Индазолы и пиразолопиридины в качестве антагонистов рецептора ccr1
Номер патента: 21015
Опубликовано: 31.03.2015
Авторы: Кузмич Даниел, Мао Кань, Кук Брайан Николас, Разави Хоссейн



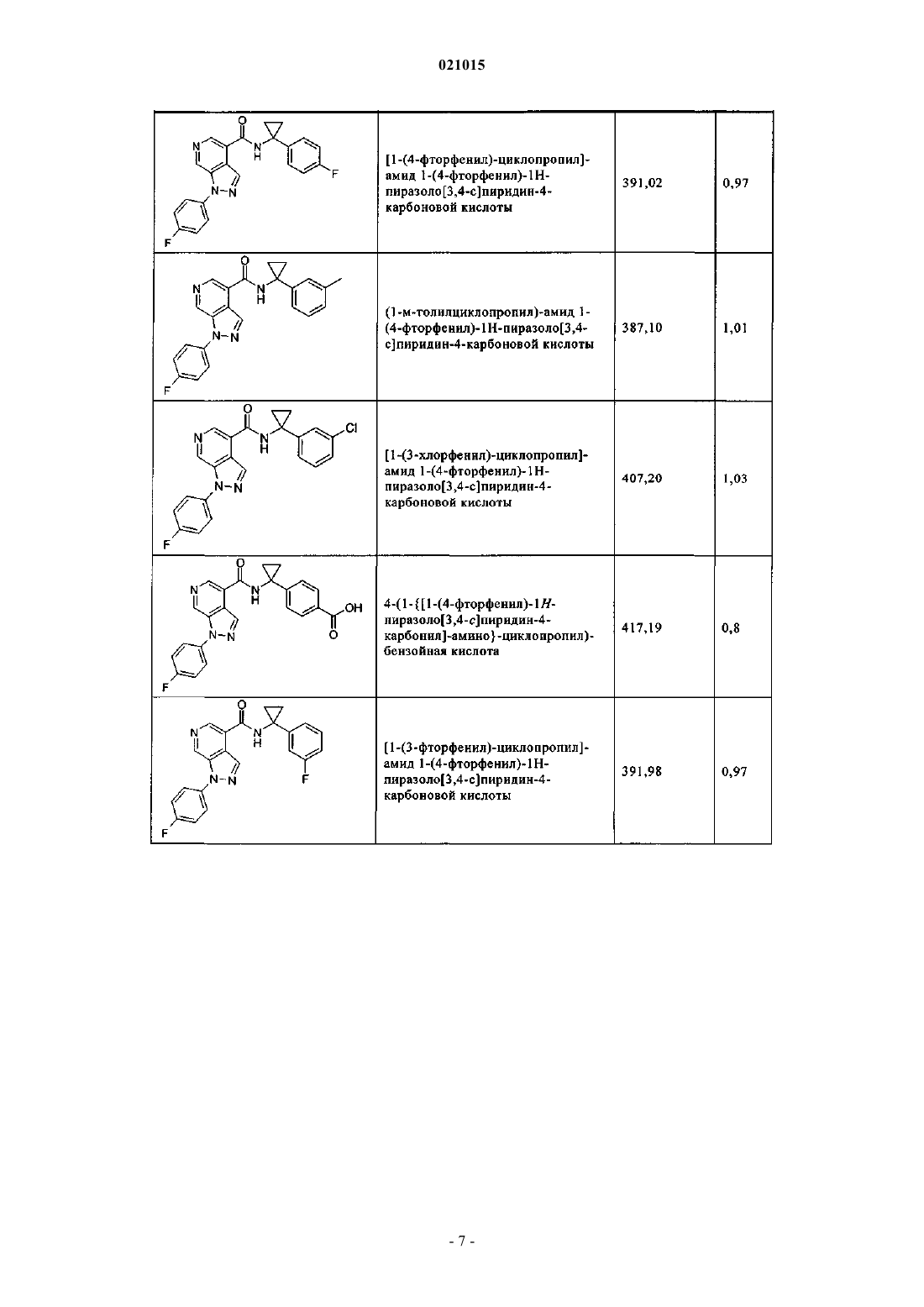
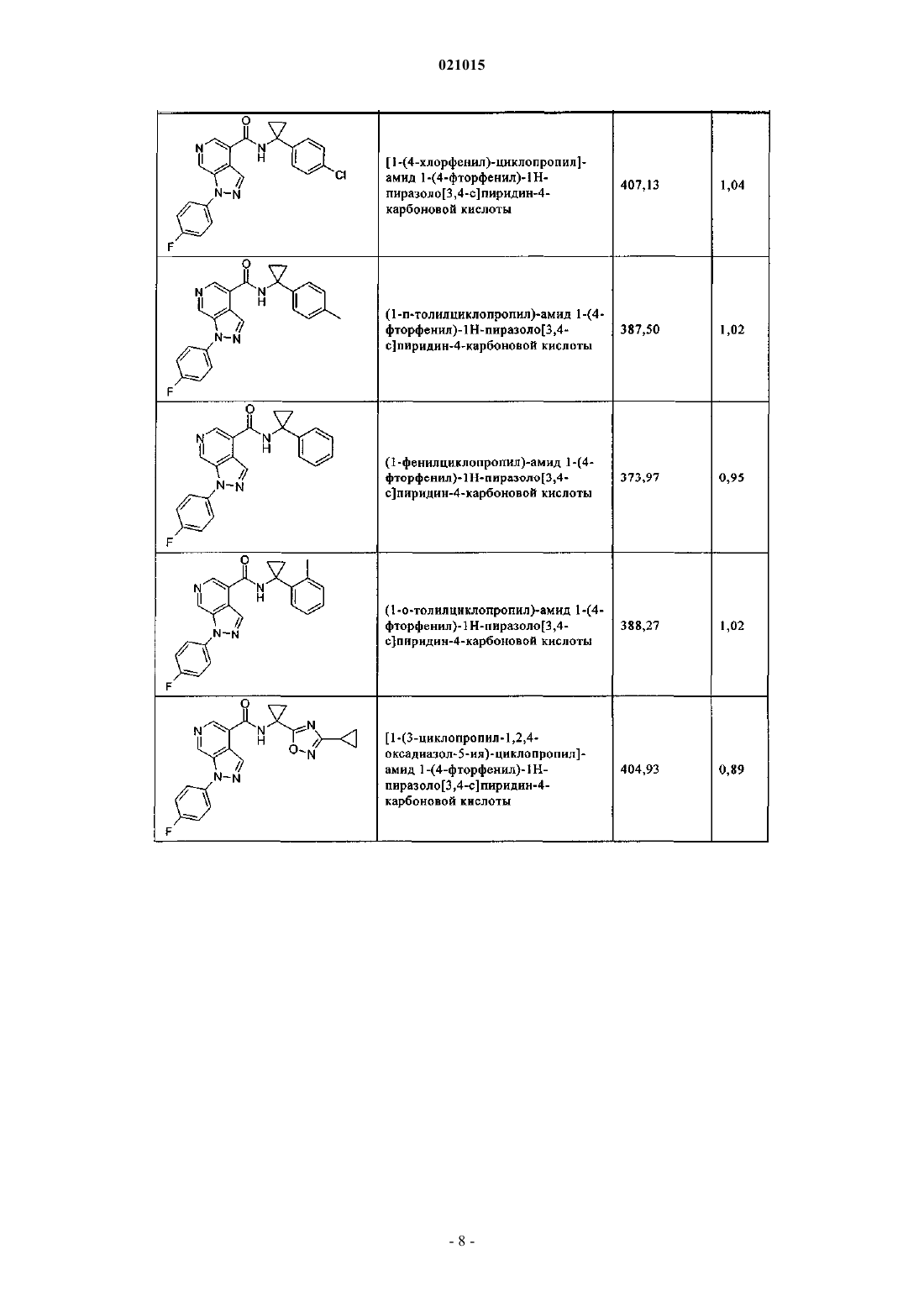
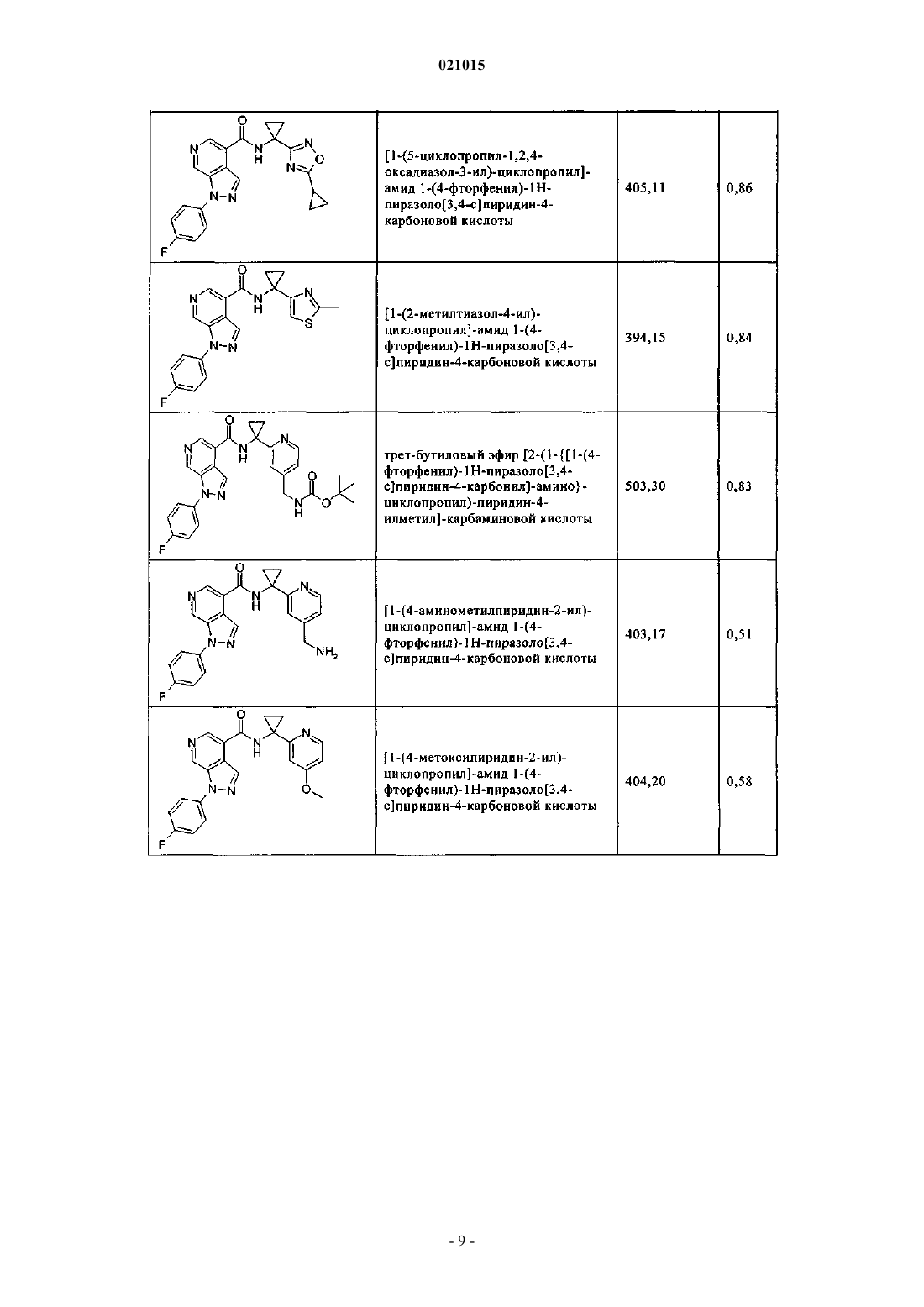
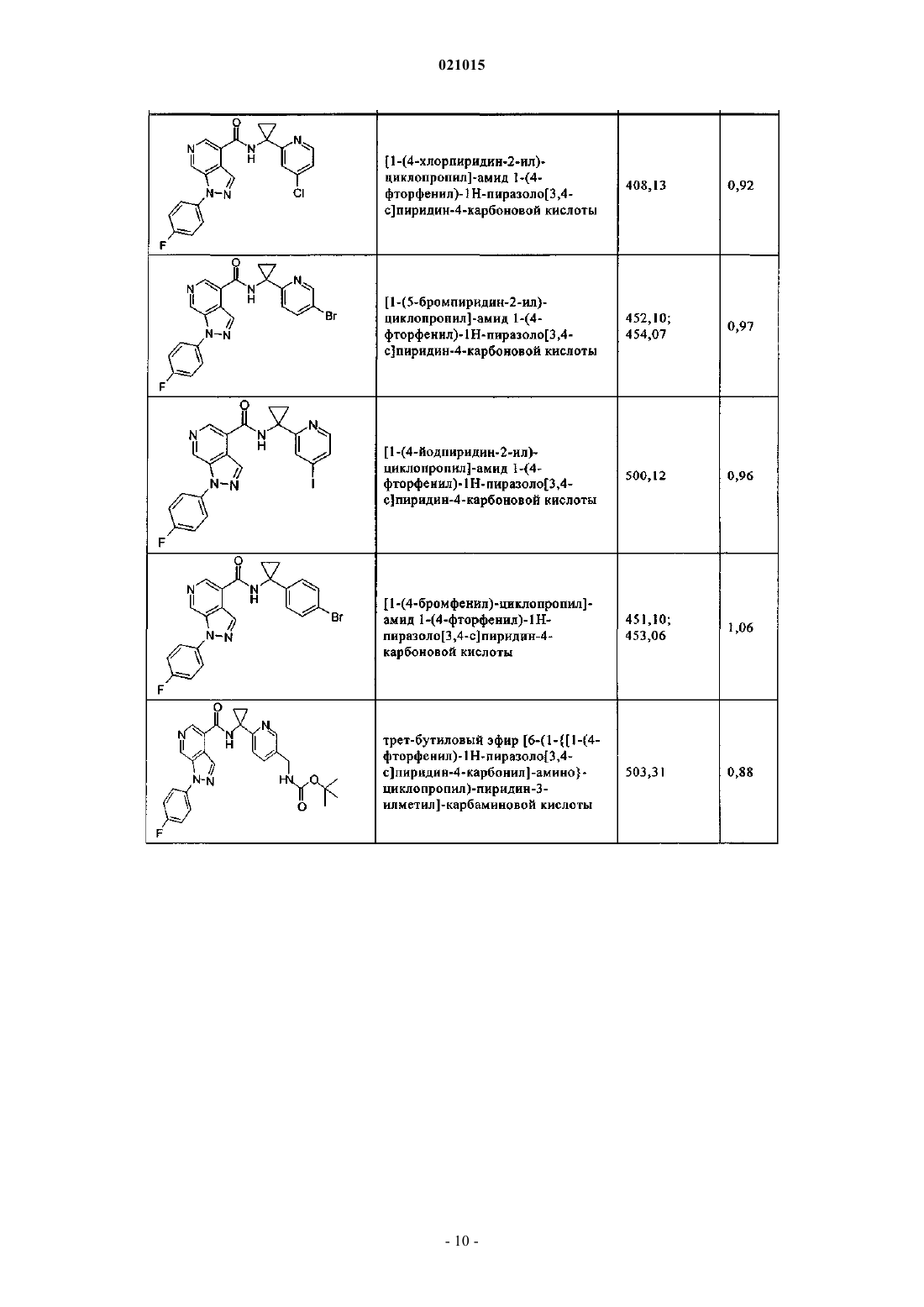
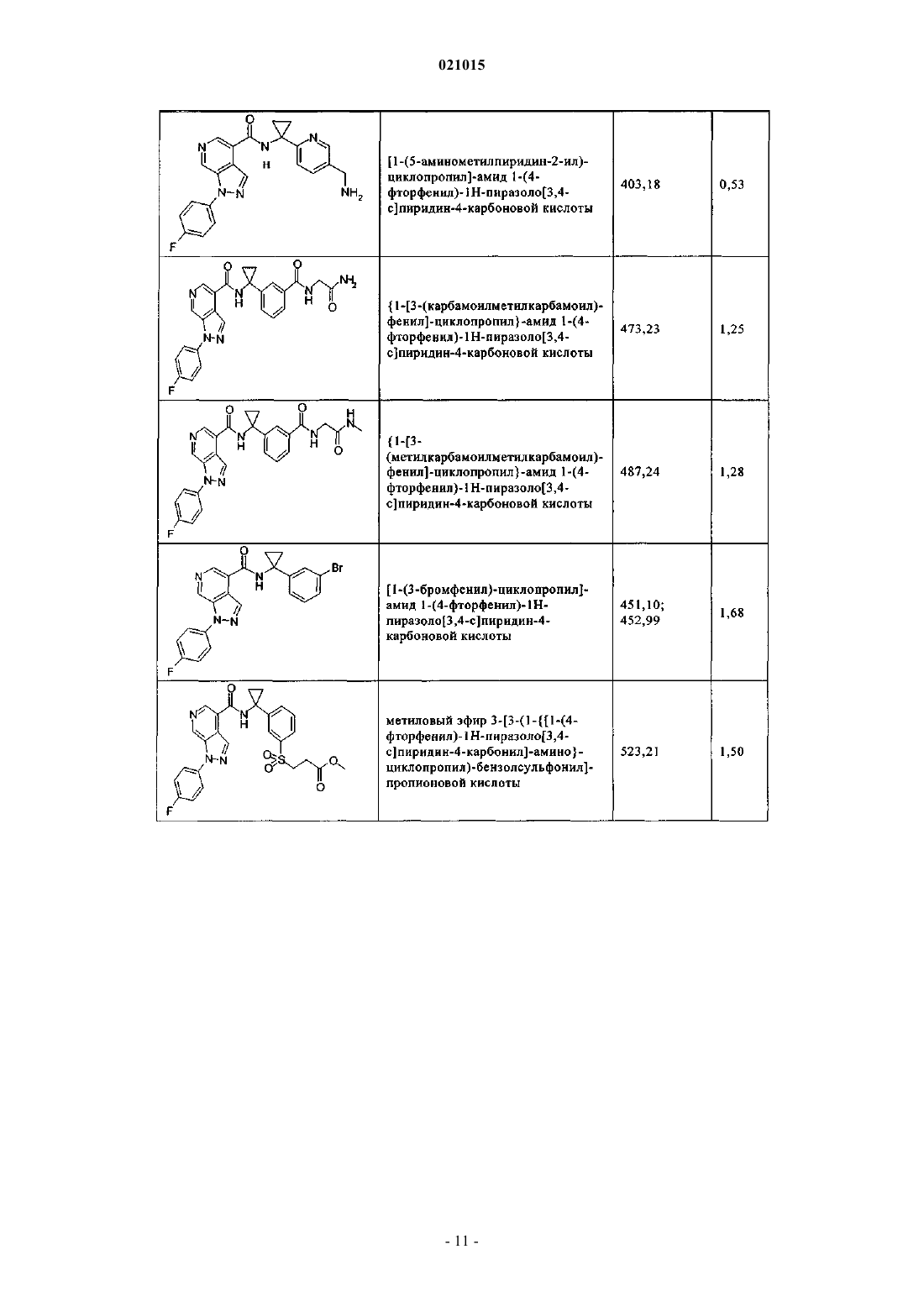
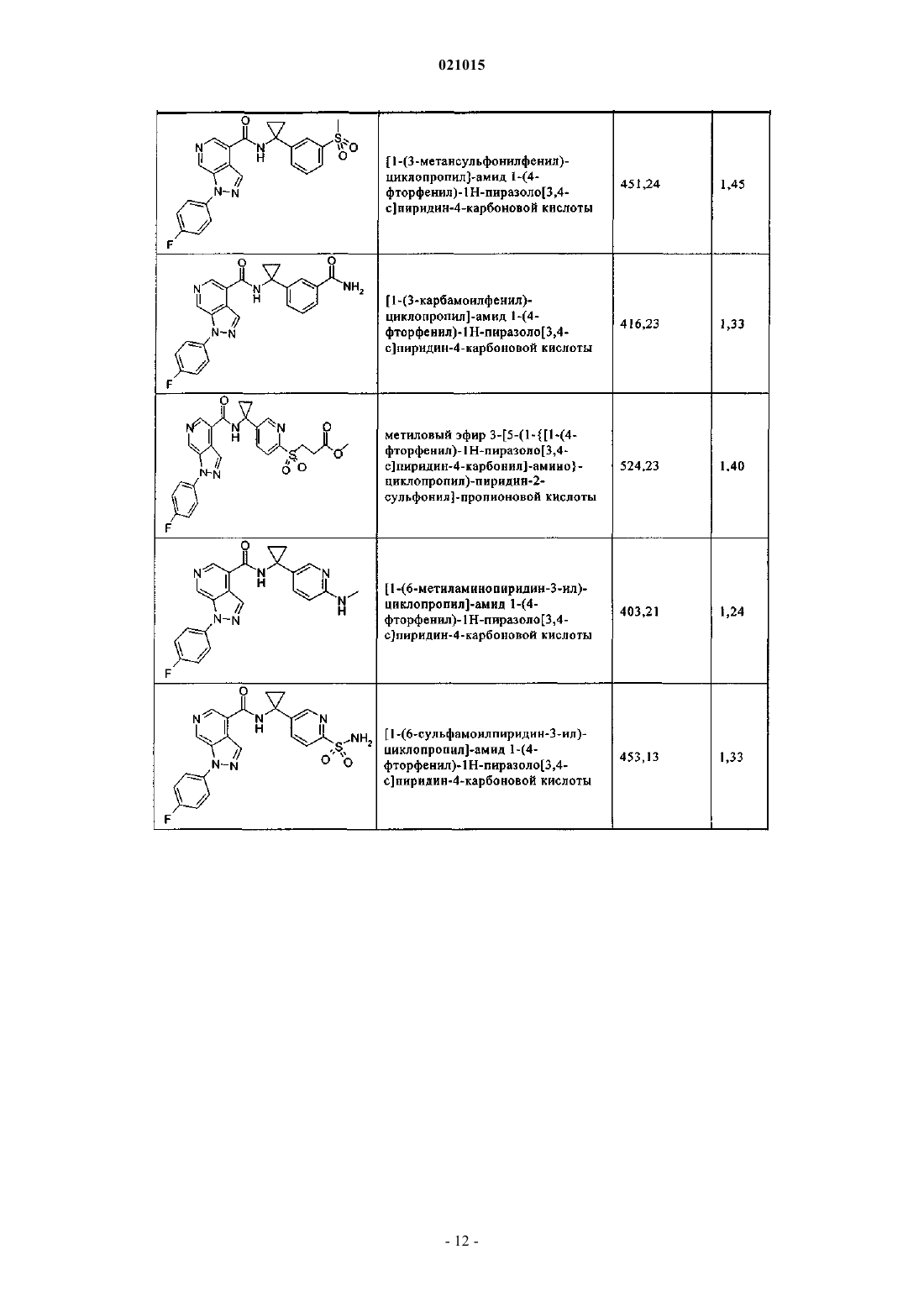
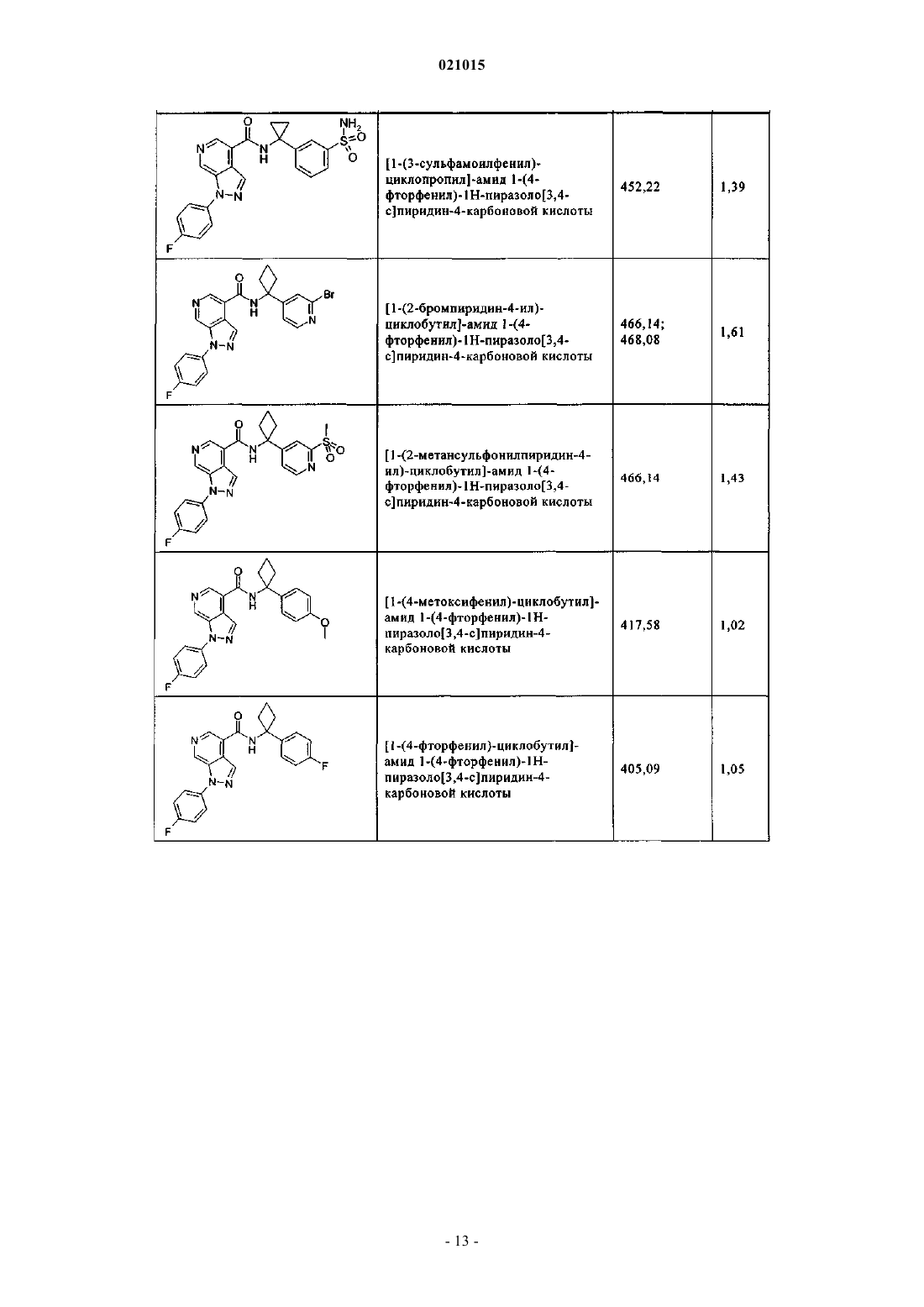
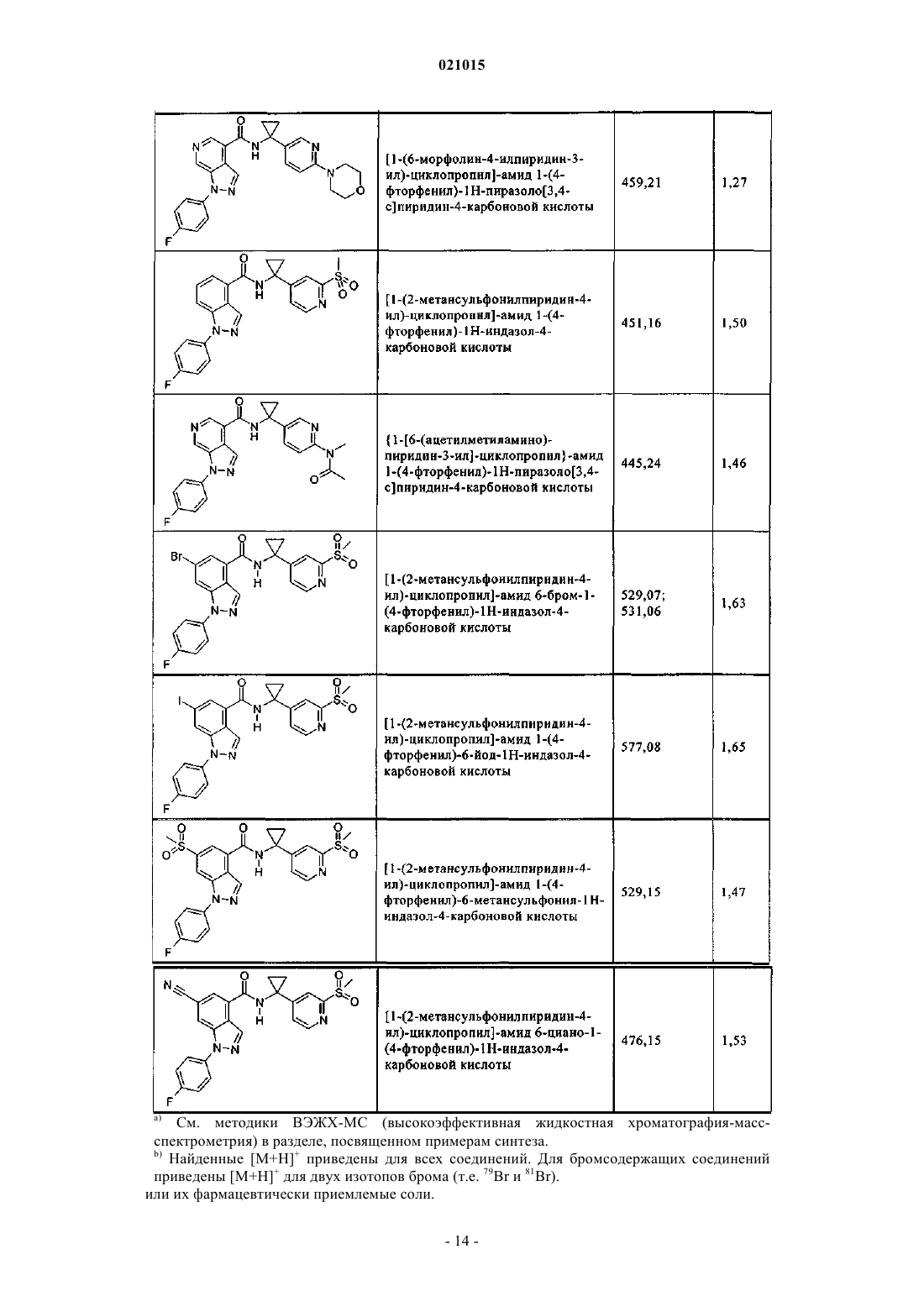












Формула / Реферат
1. Соединение формулы (I)

в которой X обозначает азот или -CR2;
Ar1 обозначает фенил, замещенный 1-2 группами Ra;
Ar2 обозначает фенил, тиадиазолил, оксадиазолил, пиримидинил, фуранил, тиазолил или пиридил, каждый из которых необязательно замещен 1-2 группами Rb;
циклический G представляет собой циклопропил или циклобутил;
R1 обозначает водород;
R2 обозначает водород или Ra;
Ra обозначает галоген, C1-С6-алкилсульфонил или цианогруппу;
Rb обозначает гидроксигруппу, карбоксигруппу, галоген, -CF3, -CN, -SO3H, C1-С3-алкил, С3-С6-циклоалкил, С1-С3-алкоксигруппу, -(СН2)n-СО2С1-С3-алкил, -(CH2)n-NRcRd, R3-S(O)m(CH2)0-1-, R3-S(O)2-NRe-, R3-NRe-S(O)2(CH2)0-1-, -NRf-C(O)-Re, -(CH2)y-C(O)-NRcRd или морфолинил;
каждый Rc, Rd независимо обозначает водород, C1-С3-алкил, С1-С3-ацил, циано-С1-С3-алкил, C1-С3-алкоксикарбонил-С0-С3-алкил, C1-С3-алкоксикарбонил-С3-С6-циклоалкил или -(CH2)n-C(O)-NReRf;
каждый Re, Rf независимо обозначает водород или С1-С3-алкил;
R3 обозначает водород или С1-С6-алкил, который необязательно содержит 1-2 следующих заместителя: C1-С6-алкоксигруппу или оксогруппу;
каждый n, у независимо равен 0-3;
каждый m независимо равен 0-2;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором
циклический G представляет собой циклопропил;
Ra обозначает -F, -Cl, метилсульфонил или цианогруппу;
Rb обозначает -СН3, карбоксигруппу, -F, -Cl, -Br, -I, -CF3, циклопропил, -ОСН3, -СО2Ме, -NRcRd, -CH2-NRcRd, R3-S(O)m-, R3-S(O)2-NRe-, R3-NRe-S(O)2-, -NRf-C(O)-Re, -C(O)NRcRd или морфолинил;
каждый Rc, Rd независимо обозначает водород, -СН3, -С(О)СН3, -CH2CN, С1-С3-алкоксикарбонил, метоксикарбонил-С1-С2-алкил-, метоксикарбонил-С3-циклоалкил- или -(CH2)-C(O)-NReRf;
каждый Re, Rf независимо обозначает водород или -СН3;
R3 обозначает водород или С1-С4-алкил, который необязательно содержит 1-2 следующих заместителя:
-ОСН3 или оксогруппу;
каждый m независимо равен 0-2;
или его фармацевтически приемлемая соль.
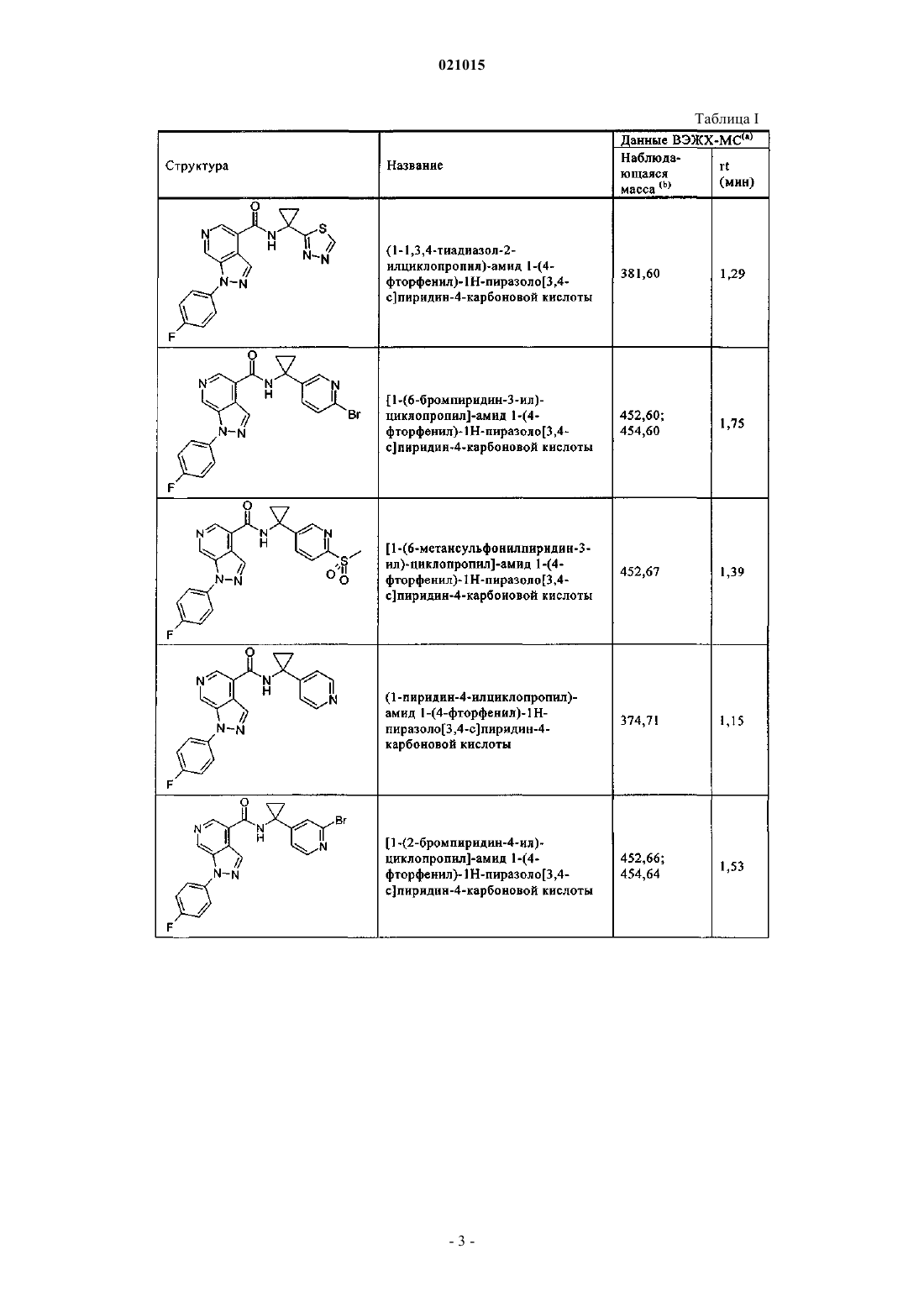
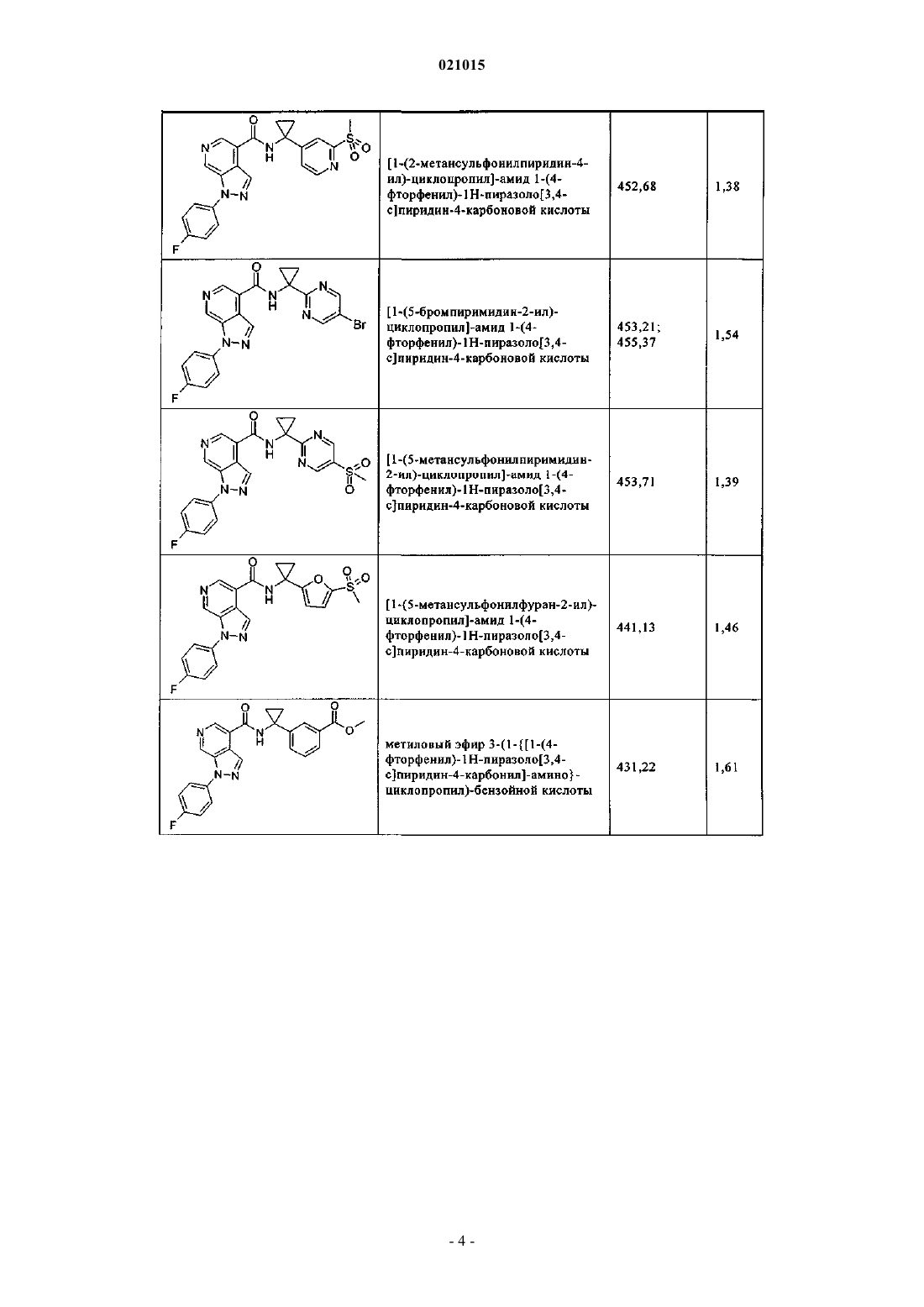
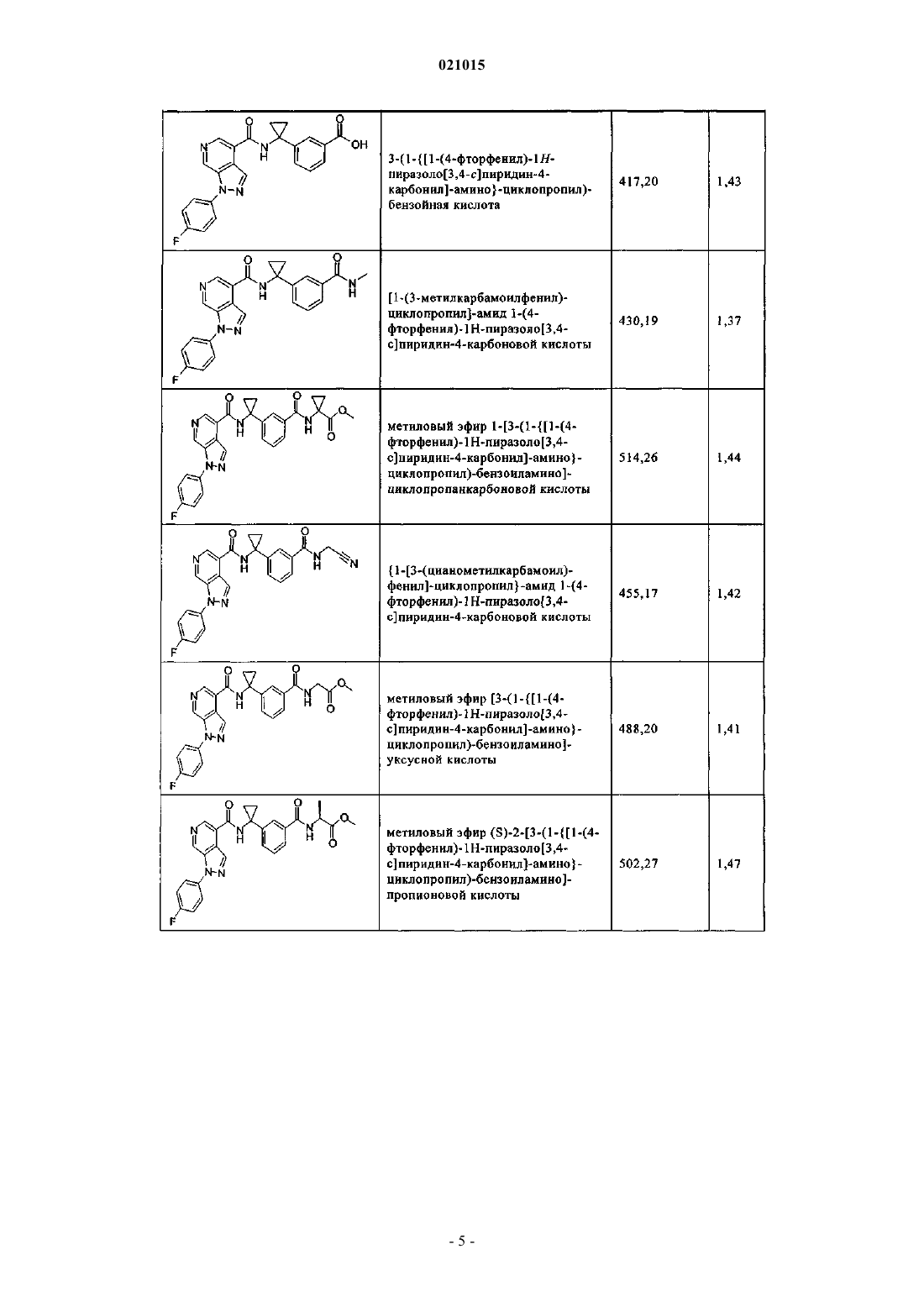
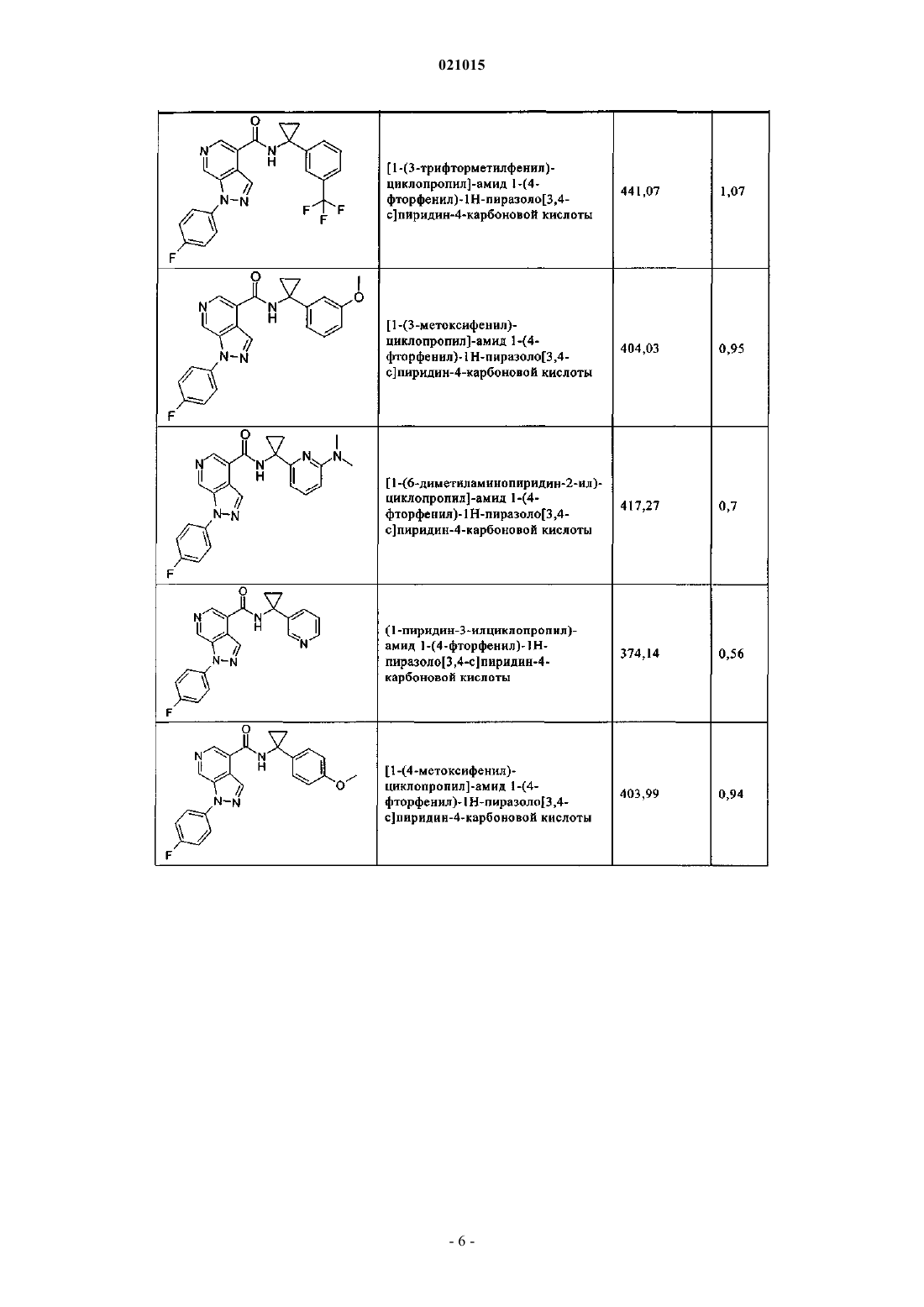
3. Соединение по п.1 или 2, выбранное из группы, включающей:







или его фармацевтически приемлемая соль.
4. Фармацевтическая композиция, содержащая соединение по любому из пп.1-3 в фармацевтически эффективном количестве и один или большее количество фармацевтически приемлемых носителей и/или вспомогательных веществ.
5. Применение соединения по любому из пп.1-3 для приготовления лекарственного средства для лечения хронического воспаления, аллергий, контактного дерматита, псориаза, ревматоидного артрита, рассеянного склероза, диабета типа 1, воспалительного заболевания кишечника, синдрома Гийена-Барре, болезни Крона, язвенного колита, заболевания трансплантат против хозяина, болезни Альцгеймера, астмы, хронической почечной недостаточности, сепсиса, аутоиммунного миокардита и системной красной волчанки.
Текст
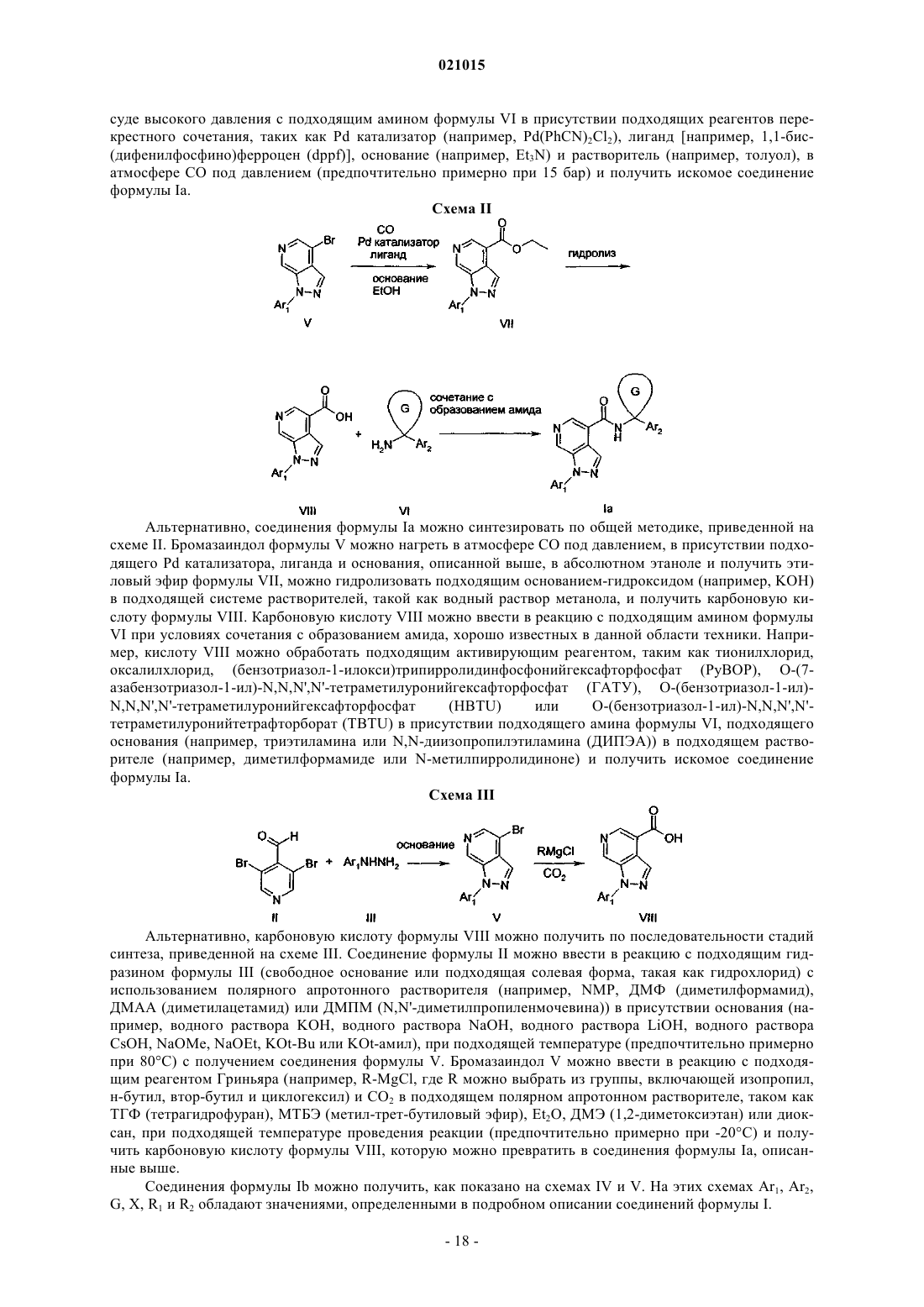
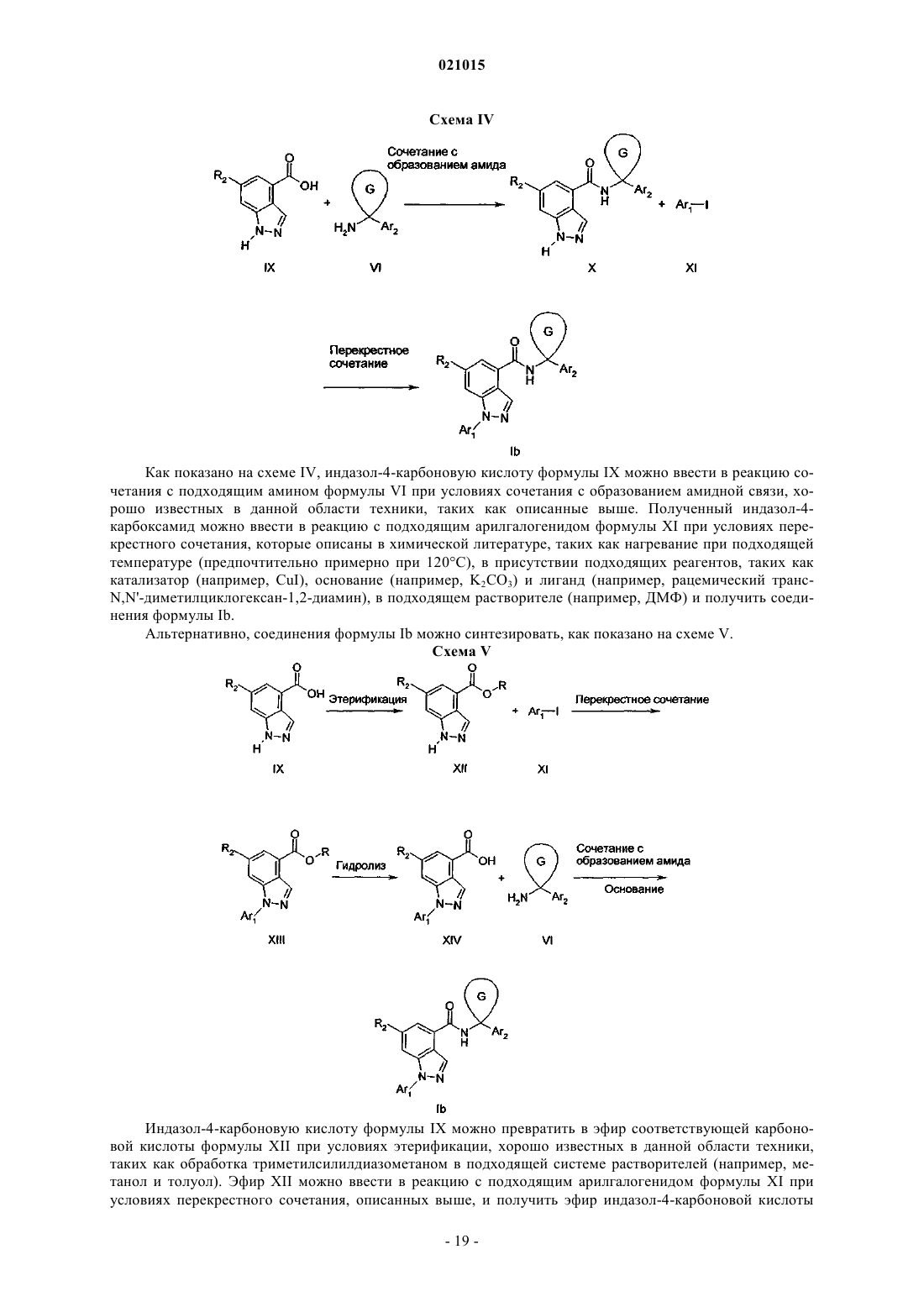
В изобретении описаны антагонисты рецептора CCR1 формулы (I) в которой X обозначает азот или -CR2; Ar1 обозначает фенил, замещенный 1-2 группами Ra; Ar2 обозначает фенил, тиадиазолил, оксадиазолил, пиримидинил, фуранил, тиазолил или пиридил,каждый из которых необязательно замещен 1-2 группами Rb; циклический G представляет собой циклопропил или циклобутил; R1 обозначает водород; R2 обозначает водород или Ra; Ra обозначает галоген, C1-С 6-алкилсульфонил или цианогруппу; Rb обозначает гидроксигруппу, карбоксигруппу,галоген, -CF3, -CN, -SO3H, C1-С 3-алкил, С 3-С 6-циклоалкил, C1-С 3-алкоксигруппу, -(СН 2)n-СО 2 С 1-С 3 алкил, -(CH2)n-NRcRd, R3-S(O)m(CH2)0-1-, R3-S(O)2-NRe-, R3-NRe-S(O)2(CH2)0-1-, -NRf-C(O)-Re, -(CH2)yC(O)-NRcRd или морфолинил; каждый Rc, Rd независимо обозначает водород, C1-С 3-алкил, C1 С 3-ацил, циано-С 1-С 3-алкил, C1-С 3-алкоксикарбонил-С 0-С 3-алкил, C1-С 3-алкоксикарбонил-С 3-С 6 циклоалкил или -(CH2)n-C(O)-NReRf; каждый Re, Rf независимо обозначает водород или C1-C3 алкил; R3 обозначает водород или C1-С 6-алкил, который необязательно содержит 1-2 следующих заместителя: C1-С 6-алкоксигруппу или оксогруппу; каждый n, у независимо равен 0-3; каждыйm независимо равен 0-2. В изобретении также описаны композиции и применение соединений формулы (I).(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) По заявке на настоящий патент испрашивается приоритет по предварительной заявке US61/253590, поданной 21 октября 2009 г. Область техники, к которой относится изобретение Настоящее изобретение относится к индазолам и пиразолопиридинам, содержащим арил- или гетероарилкарбоциклиламин, а также арил- или гетероарилгетероциклиламин, которые применимы в качестве антагонистов активности CCR1 и таким образом применимы для лечения различных заболеваний и нарушений, которые опосредуются или поддерживаются активностью CCR1, включая аутоиммунные заболевания, такие как ревматоидный артрит и рассеянный склероз. Настоящее изобретение также относится к фармацевтическим композициям, содержащим эти соединения, способам применения этих соединений для лечения различных заболеваний и нарушений, способам получения этих соединений и промежуточным продуктам, применимым в этих способах. Уровень техники Хемотаксический цитокиновый рецептор 1 (CCR1) относится к большому семейству (20) хемотаксических цитокиновых (хемокиновых) рецепторов, которые взаимодействуют с определенными хемокинами (50) и опосредуют перенос лейкоцитов, гранулярный экзоцитоз, транскрипцию гена, митогенные воздействия и апоптоз. Хорошо известно, что хемокины могут опосредовать базальный и воспалительный перенос лейкоцитов. Связывание по меньшей мере трех хемокинов (MIP-1 альфа/CCL3, MCP3/CCL7 и RANTES/CCL5) с CCR1 обеспечивает перенос моноцитов, макрофагов и ТН 1-клеток к воспаленным тканям пациентов, страдающих ревматоидным артритом (РА) и рассеянным склерозом (PC) (Trebst et al.(2001), American J. of Pathology 159, p. 1701). Воспалительный белок макрофагов 1 альфа (MIP-1 альфа),хемоаттрактантный белок макрофагов 3 (МСР-3) и белок, после активации которого он регулируется и который экспрессируется и секретируется нормальными Т-клетками (RANTES), обнаружены в центральной нервной системе (ЦНС) пациентов, страдающих PC, тогда как MIP-1 альфа и RANTES обнаружены в ЦНС в модели экспериментального аутоиммунного энцефаломиелита (ЭАЭ) для PC (обзор приведен в публикации: Gerard and Rollins (2001), Nature Immunology). Макрофаги и ТН 1-клетки в воспаленной синовиальной полости пациентов, страдающих РА, также являются основными источниками MIP-1 альфа и RANTES, которые постоянно поставляют лейкоциты в синовиальные ткани пациентов, страдающих РА, и распространяют хроническое воспаление (Volin et al. (1998), Clin. Immunol. Immunopathology; Koch et al. (1994), J. Clin. Investigation; Conlon et al. (1995), Eur. J. Immunology). Предполагают, что препятствование взаимодействиям между CCR1 его хемокиновыми лигандами блокирует хемотаксис моноцитов, макрофагов и Th1-клеток к воспаленным тканям и таким образом облегчает протекание хронического воспаления, связанного с аутоиммунными заболеваниями, такими как РА и PC. Данные о роли CCR1 в развитии и прогрессировании хронического воспаления, связанные с моделью экспериментального аутоиммунного энцефаломиелита (ЭАЭ) для рассеянного склероза, основаны на исследовании влияния делеции гена и небольших молекул-антагонистов CCR1. Показано, что мыши с дефицитом CCR1 обладают пониженной восприимчивостью (55% вместо 100%) и пониженной тяжестью протекания (1,2 вместо 2,5) активного ЭАЭ (Rottman et al. (2000), Eur. J. Immunology). Кроме того, показано, что введение небольших молекул-антагонистов CCR1 со средним сродством (Ki = 120 нМ) крысам с CCR1 при внутривенном введении задерживает появление и снижает тяжесть протекания ЭАЭ (Liang etal. (2000), J. Biol. Chemistry). Также показано, что лечение мышей антителами, специфичными по отношению к лиганду CCR1 MIP-1 альфа, является эффективным для предупреждения развития острого и рецидивирующего ЭАЭ вследствие уменьшения количества Т-клеток и макрофагов, поставляющихся в ЦНС (Karpus et al. (1995), J. Immunology; Karpus and Kennedy (1997), J. Leukocyte Biology). Таким образом, показано, что по меньшей мере один лиганд CCR1 поставляет лейкоциты в ЦНС и распространяет хроническое воспаление в ЭАЭ, что обеспечивает дополнительное подтверждение in vivo роли CCR1 в ЭАЭ и PC. Подтверждение in vivo роли CCR1 в развитии и распространении хронического воспаления, связанного с РА, также является важным. Например, показано, что введение антагониста CCR1 в модели вызванного коллагеном артрита (ВКА) мышам DBA/1, является эффективным для уменьшения синовиального воспаления и разрушения сустава (Plater-Zyberk et al. (1997), Immunology Letters). В другой публикации описаны активные антагонисты CCR1 мышей, которые при пероральном введении уменьшают тяжесть протекания (58%) ускоренного липополисахаридом (ЛПС) вызванного коллагеном артритаIb клинического испытания при пероральном введении антагониста CCR1 показали тенденцию к клиническому улучшению при отсутствии нежелательных побочных эффектов (Haringman et al. (2003), Ann.Rheum. Dis.). У трети пациентов на 18-й день на 20% уменьшились признаки и симптомы ревматоидного артрита (ACR20) и количество клеток с положительной реакцией на CCR1 в синовиальной жидкости подвергнутых лечению пациентов уменьшилось на 70% при значительном уменьшении количества определенных типов клеток, включая уменьшение количества CD4+ Т-клеток на 50%, уменьшение количества CD8+ Т-клеток на 50% и уменьшение количества макрофагов на 34%. Такие исследования, как указанные выше, подтверждают роль CCR1 в PC и РА и предоставляют терапевтическое обоснование для разработки антагонистов CCR1. Краткое изложение сущности изобретения Настоящее изобретение относится к новым соединениям, которые препятствуют взаимодействиюCCR1 и его лигандов и таким образом применимы для лечения различных заболеваний и нарушений,которые опосредуются или поддерживаются активностью CCR1, включая аутоиммунные заболевания,такие как ревматоидный артрит и рассеянный склероз. Настоящее изобретение также относится к фармацевтическим композициям, содержащим эти соединения, способам применения этих соединений для лечения различных заболеваний и нарушений. Подробное описание изобретения В наиболее широком общем варианте осуществления настоящее изобретение относится к соединению формулы (I) в которой X обозначает азот или -CR2;Ar2 обозначает фенил, тиадиазолил, оксадиазолил, пиримидинил, фуранил, тиазолил или пиридил,каждый из которых необязательно замещен 1-2 группами Rb; циклический G представляет собой циклопропил или циклобутил;R2 обозначает водород или Ra;Rb обозначает гидроксигруппу, карбоксигруппу, галоген, -CF3, -CN, -SO3H, C1-С 3-алкил, С 3-С 6 циклоалкил, C1-С 3-алкоксигруппу, -(СН 2)n-СО 2 С 1-С 3-алкил, -(CH2)n-NRcRd, R3-S(O)m(CH2)0-1-, R3-S(O)2NRe-, R3-NRe-S(O)2(CH2)0-1-, -NRf-C(O)-Re, -(CH2)y-C(O)-NRcRd или морфолинил; каждый Rc, Rd независимо обозначает водород, C1-С 3-алкил, C1-С 3-ацил, циано-C1-С 3-алкил, C1-С 3 алкоксикарбонил-С 0-С 3-алкил, C1-С 3-алкоксикарбонил-С 3-С 6-циклоалкил или -(CH2)n-C(O)-NReRf; каждый Re, Rf независимо обозначает водород или C1-С 3-алкил;R3 обозначает водород или C1-С 6-алкил, который необязательно содержит 1-2 следующих заместителя: C1-С 6-алкоксигруппу или оксогруппу; каждый n, у независимо равен 0-3; каждый m независимо равен 0-2; или его фармацевтически приемлемой соли. В другом варианте осуществления настоящее изобретение относится к соединению формулы (I),приведенной выше, в которой циклический G представляет собой циклопропил;Rb обозначает -СН 3, карбоксигруппу, -F, -Cl, -Br, -I, -CF3, циклопропил, -ОСН 3, -СО 2 Ме, -NRcRd,-CH2-NRcRd, R3-S(O)m-, R3-S(O)2-NRe-, R3-NRe-S(O)2-, -NRf-C(O)-Re, -C(O)NRcRd или морфолинил; каждый Rc, Rd независимо обозначает водород, -СН 3, -С(О)СН 3, -CH2CN, С 1-С 3-алкоксикарбонил,метоксикарбонил-С 1-С 2-алкил-, метоксикарбонил-С 3-циклоалкил- или -(CH2)-C(O)-NReRf; каждый Re, Rf независимо обозначает водород или -СН 3;R3 обозначает водород или C1-C4-алкил, который необязательно содержит 1-2 следующих заместителя: -ОСН 3 или оксогруппу; каждый n, у независимо равен 0-3; каждый m независимо равен 0-2; или его фармацевтически приемлемой соли. Ниже приведены типичные соединения, предлагаемые в настоящем изобретении, которые можно получить в соответствии с общими схемами синтеза, примерами и по методикам, известным в данной области техники.b) Найденные [М+Н]+ приведены для всех соединений. Для бромсодержащих соединений приведены [М+Н]+ для двух изотопов брома (т.е. 79Br и 81Br). или их фармацевтически приемлемые соли. Для всех соединений, раскрытых выше в настоящем изобретении, в случае, если название будет противоречить структуре, следует понимать, что соединение определяется структурой. Настоящее изобретение также относится к фармацевтическим препаратам, содержащим в качестве активного вещества одно или большее количество соединений, предлагаемых в настоящем изобретении,или их фармацевтически приемлемых производных, необязательно в комбинации с обычными инертными наполнителями и/или носителями. Соединения, предлагаемые в настоящем изобретении, также включают их изотопно-меченые формы. Изотопно-меченая форма активного агента комбинации, предлагаемой в настоящем изобретении,идентична указанному активному агенту, но отличается тем, что один или большее количество атомов указанного активного агента заменены на атом или атомы, обладающие атомной массой или массовым числом, отличающимися от атомной массы или массового числа указанного атома, который обычно обнаруживается в природе. Примеры изотопов, которые имеются в продаже и которые можно включить в активный агент комбинации, предлагаемой в настоящем изобретении, по хорошо известным методикам,включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, например 2 Н, 3 Н, 13 С,14 С, 15N, 18 О, 17 О, 31 Р, 32 Р, 35S, 18F и 36Cl соответственно. Активный агент комбинации, предлагаемой в настоящем изобретении, его пролекарство или фармацевтически приемлемая соль любого из них, которые содержат один или большее количество указанных выше изотопов и/или других изотопов других атомов,входят в объем настоящего изобретения. Настоящее изобретение включает применение любых из описанных выше соединений, содержащих один или большее количество асимметрических атомов углерода, которые могут представлять собой рацематы и рацемические смеси, отдельные энантиомеры, смеси диастереоизомеров и отдельные диастереоизомеры. Изомеры следует понимать, как означающие энантиомеры и диастереоизомеры. Все такие изомерные формы этих соединений явно включены в настоящее изобретение. Каждый стереогенный атом углерода может находиться в R или S конфигурации, а соединение может включать комбинацию конфигураций. Некоторые из соединений, предлагаемых в настоящем изобретении, могут существовать более чем в одной таутомерной форме. Настоящее изобретение включает способы, в которых применяются все такие таутомеры. Все термины при использовании в описании настоящего изобретения, если не указано иное, следует понимать в их обычных значениях, известных в данной области техники. Например, "C1-С 4 алкоксигруппа" означает С 1-С 4-алкил, включающий концевой атом кислорода, такой как метоксигруппа,этоксигруппа, пропоксигруппа, бутоксигруппа. Все алкильные, алкенильные и алкинильные группы следует понимать, как разветвленные или неразветвленные, если это возможно в структуре и если не указано иное. Другие более конкретные определения приведены ниже: Карбоциклы включают углеводородные кольца, содержащие от 3 до 12 атомов углерода. Эти карбоциклы могут представлять собой ароматические или неароматические кольцевые системы. Неароматические кольцевые системы могут быть моно- или полиненасыщенными. Предпочтительные карбоциклы включают, но не ограничиваются только ими, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогептанил, циклогептенил, фенил, инданил, инденил, бензоциклобутанил, дигидронафтил, тетрагидронафтил, нафтил, декагидронафтил, бензоциклогептанил и бензоциклогептенил. Некоторые названия циклоалкилов, такие как циклобутанил и циклобутил, будут использоваться, как взаимозаменяемые. Термин "гетероцикл" означает стабильный неароматический 4-8-членный (но предпочтительно 5 или 6-членный) моноциклический или неароматический 8-11-членный бициклический или спироциклический гетероциклический радикал, который может быть насыщенным или ненасыщенным. Каждый гетероциклил состоит из атомов углерода и одного или большего количества гетероатомов, предпочтительно от 1 до 4 гетероатомов, выбранных из группы, включающей азот, кислород и серу. Гетероцикл может быть присоединен через любой атом цикла, если это приводит к образованию стабильной структуры. Термин "гетероарил" следует понимать, как означающий ароматическое 5-8-членное моноциклическое или 8-11-членное бициклическое кольцо, содержащее 1-4 гетероатома, таких как N, О и S. Если не указано иное, гетероциклы и гетероарил включают, но не ограничиваются только ими, например, фуранил, пиранил, бензоксазолил, бензотиазолил, бензимидазолил, тетрагидропиранил, диоксанил, диоксоланил, тетрагидрофуранил, оксазолил, изоксазолил, тиазолил, пиразолил, пирролил, имидазолил, тиенил, тиадиазолил, тиоморфолинил, 1,1-диоксо-16-тиоморфолинил, морфолинил, пиридинил,пиримидинил, пиридазинил, пиразинил, триазинил, пирролидинил, пиперидинил, пиперазинил, пуринил,хинолинил, дигидро-2H-хинолинил, изохинолинил, хиназолинил, индазолил, тиено[2,3-d]пиримидинил,индолил, изоиндолил, бензофуранил, бензопиранил и бензодиоксолил. Термин "гетероатом" при использовании в настоящем изобретении следует понимать, как означающий атомы, не являющиеся атомами углерода, такие как О, N, S и Р. Во всех алкильных группах или углеродных цепях один или большее количество атомов углерода могут необязательно быть замещены гетероатомами: О, S или N, следует понимать, что если N не явля- 15021015 ется замещенным, то он представляет собой NH, также следует понимать, что в разветвленной или неразветвленной углеродной цепи гетероатомы могут заменять концевые атомы углерода или внутренние атомы углерода. Такие группы могут быть замещены, как это описано выше в настоящем изобретении,группами, такими как оксогруппы, что приводит к таким определениям, но не ограничивается только ими, как алкоксикарбонил, ацил, амидная группа и тиооксогруппа. Термин "арил" при использовании в настоящем изобретении следует понимать, как означающий ароматический карбоцикл или гетероарил, как это определено в настоящем изобретении. Каждый арил или гетероарил, если не указано иное, включает свое частично или полностью гидрированное производное. Например, хинолинил может включать декагидрохинолинил и тетрагидрохинолинил, нафтил может включать свои гидрированные производные, такие как тетрагидронафтил. Другие частично или полностью гидрированные производные арильных и гетероарильных соединений, описанные в настоящем изобретении, должны быть очевидны для специалиста с общей подготовкой в данной области техники. При использовании в настоящем изобретении "азот" и "сера" включают любую окисленную форму азота и серы и кватернизованную форму любого основного атома азота. Например, для -S-C1-С 6 алкильного радикала, если не указано иное, это следует понимать и как включение -S(О)-С 1-С 6-алкила и-S(O)2-C1-С 6-алкила. Термин "алкил" означает насыщенный алифатический радикал, содержащий от 1 до 10 атомов углерода, или моно- или полиненасыщенный алифатический углеводородный радикал, содержащий от 2 до 12 атомов углерода, если не указано иное. Моно- или полиненасыщенный алифатический углеводородный радикал содержит по меньшей мере одну двойную или тройную связь соответственно. "Алкил" означает и разветвленные, и неразветвленные алкильные группы. Следует понимать, что любой комбинированный термин, содержащий приставку "алк" или "алкил", означает аналоги, соответствующие приведенному выше определению "алкила". Например, такие термины, как "алкоксигруппа", "алкилтиогруппа", означают алкильные группы, связанные со второй группой через атом кислорода или серы. "Алканоил" означает алкильную группу, присоединенную к карбонильной группе (С=О). Термин "галоген" при использовании в настоящем описании следует понимать, как означающий бром, хлор, фтор или йод, предпочтительно фтор. Определения " галогенированный", "частично или полностью галогенированный"; "частично или полностью фторированный"; "замещенный одним или большим количеством атомов галогенов", включают, например, моно-, ди- или тригалогенпроизводные по одному или большему количеству атомов углерода. Для алкила неограничивающими примерами являются -CH2CHF2, -CF3 и т.п. Каждый алкил, карбоцикл, гетероцикл, или гетероарил, или их аналоги, описанные в настоящем изобретении, следует понимать, как необязательно частично или полностью галогенированные. Соединениями, предлагаемыми в настоящем изобретении, являются только такие, которые предполагаются "химически стабильными", что должно быть очевидно для специалистов в данной области техники. Например, соединение, которое содержит "свободную валентность", или "карбанион" не является соединением, соответствующим способам, раскрытым в настоящем изобретении. Настоящее изобретение включает фармацевтически приемлемые производные соединений формулы (I). "Фармацевтически приемлемое производное" означает любую фармацевтически приемлемую соль, или сложный эфир, или любое другое соединение, которое после введения пациенту способно образовать (прямо или косвенно) соединение, применимое в настоящем изобретении, или его фармакологически активный метаболит, или фармакологически активный остаток. Фармакологически активный метаболит следует понимать, как означающий любое соединение, предлагаемое в настоящем изобретении, способное к ферментативному или химическому метаболизму. Он включает, например, гидроксилированные или окисленные производные соединений, предлагаемых в настоящем изобретении. Фармацевтически приемлемые соли включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих кислот включают хлористоводородную, бромисто-водородную, серную, азотную, хлорную, фумаровую, малеиновую, фосфорную,гликолевую, молочную, салициловую, янтарную, толуол-п-сульфоновую, винную, уксусную, лимонную,метансульфоновую, муравьиную, бензойную, малоновую, нафталин-2-сульфоновую и бензолсульфоновую кислоты. Другие кислоты, такие как щавелевая кислота, которые сами по себе не являются фармацевтически приемлемыми, можно использовать при получении солей, использующихся в качестве промежуточных продуктов при получении соединений и их фармацевтически приемлемых солей присоединения с кислотами. Соли, полученные из подходящих оснований, включают соли щелочного металла(например, натрия), щелочно-земельного металла (например, магния), аммония и N-(C1-C4-алкил)4+. Кроме того, в объем настоящего изобретения входит применение пролекарств соединений, предлагаемых в настоящем изобретении. Пролекарства включают такие соединения, которые после простого химического превращения изменяются с образованием соединений, предлагаемых в настоящем изобретении. Простые химические превращения включают гидролиз, окисление и восстановление. Точнее, если пролекарство вводится пациенту, то пролекарство может превратиться в соединение, раскрытое выше в настоящем изобретении, и тем самым обеспечить необходимое фармакологическое воздействие. Соединения формулы I можно получить с помощью общих методик синтеза, описанных ниже, ко- 16021015 торые также являются частью настоящего изобретения. Общие методики синтеза Соединения, предлагаемые в настоящем изобретении, можно получить по общим методикам и в соответствии с примерами, приведенными ниже, и по методикам, известным специалисту с общей подготовкой в данной области техники и описанным в химической литературе. Если не указано иное, то растворители, температуры, давления и другие условия проведения реакций может легко выбрать специалист с общей подготовкой в данной области техники. Конкретные методики приведены в разделе, посвященном примерам синтеза. Промежуточные арил- или гетероарилциклоалкиламины имеются в продаже, их можно получить по общим методикам или в соответствии с приведенными ниже ссылками (которые во всей своей полноте включены в настоящее изобретение в качестве ссылки), или специалист в данной области техники может их получить по методикам, описанным в химической литературе. Арил- или гетероарилциклопропиламин можно синтезировать с помощью катализируемого алкоксидом титана восстановительного циклопропанирования соответствующих арил- или гетероарилнитрилов реагентами Гриньяра (Szymoniak, J. et al. J. Org. Chem. 2002, 67, 3965, and Bertus, P. et al. J. Org.Chem. 2003, 68, 7133) или содержащими цинк реагентами (de Meijere, A. et al. Org. Lett. 5, 2003, 753). Альтернативно, арилциклопропиламины можно синтезировать из арилнитрилов или ариловых сложных эфиров путем циклоалкилирования (например, Jonczyk, A. et al. Org. Prep. Proc. 27, 1995, 355) с последующим превращением нитрила или сложного эфира в карбоновую кислоту, с последующей перегруппировкой Курциуса полученной карбоновой кислоты в эфир карбаминовой кислоты (например, Hanano,Т. et al. Bioorg. Med. Chem. Lett. 10, 2000, 881) и удалением защитной группы из полученного эфира карбаминовой кислоты. Реакции с образованием амидной связи можно провести при стандартных условиях реакции сочетания, хорошо известных в данной области техники (см., например, публикацию Bodanszky, M. The Practice of Peptide Synthesis, Springer-Verlag, 1984, которая во всей своей полноте включена в настоящее изобретение в качестве ссылки), например, путем реакции карбоновой кислоты и амина в присутствии 1-(3 диметиламинопропил)-3-этилкарбодиимида (ЭДК) и 1-гидроксибензотриазола. За протеканием реакции можно следить с помощью обычных методик, таких как тонкослойная хроматография (ТСХ). Промежуточные продукты и конечные продукты можно очистить по методикам, известным в данной области техники, включая колоночную хроматографию, ВЭЖХ (высокоэффективная жидкостная хроматография) или перекристаллизацию. Методики, описанные ниже и приведенные в разделе, посвященном примерам синтеза, можно использовать для получения соединений формулы Ia (т.е. соединений формулы I, в которой X обозначает азот, схемы I, II и III) и соединений формулы Ib (т.е. соединений формулы I, в которой X обозначает СR2, схемы IV и V). На приведенных ниже схемах Ar1, Ar2, циклический G, X, R1 и R2 обладают значениями, определенными в подробном описании соединений формулы I. Соединения формулы Ia можно получить по схемам I-III. Схема I Как показано на схеме I, подходящий гидразин формулы III (свободное основание или подходящая солевая форма, такая как гидрохлорид), содержащий Ar1 можно ввести в реакцию с 3,5-дибром-4 пиридинкарбоксальдегидом II в присутствии ацетата натрия и в подходящем растворителе, таком какEtOH, и получить гидразон формулы IV. Соединение формулы IV можно циклизовать в присутствии подходящих реагентов, таких как диаминовый лиганд (например, транс-N,N'-диметилциклогексан-1,2 диамин), соль меди (например, CuI), основание (например, K2CO3) и растворитель (например, N-метил-2 пирролидинон), и получить соединения формулы V. Бромазаиндол V можно нагреть в герметичном со- 17021015 суде высокого давления с подходящим амином формулы VI в присутствии подходящих реагентов перекрестного сочетания, таких как Pd катализатор (например, Pd(PhCN)2Cl2), лиганд [например, 1,1-бис(дифенилфосфино)ферроцен (dppf)], основание (например, Et3N) и растворитель (например, толуол), в атмосфере СО под давлением (предпочтительно примерно при 15 бар) и получить искомое соединение формулы Ia. Схема II Альтернативно, соединения формулы Ia можно синтезировать по общей методике, приведенной на схеме II. Бромазаиндол формулы V можно нагреть в атмосфере СО под давлением, в присутствии подходящего Pd катализатора, лиганда и основания, описанной выше, в абсолютном этаноле и получить этиловый эфир формулы VII, можно гидролизовать подходящим основанием-гидроксидом (например, KOH) в подходящей системе растворителей, такой как водный раствор метанола, и получить карбоновую кислоту формулы VIII. Карбоновую кислоту VIII можно ввести в реакцию с подходящим амином формулыVI при условиях сочетания с образованием амида, хорошо известных в данной области техники. Например, кислоту VIII можно обработать подходящим активирующим реагентом, таким как тионилхлорид,оксалилхлорид, (бензотриазол-1-илокси)трипирролидинфосфонийгексафторфосфат (РуВОР), О-(7 азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (ГАТУ), O-(бензотриазол-1-ил)N,N,N',N'-тетраметилуронийгексафторфосфатO-(бензотриазол-1-ил)-N,N,N',N'тетраметилуронийтетрафторборат (TBTU) в присутствии подходящего амина формулы VI, подходящего основания (например, триэтиламина или N,N-диизопропилэтиламина (ДИПЭА в подходящем растворителе (например, диметилформамиде или N-метилпирролидиноне) и получить искомое соединение формулы Ia. Схема III Альтернативно, карбоновую кислоту формулы VIII можно получить по последовательности стадий синтеза, приведенной на схеме III. Соединение формулы II можно ввести в реакцию с подходящим гидразином формулы III (свободное основание или подходящая солевая форма, такая как гидрохлорид) с использованием полярного апротонного растворителя (например, NMP, ДМФ (диметилформамид),ДМАА (диметилацетамид) или ДМПМ (N,N'-диметилпропиленмочевина в присутствии основания (например, водного раствора KOH, водного раствора NaOH, водного раствора LiOH, водного раствораCsOH, NaOMe, NaOEt, KOt-Bu или KOt-амил), при подходящей температуре (предпочтительно примерно при 80 С) с получением соединения формулы V. Бромазаиндол V можно ввести в реакцию с подходящим реагентом Гриньяра (например, R-MgCl, где R можно выбрать из группы, включающей изопропил,н-бутил, втор-бутил и циклогексил) и СО 2 в подходящем полярном апротонном растворителе, таком как ТГФ (тетрагидрофуран), МТБЭ (метил-трет-бутиловый эфир), Et2O, ДМЭ (1,2-диметоксиэтан) или диоксан, при подходящей температуре проведения реакции (предпочтительно примерно при -20 С) и получить карбоновую кислоту формулы VIII, которую можно превратить в соединения формулы Ia, описанные выше. Соединения формулы Ib можно получить, как показано на схемах IV и V. На этих схемах Ar1, Ar2,G, X, R1 и R2 обладают значениями, определенными в подробном описании соединений формулы I. Как показано на схеме IV, индазол-4-карбоновую кислоту формулы IX можно ввести в реакцию сочетания с подходящим амином формулы VI при условиях сочетания с образованием амидной связи, хорошо известных в данной области техники, таких как описанные выше. Полученный индазол-4 карбоксамид можно ввести в реакцию с подходящим арилгалогенидом формулы XI при условиях перекрестного сочетания, которые описаны в химической литературе, таких как нагревание при подходящей температуре (предпочтительно примерно при 120 С), в присутствии подходящих реагентов, таких как катализатор (например, CuI), основание (например, K2CO3) и лиганд (например, рацемический трансN,N'-диметилциклогексан-1,2-диамин), в подходящем растворителе (например, ДМФ) и получить соединения формулы Ib. Альтернативно, соединения формулы Ib можно синтезировать, как показано на схеме V. Схема V Индазол-4-карбоновую кислоту формулы IX можно превратить в эфир соответствующей карбоновой кислоты формулы XII при условиях этерификации, хорошо известных в данной области техники,таких как обработка триметилсилилдиазометаном в подходящей системе растворителей (например, метанол и толуол). Эфир XII можно ввести в реакцию с подходящим арилгалогенидом формулы XI при условиях перекрестного сочетания, описанных выше, и получить эфир индазол-4-карбоновой кислоты формулы XIII, который можно превратить в кислоту формулы XIV при стандартных условиях гидролиза,таких как обработка подходящим основанием (например, NaOH) в подходящей водной системе растворителей (например, воде и метаноле). Как показано выше, кислоту формулы XIV можно превратить к соединению формулы Ib по реакции с амином VI при условиях сочетания с образованием амида, известной в данной области техники. Соединения формулы I, полученные по указанным выше методикам, после этого можно превратить в дополнительные соединения формулы I по методикам, известным в данной области техники и приведенным в качестве примеров в разделе, посвященном примерам синтеза. Методики, описанные ниже (т.е. на схемах VI-IX) и в разделе, посвященном примерам синтеза,можно использовать для получения промежуточных продуктов VI, которые можно использовать для получения соединений формулы I. На приведенных ниже схемах циклический G, Ar2 и Rb обладают значениями, определенными в подробном описании соединений формулы I. Промежуточные продукты формулы VIa (т.е. промежуточный продукт формулы VI, в которой Ar2 обозначает 1,3,4-тиадиазол) можно получить в соответствии со схемой VI. Схема VI Как показано на схеме VI, аминокислоту формулы XV, в которой PG обозначает подходящую защитную группу (например, Cbz), можно ввести в реакцию сочетания с содержащим защитную группуBoc гидразином XVI при условиях сочетания с образованием амидной связи, хорошо известных в данной области техники, таких как описанные выше, и получить содержащий защитную группу Boc гидразидXVII. Соединение формулы XVII можно ввести в реакцию с реагентом Лавессона в присутствии подходящего растворителя, такого как толуол, и при подходящей температуре (например, примерно при 90 С) и получить соответствующий содержащий защитную группу Boc тиогидразид, из которого можно удалить защитную группу подходящей кислотой, такой как 4 н. HCl в диоксане, и получить соответствующую солевую форму (например, гидрохлорид) тиогидразида XVIII. Соединение формулы XVIII можно ввести в реакцию с ДМФ в присутствии подходящего реагента, такого как диэтилхлорфосфат XIX, и подходящего основания (например, Et3N) и получить соответствующий 1,3,4-тиадиазол, из которого можно удалить защитную группу атома N подходящим реагентом (например, 48% HBr в уксусной кислоте) и получить промежуточный продукт формулы VIa. Кроме того, промежуточные продукты формулы VIb (т.е. промежуточный продукт формулы VI, в которой Ar2 обозначает 1,2,4-оксадиазол) можно получить в соответствии со схемой VII. Схема VII Как показано на схеме VII, содержащую подходящую защитную группу аминокислоту формулыXV (т.е. PG обозначает подходящую защитную группу, такую как Cbz) можно превратить в соответствующий амид XX при стандартных условиях сочетания с образованием амидной связи, таких как опи- 20021015 санные выше, и в присутствии подходящей соли аммония, такой как карбонат аммония, подходящего основания (например, Et3N) и подходящего растворителя (например, ДМФ). Амид формулы XX можно ввести в реакцию с подходящим дегидратирующим реагентом, таким как хлорангидрид циануровой кислоты, в присутствии подходящего растворителя (например, ДМФ) и при подходящей температуре (например, при температуре, равной примерно от 0 до 30 С) и получить нитрил формулы XXI. СоединениеXXI можно ввести в реакцию с гидроксиламингидрохлоридом в присутствии подходящего основания,такого как карбонат калия, в подходящем растворителе (например, этаноле) и при подходящей температуре (например, примерно при 79 С) с получением соединения формулы XXII. Амидоксим XXII можно ввести в реакцию с подходящей карбоновой кислотой формулы XXIII с использованием реагента сочетания с образованием амидной связи, хорошо известных в данной области техники (например, КДИ (1,1'карбонилдиимидазол, в подходящем растворителе (например, ДМФ), и при подходящих условиях (например, нагревание примерно при 100 С) и получить соответствующее производного 1,2,4-оксадиазола,из которого можно удалить защитную группу атома N, как это описано выше, и получить промежуточный продукт формулы VIb. Промежуточные продукты формулы VIc (т.е. промежуточный продукт формулы VI, в которой Ar2 обозначает другой региоизомер 1,2,4-оксадиазола) можно получить в соответствии со схемой VIII. Схема VIII Амидоксим формулы XXIV можно получить путем добавления гидроксиламина к соответствующему нитрилу при подходящих условиях, описанных выше. Как показано на схеме VIII, амидоксимXXIV можно ввести в реакцию с аминокислотой формулы XV, в которой PG обозначает подходящую защитную группу (например, Boc), с использованием подходящего реагента сочетания с образованием амида, такого как КДИ, в подходящем растворителе, таком как ДМФ, и при подходящих условиях (например, нагревание примерно при 100 С) и получить соответствующий 1,2,4-оксадиазол, из которого можно удалить защитную группу атома N при подходящих условиях (например, по реакции с подходящей кислотой, такой как 4 н. HCl в диоксане) и получить промежуточный продукт формулы VIc. Промежуточные продукты формулы VId (т.е. промежуточный продукт формулы VI, в которой Ar2 обозначает пиримидин) можно получить в соответствии со схемой IX. Схема IX Как показано на схеме IX, содержащий подходящую защитную группу аминокарбонитрил формулыXXI, в которой PG обозначает подходящую защитную группу, такую как Boc, можно превратить в соответствующий амидингидрохлорид XXV по реакции с подходящими реагентами, такими как этоксид натрия в этаноле, с последующей обработкой хлоридом аммония и аммиаком. По другой реакции синтеза 3-диметиламинопропеналь XXVI можно галогенировать подходящим реагентом (например, Br2 и NIS (Nйодсукцинимид в подходящем растворителе, таком как CHCl3 и получить 2-галогензамещенный 3- 21021015 диметиламинопропеналь формулы XXVII (т.е. Rb обозначает Br или I). Затем, амидингидрохлорид XXV можно ввести в реакцию с соединением формулы XXVII в подходящем растворителе (например, EtOH) и при подходящей температуре (например, примерно при 80 С) и получить пиримидин формулы XXVIII. Из соединения XXVIII можно удалить защитную группу атома N при условиях, хорошо известных в данной области техники и описанных выше, и получить промежуточный продукт формулы VId. Содержащая подходящую защитную группу аминокислота формулы XV, которую можно использовать для синтеза промежуточных продуктов формулы VI, имеется в продаже, ее можно получить или в соответствии с приведенной ниже ссылкой (которая во всей своей полноте включена в настоящее изобретение в качестве ссылки) или специалист в данной области техники может ее получить по методикам,описанным в химической литературе. 3-трет-Бутоксикарбониламинооксетан-3-карбоновую кислоту можно синтезировать по методике,описанной в заявке на патент WO 2009/070485 А 1. Ниже приведены примеры не являющихся природными аминокислот, которые имеются в продаже. Эти примеры приведены для описания вариантов осуществления настоящего изобретения и их не следует рассматривать в качестве каким-либо образом ограничивающих объем настоящего изобретения. Введение подходящих защитных групп в аминокислоты можно провести при стандартных условиях, хорошо известных в данной области техники (исчерпывающий перечень приведен в публикации Greene, Т.W.;Wuts, P.G.М. Protective Groups in Organic Synthesis, 3rd Ed., Wiley, New York, 1999, которая во всей своей полноте включена в настоящее изобретение в качестве ссылки). Примеры синтеза Общие методики. Если не указано иное, то все реакции проводили при комнатной температуре. Все соединения характеризовали с помощью по меньшей мере одной из следующих методик: 1 Н ЯМР (ядерный магнитный резонанс), ВЭЖХ, ВЭЖХ-МС и определение температуры плавления. Приведены данные МС для найденных [М+Н]+. Для бромсодержащих соединений приведены[М+Н]+ для двух изотопов брома (т.е. 79Br и 81Br). Времена удерживания (ВУ), приведенные в табл. I, определяли с помощью одной из приведенных ниже методик: Синтез промежуточных продуктов. Синтезы приведенных ниже промежуточных гетероарилциклопропиламинов или их соответствующих солевых форм описаны в заявке на патент WO 2009/070485 А 1: Промежуточный продукт можно получить с использованием подходящих реагентов и по методике, описанной в ссылке для родственного аналога. Пример 1. Синтез 1-(4-фторфенил)-1H-пиразоло[3,4-c]пиридин-4-карбоновой кислоты (1) В колбу объемом 1 л помещают 3,5-дибромпиридин-4-карбоксальдегид (50,0 г, 189 ммоль, 1,0 экв.) и помещают 4-фторфенилгидразингидрохлорид (31,0 г, 191 ммоль, 1,01 экв.). NMP (250 мл) и полученную взвесь перемешивают при температуре окружающей среды в течение 2 ч. Водный раствор KOH получают из гранул, содержащих 85% KOH (27,4 г, 415 ммоль, 2,2 экв.), и воды (27,4 мл) и этот растворKOH добавляют к реакционной смеси. Смесь нагревают при 80 С и выдерживают при этой температуре в течение 30-60 мин. Затем при 80 С добавляют воду (250 мл) и полученную взвесь охлаждают до температуры окружающей среды в течение 4-16 ч. Взвесь фильтруют, твердое вещество промывают водой,сушат в вакуумном сушильном шкафу и получают 4-бром-1-(4-фторфенил)-1H-пиразоло[3,4-с]пиридин в виде твердого вещества. В колбу объемом 1 л помещают 4-бром-1-(4-фторфенил)-1H-пиразоло[3,4-с]пиридин (50,0 г, 171 ммоль, 1 экв.) и ТГФ (300 мл). Взвесь охлаждают до -20 С. Добавляют раствор i-PrMgCl (128,2 мл, 256,4 ммоль, 2,0 M в ТГФ, 1,5 экв.) со скоростью, обеспечивающей поддержание температуры, равной ниже-10 С. Реакционную смесь выдерживают при -10 С в течение 3 ч. Через реакционную смесь пропускают газообразный СО 2 до достижения пика температура и затем температура начинает снижаться. Температуру устанавливают равной 22 С и добавляют i-PrOAc (325 мл). Водный раствор HCl получают из концентрированной HCl (55 мл) и воды (195 мл). Примерно 10 мл этого раствора HCl добавляют к реакционной смеси до рН 6-7. Затем смесь нагревают до 55 С и добавляют оставшиеся 240 мл раствора HCl. Реакционную смесь охлаждают до температуры окружающей среды в течение 1 ч, выдерживают при этой температуре в течение 1 ч и фильтруют. Твердое вещество промывают водой и с помощью i-PrOAc в вакуумном сушильном шкафу и получают искомое соединение в виде твердого вещества. Пример 2. Синтез 1-(4-фторфенил)-1H-индазол-4-карбоновой кислоты (2) Индазол-4-карбоновую кислоту (2,00 г, 12,3 ммоль) суспендируют в метаноле (20 мл) и толуоле (30 мл) при комнатной температуре. Медленно добавляют 2 М раствор триметилсилилдиазометана (12 мл,24 ммоль) в толуоле и смесь перемешивают при комнатной температуре, пока раствор не становится желтым. Реакцию останавливают концентрированной уксусной кислотой (5 мл) и растворитель удаляют в вакууме. Остаток очищают с помощью хроматографии на силикагеле при элюировании в градиентном режиме с помощью 0-30% этилацетата в гексанах и получают метиловый эфир 1H-индазол-4-карбоновой кислоты. Смесь метилового эфира 1 Н-индазол-4-карбоновой кислоты (5,0 г, 28 ммоль), йодида меди (5,7 г,3,0 ммоль), карбоната калия (4,15 г, 30,0 ммоль) и 4-фторйодбензола (3,47 г, 30,0 ммоль) помещают в герметизированную пробирку при комнатной температуре. Пробирку откачивают, заполняют аргоном и добавляют диметилформамид (20 мл), затем рац-транс-N,N'-диметилциклогексан-1,2-диамин (0,93 г, 6,5 ммоль). Раствор перемешивают при 120 С в течение 3 ч, затем охлаждают до комнатной температуры и разбавляют водой (50 мл) и этилацетатом (80 мл). Органический слой отделяют, промывают рассолом(30 мл) и сушат над сульфатом натрия. Неочищенный продукт фильтруют, концентрируют и очищают с помощью хроматографии на силикагеле при элюировании в градиентном режиме с помощью 0-30% этилацетата в гексанах и получают метиловый эфир 1-(4-фторфенил)-1H-индазол-4-карбоновой кислоты. При перемешивании к раствору метилового эфира 1-(4-фторфенил)-1H-индазол-4-карбоновой кислоты (2,0 г, 7,4 ммоль) в воде (20 мл) и метанола (20 мл) добавляют 2 н. раствор гидроксида натрия (10 мл). Раствор нагревают при кипячении с обратным холодильником в течение 1 ч. Раствор охлаждают до комнатной температуры и подкисляют до рН 3-4 1 н. водным раствором HCl. Смесь фильтруют и полученное твердое вещество промывают с помощью МеОН (30 мл) и сушат и получают 1-(4-фторфенил)1H-индазол-4-карбоновую кислоту. Пример 3. Синтез 1-(6-бромпиридин-3-ил)циклопропиламин-бис-трифторацетата (3) В высушенную в вакуумном сушильном шкафу круглодонную колбу объемом 2 л, снабженную механической мешалкой, в атмосфере Ar помещают безводный ТГФ (750 мл), затем Ti(Oi-Pr)4 (72,8 мл, 246 ммоль). Раствор продувают с помощью Ar и нагревают до 50 С. К смеси добавляют 6 бромникотинонитрил (30,0 г, 164 ммоль), затем по каплям добавляют (в течение 40 мин) 1 М раствор этилмагнийбромид в ТГФ (410 мл, 410 ммоль). Реакционную смесь перемешивают при 50 С. Через 3 ч реакционную смесь охлаждают до комнатной температуры и добавляют 3 М водный раствор HCl (примерно 350 мл). Смесь переносят в делительную воронку и промывают этиловым эфиром (3500 мл). Водный слой выдерживают в течение ночи. Затем водный слой подщелачивают до рН 10 2 М водным раствором NaOH. Раствор разбавляют с помощью EtOAc (500 мл) и полученный раствор энергично перемешивают в течение 5 мин. Раствор выдерживают и слои медленно разделяются. Органический слой сливают и эту же процедуру экстракции повторяют дважды. Органические слои объединяют, промывают рассолом (50 мл), сушат над MgSO4, концентрируют в вакууме и получают масло. Неочищенное масло очищают с помощью хроматографии на силикагеле в градиентном режиме 0-10% МеОН в CH2Cl2 и получают 1-(6-бромпиридин-3-ил)циклопропиламин в виде масла, которое медленно кристаллизуется (ЭР+(электрораспыление) m/z 213,3, 215,3). 1-(6-Бромпиридин-3-ил)циклопропиламин (1,16 г, 4,60 ммоль) растворяют в CH2Cl2 (20 мл). Последовательно добавляют Et3N (0,78 мл, 5,6 ммоль) и Boc2O (1,11 г, 5,10 ммоль) и реакционную смесь перемешивают при комнатной температуре. Через 20 ч реакционную смесь разбавляют с помощью CH2Cl2(20 мл) и водой (20 мл) и слои разделяют. Водный слой экстрагируют с помощью CH2Cl2 (100 мл). Объединенные слои в CH2Cl2 промывают рассолом, сушат над MgSO4, фильтруют, концентрируют и получают трет-бутиловый эфир [1-(6-бромпиридин-3-ил)циклопропил]карбаминовой кислоты в виде твердого вещества. трет-Бутиловый эфир [1-(6-бромпиридин-3-ил)циклопропил]карбаминовой кислоты (0,800 г, 2,55 ммоль) растворяют в CH2Cl2 (10 мл). По каплям добавляют ТФК (трифторуксусная кислота) (5 мл). Через 4 ч реакционную смесь концентрируют в вакууме и получают искомое соединение в виде масла (ЭР+ m/z 213,1, 215,1). 3-Диметиламинопропеналь (50 мл, 500 ммоль) растворяют в CHCl3 (400 мл) при комнатной температуре. Шприцем в течение 5 мин добавляют неразбавленный бром (25,7 мл, 0,500 моль). Через 30 мин реакционную смесь выливают в смесь 200 мл насыщенного водного раствора Na2S2O3 и 200 мл насыщенного водного раствора NaHCO3 и смесь экстрагируют с помощью CH2Cl2 (3100 мл). Объединенные органические слои сушат над MgSO4, концентрируют и получают твердое вещество. Твердое вещество растворяют в EtOAc (200 мл), нерастворимые вещества отфильтровывают, фильтрат концентрируют в вакууме, полученное твердое вещество промывают 50% раствором EtOAc в гексанах и получают 3 диметиламино-2-бромпропеналь в виде твердого вещества (ЭР+ m/z 178,28). Гидрохлорид трет-бутилового эфира (1-карбамимидоилциклопропил)карбаминовой кислоты (1,0 г,4,2 ммоль) (получен по методике, описанной в заявке на патент WO 2009/070485 А 1) и 3-диметиламино 2-бромпропеналь (1,1 г, 6,4 ммоль) добавляют к EtOH (2 мл) в пробирке высокого давления. Сосуд для проведения реакции закрывают и смесь нагревают при 80 С в течение 24 ч. Смесь охлаждают до комнатной температуры и добавляют метанол (20 мл). Полученное твердое вещество отфильтровывают и фильтрат концентрируют в вакууме. Остаток растворяют в CH2Cl2 (50 мл) и твердые вещества отфильтровывают. Фильтрат концентрируют, остаток очищают с помощью хроматографии на силикагеле в градиентном режиме 0-50% EtOAc в гексанах и получают трет-бутиловый эфир [1-(5-бромпиримидин-2 ил)циклопропил]карбаминовой кислоты в виде твердого вещества. трет-Бутиловый эфир [1-(5-бромпиримидин-2-ил)циклопропил]карбаминовой кислоты (1,18 г, 3,76 ммоль) растворяют в CH2Cl2 (5 мл) при комнатной температуре. Добавляют 4 М раствор HCl в диоксане(9,4 мл, 38 ммоль). Через 2 ч растворители удаляют потоком N2 и получают неочищенное искомое соединение в виде твердого вещества, которое используют без очистки (ЭР+m/z 216,3). Пример 5. Синтез трет-бутилового эфира [1-(5-йод-фуран-2-ил)циклопропил]карбаминовой кислоты (5) К раствору трет-бутилового эфира (1-фуран-2-илциклопропил)карбаминовой кислоты (4,30 г, 19,3 ммоль) (получен по методике, описанной в заявке на патент WO 2009/070485 А 1) в безводном ДМФ (77 мл) при комнатной температуре одной порцией добавляют твердый N-йодсукцинимид (4,77 г, 21,2 ммоль). Через 2,5 ч реакционную смесь разбавляют насыщенным водным раствором Na2S2O3 (75 мл),водой (75 мл) и этиловым эфиром (100 мл). Фазы разделяют и водный слой экстрагируют этиловым эфиром (2100 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют. Полученное твердое вещество растирают с гексанами и получают искомое соединение в виде порошкообразного вещества (ЭР+ m/z 350,5). Пример 6. Синтез метилового эфира 3-(1-аминоциклопропил)бензойной кислоты (6) Ацетилхлорид (0,600 мл, 8,46 ммоль) при 0 С добавляют к метанолу (15 мл) и раствор нагревают до комнатной температуры. После перемешивания в течение 20 мин добавляют 3 аминоциклопропилбензойную кислоту (0,500 г, 2,82 ммоль) и реакционную смесь кипятят с обратным холодильником. Через 16 ч смесь концентрируют при 65 С потоком азота. Остаток нейтрализуют насыщенным водным раствором бикарбоната натрия (50 мл) и экстрагируют этилацетатом (350 мл). Объединенные органические слои промывают рассолом (100 мл), сушат над MgSO4, фильтруют, концентрируют и получают неочищенный искомый продукт, который используют без очистки. При перемешивании при 0 С к раствору (2-бромпиридин-4-ил)метанола (3,00 г, 16,0 ммоль) и N,Nдиизопропилэтиламина (8,3 мл, 48 ммоль) в дихлорметане (30 мл) добавляют метансульфонилхлорид(1,30 мл, 16,8 ммоль). Полученную смесь нагревают до комнатной температуры. Через 1 ч смесь разбавляют дихлорметаном (20 мл) и промывают насыщенным водным раствором хлорида аммония (310 мл),насыщенным водным раствором бикарбоната натрия (10 мл), рассолом (10 мл), сушат над MgSO4, фильтруют, концентрируют и получают неочищенный 2-бромпиридин-4-илметиловый эфир метансульфоновой кислоты, который используют без очистки. 2-Бромпиридин-4-илметиловый эфир метансульфоновой кислоты (4,24 г, 15,9 ммоль) при комнатной температуре при перемешивании добавляют к раствору цианида калия (1,02 г, 15,1 ммоль) в смеси этанола (30 мл) и воды (6 мл). Через 72 ч добавляют этилацетат (80 мл) и насыщенный водный раствор бикарбоната натрия (40 мл) и фазы разделяют. Органический слой промывают водой (340 мл), сушат над MgSO4, фильтруют и концентрируют. Полученный остаток очищают с помощью хроматографии на силикагеле при элюировании в градиентном режиме с помощью 0-60% этилацетата в гептане и получаютNaH (60% дисперсия в минеральном масле, 585 мг, 14,6 ммоль) в сухом ДМСО (10 мл), не допуская повышения температуры путем охлаждения в бане с водой, и полученную смесь перемешивают при комнатной температуре. Через 18 ч добавляют воду (10 мл) и этилацетат (10 мл), фазы разделяют и водный слой экстрагируют этилацетатом (310 мл). Объединенные органические слои промывают рассолом (30 мл) и сушат над MgSO4, фильтруют и концентрируют. Остаток очищают на SiO2 при элюировании в градиентном режиме с помощью 0-60% этилацетата в гептане и получают 1-(2-бромпиридин-4 ил)циклопропанкарбонитрил в виде твердого вещества (ЭР+ m/z 223,36; 225,39). К раствору 1-(2-бромпиридин-4-ил)циклопропанкарбонитрила (1,16 г, 5,20 ммоль) в толуоле (30 мл) при -78 С добавляют ДИБАГ (диизобутилалюминийгидрид) (10,4 мл, 1 М в толуоле). Смесь перемешивают в течение 1 ч при -78 С и нагревают до комнатной температуры. Через 1 ч добавляют этилацетат(30 мл), затем 1 M раствор H2SO4 (30 мл). Фазы разделяют и водный слой экстрагируют этилацетатом(350 мл). Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют и получают неочищенный 1-(2-бромпиридин-4-ил)циклопропанкарбальдегид (ЭР+ m/z 226,48; 228,47), который используют без очистки. Раствор хлорита натрия (368 мг, 3,26 ммоль) и дигидрофосфата натрия моногидрат (449 мг, 3,26 ммоль) в 5 мл воды по каплям добавляют к раствору неочищенного 1-(2-бромпиридин-4 ил)циклопропанкарбальдегида (566 мг, 2,50 ммоль) и 2-метил-2-бутена (1,73 мл, 16,3 ммоль) в третбутаноле (12 мл) и полученную реакционную смесь перемешивают при комнатной температуре. Через 18 ч смесь концентрируют в вакууме, подкисляют до рН 2 1 M раствором HCl, разбавляют рассолом (25 мл) и экстрагируют этилацетатом (350 мл). Объединенные органические слои сушат над Na2SO4, фильтруют, концентрируют и получают неочищенную 1-(2-бромпиридин-4-ил)циклопропанкарбоновую кислоту,которую используют без очистки. К раствору неочищенной 1-(2-бромпиридин-4-ил)циклопропанкарбоновой кислоты (0,350 г, 1,45 ммоль) в трет-бутаноле (7 мл) в сосуде высокого давления добавляют дифенилфосфорилазид (0,312 мл,1,45 ммоль) и триэтиламин (0,202 мл, 1,45 ммоль). Пробирку герметизируют и реакционную смесь перемешивают при 90 С. Через 4 ч сосуд высокого давления охлаждают в бане со льдом, давление сбрасы- 27021015 вают и сосуд открывают. Реакционную смесь концентрируют в вакууме. Полученный остаток растворяют в этилацетате (70 мл), промывают насыщенным водным раствором хлорида аммония (70 мл) и насыщенным водным раствором бикарбоната натрия (70 мл), сушат над MgSO4, фильтруют и концентрируют. Остаток очищают с помощью хроматографии на силикагеле при элюировании в градиентном режиме с помощью 0-50% этилацетата в гептане и получают трет-бутиловый эфир [1-(2-бромпиридин-4 ил)циклопропил]карбаминовой кислоты. При перемешивании при комнатной температуре к раствору трет-бутилового эфира [1-(2 бромпиридин-4-ил)циклопропил]карбаминовой кислоты (0,160 г, 0,511 ммоль) в дихлорметане (3 мл) добавляют трифторуксусную кислоту (1,0 мл, 13 ммоль). Через 18 ч реакционную смесь концентрируют в вакууме и получают неочищенное искомое соединение (ЭР+ m/z 213,49, 215,40) в виде масла, которое используют без очистки. Пример 8. Синтез 1-(2-бромпиридин-4-ил)циклобутиламинтрифторацетата (8)(1,50 г, 7,61 ммоль) по методике алкилирования, описанной в примере 7, с использованием 1,3 дибромпропана вместо 1,2-дибромэтана. 1-(2-Бромпиридин-4-ил)циклобутанкарбальдегид получают из 1-(2-бромпиридин-4 ил)циклобутанкарбонитрила (1,26 г, 5,20 ммоль) по методике с использованием ДИБАГ, описанной в примере 7. 1-(2-Бромпиридин-4-ил)циклобутанкарбоновую кислоту (ЭР+ m/z 256,40, 258,38) получают из 1-(2 бромпиридин-4-ил)циклобутанкарбальдегида (532 мг, 2,22 ммоль) по методике окисления, описанной в примере 7. трет-Бутиловый эфир [1-(2-бромпиридин-4-ил)циклобутил]карбаминовой кислоты (ЭР+ m/z 327,54,329,46) получают из 1-(2-бромпиридин-4-ил)циклобутанкарбоновой кислоты (0,100 г, 0,390 ммоль) по методике перегруппировки Курциуса, описанной в примере 7, при температуре проведения реакции,равной 100 С. Искомое соединение (ЭР+ m/z 227,30, 229,27) получают из трет-бутилового эфира [1-(2 бромпиридин-4-ил)циклобутил]карбаминовой кислоты (82,0 мг, 0,262 ммоль) по методике удаления защитной группы Boc, описанной в примере 7. Пример 9. Синтез 1-(5-метансульфонилфуран-2-ил)циклопропиламингидрохлорида (9) трет-Бутиловый эфир [1-(5-метансульфонилфуран-2-ил)циклопропил]карбаминовой кислоты получают из трет-бутилового эфира [1-(5-йод-фуран-2-ил)циклопропил]карбаминовой кислоты (0,500 г, 1,43 ммоль) по методике сочетания при катализе йодидом меди(I), описанной в примере 19; однако в качестве компонента сочетания используют метансульфинат натрия вместо 3-метокси-3-оксопропан-1-сульфината натрия. трет-Бутиловый эфир [1-(5-метансульфонилфуран-2-ил)циклопропил]карбаминовой кислоты (0,430 г, 1,42 ммоль) растворяют в 4 М растворе HCl в диоксане (5,0 мл, 20 ммоль) при комнатной температуре. После перемешивания в течение 16 ч смесь выпаривают в потоке азота. Полученное маслообразное твердое вещество суспендируют в этилацетат (5 мл), добавляют этиловый эфир (25 мл), смесь фильтруют и получают неочищенный искомый продукт в виде твердого вещества, которое используют без очистки. Синтез соединений формулы I. Пример 10. Синтез [1-(5-метансульфонилфуран-2-ил)циклопропил]амида 1-(4-фторфенил)-1 Нпиразоло[3,4-с]пиридин-4-карбоновой кислоты (10)(0,310 г, 1,21 ммоль), N,N-диизопропилэтиламина (0,630 мл, 3,62 ммоль) и 1-(5-метансульфонилфуран-2 ил)циклопропиламингидрохлорида (364 мг, 1,53 ммоль) в ДМФ (30 мл) добавляют (бензотриазол-1 илокси)трипирролидинфосфонийгексафторфосфат (РуВОР) (0,650 г, 1,25 ммоль). Через 18 ч смесь разбавляют насыщенным водным раствором хлорида аммония (100 мл) и экстрагируют этилацетатом (430 мл). Объединенные органические слои промывают рассолом (50 мл), сушат над сульфатом натрия, фильтруют и концентрируют. Остаток очищают с помощью хроматографии на силикагеле при элюировании в градиентном режиме с помощью 0-100% этилацетата в гексанах и получают искомое соединение в виде твердого вещества. Указанное ниже соединение получают по методике сочетания, описанной в примере 10; однако(223 мг, 0,507 ммоль) и реакционная смесь становится гомогенной. Через 18 ч смесь концентрируют в вакууме растворяют в этилацетате (50 мл) и промывают 1 н. раствором гидроксид натрия (350 мл). Органический слой промывают насыщенным водным раствором хлорида аммония (250 мл), насыщенным водным раствором бикарбоната натрия (50 мл), рассолом (50 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток дважды очищают с помощью хроматографии на силикагеле при элюировании в градиентном режиме с помощью 0-10% метанола в метиленхлориде и получают искомое соединение в виде твердого вещества. Указанные ниже соединения получают по методике сочетания, описанной в примере 11: К суспензии 1-(4-фторфенил)-1H-пиразоло[3,4-с]пиридин-4-карбоновой кислоты (90,0 мг, 0,350 ммоль) в ДМФ (2 мл) добавляют N,N-диизопропилэтиламин (243 мкл, 1,40 ммоль) и HBTU (159 мг, 0,420 ммоль). Через 20 мин добавляют раствор 1-1,3,4-тиадиазол-2-илциклопропиламина (51,0 мг, 0,361 ммоль) в ДМФ (1 мл) и смесь перемешивают при комнатной температуре в течение ночи. Реакционную
МПК / Метки
МПК: A61K 31/4439, C07D 471/04, C07D 401/12, A61K 31/437
Метки: пиразолопиридины, антагонистов, рецептора, качестве, индазолы
Код ссылки
<a href="https://eas.patents.su/30-21015-indazoly-i-pirazolopiridiny-v-kachestve-antagonistov-receptora-ccr1.html" rel="bookmark" title="База патентов Евразийского Союза">Индазолы и пиразолопиридины в качестве антагонистов рецептора ccr1</a>
Сульфонамиды гидроксидифенилмочевины в качестве антагонистов рецептора ил-8

Номер патента: 5210
Опубликовано: 30.12.2004
Авторы: Дзин Кви, Макклеланд Брент В., Палович Майкл Р., Виддаусон Кэтрин Л.
МПК: C07C 273/00, C07D 207/10, A61K 31/17...
Метки: сульфонамиды, антагонистов, гидроксидифенилмочевины, качестве, ил-8, рецептора
Формула / Реферат:
1. Соединение формулы (I) где Rb независимо представляет собой водород, OC1-5алкил, C1-5алкил, C3алкенил, бензил, C3-5циклоалкил, C3-5циклоалкил-C1-5алкил, пиридил, 5-6-членный гетероциклил или 5-6-членный гетероциклил-C1-4алкил, содержащие в кольце атом азота и/или атом кислорода, где все указанные группы могут быть необязательно замещены одной-тремя группами, независимо выбранными из галогена, C1-4алкила, амино, моно- или...
Производные 1-аминоалкилциклогексана в качестве антагонистов рецептора nmda

Номер патента: 5684
Опубликовано: 28.04.2005
Авторы: Каусс Валерьянс, Калвиньш Иварс, Йиргенсонс Айгарс, Парсонс Кристофер Грахам Рафаэль, Даныш Войцех, Гольд Маркус
МПК: A61P 25/08, C07D 295/033, A61K 31/40...
Метки: рецептора, качестве, 1-аминоалкилциклогексана, антагонистов, производные
Формула / Реферат:
1. Циклическое производное 1-аминоалкилциклогексана формулы где R* обозначает -(CH2)n-(CR6R7)m-NR8R9, где n+m =0, 1 или 2, где R1-R7 независимо выбраны из группы, состоящей из водорода и C1-6алкила, причем, по меньшей мере R1, R4 и R5 представляют C1-6 алкил, и где R8 и R9 вместе представляют низший алкилен -(CH2)x-, где x обозначает число от 2 до 5 включительно, и их энантиомеры, оптические изомеры, гидраты и фармацевтически приемлемые...
Производные циклопентена в качестве антагонистов рецептора мотилина

Номер патента: 3252
Опубликовано: 27.02.2003
Авторы: Чен Роберт Х., Ксианг Мин, Биверз Мэри Пэт, Мур Джон Б.Мл.
МПК: A61K 31/5375, C07C 233/41, A61P 1/00...
Метки: рецептора, циклопентена, качестве, мотилина, антагонистов, производные
Формула / Реферат:
1. Соединение формулы I в которой R1 представляет H, C1-5-алкил, замещенный C1-5-алкил (где заместителями являются один или несколько галогенов), амино-C1-5-алкил, C1-5-алкиламино-C1-5-алкил, ди-C1-5-алкиламино-C1-5-алкил, RaRbN-C1-5-алкил (где Ra и Rb независимо выбраны из H и C1-5-алкила или вместе образуют морфолин, пиперазин, пиперидин или N-замещенный пиперидин, где N-заместитель представляет C1-5-алкил или фенил-C1-5-алкил),...
Производные (тио) карбамоилциклогексана в качестве антагонистов d3/d2 рецептора

Номер патента: 9022
Опубликовано: 26.10.2007
Авторы: Галамбош Янош, Ваго Иштван, Агаине Чонгор Эва, Кишш Бела, Ласловски Иштван, Дьертьян Иштван, Ласи Юдит, Ногради Каталин, Саги Каталин
МПК: A61K 31/495, A61P 25/00, C07D 243/08...
Метки: карбамоилциклогексана, производные, тио, рецептора, антагонистов, качестве
Формула / Реферат:
1. Соединение формулы (I) где R1 и R2 независимо представляют собой заместитель, выбранный из водорода, C1-6алкила, С2-7алкенила, арила, циклоалкила, ароила, или R1 и R2 могут образовывать гетероциклическое кольцо со смежным атомом азота; X представляет собой атом кислорода или серы; n является целым числом от 1 до 2, и/или его геометрические изомеры, и/или стереоизомеры, и/или диастереомеры, и/или соли, и/или гидраты, и/или сольваты. 2....
N-(2-арилэтил)бензиламины в качестве антагонистов 5-ht6- рецептора

Номер патента: 7493
Опубликовано: 27.10.2006
Авторы: Маккоуэн Джефферсон Рэй, Кохен Майкл Филип, Джиллиг Джеймс Рональд, Чен Заоген, Шаус Джон Менерт, Жиетлен Брюно, Миллер Шон Кристофер, Фишер Мэттью Джозеф
МПК: A61K 31/4045, A61K 31/4406, A61K 31/4402...
Метки: качестве, антагонистов, 5-ht6, рецептора, n-(2-арилэтил)бензиламины
Формула / Реферат:
1. Соединение формулы Формула I где X выбран из группы, состоящей из -O-, -NH-, -S-, -SO2-, -СН2-, -CH(F)-, -СН(ОН)- и -С(O)-; R1 выбран из группы, состоящей из незамещенного фенила, фенила, замещенного группами в количестве от 1 до 3, независимо выбранными из группы, состоящей из водорода, гидрокси, C1-C4алкила, C1-С4алкокси, галогена, бензилокси, карбокси, С1-С4алкоксикарбонила, амидо, N-(С1-С4алкил)амидо, сульфониламидо, циано,...
Предыдущий патент: Способ получения сетчатых полиуретанизоциануратов с заданным модулем упругости
Следующий патент: Микрополосковый диплексер
Случайный патент: Подкладка подрельсовая