до равной примерно 40 С. Смеси давали охладиться до комнатной температуры, ее перемешивали в течение 1,5 ч и фильтровали. Осадок на фильтре дважды промывали изопропилацетатом (по 243 мл для каждой промывки) и осадок на фильтре сушили на фильтре на воздухе в течение 6 ч и получали 114,4 г соединения IX. 1 Н ЯМР (400 МГц, ДМСО-d6)1,70-1,85(m, 2H), 2,14-2,34 (m, 4 Н), 2,98-3,08 (m, 1 Н), 3,09-3,19 (m, 1 Н), 3,30-3,40 (маскированный m, 1 Н), 3,503,62 (m, 1 Н), 3,65-3,77 (m, 2 Н), 4,91 (t, J=6 Гц, 1 Н), 8,63 (s, 1H); 13 С ЯМР (100 МГц, ДМСО-d6)14,5,29,6, 29,8, 32,6, 48,6, 59,2, 62,8, 119,0, 157,8, 161,4, 175,3. Другой энантиомер соединения IX можно получить при замене S-(-)-1,1'-би-2-нафтола на R-(+)-1,1'би-2-нафтол. Рацемат соединения IX можно получить по методикам, известным специалистам в данной области техники, в которых не используют хиральные реагенты и условия. Пример такой методики получения рацемических сульфоксидов описан в WO 06/111549. 3.2.4. Синтез соединения примера 1.(12,4 мл; 72,1 ммоль) перемешивали в 47 мл диоксана. Полученную смесь помещали в 3 сосуда объемом по 20 мл, которые нагревали в микроволновой печи при 120 С в течение 25 мин. После охлаждения до комнатной температуры реакционные смеси выливали в воду со льдом. Полученный осадок отфильтровывали, переносили в 500 мл этилацетата и кипятили с обратным холодильником. После кипячения с обратным холодильником смесь охлаждали в бане со льдом и полученный осадок отфильтровывали и сушили в сухой камере при пониженном давлении при 50 С и получали 7,57 г соединения примера 1. 1H ЯМР (400 МГц, ДМСО-d6)1,43-1,57 (m, 2 Н), 1,67-1,85 (m, 4 Н), 2,11-2,21 (m, 2 Н), 2,26-2,43 (m, 2 Н),2,80-3,01 (m, 5 Н), 3,17-3,47 (m, интегрированию мешало наложение пика воды), 3,67-3,76 (m, 2H), 4,744,86 (m, 3H), 7,25-7,36 (m, 5 Н). 13 С ЯМР (100 МГц, ДМСО-d6)14,3, 29,4, 29,6, 32,3, 32,5, 41,4, 44,2, 48,5,58,4, 63,6, 109,2, 128,2, 128,6, 130,5, 144,7, 157,6, 161,5, 174,7. 3.3. Получение соединения примера 2. Схема 4 В 3-горлый реактор с рубашкой объемом 500 мл, снабженный механической мешалкой, датчиком температуры и трубкой для подачи аргона, помещали 4 М раствор HCl в диоксане (225 мл, 3 экв., 900 ммоль). Раствор охлаждали до 0 С и добавляли 4-цианопиперидин (33,04 г, 300 ммоль), затем в течение 30 мин добавляли метанол (36,4 мл, 900 ммоль, 3 экв.), поддерживая температуру ниже 10 С (темпера- 14024033 тура повышалась). Полученную выше смесь перемешивали при комнатной температуре в течение 6-8 ч до завершения превращения по данным анализа раствора аликвоты в D2O с помощью 1 Н ЯМР (через 30 мин прозрачный раствор превращался в белую суспензию). Смесь охлаждали до 5 С и добавляли 25 мас.% раствор NaOMe в метаноле (129,6 г, 600 ммоль, 2 экв.), поддерживая температуру ниже 15 С. Затем смесь перемешивали в течение 1 ч. К полученной выше смеси добавляли 7,0 н. раствор аммиака в метаноле (64,2 мл, 1,5 экв., 450 ммоль) и ее перемешивали при комнатной температуре в течение 2 ч. Смесь концентрировали при пониженном давлении при 60 С до объема, равного 250 мл, и получали раствор неочищенного соединения Е, который использовали без выделения соединения. 1 Н ЯМР (400 МГц, D2O)1,80-1,95 (m, 2H), 2,15 (br d, J=4,4 Гц, 2H), 2,79-2,90 (m, 1H), 3,02 (ddd, J=13,2, 13,2, 3,0 Гц,2 Н), 3,48 (m, 2H). 3.3.1.2. Синтез соединения G. Полученный выше раствор промежуточного соединения Е охлаждали до 20 С и добавляли 25 мас.% раствор NaOMe в метаноле (162 г, 2,5 экв., 750 ммоль). Затем смесь перемешивали в течение 30 мин. К полученной выше смеси в течение -30 мин при комнатной температуре двумя порциями добавляли соединение D (=(2)-N-(2-хлор-3-(диметиламино)аллилиден)-N-метилметанаминийгексафторфосфат(V, (82,3 г, чистота 95 мас.%, 0,85 экв., 255 ммоль) и перемешивали при комнатной температуре в течение 3 ч. Смесь концентрировали при пониженном давлении при 60 С до объема, равного 200 мл. Добавляли 2-метилтетрагидрофуран (400 мл) и затем смесь концентрировали при пониженном давлении при 60 С до объема, равного 150 мл. Добавляли 2-метилтетрагидрофуран (250 мл), смесь охлаждали до 20 С, добавляли воду (150 мл) и смесь перемешивали в течение 5 мин. Слои разделяли и органический слой собирали. Органический слой промывали 30% водным раствором NaOH (120 мл) и слои разделяли. Органические вещества концентрировали до минимального объема, допускающего перемешивание (-150 мл), и добавляли н-пропанол (350 мл). К полученному выше прозрачному раствору при 65 С в течение 10 мин добавляли раствор моногидрата п-толуолсульфоновой кислоты в н-пропаноле (0,85 экв., 255 ммоль, 48,4 г в 100 мл н-пропанола). Полученную выше смесь концентрировали при пониженном давлении при 65 С и получали 350 мл при содержании воды, равном 1,0% (для предотвращения потери продукта с маточным раствором рекомендуется обеспечить содержание воды, равное менее 1,0%). Смесь при перемешивании в течение 3 ч охлаждали до 20 С. Твердые вещества отфильтровывали, промывали фильтратом и затем н-пропанолом (120 мл) и после сушки в вакуумном сушильном шкафу при 65 С в течение 12 ч получали 111 г (68% мас./мас. по данным анализа, 75,48 г) соединения G. 1 Н ЯМР (ДМСОd6, 400 МГц)1,83-1,99 (m, 2 Н), 2,13 (d, J=12 Гц, 2H), 2,97 (s, 3 Н), 3,0-3,11 (m, 2H), 3,13-3,23 (m, 1 Н),3,30-3,42 (m, 2H), 7,14 (d, J=8,0 Гц, 2H), 7,52 (d, J=8,0 Гц, 2 Н), 8,47 (br, 2 Н), 8,91 (s, 2 Н); 13 С ЯМР (ДМСОd6, 100 МГц)20,7, 27,0, 40,8, 42,8, 125,5, 128,1, 128,8, 137,9, 145,2, 155,8, 169,0. 3.3.2. Синтез соединения примера 2. В круглодонную колбу объемом 3 л в атмосфере азота помещали соединение IX (86,5 г, 291 ммоль,1 экв.), соединение G (160 г, 305 ммоль, 1,05 экв.), тетрагидрофуран (ТГФ) (484 мл), воду (121 мл) и ДИПЭА (N,N-диизопропилэтиламин, 127 мл, 727 ммоль, 2,5 экв.) и нагревали при 65 С в течение 3 ч. Затем при температуре, равной 65 С, добавляли воду (1125 мл, 13 мл/г соединения IX) и перемешивали в течение 2 ч, охлаждая до 20 С. Смесь фильтровали и осадок на фильтре дважды промывали с помощью 173 мл ацетона. Затем осадок на фильтре сушили на воронке в течение ночи и получали 116,7 г соединения примера 2. 1 Н ЯМР (400 МГц, CDCl3)1,75-1,95 (m, 4H), 2,02-2,11 (m, 2H), 2,12-2,26 (m, 2 Н), 2,38 (q,J=9,6 Гц, 2 Н), 2,93-3,12 (m, 4H), 3,12-3,22 (m, 1H), 3,28-3,39 (m, 1 Н), 3,53-3,65 (m, 1H), 3,80 (d, J=5,6 Гц,- 15024033 2H), 4,42 (t, J=5,2 Гц, 1H), 4,82 (br d, J=11,2 Гц, 2H), 6,47 (s, 1H), 8,62 (s, 2H); 13C ЯМР (100 МГц, CDCl3)14,8, 30,0, 30,1, 30,6, 32,7, 44,3, 49,4, 59,1, 68,2, 107,5, 129,1, 155,5, 159,0, 162,3, 170,5, 174,6. 3.3.2.1. Кристаллизация с получением безводной формы А соединения примера 2. Получение затравочных кристаллов (безводная форма А). Небольшие количества неочищенного соединения примера 2 (1-2 мг) суспендировали примерно в 0,1 мл следующих растворителей: этанол, ацетон, 2-бутанон, этилацетат, изопропилацетат, тетрагидрофуран, 1-пропанол, 2-бутанол и ацетонитрил. После проведения цикла нагревание/охлаждение из образцов получали суспензии кристаллической безводной формы А, на что указывали данные, полученные с помощью порошковой рентгенографии. а. Кристаллизация из уксусной кислоты, диметилсульфоксида или N-метил-2-пирролидона. Примерно 1 г неочищенного соединения примера 2 при температуре, равной 60 С, растворяли в 10 мл полярного органического растворителя, такого как уксусная кислота, диметилсульфоксид или Nметил-2-пирролидон. Раствор охлаждали до 30-40 С и добавляли антирастворитель (примерно 5-10 мл),такой как изопропиловый спирт, этиловый спирт или ацетон. В раствор добавляли затравочные кристаллы безводной формы А соединения примера 2 и охлаждали до 20 С. Для увеличения выхода добавляли дополнительное количество антирастворителя (5-10 мл). После охлаждения в течение не более 1 ч полученную суспензию фильтровали и влажный осадок на фильтре сушили в вакууме при 60 С. Безводную форму А получали в виде белого твердого вещества, что подтверждено с помощью порошковой рентгенографии (ПРРГ) имеющегося файла для стандарта безводной формы А.b. Кристаллизация из смеси тетрагидрофуран/вода. Примерно 1 г неочищенного соединения примера 2 при температуре, равной 60 С, растворяли в 10 мл смеси тетрагидрофуран/вода (8:2, об./об.). Раствор охлаждали до 40-50 С, добавляли затравочные кристаллы безводной формы А соединения примера 2 и затем в течение менее 1 ч охлаждали при 20 С. К суспензии добавляли примерно 5-10 мл антирастворителя (органический растворитель, такой как изопропиловый спирт, этиловый спирт или ацетон). Не позже чем через 1 ч после добавления антирастворителя полученную суспензию фильтровали и влажный осадок на фильтре сушили в вакууме при 60 С. Безводную форму А получали в виде белого твердого вещества, что подтверждено с помощью порошковой рентгенографии (ПРРГ) имеющегося файла для стандарта безводной формы А.c. Сушка дигидрата. Примерно 1 г дигидратной формы соединения примера 2 промывали на воронке Бюхнера с помощью примерно 5 мл безводного растворителя, такого как этанол, метанол, изопропанол или ацетон. Затем влажный осадок на фильтре сушили в вакууме при 60 С. Безводную форму А получали в виде белого твердого вещества, что подтверждено с помощью порошковой рентгенографии (ПРРГ) имеющегося файла для стандарта безводной формы А. 3.3.2.2. Кристаллизация с получением безводной формы В соединения примера 2. Получение затравочных кристаллов безводной формы В. Небольшие количества неочищенного соединения примера 2 (1-2 мг) суспендировали примерно в 0,1 мл смесей 2-пропанола и воды (одна смесь содержала 3,3% воды, другая - 6,6% воды). После проведения цикла нагревание/охлаждение из образцов получали суспензии кристаллической безводной формы В, на что указывали данные, полученные с помощью порошковой рентгенографии. Образцы в безводном 2-пропаноле обрабатывали при таких же условиях и получали смесь формы А и формы В, на что указывали данные, полученные с помощью порошковой рентгенографии. Смесь формы А и формы В суспендировали при 20 С в течение 4 дней в смесях воды и следующих растворителей: метанол, этанол, 2 пропанол, 1-пропанол и ацетон (все содержали примерно 9% воды) и получали форму В, на что указывали данные, полученные с помощью порошковой рентгенографии. а. Кристаллизация из смеси н-пропанол/вода. 10 г неочищенного соединения примера 2 при температуре, равной 65 С, растворяли в 160 мл смеси н-пропанол/вода (9:1, об./об.). Раствор охлаждали до 60 С, добавляли затравочные кристаллы безводной формы В соединения примера 2 и выдерживали в течение 0,5 ч. Суспензию в течение не менее 5 ч охлаждали при 30 С. Для уменьшения объема до равного примерно 80-100 мл с целью увеличения выхода суспензию необязательно перегоняли при пониженном давлении при 30 С. Затем суспензию охлаждали до 0 С и суспензию выдерживали в течение не менее 8 ч или до тех пор, пока не переставала обнаруживаться безводная форма А. Суспензию фильтровали и влажный осадок на фильтре сушили в вакууме при 60 С. Безводную форму В соединения примера 2 получали в виде белого твердого вещества с выходом 90%. Данные, полученные с помощью порошковой рентгенографии (ПРРГ) по данным имеющегося файла для стандарта безводной формы В, соответствовали этой форме.b. Кристаллизация из смеси тетрагидрофуран/вода. Примерно 1 г неочищенного соединения примера 2 при температуре, равной 60 С, растворяли в 10 мл смеси тетрагидрофуран/вода (8:2, об./об.). Раствор охлаждали до 40-50 С, добавляли затравочные кристаллы безводной формы В соединения примера 2 и затем в течение 2 ч охлаждали при 20 С. К суспензии добавляли примерно 10 мл антирастворителя (органический растворитель, такой как изопропиловый спирт, этиловый спирт или ацетон). Суспензию выдерживали в течение не менее 8 ч или до тех пор,- 16024033 пока не переставала обнаруживаться безводная форма А. Затем суспензию фильтровали и влажный осадок на фильтре сушили в вакууме при 60 С. Безводную форму В соединения примера 2 получали в виде белого твердого вещества. Данные, полученные с помощью порошковой рентгенографии (ПРРГ) по данным имеющегося файла для стандарта безводной формы В, соответствовали этой форме. с. Получение из дигидрата. Примерно 1 г дигидрата соединения примера 2 суспендировали в 5-10 мл безводного растворителя,такого как этанол, метанол, изопропанол, ацетон, этилацетат, изопропилацетат, тетрагидрофуран или ацетонитрил. К суспензии добавляли затравочные кристаллы безводной формы В соединения примера 2 и перемешивали при 20-40 С в течение не менее 4 ч или до тех пор, пока данные, полученные с помощью порошковой рентгенографии (ПРРГ), не указывали на завершение превращения в безводную форму В.d. Получение из безводной формы А. Примерно 1 г безводной формы А соединения примера 2 суспендировали в 5-10 мл безводного растворителя, такого как этанол, метанол, изопропанол, ацетон, этилацетат, изопропилацетат, тетрагидрофуран или ацетонитрил. К суспензии добавляли затравочные кристаллы безводной формы В соединения примера 2 и перемешивали при 20-40 С в течение не менее 4 ч или до тех пор, пока данные, полученные с помощью порошковой рентгенографии (ПРРГ), не указывали на завершение превращения в безводную форму В. 3.3.2.3. Кристаллизация с получением дигидратной формы соединения примера 2. Получение затравочных кристаллов дигидратрой формы Смесь кристаллов безводной формы А и безводной формы В соединения примера 2 суспендировали в смеси 2-бутанон/вода (содержание воды 9%) при 20 С в течение 4 дней и получали кристаллы дигидрата, на что указывали данные, полученные с помощью порошковой рентгенографии.a. Кристаллизация из смеси н-пропанол/вода. 10 г Неочищенного соединения примера 2 при температуре, равной 65 С, растворяли в 120 мл смеси н-пропанол/вода (8:2, об./об.). Раствор охлаждали до 50 С, добавляли затравочные кристаллы дигидрата соединения примера 2 и выдерживали в течение 0,5 ч. К суспензии добавляли воду (примерно 60-100 мл). Суспензию охлаждали в течение не менее 5 ч при 20 С и затем выдерживали в течение не менее 8 ч. Суспензию фильтровали и влажный осадок на фильтре промывали водой и затем сушили на воздухе.b. Кристаллизация из смеси в ТГФ/вода. Примерно 1 г неочищенного соединения примера 2 при температуре, равной 60 С, растворяли в 10 мл смеси тетрагидрофуран/вода (8:2, об./об.). Раствор охлаждали до 30-50 С, добавляли затравочные кристаллы дигидрата соединения примера 2 и затем в течение 2 ч охлаждали при 20 С. К суспензии добавляли примерною мл воды. Полученную суспензию выдерживали в течение не менее 8 ч. Суспензию фильтровали и влажный осадок на фильтре промывали водой и затем сушили на воздухе. Порошковая рентгенограмма (ПРРГ) продукта соответствовала рентгенограмме дигидрата. с. Получение из безводной формы А или из безводной формы В. Примерно 1 г безводной формы А или безводной формы В соединения примера 2 суспендировали в примерно 5-10 мл смеси, содержащей не менее 30% воды и органический растворитель, такой как этанол, метанол, изопропанол, ацетон или тетрагидрофуран. К суспензии добавляли затравочные кристаллы дигидрата соединения примера 2 и перемешивали при 20 С в течение не менее 4 ч или до тех пор, пока данные, полученные с помощью порошковой рентгенографии (ПРРГ), не указывали на завершение превращения в дигидратную форму. Суспензию фильтровали и влажный осадок на фильтре промывали водой и затем сушили на воздухе. Полиморфные формы соединения примера 2 характеризовали с помощью порошковой рентгенографии (ПРРГ), результаты приведены на фиг. 3 а-3 в, на которых представлены порошковые рентгенограммы, и в таблицах, в которых представлены все наблюдаемые пики отражений. Для проведения анализа с помощью порошковой рентгенографии использовали прибор Rigaku Miniflex II, снабженный источником рентгеновского излучения типа Power 450 W (30 кВ-15 мА) (оптическая система: щель с переменным отклонением). Диапазон гониометра составлял 3,0-35,0 2 и скорость сканирования составляла 0,02 2/мин при точности, равной более 0,01. В качестве монохроматора использовали фильтр из фольги/графита и в качестве детектора использовали сцинтилляционный счетчик Nal диаметром 23,0 мм. Образец анализировали с использованием держателя для образца Si (510), обладающего низким уровнем фона. Полиморфные формы соединения примера 2 также характеризовали с помощью дифференциальной сканирующей калориметрии (ДСК) с использованием прибора ТА Instruments DSC Q1000), результаты приведены на фиг. 4 а, 5 а и 6 а. Образцы анализировали в негерметизированном алюминиевом тигле в токе N2. Измерения проводили при температуре от 20 до 300 С при скорости нагрева, равной 10 С/мин. Полиморфные формы соединения примера 2 также характеризовали с помощью термогравиметрического анализа (ТГА) с использованием прибора ТА Instruments TGA Q500, результаты приведены на фиг. 4 б, 5 б и 6 б. Образцы анализировали в открытом платиновом тигле в токе N2. Измерения проводили при температуре от 20 до 300 С при скорости нагрева, равной 10 С/мин. Чертежи Фиг. 1 а - опорожнение желудка у крыс, которым вводили соединение примера 1; фиг. 1 б - кишечный транзит у крыс, которым вводили соединение примера 1; фиг. 2 а - опорожнение желудка у крыс, которым вводили соединение примера 2; фиг. 2 б - кишечный транзит у крыс, которым вводили соединение примера 2; фиг. 3 а - порошковая рентгенограмма безводной формы А соединения примера 2; фиг. 3 б - порошковая рентгенограмма безводной формы В соединения примера 2; фиг. 3 в - порошковая рентгенограмма дигидратной формы С соединения примера 2; фиг. 4 а - термограмма безводной формы А соединения примера 2, полученная с помощью дифференциальной сканирующей калориметрии (ДСК) (на термограмме ДСК наблюдается эндотерма плавления примерно при 235 С и последующее разложение при продолжении нагревания выше температуры плавления); фиг. 4 б - термограмма безводной формы А соединения примера 2, полученная с помощью термогравиметрического анализа (ТГА) (данные ТГА указывают на наличие несольватированной формы, о чем свидетельствует пренебрежимо малое содержание летучих веществ (минимальная потеря массы (0,145%) при нагревании до температуры плавления); фиг. 5 а - термограмма безводной формы В соединения примера 2, полученная с помощью дифференциальной сканирующей калориметрии (ДСК) (на термограмме ДСК наблюдается или переход твердое вещество-твердое вещество, или одновременно протекающие плавление/повторная кристаллизация,происходящие примерно при 218 С. Полученная форма, наиболее вероятно, является безводной формой А, на что указывает экзотерма плавления примерно при 235 С, что соответствует температуре плавления формы А. После плавления формы А соединение разлагается при нагревании выше 240 С); фиг. 5 б - термограмма безводной формы В соединения примера 2, полученная с помощью термогравиметрического анализа (ТГА) (данные ТГА указывают на пренебрежимо малое содержание летучих веществ в форме В (указывают на наличие несольватированной формы), о чем свидетельствует минимальная потеря массы (0,057%) при нагревании до температуры плавления); фиг. 6 а - термограмма дигидратной формы С соединения примера 2, полученная с помощью дифференциальной сканирующей калориметрии (ДСК) (данные ДСК указывают на протекающую при низкой температуре дегидратацию, о чем свидетельствует наличие широкой эндотермы при температуре 100 С. Дегидратированное твердое вещество, вероятнее всего, является формой А, о чем свидетельствует наличие характерной для формы А эндотермы плавления примерно при 236 С. Затем форма А разлагается при нагревании выше температуры плавления); фиг. 6 б - термограмма дигидратной формы С соединения примера 2, полученная с помощью термогравиметрического анализа (ТГА) (данные ТГА указывают на большую потерю массы формы С, о чем свидетельствует дегидратация при нагревании при температуре 100 С. У дегидратированного вещества(вероятно, форма А) практически не происходит потери массы до плавления (от 100 до 236 С), что находится в соответствии с приведенными выше данными для формы А). Примеры Приведенные ниже соединения примеров получали по методикам синтеза, аналогичным описанным выше в настоящем изобретении. Таблица А Химические структуры типичных соединений, предлагаемых в настоящем изобретении Приведенные ниже соединения предшествующего уровня техники A-D являются наиболее близкими по структуре соединениями, которые раскрыты в WO 2009/050248, который является самой родственной публикацией предшествующего уровня техники. Таблица В Химические структуры самых сходных по структуре соединений, которые раскрыты в WO 2009/050248 Биологические исследования. 5.1. Определение значений IC50 для PDE4B (in vitro). Значения IC50 соединений, предлагаемых в настоящем изобретении (соединений примеров 1 и 2), и указанных выше соединений предшествующего уровня техники A-D для их способности ингибироватьPDE4B определяли с помощью проксимального сцинтилляционного анализа (ПСА) (GE Healthcare, No.TRKQ7090). Проксимальный сцинтилляционный анализ (ПСА) основан на обнаружении разного сродства циклического 3'-5'-аденозинмонофосфата (цАМФ, низкое сродство) и линейного 5'-аденозинмонофосфата(АМФ, высокое сродство) к содержащим сцинтилляторы гранулам из силиката иттрия. Специфичная для цАМФ фосфодиэстераза (PDE) PDE4B расщепляет сложную 3'-фосфодиэфирную связь меченной тритием [3 Н]цАМФ с образованием [3 Н]5'-АМФ. Эта [3 Н]5'-АМФ связывается с содержащими сцинтилляторы гранулами вследствие их высокого сродства и вызывает сцинтилляции (вспышки света), которые можно регистрировать с использованием сцинтилляционного счетчика Wallac Microbeta. 10 мкл раствора [3 Н]цАМФ (0,05 мкКи в Н 2 О, 10-30 Ки/ммоль) добавляли к 89 мкл раствора фермента PDE4B (фрагмент активного центра, содержащий аминокислоты 152-484; 0,15-0,18 нг) в буфере для анализа (50 мМ Tris-HCl [Tris = трис-(гидроксиметиламинометан)], рН 7,5; 8,3 мМ MgCl2; 1,7 мМ этиленгликольтетрауксусной кислоты (ЭГТА); 0,25 мг/мл бычьего сывороточного альбумина (БСА и эту смесь инкубировали при 30 С в течение 1 ч:a) без добавления соединения, которое необходимо исследовать (в присутствии 1 мкл диметилсульфоксида (ДМСО), что соответствует 1% ДМСО), иb) в присутствии соединения, которое необходимо исследовать при концентрации 125, 25, 5, 1 мкМ,200, 40, 8, 1,6, 0,32, 0,064, 0,0128 нМ (серийные разведения готовили, разводя в 5 раз, начиная с концентрации, равной 125 мкМ, и до 0,0128 нМ, в присутствии 1% ДМСО). После проведения этой инкубации реакцию останавливали путем добавления 50 мкл раствора гранул (500 мг гранул/35 мл Н 2 О, 18 мМ сульфата цинка). Затем в течение 45 мин гранулам давали осадиться. Затем сцинтилляции регистрировали с использованием сцинтилляционного счетчика. Если исследуемое соединение способно ингибировать активность фермента PDE4B, то с увеличением концентрации исследуемого соединения образуется меньшее количество [3 Н]АМФ и можно зарегистрировать меньшее количество сцинтилляций. Эти результаты выражены в виде значений IC50. Значение IC50 означает концентрацию соединения, при которой активность фермента PDE4B ингибируется на значение, равное половине максимального. Поэтому чем ниже значение IC50, тем лучше ингибирование PDE4B. Таблица С Экспериментально определенные значения IC50 для ингибирования PDE4B соединениями, предлагаемыми в настоящем изобретении, и соединениями предшествующего уровня техники, раскрытыми в WO 2009/050248 Из числа соединений предшествующего уровня техники только соединения А и D обладают значениями IC50, находящимися в том же диапазоне активности, что и значения для соединений примеров 1 и 2. В соответствии с этим все последующие эксперименты проводили с использованием только соединений примеров 1 и 2 и соединений предшествующего уровня техники А и D. 5.2. Исследование зависимости реакции от дозы и расчет дозы, равной половине эффективной дозы,необходимой для ингибирования индуцируемого посредством ЛПС (липополисахарид) поступления нейтрофилов в жидкий бронхоальвеолярный лаваж у самцов крыс Wistar. Противовоспалительную активность соединений примеров 1 и 2 и соединений предшествующего уровня техники А и D оценивали с помощью модели индуцируемого посредством ЛПС воспаления легких у крыс in vivo. Для исследования фармакологической активности указанных выше соединений путем изучения зависимости реакции от дозы определяли эффективную дозу (ED50), обеспечивающую равное половине максимального ингибирование индуцируемого липополисахаридом (индуцируемого посредством ЛПС) поступления нейтрофилов в жидкий бронхоальвеолярный лаваж (ЖБАЛ). Бактериальные эндотоксины(липополисахариды [ЛПС]) являются компонентами внешней мембраны бактериальных клеток, которые играют важную роль в патогенезе инфекций, вызванных грамотрицательными бактериями. Известно, что ингаляция таких аэрозолей ЛПС индуцирует зависимое от дозы увеличение поступления нейтрофилов в легочные ткани и воздушные пространства у крыс, которое можно регистрировать путем анализа количества нейтрофилов в жидком бронхоальвеолярном лаваже (ЖБАЛ). Однако такое зависимое от дозы количество нейтрофилов в ЖБАЛ, вероятно, можно уменьшить зависимым от дозы образом в присутствии эффективного ингибитора PDE4. В этих экспериментах использовали самцов крыс Wistar (HanWistar), полученных от уполномоченного местного поставщика. Масса заказанных животных находилась в диапазоне 200-250 г. Перед проведением эксперимента животные голодали в течение ночи. В каждом эксперименте использовали всего 32 животных. Для исследования каждой дозы использовали по 8 животных (n=8) в группах, подвергавшихся лечению, 2 животных использовали в качестве контрольной группы, подвергавшейся воздействию ЛПС (положительная контрольная группа), и 2 животных использовали в качестве отрицательной контрольной группы. Животным групп, подвергавшихся воздействию ЛПС, и животным отрицательных контрольных групп вводили "только растворитель" ("только растворитель" означает 10 мл/кг массы тела в 0,5% растворе Natrosol). Животным других групп вводили разные дозы соединения примера 1, соединения примера 2, соединения предшествующего уровня техники А или соединения предшествующего уровня техники D соответственно (см. табл. D). Для каждого соединения количество соединения, необходимое для получения самой высокой концентрации, суспендировали в 10 мл 0,5% раствора Natrosol (гидоксиэтилцеллюлоза) и затем разбавляли с получением соответствующих концентраций, приведенных в табл. в табл. D. Суспензию соответствующего соединения или "только растворитель" (10 мл/кг массы тела в 0,5% растворе Natrosol) вводили перорально через желудочный зонд. Полученные дозы отдельных соединений соответствовали приведенным в табл. D. Таблица D Исследуемые соединения и их соответствующие дозы Приведенные выше дозы установлены на основании проведенных ранее исследований с помощью модели на мышах ЛПС TNF ex vivo. Через 1 ч (через 0,5 ч в случае соединения предшествующего уровня техники A и в случае соединения предшествующего уровня техники D) после введения соединения (такое время выбирали для обеспечения достаточного воздействия, как это было определено в проведенных ранее фармакокинетических экспериментах) животных подвергали воздействию ЛПС путем его распыления/опрыскивания аэрозолем. Воздействие на весь организм каждого из 12 животных проводили в камере, изготовленной из плексигласа. Животных разделяли/индивидуализировали с помощью металлических пластин с отверстиями. Аэрозоль подавали с использованием имеющегося в продаже небулайзера (PARI Master + небулайзерPARI LL (Pari GmbH). Концентрация распыляемого раствора ЛПС составляла 1 мг/мл воздуха. Время воздействия ЛПС составляло 30 мин. Через 4 ч после воздействия ЛПС животных анестезировали изофлураном и затем умерщвляли путем перелома шейного отдела позвоночника. В трахею вводили канюлю и проводили ЖБАЛ с использованием 25 мл буфера для проведения лаважа (забуференный фосфатом физиологический раствор (ЗФФ)+ 2% БСА). Определение содержания нейтрофилов в ЖБАЛ проводили с использованием гемоцитометра ADVIA 120 (Bayer Diagnostics). Количество нейтрофилов нормировали (положительный контроль (= только воздействие ЛПС) = 100%, отрицательный контроль (без воздействия ЛПС, введение "только растворителя") = 0%) и выражали в процентах от значений, полученных для контрольной группы, на которую воздействовали с помощью ЛПС. Значения ED50 определяли путем нелинейной аппроксимации (с помощью программного обеспечения Graph Pad Prism и аппроксимации сигмоидальной зависимости реакции от дозы). Значение ED50 означает эффективную дозу исследуемого соединения, обеспечивающую равное половине максимального ингибирование индуцируемого посредством ЛПС поступления нейтрофилов в ЖБАЛ. Соответственно самые низкие значения ED50 означают хорошую способность соответствующего соединения предупреждать поступление нейтрофилов в легочную ткань после воздействия ЛПС и, сле- 21024033 довательно, хорошую эффективность соответствующего соединения для предупреждения воспаления легочной ткани. Поскольку в отличие от значения IC50 значение ED50 не является результатом исследования in vitro, а является результатом проводимого на крысах исследования in vivo, и поскольку в этом случае исследуют не только непосредственное ингибирование фермента PDE4B, но и поступление нейтрофилов в легочную ткань после воздействия ЛПС, значение ED50 является весьма важным параметром,определяющим возможность применения соединения в качестве лекарственного средства для лечения воспалительных заболеваний дыхательных путей, таких как ХОЗЛ и астма (каждое из них является воспалительным заболеванием). Воздействие ЛПС на крыс приводило к явному поступлению нейтрофилов в ЖБАЛ. Предварительное введение крысам соединения примера 1, соединения примера 2, соединения предшествующего уровня техники А и соединения предшествующего уровня техники D приводило к ингибированию индуцируемого посредством ЛПС поступления нейтрофилов в ЖБАЛ. Рассчитанные значения ED50 для различных соединений приведены в табл. Е. Таблица Е Значения ED50 для исследуемых соединений, которые рассчитаны из экспериментальных данных Экспериментально определенные значения ED50 для соединений, предлагаемых в настоящем изобретении, что означает, для соединения примера 1 (ED50 = 0,31 мг/кг массы тела) и для соединения примера 2 (ED50 = 0,1 мг/кг массы тела), указывают на то, что эти соединения, предлагаемые в настоящем изобретении, соединение примера 1 и соединение примера 2, в этом исследовании обладают в 3-66 раз большей активностью, чем соединения предшествующего уровня техники А и D. Поэтому соединения, предлагаемые в настоящем изобретении, обладают более высокой эффективностью при предупреждении поступления нейтрофилов в легочную ткань и поэтому являются намного более подходящими для применения в качестве лекарственных средств, предназначенных для лечения воспалительных респираторных заболеваний, таких как астма и ХОЗЛ. 5.3. Опорожнение желудка и кишечный транзит у находящихся в сознании крыс. Чтобы идентифицировать активное средство, которое является подходящим для применения в качестве терапевтического ингибитора PDE4, необходимо определить, является ли исследуемое соединение эффективным при дозе, при которой не обнаруживаются значительные побочные эффекты, связанные с желудочно-кишечным трактом. Известно, что побочные эффекты, связанные с желудочно-кишечным трактом, являются выраженными при использовании ингибиторов PDE4 (см. Diamant, Z., Spina, D. "PDE4-inhibitors: a novel targetedtherapy for obstructive airways disease", Pulm. Pharmacol. Ther., 2011, 24 (4), p. 353-360 и Press, Banner N.J.,K.H. "PDE4 Inhibitors - A Review of the Current Field", Progress in Medicinal Chemistry, 2009, 47; p. 37-74). В приведенных выше экспериментах 1.1 и 1.2 показано, что соединения, предлагаемые в настоящем изобретении, очевидно, являются более эффективными ингибиторами PDE4B и/или более эффективно предупреждают поступление нейтрофилов в легочную ткань, и поэтому они являются более предпочтительными, чем структурно родственные соединения, раскрытые в WO 2009/050248, в частности соединения А, В, С и D. Для того чтобы определить, приводят ли соединения, предлагаемые в настоящем изобретении, к побочным эффектам, связанным с желудочно-кишечным трактом, соединения, предлагаемые в настоящем изобретении, вводили крысам за 30 мин до того, как крысам давали тестовый корм, содержащий сульфат бария. Затем исследовали влияние присутствия этих соединений на опорожнение желудка и/или кишечный транзит у этих крыс. Воздействие соединений примера 1 и примера 2 на опорожнение желудка и кишечный транзит у находящихся в сознании крыс исследовали так, как это описано ниже. Использовали крыс Wistar обоих полов, обладающих массой тела, равной 130-160 г (возраст: самцы: 7 недель, самки: 8 недель). Животных получали от уполномоченного местного поставщика, перед использованием животных требовалось держать их в изоляции в течение не менее 4 дней, в это время осуществляли стандартный уход за животными. Группы, содержащие не более 5 животных каждая, помещали в клетки в помещении с регулируемой температурой и влажностью и использовали цикл освещение/затемнение при освещении от 6 утра до 6 вечера. Животным в неограниченном количестве пре- 22024033 доставляли обычный корм для грызунов и воду. В день проведения эксперимента животных переносили в лабораторию. Опорожнение желудка, а также незначительное продвижение по кишечнику исследовали с использованием тестового корма, содержащего сульфат бария. Использовали по 5 крыс Crl:WI(Han) каждого пола (n=10). Животным не давали корм в течение 17 ч до проведения эксперимента, но обеспечивали свободный доступ к воде. Исследуемое лекарственное средство (лекарственное средство суспендировали в 0,5% растворе Natrosol до концентрации, равной 10 мл/кг массы тела) или отрицательный контроль (только растворитель вводили при концентрации 10 мл/кг массы тела) вводили (перорально) за 30 мин до тестового корма при дозах, равных 3-кратному, 10-кратному или 30-кратному значению ED50, определенному в проводимых на крысах исследованиях эффективности. Тестовый корм (суспензия 7,5 г сульфата бария в 10 мл не содержащей соли воды) вводили перорально через желудочный зонд при дозе, равной 2 мл/100 г массы тела. Через 30 мин после введения тестового корма животных умерщвляли в состоянии глубокой анастезии изофлураном путем перелома шейного отдела позвоночника. Затем проводили лапаротомию и извлекали желудок и кишечник. Извлеченный желудок взвешивали, затем разрезали, содержимое удаляли и пустой желудок повторно взвешивали. Длину кишечника, на которую переместился сульфат бария, в пересчете на полную длину кишечника определяли путем непосредственного измерения с помощью линейки. Оценка опорожнения желудка. Массу содержимого желудка определяли по разности масс полного и пустого желудка и нормировали на 100 г массы тела. Таким образом, увеличение значения разности масс указывает на ухудшенное опорожнение желудка, тогда как уменьшение значения разности масс указывает на улучшенное опорожнение желудка. Оценка кишечного транзита. Длину кишечника, на которую переместился сульфат бария (оценивали путем визуального осмотра), в пересчете на полную длину кишечника (от привратника до прямой кишки) определяли путем непосредственного измерения с помощью линейки. Кишечный транзит рассчитывали, как выраженное в процентах расстояние, на которое сульфат бария переместился в кишечнике, в пересчете на полную длину кишечника. Соответственно увеличение расстояния, на которое прошло содержимое в кишечнике, указывает на ускоренный кишечный транзит,тогда как уменьшение расстояния, на которое прошло содержимое в кишечнике, на указывает на замедленный кишечный транзит. Статистический анализ. Результаты выражали, как значениестандартное отклонение (СО). Для каждой дозы сравнения проводили с помощью дисперсионного анализа (ANOVA) и затем, если значения, полученные с помощью ANOVA, являлись статистически значимыми, с помощью критерия Даннетта для сравнения животных разных групп с животными контрольной группы. Статистически значимым считалось р 0,05. Соответственно всоответствии со статистическими данными, соединения, предлагаемые в настоящем изобретении, не приводят к существенным побочным эффектам, связанным с желудочно-кишечным трактом, даже при дозах, которые равны 30-кратному значению дозы ED50, поскольку, как показано на фиг. 1 а и 1 б, и 2 а и 2 б, у крыс, которым вводили соединение примера 1 или 2, не наблюдается ни существенно улучшенное, ни ухудшенное опорожнение желудка, ни существенно ускоренный, ни существенно замедленный кишечный транзит даже при дозах, которые равны 30-кратному значению дозы ED50. В случае соединения примера 1 нет различий опорожнения желудка при дозе, равной 3-кратному значению дозы ED50 и 10-кратному значению дозы ED50, и при дозе, равной 30-кратному значению дозыED50, обнаружено лишь несущественное увеличение разности масс в пересчете на массу тела. Однако не обнаружены существенные отличия кишечного транзита у соответствующих животных, которым вводили соединение примера 1, от результатов, полученных для животных отрицательной контрольной груп- 23024033 пы, даже при дозе, равной 30-кратному значению дозы ED50. Показано, что в случае соединения примера 2 не обнаружено существенных отличий опорожнения желудка и кишечного транзита по сравнению с результатами, полученными для отрицательной контрольной группы для всех исследуемых доз соединения примера 2, даже при дозе, равной 30-кратному значению дозы ED50. Соответственно соединения, предлагаемые в настоящем изобретении, являются не только более эффективными ингибиторами PDE4, чем соединения, раскрытые в WO 2009/050248 (как показано в экспериментах 1.1 и 1.2), но также не вызывают побочных эффектов, связанных с желудочно-кишечным трактом. Показания. Соединения формулы I перспективны для применения в разных областях терапии. Следует особо отметить случаи применения, в которых соединения формулы I, предлагаемые в настоящем изобретении,являются особенно подходящими с учетом их фармацевтической активности в качестве ингибиторовPDE4. Примеры включают респираторные или желудочно-кишечные заболевания или нарушения, воспалительные заболевания суставов, кожи или глаз, раковые заболевания, а также заболевания периферической или центральной нервной системы. Следует особо отметить предупреждение и лечение заболеваний дыхательных путей и легких, которые сопровождаются усилением образования слизи, воспалений и/или обструктивных заболеваний дыхательных путей. Примеры включают острый, аллергический или хронический бронхит, аутоиммунную гемолитическую анемию, хронический обструктивный бронхит (ХОБР), кашель, эмфизему легких,аллергический или неаллергический ринит или синусит, аллергический риноконъюнктивит, хронический ринит или синусит, астму, альвеолит, болезнь фермера, гиперреактивность дыхательных путей, инфекционный бронхит или пневмонит, астму у детей, бронхоэктаз, фиброз легких, ОРДС (острый респираторный дистресс-синдром у взрослых), отек бронхов, отек легких, бронхит, пневмонию или интерстициальную пневмонию различной этиологии, например, вызванную аспирацией, вдыханием токсичных газов или бронхитом, пневмонию или интерстициальную пневмонию, вызванную сердечной недостаточностью, радиоактивным излучением, химиотерапией, кистозный фиброз или муковисцидоз или альфа-1 антитрипсиновую недостаточность. Следует также особо отметить лечение воспалительных заболеваний желудочно-кишечного тракта. Примеры включают острые или хронические воспалительные нарушения в желчном пузыре, болезнь Крона, язвенный колит, воспалительный псевдополипоз, ювенильные полипы, глубокий кистозный колит, кистозный пневматоз кишечника, заболевания желчных протоков и желчного пузыря, например камни и конгломераты в желчном пузыре, воспалительные заболевания суставов, такие как ревматоидный артрит, или воспалительные заболевания кожи и глаз. Следует также особо отметить лечение саркоидоза. Следует также особо отметить лечение раковых заболеваний. Примеры включают все типы острых и хронических лейкозов, такие как острый лимфолейкоз и острый миелолейкоз, хронический лимфолейкоз и хронический миелолейкоз, а также опухоли костей, такие как, например, остеосаркома и все типы глиом, такие как, например, олигодендроглиома и глиобластома. Следует также особо отметить предупреждение и лечение заболеваний периферической или центральной нервной системы. Их примеры включают депрессию, биполярную или маниакальную депрессию, острые и хронические состояния тревоги, шизофрению, болезнь Альцгеймера, болезнь Паркинсона,острый и хронический рассеянный склероз или острую и хроническую боль, и повреждение мозга, вызванное ударом, гипоксией или черепно-мозговой травмой. Настоящее изобретение особенно предпочтительно относится к применению соединений формулы I для приготовления фармацевтической композиции, предназначенной для лечения воспалительных или обструктивных заболеваний верхних и нижних дыхательных путей, включая легкие, таких как, например, аллергический ринит, хронический ринит, бронхоэктаз, муковисцидоз, идиопатический фиброз легких, фиброзирующий альвеолит, ХОЗЛ, хронический бронхит, хронический синусит, астма, болезнь Крона, язвенный колит, альфа-1-антитрипсиновую недостаточность, предпочтительно ХОЗЛ, хронический бронхит и астма. Наиболее предпочтительно использовать соединения формулы I для лечения воспалительных или обструктивных заболеваний, таких как ХОЗЛ, хронический бронхит, хронический синусит, астма, болезнь Крона, язвенный колит, ревматоидный артрит, предпочтительно ХОЗЛ, хронический бронхит и астма. Также предпочтительно использовать соединения формулы I для лечения заболеваний периферической или центральной нервной системы, таких как депрессия, биполярная или маниакальная депрессия,острые и хронические состояния тревоги, шизофрению, болезнь Альцгеймера, болезнь Паркинсона, острый и хронический рассеянный склероз или острая и хроническая боль, и повреждения мозга, вызванные ударом, гипоксией или черепно-мозговой травмой. Также предпочтительно использовать соединения формулы I для лечения воспалительных заболеваний глаз, в особенности для лечения "сухого" кератита и для лечения глаукомы. Индивидуумы, стра- 24024033 дающие "сухим" кератитом, страдают от неприятных ощущений в глазах (сухость, ощущение песка; зуд; острая боль/жжение; боль/болезненное ощущение) и затуманенного зрения. Фермент фосфодиэстераза 4(PDE4) регулирует биологические процессы в организме "хозяина" посредством разрушения вторичного мессенджера цАМФ. Проведено большое количество исследований ингибиторов PDE4, как противовоспалительных лекарственных средств, поскольку известно, что повышение концентрации цАМФ ослабляет воспалительный ответ во многих типах клеток (см. Govek et al., BioorganicMed. Chem. Lett 20,(2010), p. 2928-2932). Кроме того, предпочтительно использовать соединения формулы I для лечения заболеваний глаз, в частности для лечения глаукомы, поскольку показано, что повышение концентрации цАМФ защищает ганглионарные клетки сетчатки глаза от гибели, вызванной высоким внутриклеточным (ВКД) (см. Seki Т. et al., J. Mol. Neurosci., 2011 Jan; 43(1): 30-4.), и повышение концентрации цАМФ способствует снижению ВКД (см. Naveh N. et al., Br. J. Ophthalmol., 2000 Dec.; 84(12): 1411-4), повышение которого является главной причиной развития глаукомы. Комбинации. Соединения формулы I можно использовать по отдельности или совместно с другими активными веществами формулы I, предлагаемыми в настоящем изобретении. При необходимости соединения формулы I также можно использовать в комбинации с другими фармакологически активными веществами. Для этой цели предпочтительно использовать активные вещества, выбранные, например, из группы,включающей бета-миметики, антихолинергетики, кортикостероиды, другие ингибиторы PDE4, антагонисты LTD4, ингибиторы EGFR, ингибиторы MRP4, агонисты допамина, Н 1-антигистамины, антагонистыPAF и ингибиторы киназы PI3, НСПВС, ингибиторы СОХ 2, антагонисты рецептора ЕР 4, ингибиторыDPP4 или комбинации двух или трех из них, такие как, например, комбинации соединений формулы I с одним или двумя соединениями, выбранными из группы, включающей антагонисты рецептора ЕР 4, ингибиторы DPP4, НСПВС, ингибиторы СОХ 2 и кортикостероиды,бета-миметики, кортикостероиды, ингибиторы PDE4, ингибиторы EGFR и антагонисты LTD4,антихолинергетики, бета-миметики, кортикостероиды, ингибиторы PDE4, ингибиторы EGFR и антагонисты LTD4,ингибиторы PDE4, кортикостероиды, ингибиторы EGFR и антагонисты LTD4,ингибиторы EGFR, ингибиторы PDE4 и антагонисты LTD4,ингибиторы EGFR и антагонисты LTD4,ингибиторы CCR3, ингибиторы iNOS (ингибиторы индуцируемой синтазы оксида азота), (6R)-Lэритро-5,6,7,8-тетрагидробиоптерин (ниже в настоящем изобретении называющийся "ВН 4") и его производные, такие как описанные в WO 2006/120176, и ингибиторы SYK (ингибиторы тирозинкиназы селезенки),антихолинергетики, бета-миметики, кортикостероиды, ингибиторы PDE4 и ингибиторы MRP4. Настоящее изобретение также относится к комбинациям трех активных соединений, каждое из которых выбрано из одной из указанных выше категорий соединений. Подходящие использующиеся бета-миметики предпочтительно представляют собой соединения,выбранные из группы, включающей албутерол, бамбутерол, бутолтерол, броксатерол, карбутерол, кленбутерол, фенотерол, формотерол, арформотерол, зинтерол, гексопреналин, ибутерол, изоэтарин, изопреналин, левосалбутамол, мабутерол, мелуадрин, метапротеренол, орципреналин, пирбутерол, прокатерол,репротерол, римитерол, ритодрин, салметерол, салмефамол, сотеренол, сульфонтерол, тиарамид, тербуталин,толубутерол,CHF-1035,HOKU-81,KUL-1248,3-(4-6-[2-гидрокси-2-(4-гидрокси-3 гидроксиметилфенил)этиламино]гексилоксибутил)бензилсульфонамид, 5-[2-(5,6-диэтилиндан-2-иламино)-1-гидроксиэтил]-8-гидрокси-1 Н-хинолин-2-он,4-гидрокси-7-[2-[2-[3-(2-фенилэтокси)пропил]сульфонилэтил]аминоэтил]-2(3H)-бензотиазолон,1-(2-фтор-4-гидроксифенил)-2-[4-(1-бензимидазолил)-2-метил-2-бутиламино]этанол,1-[3-(4-метоксибензиламино)-4-гидроксифенил]-2-[4-(1 бензимидазолил)-2-метил-2-бутиламино]этанол, 1-[2 Н-5-гидрокси-3-оксо-4 Н-1,4-бензоксазин-8-ил]-2-[3(4-N,N-диметиламинофенил)-2-метил-2-пропиламино]этанол,1-[2 Н-5-гидрокси-3-оксо-4 Н-1,4 бензоксазин-8-ил]-2-[3-(4-метоксифенил)-2-метил-2-пропиламино]этанол, 1-[2 Н-5-гидрокси-3-оксо-4 Н 1,4-бензоксазин-8-ил]-2-[3-(4-н-бутилоксифенил)-2-метил-2-пропиламино]этанол,1-[2 Н-5-гидрокси-3 оксо-4 Н-1,4-бензоксазин-8-ил]-2-4-[3-(4-метоксифенил)-1,2,4-триазол-3-ил]-2-метил-2-бутиламиноэтанол, 5-гидрокси-8-(1-гидрокси-2-изопропиламинобутил)-2 Н-1,4-бензоксазин-3-(4 Н)-он, 1-(4-амино-3 хлор-5-трифторметилфенил)-2-трет-бутиламино)этанол,6-гидрокси-8-1-гидрокси-2-[2(4-метоксифенил)-1,1-диметилэтиламино]этил-4 Н-бензо[1,4]оксазин-3-он, 6-гидрокси-8-1-гидрокси-2-[2-(этил-4 феноксиацетат)-1,1-диметилэтиламино]этил-4 Н-бензо[1,4]оксазин-3-он, 6-гидрокси-8-1-гидрокси 2-[2(4-феноксиацетат-1,1-Диметилэтиламино]этил-4 Н-бензо[1,4]оксазин-3-он, 8-2-[1,1-диметил-2-(2,4,6 триметилфенил)этиламино]-1-гидроксиэтил-6-гидрокси-4 Н-бензо[1,4]оксазин-3-он,6-гидрокси-8-1 гидрокси-2-[2-(4-гидроксифенил)-1,1-Диметилэтиламино]этил-4 Н-бензо[1,4]оксазин-3-он, 6-гидрокси 8-1-гидрокси-2-[2-(4-изопропилфенил)-1,1-диметилэтиламино]этил-4 Н-бензо[1,4]оксазин-3-он, 8-2[2-(4-этилфенил)-1,1-диметилэтиламино]-1-гидроксиэтил-6-гидрокси-4 Н-бензо[1,4]оксазин-3-он, 8-2[2-(4-этоксифенил)-1,1-диметилэтиламино]-1-гидроксиэтил-6-гидрокси-4 Н-бензо[1,4]оксазин-3-он 4-(4- 25024033 2-[2-гидрокси-2-(6-гидрокси-3-оксо-3,4-дигидро-2 Н-бензо[1,4]оксазин-8-ил)этиламино]-2-метилпропилфенокси)масляная кислота, 8-2-[2-(3,4-дифторфенил)-1,1-диметилэтиламино]-1-гидроксиэтил-6 гидрокси-4 Н-бензо[1,4]оксазин-3-он и 1-(4-этоксикарбониламино-3-циано-5-фторфенил)-2-(третбутиламино)этанол, необязательно в виде их рацематов, энантиомеров, диастереоизомеров и необязательно в виде их фармакологически приемлемых солей присоединения с кислотами, сольватов или гидратов. В контексте настоящего изобретения соли присоединения с кислотами бета-миметиков предпочтительно выбраны из группы, включающей гидрохлорид, гидробромид, гидройодид, гидросульфат, гидрофосфат, гидрометансульфонат, гидронитрат, гидромалеат, гидроацетат, гидроцитрат, гидрофумарат, гидротартат, гидрооксалат, гидросукцинат, гидробензоат и гидро-п-толуолсульфонат, предпочтительно гидрохлорид, гидробромид, гидросульфат, гидрофосфат, гидрофумарат и гидрометансульфонат, предпочтительно гидрохлорид, гидробромид, гидросульфат, гидрофосфат, гидрофумарат и гидрометансульфонат. Из указанных выше солей присоединения с кислотами в контексте настоящего изобретения особенно предпочтительными являются соли хлористо-водородной кислоты, метансульфоновой кислоты, бензойной кислоты и уксусной кислоты. Использующиеся антихолинергетики предпочтительно представляют собой соединения, выбранные из группы, включающей соли тиотропия, соли окситропия, соли флутропия, соли ипратропия, соли гликопиррония, соли троспия, тропенол-2,2-дифенилпропионатметобромид, скопин-2,2-дифенилпропионатметобромид, скопин-2-фтор-2,2-дифенилацетатметобромид, тропенол-2-фтор-2,2-дифенилацетатметобромид,тропенол-3,3',4,4'-тетрафторбензилатметобромид,скопин-3,3',4,4'-тетрафторбензилатметобромид, тропенол-4,4'-дифторбензилатметобромид, скопин-4,4'-дифторбензилатметобромид, тропенол-3,3'-дифторбензилатметобромид, скопин-3,3'-дифторбензилатметобромид, тропенол-9-гидроксифлуорен-9-карбоксилатметобромид, тропенол-9-фторфлуорен-9-карбоксилатметобромид, скопин-9 гидроксифлуорен-9-карбоксилатметобромид, скопин-9-фторфлуорен-9-карбоксилатметобромид, тропенол-9-метилфлуорен-9-карбоксилатметобромид,скопин-9-метилфлуорен-9-карбоксилатметобромид,циклопропилтропинбензилатметобромид, циклопропилтропин-2,2-дифенилпропионатметобромид, циклопропилтропин-9-гидроксиксантен-9-карбоксилатметобромид, циклопропилтропин-9-метилфлуорен-9 карбоксилатметобромид, циклопропилтропин-9-метилксантен-9-карбоксилатметобромид, циклопропилтропин-9-гидроксифлуорен-9-карбоксилатметобромид, метилциклопропилтропин-4,4'-дифторбензилатметобромид,тропенол-9-гидроксиксантен-9-карбоксилатметобромид,скопин-9-гидроксиксантен-9 карбоксилатметобромид, тропенол-9-метилксантен-9-карбоксилатметобромид, скопин-9-метилксантен 9-карбоксилатметобромид,тропенол-9-этилксантен-9-карбоксилатметобромид,тропенол-9 дифторметилксантен-9-карбоксилатметобромид,скопин-9-гидроксиметилксантен-9-карбоксилатметобромид, необязательно в виде их сольватов или гидратов. В указанных выше солях катионы тиотропия, окситропия, флутропия, ипратропия, гликопиррония и троспия являются фармакологически активными ингредиентами. В качестве анионов указанные выше соли могут предпочтительно содержать хлорид, бромид, йодид, сульфат, фосфат, метансульфонат, нитрат, малеат, ацетат, цитрат, фумарат, тартрат, оксалат, сукцинат, бензоат или п-толуолсульфонат, и хлорид, бромид, йодид, сульфат, метансульфонат или п-толуолсульфонат являются предпочтительными в качестве противоионов. Из всех солей хлориды, бромиды, йодиды и метансульфонат являются особенно предпочтительными. Особенно важным является тиотропийбромид. В случае использования тиотропийбромида фармацевтические комбинации, предлагаемые в настоящем изобретении, предпочтительно содержат его в форме кристаллического моногидрата тиотропийбромида, который известен из WO 02/30928. Если тиотропийбромид используют в фармацевтических комбинациях, предлагаемых в настоящем изобретении, в безводной форме, то предпочтительно использовать безводный кристаллический тиотропийбромид, который известен из WO 03/000265. Кортикостероиды, использующиеся в настоящем изобретении предпочтительно представляют собой соединения, выбранные из группы, включающей преднизолон, преднизон, бутиксокортпропионат,флунизолид, беклометазон, триамцинолон, будезонид, флутиказон, мометазон, циклезонид, рофлепонид,дексаметазон, бетаметазон, дефлазакорт, RPR-106541, NS-126, (S)-фтормегил-6,9-дифтор-17-[(2 фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботионат и(S)-(2 оксотетрагидрофуран-3S-ил)-6,9-дифтор-11-гидрокси-16-метил-3-оксо-17-пропионилоксиандроста-1,4 диен-17-карботионат, необязательно в виде их рацематов, энантиомеров или диастереоизомеров и необязательно в виде их солей и производных, сольватов и/или гидратов. Особенно предпочтительным является стероид, выбранный из группы, включающей флунизолид,беклометазон, триамцинолон, будезонид, флутиказон, мометазон, циклезонид, рофлепонид, дексаметазон,NS-126,(S)-фторметил-6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3 оксоандроста-1,4-диен-17-карботионат и (S)-(2-оксотетрагидрофуран-3S-ил)-6,9-дифтор-11-гидрокси-16 метил-3-оксо-17-пропионилоксиандроста-1,4-диен-17-карботионат, необязательно в виде их рацематов,энантиомеров или диастереоизомеров и необязательно в виде их солей и производных, сольватов и/или гидратов. Любое указание на стероиды включает указание на любые их соли или производные, гидраты или сольваты, которые могут существовать. Примерами возможных солей и производных стероидов могут быть: соли щелочных металлов, такие как, например, соли натрия или калия, их сульфобензоаты, фосфаты, изоникотинаты, ацетаты, пропионаты, дигидрофосфаты, пальмитаты, пивалаты или фуроаты. Другие ингибиторы PDE4, которые можно использовать, предпочтительно представляют собой соединения, выбранные из группы, включающей энпрофиллин, теофиллин, рофлумиласт, арифло (циломиласт), тофимиласт, пумафентрин, лиримиласт, арофиллин, атизорам, D-4396 (Sch-351591), AWD-12-281(GW-842470), NCS-613, CDP-840, D-4418, PD-168787, Т-440, Т-2585, V-11294 А, С 1-1018, CDC-801, CDC3052,D-22888,YM-58997,Z-15370,N-(3,5-дихлор-1-оксопиридин-4-ил)-4-дифторметокси-3 циклопропилметоксибензамид, (-)п-[(4aR,10bS)-9-этокси-1,2,3,4,4 а,10b-гексагидро-8-метокси-2-метилбензо[s][1.6]нафтиридин-6-ил]-N,N-диизопропилбензамид, (R)-(+)-1-(4-бромбензил)-4-[(3-циклопентилокси)-4-метоксифенил]-2-пирролидон,3-(циклопентилокси-4-метоксифенил)-1-(4-N-[N-2-циано-Sметилизотиоуреидо]бензил)-2-пирролидон, цис[4-циано-4-(3-циклопентилокси-4-метоксифенил)циклогексан-1-карбоновую кислоту], 2-карбметокси-4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-он,цис[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1 ол], (R)-(+)-этил[4-(3-циклопентилокси-4-метоксифенил)пирролидин-2-илиден]ацетат, (S)-(-)-этил[4-(3 циклопентилокси-4-метоксифенил)пирролидин-2-илиден]ацетат, 9-циклопентил-5,6-дигидро-7-этил-3-(2 тиенил)-9 Н-пиразоло[3,4-с]-1,2,4-триазоло[4,3-а]пиридин и 9-циклопентил-5,6-дигидро-7-этил-3-(третбутил)-9 Н пиразоло[3,4-с]-1,2,4-триазоло[4,3-а]пиридин, необязательно в виде их рацематов, энантиомеров или диастереоизомеров и необязательно в виде их фармакологически приемлемых солей присоединения с кислотами, сольватов и/или гидратов. Соли присоединения с фармакологически приемлемыми кислотами, которые могут образовать указанные выше ингибиторы PDE4, представляют собой, например, соли, выбранные из группы, включающей гидрохлорид, гидробромид, гидройодид, гидросульфат, гидрофосфат, гидрометансульфонат, гидронитрат, гидромалеат, гидроацетат, гидробензоат, гидроцитрат, гидрофумарат, гидротартат, гидрооксалат,гидросукцинат, гидробензоат и гидр п-толуолсульфонат, предпочтительно гидрохлорид, гидробромид,гидросульфат,' гидрофосфат, гидрофумарат и гидрометансульфонат. Антагонисты рецептора ЕР 4, которые можно использовать, предпочтительно представляют собой соединения, выбранные из группы, включающей [N-[4-(5,9-диэтокси-6-оксо-6,8-дигидро-7 Н-пирроло[3,4-g]хинолин 7-ил)-3-метилбензил]сульфонил-2-(2-метоксифенил)ацетамид]; 5-бутил-2,4-дигидро-4-2'-[N-(3-метил-2-тиофен-карбонил)сульфамоил]бифенил-4-ил]метил]-2-[(2-трифторметил)фенил]-1,2,4-триазол-3-он; (4-(1S)-1-[(5 хлор-2-[(4-фторфенил)окси]фенилкарбонил)амино]этилбензойную кислоту; N-[(2-[4-(2-этил-4,6-диметил-1 Нимидазо[4,5-с]пиридин-1-ил)фенил]этиламино)карбонил]-4-метилбензолсульфонамид; 4-4-(5-метокси-2 пиридинил)фенокси]метил]-5-метил-N-[(2-метилфенил)сульфонил]-2-фуранкарбоксамид; метиловый эфир 11 альфа, 15-альфа-дигидрокси-16-(3-метоксиметилфенил)-9-оксо-17,18,19,20-тетранор-5-тиа-13(Е)-простановой кислоты; 4-циано-2-2-(4-фтор-1-нафталинил)-1-оксопропил]амино]бензолмасляную кислоту и N-2-[4-(4,9 диэтокси-1-оксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил)фенил]ацетилбензолсульфонамид. НСПВС, которые можно использовать, предпочтительно представляют собой соединения, выбранные из группы, включающей ацеклофенак, ацеметацин, ацетилсалициловую кислоту, алклофенак, алминопрофен, амфенак, ампироксикам, амтолметин гуацил, аниролак, антрафенин, азапропазон, бенорилат,бермопрофен, биндарит, бромфенак, буклоксовую кислоту, буколом, буфексамак, бумадизон, бутибуфен,бутиксират, карбасалат кальция, карпрофен, трисалицилат холина магния, целекоксиб, цинметацин, цинноксикам, клиданак, клобузарит, дебоксамет, дексибупрофен, декскетопрофен, диклофенак, дифлунизал,дроксикам, элтенак, энфенаминовую кислоту, этерсалат, этодолак, этофенамат, эторикоксиб, феклобузон, фелбинак, фенбуфен, фенклофенак, фенопрофен, фентиазак, фепрадинол, фепразон, флобуфен,флоктафенин, флуфенаминовую кислоту, флуфенизал, флуноксапрофен, флурбипрофен, флурбипрофенаксетил, фурофенак, фурпрофен, глукаметацин, ибуфенак, ибупрофен, индобуфен, индометацин, индометацинфарнезил, индопрофен, изоксепак, изоксикам, кетопрофен, кеторолак, лобензарит, лоназолак,лорноксикам, локсопрофен, лумиракоксиб, меклофенаминовую кислоту, меклофен, менфенаминовую кислоту, мелоксикам, мезалазин, миропрофен, мофезопак, набуметон, напроксен, нифлуминовую кислоту, олзалазин, оксапрозин, оксипинак, оксифенбутазон, парекоксиб, фенилбутазон, пелубипрофен, пимепрофен, пиразолак, прироксикам, пирпрофен, пранопрофен, прифелон, приномод, проглуметацин, проквазон, протицининовую кислоту, рофекоксиб, ромазарит, салициламид, салициловую кислоту, салмистеин, салнацедин, салсалат, сулиндак, судоксикам, супрофен, талнифлумат, тенидап, тенозал, теноксикам, тепоксалин, тиапрофеновую кислоту, тарамид, тилнопрофенарбамел, тимегадин, тиноридин, тиопинак, толфенаминовую кислоту, толметин, уфенамат, валдекоксиб, ксимопрофен, залтопрофен и золипрофен. Ингибиторы СОХ 2 (коксибы), которые можно использовать, предпочтительно представляют собой соединения, выбранные из группы, включающей целекоксиб, мелоксикам, эторикоксиб, лумиракоксиб,парекоксиб, рофекоксиб и валдекоксиб. Антагонисты LTD4, которые можно использовать, предпочтительно представляют собой соединения, выбранные из группы, включающей монтелукаст, пранлукаст, зафирлукаст, МСС-847 (ZD-3523),- 27024033[2-2-(4-трет-бутил-2-тиазолил)-5 бензофуранил]оксиметил]фенил]уксусную кислоту, необязательно в виде их рацематов, энантиомеров или диастереоизомеров, необязательно в виде их фармакологически приемлемых солей присоединения с кислотами и необязательно в виде их солей и производных, сольватов и/или гидратов. Соли присоединения с фармакологически приемлемыми кислотами, которые могут образовать антагонисты LTD4, представляют собой, например, соли, выбранные из группы, включающей гидрохлорид, гидробромид, гидройодид, гидросульфат, гидрофосфат, гидрометансульфонат, гидронитрат, гидромалеат, гидроацетат, гидробензоат, гидроцитрат, гидрофумарат, гидротартат, гидрооксалат, гидросукцинат, гидробензоат и гидро-п-толуолсульфонат, предпочтительно гидрохлорид, гидробромид, гидросульфат, гидрофосфат, гидрофумарат и гидрометансульфонат. Соли или производные, которые антагонистыLTD4 могут образовать, представляют собой, например, соли щелочных металлов, такие как, например,соли натрия или калия, соли щелочно-земельных металлов, сульфобензоаты, фосфаты, изоникотинаты,ацетаты, пропионаты, дигидрофосфаты, пальмитаты, пивалаты или фуроаты. Использующиеся ингибиторы EGFR предпочтительно представляют собой соединения, выбранные из группы, включающей
МПК / Метки
МПК: C07C 211/35, C07D 495/04, A61K 31/519, C07D 401/04, A61P 29/00
Метки: лечения, хозл, астмы, пиперидинодигидротиенопиримидинсульфоксиды, применение
Код ссылки
<a href="https://eas.patents.su/30-24033-piperidinodigidrotienopirimidinsulfoksidy-i-ih-primenenie-dlya-lecheniya-hozl-i-astmy.html" rel="bookmark" title="База патентов Евразийского Союза">Пиперидинодигидротиенопиримидинсульфоксиды и их применение для лечения хозл и астмы</a>
Циклические пиримидин-4-карбоксамиды в качестве антагонистов рецептора ccr2, предназначенные для лечения воспаления, астмы и хозл

Номер патента: 20548
Опубликовано: 30.12.2014
Авторы: Хёнке Кристоф, Бюттнер Франк, Тильманн Патрик, Джованнини Риккардо, Хобби Зильке, Эбель Хайнер, Фраттини Сара, Шойерер Штефан, Тризельманн Томас
МПК: A61P 29/00, C07D 401/06, A61K 31/506...
Метки: рецептора, антагонистов, предназначенные, пиримидин-4-карбоксамиды, воспаления, астмы, хозл, лечения, циклические, ccr2, качестве
Формула / Реферат:
1. Соединения формулы (I)в которыхR1 обозначает -L1-R7,где L1 обозначает связь или группу, выбранную из метилена, этилена и этенилена,R7 обозначает кольцо, выбранное из группы, включающей циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, пирролидинил, пиперидинил, азепанил, тетрагидрофуранил, тетрагидропиранил, оксепанил, фенил, пиридил и фуранил,L1, если он отличается от связи, необязательно замещен одной или большим количеством...
Применение солей тиотропия для лечения персистирующей астмы средней тяжести

Номер патента: 15353
Опубликовано: 30.06.2011
Авторы: Хайнрихс Штефан, Энгель Михаэль
МПК: A61K 31/381, A61P 11/06
Метки: применение, солей, тиотропия, лечения, астмы, персистирующей, средней, тяжести
Формула / Реферат:
1. Применение солей тиотропия 1в которой X- обозначает однозарядный анион, предпочтительно анион, выбранный из группы, включающей хлорид, бромид, иодид, сульфат, фосфат, метансульфонат, нитрат, малеат, ацетат, цитрат, фумарат, тартрат, оксалат, сукцинат, бензоат и n-толуолсульфонат, необязательно в виде их гидратов и/или сольватов, для приготовления лекарственного средства, предназначенного для лечения пациентов, страдающих астмой со степенью...
Применение композиции, содержащей формотерол и дипропионат беклометазона, для предотвращения и/или лечения обострения астмы

Номер патента: 18589
Опубликовано: 30.09.2013
Авторы: Кьези Паоло, Поли Джанлуиджи, Ронделли Ивано, Ачерби Даниэла
МПК: A61K 31/57, A61P 11/06, A61K 31/167...
Метки: применение, дипропионат, предотвращения, обострения, беклометазона, астмы, лечения, содержащей, формотерол, композиции
Формула / Реферат:
1. Применение композиции, содержащей фиксированную комбинацию:а) формотерола, его фармацевтически приемлемых соли или сольвата либо сольвата такой соли иб) дипропионата беклометазона,для изготовления лекарственного средства для введения путем ингаляции при неотложном лечении острых приступов астмы в дополнение к поддерживающей терапии астмы тем же лекарственным средством.2. Применение по п.1, где молярное отношение (а) к (б), вычисленное как...
Способ лечения бронхиальной астмы

Номер патента: 11710
Опубликовано: 28.04.2009
Авторы: Соломатов Виктор Геннадьевич, Соломатова Галина Владимировна, Соломатов Геннадий Сергеевич, Соломатова Галина Дмитриевна, Соломатова Анна Викторовна, Соломатова Мария Викторовна
МПК: A61H 39/04, A61H 39/00, A61H 23/00...
Метки: бронхиальной, астмы, лечения, способ
Формула / Реферат:
1. Способ лечения бронхиальной астмы, включающий рефлексодиагностику известными способами и последующее воздействие на выявленные значимые биологически активные точки (БАТ) и/или зоны энергорефлексотерапии (ЭРТ), отличающийся тем, что в качестве объектов воздействия выбирают БАТ и/или зоны ЭРТ, отвечающие за нормализацию процессов тонизации и расслабления скелетных мышц шеи, грудной клетки и диафрагмы, преимущественно стимуляцией значимых БАТ...
Аэрозольная композиция для лечения астмы и copd

Номер патента: 21604
Опубликовано: 30.07.2015
Авторы: Усберти Франческа, Бонелли Сауро, Цамбелли Энрико
МПК: A61K 31/40, A61K 9/00
Метки: композиция, аэрозольная, астмы, лечения
Формула / Реферат:
1. Фармацевтическая композиция, содержащая гликопиррония бромид в дозировке 0,5-100 мкг на активацию, растворенный в HFA (гидрофторалкановом) пропелленте и сорастворителе, отличающаяся тем, что указанная композиция содержит 1 М соляную кислоту (HCl) в количестве 0,005-1,0 мкг/мкл.2. Композиция по п.1, где диапазон 1 М HCl составляет 0,18-0,32 мкг/мкл.3. Композиция по п.1 или 2, где сорастворитель представляет собой этанол.4. Композиция по любому...
Предыдущий патент: Тара для жидких продуктов
Следующий патент: Терапевтическое применение полипептидов рецептора и антител
Случайный патент: Пентафторбензолсульфонамиды и их аналоги.