Ингибиторы вируса гепатита с























Формула / Реферат
1. Соединение, выбранное из:
метил (2-((2S,5S)-2-(7-((2-((2S,5S)-1-((2S)-2-(4,4-дифторциклогексил)-2-((метоксикарбонил)амино)ацетил)-5-метил-2-пирролидинил)-1Н-бензимидазол-5-ил)этинил)-1H-нафто[1,2-d]имидазол-2-ил)-5-метил-1-пирролидинил)-2-оксо-1-(тетрагидро-2Н-пиран-4-ил)этил)карбамата;
метил ((1S)-1-(((2S,5S)-2-(5-((2-((2S,5S)-1-(((метоксикарбонил)амино)(тетрагидро-2Н-пиран-4-ил)ацетил)-5-метил-2-пирролидинил)-1Н-нафто[1,2-d]имидазол-7-ил)этинил)-1Н-бензимидазол-2-ил)-5-метил-1-пирролидинил)карбонил)-2-метилпропил)карбамата;
метил ((1S)-1-(((2S,4S)-2-(5-((2-((2S,4S)-1-((2S)-2-((метоксикарбонил)амино)-3-метилбутаноил)-4-метил-2-пирролидинил)-1Н-нафто[1,2-d]имидазол-7-ил)этинил)-1Н-бензимидазол-2-ил)-4-метил-1-пирролидинил)карбонил)-2-метилпропил)карбамата;
метил ((1S)-2-((2S,4S)-2-(7-((2-((2S,4S)-1-((2S)-2-((метоксикарбонил)амино)-2-(тетрагидро-2Н-пиран-4-ил)ацетил)-4-метил-2-пирролидинил)-1H-бензимидазол-5-ил)этинил)-1Н-нафто[1,2-d]имидазол-2-ил)-4-метил-1-пирролидинил)-2-оксо-1-(тетрагидро-2Н-пиран-4-ил)этил)карбамата;
метил ((1S)-1-(((3S,5R)-3-(5-((2-((3S,5R)-2-((2S)-2-((метоксикарбонил)амино)-3-метилбутаноил)-5-метил-2-азабицикло[3,1,0]гекс-3-ил)-1Н-нафто[1,2-d]имидазол-7-ил)этинил)-1Н-бензимидазол-2-ил)-5-метил-2-азабицикло[3,1,0]гекс-2-ил)карбонил)-2-метилпропил)карбамата и
метил ((1S)-2-((3S,5R)-3-(7-((2-((3S,5R)-2-((2S)-2-((метоксикарбонил)амино)-2-(тетрагидро-2Н-пиран-4-ил)ацетил)-5-метил-2-азабицикло[3,1,0]гекс-3-ил)-1Н-бензимидазол-5-ил)этинил)-1H-нафто[1,2-d]имидазол-2-ил)-5-метил-2-азабицикло[3,1,0]гекс-2-ил)-2-оксо-1-(тетрагидро-2Н-пиран-4-ил)этил)карбамата,
или его фармацевтически приемлемая соль.
2. Композиция для ингибирования функции белка NS5A, содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
3. Способ лечения HCV инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
Текст
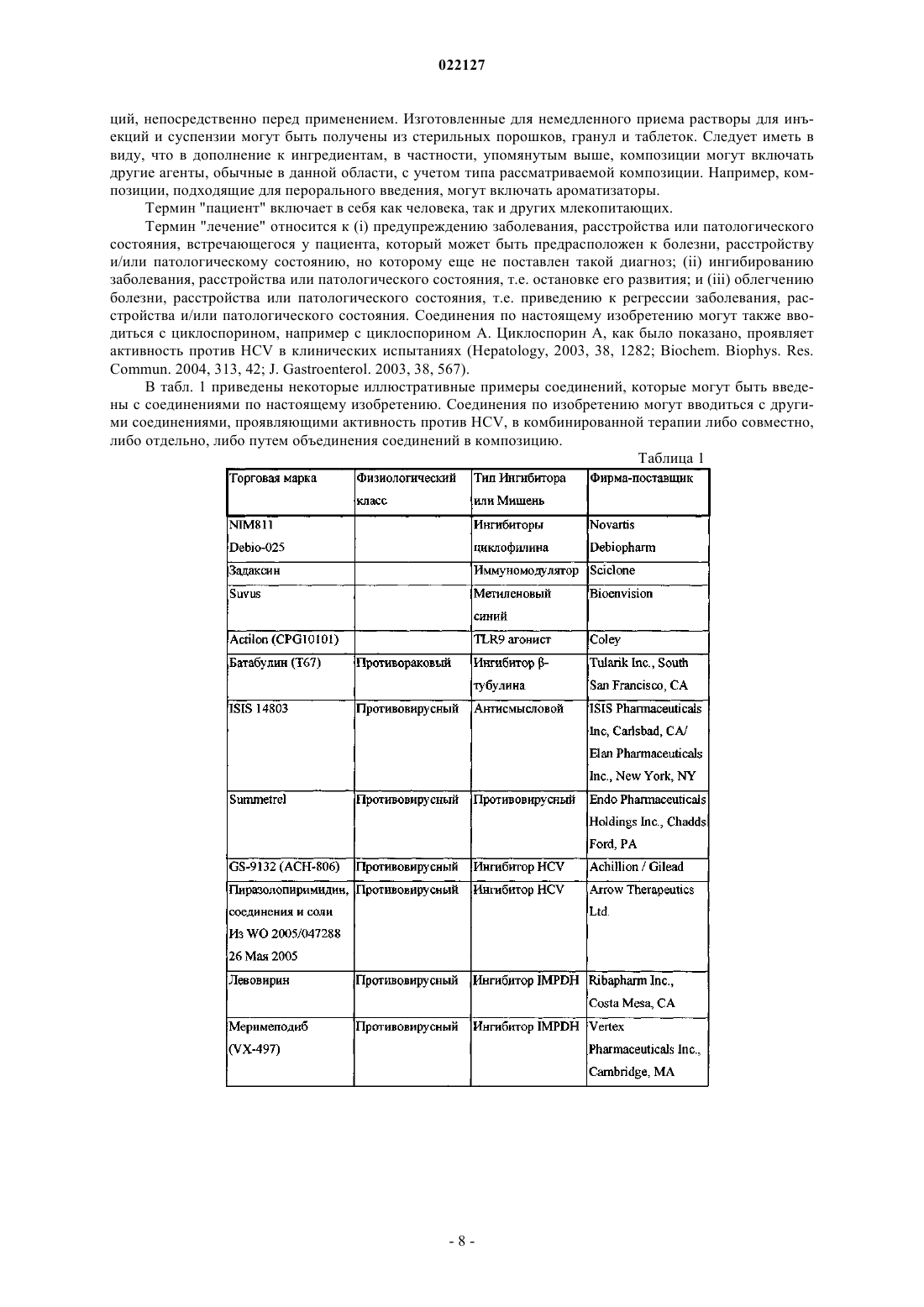
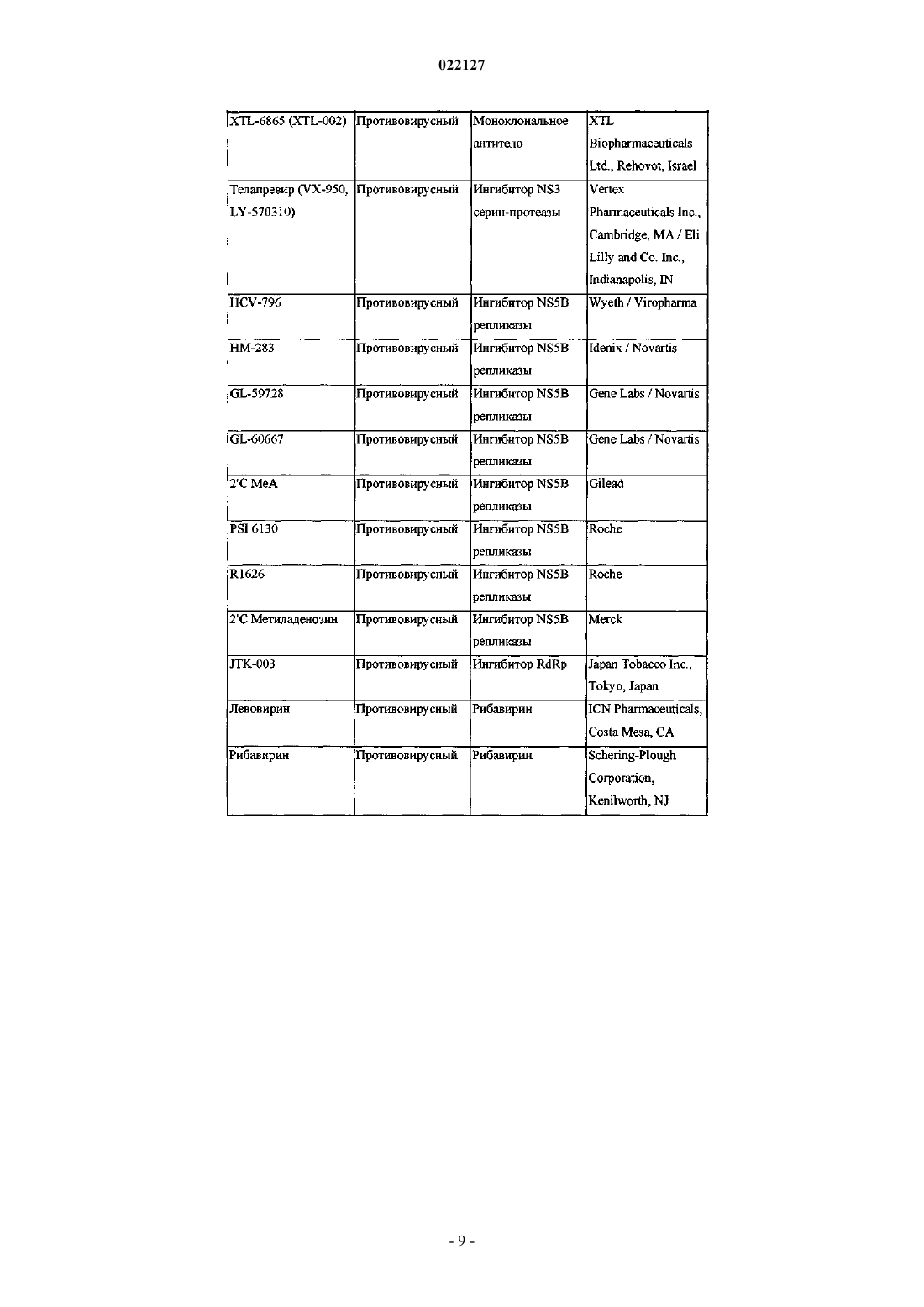
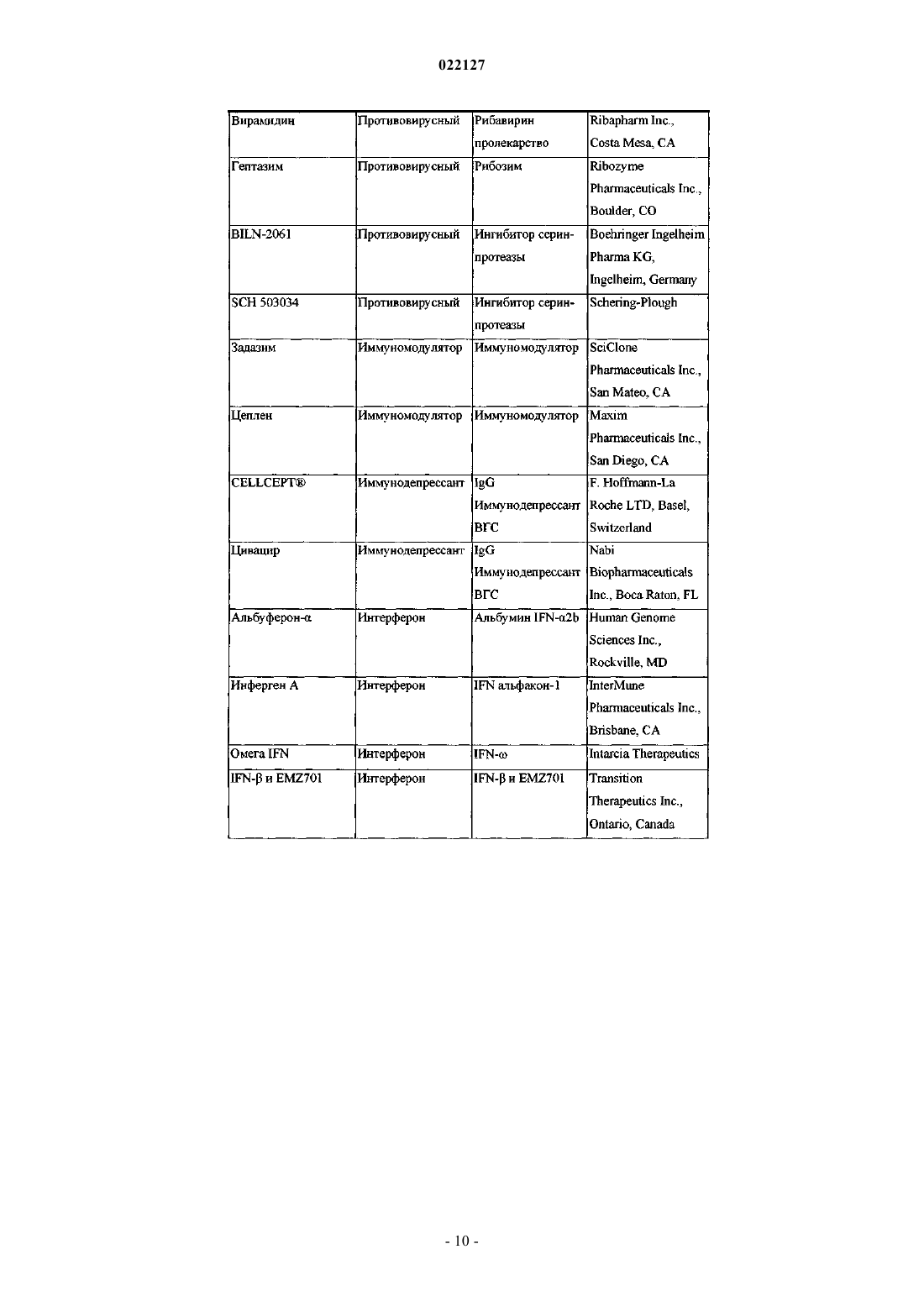
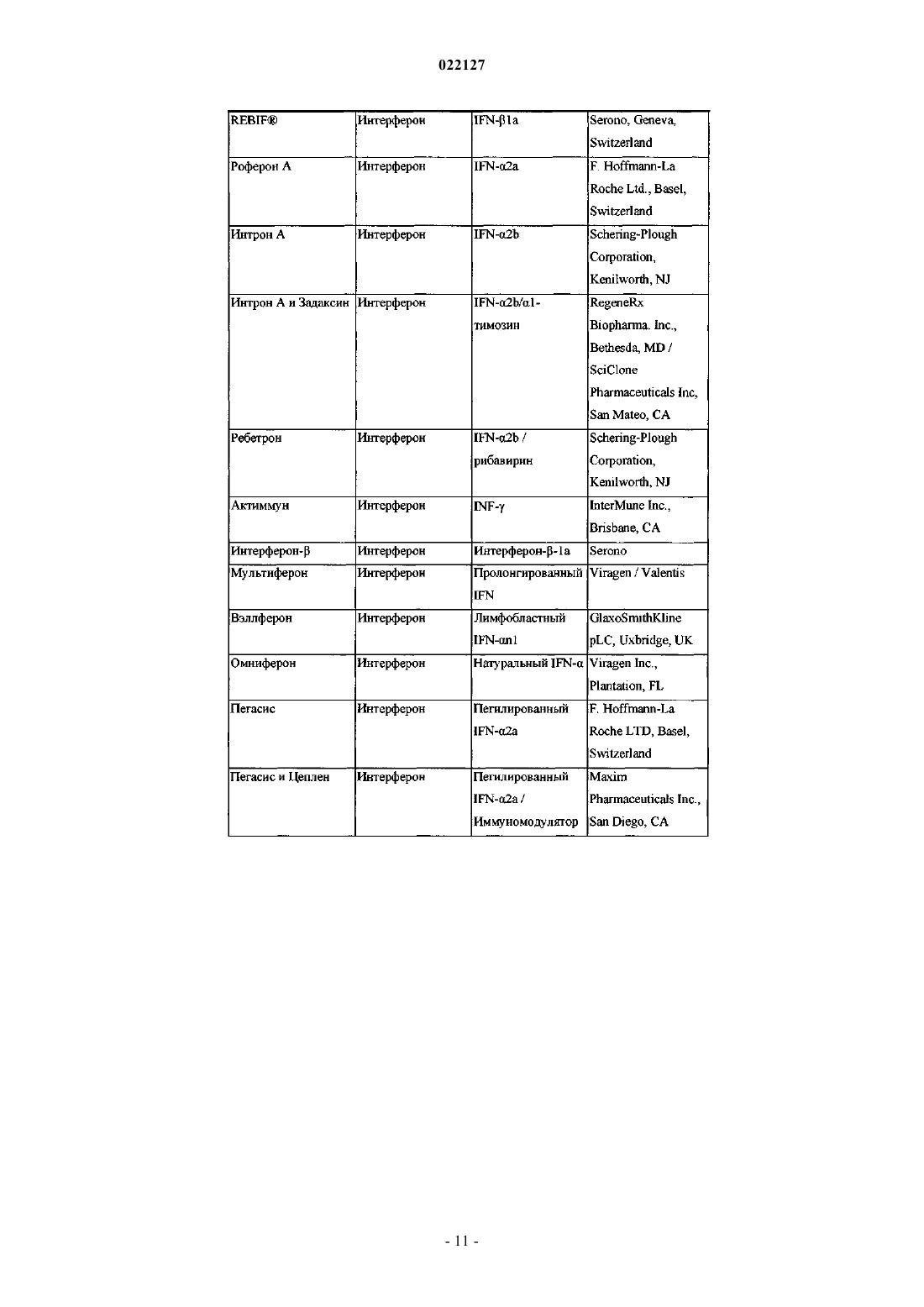
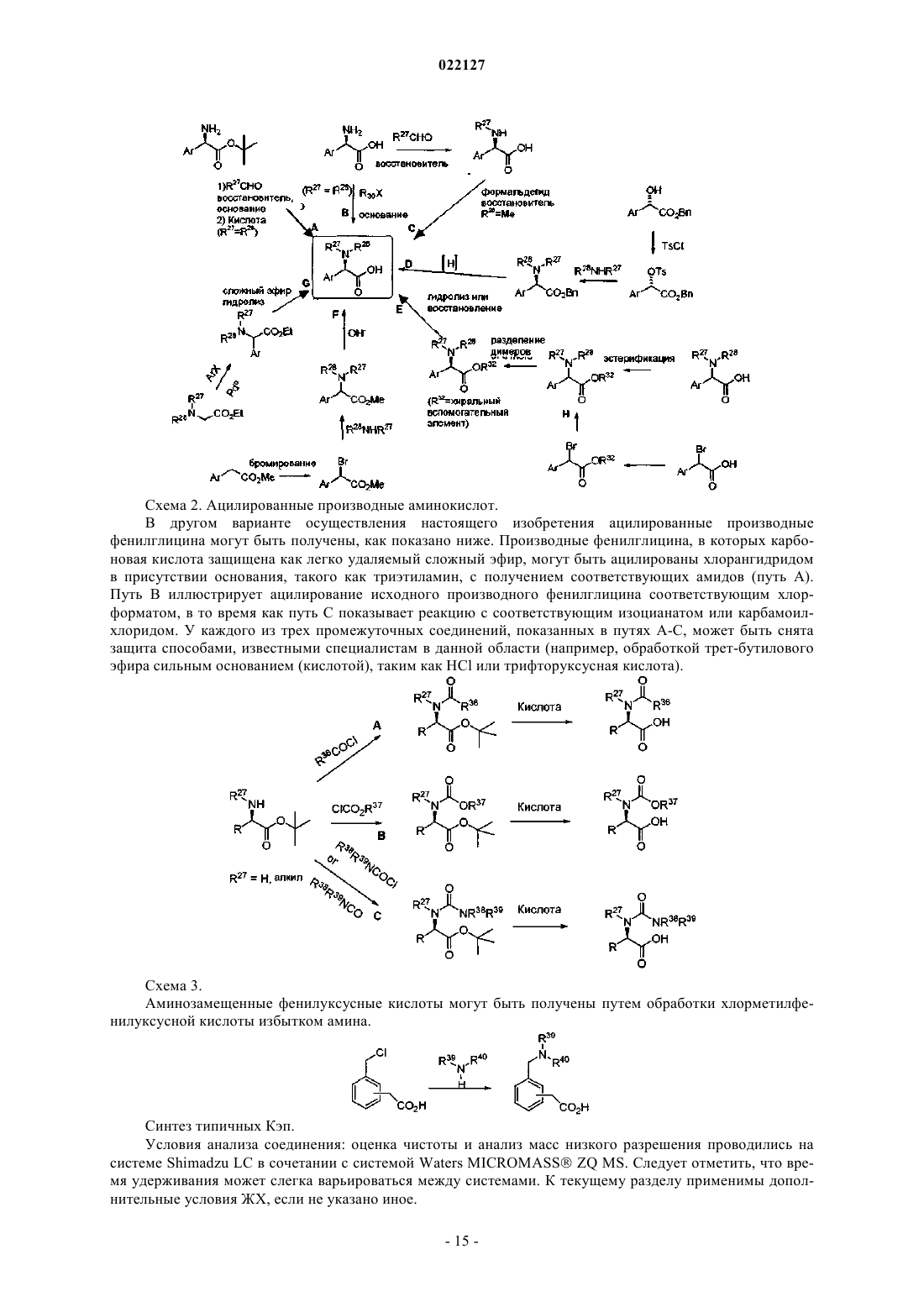
ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С Изобретение относится к соединениям, композициям и способам лечения гепатита С, вызванного вирусной инфекцией (HCV). Также описаны фармацевтические композиции, содержащие такие соединения, и способы применения этих соединений при лечении HCV инфекции. Ромайн Джеффри Ли (US) Лыу Т.Н. (RU) 022127 Настоящее изобретение в общем относится к противовирусным соединениям и, более конкретно, к соединениям, которые могут ингибировать функцию белка NS5A, кодируемого вирусом гепатита C(HCV), к композициям, содержащим такие соединения, и способам ингибирования функции белка NS5A.HCV является одним из основных человеческих патогенов, инфицирующих, по оценкам, 170 млн чел. по всему миру, что примерно в пять раз превышает число инфицированных вирусом иммунодефицита человека типа 1. У значительной части этих инфицированных HCV индивидуумов развиваются серьезные прогрессирующие заболевания печени, включая цирроз печени и гепатоклеточную карциному. В настоящее время стандартное лечение гепатита C, при котором применяется комбинация пегилированного интерферона и рибавирина, имеет неоптимальную частоту успеха в достижении устойчивого вирусологического ответа и вызывает многочисленные побочные эффекты. Так что существует очевидная и давно ощущаемая необходимость разработки эффективных методов для решения этой насущной медицинской потребности.HCV представляет собой вирус, содержащий плюс-цепь РНК. На основании сравнения расшифрованной аминокислотной последовательности и значительного сходства 5'-нетранслируемой области HCV был классифицирован как самостоятельный род в семействе Flaviviridae. Все члены семействаFlaviviridae имеют заключенные в оболочку вирионы, которые содержат геном, представленный плюсцепью РНК, кодирующий все известные вирус-специфические белки посредством трансляции одной непрерывной открытой рамки считывания. Обнаруживается значительная гетерогенность во всем геноме вируса гепатита C на уровне нуклеотидной и кодированной аминокислотной последовательности из-за высокой частоты появления ошибок кодированной РНК-зависимой РНК-полимеразы, которой не хватает способности правильного считывания. Были охарактеризованы по меньшей мере шесть основных генотипов, и более 50 подтипов были описаны как распространенные по всему миру. Клиническая значимость генетической гетерогенностиHCV проявилась в склонности к мутациям, которые возникают при монотерапевтическом лечении, так что является желательным применение дополнительных вариантов лечения. Возможное модуляторное влияние генотипов на патогенез и лечение остается трудным для понимания. Одноцепочечная РНК генома вируса гепатита C включает около 9500 нуклеотидов по длине и имеет одну открытую рамку считывания (ORF), кодирующую один большой полипротеин, состоящий из около 3000 аминокислотных остатков. В инфицированных клетках этот полипротеин расщепляется на многих сайтах посредством клеточных и вирусных протеаз с образованием структурных и неструктурных (NS) белков. В случае с гепатитом C синтез зрелых неструктурных белков (NS2, NS3, NS4A, NS4B,NS5A и NS5B) осуществляется двумя вирусными протеазами. Полагают, что первая протеаза представляет собой металлопротеазу и расщепляет по соединению NS2-NS3. Вторая является сериновой протеазой, содержащейся в N-концевом домене NS3 (также называемом здесь NS3 протеаза), которая опосредует все последующие расщепления в направлении NS3, как в цис на сайте расщепления NS3-NS4A, так и в транс на оставшихся сайтах NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. Белок NS4A, по-видимому, выполняет много функций, либо действуя как кофактор для протеазы NS3, либо способствуя локализации в мембране NS3 и других компонентов репликации вируса. Образование комплекса NS3-NS4A необходимо для нормальной активности протеазы, приводя к увеличению протеолитической эффективности расщепления. Белок NS3 также проявляет нуклеозидтрифосфатазную и РНК геликазную активности. БелокNS5B (также называемый здесь полимераза HCV) является РНК-зависимой РНК-полимеразой, которая вовлекается в репликацию вируса гепатита C с другими белками HCV, включая NS5A, в репликазном комплексе. Соединения, полезные для лечения HCV-инфицированных пациентов, являются целевыми, когда они селективно ингибируют вирусную репликацию, в частности соединения, которые являются эффективными для ингибирования функции белка NS5A. Белок HCV NS5A описан, например, в следующих источниках: S.L. Tan, et al., Virology, 284:1-12 (2001); K.-J. Park, et al., J. Biol. Chem., 30711-30718 (2003);T.L. Tellinghuisen, et al., Nature, 435, 374 (2005); R.A. Love, et al., J. Virol, 83, 4395 (2009); N. Appel, et al.,J. Biol. Chem., 281, 9833 (2006); L. Huang, J. Biol. Chem., 280, 36417 (2005); C. Rice, et al.,WO 2006/093867. В первом аспекте настоящего изобретения описано соединение, выбранное из: метил (2-2S,5S)-2-(7-(2-2S,5S)-1-2S)-2-(4,4-дифторциклогексил)-2-метоксикарбонил)амино)ацетил)-5-метил-2-пирролидинил)-1H-бензимидазол-5-ил)этинил)-1 Н-нафто[1,2-d]имидазол-2-ил)-5 метил-1-пирролидинил)-2-оксо-1-(тетрагидро-2 Н-пиран-4-ил)этил)карбамата; метил 1S)-1-2S,5S)-2-(5-2-2S,5S)-1-метоксикарбонил)амино)(тетрагидро-2 Н-пиран-4 ил)ацетил)-5-метил-2-пирролидинил)-1 Н-нафто[1,2-d]имидазол-7-ил)этинил)-1H-бензимидазол-2-ил)-5 метил-1-пирролидинил)карбонил)-2-метилпропил)карбамата; метил 1S)-1-2S,4S)-2-(5-2-2S,4S)-1-2S)-2-метоксикарбонил)амино)-3-метилбутаноил)-4 метил-2-пирролидинил)-1 Н-нафто[1,2-d]имидазол-7-ил)этинил)-1H-бензимидазол-2-ил)-4-метил-1 пирролидинил)карбонил)-2-метилпропил)карбамата; метил 1S)-2-2S,4S)-2-(7-2-2S,4S)-1-2S)-2-метоксикарбонил)амино)-2-(тетрагидро-2 Нпиран-4-ил)ацетил)-4-метил-2-пирролидинил)-1 Н-бензимидазол-5-ил)этинил)-1 Н-нафто[1,2-d]имидазол-1 022127 2-ил)-4-метил-1-пирролидинил)-2-оксо-1-(тетрагидро-2 Н-пиран-4-ил)этил)карбамата; метил 1S)-1-3S,5R)-3-(5-2-3S,5R)-2-2S)-2-метоксикарбонил)амино)-3-метилбутаноил)-5 метил-2-азабицикло[3,1,0]гекс-3-ил)-1 Н-нафто[1,2-d]имидазол-7-ил)этинил)-1H-бензимидазол-2-ил)-5 метил-2-азабицикло[3,1,0]гекс-2-ил)карбонил)-2-метилпропил)карбамата и метил 1S)-2-3S,5R)-3-(7-2-3S,5R)-2-2S)-2-метоксикарбонил)амино)-2-(тетрагидро-2 Нпиран-4-ил)ацетил)-5-метил-2-азабицикло[3,1,0]гекс-3-ил)-1 Н-бензимидазол-5-ил)этинил)-1H-нафто[1,2d]имидазол-2-ил)-5-метил-2-азабицикло[3,1,0]гекс-2-ил)-2-оксо-1-(тетрагидро-2H-пиран-4 ил)этил)карбамата,или его фармацевтически приемлемая соль. Второй аспект настоящего изобретения относится к композиции для ингибирования функции белкаNS5A, содержащей вышеуказанное соединение или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Третий вариант осуществления настоящего изобретения относится к способу лечения HCV инфекции у пациента, включающему введение пациенту терапевтически эффективного количества вышеуказанного соединения или его фармацевтически приемлемой соли. Другие варианты осуществления настоящего изобретения могут включать подходящие комбинации из двух или более вариантов осуществления изобретения и/или аспектов, описанных здесь. Еще другой вариант осуществления изобретения и аспекты изобретения будут очевидны в соответствии с описанием, представленным ниже. Соединения по настоящему изобретению также существуют в виде таутомеров, поэтому настоящее изобретение также охватывает все таутомерные формы. Описание настоящего изобретения здесь должно рассматриваться в соответствии с законами и принципами образования химической связи. Следует понимать, что соединения, охватываемые настоящим изобретением, отвечают соответствующим требованиям стабильности для использования в качестве лекарственного средства. Предполагается, что определение любого заместителя или переменной (например, R2 и R4) в конкретном месте в молекуле не зависит от их определения в другом месте в этой молекуле. Например, еслиn представляет собой 2, то каждая из двух групп R2 может быть той же самой или другой. Все патенты, патентные заявки и литературные источники, цитируемые в описании, включены сюда посредством отсылки в полном объеме. В случае несоответствия настоящее изобретение, включая определения, будет иметь приоритетное значение. Следующие термины, используемые в данном описании, имеют значения, показывающие, если не указано иное, что все арильные, циклоалкильные и гетероциклильные группы по настоящему изобретению могут быть замещены, как описано в каждом из их соответствующих определений. Например,арильный фрагмент арилалкильной группы может быть замещен, как описано в определении термина"арил". Термин "алкокси", использованный здесь, относится к алкильной группе, соединенной с исходным молекулярным фрагментом посредством атома кислорода. Термин "алкоксикарбонил", использованный здесь, относится к алкоксигруппе, соединенной с исходным молекулярным фрагментом посредством карбонильной группы. Термин "алкил", использованный здесь, относится к группе, производной от неразветвленной или разветвленной насыщенной углеводородной цепи, содержащей от 1 до 6 атомов углерода. В соединениях по настоящему изобретению, в которых m представляет собой 1 и R4 представляет собой алкил, этот алкил может необязательно образовывать 3- или 6-членное конденсированное кольцо со смежным углеродным атомом, образуя одну из структур, показанных ниже:R50 представляет собой алкил. Когда w представляет собой 2, две R50 алкильные группы могут быть одинаковыми или разными. Термин "арил", использованный здесь, относится к фенильной группе или бициклической конденсированной кольцевой системе, в которой одно или оба кольца представляют собой фенильную группу. Бициклические конденсированные кольцевые системы состоят из фенильной группы, объединенной с 4-6-членным ароматическим или неароматическим карбоциклическим соединением. Арильные группы по настоящему изобретению могут быть соединены с исходным молекулярным фрагментом посредством любого атома углерода в группе, способного к замещению. Типичные представители арильных групп-2 022127 включают, но не ограничиваются ими, инданил, инденил, нафтил, фенил и тетрагидронафтил. Арильные группы по настоящему изобретению необязательно замещены одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из алкокси, алкоксиалкила, алкоксикарбонила, алкила, алкилкарбонила, второй арильной группы, арилалкокси, арилалкила, арилкарбонила, циано, гало, галоалкокси,галоалкила, гетероциклила, гетероциклилалкила, гетероциклилкарбонила, гидрокси, гидроксиалкила,нитро, -NRxRy, (NRxRy)-алкила, оксо и -P(O)OR2, где каждый R независимо выбран из водорода и алкила; и где алкильная часть арилалкила и гетероциклилалкила является незамещенной и где вторая арильная группа, арильная часть арилалкила, арильная часть арилкарбонила, гетероциклил и гетероциклильная часть гетероциклилалкила и гетероциклилкарбонила дополнительно необязательно замещены одним,двумя или тремя заместителями, независимо выбранными из алкокси, алкила, циано, гало, галоалкокси,галоалкила и нитро. Термин "арилалкокси", использованный здесь, относится к арилалкильной группе, соединенной с исходным молекулярным фрагментом посредством кислородного атома. Термин "арилалкил", использованный здесь, относится к алкильной группе, замещенной одной,двумя или тремя арильными группами. Алкильная часть арилалкила дополнительно необязательно замещена одной или двумя дополнительными группами, независимо выбранными из алкокси, алкилкарбонилокси, гало, галоалкокси, галоалкила, гетероциклила, гидрокси и -NRcRd, где гетероциклил дополнительно необязательно замещен одним или двумя заместителями, независимо выбранными из алкокси,алкила, незамещенного арила, незамещенного арилалкокси, незамещенного арилалкоксикарбонила, гало,галоалкокси, галоалкила, гидрокси, -NRxRy и оксо. Термин "карбонил", использованный здесь, относится к -C(O)-. Термин "цианоалкил", использованный здесь, относится к алкильной группе, замещенной одной,двумя или тремя цианогруппами. Термин "циклоалкил", использованный здесь, относится к насыщенной моноциклической углеводородной кольцевой системе, имеющей от 3 до 7 углеродных атомов и не имеющей гетероатомов. Типичные представители циклоалкильных групп включают, но не ограничиваются, циклопропил, циклобутил, циклопентил и циклогексил. Циклоалкильные группы по настоящему изобретению необязательно замещены одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из алкокси, алкила, арила, циано, гало, галоалкокси, галоалкила, гетероциклила, гидрокси, гидроксиалкила, нитро и -NRxRy, где арил и гетероциклил дополнительно необязательно замещены одним, двумя или тремя заместителями, независимо выбранными из алкокси, алкила, циано, гало, галоалкокси, галоалкила, гидрокси и нитро. Термин "(циклоалкил)алкил", использованный здесь, относится к алкильной группе, замещенной одной, двумя или тремя циклоалкильными группами. Термин "циклоалкилокси", использованный здесь, относится к циклоалкильной группе, соединенной с исходным молекулярным фрагментом посредством кислородного атома. Термин "циклоалкилоксикарбонил", использованный здесь, относится к циклоалкилоксигруппе, соединенной с исходным молекулярным фрагментом посредством карбонильной группы. Термины "гало" и "галоген", использованные здесь, относятся к F, Br, Cl или I. Термин "гетероциклил", использованный здесь, относится к 4-, 5-, 6- или 7-членному кольцу, содержащему 1, 2, 3 или 4 гетероатома, независимо выбранных из азота, кислорода и серы, 4-членное кольцо не имеет двойных связей, 5-членное кольцо не имеет двух двойных связей и 6- и 7-членные кольца не имеют трех двойных связей. Термин "гетероциклил" также включает бициклические группы, в которых гетероциклильное кольцо конденсировано с другой моноциклической гетероциклильной группой или 4-6-членным ароматическим или неароматическим карбоциклическим кольцом, так же, как и связанные мостиковой связью бициклические группы, такие как 7-азабицикло[2,2,1]гепт-7-ил, 2-азабицикло[2,2.2]окт-2-ил и 2-азабицикло[2,2.2]окт-3-ил. Гетероциклильные группы по настоящему изобретению могут быть соединены с исходным молекулярным фрагментом посредством любого атома углерода или атома азота в группе. Примеры гетероциклильных групп включают, но не ограничиваются ими, бензотиенил, фурил,имидазолил, индолинил, индолил, изохинолинил, изотиазолил, изоксазолил, морфолинил, оксазолил,пиперазинил, пиперидинил, пиразолил, пиридинил. пирролидинил, пирролопиридинил, пирролил, хинолинил, тиазолил, тиенил и тиоморфолинил. Гетероциклильные группы по настоящему изобретению необязательно замещены одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из алкокси, алкоксиалкила, алкоксикарбонила, алкила, алкилкарбонила, арила, арилалкила, арилкарбонила, циано, гало, галоалкокси, галоалкила, второй гетероциклильной группы, гетероциклилалкила,гетероциклилкарбонила, гидрокси, гидроксиалкила, нитро, -NRxRy, (NRxRy)-алкила и оксо, где алкильная часть арилалкила и гетероциклилалкил являются незамещенными и где арил, арильная часть арилалкила,арильная часть арилкарбонила, вторая гетероциклильная группа и гетероциклильная часть гетероциклилалкила и гетероциклилкарбонила дополнительно необязательно замещены одним, двумя или тремя заместителями, независимо выбранными из алкокси, алкила, циано, гало, галоалкокси, галоалкила и нитро.-3 022127 Термин "гетероциклилалкил", использованный здесь, относится к алкильной группе, замещенной одной, двумя или тремя гетероциклильными группами. Алкильная часть гетероциклилалкила дополнительно необязательно замещена одной или двумя дополнительными группами, независимо выбранными из алкокси, алкилкарбонилокси, арила, гало, галоалкокси, галоалкила, гидрокси и -NRcRd, где арил дополнительно необязательно замещен одним или двумя заместителями, независимо выбранными из алкокси, алкила, незамещенного арила, незамещенного арилалкокси, незамещенного арилалкоксикарбонила, гало, галоалкокси, галоалкила, гидрокси и -NRxRy. Термин "-NRcRd", использованный здесь, относится к двум группам, Rc и Rd, которые соединены с исходным молекулярным фрагментом посредством атома азота. Rc и Rd независимо выбраны из водорода, алкенилоксикарбонила, алкоксиалкилкарбонила, алкоксикарбонила, алкила, алкилкарбонила, алкилсульфонила, арила, арилалкоксикарбонила, арилалкила, арилалкилкарбонила, арилкарбонила, арилоксикарбонила, арилсульфонила, циклоалкила, циклоалкилоксикарбонила, циклоалкилсульфонила, формила,галоалкоксикарбонила, гетероциклила, гетероциклилалкоксикарбонила, гетероциклилалкила, гетероциклилалкилкарбонила, гетероциклилкарбонила, гетероциклилоксикарбонила, гидроксиалкилкарбонила,(NReRf)-алкила, (NReRf)-алкилкарбонила, (NReRf)-карбонила, (NReRf)-сульфонила, -C(NCN)OR' и-C(NCN)NRxRy, где R' выбран из алкила и незамещенного фенила и где алкильная часть арилалкила,арилалкилкарбонила, гетероциклилалкил и гетероциклилалкилкарбонил дополнительно необязательно замещены одной -NReRf группой; где арил, арильная часть арилалкоксикарбонила, арилалкила, арилалкилкарбонила, арилкарбонила, арилоксикарбонила и арилсульфонила, гетероциклил и гетероциклильная часть гетероциклилалкоксикарбонила, гетероциклилалкила, гетероциклилалкилкарбонила, гетероциклилкарбонила, гетероциклилоксикарбонила дополнительно необязательно замещены одним, двумя или тремя заместителями, независимо выбранными из алкокси, алкила, циано, гало, галоалкокси, галоалкила и нитро. Термин "(NRcRd)-алкенил", использованный здесь, относится к где Rc и Rd являются такими, как здесь определено; и каждый Rq независимо представляет собой водород или C1-3 алкил. Термин "(NRcRd)-алкил", использованный здесь, относится к алкильной группе, замещенной одной,двумя или тремя -NRcRd группами. Алкильная часть (NRcRd)-алкила дополнительно необязательно замещена одной или двумя дополнительными группами, выбранными из алкокси, алкоксиалкилкарбонила,алкоксикарбонила, алкилсулфонила, арилалкоксикарбонила, карбокси, циклоалкила, гетероциклила, гетероциклилкарбонила, гидрокси и (NReRf)-карбонила; где гетероциклил дополнительно необязательно замещен одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из алкокси,алкила, циано, гало, галоалкокси, галоалкила и нитро. Термин "-NReRf", использованный здесь, относится к двум группам Re и Rf, которые соединены с исходным молекулярным фрагментом посредством атома азота. Re и Rf независимо выбраны из водорода, алкила, незамещенного арила, незамещенного арилалкила, незамещенного циклоалкила, незамещенного (циклоалкил)алкила, незамещенного гетероциклила, незамещенного гетероциклилалкила,(NRxRy)-алкила и (NRxRy)-карбонила. Термин "-NRxRy", использованный здесь, относится к двум группам Rx и Ry, которые соединены с исходным молекулярным фрагментом посредством атома азота. Rx и Ry независимо выбраны из водорода, алкоксикарбонила, алкила, алкилкарбонила, незамещенного арила, незамещенного арилалкоксикарбонила, незамещенного арилалкила, незамещенного циклоалкила, незамещенного гетероциклила и(NRx'Ry')-карбонила, где Rx' и Ry' независимо выбраны из водорода и алкила. В соединениях по настоящему изобретению есть асимметричные центры. Эти центры обозначены символами "R" или "S", в зависимости от конфигурации заместителей вокруг хирального атома углерода. Следует понимать, что изобретение охватывает все стереохимические изомерные формы или их смеси, которые обладают способностью ингибировать NS5A. Отдельные стереоизомеры соединений могут быть получены синтетическим путем из коммерчески доступных исходных материалов, которые имеют хиральные центры, или путем получения смесей энантиомерных продуктов с последующим разделением, таким как преобразование в смесь диастереомеров с последующим их разделением с применением техник повторной кристаллизации, хроматографии или прямого разделения энантиомеров на хиральных хроматографических колонках. Исходные соединения частных случаев стереохимии являются либо коммерчески доступными, либо могут быть получены и разделены способами, известными в данной области. Некоторые соединения по настоящему изобретению могут существовать в различных устойчивых конформационных формах, которые могут быть разделены. Торсионная асимметрия в связи с ограниченным вращением вокруг асимметричной одинарной связи, например из-за стерических препятствий-4 022127 или напряжения кольца, может позволить разделение различных конформеров. Настоящее изобретение включает каждый конформационный изомер этих соединений и их смесей. Термин "соединения по настоящему изобретению" и эквивалентные выражения предназначены,чтобы охватить соединения по настоящему изобретению и их фармацевтически приемлемые энантиомеры, диастереомеры и их соли. Подобным образом ссылки на промежуточные соединения предназначены,чтобы охватить их соли, где контекст позволяет это сделать. Настоящее изобретение предполагает включение всех изотопов атомов, встречающихся у настоящих соединений. Изотопы включают те атомы, которые имеют тот же самый атомный номер, но разные массовые числа. В качестве общего примера и без ограничения изотопы водорода включают дейтерий и тритий. Изотопы углерода включают 13 С и 14 С. Изотопно-меченые соединения по изобретению обычно могут быть получены общепринятыми способами, известными специалистам в данной области, или с помощью процессов, аналогичных описанным в настоящем документе, с использованием соответствующего изотопно-меченого реагента вместо использования в противном случае немеченного реагента. Такие соединения могут иметь множество потенциальных применений, например в качестве стандартов и реагентов при определении биологической активности. В случае стабильных изотопов такие соединения могут иметь потенциал для благоприятного изменения биологических, фармакологических или фармакокинетических свойств. Соединения по настоящему изобретению могут существовать в виде фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль", использованный здесь, представляет собой соли или цвиттер-ионные формы соединений по настоящему изобретению, которые являются водо- или маслорастворимыми или диспергируемыми, которые согласно результатам тщательной медицинской оценки пригодны для использования в контакте с тканями пациентов без чрезмерной токсичности, раздражения,аллергической реакции или другой проблемы или осложнения, соизмеримого с разумным соотношением польза/риск, и являются эффективными для их применения по назначению. Соли могут быть получены во время окончательного выделения и очистки соединений или отдельно путем взаимодействия подходящего атома азота с подходящей кислотой. Представительные примеры солей присоединения кислоты включают ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират,камфорат, камфорсульфонат; диглюконат, дигидробромид, дигидрохлорид, дигидройодид, глицерофосфат, гемисульфат, гептаноат, гексаноат, формиат, фумарат, гидрохлорид, гидробромид, гидройодид,2-гидроксиэтансульфонат, лактат, малеат, мезитиленсульфонат, метансульфонат, нафтиленсульфонат,никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, пикрат,пивалат, пропионат, сукцинат, тартрат, трихлорацетат, трифторацетат, фосфат, глутамат, бикарбонат,паратолуолсульфонат и ундеканоат. Примеры кислот, которые могут быть использованы для формирования фармацевтически приемлемых солей присоединения, включают неорганические кислоты, такие как соляная, бромисто-водородная, серная и фосфорная, и органические кислоты, такие как щавелевая,малеиновая, янтарная и лимонная. Соли присоединения основания могут быть получены во время окончательного выделения и очистки соединений путем взаимодействия карбоксильной группы с подходящим основанием, таким как гидроксид, карбонат или бикарбонат катиона металла, или с аммиаком или органическим первичным, вторичным или третичным амином. Катионы фармацевтически приемлемых солей включают литий, натрий,калий, кальций, магний и алюминий, а также нетоксичные катионы четвертичного амина, такие как аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин,диэтиламин,этиламин,трибутиламин,пиридин,N,N-диметиланилин,N-метилпиперидин,N-метилморфолин, дициклогексиламин, прокаин, дибензиламин, N,N-дибензилфенетиламин иN,N'-дибензилэтилендиамин. Другие представительные органические амины, полезные для образования солей присоединения основания, включают этилендиамин, этаноламин, диэтаноламин, пиперидин и пиперазин. Когда возможно, что для использования в терапии могут быть введены терапевтически эффективные количества соединения по настоящему изобретению, а также его фармацевтически приемлемых солей в виде исходного химического продукта, то можно представить активный ингредиент в виде фармацевтической композиции. Соответственно, изобретение дополнительно относится к фармацевтическим композициям, которые включают терапевтически эффективные количества соединения по настоящему изобретению или его фармацевтически приемлемых солей и один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ. Термин "терапевтически эффективное количество", использованный здесь, относится к общему количеству активного компонента, которого достаточно, чтобы показать значимую пользу для пациента,например снижение вирусной нагрузки. Соединения по настоящему изобретению и их фармацевтически приемлемые соли являются такими, как описано выше. Носитель(и), разбавитель(и) или вспомогательное вещество(а) должны быть приемлемыми, в смысле быть совместимыми с другими ингредиентами композиции и не вредными для реципиента. Термин "фармацевтически приемлемый", использованный здесь, относится к таким соединениям,материалам, композициям и/или лекарственным формам, которые согласно результатам тщательной ме-5 022127 дицинской оценки являются пригодными для использования в контакте с тканями пациентов без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерных с разумным соотношением польза/риск и являются эффективными для их применения по назначению. Фармацевтические композиции могут быть представлены в форме стандартной дозы, содержащей определенное количество активного ингредиента на единицу дозы. Уровни дозирования от около 0,01 до около 250 мг/кг массы тела в сутки, предпочтительно от около 0,05 до около 100 мг/кг массы тела в сутки соединений по настоящему изобретению являются типичными при монотерапии предупреждения и лечения HCV опосредованного заболевания. Как правило, фармацевтические композиции по изобретению вводятся от 1 до 5 раз в сутки или, в виде альтернативы, как непрерывная инфузия. Такое введение может применяться в качестве терапии хронического или острого состояния. Количество активного ингредиента, которое может быть соединено с материалами носителя для получения единичной лекарственной формы, будет меняться в зависимости от состояния пациента, тяжести состояния, времени введения, способа введения, скорости выведения принимаемого соединения, продолжительности лечения и возраста, пола, веса и состояния пациента. Предпочтительные единичные дозировки композиций представляют собой такие, которые содержат суточную дозу или часть дозы так, как изложено здесь выше,или подходящую часть ее как активного ингредиента. Лечение может начинаться с маленьких доз, существенно меньших, чем оптимальная доза соединения. Затем доза увеличивается небольшими порциями до тех пор, пока при данных обстоятельствах не будет достигнут оптимальный эффект. В общем соединение, наиболее желательно, вводится при уровне концентрации, который, как правило, окажет эффективные противовирусные воздействия, не вызывая никаких вредных или разрушительных побочных эффектов. Фармацевтические композиции могут быть адаптированы для введения любым подходящим способом, например, перорально (в том числе, буккально или сублингвально), ректально, назально, местно (в том числе, буккально, сублингвально или трансдермально), вагинально или парентерально (в том числе,подкожно, внутрикожно, внутримышечно, внутрисуставно, надчревно, интратекально, внутрь пораженных тканей, внутривенно или внутрикожно посредством инъекции или инфузии). Такие композиции могут быть получены любым способом, известным в области фармацевтики, например, путем введения в композицию активного ингредиента с носителем (носителями) или вспомогательным веществом (веществами). Пероральное введение или введение путем инъекции являются предпочтительными. Фармацевтические композиции для перорального введения могут быть представлены в виде дискретных единиц,таких как капсулы или таблетки; порошки или гранулы; в виде растворов или суспензий в водной или неводной жидкости; съедобных пен или взбитых масс либо жидких эмульсий масло-в-воде или эмульсий вода-в-масле. Например, для перорального введения в форме таблеток или капсул активный лекарственный компонент может быть соединен с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Порошки получают путем измельчения соединения до соответствующего тонкого размера частиц и смешивания с подобным образом измельченным фармацевтическим носителем, таким как съедобный углевод, как, например, крахмал или маннит. Ароматизатор, консервант, диспергирующий и окрашивающий агенты также могут присутствовать. Капсулы изготавливаются путем изготовления порошковой смеси, как описано выше, и наполнения ею сформированных желатиновых оболочек. К порошковой смеси до заправки капсул могут быть добавлены вещества, способствующие скольжению, и смазывающие вещества, такие как коллоидный диоксид кремния, тальк, магния стеарат, кальция стеарат или твердый полиэтиленгликоль. Дезинтегрирующий или растворяющий агент, такой как агар-агар, карбонат кальция или карбонат натрия, также может быть добавлен для улучшения доступности лекарственного средства, когда капсула проглатывается. Кроме того, при желании или необходимости, подходящие связующие, смазывающие, дезинтегрирующие агенты и окрашивающие вещества также могут быть введены в смесь. Подходящие связующие вещества включают крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические смолы, такие как камедь, трагакант или натрия альгинат, карбоксиметилцеллюлозу, полиэтиленгликоль и т.п. Смазывающие вещества, используемые в этих лекарственных формах, включают олеат натрия, хлорид натрия и т.п. Дезинтеграторы включают, без ограничения, крахмал, метилцеллюлозу, агар, бетонит,ксантановую смолу и т.п. Таблетки изготавливаются, например, путем получения порошковой смеси,гранулирования или брикетирования с добавлением скользящего вещества и разрыхлителя и прессования для таблеток. Порошковая смесь готовится путем смешивания соединения, подходящим образом измельченного, с разбавителем или основой, как описано выше, и необязательно со связующим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как парафин, ускорителем резорбции, таким как четвертичная соль, и/или поглощающим веществом, таким как бетонит, каолин или дикальцийфосфат. Порошковая смесь может быть гранулирована путем смачивания со связующим, таким как сироп, крахмальный клейстер, слизь на основе камеди, или с раствором целлюлозных или полимерных материалов и пропускания через сито. В качестве альтернативы гранулирования порошковая смесь может быть пропущена через таблеточную маши-6 022127 ну и образовать не полностью сформированные заготовки, измельчаемые в гранулы. Гранулы могут быть смазаны, чтобы предотвратить прилипание к формирующему таблетки штампу, путем добавления стеариновой кислоты, стеаратной соли, талька или минерального масла. Смазанная смесь затем прессуется в таблетки. Соединения по настоящему изобретению могут быть также объединены с сыпучим инертным носителем и спрессованы в таблетки непосредственно, минуя этапы гранулирования или получения заготовок. Прозрачное или непрозрачное защитное покрытие, состоящее из изолирующего слоя шеллака,поверхностного сахара или полимерного материала, и полирующее покрытие воском могут быть обеспечены. К этим покрытиям могут быть добавлены красящие вещества, чтобы сделать различия между лекарственными формами. Пероральные жидкости, такие как растворы, сиропы и эликсиры, могут быть изготовлены в форме единицы дозирования, так что данная величина содержит определенное количество соединения. Сиропы могут быть получены путем растворения соединения в соответствующем образом ароматизированном водном растворе, при этом эликсиры готовятся с использованием нетоксичных носителей. Также могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленовые эфиры сорбита, консерванты, ароматизирующие добавки, такие как масло мяты перечной, или натуральные подсластители, или сахарин, или другие искусственные подсластители и т.п. В случае необходимости, единица дозирования композиций для перорального введения может находиться в форме микрокапсул. Композиция может быть изготовлена таким образом, чтобы продлить или замедлить высвобождение, например, посредством покрытия или закладывания зернистого материала в полимеры, воск и т.п. Соединения по настоящему изобретению и их фармацевтически приемлемые соли могут также вводиться в форме липосомных систем доставки, таких как маленькие моноламеллярные везикулы, большие моноламеллярные везикулы и мультиламеллярные везикулы. Липосомы могут быть сформированы из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения по настоящему изобретению и их фармацевтически приемлемые соли также могут быть доставлены с использованием моноклональных антител как отдельных носителей, к которым присоединяются молекулы соединения. Соединения также могут быть присоединены к растворимым полимерам, как наводимым на цель носителям лекарственных средств. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный группами палитоила. Кроме того, соединения могут быть связаны с биологически разлагаемыми полимерами, полезными в достижении контролируемого высвобождения лекарственного средства, например с полимерами молочной кислоты, полиэпсилон-капролактоном, полигидроксимасляной кислотой, поли(орто)сложными эфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и сшитыми или амфипатичными блок-сополимерами гидрогелей. Фармацевтические композиции, адаптированные для трансдермального введения, могут быть представлены в виде дискретных пластырей, которые предназначены оставаться в тесном контакте с эпидермисом реципиента в течение продолжительного периода времени. Например, активный ингредиент может быть доставлен из пластыря посредством ионтофореза, как в общем описано в PharmaceuticalResearch. 1986, 3(6), 318. Фармацевтические композиции, адаптированные для местного введения, могут быть составлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Фармацевтические композиции, адаптированные для ректального введения, могут быть представлены как свечи или как клизмы. Фармацевтические композиции, адаптированные для назального введения, когда носитель представляет собой твердое вещество, включают курсовой порошок, имеющий размер частиц, например, в диапазоне от 20 до 500 мкм, который вводится посредством вдыхания через нос, т.е. посредством быстрого вдоха через носовой проход из контейнера с порошком, который удерживается вплотную к носу. Подходящие композиции, в которых носитель представляет собой жидкость для введения в виде назального спрея или назальных капель для носа, включают водные или масляные растворы активного ингредиента. Фармацевтические композиции, адаптированные для введения путем ингаляции, включают частицы в виде мелкой пыли или тумана, которые могут быть получены с помощью различных типов дозирующих, нагнетающих аэрозоли ингаляторов, небулайзеров или инсуфляторов. Фармацевтические композиции, адаптированные для вагинального применения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или распыляемых составов. Фармацевтические композиции, адаптированные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворы, которые переводят композицию в состояние, изотоническое с кровью назначенного реципиента; водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например в запаянных ампулах и флаконах, и могут храниться в сублимированном (лиофилизированном) состоянии, требуя лишь добавления стерильного жидкого носителя, например воды для инъек-7 022127 ций, непосредственно перед применением. Изготовленные для немедленного приема растворы для инъекций и суспензии могут быть получены из стерильных порошков, гранул и таблеток. Следует иметь в виду, что в дополнение к ингредиентам, в частности, упомянутым выше, композиции могут включать другие агенты, обычные в данной области, с учетом типа рассматриваемой композиции. Например, композиции, подходящие для перорального введения, могут включать ароматизаторы. Термин "пациент" включает в себя как человека, так и других млекопитающих. Термин "лечение" относится к (i) предупреждению заболевания, расстройства или патологического состояния, встречающегося у пациента, который может быть предрасположен к болезни, расстройству и/или патологическому состоянию, но которому еще не поставлен такой диагноз; (ii) ингибированию заболевания, расстройства или патологического состояния, т.е. остановке его развития; и (iii) облегчению болезни, расстройства или патологического состояния, т.е. приведению к регрессии заболевания, расстройства и/или патологического состояния. Соединения по настоящему изобретению могут также вводиться с циклоспорином, например с циклоспорином А. Циклоспорин А, как было показано, проявляет активность против HCV в клинических испытаниях (Hepatology, 2003, 38, 1282; Biochem. Biophys. Res.Commun. 2004, 313, 42; J. Gastroenterol. 2003, 38, 567). В табл. 1 приведены некоторые иллюстративные примеры соединений, которые могут быть введены с соединениями по настоящему изобретению. Соединения по изобретению могут вводиться с другими соединениями, проявляющими активность против HCV, в комбинированной терапии либо совместно,либо отдельно, либо путем объединения соединений в композицию. Таблица 1 Соединения по настоящему изобретению могут также применяться в качестве лабораторных реагентов. Соединения могут играть важную роль в обеспечении исследовательских инструментов при разработке анализов вирусной репликации, проверки правильности животных тест-систем и структурных биологических исследований для дальнейшего расширения знаний о механизмах заболевания вирусным гепатитом. Кроме того, соединения по настоящему изобретению полезны в установлении или определении сайтов связывания других противовирусных соединений, например, путем конкурентного ингибирования. Соединения по настоящему изобретению могут также применяться для обработки или предупреждения вирусного загрязнения материалов и, следовательно, снижать риск вирусного заражения лабораторных или медицинских работников и пациентов, которые вступают в контакт с такими материалами,например с кровью, тканями, хирургическими инструментами и одеждой, лабораторными инструментами и одеждой и аппаратами для сбора или переливания крови и материалами. Это изобретение предназначено охватить соединения по настоящему изобретению, которые получаются синтетическим способом, или в процессах обмена веществ, включая те, которые происходят в организме человека или животного (in vivo), или в процессах, происходящих in vitro. Сокращения, применяемые в настоящем описании, включая, в частности, иллюстративные схемы и примеры, которые следуют ниже, хорошо известны специалистам в данной области. Некоторые из используемых сокращений представляют собой следующие:RT - комнатная температура или время удерживания (согласно контексту);Me - метил. Соединения и способы по настоящему изобретению будут лучше поняты в связи со следующими схемами синтетических реакций, иллюстрирующими способы, которыми могут быть получены соединения по настоящему изобретению. Исходные материалы могут быть получены из коммерческих источников или изготовлены хорошо обоснованными в литературе способами, известными специалистам в данной области. Для специалистов в данной области очевидно, что соединения, описанные выше, могут быть синтезированы при замене соответствующих реагирующих веществ и агентов в синтезе, показанном ниже. Также для специалистов в данной области будет очевидно, что этапы селективной защиты и снятия защиты, а также порядок самих этапов могут осуществляться различным образом в зависимости от характера переменных, чтобы успешно завершить синтезы, приведенные ниже. Переменные являются такими, как указано выше, если иное не указано ниже. Схема 1. Замещенные производные фенилглицина. Замещенные производные фенилглицина могут быть получены с помощью ряда способов, показаннных ниже. Фенилглицин трет-бутиловый сложный эфир может быть восстановительно алкилирован(путь А) соответствующим альдегидом и восстановителем, таким как цианоборогидрид натрия в кислой среде. Гидролиз трет-бутилового сложного эфира может быть выполнен с сильной кислотой, такой какHCl или трифторуксусная кислота. Кроме того, фенилглицин может быть алкилирован алкилгалогенидом, таким как этилйодид, и основанием, таким как бикарбонат натрия или карбонат калия (путь В). Путь С иллюстрирует восстановительное алкилирование фенилглицина, как в случае (путь А), сопровождаемое вторым восстановительным алкилированием другим альдегидом, таким как формальдегид, в присутствии восстанавливающего агента и кислоты. Путь D иллюстрирует синтез замещенных фенилглицинов при посредстве соответствующих аналогов миндальной кислоты. Преобразование вторичного спирта в компетентную замещаемую группу может быть выполнено с-толуолсульфонилхлоридом. Вытеснение тозилатной группы соответствующим амином с последующим восстановительным удалением бензинового эфира может обеспечить получение замещенных производных фенилглицина. В пути Е рацемическое замещенное производное фенилглицина получают путем этерификации с энантиомерно чистым хиральным вспомогательным элементом, таким как, но не ограничиваясь, (+)-1-фенилэтанол, (-)-1-фенилэтанол, оксазолидинон Эвана или энантиомерно чистый пантолактон. Разделение диастереомеров осуществляется с помощью хроматографии (силикагель, ВЭЖХ, кристаллизация и т.д.) с последующим удалением хирального вспомогательного элемента, обеспечивающим получение энантиомерно чистых производных фенилглицина. Путь H иллюстрирует последовательность синтеза, которая пересекается с путем Е, в котором вышеупомянутый хиральный вспомогательный элемент встраивается перед добавлением амина. Альтернативно, эфир арилуксусной кислоты может быть бромирован посредством источника ионов бромония, таких как бром, N-бромсукцинимид или CBr4. Полученный бензиловый бромид может быть вытеснен различными моно- или дизамещенными аминами в присутствии основания третичного амина, такого как триэтиламин или основание Хунига. Гидролиз метилового эфира через посредство обработки гидроксидом лития при низкой температуре или 6 н. HCl при повышенной температуре обеспечивает получение замещенных производных фенилглицина. Другой способ показан как путь G. Аналоги глицина могут быть дериватизированы различными арилгалидами в присутствии источника палладия(0), такого как палладий бис-(трибутилфосфин), и основания, такого как фосфат калия. Полученный эфир затем может быть гидролизован путем обработки основанием или кислотой. Следует понимать, что в данной области существуют другие хорошо известные способы получения производных фенилглицина и могут быть внесены изменения, чтобы обеспечить получение желаемых соединений в этом изобретении. Следует также понимать, что окончательные производные фенилглицина могут быть очищены до энантиомерной чистоты более чем до 98% э.и. с помощью препаративной ВЭЖХ. Схема 2. Ацилированные производные аминокислот. В другом варианте осуществления настоящего изобретения ацилированные производные фенилглицина могут быть получены, как показано ниже. Производные фенилглицина, в которых карбоновая кислота защищена как легко удаляемый сложный эфир, могут быть ацилированы хлорангидридом в присутствии основания, такого как триэтиламин, с получением соответствующих амидов (путь А). Путь В иллюстрирует ацилирование исходного производного фенилглицина соответствующим хлорформатом, в то время как путь С показывает реакцию с соответствующим изоцианатом или карбамоилхлоридом. У каждого из трех промежуточных соединений, показанных в путях А-С, может быть снята защита способами, известными специалистам в данной области (например, обработкой трет-бутилового эфира сильным основанием (кислотой), таким как HCl или трифторуксусная кислота). Схема 3. Аминозамещенные фенилуксусные кислоты могут быть получены путем обработки хлорметилфенилуксусной кислоты избытком амина. Синтез типичных Кэп. Условия анализа соединения: оценка чистоты и анализ масс низкого разрешения проводились на системе Shimadzu LC в сочетании с системой Waters MICROMASS ZQ MS. Следует отметить, что время удерживания может слегка варьироваться между системами. К текущему разделу применимы дополнительные условия ЖХ, если не указано иное. Суспензию 10% Pd/C (2,0 г) в метаноле (10 мл) добавляли к смеси (R)-2-фенилглицина (10 г,66,2 ммоль), формальдегида (33 мл 37 мас.% в воде), 1 н. HCl (30 мл) и метанола (30 мл) и подвергали воздействию Н 2 (60 psi (фунт/кв.дюйм в течение 3 ч. Реакционную смесь фильтровали через диатомитовую землю (Целит) и фильтрат концентрировали in vacuo. Полученный в результате сырой материал перекристаллизовывали из изопропанола с получением HCl соли Кэп-1 в виде белых игл (4,0 г). Оптическое вращение: -117,1 [с=9,95 мг/мл в Н 2 О; =589 нм]. 1H ЯМР (DMSO-d6, =2,5 ppm, 500 МГц):7,43-7,34 (m, 5 Н), 4,14 (s, 1H), 2,43 (s, 6H); ЖХ (Условие I): Rt (время удерживания)=0,25; ЖХ/МС: аналитически рассчитано для [М+Н]+ C10H14NO2: 180,10; найдено: 180,17;HRMS (масс-спектрометрия высокого разрешения): аналитически рассчитано для [М+Н]+NaBH3CN (6,22 г, 94 ммоль) добавляли по частям на протяжении нескольких минут к холодной смеси (лед/вода) (R)-2-фенилглицина (6,02 г, 39,8 ммоль) и метанола (100 мл) и перемешивали в течение 5 мин. Ацетальдегид (10 мл) добавляли по каплям в течение более 10 мин и продолжали перемешивание при такой же пониженной температуре в течение 45 мин и при температуре окружающей среды в течение 6,5 ч. Реакционную смесь снова охлаждали на ледяной бане со льдом, обрабатывали водой (3 мл) и затем останавливали реакцию путем добавления по каплям концентрированной HCl на протяжении 45 мин до тех пор, пока pH смеси не составлял 1,5-2,0. Охлаждающую баню удаляли и продолжали перемешивание, добавляя в то же время концентрированную HCl, чтобы поддерживать pH смеси около 1,5-2,0. Реакционную смесь перемешивали в течение ночи, фильтровали, чтобы удалить белую суспензию, и фильтрат концентрировали in vacuo. Сырой материал перекристаллизовывали из этанола для получения HCl соли Кэп-2 в виде блестящего твердого вещества в двух порциях (порция-1: 4,16 г; порция 2: 2,19 г). 1 Н ЯМР (DMSO-d6, =2,5 ppm, 400 МГц): 10,44 (1,00, br s, 1H), 7,66 (m, 2H), 7,51 (m, 3H), 5,30 (s,1H), 3,15 (br m, 2H), 2,98 (br m, 2H), 1,20 (app br s, 6H); Порция-1: []25 -102,21 (c=0,357, H2O); порция-2: []25 -99,7 (c=0,357, H2O); ЖХ (Условие I): Rt=0,43 мин; ЖХ/МС: аналитически рассчитано для [М+Н]+ C12H18NO2: 208,13; найдено: 208,26.(4 мл/1 мл) последовательно добавляли к охлажденной смеси (15 С) (R)-2-фенилглицина (3,096 г,20,48 ммоль), 1 н. HCl (30 мл) и метанола (40 мл). Охлаждающую баню удаляли и реакционную смесь перемешивали в атмосфере баллонного H2 в течение 17 ч. Добавляли дополнительный ацетальдегид(10 мл, 178,2 ммоль) и продолжали перемешивание в атмосфере Н 2 в течение 24 ч [Примечание: подачу Н 2 пополняли по мере необходимости во время реакции]. Реакционную смесь фильтровали через диатомитовую землю (Целит) и фильтрат концентрировали in vacuo. Полученный в результате сырой материал перекристаллизовывали из изопропанола с получением HCl соли (R)-2-(этиламино)-2 фенилуксусной кислоты в виде блестящего твердого вещества (2,846 г). 1 Н ЯМР (DMSO-d6, =2,5 ppm, 400 МГц):14,15 (br s, 1 Н), 9,55 (br s, 2H), 7,55-7,48 (m, 5H), 2,88 (brm, 1 Н), 2,73 (br m, 1H), 1,20 (app t, J=7,2 Гц, 3 Н); ЖХ (Условие I): Rt=0,39 мин; индекс гомогенности 95%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C10H14NO2: 180,10; найдено: 180,18. Суспензию 10% Pd/C (536 мг) в растворе метанол/Н 2 О (3 мл/1 мл) добавляли к смеси(R)-2-(этиламино)-2-фенилуксусной кислоты/HCl (1,492 г, 6,918 ммоль), формальдегида (20 мл 37 мас.% в воде), 1 н. HCl (20 мл) и метанола (23 мл). Реакционную смесь перемешивали в атмосфере баллонного Н 2 в течение 72 ч, где Н 2 пополняли по мере необходимости. Реакционную смесь фильтровали через диатомитовую землю (Целит) и фильтрат концентрировали in vacuo. Полученный в результате сырой материал перекристаллизовывали из изопропанола (50 мл) с получением HCl соли Кэп-3 в виде белого твердого вещества (985 мг). 1br s, 2H), 2,65 (br s, 3H), 1,24 (br m, 3 Н); ЖХ (Условие I): Rt=0,39 мин; индекс гомогенности 95%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C11H16NO2: 194,12; найдено: 194,18;HRMS: аналитически рассчитано для [М+Н]+ C11H16NO2: 194,1180; найдено: 194,1181.ClCO2Me (3,2 мл, 41,4 ммоль) добавляли по каплям к охлажденному (лед/вода) THF (410 мл) полураствору (R)-трет-бутил 2-амино-2-фенилацетат/HCl (9,877 г, 40,52 ммоль) и диизопропилэтиламину(14,2 мл, 81,52 ммоль) на протяжении 6 мин и перемешивали при той же температуре в течение 5,5 ч. Летучий компонент удаляли in vacuo и остаток разделяли между водой (100 мл) и этилацетатом (200 мл). Органический слой промывали 1 н. HCl (25 мл) и насыщенным раствором NaHCO3 (30 мл), высушивали(MgSO4), фильтровали и концентрировали in vacuo. Полученное бесцветное масло растирали в порошок из гексанов, фильтровали и промывали гексанами (100 мл) с получением (R)-трет-бутил 2-(метоксикарбониламино)-2-фенилацетата в виде белого твердого вещества (7,7 г). 1 Н ЯМР (DMSO-d6, =2,5 ppm, 400 МГц): 7,98 (d, J=8,0 Гц, 1 Н), 7,37-7,29 (m, 5H), 5,09 (d, J=8,0 Гц,1H), 3,56 (s, 3H), 1,33 (s, 9H); ЖХ (Условие I): Rt=1,53 мин; индекс гомогенности 90%; ЖХ/МС: аналитически рассчитано для [M+Na]+ C14H19NNaO4: 288,12; найдено: 288,15.TFA (16 мл) добавляли по каплям к охлажденному (лед/вода) CH2Cl2 (160 мл) раствору указанного выше продукта на протяжении 7 мин, охлаждающую баню удаляли и реакционную смесь перемешивали в течение 20 ч. Поскольку снятие защитных групп еще не закончилось, добавляли дополнительную TFA(1,0 мл) и перемешивание продолжали в течение дополнительных 2 ч. Летучий компонент удаляли invacuo и полученный в результате масляный остаток обрабатывали диэтиловым эфиром (15 мл) и гексанами (12 мл) с получением осадка. Осадок фильтровали и промывали смесью диэтиловый эфир/гексаны(соотношение 1:3; 30 мл) и высушивали in vacuo с получением Кэп-4 в виде рассыпчатого белого твердого вещества (5,57 г). Оптическое вращение: -176,9 [с=3,7 мг/мл в Н 2 О; =589 нм]. 1H ЯМР (DMSO-d6, =2,5 ppm, 400 МГц):12,84 (br s, 1H), 7,96 (d, J=8,3 Гц, 1H), 7,41-7,29 (m, 5H),5,14 (d, J=8,3 Гц, 1H), 3,55 (s, 3 Н); ЖХ (Условие I): Rt=1,01 мин; индекс гомогенности 95%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C10H12NO4: 210,08; найдено: 210,17;HRMS: аналитически рассчитано для [M+H]+C10H12NO4: 210,0766; найдено: 210,0756.(2,10 г, 19,8 ммоль) в этаноле (40 мл) нагревали при 100 С в течение 21 ч. Реакционную смесь охлаждали до температуры окружающей среды и фильтровали и фильтрат концентрировали in vacuo. Остаток растворяли в этаноле и окисляли 1 н. HCl до значения pH 3-4 и летучий компонент удаляли in vacuo. Полученный в результате сырой материал очищали с помощью обращенно-фазовой ВЭЖХ (вода/метанол/ТТА) с получением TFA соли Кэп-5 в виде полувязкой белой пены (1,0 г). 1 Н ЯМР (DMSO-d6, =2,5 ppm, 500 МГц) : 10,68 (br s, 1 Н), 7,51 (m, 5H), 5,23 (s, 1H), 3,34 (app br s,2H), 3,05 (app br s, 2H), 1,95 (app br s, 4H);Rt=0,30 мин (Условие I); индекс гомогенности 98%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C12H16NO2: 206,12; найдено: 206,25.TFA соль Кэп-6 синтезировали из (R)-2-фенилглицина и 1-бром-2-(2-бромэтокси)этана, применяя способ получения Кэп-5. 1Rt=0,32 мин (Условие I); индекс гомогенности 98%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C12H16NO3: 222,11; найдено: 222,20;HRMS: аналитически рассчитано для [М+Н]+ C12H16NO3: 222,1130; найдено: 222,1121.CH2Cl2 (200 мл) раствор -толуолсульфонил хлорида (8,65 г, 45,4 ммоль) добавляли по каплям к охлажденному (-5 С) CH2Cl2 (200 мл) раствору (S)-бензил 2-гидрокси-2-фенилацетата (10,0 г,41,3 ммоль), триэтиламина (5,75 мл, 41,3 ммоль) и 4-диметиламинопиридина (0,504 г, 4,13 ммоль), поддерживая при этом температуру между -5 и 0 С. Реакционную смесь перемешивали при 0 С в течение 9 ч и затем хранили в морозильнике (-25 С) в течение 14 ч. Ее оставляли оттаивать до температуры окружающей среды и промывали водой (200 мл), 1 н. HCl (100 мл) и рассолом (100 мл), высушивали(MgSO4), фильтровали и концентрировали in vacuo с получением бензил 2-фенил-2-(тозилокси)ацетата в виде вязкого масла, которое затвердевало при стоянии (16,5 г). Хиральную чистоту продукта не проверяли, и этот продукт использовали на следующем этапе без дополнительной очистки. 1 Н ЯМР (DMSO-d6, =2,5 ppm, 500 МГц) : 7,78 (d, J=8,6, 2 Н), 7,43-7,29 (m, 10 Н), 7,20 (m, 2H), 6,12Rt=3,00 (Условие III); индекс гомогенности 90%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C22H20NaO5S: 419,09; найдено: 419,04.(3,36 мл, 30,3 ммоль) и N,N-диизопропилэтиламина (13,2 мл, 75,8 ммоль) нагревали при 65 С в течение 7 ч. Реакционную смесь оставляли охлаждаться до температуры окружающей среды и летучий компонент удаляли in vacuo. Остаток разделяли между этилацетатом и водой, и органический слой промывали водой и рассолом, высушивали (MgSO4), фильтровали и концентрировали in vacuo. Полученный в результате сырой материал очищали с помощью флэш-хроматографии (силикагель, этилацетат) с получением бензил 2-(4-метилпиперазин-1-ил)-2-фенилацетата в виде оранжево-коричневого вязкого масла(4,56 г). Анализ с помощью хиральной ВЭЖХ (CHIRALCEL OD-H) показал, что образец представляет собой смесь стереоизомеров в соотношении 38,2:58,7. Разделение стереоизомеров осуществляли следующим образом: продукт растворяли в 120 мл смеси этанол/гептан (1:1) и наносили (5 мл/нанесение) на колонку (Chiracel OJ, 5 см ID50 см L, 20 мкм) хиральной ВЭЖХ, элюируя смесью 85:15 гептан/этанол при 75 мл/мин и контролируя при 220 нм. Энантиомер-1 (1,474 г) и энантиомер-2 (2,2149 г) извлекали в виде вязкого масла. 1 Н ЯМР (CDCl3, =7,26 ppm, 500 МГц): 7,44-7,40 (m, 2 Н), 7,33-7,24 (m, 6 Н), 7,21-7,16 (m, 2 Н), 5,13Rt=2,10 (Условие III); индекс гомогенности 98%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C20H25N2O2: 325,19; найдено: 325,20. Метанольный раствор (10 мл) любого энантиомера бензил 2-(4-метилпиперазин-1-ил)-2 фенилацетата (1,0 г, 3,1 ммоль) добавляли к суспензии 10% Pd/C (120 мг) в метаноле (5,0 мл). Реакционную смесь подвергали воздействию баллонного водорода под тщательным контролем в течение 50 мин. Немедленно после завершения реакции катализатор фильтровали через диатомитовую землю (Целит) и фильтрат концентрировали in vacuo с получением Кэп-7, загрязненного фенилуксусной кислотой в виде желтовато-коричневой пены (867,6 мг; масса превышала теоретический выход). Продукт использовали на следующем этапе без дополнительной очистки. 1Rt=0,31 (Условие II); индекс гомогенности 90%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C13H19N2O2: 235,14; найдено: 235,15;HRMS: аналитически рассчитано для [М+Н]+ C13H19N2O2: 235,1447; найдено: 235,1440. Синтез Кэп-8 и Кэп-9 проводили согласно синтезу Кэп-7, применяя подходящие амины для этапа замещения SN2 (т.е. 4-гидроксипиперидин для Кэп-8 и (S)-3-фторпирролидин для Кэп-9) и измененные условия для разделения соответствующих стереоизомерных промежуточных соединений, как описано ниже. Разделение энантиомеров промежуточного соединения бензил 2-(4-гидроксипиперидин-1-ил)-2 фенилацетата осуществляли, применяя следующие условия: соединение (500 мг) растворяли в смеси этанол/гептан (5 мл/45 мл). Полученный в результате раствор наносили (5 мл/нанесение) на колонку(Chiracel OJ, 2 см ID25 см L, 10 мкм) хиральной ВЭЖХ, элюируя смесью 80:20 гептан/этанол при 10 мл/мин, контролируя при 220 нм, с получением 186,3 мг энантиомера-1 и 209,1 мг энантиомера-2 в виде светло-желтых вязких масел. Этот бензиновый сложный эфир гидролизовали согласно приготовлению Кэп-7 с получением Кэп-8. 1Rt=0,28 (Условие II); индекс гомогенности 98%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C13H18NO3: 236,13; найдено: 236,07; Разделение диастереомеров промежуточного соединения бензил 2-S)-3-фторпирролидин-1-ил)-2 фенилацетата осуществляли, применяя следующие условия: сложный эфир (220 мг) разделяли на колонке (Chiracel OJ-H, 0,46 см ID25 см L, 5 мкм) хиральной ВЭЖХ, элюируя смесью 95% СО 2/5% метанол с 0,1% TFA при давлении 10 бар, скорости потока 70 мл/мин и температуре 35 С. ВЭЖХ элюат соответствующих стереомеров концентрировали и остаток растворяли в CH2Cl2 (20 мл) и промывали водной средой (10 мл воды + 1 мл насыщенного раствора NaHCO3). Органическую фазу высушивали (MgSO4),фильтровали и концентрировали in vacuo с получением 92,5 мг фракции-1 и 59,6 мг фракции-2. Эти бензиловые сложные эфиры гидролизовали согласно приготовлению Кэп-7 для получения Кэп-9 а и Кэп-9b. Кэп-9 а (диастереомер-1; образец представляет собой TFA соль как результат очищения посредством обращенно-фазовой ВЭЖХ с использованием смеси Н 2 О/метанол/TFA растворитель): 1 Н ЯМР (DMSO-d6, =25 ppm, 400 МГц): 7,55-7,48 (m, 5H), 5,38 (d m, J=53,7 Гц, 1 Н), 5,09 (br s, 1H),3,84-2,82 (br m, 4H), 2,31-2,09 (m, 2H);Rt=0,42 (Условие I); индекс гомогенности 95%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C12H15FNO2: 224,11; найдено: 224,14; Кэп-9b (диастереомер-2): 1Rt=0,44 (Условие I); ЖХ/МС: аналитически рассчитано для [М+Н]+ C12H15FNO2: 224,11; найдено: 224,14. К раствору D-пролина (2,0 г, 17 ммоль) и формальдегида (2,0 мл 37 мас.% в Н 2 О) в метаноле (15 мл) добавляли суспензию 10% Pd/C (500 мг) в метаноле (5 мл). Смесь перемешивали в атмосфере баллонного водорода в течение 23 ч. Реакционную смесь фильтровали через диатомитовую землю (Целит) и концентрировали in vacuo с получением Кэп-10 в виде не совсем белого твердого вещества (2,15 г). 1 Н ЯМР (DMSO-d6, =2,5 ppm, 500 МГц): 3,42 (m, 1H), 3,37 (dd, J=9,4, 6,1 Гц, 1 Н), 2,85-2,78 (m, 1 Н),2,66 (s, 3 Н), 2,21-2,13 (m, 1H), 1,93-1,84 (m, 2 Н), 1,75-1,66 (m, 1 Н);Rt=0,28 (Условие II); индекс гомогенности 98%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C6H12NO2: 130,09; найдено: 129,96. Смесь (2S,4R)-4-фторпирролидин-2-карбоновой кислоты (0,50 г, 3,8 ммоль), формальдегида (0,5 мл 37 мас.% в Н 2 О), 12 н. HCl (0,25 мл) и 10% Pd/C (50 мг) в метаноле (20 мл) перемешивали в атмосфере баллонного водорода в течение 19 ч. Реакционную смесь фильтровали через диатомитовую землю(Целит) и фильтрат концентрировали in vacuo. Остаток перекристаллизовывали из изопропанола с получением HCl соли Кэп-11 в виде белого твердого вещества (337,7 мг). 1Rt=0,28 (Условие II); индекс гомогенности 98%; ЖХ/МС: аналитически рассчитано для [М+Н]+ C6H11FNO2: 148,08; найдено: 148,06.L-Аланин (2,0 г, 22,5 ммоль) растворяли в 10% водном растворе карбоната натрия (50 мл) и добавляли к нему THF (50 мл) раствор метилхлорформиата (4,0 мл). Реакционную смесь перемешивали при условиях окружающей среды в течение 4,5 ч и концентрировали in vacuo. Полученное в результате белое твердое вещество растворяли в воде и окисляли 1 н. HCl до значения pH 2-3. Полученный в результате раствор экстрагировали этилацетатом (3100 мл) и объединенную органическую фазу высушивали(Na2SO4), фильтровали и концентрировали in vacuo с получением бесцветного масла (2,58 г). 500 мг этого материала очищали с помощью обращенно-фазовой ВЭЖХ (Н 2 О/метанол/TFA) с получением 150 мг Кэп-12 в виде бесцветного масла. 1 Н ЯМР (DMSO-d6, =2,5 ppm, 500 МГц): 7,44 (d, J=7,3 Гц, 0,8H), 7,10 (br s, 0,2H), 3,97 (m, 1H), 3,53(500 мг) в метаноле (30 мл) перемешивали в атмосфере водорода (50 фунт/кв.дюйм) в течение 5 ч. Реакционную смесь фильтровали через диатомитовую землю (Целит) и фильтрат концентрировали in vacuo с получением HCl соли Кэп-13 в виде масла, которое затвердевало после выдерживания под вакуумом(4,4 г; масса была выше теоретического выхода). Продукт применяли без дополнительной очистки. 1(0,773 г, 12,3 ммоль), KOH (0,690 г, 12,3 ммоль) и уксусной кислоты (0,352 мл, 6,15 ммоль) перемешивали в метаноле при 0 С. К этой смеси по каплям добавляли глутаровый диальдегид (2,23 мл, 12,3 ммоль) на протяжении 5 мин. Реакционную смесь перемешивали так, чтобы позволить ей нагреться до температуры окружающей среды и продолжали перемешивание при такой температуре в течение 16 ч. Растворитель затем удаляли и остаток разделяли между 10% водным NaOH и этилацетатом. Органическую фазу разделяли, высушивали (MgSO4), фильтровали и концентрировали досуха с получением прозрачного масла. Этот материал очищали с помощью обращенно-фазовой препаративной ВЭЖХ (Primesphere С-18,30100 мм; CH3CN-Н 2 О-0,1% TFA) с получением промежуточного сложного эфира (2,70 г, 56%) в виде прозрачного масла. 1- 24022127 ЖХ/МС: аналитически рассчитано для C17H25NO2: 275; найдено: 276 (М+Н)+. Этап 2. К перемешанному раствору промежуточного сложного эфира (1,12 г, 2,88 ммоль) в дихлорметане (10 мл) добавляли TFA (3 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч и затем ее концентрировали досуха с получением светло-желтого масла. Масло очищали с помощью обращенно-фазовой препаративной ВЭЖХ (Primesphere C-18, 30100 мм; CH3CN-H2O0,1% TFA). Подходящие фракции объединяли и концентрировали досуха in vacuo. Остаток затем растворяли в минимальном количестве метанола и применяли для помещения в экстракционные картриджи МСХ LP (26 г). Картриджи промывали метанолом (40 мл) и затем целевое соединение элюировали с применением 2 М аммиака в метаноле (50 мл). Фракции, содержащие продукт, объединяли и концентрировали, остаток помещали в воду. Лиофилизация этого раствора обеспечивала указанное в заголовке соединение (0,492 г, 78%) в виде светло-желтого твердого вещества. 1 Н ЯМР (DMSO-d6) : 7,50 (s, 5 Н), 5,13 (s, 1 Н), 3,09 (br s, 2H), 2,92-2,89 (m, 2H), 1,74 (m, 4H), 1,48(br s, 2H); ЖХ/МС: аналитически рассчитано для C13H17NO2: 219; найдено: 220 (M+H)+. Этап 1. (S)-1-Фенилэтил 2-бром-2-фенилацетат. К смеси -бромфенилуксусной кислоты (10,75 г, 0,050 моль), (S)-(-)-1-фенилэтанола (7,94 г, 0,065 моль) и DMAP (0,61 г, 5,0 ммоль) в сухом дихлорметане (100 мл) добавляли твердое вещество EDCI(12,46 г, 0,065 моль), все сразу. Полученный в результате раствор перемешивали при комнатной температуре в атмосфере аргона в течение 18 ч и затем его разбавляли этилацетатом, промывали (Н 2 О 2, рассол), высушивали (Na2SO4), фильтровали и концентрировали с получением бледно-желтого масла. Флэшхроматография (SiO2/гексан-этилацетат, 4:1) этого масла обеспечивала указанное в заголовке соединение(11,64 г, 73%) в виде белого твердого вещества. 1(s, 0,5H), 5,39 (s, 0,5H), 1,58(d, J=6,6 Гц, 1,5 Н), 1,51 (d, J=6,6 Гц, 1,5H). Этап 2. (S)-1-Фенилэтил (R)-2-(4-гидрокси-4-метилпиперидин-1-ил)-2-фенилацетат. К раствору (S)-1-фенилэтил 2-бром-2-фенилацетата (0,464 г, 1,45 ммоль) в THF (8 мл) добавляли триэтиламин (0,61 мл, 4,35 ммоль), затем тетрабутиламмония йодид (0,215 г, 0,58 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 мин и затем добавляли раствор 4-метил-4 гидроксипиперидина (0,251 г, 2,18 ммоль) в THF (2 мл). Смесь перемешивали в течение 1 ч при комнатной температуре и затем ее нагревали при 55-60 С (температура масляной бани) в течение 4 ч. Охлажденную реакционную смесь затем разбавляли этилацетатом (30 мл), промывали (Н 2 О 2, рассол), высушивали (MgSO4), фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (0-60% этилацетат-гексан) с получением первого (S,R)-изомера указанного в заголовке соединения (0,306 г, 60%) в виде белого твердого вещества и затем соответствующего (S,S)-изомера (0,120 г,23%), также в виде белого твердого вещества.(m, 2H), 2,41-2,29 (m, 2 Н), 1,71-1,49 (m, 4 Н), 1,38 (d, J=6,6 Гц, 3 Н), 1,18 (s, 3H); ЖХ/МС: аналитически рассчитано для C22H27NO3: 353; найдено: 354 (М+Н)+.(m, 3H), 1,50 (d, J=6,8 Гц, 3 Н), 1,20 (s, 3 Н); ЖХ/МС: аналитически рассчитано для C22H27NO3: 353; найдено: 354 (М+Н)+. Этап 3. (R)-2-(4-Гидрокси-4-метилпиперидин-1-ил)-2-фенилуксусная кислота. К раствору (S)-1-фенилэтил (R)-2-(4-гидрокси-4-метилпиперидин-1-ил)-2-фенилацетата (0,185 г,0,52 ммоль) в дихлорметане (3 мл) добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Летучие компоненты затем удаляли in vacuo и остаток очищали с помощью обращенно-фазовой препаративной ВЭЖХ (Primesphere C-18, 20100 мм; CH3CN-H2O0,1% TFA) с получением указанного в заголовке соединения (в виде TFA соли) в виде бледно-синеватого твердого вещества (0,128 г, 98%). ЖХ/МС: аналитически рассчитано для C14H19NO3: 249; найдено: 250 (М+Н)+. Этап 1. (S)-1-Фенилэтил 2-(2-фторфенил)ацетат. Смесь 2-фторфенилуксусной кислоты (5,45 г, 35,4 ммоль), (S)-1-фенилэтанола (5,62 г, 46,0 ммоль),EDCI (8,82 г, 46,0 ммоль) и DMAP (0,561 г, 4,60 ммоль) в CH2Cl2 (100 мл) перемешивали при комнатной температуре в течение 12 ч. Растворитель затем концентрировали и остаток разделяли между Н 2 Оэтилацетатом. Фазы разделяли и водный слой снова экстрагировали этилацетатом (2). Объединенные органические фазы промывали (Н 2 О, рассол), высушивали (Na2SO4), фильтровали и концентрировали invacuo. Остаток очищали с помощью хроматографии на силикагеле (Biotage/0-20% этилацетат-гексан) с получением указанного в заголовке соединения в виде бесцветного масла (8,38 г, 92%). 1H ЯМР (400 МГц, CD3OD) : 7,32-7,23 (m, 7H), 7,10-7,04 (m, 2), 5,85 (q, J=6,5 Гц, 1H), 3,71 (s, 2H),1,48 (d, J=6,5 Гц, 3 Н). Этап 2. (R)-S)-1-Фенилэтил) 2-(2-фторфенил)-2-(пиперидин-1-ил)ацетат. К раствору (S)-1-фенилэтил 2-(2-фторфенил)ацетата (5,00 г, 19,4 ммоль) в THF (1200 мл) при 0 С добавляли DBU (6,19 г, 40,7 ммоль) и раствор оставляли нагреваться до комнатной температуры, при этом перемешивая, в течение 30 мин. Раствор затем охлаждали до -78 С, добавляли раствор CBr4 (13,5 г,40,7 ммоль) в THF (100 мл) и смесь оставляли нагреваться до -10 С и перемешивали при этой температуре в течение 2 ч. Реакцию останавливали насыщенным водным NH4Cl и слои разделяли. Водный слой снова экстрагировали этилацетатом (2) и объединенные органические фазы промывали (Н 2 О, рассол),высушивали (Na2SO4), фильтровали и концентрировали in vacuo. К остатку добавляли пиперидин(5,73 мл, 58,1 ммоль) и раствор перемешивали при комнатной температуре в течение 24 ч. Летучие компоненты затем концентрировали in vacuo и остаток очищали с помощью хроматографии на силикагеле(Biotage/0-30% диэтиловый эфир-гексан) с получением чистой смеси диастереоизомеров (2:1 соотношение согласно 1H ЯМР) в виде желтого масла (2,07 г, 31%), вместе с непрореагировавшим исходным продуктом (2,53 г, 51%). Дополнительное хроматографирование диастереоизомерной смеси (Biotage/0-10% диэтиловый эфир-толуол) обеспечивало указанное в заголовке соединение в виде бесцветного маслаH ЯМР (400 МГц, CD3OD) : 7,52 (ddd, J=9,4, 7,6, 1,8 Гц, 1 Н), 7,33-7,40 (m, 1), 7,23-7,23 (m, 4H),7,02-7,23 (m, 4H), 5,86 (q, J=6,6 Гц, 1 Н), 4,45 (s, 1H), 2,39-2,45 (m, 4H), 1,52-1,58 (m, 4H), 1,40-1,42 (m,1H), 1,38 (d, J=6,6 Гц, 3 Н); ЖХ/МС: аналитически рассчитано для C21H24FNO2: 341; найдено: 342 (М+Н)+. Этап 3. (R)-2-(2-Фторфенил)-2-(пиперидин-1-ил)уксусная кислота. Смесь (R)-S)-1-фенилэтил) 2-(2-фторфенил)-2-(пиперидин-1-ил)ацетата (0,737 г, 2,16 ммоль) и 20% Pd(OH)2/C (0,070 г) в этаноле (30 мл) гидрировали при комнатной температуре и атмосферном давлении (Н 2 баллонный) в течение 2 ч. Раствор затем продували аргоном, фильтровали через диатомитовую землю (Целит) и концентрировали in vacuo. Это обеспечивало указанное в заголовке соединение в виде бесцветного твердого вещества (0,503 г, 98%). 1H ЯМР (400 МГц, CD3OD) : 7,65 (ddd, J=9,1, 7,6, 1,5 Гц, 1 Н), 7,47-7,53 (m, 1H), 7,21-7,30 (m, 2 Н),3,07-3,13 (m, 4 Н), 1,84 (br s, 4H), 1,62 (br s, 2H); ЖХ/МС: аналитически рассчитано для C13H16FNO2: 237; найдено: 238 (М+Н)+. Этап 1. (S)-1-Фенилэтил (R)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-2-фенилацетат. К раствору (S)-1-фенилэтил 2-бром-2-фенилацетата (1,50 г, 4,70 ммоль) в THF (25 мл) добавляли триэтиламин (1,31 мл, 9,42 ммоль) с последующим добавлением тетрабутиламмония йодида (0,347 г,0,94 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 мин и затем добавляли раствор 4-фенил-4-гидроксипиперидина (1,00 г, 5,64 ммоль) в THF (5 мл). Смесь перемешивали в течение 16 ч и затем ее разбавляли этилацетатом (100 мл), промывали (Н 2 О 2, рассол), высушивали(MgSO4), фильтровали и концентрировали. Остаток очищали на силикагельной колонке (0-60% этилаце- 26022127 тат-гексан) с получением приблизительно 2:1 смеси диастереоизомеров согласно данным 1H ЯМР. Разделение этих изомеров выполняли с помощью сверхкритической флюидной хроматографии (СКФХ)(CHIRALCEL OJ-H, 30250 мм; 20% этанол в СО 2 при 35 С) с получением первого (R)-изомера указанного в заголовке соединения (0,534 г, 27%) в виде желтого масла и затем соответствующего (S)-изомера(dt, J=11,1, 2,5 Гц, 1H), 2,20 (dt, J=12,1, 4,6 Гц, 1 Н), 2,10 (dt, J=12,1, 4,6 Гц, 1H), 1,72-1,57 (m, 2 Н), 1,53 (d,J=6,5 Гц, 3 Н); ЖХ/МС: аналитически рассчитано для C27H29NO3: 415; найдено: 416 (М+Н)+;(S,S)-изомер: 1 Н ЯМР (400 МГц, CD3OD) : 7,55-7,48 (m, 2H), 7,45-7,39 (m, 2H), 7,38-7,30 (m, 5H),7,25-7,13 (m, 4H), 7,08-7,00 (m, 2H), 5,88 (q, J=6,6 Гц, 1H), 4,12 (s, 1H), 2,95-2,85 (m, 1H), 2,68 (dt, J=11,1,2,5 Гц, 1H), 2,57-2,52 (m, 1H), 2,42 (dt, J=11,1, 2,5 Гц, 1 Н), 2,25 (dt, J=12,1, 4,6 Гц, 1 Н), 2,12 (dt, J=12,1,4,6 Гц, 1H), 1,73 (dd, J=13,6, 3,0 Гц, 1 Н), 1,64 (dd, J=13,6, 3,0 Гц, 1 Н), 1,40 (d, J=6,6 Гц, 3 Н); ЖХ/МС: аналитически рассчитано для C27H29NO3: 415; найдено: 416 (М+Н)+. Следующие сложные эфиры приготавливали подобным образом. Условия определения времени удерживания посредством хиральной СКФХ (SFC): Кэп 17, этап 2: (R)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-2-фенилуксусная кислота. К раствору (S)-1-фенилэтил (R)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-2-фенилацетата (0,350 г,0,84 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Летучие компоненты затем удаляли in vacuo и остаток очищали с помощью обращенно-фазовой препаративной ВЭЖХ (Primesphere С-18, 20100 мм; CH3CN-H2O0,1% TFA) с получением указанного в заголовке соединения (в виде TFA соли) в виде белого твердого вещества (0,230 г, 88%); ЖХ/МС: аналитически рассчитано для C19H21NO3: 311,15; найдено: 312 (М+Н)+.- 28022127 Следующие карбоновые кислоты изготавливали в оптически чистой форме подобным образом:
МПК / Метки
МПК: C07D 405/14, C07D 403/14, A61K 31/4184, A61P 31/12
Метки: вируса, ингибиторы, гепатита
Код ссылки
<a href="https://eas.patents.su/30-22127-ingibitory-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы вируса гепатита с</a>
Ингибиторы вируса гепатита с

Номер патента: 20815
Опубликовано: 30.01.2015
Авторы: Сейнт Лорен Денис Р., Истер Джон А., Лопез Омар Д., Ромине Джефри Ли, Ван Гань, Белема Маконен, Ян Чжун, Нгуен Ван Н., Хевавасам Пиясена, Бендер Джон А., Сюй Ниннин, Минвелл Николас А., Смит Майкл Дж., Чэнь Ци, Су Бао-Нин
МПК: C07D 403/14, C07D 405/14
Метки: ингибиторы, вируса, гепатита
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеn равно 0,1 или 2;X выбирают из водорода, циано, галогена циклопропила, циклопропилметила, 1-метилциклопропила, аллила, 1-метилпиразол-5-ила, 1-метилимидазол-5-ила и изоксазол-4-ила;R1 представляет собой водород;R2 выбирают из водорода, циклопропила и галогена; илиR1 и R2 вместе образуют -CH=CH- или -CH=CCl-;при условии, что по меньшей мере один из X и R2 не обозначает...
Ингибиторы вируса гепатита с

Номер патента: 21194
Опубликовано: 30.04.2015
Авторы: Кеннет Дж., Белема Маконен, Тимонко Стивен, Натали Джр., Пател Бхарат П., Пак Шон К.
МПК: C07D 209/54
Метки: вируса, гепатита, ингибиторы
Формула / Реферат:
1. Способ получения соединения формулы (III)или его фармацевтически приемлемой соли,где R4 и R5 независимо выбраны из атома водорода, (С1-С6)алкила, (C6-C10)арила, (С6-С10)арил(С1-С6)алкила, гетероциклила и гетероциклил(С1-С2)алкила, где гетероциклил представляет собой 5-6-членный гетероциклил, включающий 1-2 гетероатома, выбранных из N, S и О,включающий обработку соединения формулы (IV)ацетатом аммония в присутствии основания.2. Способ по п.1,...
Ингибиторы вируса гепатита с (hcv)

Номер патента: 15415
Опубликовано: 31.08.2011
Авторы: Линк Джон О., Граупе Михаель, Венкатарамани Чандрэсикар
МПК: A61K 31/00, A61K 47/00, A61K 31/74...
Метки: вируса, hcv, ингибиторы, гепатита
Формула / Реферат:
1. Соединение формулы (I)где Е представляет собой -COCONHR6, где R6представляет собой водород, алкил, циклоалкил, аралкил или гетероаралкил, где ароматическое кольцо необязательно замещено одним или двумя галоидами;R1 представляет собой алкил, циклоалкилалкил, где алифатические или алициклические группы в R1необязательно замещены одним или двумя радикалами Rb, которые независимо выбирают из гидрокси, алкокси, арилокси, гетероарилокси, алкилтио,...
Ингибиторы вируса гепатита с

Номер патента: 18782
Опубликовано: 30.10.2013
Авторы: Снайдер Лоуренс Б., Ст.Лоран Денис Р., Гуд Эндрю К., Гудрих Джейсон, Лопез Омар Д., Рюдигер Эдвард Х., Дэон Даниэль Х., Минвелл Николас А., Лэнгли Дэвид Р., Нгуен Ван Н., Ванг Гэн, Джеймс Клинт А., Лавуа Рико, Мартел Алан, Белема Маконен, Ромин Джеффри Ли, Янг Фуканг, Бачанд Кэрол, Хаманн Лоуренс Г.
МПК: A61P 31/22, A61K 31/4164, C07D 403/10...
Метки: ингибиторы, вируса, гепатита
Формула / Реферат:
1. Соединение, выбранное издиметил (4,4'-бифенилдиилбис(1H-имидазол-4,2-диил((1S)-2-метил-1,1-пропандиил)имино((2S)-1-оксо-1,2-пропандиил)))бискарбамата;диметил (4,4'-бифенилдиилбис(1H-имидазол-4,2-диил((1S)-2-метил-1,1-пропандиил)имино((2S,3R)-3-метокси-1-оксо-1,2-бутандиил)))бискарбамата;диметил (4,4'-бифенилдиилбис(1H-имидазол-4,2-диил((1S)-2-метил-1,1-пропандиил)имино((2S)-3-метил-1-оксо-1,2-бутандиил)))бискарбамата;диметил...
Ингибиторы вируса гепатита с

Номер патента: 18793
Опубликовано: 30.10.2013
Авторы: Кэдоу Джон Ф., Белема Маконен
МПК: A61K 31/439, A61P 31/14, A61K 31/4178...
Метки: ингибиторы, гепатита, вируса
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, где L выбран из связи,R1 и R2 представляют собойили R1 представляет собойи R2 выбран изгде обозначает точку присоединения к исходной молекуле;R3 и R4 независимо выбраны из водорода и гало;каждый R5 независимо выбран из водорода;каждый R6 независимо выбран из алкила;R6a представляет собой алкил, где алкил необязательно может образовывать конденсированное 3-членное кольцо со смежным...