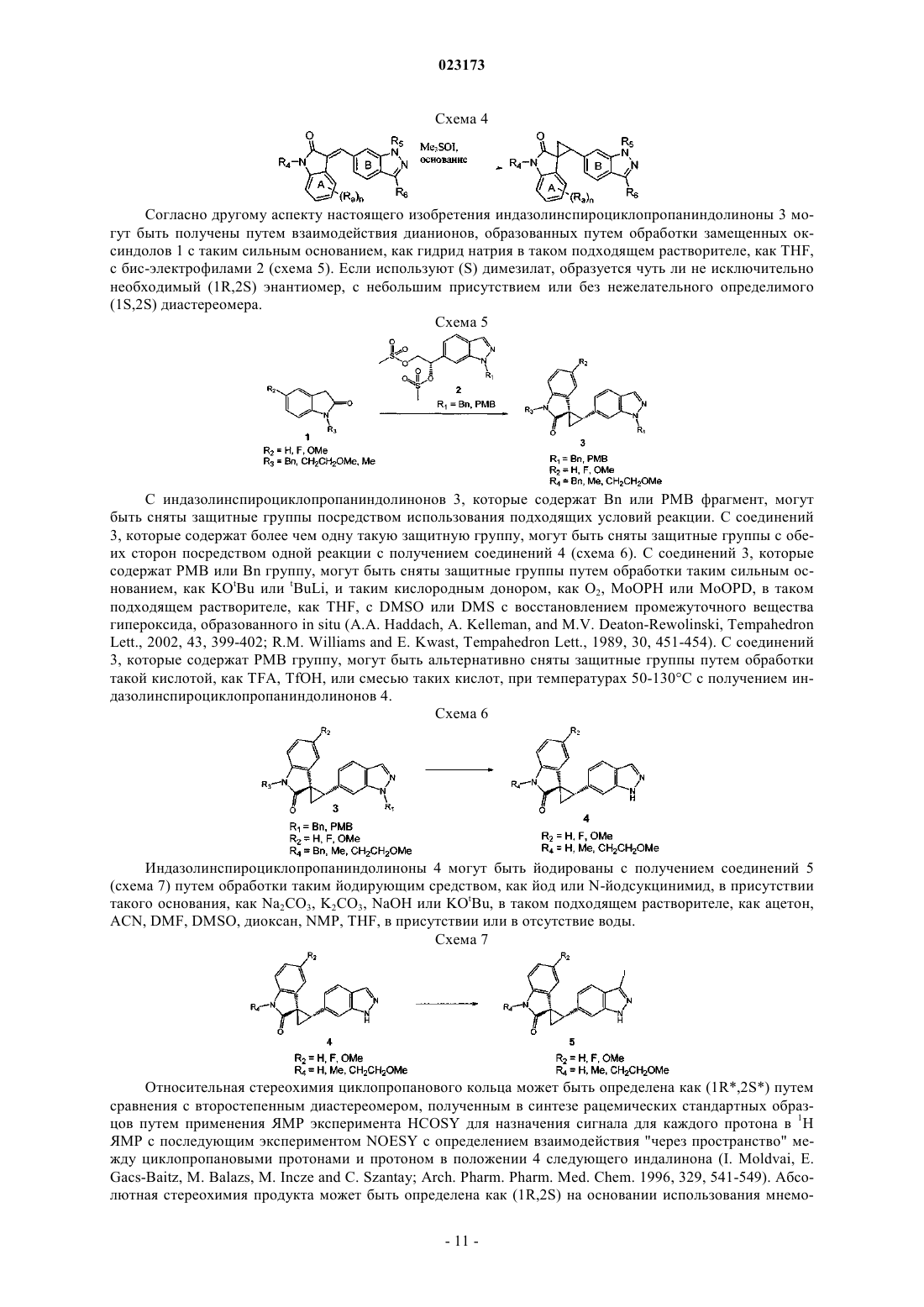
Ингибиторы киназы и способ лечения злокачественной опухоли с их помощью
Номер патента: 23173
Опубликовано: 29.04.2016
Авторы: Сампсон Питер Брент, Лауфер Радослав, Фехер Миклос, Пател Нарендра Кумар Б., Пан Гуохуа, Паулс Хайнц В., Ли Сцзе-Вань, Форрест Брайан Т., Лю Юн, Эдвардс Луис Г.





























Формула / Реферат
1. Соединение, представленное следующей структурной формулой:

или его фармацевтически приемлемая соль, где
Ra представляет собой -F, метокси, метил или этил;
R4 представляет собой H или метил и
R6 представляет собой -CH=CH-(необязательно замещенный фенил); где
фенил в -CH=CH-(фенил) является необязательно замещенным одним или несколькими заместителями, независимо выбранными из группы, состоящей из галогена, -(CH2)0-3-N-пиперидинила, -(CH2)0-3-N-морфолинила, -(CH2)0-3-N-пирролидинила, -(CH2)0-3-N-пиперазинила и -(CH2)0-3-N-оксазепанила, где N-пиперазинил необязательно N'-замещен C1-6 алкилом.
2. Соединение по п.1, причем соединение представлено следующей структурной формулой:

или его фармацевтически приемлемая соль.
3. Соединение по п.1, причем соединение представлено следующей структурной формулой:

или его фармацевтически приемлемая соль.
4. Соединение, представленное следующей структурной формулой:

или его фармацевтически приемлемая соль.
5. Соединение, представленное следующей структурной формулой:

или его фармацевтически приемлемая соль.
6. Фармацевтическая композиция для ингибирования polo-подобных киназ 4 (PLK4) и Aurora киназ, содержащая фармацевтически приемлемый носитель и соединение по любому из пп.1-5 или его фармацевтически приемлемую соль.
7. Способ лечения субъекта, страдающего злокачественной опухолью, включающий введение эффективного количества соединения по любому из пп.1-5 или его фармацевтически приемлемой соли.
8. Способ по п.7, при котором злокачественная опухоль выбрана из группы, состоящей из злокачественной опухоли легкого, злокачественной опухоли молочной железы, злокачественной опухоли толстой кишки, злокачественной опухоли мозга, нейробластомы, злокачественной опухоли предстательной железы, меланомы, полиморфной глиобластомы, злокачественной опухоли яичников, лимфомы, лейкоза, меланомы, саркомы, паранеоплазии, остеосаркомы, герминомы, глиомы и мезотелиомы.
9. Способ по п.8, при котором злокачественная опухоль выбрана из группы, состоящей из злокачественной опухоли легкого, злокачественной опухоли молочной железы или злокачественной опухоли толстой кишки.
10. Способ по п.9, при котором злокачественная опухоль представляет собой базальный подтип рака молочной железы или люминальный В подтип рака молочной железы.
11. Способ по п.10, при котором злокачественная опухоль представляет собой базальный подтип рака молочной железы, который избыточно экспрессирует PLK4.
12. Способ по п.9, при котором злокачественная опухоль представляет собой базальный подтип рака молочной железы, который представляет собой ER-, HER2- и PR-негативный рак молочной железы.
13. Способ по п.7, при котором злокачественная опухоль представляет собой злокачественную опухоль мягких тканей, представляющую собой саркому, выбранную из группы, состоящей из фибросаркомы, желудочно-кишечной саркомы, лейомиосаркомы, дедифференцированной липосаркомы, плеоморфной липосаркомы, злокачественной фиброзной гистиоцитомы, круглоклеточной саркомы и синовиальной саркомы.
Текст
ИНГИБИТОРЫ КИНАЗЫ И СПОСОБ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛИ С ИХ ПОМОЩЬЮ Изобретение относится к соединению, представленному следующей структурной формулой, и его фармацевтически приемлемым солям: Сампсон Питер Брент, Лю Юн, Ли Сцзе-Вань, Форрест Брайан Т., Паулс Хайнц В., Эдвардс Луис Г., Фехер Миклос, Пател Нарендра Кумар Б.,Лауфер Радослав, Пан Гуохуа (CA) Указанные соединения являются ингибиторами polo-подобных киназ 4 (PLK4) и Aurora киназ и могут быть использованы для лечения злокачественной опухоли. Ссылка на родственные заявки По настоящей заявке испрашивается приоритет на основании предварительных заявок на выдачу патента США 61/321332, поданной 6 апреля 2010 года и,61/321329, поданной 6 апреля 2010 года. По настоящей заявке также испрашивается приоритет на основании международной заявкиРСТ/СА 2010/000518, поданной 6 апреля 2010 года. Все содержание этих трех заявок включено в настоящий документ посредством ссылки. Уровень техники Протеинкиназы были объектом широкомасштабных исследований в ходе поиска новых терапевтических средств при разнообразных заболеваниях, например злокачественных опухолях. Известно, что протеинкиназы опосредуют внутриклеточную передачу сигналов путем осуществления переноса фосфорила с нуклеозидтрифосфата на белок-акцептор, т.е. участвуют в пути передачи сигнала. Существует ряд киназ и путей, посредством которых внеклеточные и другие стимулы вызывают возникновение разнообразных клеточных ответов внутри клетки. Семейство серин/треониновых polo-подобных киназ (PLK) содержит по меньшей мере четыре известных члена: PLK1, PLK2 (также известную как Snk), PLK3 (также известную как Fnk или Prk) и PLK4(также известную как Sak). PLK4 является наименее изученным и наиболее отличающимся членом семейства PLK. N-концевой каталитический домен PLK4 характеризуется субстратной специфичностью,отличающейся от таковой PLK1-3. PLK4 также характеризуется отличающимся С-концом, содержащим лишь единственную последовательность polo-box, а не тандемные последовательности РВ, как у PLK1-3,которая, видимо, действует, скорее, как домен гомодимеризации, нежели как домен локализации (Loweryet al., (2005) Oncogene 24: 248-259). Известно, что PLK4 вовлечена в контроль вхождения в митоз и выхода из него и является регулятором удвоения центросомы (Habedanck et al. Nature Cell Biology 7: 1140-1146, 2005). Содержание транскриптов PLK4 возрастает от S- к М-фазе, и белок убиквитинилируется и разрушается комплексом, стимулирующим анафазу, (АРС) (Hudson et al. Curr. Biol. 11: 441-446, 2001; Fode et al. Mol. Cell. Biol. 16: 4665-4672, 1996). PLK4 необходима для протекания конечных стадий митоза (Fode et al. PNAS. 91: 63886392, 1994; Hudson et al. Curr. Biol. 11: 441-446, 2001), выживания клетки и постгаструляционного эмбрионального развития (Hudson et al. Curr. Biol. 11: 441-446, 2001). Нокаутные по PLK4 мыши имели эмбриональную леталь (Е 7.5) с заметным увеличением количества митотических и апоптотических клеток(Hudson et al. Curr. Biol. 11: 441-446, 2001). PLK4 транскрипционно репрессируется p53 (Li et al. Neoplasia 7: 312-323, 2005). Эта репрессия, вероятно, опосредована рекрутингом гистондезацетилазных репрессоров (HDAC), и репрессия, очевидно, участвует в индуцируемом р 53 апоптозе (Li et al. Neoplasia 7: 312323, 2005). Сообщалось, что PLK4 избыточно экспрессируется в колоректальных опухолях при слабой экспрессии в прилегающей нормальной слизистой кишечника (Macmillian et al. Ann. Surg. Oncol. 8: 729-740,2001). К тому же сообщалось, что мРНК PLK4 избыточно экспрессируется в некоторых линиях опухолевых клеток (Hitoshi et al., заявка на выдачу патента США 2003/0027756). Кроме того, заявители описали избыточную экспрессию PLK4 в базальноподобных опухолях в совместно рассматриваемой предварительной заявке на выдачу патента США 61003825, поданной 20 ноября 2007 года (полное содержание которой включено в настоящий документ посредством ссылки). Сообщалось, что PLK4 избыточно экспрессируется в колоректальных опухолях при слабой экспрессии в прилегающей нормальной слизистой кишечника (Macmillian et al. Ann. Surg. Oncol. 8: 729-740,2001). К тому же сообщалось, что мРНК PLK4 избыточно экспрессируется в некоторых линиях опухолевых клеток (Hitoshi et al., заявка на выдачу патента США 2003/0027756). Кроме того, заявители описали избыточную экспрессию PLK4 в базальноподобных опухолях в совместно рассматриваемой предварительной заявке на выдачу патента США 61003825, поданной 20 ноября 2007 года (полное содержание которой включено в настоящий документ посредством ссылки). Онкобелки Е 7 вируса папилломы человека (HPV-16) избыточно экспрессируются при HPVассоциированном аногенитальном и ротоглоточном раке. Онкобелок Е 7 запускает чрезмерную дупликацию центросомы через путь, который включает одновременное образование множества дочерних центриолей на единичных материнских. Онкобелок Е 7 HPV-16 применяли в качестве инструмента для анализа аномального биогенеза центриолей, и имеются некоторые доказательства ключевой роли PLK4 в этом процессе (Duensing et al Environ. Mol. Mutagen. 50: 741-747, 2009). К тому же повышенный уровень транскрипции PLK4 обнаружен в кератиноцитах, устойчиво экспрессирующих Е 7 HPV-16. Было обнаружено, что способность Е 7 HPV-16 положительно регулировать транскрипцию мРНК PLK4 зависит от его способности разрушать ретинобластомный белок (pRb), что позволяет предположить роль опосредованной E2F генной транскрипции в дерегуляции PLK4 (Korzeniewski et al, AACR Meeting, Washington,2010, Abstr. 5354). Эти результаты позволяют идентифицировать PLK4 как мишень для ингибирования низкомолекулярным соединением для предотвращения центриольных аномалий, ошибок при митозе и злокачественной прогрессии HPV-ассоциированных злокачественных опухолей. Следовательно, средства, которые ингибируют протеинкиназу, в частности PLK4, обладают потенциалом для лечения злокачественных опухолей. Существует потребность в дополнительных средствах,-1 023173 которые могут действовать как ингибиторы протеинкиназы, в частности ингибиторы PLK4. Сущность изобретения Заявителями было открыто, что определенные спироциклопропилиндолиноновые соединения являются сильными ингибиторами киназ, таких как polo-подобные киназы 4 (PLK4) и киназы Aurora (см. пример B и F). Заявителями также было открыто, что эти спироциклопропилиндолиноновые соединения обладают сильной противораковой активностью (см. пример J) и проявляют антиангиогенную активность (пример K). На основании этих открытий в настоящем документе раскрыты спироциклопропилиндолиноновые соединения, их фармацевтические композиции и способы лечения злокачественной опухоли спироциклопропилиндолиноновыми соединениями. Согласно одному варианту осуществления настоящее изобретение относится к соединению, представленному структурной формулой или его фармацевтически приемлемой соли, гдеR6 представляет собой -CH=CH-(необязательно замещенный фенил); где фенил в -CH=CH-(фенил) является необязательно замещенным одним или несколькими заместителями, независимо выбранными из группы, состоящей из галогена, (CH2)0-3 N-пиперидинила, (CH2)0-3 N-морфолинила, (CH2)0-3 Nпирролидинила, (CH2)0-3 N-пиперазинила и -(CH2)0-3 N-оксазепанила, где N-пиперазинил необязательноN'-замещен C1-6 алкилом. В одном частном варианте изобретения соединение представляет собой соединение, характеризующееся следующей структурной формулой: или его фармацевтически приемлемую соль. В другом частном варианате соединение представлено следующими структурными формулами: или их фармацевтически приемлемыми солями. В следующем варианте изобретение относится к фармацевтической композиции для ингибированияpolo-подобных киназ 4 (PLK4) и Aurora киназ, содержащей фармацевтически приемлемый носитель и любое из вышеперечисленных соединений или фармацевтически приемлемую соль любого из указанных соединений. Изобретение также относится к способу лечения субъекта, страдающего злокачественной опухолью, включающему введение эффективного количества соединения по изобретению или его фармацевтически приемлемой соли. При этом злокачественная опухоль выбрана из группы, состоящей из злокачественной опухоли легкого, злокачественной опухоли молочной железы, злокачественной опухоли толстой кишки, злокачественной опухоли мозга, нейробластомы, злокачественной опухоли предстательной железы, меланомы,полиморфной глиобластомы, злокачественной опухоли яичников, лимфомы, лейкоза, меланомы, саркомы, паранеоплазии, остеосаркомы, герминомы, глиомы и мезотелиомы. Краткое описание чертежей На фигуре проиллюстрирован антиангиогенный эффект соединений А 23. Подробное описание изобретения Конкретные примеры соединений согласно настоящему изобретению включают в себя соединения,представленные в примерах ниже, их стереоизомеры и их фармацевтически приемлемые соли. В описанных в настоящем документе структурных формулах, если атом(ы) водород показан(ы) в конкретном(ых) положении(ях) ароматического(их) кольца(колец) структурной(ых) формулы(формул), в этим(этих) конкретном(ых) положении(ях) замещения не допускаются. Таутомерные формы существуют, если соединение представляет собой смесь двух или нескольких структурно несовпадающих соединений, которые приходят быстро в равновесие. Определенные соединения согласно настоящему изобретению существуют в таутомерных формах. Например, следующее соединение, которое охватывается структурной формулой (I), включает в себя, по меньшей мере, следующие таутомерные формы: Следует понимать, что, если одна таутомерная форма соединения представлена по названию или структуре, включены все таутомерные формы соединения. Соединения согласно настоящему изобретению содержат по меньшей мере два хиральных центра и циклопропан и, следовательно, существуют в качестве стереоизомеров, например изомеров циклопропанового типа (т.е. цис/транс изомеры), энантиомеров и/или диастереомеров. Если соединения согласно настоящему изобретению показаны или названы без указания стереохимии, следует понимать, что охватываются обе стереомерно чистые формы (например чистая цис или чистая транс, энантиомерно чистая или диастереомерно чистая) и стереоизомерные смеси. Например, соединения, представленные структурной формулой (I), содержат "цис" и "транс" изомеры, показанные ниже Кольцо А и кольцо В представляют собой транс-изомер. Формулировка "кольцо А и кольцо В представляет собой цис-изомер" означает, что и кольцо А, и кольцо В находятся на одной стороне циклопропана, тогда как формулировка "кольцо А и кольцо В представляет собой транс-изомер" означает, что кольцо А и кольцо В находятся на различных сторонах циклопропана. Стереоизомеры цис/транс модификации также называются геометрические изомеры. Соответственно, соединения согласно настоящему изобретению, показанные структурной формулой (I),включают чистый цис-изомер, чистый транс-изомер и их смеси, включая цис/транс смеси, обогащенные цис-геометрическим изомером, и цис/транс смеси, обогащенные транс-геометрическим изомером. Например, на структурной формуле (II) изображено цис взаимодействие между кольцом А и В. Также следует понимать, что и цис-, и транс-формы структурных формул (I) по отношению к кольцам А и В охватываются в пределах настоящего изобретения. Если геометрический изомер представлен по названию или структуре, следует понимать, что геометрическая изомерная чистота названного или показанного геометрического изомера, по меньшей мере,равна 60, 70, 80, 90, 99 или 99,9 мас.% чистоты. Геометрическая изомерная чистота определена путем разделения массы названного показанного геометрического изомера в смеси на общую массу обоих геометрических изомеров в смеси. Рацемическая смесь представляет собой 50% одного энантиомера и 50% его соответствующего энантиомера. Настоящее изобретение охватывает все энантиомерно чистые, энантиомерно обогащенные,диастереомерно чистые, диастереомерно обогащенные и рацемические смеси, и диастереомерные смеси соединений согласно настоящему изобретению. Энантиомерные и диастереомерные смеси могут быть повторно растворены в их составляющих энантиомерах или стереоизомерах хорошо известными способами, такими как газовая хроматографияс хиральной фазой, высокоэффективная жидкостная хроматография с хиральной фазой, кристаллизацией соединения в виде сложной хиральной соли или кристаллизацией соединения в хиральном растворителе. Энантиомеры и диастереомеры также могут быть получены из диастереомерно или энантиомерно чистых промежуточных соединений, реагентов и катализаторов хорошо известными ассиметричными способами синтеза. Если соединение определено названием или структурой, которая обозначает один энантиомер, если не указано иное, соединение является по меньшей мере на 60, 70, 80, 90, 99 или 99,9% оптически чистым(также называется "энантиомерно чистым"). Оптическая чистота равна массе в смеси названного или показанного энантиомера, разделенной на общую массу в смеси обоих энантиомеров. Если стереохимия раскрытого соединения названа или показана при помощи структуры, и названная или показанная структура охватывает более чем один стереооизомер (например, в диастереомерной паре), следует понимать,что включен один из охватываемых стереоизомеров или любая смесь охватываемых стереоизомеров. Кроме того, следует понимать, что стереоизомерная чистота названных или показанных стереоизомеров,по меньшей мере, равна 60, 70, 80, 90, 99 или 99,9 мас.%. Стереоизомерная чистота в данном случае определена делением общей массы в смеси стереоизомеров, охватываемых названием или структурой, на общую массу в смеси всех стереоизомеров. В настоящее изобретение включены фармацевтически приемлемые соли раскрытых в настоящем документе соединений. Раскрытые соединения содержат основные аминовые группы и, следовательно,могут образовывать фармацевтически приемлемые соли с фармацевтически приемлемой(ыми) кислотой(амии). Подходящие фармацевтически приемлемые кислотно-аддитивные соли соединений согласно настоящему изобретению содержат соли неорганических кислот (таких как хлористоводородная кислота,бромистоводородная, фосфорная, метафосфорная, азотная и серная кислоты) и органических кислот (таких как уксусная кислота, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изэтиновая, молочная. лактобионовая, малеиновая, яблочная, метансульфоновая,янтарная, р-толуолсульфоновая и винная кислоты). Соединения согласно настоящему изобретению с кислотными группами, такие как карбоновые кислоты, могут образовывать фармацевтически приемлемые соли с фармацевтически приемлемым(и) основанием(ями). Подходящие фармацевтически приемлемые основные соли включают соли аммония, соли щелочных металлов (такие как соли натрия и калия) и соли щелочно-земельных металлов (такие как соли магния и кальция). Соединения с четвертичной аммонийной группой также содержат такой противоион, как хлорид, бромид, йодид, ацетат, перхлорат и т.п. Другие примеры таких солей включают гидрохлориды, гидробромиды, сульфаты, метансульфонаты,нитраты, малеаты, ацетаты, цитраты, фумараты, тартраты [например, (+)-тартраты. (-)-тартраты или их смеси, включая рацемические смеси], сукцинаты, бензоаты и соли с аминокислотами, такими как глутаминовая кислота. Используемый в настоящем документе термин "галоген" означает галоген и включает хлор, фтор,бром и йод."Алифатическая группа" является ациклической, неароматической, состоит только из углерода и водорода и может необязательно содержать один или несколько элементов ненасыщения, например двойные и/или тройные связи. Алифатическая группа может быть с неразветвленной цепью или с разветвленной. Алифатическая группа типично содержит от приблизительно одного до приблизительно двадцати атомов углерода, типично от приблизительно одного до приблизительно десяти атомов углерода, более типично от приблизительно одного до приблизительно шести атомов углерода. "Замещенная алифатическая группа" является замещенной на любом одном или нескольких "подходящих атомах углерода". "Подходящий атом углерода" в алифатической группе представляет собой углерод в алифатической группе, которая связана с одним или несколькими атомами водорода Один или несколько атомов водорода могут быть необязательно замещены подходящей группой заместителей."Галогеналифатическая группа" также является определенной выше алифатической группой, замещенной одним или несколькими атомами галогена. Используемый отдельно или как часть большего фрагмента термин "алкил", такой как "алкокси","галогеналкил", "арилалкил", "алкиламин", "диалкиламин", "алкиламино", "диалкиламино" "алкилкарбонил", "алкоксикарбонил" и т.п., означает насыщенную с неразветвленной или разветвленной цепью алифатическую группу. Используемая настоящем документе C1-C6 алкильная группа относится к "низшему алкилу". Подобным образом, выражения "низший алкокси", "низший галогеналкил", "низший арилалкил", "низший алкиламин", "низший диалкиламин", "низший алкиламино", "низший диалкиламино""низший алкилкарбонил", "низший алкоксикарбонил", включают в себя неразветвленные и разветвленные, насыщенные цепочки, содержащие от одного до шести атомов углерода. Термин "алкенил" означает алифатическую группу с неразветвленной или разветвленной цепью,содержащей по меньшей мере одну двойную связь. Термин "алкинил" означает алифатическую группу с неразветвленной или разветвленной цепью,содержащей по меньшей мере одну тройную связь. Термин "алкокси" означает -О-алкил; "гидроксиалкил" означает алкил, замещенный гидрокси;"аралкил" означает алкил, замещенный арильной группой; "алкоксиалкил" означает алкил, замещенный алкоксигруппой; "алкиламин" означает амин, замещенный алкильной группой; "циклоалкилалкил" означает алкил, замещенный циклоалкилом; "диалкиламин" означает амин, замещенный двумя алкильными группами; "алкилкарбонил" означает -C(O)-R, где R представляет собой алкил; "алкоксикарбонил" означает -C(O)-OR, где R представляет собой алкил; и где алкил определен выше. Термины "галогеналкил" и "галогеналкокси" означает алкил или алкокси, в зависимости от обстоятельств, который может быть замещен одним или несколькими атомами галогена. Термин "галоген" означает F, Cl, Br или I. Предпочтительно, галоген в галогеналкиле или галогеналкокси представляет собойF. Термин "ацильная группа" означает -C(O)R, где R представляет собой необязательно замещенную алкильную группу или арильную группу (например, необязательно замещенную фенилом). R предпочтительно представляет собой незамещенную алкильную группу или фенил."Алкиленовая группа" представлена -[CH2]z-, где z представляет собой положительное целое число,предпочтительно от одного до восьми, более предпочтительно от одного до четырех."Алкенилен" представляет собой алкиленовую группу, в которой один метилен был замещен двойной связью. Используемый отдельно или как часть большего фрагмента термин "арильная группа", как в "аралкиле", "аралкокси" или "арилоксиалкил", означает карбоциклическое ароматическое кольцо. Термин"арил" может быть использован взаимозаменяемо с терминами "арильное кольцо" "карбоциклическое ароматическое кольцо", "арильная группа" и "карбоциклическая ароматическая группа". Арильная группа типично содержит от шести до четырнадцати кольцевых атомов. Примеры включают в себя фенил,нафтил, антраценил, 1,2-дигидронафтил, 1,2,3,4-тетрагидронафтил, фторенил, инданил, инденил и т.п."Замещенная арильная группа" замещена на любом одном или нескольких подходящих атомах углерода,которые представляют собой кольцевой атом углерода, связанный с водородом. Термин "циклоалкил" относится к моноциклической или полициклической насыщенной углеводородной кольцевой системе. Например, С 5-7 циклоалкил включает в себя без ограничения циклопентил,циклогексил или циклопентил, каждый из которых необязательно замещенный. Используемый отдельно или как часть большего фрагмента термин "гетероарил", "гетероароматический", "гетероарильное кольцо", "гетероарильная группа", "гетероароматическое кольцо" и "гетероароматическая группа", как в "гетероаралкиле" или "гетероарилалкокси", относится к ароматическим кольцевым группам, содержащим от пяти до четырнадцати кольцевых атомов, выбранных из углерода и по меньшей мере одного (типично, 1-4, более типично, 1 или 2) из гетероатомов (например, кислорода,азота или серы). "Гетероарил" включает в себя моноциклические кольца и полициклические кольца, в которых моноциклическое гетероароматическое кольцо конденсировано с одним или несколькими другими карбоциклическими ароматическими или гетероароматическими кольцами. По сути, "5-14-членный гетероарил" включает в себя моноциклические, бициклические или трициклические кольцевые системы. Примеры моноциклический 5-6-членных гетероарильных групп включают в себя фуранил (например, 2-фуранил, 3-фуранил), имидазолил (например, N-имидазолил, 2-имидазолил, 4-имидазолил, 5 имидазолил), изоксазолил (например, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил), оксадиазолил (например, 2-оксадиазолил, 5-оксадиазолил), оксазолил (например, 2-оксазолил, 4-оксазолил, 5-оксазолил),пиразолил (например, 3-пиразолил, 4-пиразолил), пирролил (например, 1-пирролил, 2- пирролил, 3-5 023173 пирролил), пиридил (например, 2-пиридил, 3-пиридил, 4-пиридил), пиримидинил (например, 2 пиримидинил, 4-пиримидинил, 5-пиримидинил), пиридазинил (например, 3-пиридазинил), тиазолил (например, 2-тиазолил, 4-тиазолил, 5-тиазолил), триазолил (например, 2-триазолил, 5-триазолил), тетразолил (например, тетразолил), тиенил (например, 2-тиенил, 3-тиенил), пиримидинил, пиридинил и пиридазинил. Примеры полициклических ароматических гетероарильных групп включают в себя карбазолил,бензимидазолил, бензотиенил, бензофуранил, индолил, хинолинил, бензотриазолил, бензотиазолил, бензоксазолил, бензимидазолил, изохинолинил, индолил, изоиндолил, акридинил или бензизоксазолил. "Замещенная гетероарильная группа" замещена на любом одном или нескольких подходящих кольцевых атомах, которые представляют собой кольцевой углеродный или кольцевой азотный атом, связанный с водородом. Термин "гетероциклильная группа" или "гетероциклическая группа" означает моноциклическое,неароматическое кольцо с 3-10-членами, содержащее 1-3 кольцевых гетероатома, или полициклическое кольцо с кольцом с 7-20-членами и 1-4 кольцевыми гетероатомами, где полициклическое кольцо содержит один или несколько моноциклических неароматических нетероциклических колец, конденсированных с одним или несколькими ароматическими или гетероароматическими кольцами. Согласно одному варианту осуществления гетероциклильная группа представляет собой бициклическое кольцо, содержащее моноциклическое неароматическое гетероциклическое кольцо, конденсированное с фенильной группой. Для иллюстрации полициклическая гетероциклическая группа включает в себя тетрагидроизохинолинил (такой как 1,2,3,4-тетрагидроизохинолин-7-ил, 2-метил-1,2,3,4-тетрагидроизохинолин-7-ил,1,2,3,4-тетрагидроизохинолин-6-ил и 2-метил-1,2,3,4-тетрагидроизохинолин-6-ил), изоиндолинил (такой как 2-этилизоиндолин-5-ил, 2-метилизоиндолин-5-ил), индолинил, тетрагидробензо[f]оксазепинил (такой как 2,3,4,5-тетрагидробензо[f][1,4]оксазепин-7-ил). Термин "неароматическая гетероциклическая группа" означает моноциклическое, неароматическоое кольцо с 3-10-членами, содержащее 1-3 кольцевых гетероатома, или полициклическое неароматическое кольцо с 7-20 членами и с 1-4 кольцевыми гетероатомами. Каждый гетероатом независимо выбран из азота, четвертичного азота, окисленного азота (например, NO); кислорода и серы, включая в себя сульфоксид и сульфон. Замещенная неароматическая гетероциклическая группа может быть присоединена через подходящий гетероатом или атом углерода Иллюстративные неароматические гетероциклические группы включают в себя морфолинил, тиоморфолинил, пирролидинонил, пирролидинил, пиперидинил, пиперазинил, гидантоинил. валеролактамил, оксиранил, оксэтанил, тетрагидрофуранил, тетрагидропиранил, тетрагидропириндинил, тетрагидропиримидинил, тетрагидротиофенил, тетрагидротиоруганил и т.п. "Замещенная неароматическая гетероциклическая группа" замещена на любом одном или нескольких подходящих кольцевых атомах, которые представляют собой кольцевой углеродный или кольцевой азотный атом, связанный с водородом. Спироциклопропилиндолиноновые соединения согласно настоящему изобретению могут ингибировать различные киназы, включая PLK4, PLK1, PLK2, Aurora A, Aurora В и FLT-3 (см. примеры B-G). Таким образом, обычно спироциклопропилиндолиноновые соединения согласно настоящему изобретению применимы при лечении связанных с такими киназами заболеваний или состояний. Например, полагали, что PLK4, PLK1, Aurora А и Aurora В были включены в клеточное миотическое прогрессирование. Таким образом, низкомолекулярные ингибиторы этих ферментов могут быть сильными противоопухолевыми агентами. Согласно конкретному варианту осуществления соединения согласно настоящему изобретению представляют собой PLK, Aurora A, Aurora В и/или FLT-3 ингибиторы, и они применимы для лечения таких заболеваний, как злокачественная опухоль, связанная с такой(такими) киназой(ами). Согласно другому конкретному варианту осуществления соединения согласно настоящему изобретению представляют собой PLK ингибиторы и они применимы для лечения заболеваний, связанных с PLK, таких как злокачественная опухоль. Типично, PLK представляет собой PLK4, PLK2 и PLK1. В одном примере PLK представляет собой PLK1 и PLK4. В другом примере PLK представляет собой PLK4. Согласно другому конкретному варианту осуществления соединения согласно настоящему изобретению представляют собой ингибиторы Aurora А и/или В, и они применимы при ингибировании активности Aurora А и/или В для лечения таких различных состояний, как злокачественные опухоли. Согласно другому конкретному варианту осуществления соединения согласно настоящему изобретению представляют собой FLT-3 ингибиторы и являются применимыми при ингибировании активности FLT-3 для лечения таких различных состояний, как злокачественные опухоли. Согласно другому аспекту настоящее изобретение относится к способу лечения субъекта, страдающего злокачественной опухолью, предусматривающий введение субъекту эффективного количества соединения согласно настоящему изобретению. Согласно одному варианту осуществления соединения согласно настоящему изобретению ингибируют рост опухоли. В частности, соединения согласно настоящему изобретению ингибируют рост опухоли, которая избыточно экспрессирует по меньшей мере одно из PLK, Aurora A, Aurora В и FLT-3. Более конкретно, соединения согласно настоящему изобретению ингибируют рост опухоли, которая избыточно экспрессирует PLK, например, PLK1, PLK2 и/или PLK4. Даже более конкретно, соединения согласно настоящему изобретению ингибируют рост опухоли, кото-6 023173 рая избыточно экспрессирует PLK4. Согласно другому варианту осуществления соединения согласно настоящему изобретению ингибируют рост опухоли путем индуцирования апоптоза опухолевых клеток или путем ингибирования пролиферации опухолевых клеток. Злокачественные опухоли, которые можно лечить или предупреждать при помощи способов согласно настоящему изобретению, включают в себя злокачественную опухоль легкого, злокачественную опухоль молочной железы, злокачественную опухоль толстой кишки, злокачественную опухоль мозга, нейробластому, злокачественную опухоль предстательной железы, меланому, полиморфную глиобластому, злокачественную опухоль яичников, лимфому, лейкоз, меланому, саркому, паранеоплазию, остеосаркому, герминому, глиому и мезотелиому. Согласно одному конкретному варианту осуществления злокачественная опухоль представляет собой злокачественную опухоль легкого, злокачественную опухоль толстой кишки, злокачественную опухоль мозга, нейробластому, злокачественную опухоль предстательной железы, меланому, полиморфную глиобластому или злокачественную опухоль яичников. Согласно другому конкретному варианту осуществления злокачественная опухоль представляет собой злокачественную опухоль легкого, злокачественную опухоль молочной железы, злокачественную опухоль толстой кишки, злокачественную опухоль мозга, нейробластому, злокачественную опухоль предстательной железы, меланому, полиморфную глиобластому или злокачественную опухоль яичников. Согласно другому конкретному варианту осуществления злокачественная опухоль представляет собой злокачественную опухоль молочной железы. Согласно другому конкретному варианту осуществления злокачественная опухоль представляет собой базальный подтип рака молочной железы или люминальный В подтип рака молочной железы. Согласно одному варианту осуществления базальный подтип рака молочной железы представляет собой ER (рецептор эстрогена),HER2 и PR (прогестероновый рецептор) негативный рак молочной железы. Согласно другому конкретному варианту осуществления злокачественная опухоль представляет собой злокачественную опухоль мягких тканей. "Злокачественная опухоль мягких тканей" представляет собой признанный в настоящей области техники термин, который охватывает опухоли, полученные из любой мягкой ткани организма. Такие мягкие ткани соединяют, поддерживают или окружают различные структуры и органы организма,включая без ограничения гладкие мышцы, скелетные мышцы, сухожилия, волокнистые ткани, жировые ткани, кровеносные и лимфатические сосуды, периваскулярные ткани, нервы, мезенхимальные клетки и синовиальные ткани. Таким образом, злокачественная опухоль мягких тканей может представлять собой злокачественную опухоль жировой ткани, мышечной ткани, нервной ткани, соединительной ткани, кровеносных сосудов, лимфатических сосудов и волокнистых тканей. Злокачественные опухоли мягких тканей могут характеризоваться более доброкачественным или более злокачественным течением. Обычно,злокачественную опухоль мягких тканей более злокачественного течения называют саркомой, или саркомой мягких тканей. Существует много типов опухолей мягких тканей, которые включают в себя липому, липосаркому, гиберному, липосаркому, лейомиому, лейомиосаркому, рабдомиому, рабдомиосаркому, нейрофиброму, шванному (невриному), неврому, злокачественную шванному, нейрофибросаркому,неврогенную саркому, узловатый тендосиновит, синовиальную саркому, гемангиому, гломусную опухоль, гемангиоперицитому, гемангиоэндотелиому, ангиосаркому, саркому Капоши, лимфангиому, фиброму, эластофиброму, поверхностный фиброматоз, фиброзную гистиоцитоксантому, фибросаркому,фиброматоз, возвышающуюся дерматофибросаркому (DFSP), злокачественную фиброзную гистиоцитоксантому (MFH), миксому, зернисто-клеточную миобластому, злокачественную мезенхимому, альвеолярную саркому мягких тканей, эпителиоидную саркому, саркому главной клетки и десмопластическую мелкоклеточную опухоль. Согласно конкретному варианту осуществления злокачественная опухоль мягких тканей представляет собой саркому, выбранную из группы, состоящей из фибросаркомы, желудочно-кишечной саркомы, лейомиосаркомы, дедифференцированной липосаркомы, плеоморфной липосаркомы, злокачественной фиброзной гистиоцитомы, круглоклеточной саркомы и синовиальной саркомы. Кроме того, настоящее изобретение относится к способу лечения субъекта с опухолевыми клетками, предусматривающему введение субъекту количество раскрытого в настоящем документе соединения, которое эффективно для эффективного снижения активности PLK, например активности PLK 2 илиPLK4, у субъекта. Согласно специфическому варианту осуществления PLK представляет собой PLK4. Термин "эффективное количество" означает введенное количество субъекту, которое приводит к благоприятным или необходимым результатам, включая клинические результаты, например снижение вероятности развития злокачественно опухоли или ингибирование, подавление или ослабление злокачественной опухоли (например, как определено клиническими симптомами или количеством злокачественных клеток) у субъекта по сравнению с контролем. В частности, "лечение субъекта, страдающего злокачественной опухолью" предусматривает обеспечение, частично или в значительной степени, одного или более из следующего: подавление роста или распространения злокачественной опухоли, снижение степени выраженности злокачественной опухоли (например, уменьшение размера опухоли или снижение количества пораженных участков), ингибирование скорости роста злокачественной опухоли и улучшение или усовершенствование клинического симптома или показателя, связанного со злокачественной опухолью(например, компоненты ткани или сыворотки). Это также понижает вероятность повторения злокачественной опухоли. Обычно, эффективное количество соединения согласно настоящему изобретению изме-7 023173 няется в зависимости от различных факторов, таких как данное лекарственное средство или соединение,фармацевтический состав, путь введения, тип заболевания или нарушения, идентичностьи субъекта или хозяина, которых лечили и т.п., но может тем не менее обычно определяться специалистом настоящей области. Эффективное количество соединения согласно настоящему изобретению может быть легко определено одним из специалистов обычными способами, известными в настоящем уровне техники. Согласно варианту осуществления эффективное количество соединение настоящего изобретения находится в диапазоне от 0,01 до приблизительно 1000 мг/кг на массу тела, альтернативно от приблизительно 0,05 до приблизительно 500 мг/кг на массу тела, альтернативно от приблизительно 0,1 до приблизительно 100 мг/кг на массу тела, альтернативно от приблизительно 0,1 до приблизительно 15 мг/кг на массу тела, альтернативно от приблизительно 1 до приблизительно 5 мг/кг на массу тела и согласно другому альтернативному варианту от приблизительно 2 до приблизительно 3 мг/кг на массу тела. Специалист настоящей области отметит, что определенные факторы могут воздействовать на дозировку, необходимую для эффективного лечения субъекта, который страдает от злокачественной опухоли, и эти факторы включают без ограничения серьезность заболевания или нарушения, предшествующее лечение,общее состояние здоровья и/или возраст субъекта и другие присутствующие заболевания. Более того,режим "лечения" субъекта эффективным количеством соединения согласно настоящему изобретению может состоять из одного введения или альтернативно содержать серию применений. Например, соединение согласно настоящему изобретению может быть введено по меньшей мере раз в неделю. Однако согласно другому варианту осуществления соединение может быть введено субъекту от приблизительно одного раза в неделю до одного раза в день для данного лечения. Длительность периода лечения зависит от разнообразных факторов, таких как серьезность заболевания, возраст пациента, концентрация и активность соединения согласно настоящему изобретению или их комбинации. Также будет отмечено, что эффективная дозировка соединения, используемого для лечения или профилактики, может повышаться или понижаться в течение курса особого лечения или профилактического режима. В результате могут быть изменения в дозировке и они могут становиться очевидными при помощи стандартных диагностических анализов, известных в настоящем уровне техники. В некоторых случаях может быть необходимо постоянное введение. Используемый в настоящем документе термин "лечение" является подходом для получения благоприятных или необходимых результатов, включая в себя клинические результаты. Благоприятные или необходимые клинические результаты могут включать без ограничения смягчение или улучшение состояния одного или нескольких симптомов или состояний, сокращение степени заболевания, стабилизацию (т.е. не ухудшение) состояния заболевания, сокращение вероятности распространения заболевания,задержку или замедление развития заболевания, улучшение состояния или временное ослабление состояния заболевания и ремиссию (или частичную, или полную), где определимо или не определимо. "Лечение" также может означать более длительное жизнеобеспечение по сравнению с ожидаемым жизнеобеспечением, если нет лечения. "Лечение" также включает снижение вероятности развития заболевания или снижение вероятности повторения заболевания."Субъект" представляет собой млекопитающее, предпочтительно человека, но также может быть животным при потребности ветеринарного лечения, например домашние животные (например, собаки,кошки и т.п.), сельскохозяйственные животное (например, коровы, овцы, свиньи, лошади и т.п.) и лабораторные животные (например, крысы, мыши, морские свинки и т.п.). Согласно одному варианту осуществления способ согласно настоящему изобретению представляет собой монотерапию, при которой фармацевтические композиции согласно настоящему изобретению вводят отдельно. Соответственно, согласно настоящему варианту осуществления соединение согласно настоящему изобретению является только фармацевтически активным ингредиентом в фармацевтических композициях или только фармацевтически активным ингредиентом, введенным субъекту. Согласно другому варианту осуществления способ согласно настоящему изобретению представляет собой совместную терапию одним или несколькими другими терапевтически активными лекарственными средствами или терапии, известные в настоящем уровне техники для лечения требуемых заболеваний или признаков. В одном примере одну или несколько других антипролиферативных или противораковых терапий объединяли с соединениями согласно настоящему изобретению. В другом примере раскрытые в настоящем документе соединения совместно вводили с одним или несколькими другими противораковыми лекарственными средствами, известными в настоящем уровне техники. Противораковые терапии,которые могут быть использованы в комбинации с соединением согласно настоящему изобретению,включают в себя хирургию, радиотерапию (включая без ограничения гамма-излучение, нейтроннолучевую радиотерапию, электронно-лучевую радиотерапию, протонную терапию, брахитерапию и системные радиоактивные изотопы) и терапию препаратами желез внутренней секреции. Противораковые средства, которые могут быть использованы в комбинации с соединениями согласно настоящему изобретению, включают в себя модификаторы биологического отклика (включая без ограничения интерфероны, интерлейкины и фактор некроза опухолей (TNF, гипертермию и криотерапию, средства для уменьшения любых неблагоприятных эффектов (например, противорвотные средства) и другие одобренные химиотерапевтические лекарственные средства (например, таксол и его аналоги). Если соединения согласно настоящему изобретению объединены с другими противораковыми лекарственными средствами, они могут быть введены одновременно. Используемый в настоящем документе термин "одновременно введенные" означает, что два вещества вводят субъекту так, чтобы они оба были биологически активными у субъекта в одно и то же время. Точные подробности введения будут зависеть от фармакокинетики двух веществ в присутствии друг друга и могут включать введение одного вещества за период времени друг друга, например 24 ч введения другого, если фармакокинетика является подходящей. Структура подходящих режимов дозировки является типичной для специалиста настоящей области. Согласно конкретным вариантам осуществления два вещества будут введены в основном одновременно, т.е. в пределах минут друг за другом, или в одной композиции, которая содержит оба вещества. Альтернативно, два средства могут быть введены отдельно, так чтобы у субъекта в одно и то же время присутствовало только одно биологически активное средство. Соединения согласно настоящему изобретению могут быть введены пациенту в разнообразных формах в зависимости от выбранного пути введения, как будет понятно специалистам настоящего уровня техники. Соединения согласно настоящему изобретению могут быть введены, например, пероральным, парентеральным, буккальным, сублингвальным, назальным, ректальным, через пластырь, насос или трансдермальным введением, и фармацевтические композиции составлены соответственно. Парентеральное введение включает внутривенный, внутрибрюшинный, подкожный, внутримышечный, трансэпителиальный, назальный, внутрилегочный, интратекальный, ректальный и местный пути введения. Парентеральное введение может быть при помощи непрерывной инфузии за выбранный период времени. Соединения согласно настоящему изобретению могут быть подходяще сформулированы в фармацевтические композиции для введения субъекту. Фармацевтические композиции согласно настоящему изобретению необязательно включают один или несколько фармацевтически приемлемых носителей и/или их разбавителей, такие как лактоза, крахмал, целлюлоза и декстроза Другиенаполнители, такие как ароматизирующие средства, подсластители и консерванты, такие как метил, этил, пропил и бутилпарабены, также могут быть включены. Более полные перечни подходящих наполнителей могут быть обнаружены в Handbook of Pharmaceutical Excipients (5th Ed., Pharmaceutical Press (2005. Специалист настоящего уровня техники будет понимать, как получить составы, подходящие для различных типов путей введения. Обычные методики и ингредиенты для выбора и получения подходящих составов описаны,например, в Remington's Pharmaceutical Sciences (2003 - 20 е издание) и в The United States Pharmacopeia:The National Formulary (USP 24 NF19), опубликованном в 1999. Носители, разбавители и/или наполнители "приемлемы" в смысле совмещения с другими ингредиентами фармацевтической композиции и не вредны для получателя. Типично, для перорального терапевтического введения соединение согласно настоящему изобретению может быть включено с инертным наполнителем и применено в форме проглатываемых таблеток,буккальных таблеток, лепешек, капсул, эликсиров, суспензии, сиропов, облаток и т.п. Типично, растворы соединения для паретнерального введения согласно настоящему изобретению обычно могут быть получены в воде, подходяще смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии также могут быть получены в глицерине, жидких полиэтиленгликолях, DMSO и их смесях с или без спирта, и в маслах. При обычных условиях хранения и применения эти препараты содержат консервант для предотвращения роста микроорганизмов. Типично, для инъецируемого применения используют стерильные водные растворы или дисперсии, и стерильные порошки, соединений согласно настоящему изобретению для непредусмотренного получения стерильных инъецируемых растворов или дисперсий. Для назального введения соединения согласно настоящему изобретению могут быть составлены в виде аэрозолей, капель, гелей и порошков. Аэрозольные составы типично содержат раствор или высокодисперсную суспензию активного вещества в физиологически приемлемом водном или неводном растворителе, и они обычно представлены в количествах одной дозы или многих дозах в стерильной форме в герметизированном контейнере, который может принимать форму картриджа или повторно заполняться для применения с распыляющим устройством. Альтернативно, герметизированный контейнер может быть таким цельным устройством для распределения, как назальный ингалятор с однократной дозой или аэрозольный прибор для распыления, оборудованный дозировочным клапаном, который положено удалить после применения. Если лекарственная форма содержит аэрозольный прибор для распыления, она будет содержать пропеллент, который может представлять собой сжатый газ, такой как сжатый воздух,или органический пропеллент, такой как фторхлоргидроуглерод. Аэрозольные лекарственные формы также могут принимать форму атомизатора с насосом. Для буккального или сублингвального введения соединения согласно настоящему изобретению могут быть составлены с таким носителем, как сахар, гуммиарабик, трагакант или желатин и глицерин, в виде таблеток, лепешек или пастилок. Для ректального введения соединения согласно настоящему изобретению могут быть составлены в форме суппозиториев, содержащих обычное основание суппозтория,такое как масло какао. Соединения согласно настоящему изобретению могут быть составлены отдельно или для одновременного введения с другими средствами для лечения злокачественной опухоли. Следовательно, согласно другому аспекту фармацевтическая композиция согласно настоящему изобретению содержит фармацевтически приемлемый носитель или разбавитель, раскрытое в настоящем документе соединение или его фармацевтически приемлемую соль, и другое противораковое средство, например без ограничения ингибитор метаболизма глюкозы или таксол. В соответствии с другим аспектом настоящего изобретения соединения согласно настоящему изобретению могут быть получены способом, аналогичным установленным в настоящем уровне техники способам. С целью иллюстрации соединения формулы (I), где кольца А и В таковы, как определено в настоящем документе, могут быть получены способами, отмеченными в схеме 1. Соответствующе замещенный индазолилметилениндолинон 1 (где кольцо А определено в настоящем документе и кольцо В и С вместе представляют собой индазол) взаимодействовал с таким подходящим метиленовым источником, как триметилсульфониййодид или триметилсульфоксониййодид в присутствии основания (такого как гидрид натрия, LDA или NaHMDS), в полярном растворителе (таком как DMF, THF или DMSO). Взаимодействие обычно выполняют при подходящей температуре (обычно в диапазоне от 20 до 60C). При типичных условиях реакции Хека может быть установлена виниловая связь, где Ar представляет собой определенную в настоящем документе фенильную и гетероарильную группу, и Р 1 представляет собой подходящую индазольную защитную группу (такую как Boc, ацетил или SEM), и X представляет собой галогенид. Альтернативно, при типичных условиях реакции Сузуки она может быть установлена с применением или бороновой кислоты, или боронатного сложного эфира, где Ar представляет собой определенную выше фенильную и гетероарильную группу, и Р 1 представляет собой подходящую индазольную защитную группу (такой как Boc, ацетил или SEM), и X представляет собой галогенид. Взаимодействие обычно выполняют при условиях микроволнового облучения, преимущественно в диапазоне, например, от 100 до 150C, или обычно при приблизительно 120C. Удаление защитной группы могут выполнять при помощи любой известной методики для выполнения такого преобразования. Например, если защитная группа Р 1 представляет собой SEM, преобразование может быть выполнено путем обработки фторидом тетрабутиламмония в таком полярном растворителе, как THF, с обратным холодильником, или путем постадийной обработки эфиратом трехфтористого бора и 2 н HCl в этаноле. Схема 1 Согласно другому аспекту настоящего изобретения (схема 2), арильная связь, где R6 таково, как определено в настоящем документе, Р 1 представляет собой или Н, или подходящую индазольную защитную группу (такую как Boc, ацетил или SEM) и X представляет собой галогенид, может быть установлена при типичных условиях реакции Сузуки с применением или бороновой кислоты, или боронатного сложного эфира. Взаимодействие обычно выполняют при условиях микроволнового облучения, преимущественно в диапазоне, например, от 100 до 150C или обычно при приблизительно 120C. Удаление защитной группы могут выполнять при помощи любой известной методики для выполнения такого преобразования. Например, если защитная группа Р 1 представляет собой SEM, преобразование может быть выполнено путем обработки фторидом тетрабутиламмония в таком полярном растворителе, как THF, с обратным холодильником, или путем постадийной обработки эфиратом трехфтористого бора и 2 н. HCl в этаноле. Схема 2 Схема 3. (Опущена). Согласно другому аспекту настоящего изобретения (схема 4) циклопропанирование может быть выполнено на таких соединениях, как (I), где R5 определено в настоящем документе, и R6 определено в настоящем документе путем осуществления взаимодействия такого подходящего метиленового источника, как триметилсульфониййодид или триметилсульфоксониййодид в присутствии основания (такого как гидрид натрия, LDA или NaHMDS), в полярном растворителе (таком как DMF, THF или DMSO). Взаимодействие обычно выполняли при подходящей температуре (обычно в диапазоне от 20 до 60C). Согласно другому аспекту настоящего изобретения индазолинспироциклопропаниндолиноны 3 могут быть получены путем взаимодействия дианионов, образованных путем обработки замещенных оксиндолов 1 с таким сильным основанием, как гидрид натрия в таком подходящем растворителе, как THF,с бис-электрофилами 2 (схема 5). Если используют (S) димезилат, образуется чуть ли не исключительно необходимый (1R,2S) энантиомер, с небольшим присутствием или без нежелательного определимого С индазолинспироциклопропаниндолинонов 3, которые содержат Bn или РМВ фрагмент, могут быть сняты защитные группы посредством использования подходящих условий реакции. С соединений 3, которые содержат более чем одну такую защитную группу, могут быть сняты защитные группы с обеих сторон посредством одной реакции с получением соединений 4 (схема 6). С соединений 3, которые содержат РМВ или Bn группу, могут быть сняты защитные группы путем обработки таким сильным основанием, как KOtBu или tBuLi, и таким кислородным донором, как O2, МоОРН или MoOPD, в таком подходящем растворителе, как THF, с DMSO или DMS с восстановлением промежуточного вещества гипероксида, образованного in situ (A.A. Haddach, A. Kelleman, and M.V. Deaton-Rewolinski, TempahedronLett., 2002, 43, 399-402; R.M. Williams and E. Kwast, Tempahedron Lett., 1989, 30, 451-454). С соединений 3, которые содержат РМВ группу, могут быть альтернативно сняты защитные группы путем обработки такой кислотой, как TFA, TfOH, или смесью таких кислот, при температурах 50-130C с получением индазолинспироциклопропаниндолинонов 4. Схема 6 Индазолинспироциклопропаниндолиноны 4 могут быть йодированы с получением соединений 5(схема 7) путем обработки таким йодирующим средством, как йод или N-йодсукцинимид, в присутствии такого основания, как Na2CO3, K2CO3, NaOH или KOtBu, в таком подходящем растворителе, как ацетон,ACN, DMF, DMSO, диоксан, NMP, THF, в присутствии или в отсутствие воды. Схема 7 Относительная стереохимия циклопропанового кольца может быть определена как (1R,2S) путем сравнения с второстепенным диастереомером, полученным в синтезе рацемических стандартных образцов путем применения ЯМР эксперимента HCOSY для назначения сигнала для каждого протона в 1H ЯМР с последующим экспериментом NOESY с определением взаимодействия "через пространство" между циклопропановыми протонами и протоном в положении 4 следующего индалинона (I. Moldvai, E.Gacs-Baitz, M. Balazs, M. Incze and С. Szantay; Arch. Pharm. Pharm. Med. Chem. 1996, 329, 541-549). Абсолютная стереохимия продукта может быть определена как (1R,2S) на основании использования мнемо- 11023173 нической схемы Sharpless для предсказания конфигурации диола как (S) и допуская процесс соединенияSN2 С образованием циклопропана, приводя к преобразованию (S) стереоцентра диола с сохранением э.и. Изобретение представлено при помощи следующих примеров, которые не предназначены для ограничения каким-либо образом. Следует отметить, что примеры не пронумерованы последовательно. Примеры А. Синтезы соединений согласно настоящему изобретению. Общие способы экспериментов. Коммерчески доступные исходные материалы, реагенты и растворители применяли непосредственно после получения, за исключением N,N-диметил-1-(4-винилфенил)метанамина, который очищали посредством хроматографии на силикагеле перед применением в реакциях Хека. В основном, безводные реакции выполняли в такой инертной атмосфере, как азот или аргон. Взаимодействия под микроволновым излучением проводили в микроволновом реакторе Biotage Initiator. Ход реакции обычно наблюдали при помощи ТСХ с применением силикагелевых планшетов Merck с визуальным отображением при помощи УФ при 254 нм, при помощи аналитической ВЭЖХ или при помощи LCMS (Bruker Exquire 4000). Очистку колоночной флеш- хроматографией промежуточных соединений или готовых продуктов выполняли с применением силикагеля 60 230-400 меш из химикатов EMD. Готовые продукты время от времени очищали препаративной ВЭЖХ с обращенной фазой Очистку выполняли на Varian PrepStar модели SD-1 системы ВЭЖХ с Varian Monochrom 10u C-18 колонкой с обращенной фазой с применением градиента приблизительно от 5-30% ацетонитрила/0,05% TFA воды до 70-100% ацетонитрила/0,05% TFA воды в течение периода 20-40 мин при скорости потока 30-50 мл/мин. Фракции, содержащие необходимые вещества, концентрировали и лиофилизировали с получением готовых продуктов. Протоны ЯМР записывали на спектрометре Bruker 400 МГц и получали массовый спектр с применением спектрометраBraker Esquire 4000. Оптические вращения измеряли при натрия D-линии (589,44 нМ) с применением поляриметра АА-55 от Optical Activity Ltd с 2,5100 мм безоболочечной трубкой из нержавеющей стали при данных концентрациях образца (с, единицы г/100 мл). Названия соединений образовывали с применением программного обеспечения, встроенного вChemBioDraw Ultra версии 11,0 со следующим исключением. Рацемические соединения с известной относительной стереохимией называли с применением системы R/S, как описано в North (Principles andApplications of Stereochemistry, CRC Press, 1998), где ниже пронумерованный атом произвольно обозначен как R, и выше пронумерованные атомы определены относительно того центра. Таким образом, рацемическая смесь энантиомеров соединения с двумя хиральными центрами обозначена как (1R, 2S) или (1R, 2S) в зависимости от известной относительной стереохимии. Стандартную R и S номенклатуру или "abs", являющуюся абсолютной, использовали для описания простых энантиомеров или энантиомерно обогащенных соединений более чем 95% э.и. Аббревиатуры вод. - водныйEtOH - этанол Ч - часы гекс. - гексан АсОН - уксусная кислота ВЭЖХ - высокоэффективная жидкостная хроматографияPPh3 - трифенилфосфин Преп. ВЭЖХ - препаративная шкала высокоэффективной жидкостной хроматографии Преп. ТСХ - препаративная шкала тонкослойной хроматографии КДК - круглодонная колба к.т. - комнатная температура нас. - насыщенныйTHF - тетрагидрофуран мас.% - массовый процент Получение исходных материалов Синтез 5-метоксиоксиндола В раствор 5-метоксиизатина (10,62 г, 60 ммоль) в DMSO (30 мл) по каплям добавляли N2H4xH2O(гидрат гидразина, 6 мл, 120 ммоль) в течение 5 мин (экзотермический). После добавления полученную смесь нагревали при 140C (темп. масла) в течение 2 ч и затем охлаждали до к.т. После разбавления Н 2 О (30 мл) добавляли 6 М HCl (12 мл, 72 ммоль) и полученную смесь перемешивали в течение 1 ч при к.т. Добавляли лед (30 мл) и реакционную смесь перемешивали O/N при к.т. Образованный осадок собирали фильтрованием с отсасыванием, промывали H2O, затем сушили с получением 5-метоксиоксиндола (6,523 г) в виде коричневого твердого вещества (приблизительно 10% примесей представляло собой оксим из исходного вещества 5-метоксиизатин). 1 В смесь 3-йод-1 Н-индазол-6-карбальдегида (1,360 г, 5 ммоль) и 5-метоксиоксиндола (1,06 г, 6,5 ммоль) в метаноле (50 мл) добавляли пиперидин (0,1 мл, 1 ммоль). Полученную смесь кипятили с обратным холодильником (темп. масла 75C) в течение 3 ч, затем охлаждали до к.т. и перемешивали в течение 2 ч при к.т. Полученные осадки собирали фильтрованием с отсасыванием и сушили с получением (E/Z)3-3-йод-1 Н-индазол-6-ил)метилен)-5-метоксииндолин-2-она (E/Z = 2:1) в виде темного кирпичнооранжевого твердого вещества (1,966 г, 94%). Смесь использовали как промежуточное вещество без очистки изомеров. Это промежуточное вещество также получали с применением следующих условий: Круглодонную колбу загружали 5-метоксиоксиндолом (коммерческий реагент от Prime Organics, 300 мг, 1,84 ммоль), 3 йод-1H-индазол-6-карбальдегидом (500 мг, 1,84 ммоль), пиперидином (20 мкл, 0,18 ммоль) и МеОН (7 мл). Реакционную смесь затем нагревали до 60C в течение 4 ч. Образовался ярко-красный осадок, который далее осаждали посредством охлаждения до комнатной температуры. Красный порошок затем фильтровали и промывали МеОН с получением 658 мг, 86% названного соединения. Получали смесь (E)- и В смесь 3-йод-1 Н-индазол-6-карбальдегида (1,360 г, 5 ммоль) и 2-оксиндола (732 г, 5,5 ммоль) в МеОН (25 мл) добавляли пиперидин (0,1 мл, 1 ммоль). Полученную смесь кипятили с обратным холодильником (темп. масла 75C) в течение 90 мин, затем охлаждали до к.т. Полученные осадки собирали фильтрованием с отсасыванием и сушили с получением (E/Z)-3-3-йод-1 Н-индазол-6-ил)метилен)индолин-2-она в виде желтого твердого вещества (E:Z = 5:1, 1,86 г). Смесь использовали в виде промежуточного вещества без очистки изомеров, или альтернативно чистый Е изомер может быть очищен посредством растворения в THF (1,57 г в 46,85 мл) при комнатной температуре. В прозрачный раствор с перемешиванием добавляли гексан (146,8 мл) с получением желтого осадка. Твердую суспензию нагревали до 70C в течение 30 мин и затем охлаждали до комнатной температуры. Желтое твердое вещество фильтровали и промывали гексаном (3,14 мл) с получением названного соединения (1,22 г, 79%). 1 Оксиндол (665 мг, 5 ммоль) и 3-йод-1-2-(триметилсилил)этокси)метил)-1 Н-индазол-6 карбальдегид (2 г, 5 ммоль) растворяли в этаноле (25 мл). Добавляли пиперидин (0,1 мл) и раствор нагревали до 70C в течение 2 ч, охлаждали до к.т. и перемешивали в течение ночи. Растворитель удаляли invacuo с получением оранжевого твердого вещества, которое перетирали в порошок с этанолом с получением названного соединения в количественном выходе. 1 В раствор триметилсульфоксониййодида (1,89 г, 8,6 ммоль) в безводном DMF (40 мл) добавляли гидрид натрия (60% дисперсия в масле) (1,03 г, 25,8 ммоль) при 0C. Смесь перемешивали в течение 15 мин,после чего добавляли(Е)-3-3-йод-1-2-(триметилсилил)этокси)метил)-1H-индазол-6 ил)метилен)индолин-2-он (2,2 г, 4,3 ммоль). Раствор перемешивали в течение ночи при к.т. Реакцию гасили нас. раствором NH4Cl (50 мл), экстрагировали EtOAc (4100 мл), сушили над MgSO4 и концентрировали досуха Названное соединение выделяли хроматографией на силикагеле (CH2Cl2/МеОН 98:2) в виде желтого твердого вещества (1,5 г, 66%). 1 Круглодонную колбу загружали 5-фториндолин-2-оном (100 мг, 0,661 ммоль), 3-йод-1-2(триметилсилил)этокси)метил)-1H-индазол-6-карбальдегидом (266,18 мг, 0,661 ммоль), пиперидином (13 мкл, 0,013 ммоль) и метанолом (7,5 мл). Реакционную смесь затем нагревали до 55C в течение 4 ч перед охлаждением реакционной массы до комнатной температуры. При помощи фильтрации и промывания метанолом (0,50 мл 2) получали названное соединение в виде желтого твердого вещества (273 мг, 77%). 1 Триметилсульфоксониййодид (164,4 мг, 0,747 ммоль) добавляли в суспензию гидрида натрия (89,6 мг, 2,24 ммоль) (60% дисперсия в масле) в DMF (2,0 мл) при комнатной температуре. Смесь перемешивали в течение 15 мин, после чего добавляли раствор (Е)-5-фтор-3-3-йод-1-2-(триметилсилил)этокси) метил)-1H-индазол-6-ил)метилен)индолин-2-она (200 мг, 0,373 ммоль) в DMF (1,25 мл). Раствор перемешивали при 55C в течение 7,0 ч перед гашением реакционной массы избытком раствора 25% NH4Cl(10 мл) при комнатной температуре. Продукт экстрагировали с применением этилацетата (15 мл 2) и органический слой сушили над MgSO4 и выпаривали in vacuo. Неочищенный продукт очищали с применением колоночной хроматографии с силикагелем (гексан : ацетон 80:20 в качестве элюента) с получением кремового полутвердого вещества, которое затем перетирали в порошок с гексанами (2,0 мл) с получением названного соединения в виде беловатого порошка (94 мг, 46%). 1 В раствор 1 Н-индазол-6-карбальдегида (2,00 г, 13,7 ммоль), K2CO3 (3,79 г, 27,4 ммоль) в DMF (15 мл) добавляли по каплям раствор I2 (5,91 г, 23,3 ммоль) в DMF (15 мл) и реакционную смесь оставляли перемешиваться в течение двух часов. Затем добавляли водный раствор, состоящий из Na2S2O4 (3,30 г)/K2CO3 (0,20 г)/H2O (30 мл), и раствор перемешивали в течение одного часа. Продукт затем осаждали посредством выливания раствора в ледяную воду (300 мл) и собирали посредством вакуумной фильтрации с получением после сушки 3,02 г, 81% бежевого порошка. 1 В суспензию 3-йод-1 Н-индазол-6-карбальдегида (3,01 г, 11,1 ммоль) в CH2Cl2 (70 мл) и 50 % водн. КОН (20 мл) добавляли бромид тетрабутиламмония (36 мг, 0,111 ммоль), и раствор охлаждали до 0C. Затем по каплям добавляли (2-(хлорметокси)этил)триметилсилан (2,3 мл, 13,3 ммоль) и реакционную смесь перемешивали при 0C в течение 3 ч. Раствор затем переносили в отдельную воронку, содержащую CH2Cl2 (200 мл) и органический слой промывали рассолом (2 100 мл), сушили (MgSO4) и растворитель удаляли in vacuo. Полученный остаток очищали колоночной хроматографией (100% CH2Cl2) с получением 2,88 г, 65% N-1 изомера (высокая точка элюирования) и 757 мг, 17% N-2 изомера (низшая точка элюирования). В раствор NaNO2 (11,04 г. 160 ммоль) в H2O (200 мл) медленно добавляли одну порцию 6 цианоиндола (5,68 г, 40 ммоль). Полученную суспензию перемешивали в течение 5 мин при к.т. в тече- 15023173 ние 30 мин, через капельную воронку по каплям добавляли HCl (32 мл, 192 ммоль, 6 н.), и рН было равно приблизительно 1. Полученную суспензию перемешивали в течение 4,5 ч при к.т. перед добавлениемEtOAc (400 мл). После перемешивания дополнительно 10 мин для растворения осадка два слоя разделяли и водный слой экстрагировали EtOAc (150 мл). Объединенные экстракты сушили над Na2SO4. Путем удаления растворителей получали 6,864 г (100%) названного соединения в виде коричневого (кофейного цвета) твердого вещества. 1 Названное соединение синтезировали согласно способу, описанному для (Е)-3-1 Н-индазол-5 ил)метилен)индолин-2-она, за исключением взаимодействия оксиндола (67 мг, 0,216 ммоль) с 1 Ниндазол-6-карбальдегидом (73 мг, 0,238 ммоль) с получением 32 мг, 51%. 1 В раствор изатина (2,94 г, 20 ммоль) в DMF (40 мл) при 0C порционно добавляли 60% NaH (1,00 г,25 ммоль). После добавления полученную смесь перемешивали в течение 15 мин при 0C и добавляли по каплям 1-бром-2-метоксиэтан (2,35 мл, 25 ммоль) в течение 2 мин. Полученную смесь перемешивали в течение 10 мин при 0C, нагревали до к.т. и перемешивали O/N. Реакционную смесь затем охлаждали до 0C, гасили нас. NH4Cl, льдом, Н 2 О, экстрагировали EtOAc (150 мл 2), сушили над Na2SO4 и концентрировали с получением темной оранжево-красной жидкости, которую повторно растворяли в DMSO (10 мл). N2H4-xH2O (2 мл) добавляли по каплям в течение 7 мин. После добавления реакционную смесь перемешивали в течение 5 мин при к.т., затем 2 ч при 140C (темп. масла) перед охлаждением до к.т. Добавляли лед/Н 2 О (20 мл), а затем 6 М HCl (7 мл, 42 ммоль) и полученную смесь перемешивали в течение 30 мин при к.т. Добавляли дополнительное количество льда/Н 2 О (40 мл) и смесь экстрагировали EtOAc(50 мл 3). Неочищенный продукт очищали флеш-хроматографией (градиент: EtOAc/гекс. 0-40%) с получением названного соединения в виде оранжевой жидкости (2,32 г, 61% за 2 стадии). 1 В смесь изатина (5,0 г, 35 ммоль), K2CO3 (5,5 г, 40 ммоль) и хлорацетамида (3,74 г, 40 ммоль) в 100 мл колбы добавляли DMF (25 мл). Полученную смесь нагревали при 90C (темп. масла) в течение 2 ч. После охлаждения до к.т. ее выливали в лед/H2O (200 мл) и полученный осадок собирали фильтрованием с отсасыванием с получением 2-(2,3-диоксоиндолин-1-ил)ацетамида (4,32 г) после сушки. 1H ЯМР (400 МГц, DMSO-d6)7,72 (s, 1H), 7,65 (t, J = 7,6 Гц, 1H), 7,58 (d, J = 7,2 Гц, 1H), 7,30 (s,1H), 7,14 (t, J = 7,4 Гц, 1H), 7,02 (d, J = 8,0 Гц, 1H), 4,25 (s, 2H). Вышеуказанный 2-(2,3-диоксоиндолин-1-ил)ацетамид (4,32 г) повторно растворяли в DMSO (20 мл), и N2H4-xH2O (2,5 мл) добавляли по каплям в течение 10 мин. После добавления полученную смесь перемешивали в течение 5 мин при к.т., затем 2 ч при 140C перед охлаждением до к.т. Реакцию гасили льдом (20 мл) и 6 М HCl (8 мл), затем перемешивали в течение 30 мин при к.т. Фильтрованием с отсасыванием получали неочищенное названное соединение (2,92 г) в виде светло-желтого твердого вещества. Продукт суспендировали в EtOAc (120 мл) и добавляли Н 2 О (60 мл), с последующим добавлением 2 МHCl (30 мл). Смесь разделяли и фильтровали с отсасыванием водного слоя с получением названного соединения в виде светло-желтого твердого вещества (1,78 г, 27% за 2 стадии) после сушки. В смесь (1R,2S)-2-(3-йод-1H-индазол-6-ил)-спиро[циклопропан-1,3'-индолин]-2'-она (802 мг, 2 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (462 мг, 3 ммоль) в 20 мл пробирку для микроволнового нагревания добавляли PhCH3/EtOH (8 мл/4 мл), а затем добавляли 1 М Na2CO3 (3 мл, 3 ммоль) и Ph(pph3)4 (46 мг, 0,04 ммоль, 2 моль.%), и полученную смесь продували аргоном, затем облучали микроволновым излучением 3 ч при 120C. После водной обработки раствор экстрагировали EtOAc и очищали флеш- хроматографией (гекс./EtOAc 1:1) с получением неочищенного названного соединения в виде светло-желтой пены (512 мг), которую использовали без дополнительной очистки. 1(1,00 г, 2,32 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (500 мг, 3,25 ммоль) в 20 мл пробирку для микроволнового нагревания добавляли PhCH3/EtOH (7 мл/3,5 мл), а затем 1 М Na2CO3 (3 мл, 3 ммоль). После перемешивания в течение 1 мин при к.т. добавляли Ph(pph3)4 (50 мг, 0,043 ммоль, 1,9 моль.%) и полученную смесь продували аргоном и облучали микроволновым излучением 3 ч при 120C. Эту реакцию повторяли дважды в таком же масштабе и полученные смеси объединяли. При помощи водной обработки получали неочищенное названное соединение в виде темно-оранжевого твердого вещества/пены (3,10 г), которое использовали без дополнительной очистки. Образец чистого соединения может быть получен при помощи флеш-хроматографии (гекс./EtOAc 1:1). 1(d, J = 18,0 Гц, 1H), 5,56 (d, J = 1,6 Гц, 1H), 5,52 (d, J = 11,6 Гц, 1H), 3,56 (t, J = 7,6 Гц, 1 Н, частичное перекрывание остатком MeOH), 3,26 (s, 3H), 2,24 (dd, J = 7,8 Гц, J = 5,0 Гц, 1H), 2,18 (dd, J = 9,2 Гц, J = 4,8 Гц,1H); MS ESI 332,0 [M + H]+, рассчитано для [C20H17N3O2 + H]+ 332,1. Синтез (1R,2S)-5'-этил-2-(3-йод-1 Н-индазол-6-ил)спиро[циклопропан-1,3'-индолин]-2'-она Названное соединение в качестве отдельного диастереомера (710 г, 33% за 2 стадии, перетирали в порошок с гекс./МеОН) получали в виде светло-оранжевого твердого вещества из 5-этилиндолин-2-она(885 мг, 5,5 ммоль) и 3-йод-1H-индазол-6-карбальдегида (1,36 г, 5 ммоль) с применением способа получения (1R,2S)-2-(3-винил-1 Н-индазол-6-ил)спиро[циклопропан-1,3'-индолин]-2'-она. 1 Неочищенное названное соединение (2,06 г, 99% за 2 стадии) получали в виде желтого твердого вещества из 5-метилиндолин-2-она (772 мг, 5,25 ммоль) и 3-йод-1H-индазол- 6-карбальдегида (1,36 г, 5 ммоль) с применением способа для получения(1R,2S)-2-(3-винил-1H-индазол-6 ил)спиро[циклопропан-1,3'-индолин]-2'-она ЯМР указывало на смесь 6:1 названного соединения и незначительного количества диастереомера. 1(544 мг, 2 ммоль) в МеОН (20 мл) добавляли пиперидин (0,04 мл). Полученную смесь нагревали при 75C (темп, масла) в течение 90 мин. После охлаждения до к.т. полученный осадок собирали фильтрованием с отсасыванием с получением желтого твердого вещества (850 мг). В смесь триметилсульфоксониййодида (880 мг, 4 ммоль) и 60% NaH (486 мг, 12 ммоль) в 100 мл колбе добавляли DMF (5 мл). Полученную смесь перемешивали в течение 5 мин при к.т. перед тем, как суспензию вышеуказанного желтого твердого вещества (850 мг) в DMF (20 мл) добавляли через пипетку. После добавления полученную розовую смесь перемешивали в течение 2 ч при к.т. и охлаждали до 0C. Реакцию гасили льдом/Н 2 О, нас. NH4Cl (15 мл), а затем льдом/H2O до общего объема 100 мл. После перемешивания в течение 2 мин при к.т. полученный осадок собирали фильтрованием с отсасыванием с получением неочищенного названного соединения в виде розового твердого вещества (805 мг, 88% за 2 стадии) после сушки. 1H ЯМР (400 МГц, DMSO-d6)13,50 (s, 1H), 7,94 (s, 1H), 7,70 (s, 1H, NH), 7,49 (s, 1H), 7,31 (d, J = 8,4 Гц, 1H), 7,26 (s, 1H), 7,06 (t, J = 7,6 Гц, 1 Н, частичное перекрывание пика при 7,03 ч./млн), 7,03 (d, J = 8,8 Гц, 1H, частичное перекрывание пика при 7,06 ч./млн), 6,86 (d, J = 7,6 Гц, 1H), 6,60 (t, J = 7,6 Гц, 1H),4,38 (t, J = 18,4 Гц, 2H), 3,26 (t, J = 8,8 Гц, 1H), 2,40-2,35 (m, 1H), 2,10-2,04 (m, 1H); MS ESI 459,1 [M + Неочищенное названное соединение (750 мг, 82% за 2 стадии) получали в виде светло-бежевого твердого вещества из 1-(2-метоксиэтил)индолин-2-она (382 мг, 2 ммоль) и 3-йод-1 Н-индазол-6 карбальдегида (544 мг, 2 ммоль) с применением способа для получения (1R,2S)-2-(3-винил-1 Ниндазол-6-ил)спиро[циклопропан-1,3'-индолин]-2'-она. ЯМР указывало на смесь 6:1 названного соединения и незначительное количество диастереомера. 1 Ледяную уксусную кислоту (0,2 мл) добавляли в смесь 4-этинилбензальдегида (1 г, 7,5 ммоль),пирролидина (1,2 мл, 15 ммоль) и NaBH(OAc)3 (2,5 г, 11,5 ммоль) в DCE (35 мл). Полученную смесь перемешивали в течение 2 ч при к.т. Реакцию гасили насыщенным водным NaHCO3 (50 мл). Продукт экстрагировали в CH2Cl2 (2100 мл), и объединенный органический слой промывали насыщенным солевым раствором (25 мл), сушили (MgSO4) и выпаривали in vacuo с получением 1-(4-этинилфенил)пирролидина в количественном выходе. 1(4-этинилфенил)пирролидин (1 г, 5 ммоль) и HRuCl(СО)(PPh3)3 (120 мг, 0,11 ммоль) в атмосфере аргона. Полученную смесь нагревали при 50C в течение 4 ч. Продукт экстрагировали в EtOAc (250 мл), и органический слой промывали последовательно водой (320 мл) и насыщенным солевым раствором (20 мл),сушили (MgSO4) и выпаривали in vacuo. Очисткой колоночной хроматографией (силикагель, 0-20% МеОН/EtOAc) получали названное соединение (1,2 г, 77%). 1 Названное соединение (4,35 г, 71%) получали в виде от белого до желтого твердого вещества из 4(4-бромбензил)морфолина (4,18 г, 16,3 ммоль) и 4,4,5,5-тетраметил-2- винил-1,3,2-диоксаборолана (3 мл,17,7 ммоль, 1,1 экв.) с применением способа для получения соединения согласно примеру А 51 А (PhCH3 Названное соединение (498 мг, 71%) получали в виде светло-желтого твердого вещества из 4 бромбензальдегида (500 мг, 2,71 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (0,5 мл, 2,95 ммоль, 1,1 экв.) с применением способа для получения соединения согласно примеру А 51 А (PhCH3 = 8 мл, 2 мол.% Pd(PtBu3)2, 80C, O/N). 1 Н ЯМР (400 МГц, CDCl3)10,01 (s, 1H), 7,88 (d, J = 8,0 Гц, 2H), 7,64 (d, J = 8,4 Гц, 2H), 7,43 (d, J = 18,4 Гц, 1H), 6,34 (d, J = 18,4 Гц, 1H), 1,34 (s, 12H); MS ESI 258,9 [M + H]+, рассчитано для [C15H19BO3 + В смесь 3-этинилбензальдегида (650 мг, 5 ммоль) и морфолина (0,87 мл, 10 ммоль) в DCE (15 мл) добавляли NaBH(OAc)3 (1,325 г, 6,25 ммоль), а затем АсОН (0,2 мл). Полученную смесь перемешивали в течение 2 ч при к.т. Водной обработкой с последующей экстракцией EtOAc, получали неочищенный 4(3-этинилбензил)морфолин (0,98 г) в виде светло-коричневого масла. Названное соединение (1,75 г, количественный выход за 2 стадии) получали в виде светло-коричневого масла с применением способа(PhCH3 = 12 мл, 1 мол.% HRuCl(СО)(PPh3)3, 50C, 2 ч) для получения соединения согласно примеру А 42 А. 1 Названное соединение (267 мг, 68%) получали в виде желтого твердого вещества из 1-(4 бромфенил)пиперазина (653 мг, 2,71 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (0,5 мл,2,95 ммоль, 1,1 экв.q.) с применением способа для получения соединения согласно примеру А 51 А Названное соединение (2,52 г, 71%) получали в виде белого твердого вещества из 4-(4-бромбензил)цис-2,6-диметилморфолина (2,82 г, 10 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (1,85 мл, 11 ммоль, 1,1 экв.) с применением способа для получения соединения согласно примеру А 51 А Названное соединение (674 мг, 76%) получали в виде светло-желтого твердого вещества из 1-(4 бромфенил)-4-метилпиперазина (691 мг, 2,71 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана(0,5 мл, 2,95 ммоль, 1,1 экв.) с применением способа для получения соединения согласно примеру А 51 А Названное соединение (601 мг, 65%) получали в виде светло-желтого твердого вещества из 1-(4 бромфенил)-4-этилпиперазина (729 мг, 2,71 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана(0,5 мл, 2,95 ммоль, 1,1 экв.) с применением способа для получения соединения согласно примеру А 51 А Названное соединение (504 мг, 52%) получали в виде светло-оранжевого твердого вещества из 1-(4 йодфенил)-4-изопропилпиперазина (894 мг, 2,71 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2 диоксаборолана (0,5 мл, 2,95 ммоль, 1,1 экв.) с применением способа для получения соединения согласно примеру А 51 А (PhCH3 = 10 мл, 2 мол.% Pd(PtBu3)2, 80C, O/N). 1 Названное соединение (902 г, 72%) получали в виде белого твердого вещества из 4-(2-(4 бромфенокси)этил)морфолина (1 г, 3,50 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (0,6 мл, 3,58 ммоль, 1,02 экв.) с применением способа для получения соединения согласно примеру А 51 А Названное соединение (610 мг, 55%) получали в виде желтого твердого вещества из 4-бром-2 фторбензальдегида (812 мг, 4 ммоль) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (0,8 мл, 4,8 ммоль) с применением способа для получения соединения согласно примеру А 51 А (PhCH3 = 10 мл, 1 мол.% Pd(PtBu3)2, 80C, 1,5 ч). 1H ЯМР (400 МГц, CDCl3)10,34 (s, 1H), 7,85 (t, J = 7,6 Гц, 1H), 7,36 (d, J = 8,8 Гц, 1H, частичное перекрывание пика при 7,35 ч./млн), 7,35 (d, J = 17,2 Гц, 1H, частичное перекрывание пика при 7,36 ч./млн), 7,26 (d, J = 11,2 Гц, 1 Н, частичное перекрьшание CDCl3 остатка), 6,32 (d, J = 18,4 Гц, 1H), 1,33 (s,12H). Синтез (Е)-4-(2-фтор-4-(2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)винил)бензил)морфолина(0,5 мл). Полученную смесь перемешивали в течение 2 ч при к.т. Реакцию гасили нас. NaHCO3 (10 мл),H2O (10 мл), и экстрагировали EtOAc (230 мл). Растворители удаляли in vacuo с получением названного соединения в виде белого твердого вещества (0,72 г, 94%). 1(0,44 мл, 2,6 ммоль) с применением способа для получения соединения согласно примеру А 51 А (PhCH3 = 10 мл, 2,5 мол.% Pd(PtBu3)2, 80C, 75 мин). 1H ЯМР (400 МГц, CDCl3)7,43 (d, J = 8,0 Гц, 2 Н, частичное перекрывание пика при 7,40 ч./млн),7,40 (d, J = 18,8 Гц, 1H, частичное перекрывание пиков при 7,43 и 7,36 ч./млн), 7,36 (d, J = 8,4 Гц, 2 Н, частичное перекрывание пика при 7,40 ч./млн), 6,14 (d, J = 18,4 Гц, 1H), 3,78 (s, 2H), 2,53-2,44 (m, 2H), 1,681,55 (m, 3H), 1,40-1,28 (m, 3H), 1,05 (d, J = 6,4 Гц, 6 Н); MS ESI 356,2 [М + Н]+, рассчитано для Получали согласно методике из литературы (С. Martin and E. M. Carreira, J. Am. Chem. Soc, 2005,127, 11505-11515). Перемешанный раствор изатина (10,0 г, 68 ммоль) в сухом DMF (125 мл) охлаждали в ледяной бане перед добавлением 10 порций гидрида натрия (60 мас.% в минеральном масле, 2,86 г, 71,5 ммоль), оранжевый раствор быстро становился пурпурным. Когда не наблюдалось никакого дополнительного выделения газа при помощи шприца добавляли бензилбромид (13,4 г, 78,0 ммоль). В течение 20 мин наблюдали изменение цвета обратно на оранжевый. С перемешиванием добавляли воду (300 мл) и полученный оранжево-красный осадок собирали фильтрацией и промывали водой и небольшим количеством холодного этанола. Твердое вещество затем перекристаллизовывали из кипящего этанола (300 мл) с получением N-бензилизатина (13,7 г, 85%) в виде длинных красных иголок. N-бензилизатин (13,0 г, 55 ммоль) смешивали с гидратом гидразина (60 мл) и помещали в масляную баню. Смесь постадийно нагревали до 125C, на первой стадии она становилась зеленым илистым отложением, затем желтым с кусками липкого твердого вещества. После всех 5 ч при 125C смесь охлаждали и экстрагировали EtOAc(2100 мл). Объединенные органические порции промывали дважды 1,0 М водн. H2SO4, и раз каждый слой наполовину насыщенным солевым раствором, затем насыщенным солевым раствором, сушили надMgSO4, фильтровали и концентрировали с получением бледно-желтого твердого вещества. Повторным осаждением из эфира/пентана получали названное соединение в виде беловатого твердого вещества (9,6 г, 75%). Спектральные данные совпадали с литературными значениями (С. Martin and E. M. Carreira, J. Способом, подобным способу получения N-бензилизатина, 5-фторизатина (10,0 г, 60,5 ммоль) получали 5-фтор-N-бензилизатин в виде оранжево-красного порошка (14,5 г, 93%). Неочищенный продукт использовали на следующей стадии без очистки. 1(s, 2H); MS ESI 255,9 [M + H]+, рассчитано для [C15H10FNO2+ H]+ 255,07. Названное соединение получали способом, подобным способу для N-бензилоксиндола с использованием 5-фтор-N-бензилизатина (14,5 г,56,8 ммоль). Перетиранием в порошок с применением Et2O : гексана получали названное соединение в виде светло-желтого твердого вещества (10,3 г, 75%). 1BnBr (6,5 мл, 55 ммоль) по каплям в течение 2 мин. После добавления полученную смесь нагревали в масляной бане при 75C в течение 1,5 ч. После охлаждения до к.т. реакционную смесь выливали в лед/холодную воду (250 мл), промывали Н 2 О (50 мл) и перемешивали в течение 5 мин. Полученные осадки собирали фильтрованием с отсасыванием и сушили на воздухе с получением 1-бензил-5 метилизатина в виде темно-красного твердого вещества MS ESI 252,0 [М + Н]+, рассчитано для(5 мл) добавляли по каплям в течение 5 мин. После добавления полученный прозрачный красный раствор нагревали при 120C в течение 2 ч, затем при 140C в течение 5 ч. После охлаждения до к.т. ее выливали в 1 л колбу Эрленмейера, промывали H2O (50 мл) и лед добавляли до общего объема приблизительно 300 мл. Добавляли 2 М HCl (50 мл), и смесь экстрагировали EtOAc (200 мл 2, затем 100 мл), и органический слой сушили (Na2SO4). Путем удаления растворителей с последующей сушкой под высоким вакуумом в течение 2 дней получали названное соединение в виде темно-коричневого твердого вещества (12,53 г, количественный выход за 2 стадии, содержащий тот же остаток DMSO). 1 Перемешанный раствор 5-метоксиизатина (5,0 г, 28 ммоль) в сухом DMF (40 мл) медленно охлаждали в ледяной бане перед добавлением гидрида натрия (60 мас.% в минеральном масле, 1,7 г, 42 ммоль),темно-красный раствор быстро превратился в черный. После перемешивания в течение 20 мин в реакционную смесь при помощи шприца добавляли BnBr (3,7 мл, 31 ммоль) и полученную смесь перемешивали в течение 1 ч. С перемешиванием добавляли воду (150 мл) и полученный темно-красный осадок собирали фильтрацией и промывали водой с получением 1-бензил-5-метоксииндолин-2,3-диона в виде темнокрасного твердого вещества (6,1 г, 81%). 1H ЯМР (400 МГц, CDCl3)7,39-7,31 (m, 5H), 7,17 (s, 1H), 7,03 (d, J = 8,1 Гц, 1H), 6,68 (d, J = 8,6 Гц,1H), 4,92 (s, 2H), 3,79 (s, 3H). MS ESI 268,1 [М + Н]+, рассчитано для [C16H13NO3+H]+ 268,09. Раствор 1-бензил-5-метоксииндолин-2,3-диона (6,1 г, 23 ммоль) и гидрат гидразина (50- 60% содержания, 2,9 мл, прибл. 2 экв.) в DMSO (15 мл) нагревали до 140C в масляной бане. Через 3 ч смесь охлаждали, разбавляли водой и EtOAc, слои разделяли и водный трижды экстрагировали EtOAc (30 мл). Объединенные органические порции промывали 2 М H2SO4, насыщенным солевым раствором и сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта в виде вязкого коричневого масла. Неочищенный продукт очищали хроматографией на силикагеле (20-50% EtOAc в гексане) с выходом названного соединения в виде коричневого масла (5,0 г, 85%). 1H ЯМР (400 МГц, CDCl3)7,34-7,23 (m, 5H), 6,89 (s, 1H), 6,69 (d, J = 8,3 Гц, 1H), 6,61 (d, J = 8,5 Гц, 1H), 4,91 (s, 2H), 3,76 (s, 3H),- 22023173 3,62 (s, 2H). MS ESI 254,0 [M + H]+, рассчитано для [C16H15NO2+ H]+ 254,1. Синтез N1-бензил-6-винил-1H-индазолаTHF/воде (9:1, 350 мл) продували азотом. В отдельной колбе перемешивали вместе Pd(OAc)2 (0,16 г, 0,7 ммоль, 2 мол.%) и PPh3 (0,37 г, 1,4 ммоль, 4 мол.%) в продуваемом азотом сухом THF (35 мл) в течение 10 мин с образованием красного раствора с некоторыми суспендированными твердыми веществами. Пинаколовый сложный эфир винилбороновой кислоты (7,5 мл, 44,4 ммоль) и каталитический раствор добавляли в реакционную смесь и полученный раствор снова продували азотом. Смесь нагревали в масляной бане, установленной на 65C; при помощи ТСХ показали расход исходного вещества за 7 ч. Смесь концентрировали при пониженном давлении с удаление большей части THF, затем разбавляли водой (50 мл), насыщенным солевым раствором (50 мл) и EtOAc (250 мл). Слои разделяли и водную фазу дополнительно экстрагировали EtOAc (450 мл). Объединенные органические части промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали (при 70C/20 мбар) с получением неочищенного продукта. Их хроматографировали на диоксиде кремния с применением 10-20%EtOAc в циклогексане с получением названного соединения (7,5 г, 90%) в виде желтого масла, которое отвердевало при отстаивании. 1= 7,0 Гц, , 2H), 6,75 (dd, J = 17,6, 10,9 Гц, 1H), 5,76 (d, J = 17,5 Гц, 1H), 5,53 (s, 2H), 5,26 (d, J = 10,9 Гц,1H). MS (ES+): 235 ([M+H]+); рассчитано для [C16H14N2+H]+ 235,1. Способ 2. С применением 4,4,6-триметил-2-винил-1,3,2-диоксаборинана. Смесь N1-бензил-6-бром 1 Н-индазола (1,44 г, 5,0 ммоль) и NaOH (0,4 г, 10,0 ммоль) в THF/воде (5:1, 15 мл) продували азотом. В отдельной колбе перемешивали вместе Pd(OAc)2 (11 мг, 0,05 ммоль, 1 мол.%) и PPh3 (26 мг, 0,1 ммоль, 2 мол.%) в продутом азотом THF (2,5 мл) в течение 10 мин с образованием красного раствора с некоторым количеством суспендированных твердых частиц. Используемый THF представлял собой квалификацию для ВЭЖХ и без ингибитора; эффект более низкой квалификации или стабилизированного THF не известен. 4,4,6-Триметил-2-винил-1,3,2-диоксаборинан (1,12 мл, 6,5 ммоль) и каталитический раствор добавляли в реакционную смесь и полученный раствор снова продували азотом. Смесь нагревали в масляной бане, установленной на 65C; нагревание продолжали в течение 24 ч, но реакция вероятно заканчивалась за менее чем 8 ч. Неочищенную смесь затем объединяли со второй, параллельной реакцией того же масштаба, при которой использовачи большее разбавление. Смесь концентрировали при пониженном давлении для удаления большего количества THF, затем разбавляли водой, насыщенным солевым раствором и циклогексаном. Слои разделяли и водную фазу дополнительно экстрагировали циклогексаном, пока ТСХ не показало, что весь желаемый продукт был экстрагирован (3-4 экстракта). Объединенные органические порции промывали насыщенным солевым раствором, сушили над MgSO4, и затем пропускали через 1 см прокладку диоксида кремния для удаления исходного вещества. Любой оставшийся на диоксиде кремния продукт элюировали с применением 10% EtOAc в циклогексане (Rf. 0,15 в этом элюенте). Объединенный элюат концентрировали с получением названного соединения (2,05 г, 88%) в виде желтого масла,которое отвердевало при отстаивании и было достаточной чистым для использования в последующих реакциях. Способ 3. N1-бензил-6-бром-1H-индазол (половина неочищенного вещества, полученного в способе 3 выше) обрабатывали в две серии следующим образом: смесь неочищенного N1-бензил-6-бром-1Hиндазола (153 г, содержащего максимум 0,5 моль, предполагая 100% выхода при бензилировании/равновесии) и NaOH (40 г, 1,0 моль) в THF/вода (5:1, 1,5 л; квалификации при ВЭЖХ без ингибитораTHF) продували азотом. В отдельной колбе вместе перемешивали Pd(OAc)2 (1,13 г, 5,0 ммоль, 1 мол.%) иPPh3 (2,6 г, 10,0 ммоль, 2 мол.%) в продуваемом азотом THF (250 мл) в течение 10 мин с образованием красного раствора с некоторым количеством суспендированных твердых частиц. 4,4,6-Триметил-2 винил-1,3,2-диоксаборинан (112 мл, 0,65 моль) и каталитический раствор добавляли в реакционную смесь, и полученный раствор снова продували азотом. Смесь нагревали в течение ночи в масляной бане,установленной на 60C. 1 Н ЯМР образца указывал на то, что осталось некоторого количество исходного вещества, и, таким образом, дополнительный винильный донор (30 мл) добавляли для ускорения завершения. Обе партии смеси объединяли и смесь концентрировали при пониженном давлении с удалением большего количества THF, затем разбавляли водой, насыщенным солевым раствором и циклогексаном. Слои разделяли и водную фазу экстрагировали дополнительным количеством циклогексана, пока ТСХ не показало, что весь необходимый продукт был экстрагирован (всего 3,5 л циклогексана). Объединенные органические порции промывали насыщенным солевым раствором, сушили над MgSO4 и затем пропускали через 2 см прокладку доиксида кремния с удалением исходного вещества. Любой оставшийся продукт на диоксиде кремния элюировали с применением 10% EtOAc в циклогексане (Rf. 0,15 в этом элюенте). Объединенный элюат концентрировали с получением 309 г неочищенного масла, содержащего названное соединение, небольшое количество диола, полученного из винильного донора, и некоторое количество содержащих бензил примесей. Способ 4. Дополнительное взаимодействие проводили с применением дистиллированного N1 бензил-6-бром-1H-индазола (64,3 г, 0,144 моль) с получением полного преобразования без потребности в дополнительной порции винильного донора и получали полуочищенный N1-бензил-6-винил-1H-индазол(55,5 г, количественно), который использовали без дополнительной очистки ниже. Синтез (S)-1-(N1-бензил-1 Н-индазол-6-ил)-этан-1,2-диола Способ 1. K3Fe(CN)6 (16,7 г, 51,0 ммоль), K2CO3 (7,05 г, 51,0 ммоль), (DHQ)2PHAL (0,13 г, 0,17 ммоль, 1 мол.%) и K2OsO4,2H2O (12,8 мг, 0,034 ммоль, 0,2 мол.%) помещали в колбу с округленным дном. Добавляли смесь tBuOH и воды (1:1, 160 мл) с образованием прозрачной, двухфазной смеси при перемешивании. Смесь охлаждали в ледяной бане, приводя к частичному осаждению перед добавлением порошкообразного N1-бензил-6-винил-1H-индазола (4,0 г, 17,1 ммоль). Полученную смесь энергично перемешивали в ледяной бане в течение 5 ч, при этом не наблюдали дополнительное количество твердых веществ и при помощи ТСХ показывали расход исходного вещества Реакцию гасили путем добавления метабисульфита натрия (40 г) с образованием газовыделения, что вызывает переливание реакционной смеси в ледяную баню. В ледяную баню добавляли оставшееся вещество и полученную смесь (содержащую приблизительно 1 л воду и лед) перемешивали в течение ночи, медленно нагревали. Добавляли целит и CH2Cl2 (200 мл), смесь тщательно перемешивали и затем фильтровали. Твердые вещества тщательно промывали дополнительным количеством CH2Cl2 (250 мл). Двухфазный фильтрат отделяли и водный слой экстрагировали CHCl3 (450 мл). Объединенные органические порции промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Остаток переносили вEtOAc и фильтровали через прокладку диоксида кремния (1 см глубиной 8 см диаметром) с элюированием дополнительным количеством EtOAc с удалением основного вещества. Элюат концентрировали и десорбировали толуолом с удалением следов tBuOH. В заключение, остаток перекристаллизовывали из горячего толуола (10 мл/г) с получением названного соединения в виде белых иголок (3,87 г, 84%, 98,8% э.и.) с основным (S) энантиомером с элюированием приблизительно 16,8 мин (Daicel Chiralpak IB(2504,6 мм); изократический 10% EtOH в н-гептане; 1 мл/мин; подходящая температура (прибл. 22C); Определение: 254, 230, 210 нм); от рацемического стандартного образца время удержания (R) энантиомера было 14,8 мин с применением этого способа и N1-бензил-6-винил-1H-индазол элюировали приблизительно 5,4 мин. 1H ЯМР и массовые спектральные данные были идентичными рацемическому 1-(1 бензил-1 Н-индазол-6-ил)-этан-1,2-диолу, полученному выше. Оптическое вращение: []22n = 13 (с 1,018,MeOH). Способ 2. Полуочищенный N1-бензил-6-винил-1H-индазол (способ 4 выше, 55,5 г) дигидроксилировали подобным способом с получением, после перекристаллизации, 2 порций твердого вещества, чистого (S)-1-(N1-бензил-1H-индазол-6-ил)-этан-1,2-диола (38 г, количественно). Способ 3. K3Fe(CN)6 (0,98 кг, 3 моль), K2CO3 (0,55 кг, 3 моль), (DHQ)2PHAL (3,9 г, 5,0 ммоль) иK2OsO4,2H2O (0,37 г, 1 ммоль) помещали в 10 л реакционный сосуд с верхним зажимом, оборудованный мешалкой сверху. Добавляли смесь tBuOH и воды (1:1, 7,5 л) с образованием прозрачной, двухфазной смеси при перемешивании. Смесь охлаждали с применением холодильника Haake EK90, приводя к частичному осаждению, перед добавлением неочищенного N1-бензил-6-винил-1H-индазола (прибл. 0,7-0,8 моль). Полученную смесь энергично перемешивали, но отвердевшее твердое вещество создавало недостаточное пространство для характерной циркуляции в охлаждающей бане и истинная температура падала до приблизительно -20C, когда ее оставляли на выходные. Наблюдали небольшое преобразование. Для ускорения реакции дополнительно добавляли (DHQ)2PHAL (2,5 ммоль) и K2OsO4,2H2O (0,5 ммоль) и смесь оставляли нагреваться до прибл. 10C; реакция затем протекала удовлетворительно. Реакцию гасили порционным добавлением метабисульфита натрия (1,5 кг). Смесь перемешивали в течение 1 ч при к.т., она становилась почти прозрачной, затем фильтровали через прокладку целита с удалением осадкаOsO2. Фильтрат экстрагировали CH2Cl2 (4 экстракта, конечный объем 7 л), и объединенные органические порции сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт перекристаллизовывали из горячего толуола (10 мл/г); собирали две части названного соединения, 98,7% и 98,0% э.и.,всего 163,7 г (55% от 6-бром-1H- индазола). Синтез 2-(N1-бензил-1 Н-индазол-6-ил)-2-метансульфонилоксиэтилового сложного эфира (S)метансульфоновой кислоты Способ 1. Раствор (S)-1-(N1-бензил-1 Н-индазол-6-ил)-этан-1,2-диола (3,75 г, 14,0 ммоль, 98,8% э.и.) и Et3N (4,9 мл, 35,0 ммоль) в сухом CH2Cl2 (350 мл) охлаждали в ледяной бане перед добавлением по каплям MsCl (2,17 мл, 28.0 ммоль) в течение 10 мин. Полученную смесь оставляли перемешиваться в течение 30 мин. После разбавления дополнительным количеством CH2Cl2 (250 мл) раствор промывали холодной 1,0 М водн. HCl (250 мл), нас. водн. NaHCO3 (50 мл) и насыщенным солевым раствором (50 мл), затем сушили над Na2SO4. Раствор выливали на короткую прокладку их диоксида кремния (1 см глубины 8 см диаметром) под вакуумом. Начальный фильтрат не содержал никакого продукта; его впоследствии элюировали 1:1 Et2O/CH2Cl2. Элюат концентрировали при пониженном давлении с получением названного соединения (5,98 г, колич.) в виде белого твердого вещества. 1H ЯМР и массовые спектральные данные были идентичны полученному выше рацемическому 2-(1-бензил-1H-индазол-6-ил)-2 метансульфонилоксиэтиловому сложному эфиру метансульфоновой кислоты. Э.и. этой партии вещества не определяли на этой стадии, но ее проводили перед следующей стадией. Оптическое вращение: []22DEt3N (174 мл, 1,25 моль) в CH2Cl2 (2,5 L) охлаждали в ледяной бане перед медленным добавлением MsCl(81,3 мл, 1,05 моль) за прибл. 1 ч. Внутреннюю температуру повышали до максимум 11C. Полученную смесь оставляли перемешиваться в течение 30 мин. Реакцию гасили холодным 1,0 М водн. HCl (400 мл),фазы разделяли и органическую фазу промывали дополнительным холодным 1,0 М водн. HCl, водн.NaHCO3 и насыщенным солевым раствором, затем сушили над MgSO4. Раствор выливали на короткую прокладку из диоксида кремния под вакуумом. Некоторое количество продукта элюировали из диоксида кремния во время этой фильтрации и остаток элюировали с применением 1:1 Et2O/CH2Cl2 (2 л). Элюат концентрировали при пониженном давлении с получением твердого белого вещества. Его перетирали в порошок с Et2O (800 мл) в течение ночи. Мелкий белый порошок собирали фильтрацией и промывали дополнительным количеством Et2O (2100 мл) с получением названного соединения (184,2 г, 87%, 99% э.и.) с основным (S) энантиомером с элюированием приблизительно 13,4 мин (Daicel Chiralpak IB(2504,6 мм); изократический 30% EtOH в н-гептане; 1 мл/мин; температура окружающей среды (прибл. 22C); Определение: 254, 230, 210 нм); Из рацемического стандартного образца время удержания (R) энантиомера было 14,4 мин с применением этого способа Фильтрат содержал только небольшое количество продукта низкого э.и. и он был отброшен. Синтез (1R,2S)-1'-бензил-2-(1-бензил-1H-индазол-6-ил)спиро[циклопропан-1,3'-индолин]-2'-она Способ 1. Раствор N-бензилоксиндола (3,57 г) в сухом THF (120 мл) охлаждали в ледяной бане перед добавлением NaH (60 мас.% в минеральном масле, 1,92 г, 48,0 ммоль) в четырех порциях; раствор быстро становился сильно пурпурным. Через 30 мин раствор 2-(N1-бензил-1 Н-индазол-6-ил)-2 метансульфонилоксиэтилового сложного эфира (S)-метансульфоновой кислоты (6,79 г, 16,0 ммоль,98,5% э.и., ранее десорбированного дважды сухим THF) в сухом THF (80 мл) добавляли шприцевым насосом за 1 ч. При помощи ТСХ указывали быстрое превращение с отдельное соединение с Rf 0,45(25% EtOAc в циклогексане, элюировали дважды; исходные вещества Rf 0,5 и Rf 0,2). После перемешивания в течение 2 ч смесь выливали в нас. водн. NH4Cl (50 мл), разбавляли водой (50 мл), и EtOAc (100 мл). Фазы разделяли и водный слой экстрагировали дополнительными порциями EtOAc (450 мл). Объединенные органические порции промывали насыщенным солевым раствором, сушили над Na2SO4,фильтровали и концентрировали с получением неочищенного продукта, который при помощи 1 Н ЯМР состоял почти исключительно из названного соединения. Неочищенный продукт пропускали через короткую прокладку диоксида кремния (1 см глубиной 5 см диаметром) с элюированием 1:1 EtOAc в циклогексане. Остаток перетирали в порошок с н-гептаном (350 мл) с удалением минерального масла, и десорбировали толуолом с получением названного соединения (7,0 г, до 90% выхода) в виде стеклообразного твердого вещества, которое содержало некоторое количество растворителя. ВЭЖХ указывала оптическую чистоту 98% э.и. с основным (1R,2S) энантиомером с элюированием приблизительно 13,3 мин (Daicel Chiralpak IA, 2504,6 мм; изократический 10% EtOH в н-гептане; 1 мл/мин; температура окружающей среды (прибл. 22C); Определение: 254, 230, 210 нм); Из рацемического стандартного образца время удержания (1S,2R) энантиомера 12,1 мин с применением этого способа.(t, J = 7,4 Гц, 1H), 5,76 (d, J = 7,3 Гц, 1H), 5,61 (d, J = 15,8 Гц, 1H), 5,53 (d, J = 15,8 Гц, 1H), 5,08 (d, J = 15,6 Гц, 1H), 4,97 (d, J = 15,7 Гц, 1H), 3,48 (t, J = 8,5 Гц, 1H), 2,28 (dd, J = 9,0, 4,5 Гц, 1H), 2,02 (dd, J = 8,0, 4,6 Гц, 1H). MS (ES+): 456 ([M+H]4), рассчитано для [C31H25N3O + H]+ 456,2. Способ 2. В отдельном наборе отдельных экспериментов, которые проводили подобным способом как способ 1, но без выполнения колоночной хроматографии, с использованием 20-45 г 2-(N1-бензил-1 Ниндазол-6-ил)-2-метансульфонилоксиэтилового сложного эфира (S)-метансульфоновой кислоты на партию, далее использовали всего 133,8 г, 315 ммоль. Некоторые партии объединяли и пропускали через прокладку из диоксида кремния с удалением следов основного вещества перед использованием, но, казалось, это не имеет никакого значения в следующих реакциях. Неочищенный продукт выделяли в виде пенистого твердого вещества (174,1 г, содержащего минеральное масло из гидрида натрия, составляющего приблизительно 10% на каждый неочищенный продукт, а также переменные количества EtOAc, оценивали следующий выход 80% на основе установленной отдельной чистоты партии). Вещество использовали без дополнительной очистки. Синтез (1R,2S)-1'-бензил-2-(1-бензил-1 Н-индазол-6-ил)спиро[циклопропан-1,3'-индолин]-2'-она Способ 1. Раствор N-бензилоксиндола (3,57 г) в сухом THF (120 мл) охлаждали в ледяной бане перед добавлением NaH (60 мас.% в минеральном масле, 1,92 г, 48,0 ммоль) за четыре порции; раствор быстро становился сильно пурпурным. Через 30 мин добавляли раствор 2-(N1-бензил-1 Н-индазол-6-ил)-2 метансульфонилоксиэтилового сложного эфира (S)-метансульфоновой кислоты (6,79 г, 16,0 ммоль,98,5% э.и., ранее десорбированный дважды сухим THF) в сухом THF (80 мл) шприцевым насосом за 1 ч. ТСХ указывала быстрое преобразование в отдельное соединение с Rf 0,45 (25% EtOAc в циклогексане,элюировали дважды; исходные вещества Rf 0,5 и Rf 0,2). После перемешивания в течение 2 ч, смесь выливали в нас. водн. NH4Cl (50 мл), разбавляли водой (50 мл) и EtOAc (100 мл). Фазы разделяли и водный слой экстрагировали дополнительными порциями EtOAc (450 мл). Объединенные органические порции промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который при помощи определения 1H ЯМР состоял почти исключительно из названного соединения. Неочищенный продукт пропускали через короткую прокладку диоксида кремния (1 см глубиной 5 см диаметром), с элюированием 1:1 EtOAc в циклогексане. Остаток перетирали в порошок с н-гептаном (350 мл) с удалением минерального масла, и десорбировали толуолом с получением названного соединения (7,0 г, до 90% выхода) в виде стеклообразного твердого вещества,которое содержало некоторое количество растворителя. ВЭЖХ указывала оптическую чистоту 98% э.и. с основным (1R,2S) энантиомером с элюированием приблизительно 13,3 мин (Daicel Chiralpak IA, 2504,6 мм; изократический 10% EtOH в н-гептане; 1 мл/мин; температура окружающей среды (прибл. 22C); Определение: 254, 230, 210 нм); из рацемического стандартного образца время удержания (1S,2R) энантиомера было 12,1 мин с применением этого способа. 1 Названное соединение получали способом, подобным способу получения (1R,2S)-1'- бензил-2-(1 бензил-1 Н-индазол-6-ил)спиро[циклопропан-1,3'-индолин]-2'-она с применением (S)-1-(1-бензил-1Hиндазол-6-ил)этан-1,2-диилдиметансульфоната (501,4 мг, 1,181 ммоль) и 1-бензил-5-фториндолин-2-она(285,0 мг, 1,181 ммоль). Очисткой с применением Biotage Isolera (SNAP 25g колонка, 25-100% EtOAc в гексане) получали названное соединение в виде кремового твердого вещества (352 мг, 63%; 97% э.и.) с основным (1R,2S) энантиомером с элюированием приблизительно 7,03 мин (Phenomenex Lux 5 Cellulose1 (1504,6 мм), 1,0 мл/мин изократический при 80% EtOH в гексане в течение 1,0 мин, затем градиент 8090% EtOH в гексане в течение 10 мин). Из рацемического стандартного образца время удержания (1S,2R) энантиомера было 5,95 мин с применением этого способа. 1 Названное соединение получали способом, подобным способу хирального синтеза соединения согласно примеру А 4 с применением(1R,2S)-1'-бензил-2-(1-бензил-1 Н-индазол-6-ил)-5'фторспиро[циклопропан-1,3'-индолин]-2'-она (560 мг, 1,18 ммоль). Очисткой с использованием колоночной хроматографии с силикагелем с 5-95% EtOAc в гексане получали названное соединение в виде кремового твердого вещества (179 мг, 52%). 1 Названное соединение получали способом, подобным способу хирального синтеза соединения согласно примеру А 10 с применением (1R,2S)-5'-фтор-2-(1 Н-индазол-6-ил)спиро[циклопропан-1,3'индолин]-2'-она (240 мг, 0,818 ммоль). Очисткой с применением Biotage Isolera с SNAP 25g колонки с 590% EtOAc в гексане получали названное соединение в виде кремового твердого вещества (195 мг, 57%; 97% э. и.) с основным (1R,2S) энантиомером с элюированием приблизительно 3,7 мин (Phenomenex Lux 5Cellulose-2 (1504,6 мм); изократический 25% EtOH в н-гексане; 1,5 мл/мин; 24C; определение: 254 нм). Из рацемического стандартного образца время удержания (1S,2R) энантиомера было 3,2 мин с применением этого способа. 1(20 мл) и полученную смесь охлаждали до 0C. Раствор 1-бензил-5- метилиндолин-2-она (2,37 г, 10 ммоль) в сухом THF (25 мл) добавляли в течение 2 мин, затем промывали THF (5 мл). После перемешивания в течение 20 мин при 0C по каплям добавляли раствор (S)-1-(1-бензил-1 Н-индазол-6-ил)этан-1,2 диилдиметансульфоната (4,24 г, 10 ммоль) в сухом THF (45 мл) через капельную воронку в течение 40 мин, с последующим промыванием THF (5 мл). После добавления полученную смесь перемешивали в течение 30 мин при 0C (ТСХ показала завершение), затем оставляли на O/N при к.т. После охлаждения до 0C реакционную смесь выливали в колбу Эрленмейера, содержащую лед (100 мл) и нас. NH4Cl (30 мл) и экстрагировали EtOAc (150 мл 2), сушили (Na2SO4). После удаления растворителей остаток переносили в 100 мл КДК с применением 30 мл EtOAc и образовывались кристаллы. Фильтрованием с отсасыванием получали названное соединение в виде бежевого твердого вещества (1,537 г). Фильтрат кон- 27023173EtOAc/гексаном с получением 2-го выхода в виде беловатого твердого вещества (1,560 г). Фильтрат очищали с применением вышеуказанной методики с получением 3-го выхода в виде бежевого твердого вещества (115 мг). Всего 3,212 г (68%). 1(1R,2S)-1'-бензил-2-(1-бензил-1 Н-индазол-6-ил)-5'метилспиро[циклопропан-1,3'-индолин]-2'-оном (469 мг, 1 ммоль), добавляли сухой THF (2 мл) и полученную смесь перемешивали при 0C перед добавлением KOtBu (1 М в THF. 18 мл, 18 ммоль) в течение 2 мин. После добавления полученную смесь перемешивали в течение 15 мин при 0C и добавляли DMSO(1,85 мл). Кислород барботировали через нее в течение 1 ч и реакция превратилась из гомогенной в гетерогенную. LC-MS показали хорошее преобразование за 50 мин. Ее гасили нас. NH4Cl. Вышеуказанную реакцию повторяли при большем масштабе с применением (1R,2S)-1'-бензил-2-(1 бензил-1H-индазол-6-ил)-5'-метилспиро[циклопропан-1,3'-индолин]-2'-она (1,41 г, 3 ммоль). После гашения насыщенным NH4Cl объединяли две реакции, разбавляли H2O и экстрагировали EtOAc (100 мл 2). Очисткой Biotage Isolera (10-95% EtOAc в гексане) получали названное соединение в виде светложелтого твердого вещества (680 мг, 53%). 1 В раствор (1R,2S)-2-(N1-индазол-6-ил)-5'-метилспиро[циклопропан-1,3'-индолин]-2'-она (680 мг,2,35 ммоль) в DMF (16 мл) добавляли K2CO3 (544 мг, 4 ммоль), а затем йод (851 мг, 3,2 ммоль). Полученную смесь перемешивали в течение 3 ч при к.т., охлаждали до 0C, гасили нас. Na2S2O3, разбавляли Н 2 О, экстрагировали EtOAc (50 мл 3) и сушили (Na2SO4). Путем выпаривания растворителей и очисткойBiotage Isolera (градиент EtOAc/гексан: 10-90%) получали названное соединение в виде светло-желтого твердого вещества (794 мг, 81%; 98% э.и.). Основной (1R,2S)-энантиомер элюировали приблизительно 9,6 мин (Phenomenex Lux 5u Cellulose-2 (1504,6 мм); изократический 10% EtOH в н-гексане 1,75 л/мин; температура окружающей среды; Определение: 254, 214 нм). Из рацемического стандартного образца время удержания (1R,2S)-энантиомера было 7,7 мин с применением этого способа. 1 Названное соединение получали способом, подобным способу получения (1R,2S)-1'-бензил-2-(1 бензил-1H-индазол-6-ил)спиро[циклопропан-1,3'-индолин]-2'-она с применением (S)-1-(1-бензил-1 Ниндазол-6-ил)этан-1,2-диилдиметансульфоната (6,70 г, 15,8 ммоль) и 1-метилиндолин-2-она (2,33 г, 15,8 ммоль). Очисткой колоночной хроматографией (силикагель, 25-50% EtOAc в гексане) получали названное соединение в виде бледно-оранжевого кристаллического твердого вещества (5,01 г, 84%). Названное соединение получали способом, подобным способу хирального синтеза соединения согласно примеру А 4 с применением (1R,2S)-2-(1-бензил-1 Н-индазол-6-ил)-1'-метилспиро[циклопропан 1,3'-индолин]-2'-она (1,16 г, 3,06 ммоль). Очисткой колоночной хроматографией (силикагель, 3-6% МеОН в CH2Cl2) получали названное соединение в виде бледно-желтого твердого вещества (656 мг,74%). 1 Н ЯМР (400 МГц, CDCl3)10,06 (br. s, 1H), 8,05 (s, 1H), 7,64 (d, 1H, J = 7,6 Гц), 7,36 (s, 1H), 7,14 (t,J = 8,7 Гц, 1H), 6,97 (d, J = 8,7 Гц, 1H), 6,87 (d, J = 8,2 Гц, 1H), 6,62 (t, J = 7,6 Гц, 1H). 5,91 (d, J = 7,9 Гц,1H), 3,46 (t, J = 7,8 Гц, 1H), 3,34 (s, 3H), 2,26-2,23 (m, 1H), 2,08-2,04 (m, 1 Н); MS ESI 290,1 [М + Н]+, рассчитано для [C18H15N3O+ H]+ 290,13. Синтез (1R,2S)-2-(3-йод-1H-индазол-6-ил)-1'-метилспиро[циклопропан-1,3'-индолин]-2'-она Названное соединение получали способом, подобным способу хирального синтеза соединения согласно примеру А 10 с применением (1R,2S)-2-(1 Н-индазол-6-ил)-1'-метилспиро[циклопропан-1,3'индолин]-2'-она (930 мг, 3,21 ммоль). Осаждением с EtOAc с последующей фильтрацией и промываниемEtOAc получали названное соединение (970 мг, 73%; 98 % э.и.) с основным энантиомером с элюированием приблизительно 2,4 мин (Phenomenex Lux 5 Amylose-2 1504,6 мм, 2,5 мл/мин с изократическим при 20% EtOH в гексане в течение 0,5 мин, затем градиент 20-50% EtOH в гексане в течение 2,5 мин, затем с изократическим при 50% в течение 1 мин). Из рацемического стандартного образца время удержания (1S,2R) энантиомера было 3,0 мин с применением этого способа. 1 Названное соединение получали способом, подобным способу получения (1R,2S)-1'-бензил-2-(1 бензил-1H-индазол-6-ил)спиро[циклопропан-1,3'-индолин]-2'-она с применением (S)-1-(1-бензил-1Hиндазол-6-ил)этан-1,2-диилдиметансульфоната (1,44 г, 3,39 ммоль) и 5-метокси-1-метилиндолин-2-она(0,601 г, 3,39 ммоль). Очисткой с применением Biotage Isolera (1-50% EtOAc в гексане, SNAP 25g колонка) получали названное соединение (светло-коричневое твердое вещество, 1,05 г, 76%). 1(d, J = 7,6 Гц, 2H), 6,95 (d, J = 8,4 Гц, 1H), 6,89 (d, J = 8,4 Гц, 1H), 6,67 (d, J = 8,4 Гц, 1H), 5,63 (d, J = 16,4 Гц, 1H), 5,58 (d, J = 16,0 Гц, 1H), 5,41 (s, 1H), 3,37 (t, J = 8,8 Гц, 1H), 3,15 (s, 3H), 2,23-2,19 (m, 1H), 2,182,14 (m, 1H), -OCH3 протон затемнен пиком метанола. MS ESI 410,2 [M + H]+, рассчитано для
МПК / Метки
МПК: C07D 403/04, A61K 31/5377, A61P 35/00, C07D 413/14
Метки: киназы, способ, злокачественной, опухоли, помощью, лечения, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-23173-ingibitory-kinazy-i-sposob-lecheniya-zlokachestvennojj-opuholi-s-ih-pomoshhyu.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы киназы и способ лечения злокачественной опухоли с их помощью</a>
Ингибиторы jak2 и их применение для лечения миелопролиферативных заболеваний и злокачественной опухоли

Номер патента: 22488
Опубликовано: 29.01.2016
Авторы: Гребински Джеймс В., Харт Эми, Пьюрандэер Ашок В., Ингрим Дженнифер, Ван Хунхе, Шрёдер Гретчен
МПК: A61P 35/00, A61K 31/437, C07D 471/14...
Метки: ингибиторы, лечения, заболеваний, злокачественной, опухоли, применение, миелопролиферативных
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль,где X представляет собой С1-6алкил;Y представляет собой С1-4алкил;R представляет собойкаждый из которых необязательно конденсирован с 5- или 6-членным углеродным кольцом, которое является насыщенным, частично ненасыщенным или полностью ненасыщенным или гетероциклом, причем указанный гетероцикл содержит один гетероатом, выбранный из NR3 или S, указанные конденсированные углеродное...
Способ лечения злокачественной опухоли с использованием анти-erbb3 антитела

Номер патента: 22884
Опубликовано: 31.03.2016
Авторы: Хармс Брайан, Кубасек Уильям, Гиббонс Фрэнсис Дэвид, Шёберл Бирджит, Фицджеральд Джонатан Бэзил, Онсум Мэтью Дэвид, Нильсен Ульрик
МПК: C07K 16/32, C12Q 1/68
Метки: использованием, лечения, опухоли, способ, анти-erbb3, злокачественной, антитела
Формула / Реферат:
1. Способ лечения пациента, имеющего злокачественную опухоль, включающий введение анти-ErbB3 антитела пациенту, при условии что уровень фосфорилированного ErbB3 (pErbB3) в образце опухоли, полученном от пациента, составляет не ниже, чем 0,064 пг/мкг общего белка.2. Способ по п.1, в котором опухоль представляет собой злокачественную опухоль органа, выбранного из толстой кишки, легкого, прямой кишки, желчного пузыря, головного мозга, спинного...
Замещенные производные бензимидазола и их применение для лечения злокачественной опухоли

Номер патента: 6802
Опубликовано: 28.04.2006
Авторы: Карре Шанталь, Депати Изабелль, Клерк Франсуа, Реснер Манфред, Депре Стефани, Ангуйан-Бонифас Одиль, Ами Франсуа
МПК: C07D 235/30, A61P 35/00, A61K 31/4184...
Метки: замещенные, злокачественной, производные, опухоли, применение, лечения, бензимидазола
Формула / Реферат:
1. Соединения формулы (I) где А представляет собой фенил, где R1 выбран из одной или нескольких аналогичных групп, выбранных из алкила, возможно, замещенного алкокси, гетероалкилом, арилом, ацилом, группой ацильного производного, галогеном; алкокси, возможно, замещенного алкилом, гетероалкилом, арилом, гетероарилом, алкоксиалкилом, гидроксиалкиламидом или перфторалкоксигруппой или алкилтио, возможно, замещенного амидом или перфторалкилтио;...
Композиции для доставки лекарственных средств с высокой водорастворимостью (варианты) и их применение в лечении злокачественной опухоли

Номер патента: 12522
Опубликовано: 30.10.2009
Автор: Чэнь Эндрю Сянь
МПК: A61K 9/00, A61K 31/475, A61K 36/24...
Метки: лекарственных, средств, применение, высокой, варианты, лечении, водорастворимостью, композиции, злокачественной, опухоли, доставки
Формула / Реферат:
1. Композиция, содержащая триглицеридное масло, эмульгатор, стабилизатор, воду и алкалоид барвинка или его фармацевтически приемлемую соль, где: (a) композиция представляет собой эмульсию, содержащую масляную и водную фазу, и (b) не менее 80% алкалоида барвинка или его фармацевтически приемлемой соли присутствует в масляной фазе. 2. Композиция по п.1, где алкалоид барвинка представляет собой винкристин, винбластин или винорелбин. 3. Композиция...
Однородная вакцинная композиция для лечения опухоли и способ ее получения

Номер патента: 21905
Опубликовано: 30.09.2015
Авторы: Винья Родригес Лисель, Чико Велис Эрнесто, Кальво Гонсалес Лоани, Лахе Давила Агостин Бьенвенидо, Албиса Ново Айрама, Кромбет Рамос Таниа, Гонсалес Маринелло Гисела Мария, Куэвас Фиалло Ариадна, Родригес Мартинес Грисселль Мария
МПК: A61K 39/385
Метки: лечения, однородная, получения, композиция, способ, вакцинная, опухоли
Формула / Реферат:
1. Способ получения конъюгата hrEGF-P64K, включающий ковалентное сопряженное связывание белка hrEGF с белком-носителем P64K и очистку молекулярного комплекса методом ультрафильтрации/диафильтрации с использованием мембраны, имеющей размер пор, пропускающих белки с молекулярной массой от 50 до 100 кДа согласно следующим стадиям:i) реакция сопряженного связывания, которая включает смешивание белка hrEGF и белка P64K в реакторе сопряженного...
Предыдущий патент: Способ интерпретации данных электромагнитной разведки
Следующий патент: Криогенная система для удаления кислотных газов из потока газообразных углеводородов с удалением сероводорода
Случайный патент: Нагревательный элемент с резистивной поверхностью