Антагонисты рецепторов простагландина d2
Номер патента: 19351
Опубликовано: 31.03.2014
Авторы: Сейдерс Томас Ён, Ванг Бовей, Стирнс Брайан Эндрю, Хатчинсон Джон Говард, Арруда Дженни М.







Формула / Реферат
1. Соединение формулы (I) или его фармацевтически приемлемая соль
где RA представляет собой Н или C1-C6-алкил;
R4 представляет собой Н, галоген, -CN, -OH, C1-C4-алкил, -CH=CH2, C1-C4-фторалкил, C1-C4-фторалкокси или C1-C4-алкокси;
R6 представляет собой -NR13S(=O)2R12, -S(=O)2N(R12)(R13), -N(R12)(R13), -C(=O)N(R12)(R13),
-NHC(=O)N(R12)(R13), -NRI3C(=O)R12 или -NR13C(=O)OR12;
R11 представляет собой C1-C6-алкил, C1-C6-галогеналкил, C3-C6-циклоалкил, замещенный или незамещенный фенил, замещенный или незамещенный нафтил, замещенный или незамещенный 5-звенный гетероарил, замещенный или незамещенный 6-звенный гетероарил или -C1-C4-алкил-(замещенный или незамещенный фенил);
R12 представляет собой C1-C6-алкил, C1-C6-фторалкил, C3-C6-циклоалкил, замещенный или незамещенный фенил, замещенный или незамещенный нафтил, замещенный или незамещенный 6-звенный гетероарил или -C1-C4-алкил-(замещенный или незамещенный фенил);
R13 представляет собой Н или C1-C6-алкил или
R12 и R13 вместе с атомом N, к которому они присоединены, образуют замещенный или незамещенный 5-6-членный гетероциклоалкил, содержащий по меньшей мере один атом N в кольце;
х равен 0, 1 или 2;
где замещенные группы замещены 1 или 2 группами, индивидуально и независимо выбранными из галогена, -OH, -CN, C1-C4-алкила, C1-C4-фторалкила, C1-C4-алкокси, C1-C4-фторалкокси, -NH2, -NH(C1-C4-алкила) и -N(C1-C4-алкил)2;
5-членный гетероарил представляет собой имидазолил, пиразолил, триазолил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, оксадиазолил, тиадиазолил или фуразанил;
6-членный гетероарил представляет собой пиридинил, пиридазинил, пиримидинил, пиразинил или тиазинил.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где
RA представляет собой Н, -CH3 или -CH2CH3;
R4 представляет собой Н, F, Cl, Br, -OH, C1-C4-алкил, C1-C4-фторалкил, C1-C4-фторалкокси или C1-C4-алкокси.
3. Соединение по п.1 или 2 или его фармацевтически приемлемая соль, где R11 представляет собой C1-C6-алкил, C1-C6-галогеналкил, C3-C6-циклоалкил, замещенный или незамещенный фенил или -C1-C4-алкил-(замещенный или незамещенный фенил).
4. Соединение по любому из пп.1-3 формулы (II) или его фармацевтически приемлемая соль
5. Соединение по любому из пп.1-4 или его фармацевтически приемлемая соль, где R4 представляет собой Н, F, Cl, Br, -OCH3, -CH3, -CH2CH3, -CH=CH2, -CHF2, -CF3, -OCHF2 или -OCF3.
6. Соединение по любому из пп.1-5 или его фармацевтически приемлемая соль, где
R12 представляет собой C1-C6-алкил, C1-C6-фторалкил, C3-C6-циклоалкил, замещенный или незамещенный фенил или -C1-C4-алкил-(замещенный или незамещенный фенил);
R13 представляет собой Н или -CH3.
7. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль, где R6 представляет собой
-NR13S(=O)2R12, -N(R12)(R13), -C(=O)N(R12)(R13), -NHC(=O)N(R12)(R13), -NR13C(=O)R12 или -NR13C(=O)OR12.
8. Соединение по любому из пп.1-7 или его фармацевтически приемлемая соль, где R11 представляет собой C1-C6-алкил, C1-C6-галогеналкил, замещенный или незамещенный фенил или -C1-C4-алкил-(замещенный или незамещенный фенил).
9. Соединение по любому из пп.1-8 или его фармацевтически приемлемая соль, где R4 представляет собой F, Cl, Br, -OCH3, -CH3, -CH2CH3, -CH=CH2, -CHF2, -CF3, -OCHF2 или -OCF3.
10. Соединение по любому из пп.1-9 или его фармацевтически приемлемая соль, где R6 представляет собой
-NR13C(=O)R12.
11. Соединение по любому из пп.1-10 или его фармацевтически приемлемая соль, где R12 представляет собой C1-C6-алкил, C3-C6-циклоалкил, замещенный или незамещенный фенил или замещенный или незамещенный бензил.
12. Соединение по любому из пп.1-11 или его фармацевтически приемлемая соль, где R11 представляет собой -CH2CH3, -CH(CH3)2, -C(CH3)3, -CH2CF3, замещенный или незамещенный фенил, -C1-C2-алкил-(замещенный или незамещенный фенил).
13. Соединение по любому из пп.1-12 или его фармацевтически приемлемая соль, где R12 представляет собой -CH(CH3)2, -С(CH3)3, -CH2CH(CH3)2, -CH2C(CH3)3, циклопропил, циклобутил, циклопентил, циклогексил, замещенный или незамещенный фенил или замещенный или незамещенный бензил.
14. Соединение по любому из пп.1-13 формулы (III) или его фармацевтически приемлемая соль
15. Соединение по п.14 или его фармацевтически приемлемая соль, где
R11 представляет собой -CH2CH3, -CH(CH3)2, -С(CH3)3 или -CH2CF3;
R12 представляет собой -CH(CH3)2, -С(CH3)3, -CH2CH(CH3)2, -CH2C(CH3)3 или замещенный или незамещенный фенил;
R13 представляет собой Н.
16. Соединение по п.15 или его фармацевтически приемлемая соль, где
R4 представляет собой F, Cl, -OCH3, -CF3 или -OCF3;
R11 представляет собой -С(CH3)3;
R12 представляет собой -С(CH3)3;
R13 представляет собой Н.
17. Соединение, выбранное из
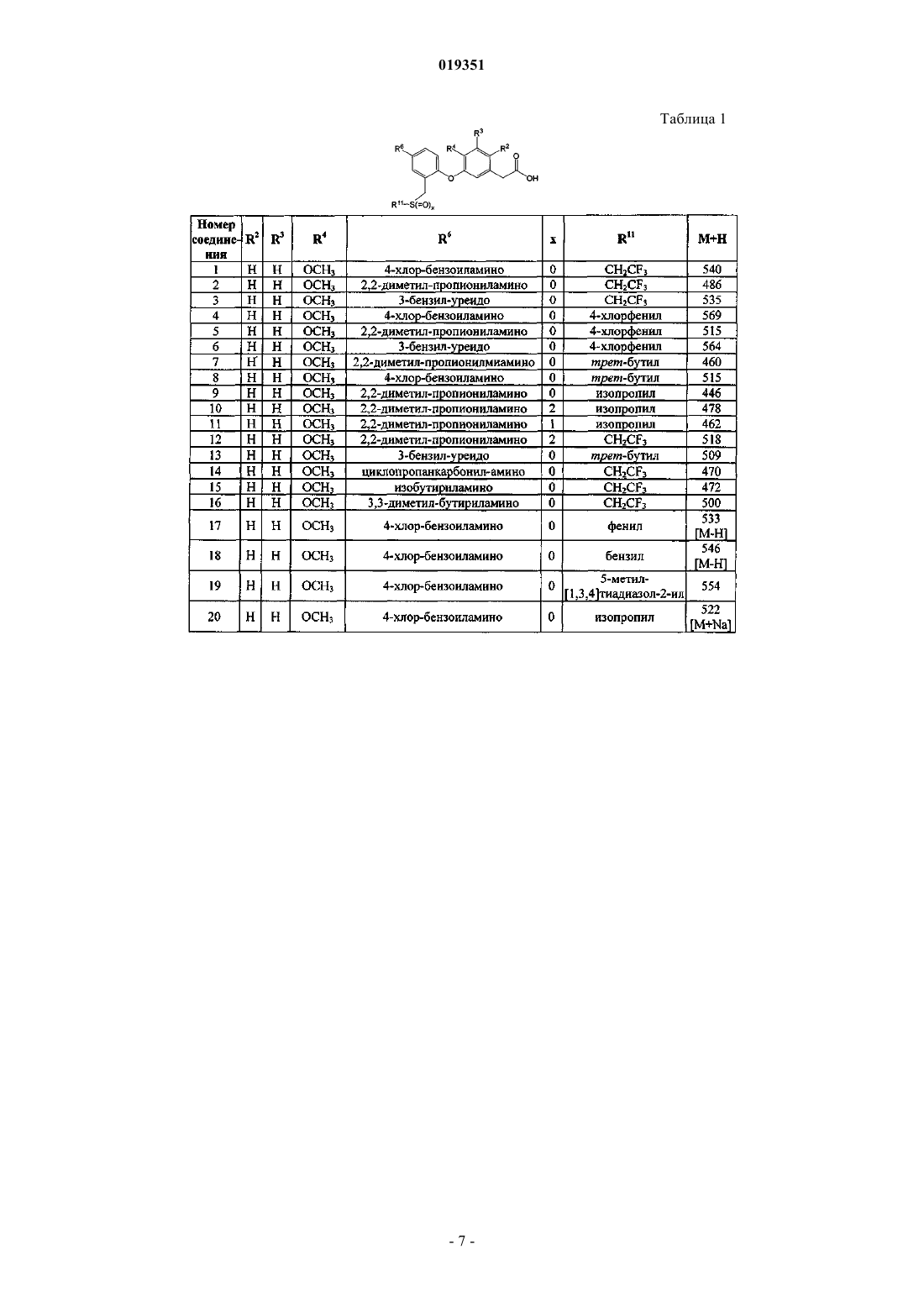
{3-[4-(4-хлорбензоиламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 1);
{3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 2);
{3-[4-(3-бензилуреидо)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 3);
{3-[4-(4-хлорбензоиламино)-2-(4-хлорфенилсульфанилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 4);
{3-[2-(4-хлорфенилсульфанилметил)-4-(2,2-диметилпропиониламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 5);
{3-[4-(3-бензилуреидо)-2-(4-хлорфенилсульфанилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 6);
{3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 7);
{3-[2-трет-бутилсульфанилметил-4-(4-хлорбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 8);
{3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]-4-метоксифенил}уксусной кислоты (соединение 9);
{3-[4-(2,2-диметилпропиониламино)-2-(пропан-2-сульфонилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 10);
{3-[4-(2,2-диметилпропиониламино)-2-(пропан-2-сульфинилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 11);
{3-[4-(2,2-диметилпропиониламино)-2-(трифторэтансульфонилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 12);
{3-[4-(3-бензилуреидо)-2-трет-бутилсульфанилметилфенокси]-4-метоксифенил}уксусной кислоты (соединение 13);
{3-[4-(циклопропанкарбониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]4-метоксифенил}уксусной кислоты (соединение 14);
{3-[4-изобутириламино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 15);
{3-[4-(3,3-диметилбутириламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 16);
{3-[4-(4-хлорбензоиламино)-2-фенилсульфанилметилфенокси]-4-метоксифенил}уксусной кислоты (соединение 17);
{3-[2-бензилсульфанилметил-4-(4-хлорбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 18);
{3-[4-(4-хлорбензоиламино)-2-(5-метил[1,3,4]тиадиазол-2-илсульфанилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 19);
{3-[4-(4-хлорбензоиламино)-2-изопропилсульфанилметилфенокси]-4-метоксифенил}уксусной кислоты (соединение 20);
{3-[4-(4-хлорбензоиламино)-2-(пропан-2-сульфинилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 21);
{3-[4-(4-хлорбензоиламино)-2-(пропан-2-сульфонилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 22);
{3-[2-бензенсульфинилметил-4-(4-хлорбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 23);
{3-[2-бензенсульфонилметил-4-(4-хлорбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 24);
[3-(4-этилкарбамоил-2-изопропилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты (соединение 25);
{3-[4-(4-хлорбензилкарбамоил)-2-изопропилсульфанилметилфенокси]-4-метоксифенил}уксусной кислоты (соединение 26);
(3-{4-[2-(4-фторфенил)этилкарбамоил]-2-изопропилсульфанилметилфенокси}-4-метоксифенил)уксусной кислоты (соединение 27);
{3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенил}уксусной кислоты (соединение 28);
{3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(трифторэтансульфонилметил)фенокси]фенил}уксусной кислоты (соединение 29);
{3-хлор-5-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]фенил}уксусной кислоты (соединение 30);
{3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(пропан-2-сульфонилметил)фенокси]фенил}уксусной кислоты (соединение 31);
{3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-5-трифторметилфенил}уксусной кислоты (соединение 32);
{3-[4-(2,2-диметилпропиониламино)-2-(трифторэтансульфонилметил)фенокси]-5-трифторметилфенил}уксусной кислоты (соединение 33);
{3-[4-[(2,2-диметилпропионил)метиламино]-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 34);
{3-[2-трет-бутилсульфанилметил-4-(циклопропанкарбониламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 35);
[3-(2-трет-бутилсульфанилметил-4-изобутириламинофенокси)-4-метоксифенил]уксусной кислоты (соединение 36);
{3-[2-трет-бутилсульфанилметил-4-(3,3-диметилбутириламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 37);
{4-метокси-3-[4-(2-оксооксазолидин-3-ил)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенил}уксусной кислоты (соединение 38);
[3-(4-трет-бутилкарбамоил-2-трет-бутилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты (соединение 39);
{3-[2-трет-бутилсульфанилметил-4-(2-оксо-2-фенилэтилкарбамоил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 40);
(3-{2-трет-бутилсульфанилметил-4-[2-(4-фторфенил)-1,1-диметилэтилкарбамоил]фенокси}-4-метоксифенил)уксусной кислоты (соединение 41);
{3-[2-трет-бутилсульфанилметил-4-(пиперидин-1-карбонил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 42);
{3-[2-трет-бутилсульфанилметил-4-(6-метоксипиридин-3-илкарбамоил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 43);
{3-[2-трет-бутилсульфанилметил-4-(2,2,2-трифторэтилкарбамоил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 44);
{3-[2-трет-бутилсульфанилметил-4-(изопропилметилкарбамоил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 45);
{3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропилкарбамоил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 46);
{3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-5-хлорфенил}уксусной кислоты (соединение 47);
{3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(2-метилпропан-2-сульфонилметил)фенокси]фенил}уксусной кислоты (соединение 48);
{4-дифторметокси-3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенил}уксусной кислоты (соединение 49);
{4-дифторметокси-3-[4-(2,2-диметилпропиониламино)-2-(трифторэтансульфонилметил)фенокси]фенил}уксусной кислоты (соединение 50);
{3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метилфенил}уксусной кислоты (соединение 51);
{4-хлор-3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]фенил}уксусной кислоты (соединение 52);
{4-хлор-3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенил}уксусной кислоты (соединение 53);
{3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]-5-трифторметилфенил}уксусной кислоты (соединение 54);
{3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-винилфенил}уксусной кислоты (соединение 55);
{3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-этилфенил}уксусной кислоты (соединение 56);
{4-метокси-3-[4-(2-оксоимидазолидин-1-ил)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенил}уксусной кислоты (соединение 57);
[3-(4-бензоиламино-2-трет-бутилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты (соединение 58);
{3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4-хлорфенил}уксусной кислоты (соединение 59);
{3-[2-трет-бутилсульфанилметил-4-(4-хлорбензоиламино)фенокси]-4-хлорфенил}уксусной кислоты (соединение 60);
[3-(2-трет-бутилсульфанилметил-4-изобутириламинофенокси)-4-хлорфенил]уксусной кислоты (соединение 61);
{3-[2-трет-бутилсульфанилметил-4-(3-фторбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 62);
{3-[2-трет-бутилсульфанилметил-4-(4-фторбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 63);
{3-[2-трет-бутилсульфанилметил-4-(2-фторбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 64);
{3-[2-трет-бутилсульфанилметил-4-(2,4-дихлорбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 65);
{3-[2-трет-бутилсульфанилметил-4-(3,5-дихлорбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 66);
{3-[2-трет-бутилсульфанилметил-4-(3,5-дифторбензоиламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 67);
{4-метокси-3-[2-(2,2,2-трифторэтилсульфанилметил)-4-(3-трифторметилбензоиламино)фенокси]фенил}уксусной кислоты (соединение 68);
{4-метокси-3-[2-(2,2,2-трифторэтилсульфанилметил)-4-(4-трифторметилбензоиламино)фенокси]фенил}уксусной кислоты (соединение 69);
{4-метокси-3-[4-[(пиридин-3-карбонил)амино]-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенил}уксусной кислоты (соединение 70);
{4-метокси-3-[4-[(пиридин-4-карбонил)амино]-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенил}уксусной кислоты (соединение 71);
{3-[2-трет-бутилсульфанилметил-4-(5-метиламинонафтален-1-сульфониламино)фенокси]-4-метоксифенил}уксусной кислоты (соединение 72);
{3-[4-(2,2-диметилпропиониламино)-2-(2-метилпропан-2-сульфинилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 73);
{3-[4-(2,2-диметилпропиониламино)-2-(2-метилпропан-2-сульфонилметил)фенокси]-4-метоксифенил}уксусной кислоты (соединение 74);
{3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4-гидроксифенил}уксусной кислоты (соединение 75) и
{3-[4-(2,2-диметилпропиониламино)-2-(2-метилпропан-2-сульфинилметил)фенокси]-4-гидроксифенил}уксусной кислоты (соединение 76)
или их фармацевтически приемлемых солей.
18. Соединение по любому из пп.1-17, где фармацевтически приемлемая соль представляет собой натриевую соль.
19. Соединение по п.1, представленное следующей структурой:
или его фармацевтически приемлемая соль.
20. Соединение по п.19, где фармацевтически приемлемая соль представляет собой натриевую соль.
21. Фармацевтическая композиция для применения в лечении заболевания или состояния, опосредованных простагландином D2, включающая терапевтически эффективное количество соединения по одному из пп.1-20 или фармацевтически приемлемую соль этого соединения, а также как минимум один фармацевтически неактивный ингредиент, выбранный из следующей группы: фармацевтически приемлемые растворители, фармацевтически приемлемые формообразующие вещества и фармацевтически приемлемые носители.
22. Фармацевтическая композиция по п.21, выполненная в виде лекарственной формы для внутривенного введения, перорального применения, ингаляции, интраназального применения, местного применения, субконъюнктивального применения или ушного применения.
23. Фармацевтическая композиция по п.21, выполненная в виде таблетки, пилюли, капсулы, жидкости, ингалянта, интраназального спрея, суппозитория, суспензии, геля, коллоида, дисперсии, суспензии, раствора, эмульсии, мази, лосьона, глазных капель или ушных капель.
24. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли в лечении заболевания или состояния, опосредованных простагландином D2.
25. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли в лечении заболевания или состояния, связанных с поражением дыхательной системы, аллергического заболевания или состояния или воспалительного заболевания или состояния у млекопитающего.
26. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли в лечении бронхиальной астмы у млекопитающего.
27. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли в лечении хронической обструктивной болезни легких (ХОБЛ) у млекопитающего.
28. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли в лечении аллергического ринита у млекопитающего.
29. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли в лечении атопического дерматита у млекопитающего.
30. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли в лечении аллергического конъюнктивита у млекопитающего.
31. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли в лечении эозинофильного эзофагита у млекопитающего.
Текст
АНТАГОНИСТЫ РЕЦЕПТОРОВ ПРОСТАГЛАНДИНА D2 В изобретении приведено описание соединений формулы (I), являющихся антагонистами рецепторов PGD2. Кроме того, в изобретении приведены фармацевтические композиции, которые включают описанные соединения, и методы использования антагонистов рецепторов PGD2 в виде монотерапии или комбинированной терапии для лечения состояний или заболеваний с поражением дыхательной, сердечно-сосудистой систем и других PGD2-зависимых и PGD2-опосредованных заболеваний и состояний. Хатчинсон Джон Говард, Сейдерс Томас н, Ванг Бовей, Арруда Дженни М., Стирнс Брайан Эндрю (US) Дементьев В.Н. (RU) Область применения изобретения Предметом настоящего изобретения являются соединения, связанные с простагландином D2, методы получения таких соединений, фармацевтические композиции, включающие такие соединения, а также применение таких соединений для лечения заболеваний или состояний. Предпосылки создания изобретения Простагландины обладают широким спектром активности и играют важную роль в механизмах боли и воспаления. Простагландин D2 (PGD2) выделяется тучными клетками, макрофагами и T-хелперами 2 в ответ на местное повреждение тканей, а также при аллергическом воспалении при таких заболеваниях, как астма, аллергический ринит и атопический дерматит. PGD2 связывается с различными рецепторами, а именно: с простаноидным рецептором тромбоксанового типа (ТР), рецептором PGD2 (DP илиDP2). Сущность изобретения В настоящем изобретении раскрыты соединения, фармацевтические композиции и способы их применения для (а) профилактики или лечения аллергического и неаллергического воспаления, (b) уменьшения неблагоприятных симптомов, связанных с воспалением, и/или (с) контроля иммунологических,пролиферативных расстройств. Этиология этих расстройств связана с PGD2 и может быть генетической,ятрогенной, иммунологической, инфекционной, онкологической, токсической, хирургической и/или травматической. В одном аспекте настоящего изобретения соединения, фармацевтические композиции,описанные далее, включают антагонисты рецепторов PGD2. В одном аспекте настоящего изобретения методы, соединения, фармацевтические композиции, описанные далее, включают антагонисты DP2. В одном аспекте настоящего изобретения указанные соединения описываются формулой (I), являются фармацевтически приемлемыми солями. Эти соединения являются антагонистами DP2, используются для лечения млекопитающих, страдающих от одного или нескольких состояний или заболеваний,зависимых от PGD2. Эти заболевания и состояния включают бронхиальную астму, ринит, аллергический конъюнктивит, атопический дерматит, хроническую обструктивную болезнь легких (ХОБЛ), легочную гипертензию, интерстициальный легочный фиброз, артрит, аллергию, псориаз, воспалительные заболевания кишечника, острый респираторный дистресс-синдром, инфаркт миокарда, аневризму, инсульт, рак,нарушение заживления раны, эндотоксиновый шок, боль, воспалительные состояния, эозинофильный эзофагит, эозинофильные желудочно-кишечные расстройства (ЭЖКР), идиопатический гиперэозинофильный синдром, отит, сужение дыхательных путей, слизистую секрецию, заложенность носа, увеличенную микрососудистую проницаемость и активизацию эозинофилов, крапивницу, синусит, ангионевротический отек, анафилаксию, хронический кашель и синдром Черджа-Стросса. В одном аспекте настоящего изобретения указанное соединение описывается формулой (I) или является фармацевтически приемлемой солью соединения формулы (I)R11 представляет собой C1-C6-алкил, C1-C6-галогеналкил, C3-C6-циклоалкил, замещенный или незамещенный фенил, замещенный или незамещенный нафтил, замещенный или незамещенный 5-звенный гетероарил, замещенный или незамещенный 6-звенный гетероарил или -C1-C4-алкил-(замещенный или незамещенный фенил);R12 представляет собой C1-C6-алкил, C1-C6-фторалкил, C3-C6-циклоалкил, замещенный или незамещенный фенил, замещенный или незамещенный нафтил, замещенный или незамещенный 6-звенный гетероарил или -C1-С 4-алкил-(замещенный или незамещенный фенил);R12 и R13 вместе с атомом N, к которому они присоединены, образуют замещенный или незамещенный 5-6-членный гетероциклоалкил, содержащий по меньшей мере один атом N в кольце; и х равно 0, 1 или 2; где замещенные группы замещены 1 или 2 группами, индивидуально и независимо выбранными из галогена, -OH, -CN, C1-C4-алкила, C1-C4-фторалкила, C1-C4-алкокси, C1-C4-фторалкокси, -NH2,-NH(C1-C4-алкила) и -N(C1-C4-алкил)2; 5-членный гетероарил представляет собой имидазолил, пиразо-1 019351 лил, триазолил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил,оксадиазолил, тиадиазолил или фуразанил и 6-членный гетероарил представляет собой пиридинил, пиридазинил, пиримидинил, пиразинил или тиазинил. Соединения, описываемые формулой (I), являются антагонистами DP2. В другом аспекте настоящего изобретения указанные соединения представлены в табл. 1 или являются фармацевтически приемлемыми солями этих соединений. В одном аспекте настоящего изобретения указанные фармацевтические композиции содержат терапевтически эффективное количество соединения, описываемого формулой (I). В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции включают как минимум один фармацевтически приемлемый неактивный ингредиент: формообразующее вещество, растворитель, носитель. В определенных вариантах осуществления настоящего изобретения указанные методы леченияPGD2-зависимых состояний или заболеваний у млекопитающих включают назначение млекопитающим терапевтически эффективного количества соединения, описываемого формулой (I). В другом аспекте настоящего изобретения соединения, описываемые формулой (I), используются для лечения или профилактики воспалительных заболеваний или состояний. Воспалительные состояния включают бронхиальную астму, ринит, хроническую обструктивную болезнь легких, легочную гипертензию, интерстициальный легочный фиброз, атеросклероз, аневризму аорты, инфаркт миокарда, инсульт. В отдельном аспекте настоящего изобретения применение для лечения бронхиальной астмы у млекопитающих включает назначение этому млекопитающему терапевтически эффективного количества описываемого соединения. В другом аспекте настоящего изобретения соединения, описываемые формулой (I), используются для лечения или профилактики иммунологических расстройств, включающих аллергию, избыточный или неадекватный ответ на эндогенный или экзогенный антиген, а также другую патологию. В определенных вариантах реализации настоящего изобретения иммунологическое расстройство, которое характеризуется нарушением иммунной регуляции, не сопровождается воспалением. В некоторых аспектах настоящего изобретения такие состояния или заболевания носят ятрогенный характер, и повышение уровня PGD2 или патологическая его локализация вызываются другими методами лечения, приемом лекарств или хирургическими процедурами. В других вариантах реализации настоящего изобретения PGD2-зависимые или PGD2-опосредованные состояния или заболевания вызываются хирургическими операциями. В другом аспекте настоящего изобретения описанные соединения применяются для лечения ринита у млекопитающих. В дальнейших вариантах данного аспекта описанные здесь соединения применяются для лечения аллергического (экзогенного) ринита, неаллергического (эндогенного) ринита, хронического ринита, аллерген-индуцированного ринита, аспиринового ринита, ринита у детей, ринита у взрослых,профессионального ринита, стероид-резистентного ринита, сезонного ринита, круглогодичного ринита,риносинусита и полипоза носа. В другом аспекте настоящего изобретения указанные соединения могут применяться для лечения хронической обструктивной болезни легких и включают как минимум однократное назначение млекопитающим эффективного количества соединения, описываемого формулой (I). В дальнейших вариантах данного аспекта настоящего изобретения хроническая обструктивная болезнь легких включает хронический бронхит и/или эмфизему, легочную гипертензию, интерстициальный легочный фиброз и/или воспаление дыхательных путей, муковисцидоз. Соединения могут также применяться для профилактики повышенной слизистой секреции и/или отека у млекопитающих при однократном назначении млекопитающим их эффективного количества. Соединения, описываемые формулой (I), могут назначаться млекопитающим при вовлечении в процесс эозинофилов, базофилов, дендритных клеток, нейтрофилов, моноцитов и/или T-хелперов-2. В другом аспекте настоящего изобретения соединения, описываемые формулой (I), применяются для лечения и профилактики воспаления глаз, конъюнктивита, ретинита, склерита, увеита, аллергического конъюнктивита, весеннего кератоконъюнктивита, папиллярного конъюнктивита и включают как минимум однократное назначение млекопитающим их эффективного количества. В другом аспекте настоящего изобретения лечение острых или хронических расстройств, характеризующихся вовлечением в процесс или активацией эозинофилов, может включать минимум однократное назначение млекопитающим эффективного количества соединения, описываемого формулой (I). Лечение воспалительных заболеваний кожи включает как минимум однократное назначение млекопитающим эффективного количества как минимум одного соединения, описываемого формулой (I). К таким воспалительным заболеваниям кожи относятся, например, псориаз, дерматит, атопический дерматит, контактный дерматит, экзема, крапивница, розацеа, буллезные расстройства, коллагенозы, болезнь Кавасаки, синдром Шегрена-Ларссона, нарушение заживления ран и образование рубцов. Таким образом можно достичь снижения выраженности псориатических поражений кожи, суставов и других тканей и органов. В дальнейших аспектах настоящего изобретения указанные соединения применяются для модулирования иммунного ответа на эндогенные или экзогенные антигены. В дальнейших аспектах изобретения указанные соединения применяются для лечения острых и хронических аллергических ответов на экзогенные вещества, которые поступают в организм как продукты питания (например, арахис) или как лекарственные препараты (например, пенициллин, нестероидные противовоспалительные препараты и другие лекарства). В другом аспекте настоящего изобретения предполагается использование соединения, описываемого формулой (I), для производства лекарственных средств для лечения воспалительных заболеваний или состояний у млекопитающих, при которых активность как минимум одного PGD2-связанного белка способствует развитию патологического процесса и/или симптомов заболевания или состояния. В одном варианте данного аспекта настоящего изобретения белок пути PGD2 - это DP2. В другом или дальнейшем варианте данного аспекта настоящего изобретения воспалительные заболевания или состояния - это заболевания дыхательной, сердечно-сосудистой систем или пролиферативные заболевания. Заболевания или состояния сердечно-сосудистой системы - это заболевания, поражающие сердце или кровеносные сосуды. К таким заболеваниям относятся аритмии (предсердные, желудочковые или смешанные); атеросклероз и его последствия; стенокардия; нарушения ритма сердца; ишемия миокарда; инфаркт миокарда; аневризма сердца или сосуда; васкулит; инсульт; облитерирующее заболевание периферических артерий нижних конечностей, органов или тканей; реперфузионное поражение после ишемии головного мозга, сердца или другого органа или ткани; эндотоксический, хирургический или травматический шок; артериальная гипертензия; заболевания клапанов сердца, сердечная недостаточность, аномальное артериальное давление; шок; вазоконстрикция (в том числе связанная с мигренью); сосудистая аномалия, воспаление, недостаточность, ограниченная определенным органом или тканью; а также другие заболевания. Во всех указанных выше аспектах настоящего изобретения имеются дальнейшие варианты осуществления изобретения, в которых: (а) эффективное количество соединения назначается млекопитающему систематически; (b) эффективное количество соединения назначается млекопитающему перорально; (с) эффективное количество соединения назначается млекопитающему внутривенно; (d) эффективное количество соединения назначается млекопитающему ингаляционно; (е) эффективное количество соединения назначается млекопитающему интраназально; (f) эффективное количество соединения назначается млекопитающему в виде инъекции; (g) эффективное количество соединения назначается млекопитающему местно (накожно); (h) эффективное количество соединения назначается млекопитающему субконъюнктивально и/или (i) эффективное количество соединения назначается млекопитающему ректально. Во всех указанных выше аспектах настоящего изобретения имеются дальнейшие варианты осуществления изобретения, предполагающие однократное применение эффективного количества соединения,которые включают дальнейшие варианты, при которых (i) соединение назначается однократно; (ii) соединение назначается млекопитающему несколько раз в течение одного дня; (iii) соединение назначается длительно и регулярно или (iv) длительно и постоянно. Во всех указанных выше аспектах настоящего изобретения имеются дальнейшие варианты осуществления изобретения, предполагающие многократное применение эффективного количества соединения, которые включают дальнейшие варианты, при которых (i) соединение назначается один раз в день;(ii) соединение назначается два раза в день; (iii) соединение назначается в виде циклов, которые включают ежедневное назначение в течение некоторого периода времени с последующим перерывом как минимум на один день; (iv) соединение назначается в виде циклов, которые включают ежедневное назначение в течение некоторого периода времени с последующим периодом длительностью как минимум один день, характеризующимся снижением дозы соединения. Во всех указанных выше аспектах настоящего изобретения, включающих лечение PGD2-зависимых заболеваний или состояний, имеются дальнейшие варианты осуществления изобретения, предполагающие назначение как минимум одного дополнительного фармацевтически неактивного агента в дополнение к соединению, структура которого описывается формулой (I). Другие характеристики, свойства и преимущества соединений, применения и композиций, описанных в этом изобретении, будут освещены в нижеследующем подробном описании. Однако следует понимать, что подробное описание и конкретные примеры указывают на отдельные варианты осуществления настоящего изобретения и приводятся исключительно с иллюстративной целью. Поэтому на основании информации, содержащейся в этом подробном описании, специалистам в данной области будут понятны возможные различные изменения и модификации в рамках настоящего описания изобретения. Краткое описание фигур Фиг. 1. Иллюстративные примеры описанных здесь соединений. Фиг. 2. Иллюстративные примеры описанных здесь соединений. Фиг. 3. Иллюстративные примеры описанных здесь соединений. Фиг. 4. Иллюстративные примеры описанных здесь соединений. Фиг. 5. Иллюстративные примеры описанных здесь соединений. Фиг. 6. Иллюстративные примеры описанных здесь соединений. Фиг. 7. Иллюстративные примеры описанных здесь соединений. Фиг. 8. Иллюстративные примеры описанных здесь соединений. Подробное описание изобретения Простагландин D2 (PGD2) - это кислый липид, получающийся в результате метаболизма арахидоновой кислоты с участием циклооксигеназ и синтаз PGD2. PGD2 выделяется тучными клетками, макрофагами и T-хелперами-2 в ответ на местное повреждение тканей, а также при аллергическом воспалении при таких заболеваниях, как бронхиальная астма, аллергический ринит и атопический дерматит. Экзогенный PGD2 при воздействии на бронхиальные дыхательные пути вызывает ответ, характерный для обострения бронхиальной астмы. Активация DP2 связана с хемотаксисом и активацией T-хелперов-2, эозинофилов и балофилов.PGD2 связывается с DP2 и опосредует многие его эффекты через Gi-зависимое повышение уровня внутриклеточного кальция и снижение количества цикло-АМФ. Образование ИЛ-4, ИЛ-5 и ИЛ-13 вT-хелперах-2 также стимулируется путем активации DP2. Эти цитокины обладают различными биологическими эффектами, например участвуют в образовании иммуноглобулина Е, ответе дыхательных путей,секреции слизи, вовлечении в процесс эозинофилов. В головном мозге и во всей центральной нервной системе также происходит образование PGD2; считается, что он участвует в восприятии боли и регуляции сна. В других тканях PGD2 образуется, главным образом, в активизированных иммуноглобулином Е тучных клетках и, в меньшей степени, в макрофагах, дендритных клетках, Т-хелерах-2 (Th-2) и других лейкоцитах. В клетке PGD2 быстро метаболизируется и превращается в нижестоящие эффекторы, к которым относятся 12PGJ2, 911PGF2,13,14-дигидро-15-кето-PGD2 и 15-дезокси-12,14PGD2.PGD2 образуется в тучных клетках в больших количествах в ответ на стимуляцию аллергеном. Исследования на доклинических видах показали, что при введении PGD2 в препараты in vivo или при его избыточной продукции, достигнутой методами генной инженерии, наблюдается следующее: вазодилатация, ведущая к эритеме (покраснению) и отеку (образованию волдырей), вовлечение в процесс эозинофилов и T-хелперов-2, модуляция образования цитокинов Th-2, бронхоконстрикция. Внутрикожная инъекция PGD2 у человека, как было показано, приводит к образованию эритемы,сохраняющейся в течение длительного времени, потенцирует эффекты других препаратов в отношении уплотнения и лейкоцитарной инфильтрации кожи человека и способствует образованию отека кожи у крыс. Вероятнее всего, что эти эффекты PGD2, как и других вазодилатирующих простагландинов, обусловлены повышением кровотока в очагах воспалительного поражения и, следовательно, опосредуются,главным образом, рецептором DP1. И хотя эти наблюдения указывают на то, что DP1 опосредует сосудистые эффекты PGD2, способность PGD2 приводить к клеточным изменениям, связанным с воспалением,опосредуется воздействием на другой рецептор. Многие грани провоспалительной активности PGD2 связаны со взаимодействием с DP2. DP2 - это рецептор, связанный с G-белком. Обычно он в больших количествах экспрессируется на Th-2, эозинофилах и базофилах. Активация DP2 приводит к прямой активации и вовлечению в процесс T-хелперов-2 и эозинофилов. Активированные T-хелперы-2 образуют и выделяют воспалительные цитокины, к которым относятся ИЛ-4, ИЛ-5 и ИЛ-13. Несмотря на то что PGD2 связывается с DP1 и DP2 с одинаковой аффинностью, структурно DP2 не связан с DP1 и передает сигнал по другому механизму. Эффекты DP2 опосредованы Gi-зависимым повышением уровня внутриклеточного кальция и снижением внутриклеточного уровня циклического АМФ. Активация DP2 важна для вовлечения в процесс эозинофилов в ответ на аллергическую стимуляцию в таких тканях, как слизистая оболочка носа, бронхов и в коже. Введение PGD2 или селективных агонистов DP2 обостряет и усиливает аллергический ответ в легких и коже. АктивацияDP2, по-видимому, играет ключевую роль в реализации аллергического ответа. Использование антагонистов активации рецептора DP2 простагландином D2 является одним из подходов к лечению воспалительного компонента воспалительных заболеваний и состояний, заболеваний или состояний дыхательной системы, таких как бронхиальная астма, ринит, дерматит, а также других заболеваний.R11 представляет собой C1-C6-алкил, C1-C6-галогеналкил, C3-C6-циклоалкил, замещенный или незамещенный фенил, замещенный или незамещенный нафтил, замещенный или незамещенный 5-звенный гетероарил, замещенный или незамещенный 6-звенный гетероарил или -C1-C4-алкил-(замещенный или незамещенный фенил);R12 представляет собой C1-C6-алкил, C1-C6-фторалкил, C3-C6-циклоалкил, замещенный или незамещенный фенил, замещенный или незамещенный нафтил, замещенный или незамещенный 6-звенный гетероарил или -C1-C4-алкил-(замещенный или незамещенный фенил);R12 и R13 прикреплены к одному атому N и вместе с этим атомом образуют замещенный или незамещенный 5-6-членный гетероциклоалкил, содержащий по меньшей мере один атом N в кольце; х равно 0, 1 или 2; где замещенные группы замещены 1 или 2 группами, индивидуально и независимо выбранными из галогена, -OH, -CN, C1-C4-алкила, C1-C4-фторалкила, C1-C4-алкокси, С 1-С 4-фторалкокси, -NH2,-NH(C1-C4-алкила) и -N(C1-C4-алкил)2; 5-членный гетероарил представляет собой имидазолил, пиразолил, триазолил, тетразолил, фурил,тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, оксадиазолил, тиадиазолил или фуразанил и 6-членный гетероарил представляет собой пиридинил, пиридазинил, пиримидинил, пиразинил или тиазинил. Для всех вариантов осуществления настоящего изобретения заместители могут выбираться из множества указанных альтернатив. Например, в некоторых вариантах осуществления изобретения RA - это Н, -CH3 или -CH2CH3 и R4 представляет собой Н, F, Cl, Br, -OH, C1-C4-алкил, C1-C4-фторалкил, С 1-С 4-фторалкокси илиC1-C4-алкокси. В некоторых вариантах осуществления изобретения R11 представляет собой C1-C6-алкил,C1-C6-галогеналкил, C3-C6-циклоалкил, замещенный или незамещенный фенил или -C1-C4-алкил(замещенный или незамещенный фенил). В некоторых вариантах осуществления изобретения соединения, описываемые формулой (I), характеризуются структурой формулы (II) В некоторых вариантах осуществления изобретения R4 представляет собой H, F, Cl, Br, -OCH3,-CH3, -CH2CH3, -CH=CH2, -CHF2, -CF3, -OCHF2 или -OCF3. В некоторых вариантах осуществления изобретения R12 представляет собой C1-C6-алкил,C1-C6-фторалкил, C3-C6-циклоалкил, замещенный или незамещенный фенил или -C1-C4-алкил(замещенный или незамещенный фенил) и R13 представляет собой Н или -CH3. В некоторых вариантах осуществления изобретения R6 представляет собой -NR13S(=O)2R12,12-N(R )(R13), -C(=O)N(R12)(R13), -NHC(=O)N(R12)(R13), -NR13C(=O)R12 или -NR13C(=O)OR12. В некоторых вариантах осуществления изобретения R11 представляет собой C1-C6-алкил, C1-C6 галогеналкил, замещенный или незамещенный фенил или -C1-C4-алкил-(замещенный или незамещенный фенил).-5 019351 В некоторых вариантах осуществления изобретения R4 представляет собой F, Cl, Br, -OCH3, -CH3,-CH2CH3, -CH=CH2, -CHF2, -CF3, -OCHF2 или -OCF3. В некоторых вариантах осуществления изобретения R6 представляет собой -NR13C(=O)R12. В некоторых вариантах осуществления изобретения R12 представляет собой C1-C6-алкил,C3-C6-циклоалкил, замещенный или незамещенный фенил или замещенный или незамещенный бензил. В некоторых вариантах осуществления изобретения R11 представляет собой -CH2CH3, -CH(CH3)2,-С(CH3)3, -CH2CF3, замещенный или незамещенный фенил, -C1-C2-алкил-(замещенный или незамещенный фенил). В некоторых вариантах осуществления изобретения R12 представляет собой -CH(CH3)2, -С(CH3)3,-CH2CH(CH3)2, -CH2C(CH3)3, циклопропил, циклобутил, циклопентил, циклогексил, замещенный или незамещенный фенил или замещенный или незамещенный бензил. В некоторых вариантах осуществления изобретения соединения, описываемые формулами (I) и (II),характеризуются структурой формулы (III) В некоторых вариантах осуществления изобретенияR12 представляет собой -CH(CH3)2, -С(CH3)3, -CH2CH(CH3)2, -CH2C(CH3)3 или замещенный или незамещенный фенил иR13 представляет собой Н. В некоторых вариантах осуществления изобретенияR13 представляет собой Н. В одном аспекте настоящего изобретения R4 - это заместитель, описанный в табл.1. В одном аспекте изобретения R5 - это заместитель, описанный в табл.1. В одном аспекте изобретения R20 - это заместитель, описанный в табл.1. В одном аспекте изобретения R11 - это заместитель, описанный в табл.1. В рамках настоящего изобретения возможны любые комбинации групп, описанных выше. Согласно спецификации группы и их заместители выбираются специалистом в данной области, способным синтезировать стабильные заместители и соединения. В одном аспекте настоящего изобретения соединения, описываемые формулой (I), включают соединения, перечисленные в табл.1, а также другие вещества. Дополнительные формы соединений. В определенных вариантах осуществления изобретения соединения, описываемые формулой (I),имеют вид фармацевтически приемлемых солей. Эти соли получаются в результате реакции этого соединения в форме свободного основания с фармацевтически приемлемой неорганической или органической кислотой, например с такой неорганической кислотой, как хлористо-водородная (соляная), бромистоводородная, серная, азотная, фосфорная, метафосфорная, а также другие подобные кислоты; или с такой органической кислотой, как уксусная, пропионовая, капроновая, циклопентанпропионовая, гликолевая,пировиноградная,молочная,малоновая,янтарная,яблочная,малеиновая,фумаровая,-8 019351 р-толуенсульфоновая, винная, трифторуксусная, лимонная, бензоевая, 3-(4-гидроксибензоил)бензоевая,коричная, миндальная, арилсульфоновая, метансульфоновая, этансульфоновая, 1,2-этандисульфоновая,2-гидроксиэтансульфоновая, бензенсульфоновая, 2-нафталенсульфоновая, 4-метилбицикло-[2.2.2]окт-2 ен-1-карбоновая, глюкогептоновая, 4,4'-метилен-бис-(3-гидрокси-2-ен-1-карбоновая), 3-фенилпропионовая, триметилуксусная, третичная бутилуксусная, лаурилсульфоновая, глюконовая, глутаминовая,гидроксинафтойная, салициловая, стеариновая и муконовая кислоты. Фармацевтически приемлемые соли также получаются в результате реакции соединения, описываемого формулой (I), с основанием, в результате чего получаются такие соли, как соль аммония, соль щелочного металла, например соль натрия или калия, соль щелочно-земельного металла, например соль кальция или магния, соль органического основания, такого как дициклогексиламин,N-метил-D-глюкамин, трет-(гидроксиметил)метиламин, а также соли аминокислот, таких как аргинин,лизин и другие подобные аминокислоты. В других вариантах осуществления настоящего изобретения соединения, описываемые формулой(I), имеют вид фармацевтически приемлемых солей, которые получаются в результате реакции описываемого соединения в форме свободной кислоты с фармацевтически приемлемым органическим или неорганическим основанием, например с таким органическим основанием, как этаноламин, диэтаноламин,триэтаноламин, трометамин, N-метилглюкамин и другие подобные основания, или с таким неорганическим основанием, как гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия, гидроксид натрия и другие подобные основания. Фармацевтически приемлемые соли включают также комплексы с растворителями и их кристаллические формы, в частности сольваты или полиморфы. Сольваты содержат стехиометрические или нестехиометрические количества растворителя и могут, например, получаться в результате процесса кристаллизации с фармацевтически приемлемым растворителем, таким как вода, этанол и другие подобные растворители. Если растворителем является вода, то полученные сольваты будут называться гидратами; если растворителем является спирт, то полученные сольваты будут называться алкоголятами. Сольваты соединений, описываемых формулой (I), легко получаются в соответствии с описанной в этом документе методикой. В качестве примера можно указать, что гидраты соединений, описываемых формулой (I),легко получаются в результате рекристаллизации из смеси водного и органического растворителя; при этом используются такие органические растворители, как диоксан, тетрагидрофуран, этанол или метанол. Кроме того, указанные соединения могут существовать в растворенной или нерастворенной форме. В целом, для описываемых в настоящем документе соединений и методов растворенные формы считаются эквивалентами нерастворенных форм. В другом варианте осуществления настоящего изобретения соединения, описываемые формулой (I),характеризуются наличием одного или нескольких стереоцентров. Эти центры независимо друг от друга могут существовать в R- и S-конфигурации. Представленные в настоящем изобретении соединения включают все диастереомерные, энантиомерные и эпимерные формы, а также их соответствующие смеси. Стереоизомеры получаются по таким методикам, как разделение стереоизомеров на хиральных хроматографических колонках или стереоселективный синтез. Методы и лекарственные формы, описываемые в настоящем изобретении, включают использованиеN-оксидов (если это уместно), кристаллических форм (также известных как полиморфы) или фармацевтически приемлемых солей соединений, описываемых формулой (I), a также активных метаболитов этих соединений, характеризующихся таким же типом активности. В некоторых случаях соединения существуют в виде таутомеров. В некоторых вариантах осуществления настоящего изобретения описанные соединения существуют в виде таутомеров. Все таутомеры, как предполагается, соответствуют описанным в настоящем изобретении молекулярным формулам. Соединения, описанные в настоящем документе, могут включать изотопные метки. Эти соединения соответствуют представленным различным формулам и характеризуются такой же структурой, за исключением того, что некоторые атомы замещены атомами с другой атомной массой или другим массовым числом, которые отличаются от атомной массы и массового числа атомов, встречающихся в природе. При этом один или несколько атомов водорода замещены дейтерием. В некоторых вариантах осуществления изобретения метаболические области описанных соединений дейтерированы. Замещение дейтерием предоставляет определенные терапевтические преимущества вследствие большей метаболической активности, например вследствие повышенного периода полувыведения in vivo или снижения необходимой дозы препарата. Определения Если не указано иное, приведенные ниже термины, применяемые в настоящем описании, в том числе в спецификации и пунктах изобретения, имеют указанные значения. Необходимо отметить, что понятия, используемые в спецификации и прилагаемых пунктах изобретения в единственном числе, подразумевают множественную форму, если контекст явно не указывает на иное. В настоящем описании союзы"или" и "и" означают "и/или", если не указано иное. Кроме того, использование термина "включая", а также других его форм, таких как "включать", "включают", "включает", "включали", не является исчерпывающим."Алкокси" означает (алкил)О-, где "алкил" соответствует приведенному ниже определению."Алкил" означает алифатическую углеводородную группу. Алкил может быть насыщенным или ненасыщенным. В одном аспекте настоящего изобретения алкильные группы - это группы из следующего перечня: метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил и неопентил."Циклоалкил" означает моноциклический алифатический неароматический радикал, в котором все атомы, формирующие кольцо (т.е. скелетные атомы), являются атомами углерода. Циклоалкильные группы включают, например, циклопропан, циклобутил, циклопентил и циклогексил."Фторалкил" означает алкил, в котором один или несколько атомов водорода замещены атомом фтора. В одном аспекте настоящего изобретения фторалкил - это заместитель из следующей группы: CF3, -CHF2, -CH2F, -CH2CF3 и -CF2CF3."Фторалкокси" означает (фторалкил)О-, где "фторалкил" соответствует приведенному выше определению."Гетероалкил" означает алкильную группу, в которой один или несколько скелетных атомов алкила- это атомы, отличающиеся от углерода, например кислород, азот, сера, фосфор или их комбинации. В одном аспекте настоящего изобретения "гетероалкил" означает алкильную группу, в которой один из скелетных атомов алкила - это кислород, азот или сера. В другом аспекте настоящего изобретения "гетероалкил" означает алкильную группу, в которой один из скелетных атомов алкила - это кислород."Гетероциклоалкил" означает циклоалкильную группу, которая включает как минимум один гетероатом из следующей группы: азот, кислород, сера. Гетероциклоалкил представлен 5-звенным кольцом или 6-звенным кольцом. В некоторых вариантах осуществления изобретения гетероцилкоалкил включает как минимум один атом азота в кольце. Гетероциклоалкилы включают оксазолидинонил, пирролидинил,тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, тетрагидротиопиранил, пиперидинил, морфолинил, тиоморфолинил и пиперазинил."5-звенный гетероарил" включает имидазолил, пиразолил, триазолил, тетразолил, фурил, тиенил,изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, оксадиазолил, тиадиазолил и фуразанил. В одном аспекте настоящего изобретения гетероарил содержит 0-3 атома азота."6-звенный гетероарил" включает пиридинил, пиридазинил, пиримидинил, пиразинил и триазинил. Термин "необязательно замещенный" или "замещенный" означает, что указанная группа может быть замещена одной или несколькими дополнительными группами, индивидуально и независимо выбираемыми из следующего перечня: галоген, -OH, -CN, C1-C4-алкил, C1-C4-фторалкил, C1-C4-алкокси,C1-C4-фторалкокси, -NH2, -NH(C1-C4-алкил), -N(C1-C4-алкил)2 и C1-C4-гетероалкил. В некоторых случаях указанная замещенная группа замещается одной или двумя указанными выше группами. Например, в некоторых вариантах осуществления изобретения указанная замещенная группа замещается как минимум одной группой из следующего перечня: галоген, -OH, -CN, -CH3, -CH2CH3, -CF3, -OCH3, -OCH2CH3 и"Метаболит" соединения, описанного в настоящем документе, - это производное этого соединения,которое образуется при метаболизме данного соединения. Термин "активный метаболит" означает биологически активное производное соединения, которое образуется при метаболизме данного соединения. Термин "PGD2-зависимый" относится к состояниям или расстройствам, которые не развились бы вообще или не развились бы в такой степени в отсутствие PGD2. Термин "PGD2-опосредованный" относится к состояниям или расстройствам, которые могли развиться в отсутствие PGD2, но могут развиваться в присутствии PGD2. Термины "эффективное количество" или "терапевтически эффективное количество", использующиеся в настоящем документе, означают достаточное количество агента или соединения, которое при назначении облегчит в некоторой степени один или несколько симптомов целевого заболевания или состояния. Соответствующее эффективное количество в каждом индивидуальном случае может быть определено с помощью специальных методов, таких как исследование с эскалацией доз. Термины "лечить" или "лечение", которые используются в настоящем документе, включают облегчение или купирование симптомов заболевания или состояния; профилактику дополнительных симптомов; облегчение или профилактику основных метаболических причин симптомов; подавление заболевания или состояния, например остановку прогрессирования заболевания или состояния; облегчение состояния, вызванного заболеванием или состоянием; а также остановку развития симптомов заболевания или состояния (профилактически или терапевтически). Термины "субъект" или "пациент" охватывают млекопитающих и не млекопитающих. В одном аспекте настоящего изобретения "субъект" или "пациент" - это млекопитающее. В одном варианте осуществления изобретения млекопитающее - это человек. Фармацевтическая композиция и лекарственная форма. К соответствующим способам назначения относятся пероральный, внутривенный, ректальный,аэрозольный, парентеральный, глазной (субконъюнктивальный), легочный, чресслизистый, чрескожный,вагинальный, ушной, назальный, внутримышечный, подкожный и местный, а также другие способы назначения. Кроме того, к примеру, парентеральное введение включает внутримышечные, подкожные,внутривенные и внутрикостные инъекции, а также интратекальные, прямые внутрижелудочковые, внутрибрюшинные, внутрилимфатические и внутриносовые инъекции. В определенных вариантах осуществления настоящего изобретения описанное соединение назначается не системно, а местно. В других вариантах осуществления настоящего изобретения описанное соединение назначается в лекарственной форме с быстрым высвобождением, в лекарственной форме с замедленным высвобождением или в лекарственной форме с промежуточным высвобождением. В других вариантах осуществления настоящего изобретения описанные соединения назначаются местно. В некоторых вариантах осуществления настоящего изобретения описанные соединения входят в состав фармацевтических композиций. В определенных вариантах осуществления настоящего изобретения фармацевтические композиции облечены в традиционную форму с помощью одного или нескольких физиологически приемлемых носителей, к которым относятся формообразующие и вспомогательные вещества, облегчающие включение активных соединений в фармацевтические препараты, готовые к использованию. Соответствующие лекарственные формы зависят от избранного способа назначения препаратов. Все фармацевтически приемлемые методики, носители и формообразующие вещества применяются для получения лекарственных форм в соответствии с приведенной в нижеуказанных источниках информацией: Remington: The Science and Practice of Pharmacy, Nineteenth Ed. (Easton, Pa.: Mack Publishing Company, 1995); Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton,Pennsylvania, 1975; Liberman, H.A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker,New York, N.Y., 1980 и Pharmaceutical Dosage Forms and Drug Delivery Systems, Seventh Ed. (LippincottWilliamsWilkins, 1999). Фармацевтические композиции - это смеси соединения, описываемого формулой (I), с другими химическими соединениями, такими как носители, стабилизаторы, растворители, диспергирующие вещества, суспендирующие вещества, загустители и/или формообразующие вещества. В определенных вариантах осуществления настоящего изобретения фармацевтическая композиция облегчает назначение описываемого соединения млекопитающему. В некоторых вариантах осуществления настоящего изобретения описанные соединения входят в состав фармацевтических композиций для перорального применения. Соединения, описанные в настоящем документе, входят в состав лекарственных форм для перорального приема. Эти формы включают,например, таблетки, порошки, пилюли, драже, капсулы, жидкости, гели, сиропы, эликсиры, взвеси, суспензии и другие подобные лекарственные формы. В одном варианте реализации настоящего изобретения соединения, описываемые формулой (I),входят в состав лекарственной формы в виде водного раствора. В отдельных вариантах осуществления настоящего изобретения водный раствор - это, например, раствор из следующей группы: раствор физиологически совместимого буфера, такого как раствор Хэнка, раствор Рингера или физиологический солевой буфер. В других вариантах реализации настоящего изобретения соединения, описываемые формулой (I),входят в состав формы для чресслизистого применения. В отдельных вариантах реализации настоящего изобретения лекарственные формы для чресслизистого применения включают проникающие вещества,которые способны проходить через слизистый барьер. В других вариантах осуществления настоящего изобретения, в которых описываемые соединения входят в состав лекарственных форм для парентеральных инъекций, соответствующие лекарственные формы включают водные и неводные растворы. В определенных вариантах реализации настоящего изобретения фармацевтические препараты для перорального применения получаются путем смешивания одного или нескольких твердых формообразующих веществ с одним или несколькими описанными соединениями с последующим возможным измельчением получившейся смеси и дальнейшим использованием смеси гранул (после добавления соответствующих вспомогательных веществ) для получения таблеток или пилюль. К соответствующим формообразующим веществам, в частности, относятся наполнители, такие как сахара, в том числе лактоза,сахароза, маннитол или сорбитол; препараты целлюлозы, такие как, например, кукурузный крахмал,пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, микрокристаллическая целлюлоза, гидроксипропилметилцеллюлоза, натрия карбоксиметилцеллюлоза; а также другие вещества, такие как поливинилпирролидон (ПВП, или повидон) или кальция фосфат. В отдельных вариантах осуществления настоящего изобретения возможно также добавление разрыхляющих веществ. К разрыхляющим веществам относятся, например, поперечно-связанная кроскармеллоза натрия, поливинилпирролидон, агар, альгиновая кислота или ее соль, такая как альгинат натрия. Лекарственные формы для перорального применения также включают плотно укупоренные капсулы из желатина и мягкие герметичные капсулы из желатина и пластификатора, такого как глицерин или сорбитол. В отдельных вариантах реализации настоящего изобретения плотно укупоренные капсулы содержат активные ингредиенты с добавлением одного или нескольких дополнительных веществ. К наполнителям относятся, например, лактоза, связующие вещества, такие как крахмалы и/или смазки, такие как тальк или стеарат магния и, как вариант, стабилизаторы. В других вариантах реализации настоящего изобретения мягкие капсулы содержат одно или несколько активных соединений, которые растворяются или суспендируются в соответствующей жидкости. К соответствующей жидкости относятся, например,одно или несколько жидких масел, жидкий парафин и жидкий полиэтиленгликоль. В дополнение к этому возможно также добавление стабилизаторов. В других вариантах осуществления настоящего изобретения соединения, описываемые формулой(I), назначаются местно. Композиции, которые можно применять местно, включают растворы, суспензии,лосьоны, гели, пасты, медикаментозные пластыри, бальзамы, кремы или мази. В других вариантах реализации настоящего изобретения соединения, описываемые формулой (I),входят в состав форм для ингаляции. К формам, которые можно применять ингаляционно, относятся аэрозоли, туманы или порошки, а также другие формы. Активный ингредиент в фармацевтической композиции может находиться в форме свободной кислоты, свободного основания или в форме фармацевтически приемлемой соли. В дополнение к этому методы и описанные в этом изобретении фармацевтические композиции включают использованиеN-оксидов, кристаллических форм (также называемых полиморфами), а также активных метаболитов этих соединений, обладающих таким же типом активности. Представленные в этом документе соединения включают также все таутомеры описанных в настоящем изобретении соединений. Кроме того, описанные в этом изобретении соединения охватывают нерастворенные и растворенные в фармацевтически приемлемых растворителях формы. К таким растворителям относятся вода, этанол и другие подобные растворители. Следует считать, что в этом документе также представлены сольватированные формы представленных в этом изобретении соединений. В дополнение к этому фармацевтические композиции также могут включать другие лекарственные или фармацевтические агенты, носители, адъюванты, такие как консерванты; стабилизаторы; увлажняющие и эмульгирующие агенты; вещества, способствующие растворению; соли для регулирования осмотического давления; буферы и/или другие терапевтически полезные вещества. В определенных вариантах реализации настоящего изобретения композиции, содержащие описанные в этом изобретении вещества, назначаются с профилактическими и/или лечебными целями. Описываемые композиции по терапевтическим показаниям могут назначаться пациентам, которые страдают каким-либо заболеванием или состоянием, в количестве, достаточном для лечения или, по меньшей мере,частичной ликвидации заболевания или состояния. Эффективное количество, необходимое для этого,зависит от тяжести и характера течения заболевания или состояния, предшествовавшей терапии, общего состояния здоровья пациента, массы тела и ответа на лечение, а также от мнения лечащего врача. Терапевтически эффективное количество может определяться с помощью различных методов, например клинического исследования с эскалацией доз. Композиции, включающие описанное в этом изобретении соединение, по профилактическим показаниям могут назначаться пациенту, у которого подозревается определенное заболевание, повышен риск развития определенного заболевания, расстройства или состояния. В этом случае точное количество соединения также зависит от общего состояния здоровья пациента, массы тела и т.д. Соответствующее количество назначаемого агента зависит от различных факторов: назначаемого соединения, характера и тяжести заболевания или состояния, особенностей (например, массы тела) млекопитающего, которому показано лечение. Кроме того, доза агента также зависит от других обстоятельств, к которым относятся, например, вид назначаемого агента; способ назначения; состояние, которое планируется лечить; а также вид млекопитающего, которому назначается лечение. Дозы, которые используются для лечения взрослого человека, обычно находятся в диапазоне от 0,02 до 5000 мг в сутки,от 0,5 до 1500 мг в сутки или от 1 до 500 мг в сутки. В одном варианте осуществления изобретения такая доза вводится один раз в сутки или назначается в виде нескольких доз через соответствующие интервалы времени, например по два, три, четыре или большее число раз в сутки. В одном варианте реализации этого изобретения суточные дозы соединения, описываемого формулой (I), составляют примерно от 0,01 до 10 мг/кг массы тела. В определенных вариантах реализации настоящего изобретения соответствующие лекарственные формы для перорального приема содержат примерно от 1 до 500 мг активного ингредиента. В других вариантах реализации настоящего изобретения суточные дозы или количества активного ингредиента в лекарственной форме выше или ниже указанных в этом документе пределов. В некоторых случаях возможно назначение как минимум одного соединения, описываемого формулой (I), в комбинации с другими лекарственными средствами. При комбинированной терапии различные лекарственные средства (одно из которых является описываемым в этом документе соединением) назначаются в любом порядке или одновременно. При одновременном введении различные лекарственные средства могут содержаться, например, в одной лекарственной форме или в различных лекарственных формах (например, в одной таблетке или в двух отдельных таблетках). В некоторых вариантах реализации настоящего изобретения соединения, описываемые формулой(I), назначаются в течение длительного времени. В некоторых вариантах реализации настоящего изобретения соединения, описываемые формулой (I), назначаются периодически (например, устанавливаются"выходные", в течение которых описываемое соединение не назначается или назначается в уменьшенном количестве). В некоторых вариантах реализации настоящего изобретения соединения, описываемые формулой (I), назначаются в виде циклов: (а) первый период, включающий ежедневное назначение соединения, описываемого формулой (I); затем (b) второй период, во время которого суточное количество соединения, описываемого формулой (I), уменьшается. В некоторых вариантах реализации настоящего изобретения соединение, описываемое формулой (I), во время второго периода не назначается. В некоторых вариантах реализации настоящего изобретения длительность первого и второго периодов, а также дозы соединений определяются с помощью методов, хорошо известным специалистам в этой области. Примеры Приведенные ниже примеры представлены в иллюстративных целях и не ограничивают рамок пунктов настоящего изобретения. Пример 1. Общая процедура окисления для получения сульфонов. К сульфиду (0,06 ммоль) в CH2Cl2 (2 мл) добавлялась 3-хлорпербензоевая кислота (0,027 г,0,12 ммоль) и реакционная смесь перемешивалась при комнатной температуре в течение 20 мин. Полученная смесь концентрировалась и очищалась с помощью препаративной ВЭЖХ; в результате получался сульфон. Пример 2. Общая процедура окисления для получения сульфоксидов. К сульфиду (0,06 ммоль) в CH2Cl2 (2 мл) добавлялась 3-хлорпербензоевая кислота (0,014 г,0,06 ммоль) и реакционная смесь перемешивалась при комнатной температуре в течение 20 мин. Полученная смесь концентрировалась и очищалась с помощью препаративной ВЭЖХ; в результате получался сульфоксид. Пример 3. Синтез 3-[4-(4-хлорбензоиламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты (соединение 1). Этап 1. Этиловый эфир [3-(2-формил-4-нитрофенокси)-4-метоксифенил]уксусной кислоты. К 3-гидрокси-4-метоксифенилуксусной кислоте (5,0 г, 27,4 ммоль) в EtOH (100 мл) добавлялась серная кислота (1 мл), и полученная смесь перемешивалась в течение ночи при комнатной температуре. Отсутствие исходного вещества устанавливалось с помощью тонкослойной хроматографии. После этого раствор концентрировался и высушивался под сильным разрежением; в результате получался этиловый эфир (3-гидрокси-4-метоксифенил)уксусной кислоты. Раствор этилового эфира (3-гидрокси-4 метоксифенил)уксусной кислоты (1 экв.), 2-фтор-5-нитробензальдегида (1 экв.) и карбоната калия(2 экв.) в 1,4-диоксане нагревался при 70 С в течение ночи. Смесь разделялась между EtOAc и H2O и закислялась 1 н. водным раствором HCl до pH 1, а затем экстрагировалась EtOAc. Комбинированные органические слои высушивались над MgSO4, отфильтровывались и концентрировались; получившийся остаток очищался с помощью хроматографии на силикагеле (градиентное элюирование EtOAc в гексане); в результате получалось титульное соединение. Этап 2. Этиловый эфир [3-(2-гидроксиметил-4-нитрофенокси)-4-метоксифенил]уксусной кислоты. К этиловому эфиру [3-(2-формил-4-нитрофенокси)-4-метоксифенил]уксусной кислоты (1 экв.) в метаноле добавлялся боргидрид натрия (1,2 экв.), и реакционная смесь перемешивалась при комнатной температуре в течение 15 мин. Затем смесь концентрировалась и разделялась между EtOAc и Н 2 О. Водный слой разделялся и экстрагировался EtOAc; комбинированные органические слои высушивались надMgSO4, отфильтровывались и концентрировались; в результате получалось титульное соединение. Этап 3. Этиловый эфир [3-(2-бромметил-4-нитрофенокси)-4-метоксифенил]уксусной кислоты. К раствору этилового эфира [3-(2-гидроксиметил-4-нитрофенокси)-4-метоксифенил]уксусной кислоты (15,14 г, 41,9 ммоль) в ДМЭ добавлялся трибромид фосфора (5,92 мл, 62,8 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение ночи. После охлаждения до 0 С смесь гасилась насыщенным водным раствором NaHCO3 и экстрагировалась EtOAc. Комбинированные органические слои высушивались над MgSO4, отфильтровывались и концентрировались; в результате получалось титульное соединение. Этап 4. Этиловый эфир 4-метокси-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. К раствору этилового эфира [3-(2-бромметил-4-нитрофенокси)-4-метоксифенил]уксусной кислоты лялся гидрид натрия (60% дисперсия в минеральном масле; 0,207 г, 5,18 ммоль), и реакционная смесь перемешивалась при 0 С в течение 30 мин. Смесь разделялась между EtOAc и Н 2 О. Водный слой отделялся и закислялся, а затем экстрагировался EtOAc. Комбинированные органические слои высушивались над MgSO4, отфильтровывались и концентрировались; получившийся остаток очищался с помощью хроматографии на силикагеле; в результате получалось титульное соединение. Этап 5. Этиловый эфир 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты. Раствор этилового эфира 4 м-етокси-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (1,46 г, 3,18 ммоль), хлорида железа (0,026 г, 0,16 ммоль),1,1-диметилгидразина (1,69 мл, 22,27 ммоль) и DARCO (0,300 г) в EtOH (30 мл) перемешивался в течение ночи при 65 С. Смесь разделялась между EtOAc и Н 2 О и трижды экстрагировалась EtOAc. Комбинированные органические слои промывались водой и солевым раствором, а затем высушивались надMgSO4, отфильтровывались и концентрировались. Остаток очищался с помощью хроматографии на силикагеле (градиентное элюирование EtOAc в гексанах); в результате получалось титульное соединение. Этап 6. Этиловый эфир 3-[4-(4-хлорбензоиламино)-2-(2,2,2-трифтор-этилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты. К раствору этилового эфира 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты (0,200 г, 0,47 ммоль) и триэтиламина (0,08 мл, 0,56 ммоль) в CH2Cl2 добавлялся 4-хлорбензоилхлорид (0,07 мл, 0,56 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение 1 ч. Смесь концентрировалась и использовалась в таком виде на этапе гидролиза. Этап 7. Этиловый эфир 3-[4-(4-хлорбензоиламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты. К раствору этилового эфира 3-[4-(4-хлорбензоиламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты (0,47 ммоль) в MeOH и H2O добавлялся 1 н. водный раствор лития гидроксида. Реакционная смесь перемешивалась в течение ночи при 65 С, а затем закислялась и экстрагировалась EtOAc. Комбинированные органические слои высушивались над MgSO4, отфильтровывались и концентрировались; получившийся остаток очищался с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. После процедур, описанных в примере 3, этиловый эфир 3-[4-амино-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты реагировал со следующими соединениями: с пивалоилхлоридом с получением этилового эфира 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался с получением 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты (соединение 2); с бензилизоцианатом с получением этилового эфира 3-[4-(3-бензилуреидо)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[4-(3-бензилуреидо)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты (соединение 3); с циклопропанкарбонилхлоридом с получением этилового эфира 3-[4(циклопропанкарбониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты,который гидролизовался до 3-[4-(циклопропанкарбониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты (соединение 14); с изобутирилхлоридом с получением этилового эфира 3-[4-изобутириламино-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[4-изобутириламино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты (соединение 15); с трет-бутилацетилхлоридом с получением этилового эфира 3-[4-(3,3-диметилбутириламино)-2(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[4-(3,3-диметилбутириламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты (соединение 16); с 3-(трифторметил)бензоилхлоридом с получением этилового эфира 4-метокси-3-[2-(2,2,2 трифторэтилсульфанилметил)-4-(3-трифторметилбензоиламино)фенокси]фенилуксусной кислоты, который гидролизовался до 4-метокси-3-[2-(2,2,2-трифторэтилсульфанилметил)-4-(3-трифторметилбензоиламино)фенокси]фенилуксусной кислоты (соединение 68); с 4-(трифторметил)бензоилхлоридом с получением этилового эфира 4-метокси-3-[2-(2,2,2 трифторэтилсульфанилметил)-4-(4-трифторметилбензоиламино)фенокси]фенилуксусной кислоты, который гидролизовался до 4-метокси-3-[2-(2,2,2-трифторэтилсульфанилметил)-4-(4-трифторметилбензоиламино)фенокси]фенилуксусной кислоты (соединение 69); с никотиноилхлорида гидрохлоридом с получением этилового эфира 4-метокси-3-[4-[(пиридин-3 карбонил)амино]-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты, который гидролизовался до 4-метокси-3-[4-[(пиридин-3-карбонил)амино]-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (соединение 70); с изоникотиноилхлорида гидрохлоридом с получением этилового эфира 4-метокси-3-[4-[(пиридин 4-карбонил)амино]-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты, который гидролизовался до 4-метокси-3-[4-[(пиридин-4-карбонил)амино]-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (соединение 71). Пример 4. Синтез 3-[4-(4-хлорбензоиламино)-2-(4-хлорфенилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты (соединение 4). После процедуры, описанной для этапа 4, примера 3, из этилового эфира [3-(2-бромметил-4 нитрофенокси)-4-метоксифенил]уксусной кислоты и 4-хлорбензентиола получался 3-[2-(4-хлорфенилсульфанилметил)-4-нитрофенокси]-4-метоксифенилуксусной кислоты. Затем этиловый эфир 3-[2-(4-хлорфенилсульфанилметил)-4-нитрофенокси]-4-метоксифенилуксусной кислоты восстанавливался до этилового эфира 3-[4-амино-2-(4-хлорфенилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты, как это описано для этапа 5, примера 3. Обработка амина 4-хлорбензоилом в соответствии с процедурой, описанной для этапа 6, примера 3, приводила к получению этилового эфира 3-[4-(4-хлорбензоиламино)-2-(4-хлорфенилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 7, примера 3, приводил к получению кислоты. После процедур,описанных в примере 3,этиловый эфир 3-[4-амино-2-(4 хлорфенилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты реагировал со следующими соединениями: с пивалоилхлоридом с получением этилового эфира 3-[2-(4-хлорфенилсульфанилметил)-4-(2,2 диметилпропиониламино)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[2-(4-хлорфенилсульфанилметил)-4-(2,2-диметилпропиониламино)фенокси]-4 метоксифенилуксусной кислоты (соединение 5); с бензилизоцианатом с получением этилового эфира 3-[4-(3-бензилуреидо)-2-(4 хлорфенилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[4-(3-бензилуреидо)-2-(4-хлорфенилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты[3-(2-трет-бутилсульфанилметил-4-нитрофенокси)-4 метоксифенил]уксусной кислоты. Получался в соответствии с процедурой, описанной для этапа 4, примера 3, с использованием следующих исходных веществ: этилового эфира[3-(4-амино-2-трет-бутилсульфанилметилфенокси)-4 метоксифенил]уксусной кислоты. Получался в соответствии с процедурой, описанной для этапа 5, примера 3, с использованием следующего исходного вещества: этилового эфира [3-(2-трет-бутилсульфанилметил-4-нитрофенокси)-4 метоксифенил]уксусной кислоты. Этап 3. Этиловый эфир 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]4-метоксифенилуксусной кислоты Получался в соответствии с процедурой, описанной для этапа 6, примера 3, с использованием следующих исходных веществ: этилового эфира [3-(4-амино-2-трет-бутилсульфанилметилфенокси)-4 метоксифенил]уксусной кислоты и пивалоилхлорида. Этап 4. 3-[2-трет-Бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4 метоксифенилуксусная кислота. Получалась в соответствии с процедурой, описанной для этапа 7, примера 3, с использованием следующего исходного вещества: этилового эфира 3-[2-трет-бутилсульфанилметил-4-(2,2 диметилпропиониламино)фенокси]-4-метоксифенилуксусной кислоты. После процедур,описанных в примере 3,этиловый эфир[3-(4-амино-2-третбутилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты реагировал со следующими соединениями: с 4-хлорбензоилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4-(4 хлорбензоиламино)фенокси]-4-метоксифенилуксусной кислоты,который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(4-хлорбензоиламино)фенокси]-4-метоксифенилуксусной кислоты(соединение 8); с бензилизоцианатом с получением этилового эфира 3-[4-(3-бензилуреидо)-2-третбутилсульфанилметилфенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[4-(3-бензилуреидо)-2-трет-бутилсульфанилметилфенокси]-4-метоксифенилуксусной кислоты (соединение 13); с циклопропанкарбонилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил 4-(циклопропанкарбониламино)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(циклопропанкарбониламино)фенокси]-4-метоксифенилуксусной кислоты (соединение 35); с изобутирилхлоридом с получением этилового эфира [3-(2-трет-бутилсульфанилметил-4 изобутириламинофенокси)-4-метоксифенил]уксусной кислоты,который гидролизовался до[3-(2-трет-бутилсульфанилметил-4-изобутириламинофенокси)-4-метоксифенил]уксусной кислоты (соединение 36); с трет-бутилацетилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4(3,3-диметилбутириламино)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(3,3-диметилбутириламино)фенокси]-4-метоксифенилуксусной кислоты (соединение 37); с бензоилхлоридом с получением этилового эфира[3-(4-бензоиламино-2-трет-бутилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты (соединение 58); с 3-фторбензоилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4-(3 фторбензоиламино)фенокси]-4-метоксифенилуксусной кислоты,который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(3-фторбензоиламино)фенокси]-4-метоксифенилуксусной кислоты(соединение 62); с 4-фторбензоилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4-(4 фторбензоиламино)фенокси]-4-метоксифенилуксусной кислоты,который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(4-фторбензоиламино)фенокси]-4-метоксифенилуксусной кислоты(соединение 63); с 2-фторбензоилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4-(2 фторбензоиламино)фенокси]-4-метоксифенилуксусной кислоты,который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(2-фторбензоиламино)фенокси]-4-метоксифенилуксусной кислоты(соединение 64); с 2,4-дихлорбензоилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4(2,4-дихлорбензоиламино)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3[2-трет-бутилсульфанилметил-4-(2,4-дихлорбензоиламино)фенокси]-4-метоксифенилуксусной кислоты(соединение 65); с 3,5-дихлорбензоилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4(3,5-дихлорбензоиламино)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(3,5-дихлорбензоиламино)фенокси]-4-метоксифенилуксусной кислоты (соединение 66); с 3,5-дифторбензоилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4(3,5-дифторбензоиламино)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3[2-трет-бутилсульфанилметил-4-(3,5-дифторбензоиламино)фенокси]-4-метоксифенилуксусной кислоты(соединение 67); с дансилхлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4-(5 диметиламинонафтален-1-сульфониламино)фенокси]-4-метоксифенилуксусной кислоты (неочищенный продукт реакции очищался с помощью хроматографии на силикагеле), который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(5-диметиламинонафтален-1-сульфониламино)фенокси]-4 метоксифенилуксусной кислоты (соединение 72); Пример 6. Синтез 3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]-4 метоксифенил]уксусной кислоты (соединение 9). Этиловый эфир [3-(2-бромметил-4-нитрофенокси)-4-метоксифенил]уксусной кислоты (0,4 г,0,94 ммоль), 2-пропантиол (0,11 мл, 1,13 ммоль) и гидрид натрия (60% дисперсия в минеральном масле; 0,05 г, 1,13 ммоль) смешивались в 1,4-диоксане и перемешивались при комнатной температуре в течение 30 мин. Из смеси получался этиловый эфир [3-(2-изопропилсульфанилметил-4-нитрофенокси)-4 метоксифенил]уксусной кислоты, который затем восстанавливался до амина, как это описано для этапа 5, примера 3, с получением этилового эфира [3-(4-амино-2-изопропилсульфанилметилфенокси)-4 метоксифенил]уксусной кислоты. Затем амин обрабатывался пивалоилхлоридом, как это описано для этапа 6, примера 3, с получением этилового эфира 3-[4-(2,2-диметилпропиониламино)-2 изопропилсульфанилметилфенокси]-4-метоксифенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 7, примера 3, приводил к получению кислоты. Пример 7. Синтез 3-[4-(2,2-диметилпропиониламино)-2-(пропан-2-сульфонилметил)фенокси]-4 метоксифенилуксусной кислоты (соединение 10). Получалась в соответствии с процедурой, описанной в примере 1, с использованием 3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]-4-метоксифенилуксусной кислоты. Пример 8. Синтез 3-[4-(2,2-диметилпропиониламино)-2-(пропан-2-сульфинилметил)фенокси]-4 метоксифенилуксусной кислоты (соединение 11). Получалась в соответствии с процедурой, описанной в примере 2, с использованием 3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]-4-метоксифенилуксусной кислоты. Пример 9. Синтез 3-[4-(22-диметилпропиониламино)-2-(трифторэтансульфонилметил)фенокси]4-метоксифенилуксусной кислоты (соединение 12). Получалась в соответствии с процедурой, описанной в примере 1, с использованием 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты. Пример 10. Синтез [3-(2-бензилсульфанилметил-4-хлорфенокси)-4-метоксифенил]уксусной кислоты. Этап 1. Этиловый эфир [3-(4-хлор-2-формилфенокси)-4-метоксифенил]уксусной кислоты. Этиловый эфир (3-гидрокси-4-метоксифенил)уксусной кислоты (0,75 г, 4,8 ммоль),5-хлор-2-фторбензальдегид (1,0 г, 4,8 ммоль) и карбонат калия (1,0 г, 7,5 ммоль) смешивались в 1,4-диоксане (30 мл) и нагревались при 100 С в течение 3 дней. После этого неочищенный материал очищался с помощью хроматографии на силикагеле (элюирование 0-20% EtOAc в гексанах); в результате получалось титульное соединение. Этап 2. Этиловый эфир [3-(4-хлор-2-гидроксиметилфенокси)-4-метоксифенил]уксусной кислоты. К этиловому эфиру [3-(4-хлор-2-формилфенокси)-4-метоксифенил]уксусной кислоты (0,9 г,2,6 ммоль) в MeOH (30 мл) добавлялся боргидрид натрия (0,11 г, 2,9 ммоль), и смесь перемешивалась при комнатной температуре в течение 10 мин. Из получившейся смеси выделялось титульное соединение. Этап 3. Этиловый эфир [3-(2-бромметил-4-хлорфенокси)-4-метоксифенил]уксусной кислоты. К этиловому эфиру [3-(4-хлор-2-гидроксиметилфенокси)-4-метоксифенил]уксусной кислоты (0,9 г,2,6 ммоль) в ДМЭ добавлялся трибромид фосфора (0,37 мл, 3,9 ммоль), и реакционная смесь перемешивалась в течение 3 ч при комнатной температуре. Из получившейся смеси выделялось титульное соединение. Этап 4. Этиловый эфир [3-(2-бензилсульфанилметил-4-хлорфенокси)-4-метоксифенил]уксусной кислоты. К этиловому эфиру [3-(2-бромметил-4-хлорфенокси)-4-метоксифенил]уксусной кислоты (0,15 г,0,36 ммоль) и бензилмеркаптану (0,06 мл, 0,4 ммоль) в 1,4-диоксане (10 мл) добавлялся гидрид натрия(60% дисперсия в минеральном масле; 0,05 г, 1,25 ммоль), и реакционная смесь перемешивалась в течение 1 ч при комнатной температуре. После этого неочищенный материал очищался с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Этап 5. Этиловый эфир [3-(2-бензилсульфанилметил-4-хлорфенокси)-4-метоксифенил]уксусной кислоты. Этиловый эфир [3-(2-бензилсульфанилметил-4-хлорфенокси)-4-метоксифенил]уксусной кислоты гидролизовался с гидроксидом лития в MeOH и Н 2 О; в результате получалось титульное соединение. Пример 11. Синтез 3-[4-(4-хлорбензоиламино)-2-фенилсульфанилметилфенокси]-4 метоксифенилуксусная кислота (соединение 17). Этап 1. Этиловый эфир [4-метокси-3-(4-нитро-2-фенилсульфанилметилфенокси)фенил]уксусной кислоты. Получался в соответствии с процедурой, описанной для этапа 4, примера 10, с использованием следующих исходных веществ: этилового эфира(0,4 г, 0,9 ммоль) и хлорид олова(II) (0,6 г, 2,7 ммоль) смешивались в EtOH (20 мл) и перемешивались в течение ночи при 70 С. К раствору добавлялись CH2Cl2, H2O и натрия бикарбонат; полученная смесь отфильтровывалась через целит (диатомовую землю). Органический слой отделялся и концентрировался; в результате получалось титульное соединение.(0,29 мл, 2,3 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение 20 мин. Смесь концентрировалась; в результате получалось титульное соединение, которое непосредственно использовалось на этапе гидролиза. Этап 4. 3-[4-(4-Хлорбензоиламино)-2-фенилсульфанилметилфенокси]-4-метоксифенилуксусная кислота. К раствору этилового эфира 3-[4-(4-хлорбензоиламино)-2-фенилсульфанилметилфенокси]-4 метоксифенилуксусной кислоты (0,9 ммоль) в MeOH и H2O добавлялись лития гидроксид (0,3 г), Н 2 О(5 мл) и MeOH (20 мл). Реакционная смесь перемешивалась при 60 С, разделялась и очищалась с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Пример 12. Синтез 3-[2-бензилсульфанилметил-4-(4-хлорбензоиламино)фенокси]-4 метоксифенилуксусной кислоты (соединение 18). Как это было описано в примере 10, этиловый эфир [3-(2-бромметил-4-нитрофенокси)-4 метоксифенил]уксусной кислоты и бензилмеркаптан реагировали с получением этилового эфира[3-(2-бензилсульфанилметил-4-нитрофенокси)-4-метоксифенил]уксусной кислоты, который восстанавливался до этилового эфира [3-(4-амино-2-бензилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты. Это вещество обрабатывалось 4-хлорбензоилхлоридом, что приводило к получению этилового эфира 3-[2-бензилсульфанилметил-4-(4-хлорбензоиламино)фенокси]-4-метоксифенилуксусной кислоты. Гидролиз эфира приводил к получению соединения 18. Пример 13. Синтез 3-[4-(4-хлорбензоиламино)-2-(5-метил-[1,3,4]тиадиазол-2 илсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты (соединение 19). Как это описано в примере 10, этиловый эфир [3-(2-бромметил-4-нитрофенокси)-4 метоксифенил]уксусной кислоты и 5-метил-1,3,4-тиадиазол-2-тиол реагировали с получением этилового эфира 4-метокси-3-[2-(5-метил[1,3,4]тиадиазол-2-илсульфанилметил)-4-нитрофенокси]фенилуксусной кислоты, который восстанавливался до этилового эфира 3-[4-амино-2-(5-метил[1,3,4]тиадиазол-2 илсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты. Это вещество обрабатывалось 4-хлорбензоилхлоридом, что приводило к получению этилового эфира 3-[4-(4-хлорбензоиламино)-2-(5 метил[1,3,4]тиадиазол-2-илсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты. Гидролиз эфира приводил к получению кислоты - соединения 19. Пример 14. Синтез 3-[4-(4-хлорбензоиламино)-2-изопропилсульфанилметилфенокси]-4 метоксифенилуксусной кислоты (соединение 20). Этиловый эфир [3-(2-изопропилсульфанилметил-4-нитрофенокси)-4-метоксифенил]уксусной кислоты в соответствии с процедурой, описанной для этапа 2, примера 11, восстанавливался до амина. Амин обрабатывался 4-хлорбензоилхлоридом, что приводило к получению этилового эфира 3-[4-(4-хлорбензоиламино)-2-изопропилсульфанилметилфенокси]-4-метоксифенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 4, примера 11, приводил к получению кислоты. Пример 15. Синтез этилового эфира [3-(4-бром-2-бромметилфенокси)-4-метоксифенил]уксусной кислоты. Этап 1. Этиловый эфир (3-гидрокси-4-метоксифенил)уксусной кислоты. К 3-гидрокси-4-метоксифенилуксусной кислоте (5,0 г, 23,8 ммоль) в EtOH (100 мл) добавлялась серная кислота (1 мл), и реакционная смесь перемешивалась в течение ночи при комнатной температуре. После исчезновения исходного вещества (по данным тонкослойной хроматографии) смесь концентрировалась, что приводило к получению искомого продукта реакции. Этап 2. Этиловый эфир [3-(4-бром-2-формилфенокси)-4-метоксифенил]уксусной кислоты. Этиловый эфир (3-гидрокси-4-метоксифенил)уксусной кислоты (2,0 г, 9,5 ммоль),5-бром-2-фторбензальдегид (2,0 г, 9,5 ммоль) и карбонат калия (2,0 г, 14,3 ммоль) смешивались в 1,4 диоксане и нагревались при 90 С в течение ночи. После этого неочищенный материал очищался с помощью хроматографии на силикагеле (элюирование 0-25% EtOAc в гексанах); в результате получался искомый продукт реакции (1,8 г). Этап 3. Этиловый эфир [3-(4-бром-2-гидроксиметилфенокси)-4-метоксифенил]уксусной кислоты. Получался в соответствии с процедурой, описанной для этапа 2, примера 10, с использованием следующего исходного вещества: этилового эфира [3-(4-бром-2-формилфенокси)-4-метоксифенил]уксусной кислоты. Этап 4. Этиловый эфир [3-(4-бром-2-бромметилфенокси)-4-метоксифенил]уксусной кислоты. Получался в соответствии с процедурой, описанной для этапа 3, примера 10, с использованием следующего исходного вещества: этилового эфира Этап 5. Этиловый эфир [3-(4-бром-2-изопропилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты. Этиловый эфир [3-(4-бром-2-бромметилфенокси)-4-метоксифенил]уксусной кислоты (0,4 г,0,87 ммоль) и 2-пропантиол (0,08 г, 1,0 ммоль) смешивались в 1,4-диоксане (20 мл). К реакционной смеси добавлялся гидрид натрия (60% дисперсия в минеральном масле; 0,04 г, 1,0 ммоль), и полученная смесь перемешивалась при комнатной температуре в течение 20 мин. Получившаяся смесь разделялась с получением титульного соединения. Пример 16. Синтез 3-[4-(4-хлорбензоиламино)-2-(пропан-2-сульфинилметил)фенокси]-4 метоксифенилуксусной кислоты (соединение 21) и 3-[4-(4-хлорбензоиламино)-2-(пропан-2 сульфонилметил)фенокси]-4-метоксифенилуксусной кислоты (соединение 22). К 3-[4-(4-хлорбензоиламино)-2-изопропилсульфанилметилфенокси]-4-метоксифенилуксусной кислоте (0,17 г, 0,34 ммоль) в CH2Cl2 (20 мл) добавлялась 3-хлорпербензоевая кислота (77%; 0,076 г,0,34 ммоль), и реакционная смесь перемешивалась в течение 10 мин при комнатной температуре. В реакционную смесь вносилось дополнительное количество 3-хлорпербензоевой кислоты (77%; 0,025 г,0,11 ммоль), чтобы увеличить выход сульфона. Затем смесь концентрировалась и очищалась с помощью препаративной ВЭЖХ; в результате получались титульные сульфоксид и сульфон. Пример 17. Синтез 3-[2-бензенсульфинилметил-4-(4-хлорбензоиламино)фенокси]-4 метоксифенилуксусной кислоты(соединение 23) и 3-[2-бензенсульфонилметил-4-(4 хлорбензоиламино)фенокси]-4-метоксифенилуксусной кислоты (соединение 24). К 3-[4-(4-хлорбензоиламино)-2-фенилсульфанилметилфенокси]-4-метоксифенилуксусной кислоте (0,16 ммоль) в CH2Cl2 (3 мл) добавлялась 3-хлорпербензоевая кислота (77%; 0,036 г, 0,16 ммоль) и реакционная смесь перемешивалась при комнатной температуре в течение 1 ч. Полученная смесь концентрировалась и очищалась с помощью препаративной ВЭЖХ; в результате получались титульные сульфоксид и сульфон. Пример 18. Синтез[3-(4-этилкарбамоил-2-изопропилсульфанилметилфенокси)-4 метоксифенил]уксусной кислоты (соединение 25). Этап 1. Этиловый эфир [3-(4-бром-2-изопропилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты. К этиловому эфиру [3-(4-бром-2-бромметилфенокси)-4-метоксифенил]уксусной кислоты и 2-пропантиолу в 1,4-диоксане добавлялся натрия гидрид (60% дисперсия в минеральном масле), и реакционная смесь перемешивалась в течение 1 ч при комнатной температуре. После этого неочищенный материал очищался с помощью хроматографии на силикагеле (элюирование 0-20% EtOAc в гексанах); в результате получалось титульное соединение. Этап 2. Метиловый эфир 4-(5-этоксикарбонилметил-2-метоксифенокси)-3 изопропилсульфанилметилбензоевой кислоты. Этиловый эфир [3-(4-бром-2-изопропилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты (0,5 г, 1,1 ммоль) и триэтиламин (0,5 мл) растворялись в ДМФ (10 мл) и MeOH (10 мл). Полученный раствор дегазировался в течение 10 мин в атмосфере N2. К раствору добавлялся[1,1'-бис-(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,09 г, 0,11 ммоль), а затем через раствор в течение 10 мин пропускался монооксид углерода. Реакционная смесь перемешивалась под баллоном с монооксидом углерода при 65 С в течение ночи, а затем концентрировалась и очищалась с помощью хроматографии на силикагеле; в результате получалось титульное соединение. Этап 3. 4-(5-Этоксикарбонилметил-2-метоксифенокси)-3-изопропилсульфанилметилбензоевая кислота. Метиловый эфир 4-(5-этоксикарбонилметил-2-метоксифенокси)-3-изопропилсульфанилметилбензоевой кислоты (0,5 г, 1,16 ммоль) и метилтиолат натрия (0,16 г, 2,3 ммоль) смешивались в ДМФ(20 мл) и перемешивались при 65 С в течение 1 ч. После обработки кислотой неочищенный материал очищался с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Этап 4. Этиловый эфир(0,07 г, 0,17 ммоль) в CH2Cl2 (10 мл) и ДМФ (1 капля) добавлялся оксалилхлорид (0,04 мл, 0,5 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение 30 мин. После концентрирования до сухого остатка к нему добавлялся этиламин (2 М раствор в ТГФ; 0,25 мл, 0,5 ммоль), а затем - CH2Cl2 (10 мл) и диизопропилэтиламин (0,5 мл); реакционная смесь перемешивалась при комнатной температуре в течение 15 мин. Смесь разделялась, что приводило к получению титульного соединения. Этап 5. [3-(4-Этилкарбамоил-2-изопропилсульфанилметилфенокси)-4-метоксифенил]уксусная кислота. Получалась в соответствии с процедурой, описанной для этапа 4, примера 11, с использованием следующего исходного вещества: этилового эфира [3-(4-этилкарбамоил-2-изопропилсульфанил- 19019351 метилфенокси)-4-метоксифенил]уксусной кислоты. Пример 19. Синтез 3-[4-(4-хлорбензилкарбамоил)-2-изопропилсульфанилметилфенокси]-4 метоксифенилуксусной кислоты (соединение 26). После процедуры, описанной для этапа 4, примера 18, из 4-(5-этоксикарбонилметил-2 метоксифенокси)-3-изопропилсульфанилметилбензоевой кислоты и 4-хлорбензиламина получался этиловый эфир 3-[4-(4-хлорбензилкарбамоил)-2-изопропилсульфанилметилфенокси]-4 метоксифенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 4, примера 11, приводил к получению кислоты. Пример 20. Синтез (3-4-[2-(4-фторфенил)этилкарбамоил]-2-изопропилсульфанилметилфенокси-4 метоксифенил)уксусной кислоты (соединение 27). После процедуры, описанной для этапа 4, примера 18, из 4-(5-этоксикарбонилметил-2 метоксифенокси)-3-изопропилсульфанилметилбензоевой кислоты и 4-фторфенэтиламина получался этиловый эфир(3-4-[2-(4-фторфенил)этилкарбамоил]-2-изопропилсульфанилметилфенокси-4 метоксифенил)уксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 4, примера 11, приводил к получению кислоты. Пример 21. Синтез(3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (соединение 28). Этап 1. 1-Бензилокси-3-бром-5-хлорбензен. К раствору 1-бром-3-хлор-5-фторбензена (25 г, 112 ммоль) и бензилового спирта (25 мл, 239 ммоль) в N-метилпирролидоне (N-МП) (200 мл) при комнатной температуре добавлялся гидрид натрия (60% дисперсия в минеральном масле; 10,5 г, 263 ммоль); реакционная смесь нагревалась при 120 С в течение 10 ч. Смесь закислялась 10% водным раствором HCl и экстрагировалась EtOAc; в результате получалось титульное соединение. Этап 2. Диметиловый эфир 2-(3-бензилокси-5-хлорфенил)малоновой кислоты. К раствору 1-бензилокси-3-бром-5-хлорбензена (31 г, 101,7 ммоль), диметилмалоната (25,7 мл,223,8 ммоль) и бромида меди(I) (32 г, 223,8 ммоль) в 1,4-диоксане (300 мл) при 0 С в атмосфере N2 добавлялся гидрид натрия (60% дисперсия в минеральном масле; 9 г, 223,9 ммоль) по порциям. Через 10 мин реакционная смесь нагревалась до 100 С и перемешивалась в течение 6 ч. Аналитическая ЖХМС указала на наличие оставшегося исходного вещества, поэтому реакционная смесь перемешивалась при 100 С еще в течение 24 ч. К реакционной смеси добавлялся 50% водный раствор NH4OH (1 л), чтобы разделить на части твердый остаток; после этого смесь трижды экстрагировалась CH2Cl2 (всего 3 л). Органический слой промывался солевым раствором, высушивался над MgSO4, отфильтровывался и концентрировался; получившийся остаток очищался с помощью хроматографии на силикагеле; в результате получалось титульное соединение. Этап 3. Метиловый эфир (3-бензилокси-5-хлорфенил)уксусной кислоты. Смесь диметилового эфира 2-(3-бензилокси-5-хлорфенил)малоновой кислоты (21 г, 60,2 ммоль) и хлорида лития (3,06 г, 72,2 ммоль) в ДМСО: Н 2 О (10:1; 200 мл) перемешивалась при 150 С в течение 5 ч. После охлаждения реакционной смеси до комнатной температуры к ней добавлялось 500 мл Н 2 О; получившаяся смесь экстрагировалась EtOAc с гексанами (1:10; всего 1,5 л), что приводило к получению титульного соединения. Этап 4. Метиловый эфир (3-хлор-5-гидроксифенил)уксусной кислоты. К метиловому эфиру (3-бензилокси-5-хлорфенил)уксусной кислоты (8 г, 28 ммоль) в CH2Cl2(100 мл) при 0 С добавлялся трибромид бора (1 М раствор в CH2Cl2; 56 мл, 56 ммоль). Реакционная смесь согревалась до комнатной температуры за 30 мин и перемешивалась в течение 2 ч. После исчезновения исходного вещества (по данным ЖХМС и тонкослойной хроматографии) смесь охлаждалась до 0 С и гасилась Н 2 О (50 мл). Смесь концентрировалась; полученный остаток разбавлялся EtOAc (500 мл). Твердый остаток отфильтровывался, а органический слой разделялся и концентрировался. Неочищенный материал очищался с помощью хроматографии на силикагеле; в результате получалось титульное соединение. Этап 5. Метиловый эфир [3-хлор-5-(2-формил-4-нитрофенокси)фенил]уксусной кислоты. Метиловый эфир (3-хлор-5-гидроксифенил)уксусной кислоты (2,9 г, 14,5 ммоль),2-фтор-5-нитробензальдегид (2,7 г, 15,9 ммоль) и карбонат калия (4,0 г, 28,9 ммоль) растворялись в 1,4-диоксане (20 мл); полученный раствор перемешивался при 100 С в течение ночи. После исчезновения из реакционной смеси исходного вещества (по данным аналитической ЖХМС и тонкослойной хроматографии) смесь обрабатывалась EtOAc и 10% водным раствором HCl. Неочищенный материал очищался с помощью хроматографии на силикагеле; в результате получалось титульное соединение. Этап 6. Метиловый эфир [3-хлор-5-(2-гидроксиметил-4-нитрофенокси)фенил]уксусной кислоты. К метиловому эфиру [3-хлор-5-(2-формил-4-нитрофенокси)фенил]уксусной кислоты (3,7 г,10,6 ммоль) в MeOH (30 мл) добавлялся боргидрид натрия (0,52 г, 13,8 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение 20 мин. После разделения между EtOAc и Н 2 О неочищенный материал очищался с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Этап 7. Метиловый эфир [3-(2-бромметил-4-нитрофенокси)-5-хлорфенил]уксусной кислоты. К раствору метилового эфира [3-хлор-5-(2-гидроксиметил-4-нитрофенокси)фенил]уксусной кислоты (2,4 г, 6,8 ммоль) в ДМЭ (20 мл) добавлялся трибромид фосфора (0,97 мл, 10,2 ммоль), и реакционная смесь перемешивалась в течение 1 ч при комнатной температуре. После разделения между EtOAc и Н 2 О неочищенный материал очищался с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Этап 8. Метиловый эфир 3-хлор-5-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. К раствору метилового эфира [3-(2-кромометил-4-нитрофенокси)-5-хлорфенил]уксусной кислоты(0,5 г, 1,25 ммоль) и 2,2,2-трифторэтантиол (0,13 мл, 1,38 ммоль) в 1,4-диоксане (6 мл) добавлялся гидрид натрия (60% дисперсия в минеральном масле; 0,055 г, 1,38 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение ночи. После разделения между EtOAc и 10% водным раствором HCl неочищенный материал очищался с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Этап 9. Метиловый эфир 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-5 хлорфенилуксусной кислоты. Метиловый эфир 3-хлор-5-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (0,07 г, 0,16 ммоль), 1,1-диметилгидразин (0,08 мл, 1,09 ммоль), хлорид железа(0,003 г, 0,02 ммоль) и DARCO (0,016 г) растворялись в EtOH; полученный раствор перемешивался при 65 С в течение 30 ч. После разделения между EtOAc и Н 2 О неочищенный материал очищался с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Этап 10. Метиловый эфир 3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. К раствору метилового эфира 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-5 хлорфенилуксусной кислоты (0,058 г, 0,14 ммоль) в CH2Cl2 (2 мл) добавлялся триэтиламин (0,04 мл,0,28 ммоль), а затем - пивалоилхлорид (0,02 мл, 0,17 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение 10 мин. После разделения между EtOAc и Н 2 О неочищенный материал очищался с помощью хроматографии на силикагеле; в результате получалось титульное соединение. Этап 11. 3-Хлор-5-(4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусная кислота. К раствору метилового эфира 3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (0,038 г, 0,08 ммоль) в ТГФ:Н 2 О:MeOH(2:1:2; 2 мл) добавлялся 1 н. водный раствор гидроксида лития; смесь перемешивалась при комнатной температуре в течение ночи. Смесь закислялась до pH 5 10% водным раствором HCl и экстрагироваласьEtOAc. Неочищенный материал очищался с помощью препаративной хроматографии; в результате получалось титульное соединение. Пример 22. Синтез 3-хлор-5-[4-(2,2-диметилпропиониламино)-2(трифторэтансульфонилметил)фенокси]фенилуксусной кислоты (соединение 29). К раствору 3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (0,02 г, 0,04 ммоль) в CH2Cl2 (5 мл) добавлялась 3-хлорпербензоевая кислота (77%; 0,046 г, 0,2 ммоль), и реакционная смесь перемешивалась в течение 3 ч при комнатной температуре. После разделения между CH2Cl2 и Н 2 О неочищенный материал очищался с помощью хроматографии на силикагеле; в результате получалось титульное соединение. Пример 23. Синтез 3-хлор-5-[4-(2,2-диметилпропиониламино)-2 изопропилсульфанилметилфенокси]фенилуксусной кислоты (соединение 30). После процедур, описанных для этапа 8, примера 21 (с метиловым эфиром [3-(2-бромметил-4 нитрофенокси)-5-хлорфенил]уксусной кислоты и 2-пропантиолом в качестве исходных веществ), для этапа 9, примера 21 и этапа 10, примера 21 (с использованием пивалоилхлорида), получался метиловый эфир 3-хлор-5-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]фенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 11, примера 21, приводил к получению кислоты. Пример 24. Синтез 3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(пропан-2 сульфонилметил)фенокси]фенилуксусной кислоты (соединение 31). К раствору метилового эфира 3-хлор-5-[4-(2,2-диметилпропиониламино)-2 изопропилсульфанилметилфенокси]фенилуксусной кислоты (0,05 г, 0,29 ммоль) в CH2Cl2 (3 мл) добавлялась 3-хлорпербензоевая кислота (77%; 0,12 г, 1,45 ммоль), и реакционная смесь перемешивалась в течение 1 ч при комнатной температуре. После обработки EtOAc и солевым раствором неочищенный материал очищался с помощью хроматографии на силикагеле, что приводило к получению метилового эфира 3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(пропан-2-сульфонилметил)фенокси]фенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 11, примера 21, приводил к получению кислоты. Пример 25. Синтез 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-5-трифторметилфенилуксусной кислоты (соединение 32). К раствору бензилового спирта (1,1 г, 10 ммоль) в N-МП (20 мл) добавлялся гидрид натрия (60% дисперсия в минеральном масле; 0,44 г, 11 ммоль), и реакционная смесь перемешивалась в течение 30 мин при комнатной температуре. Затем смесь вносилась во флакон, содержащий 3-фтор-5(трифторметил)фенилуксусную кислоту (1 г, 4,5 ммоль), и реакционная смесь перемешивалась при 120 С в течение 3 ч. Обработка кислотой приводила к получению титульного соединения - (3-бензилокси-5-трифторметилфенил)уксусной кислоты. К раствору (3-бензилокси-5-трифторметилфенил)уксусной кислоты (1,5 г, 5,4 ммоль) в EtOH(30 мл) добавлялась серная кислота (1 мл), и смесь перемешивалась в течение ночи при комнатной температуре. Отсутствие исходного вещества устанавливалось с помощью аналитической ЖХМС. После этого реакционная смесь разделялась с получением этилового эфира (3-бензилокси-5 трифторметилфенил)уксусной кислоты. Этиловый эфир (3-бензилокси-5-трифторметилфенил)уксусной кислоты (1,7 г, 5,6 ммоль) растворялся в EtOH (30 мл); полученный раствор дегазировался в атмосфере азота. К реакционной смеси добавлялся палладий (5%) на угле, и реакционная смесь продувалась Н 2, а затем перемешивалась в атмосфере Н 2 при 50 С в течение ночи. Смесь отфильтровывалась и концентрировалась; в результате получился этиловый эфир (3-гидрокси-5-трифторметилфенил)уксусной кислоты. В результате реакции этилового эфира (3-гидрокси-5-трифторметилфенил)уксусной кислоты и 2-фтор-5-нитробензальдегида (процедура описана для этапа 5, примера 21) получался этиловый эфир[3-(2-формил-4-нитрофенокси)-5-трифторметилфенил]уксусной кислоты. Этиловый эфир [3-(2-формил-4-нитрофенокси)-5-трифторметилфенил]уксусной кислоты восстанавливался до этилового эфира [3-(2-гидроксиметил-4-нитрофенокси)-5-трифторметилфенил]уксусной кислоты (в соответствии с процедурой, описанной для этапа 6, примера 21), а затем бромировался, как это описано для этапа 7, примера 21. Продукт реакции обрабатывался 2,2,2-трифторэтантиолом, что приводило к получению этилового эфира 3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-5 трифторметилфенилуксусной кислоты. Амин восстанавливался, как это было описано для этапа 9, примера 21, а затем обрабатывался пивалоилхлоридом, как это описано для этапа 10, примера 21; в результате этого получался этиловый эфир 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-5-трифторметилфенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 11, примера 21, приводил к получению кислоты. Пример 26. Синтез 3-[4-(2,2-диметилпропиониламино)-2-(трифторэтансульфонилметил)фенокси]5-трифторметилфенилуксусной кислоты (соединение 33). Получалась в соответствии с процедурой, описанной в примере 24, с использованием этилового эфира 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-5 трифторметилфенилуксусной кислоты. Пример 27. Синтез 3-[4-[(2,2-диметилпропионил)метиламино]-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты (соединение 34). К раствору 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 метоксифенилуксусной кислоты (0,128 г, 0,26 ммоль) в MeCN (3 мл) добавлялся йодметан (0,03 мл,0,53 ммоль/л), а затем - гидрид натрия (60% дисперсия в минеральном масле; 0,021 г, 0,53 ммоль). Реакционная смесь перемешивалась при комнатной температуре. Аналитическая ЖХМС указывала на наличие только исходного вещества, поэтому реакционная смесь нагревалась до 60 С в течение 2 ч, охлаждалась до комнатной температуры и перемешивалась в атмосфере N2 в течение 2 дней. Затем смесь в таком виде подвергалась гидролизу. К раствору метилового эфира 3-[4-[(2,2-диметилпропионил)метиламино]2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты (0,26 ммоль) вMeCN, добавлялись Н 2 О, MeOH и гидроксид лития; реакционная смесь нагревалась до 65 С в течение 1 ч; в результате получалось титульное соединение. Пример 28. Синтез 4-метокси-3-[4-(2-оксооксазолидин-3-ил)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (соединение 38). Этап 1. Этиловый эфир 3-[4-(2-хлорэтоксикарбониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты. Получался в соответствии с процедурой, описанной для этапа 6, примера 3, с использованием следующих исходных веществ: этилового эфира 3-[4-амино-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты и 2-хлорэтилхлорформиата. Этап 2. 4-Метокси-3-[4-(2-оксооксазолидин-3-ил)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусная кислота. К раствору этилового эфира 3-[4-(2-хлорэтоксикарбониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты (0,100 г, 0,19 ммоль) в EtOH (5 мл) добавлялся этоксид натрия (21% (по массе) в EtOH; 4,61 мл, 0,37 ммоль), и реакционная смесь перемешивалась при 65 С в течение ночи. Смесь разделялась между EtOAc и Н 2 О, водный слой экстрагировался EtOAc. Комбинированные органические слои высушивались над MgSO4, отфильтровывались и концентрировались; получившийся остаток очищался с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Пример 29. Синтез[3-(4-бром-2-трет-бутилсульфанилметилфенокси)-4 метоксифенил]уксусной кислоты. Получался в соответствии с процедурой, описанной для этапа 5, примера 15, с использованием следующих исходных веществ: этилового эфира[3-(4-бром-2-бромметилфенокси)-4 метоксифенил]уксусной кислоты и 2-метил-2-пропантиола. Этап 2. 3-трет-Бутилсульфанилметил-4-(5-этоксикарбонилметил-2-метоксифенокси)бензоевая кислота. Этиловый эфир [3-(4-бром-2-трет-бутилсульфанилметилфенокси)-4-метоксифенил]уксусной кислоты (2,0 г, 4,3 ммоль) и триэтиламин (5,9 мл, 43 ммоль) растворялись в Н 2 О (5 мл) и ДМФ (50 мл); полученный раствор дегазировался с помощью монооксида углерода в течение 20 мин. Добавлялся [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,35 г, 0,43 ммоль), и реакционная смесь нагревалась при 80 С в течение 4 ч. Смесь закислялась и экстрагировалась EtOAc; в результате получалось титульное соединение. Этап 3. Этиловый эфир [3-(4-трет-бутилкарбамоил-2-трет-бутилсульфанилметилфенокси)-4 метоксифенил]уксусной кислоты. 3-трет-Бутилсульфанилметил-4-(5-этоксикарбонилметил-2-метоксифенокси)бензоевая кислота(0,15 мл,13,9 ммоль),1-этил-3-(3'диметиламинопропил)карбодиимид (0,11 г, 0,55 ммоль) и N-гидроксибензотриазол (0,074 г, 0,55 ммоль) растворялись в CH2Cl2 (8 мл); полученный раствор перемешивался в течение ночи. Полученная смесь концентрировалась и очищалась с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Этап 4.[3-(4-трет-бутилкарбамоил-2-трет-бутилсульфанилметилфенокси)-4 метоксифенил]уксусной кислоты (0,46 ммоль) обрабатывался гидроксидом лития в MeOH и Н 2 О при 60 С в течение 20 мин; в результате получалось титульное соединение. После процедур,описанных для примера 29,3-трет-бутилсульфанилметил-4-(5 этоксикарбонилметил-2-метоксифенокси)бензоевая кислота вступала в реакцию со следующими веществами: с 2-аминоацетофенона гидрохлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4-(2-оксо-2-фенилэтилкарбамоил)фенокси]-4-метоксифенилуксусной кислоты,который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(2-оксо-2 фенилэтилкарбамоил)фенокси]-4-метоксифенилуксусной кислоты (соединение 40); с 1-(4-фторфенил)-2-метил-2-пропиламином с получением этилового эфира(3-2-трет-бутилсульфанилметил-4-[2-(4-фторфенил)-1,1-диметилэтилкарбамоил]фенокси-4 метоксифенил)уксусной кислоты, который гидролизовался до (3-2-трет-бутилсульфанилметил-4-[2-(4 фторфенил)-1,1-диметилэтилкарбамоил]фенокси-4-метоксифенил)уксусной кислоты (соединение 41); с пиперидином с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4-(пиперидин-1 карбонил)фенокси]-4-метоксифенилуксусной кислоты,который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(пиперидин-1-карбонил)фенокси]-4-метоксифенилуксусной кислоты (соединение 42); с 5-амино-2-метоксипиридином с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4(6-метоксипиридин-3-илкарбамоил)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(6-метоксипиридин-3-илкарбамоил)фенокси]-4 метоксифенилуксусной кислоты (соединение 43); с 2,2,2-трифторэтиламина гидрохлоридом с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4-(2,2,2-трифторэтилкарбамоил)фенокси]-4-метоксифенилуксусной кислоты,который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(2,2,2 трифторэтилкарбамоил)фенокси]-4-метоксифенилуксусной кислоты (соединение 44); с N-метилизопропиламином с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4(изопропилметилкарбамоил)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(изопропилметилкарбамоил)фенокси]-4-метоксифенилуксусной кислоты (соединение 45); с неопентиламином с получением этилового эфира 3-[2-трет-бутилсульфанилметил-4-(2,2 диметилпропилкарбамоил)фенокси]-4-метоксифенилуксусной кислоты, который гидролизовался до 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропилкарбамоил)фенокси]-4-метоксифенилуксусной кислоты (соединение 46). Пример 30. Синтез 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-5 хлорфенилуксусной кислоты (соединение 47). В соответствии с процедурой, описанной для этапа 8, примера 21, получался метиловый эфир[3-(2-трет-бутилсульфанилметил-4-нитрофенокси)-5-хлорфенил]уксусной кислоты. Для этого использовались метиловый эфир [3-(2-бромметил-4-нитрофенокси)-5-хлорфенил]уксусной кислоты и 2-метил-2 пропантиол. Полученное вещество восстанавливалось до метилового эфира [3-(4-амино-2-третбутилсульфанилметилфенокси)-5-хлорфенил]уксусной кислоты, как это описано для этапа 9, примера 21. Амин обрабатывался пивалоилхлоридом, как это описано для этапа 10, примера 21; это приводило к получению метилового эфира 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]5-хлорфенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 11, примера 21, приводил к получению кислоты. Пример 31. Синтез 3-хлор-5-[4-(2,2-диметилпропиониламино)-2-(2-метилпропан-2 сульфонилметил)фенокси]фенилуксусной кислоты (соединение 48). Получалась в соответствии с процедурой, описанной в примере 22, с использованием 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-5-хлорфенилуксусной кислоты. Пример 32. Синтез 4-дифторметокси-3-[4-(2,2-диметилпропиониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (соединение 49). Этиловый эфир 4-метокси-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (1,5 г, 3,6 ммоль) обрабатывался 48% раствором бромида водорода в уксусной кислоте (1:1; 20 мл) при 100 С в течение ночи. После обработки EtOAc и Н 2 О неочищенный материал очищался с помощью препаративной ВЭЖХ, что приводило к получению 4-гидрокси-3-[4-нитро-2(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. 4-Гидрокси-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусная кислота(3,6 ммоль) и хлороводород (4 н. раствор в 1,4-диоксане) растворялись в EtOH; полученный раствор перемешивался при 80 С в течение 3 ч. После концентрирования сухой остаток очищался с помощью хроматографии на силикагеле, что приводило к получению метилового эфира 4-гидрокси-3-[4-нитро-2(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. Метиловый эфир 4-гидрокси-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (0,40 г, 0,92 ммоль), хлордифторацетат натрия (0,282 г, 1,86 ммоль) и карбонат калия (0,14 г, 1,02 ммоль) растворялись в ДМФ:Н 2 О (8,5:1; 4,6 мл); полученный раствор дегазировался с помощью N2 в течение 15 мин. Реакционная смесь перемешивалась при 100 С в течение 4 ч. После обработки EtOAc и 1 н. водным раствором HCl остаток очищался с помощью хроматографии на силикагеле,что приводило к получению метилового эфира 4-дифторметокси-3-[4-нитро-2-(2,2,2 трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. Метиловый эфир 4-дифторметокси-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметилфенокси]фенилуксусной кислоты восстанавливался до метилового эфира 3-[4-амино-2-(2,2,2 трифторэтштсульфанилметил)фенокси]-4-дифторметоксифенилуксусной кислоты, как это описано для этапа 9, примера 21. Полученное соединение обрабатывалось пивалоилхлоридом, как это описано для этапа 10, примера 21; в результате получался метиловый эфир 4-дифторметокси-3-[4-(2,2 диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 11, примера 21, приводил к получению кислоты. Пример 33. Синтез 4-дифторметокси-3-[4-(2,2-диметилпропиониламино)-2-(трифторэтансульфонилметил)фенокси]фенилуксусной кислоты (соединение 50). Получалась в соответствии с процедурой, описанной в примере 22, с использованием 4-дифторметокси-3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. Пример 34. Синтез(3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метилфенилуксусной кислоты (соединение 51). К раствору метилового эфира 4-гидрокси-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (0,50 г, 1,16 ммоль) в ДМФ (10 мл) добавлялисьN-фенил-бис-(трифторметансульфонимид) (0,455 г, 1,27 ммоль) и карбонат цезия (0,755 г, 2,32 ммоль); и реакционная смесь перемешивалась при комнатной температуре в течение 2 ч. После обработки EtOAc и Н 2 О неочищенный материал очищался с помощью хроматографии на силикагеле, что приводило к получению метилового эфира 3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 трифторметансульфонилоксифенилуксусной кислоты. Метиловый эфир 3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-трифторметансульфонилоксифенилуксусной кислоты (0,20 г, 0,35 ммоль), триметилбороксин (0,07 мл, 0,53 ммоль),карбонат калия (0,123 г, 0,89 ммоль) и тетракис-(трифенилфосфин)палладий(0) (0,041 г, 0,035 ммоль) растворялись в ДМЭ:Н 2 О (2:1; 4 мл); полученный раствор дегазировался с помощью N2 в течение 8 мин. Реакционная смесь перемешивалась при 90 С в течение 2 ч, а затем разделялась с помощью EtOAc и 10% водного раствора HCl. Остаток очищался с помощью хроматографии на силикагеле, что приводило к получению метилового эфира 4-метил-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. После процедур, описанных для примера 21, метиловый эфир 4-метил-3-[4-нитро-2-(2,2,2 трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты восстанавливался до метилового эфира 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метилфенилуксусной кислоты; полученное соединение обрабатывалось пивалоилхлоридом, что приводило к получению метилового эфира 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 метилфенилуксусной кислоты. Гидролиз эфира приводил к получению кислоты. Пример 35. Синтез 4-хлор-3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]фенилуксусной кислоты (соединение 52). К раствору 4-хлор-3-фторфенилуксусной кислоты (5 г, 26,5 ммоль) и бензилового спирта (5,5 мл,53,0 ммоль) в N-МП (50 мл) при 0 С добавлялся гидрид натрия (60% дисперсия в минеральном масле; 2,3 г, 58,3 ммоль); реакционная смесь нагревалась до 120 С и перемешивалась в течение ночи. Смесь закислялась до pH 4 и экстрагировалась EtOAc; в результате получалось (3-бензилокси-4 хлорфенил)уксусная кислота.(3-Бензилокси-4-хлорфенил)уксусная кислота (8 г) нагревалась с хлороводородом (4 н. раствор в 1,4-диоксане; 6 мл) в EtOH (100 мл) при 80 С в течение 3 ч. После концентрирования сухой остаток очищался с помощью хроматографии на силикагеле, что приводило к получению этилового эфира(3-бензилокси-4-хлорфенил)уксусной кислоты. Этиловый эфир (3-бензилокси-4-хлорфенил)уксусной кислоты вступал в реакцию, описанную для этапа 4, примера 21; в результате получался этиловый эфир (4-хлор-3-гидроксифенил)уксусной кислоты. Этиловый эфир(4-хлор-3-гидроксифенил)уксусной кислоты реагировал с 2-фтор-5-нитробензальдегидом, как это описано для этапа 5, примера 21; в результате получался этиловый эфир [4-хлор-3-(2-формил-4-нитрофенокси)фенил]уксусной кислоты. Этиловый эфир [4-хлор-3-(2-формил-4-нитрофенокси)фенил]уксусной кислоты восстанавливался до этилового эфира [4-хлор-3-(2-гидроксиметил-4-нитрофенокси)фенил]уксусной кислоты, как это описано для этапа 6, примера 21. Полученное соединение, как это описано для этапа 7, примера 21, бромировалось с получением этилового эфира [3-(2-бромметил-4-нитрофенокси)-4-хлорфенил]уксусной кислоты. Этиловый эфир [3-(2-бромметил-4-нитрофенокси)-4-хлорфенил]уксусной кислоты обрабатывался 2-пропантиолом, как это описано для этапа 8, примера 21. В результате получался этиловый эфир[4-хлор-3-(2-изопропилсульфанилметил-4-нитрофенокси)фенил]уксусной кислоты. Это соединение восстанавливалось до этилового эфира[3-(4-амино-2-изопропилсульфанилметилфенокси)-4 хлорфенил]уксусной кислоты, как это описано для этапа 9, примера 21. Обработка амина пивалоилхлоридом, как это описано для этапа 10, примера 21, приводила к образованию этилового эфира 4-хлор-3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]фенилуксусной кислоты. Гидролиз этого эфира в соответствии с процедурой, описанной для этапа 11, примера 21, приводил к получению кислоты. Пример 36. Синтез 4-хлор-3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (соединение 53). Этиловый эфир[3-(2-бромметил-4-нитрофенокси)-4-хлорфенил]уксусной кислоты и 2,2,2-трифторэтантиол вступали в реакцию, как это описано для примера 21; в результате получался этиловый эфир 4-хлор-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. Это соединение восстанавливалось до амина, который обрабатывался пивалоилхлоридом, что приводило к получению этилового эфира 4-хлор-3-[4-(2,2-диметилпропиониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. Гидролиз эфира приводил к получению кислоты. Пример 37. Синтез 3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]-5 трифторметилфенилуксусной кислоты (соединение 54). Этиловый эфир [3-(2-бромметил-4-нитрофенокси)-5-трифторметилфенил]уксусной кислоты и 2-пропантиол вступали в реакцию, как это описано для примера 21. В результате получался этиловый эфир [3-(2-изопропилсульфанилметил-4-нитрофенокси)-5-трифторметилфенил]уксусной кислоты. Это соединение восстанавливалось до амина, который обрабатывался пивалоилхлоридом; в результате получался этиловый эфир 3-[4-(2,2-диметилпропиониламино)-2-изопропилсульфанилметилфенокси]-5 трифторметилфенилуксусной кислоты. Гидролиз эфира приводил к получению кислоты. Пример 38. Синтез 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-винилфенилуксусной кислоты ты (соединение 56). Метиловый эфир 3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-трифторметансульфонилоксифенилуксусной кислоты (0,920 г, 1,63 ммоль), (триметилсилил)ацетилен (0,34 мл,2,45 ммоль), дихлор-бис-(трифенилфосфин)палладий(II) (0,1155, 0,16 ммоль) и йодид меди (0,031 г,0,16 ммоль) растворялись в триэтиламине (8 мл); полученный раствор дегазировался в течение 5 мин. Реакционная смесь нагревалась в течение 8 ч, а затем разделялась между CH2Cl2 и H2O. Неочищенный материал очищался с помощью хроматографии на силикагеле, что приводило к получению метилового эфира 3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-триметилсиланилэтинилфенилуксусной кислоты. К раствору метилового эфира 3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 триметилсиланилэтинилфенилуксусной кислоты (0,410 г, 0,8 ммоль) в ТГФ (3 мл) добавлялся тетрабутиламмония фторид (1 М раствор в ТГФ; 1,2 мл, 1,2 ммоль), и реакционная смесь перемешивалась в течение 30 мин при комнатной температуре. Отсутствие исходного вещества устанавливалось с помощью аналитической тонкослойной хроматографии. После этого смесь разделялась между EtOAc и H2O; остаток очищался с помощью хроматографии на силикагеле, что приводило к получению метилового эфира 4-этинил-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты. Метиловый эфир 4-этинил-3-[4-нитро-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (0,170 г, 0,39 ммоль) гидрогенизовался в присутствии 10% палладия на угле в EtOH под давлением 50 фкд в атмосфере Н 2 (с использованием аппарата Парра) в течение ночи. Смесь отфильтровывалась через целит, фильтрат концентрировался с получением смеси (1:1) метилового эфира 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-винилфенилуксусной кислоты и метилового эфира 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-этилфенилуксусной кислоты. После процедур, описанных для примера 21, в результате реакции между смесью (1:1) метилового эфира 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-винилфенилуксусной кислоты и метилового эфира 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-этилфенилуксусной кислоты с пивалоилхлоридом получались метиловый эфир 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2 трифторэтилсульфанилметил)фенокси]-4-винилфенилуксусной кислоты и метиловый эфир 3-[4-(2,2-диметилпропиониламино)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4 этилфенилуксусной кислоты. Амиды разделялись с помощью хроматографии. Гидролиз эфиров приводил к получению кислот. Пример 39. Синтез 4-метокси-3-[4-(2-оксоимидазолидин-1-ил)-2-(2,2,2-трифторэтилсульфанилметил)фенокси]фенилуксусной кислоты (соединение 57). Этиловый эфир 3-[4-амино-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты вступал в реакцию с 2-хлорэтилизоцианатом, как это описано для этапа 6, примера 3; неочищенный материал очищался с помощью хроматографии на силикагеле. К раствору этилового эфира 3-[4-[3-(2-хлорэтил)уреидо]-2-(2,2,2-трифторэтилсульфанилметил)фенокси]-4-метоксифенилуксусной кислоты (0,050 г, 0,094 ммоль) в EtOH (5 мл) добавлялся этоксид натрия (20% [мас./об.]; 2,27 мл, 0,187 ммоль). Реакционная смесь перемешивалась при 65 С в течение ночи и разделялась между EtOAc и Н 2 О. Водный слой разделялся и экстрагировалсяEtOAc; комбинированные органические слои высушивались над MgSO4, отфильтровывались и концентрировались. Остаток очищался с помощью препаративной хроматографии; в результате получалось соединение 57. Пример 40. Синтез 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4 хлорфенилуксусной кислоты (соединение 59). Как описано для примера 21, этиловый эфир [3-(2-бромметил-4-нитрофенокси)-4 хлорфенил]уксусной кислоты и 2-метил-2-пропантиол вступали в реакцию с получением этилового эфира [3-(2-трет-бутилсульфанилметил-4-нитрофенокси)-4-хлорфенил]уксусной кислоты; это соединение восстанавливалось до этилового эфира[3-(4-амино-2-трет-бутилсульфанилметилфенокси)-4 хлорфенил]уксусной кислоты. Амин обрабатывался триметилацетилхлоридом, что приводило к получению этилового эфира 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4 хлорфенилуксусной кислоты. Гидролиз эфира приводил к получению кислоты. Пример 41. Синтез 3-[2-трет-бутилсульфанилметил-4-(4-хлорбензоиламино)фенокси]-4 хлорфенилуксусной кислоты (соединение 60). После процедуры, описанной для примера 21, из этилового эфира [3-(4-амино-2-третбутилсульфанилметилфенокси)-4-хлорфенил]уксусной кислоты и 4-хлорбензоилхлорида получался этиловый эфир 3-[2-трет-бутилсульфанилметил-4-(4-хлорбензоиламино)фенокси]-4-хлорфенилуксусной кислоты. Гидролиз эфира приводил к получению кислоты. Пример 42. Синтез[3-(2-трет-бутилсульфанилметил-4-изобутириламинофенокси)-4 хлорфенил]уксусной кислоты (соединение 61). После процедуры, описанной для примера 21, из этилового эфира [3-(4-амино-2-третбутилсульфанилметилфенокси)-4-хлорфенил]уксусной кислоты и изобутирилхлорида получался этиловый эфир [3-(2-трет-бутилсульфанилметил-4-изобутириламинофенокси)-4-хлорфенил]уксусной кислоты. Гидролиз эфира приводил к получению кислоты. Пример 43. Синтез 3-[4-(2,2-диметилпропиониламино)-2-(2-метилпропан-2 сульфинилметил)фенокси]-4-метоксифенилуксусной кислоты (соединение 73). Получалась в соответствии с процедурой, описанной в примере 2, с использованием 3-[2-третбутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4-метоксифенилуксусной кислоты и 3 хлорпербензоевой кислоты (1 экв.). Пример 44. Синтез 3-[4-(2,2-диметилпропиониламино)-2-(2-метилпропан-2 сульфонилметил)фенокси]-4-метоксифенилуксусной кислоты (соединение 74). Получалась в соответствии с процедурой, описанной в примере 1, с использованием 3-[2-третбутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4-метоксифенилуксусной кислоты и 3 хлорпербензоевой кислоты (2 экв.). Пример 45. Синтез 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4 гидроксифенилуксусной кислоты (соединение 75). Раствор 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4 метоксифенилуксусной кислоты (0,953 г, 2,07 ммоль) в CH2Cl2 охлаждался до 0 С. К раствору добавлялся трибромид бора (1 М раствор в CH2Cl2; 6,21 мл, 6,21 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение 2 ч. Реакция гасилась и после обработки кислотой неочищенный материал очищался с помощью препаративной ВЭЖХ; в результате получалось титульное соединение. Пример 46. Синтез 3-[4-(2,2-диметилпропиониламино)-2-(2-метилпропан-2 сульфинилметил)фенокси]-4-гидроксифенилуксусной кислоты (соединение 76). Получалась в соответствии с процедурой, описанной в примере 2, с использованием 3-[2-трет-бутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4-гидроксифенилуксусной кислоты и 3-хлорпербензоевой кислоты (1 экв.). Пример 47. СинтезN-метилморфолин (0,1 мл, 0,7 ммоль) растворялись в MeCN, и реакционная смесь перемешивалась в течение ночи при комнатной температуре. Смесь концентрировалась и остаток очищался с помощью препаративной ВЭЖХ с получением бензилового эфира(2R,3R,4R,5S,6S)-6-(2-3-[2-третбутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4-метоксифенилацетокси)-3,4,5 тригидрокситетрагидропиран-2-карбоновой кислоты, который затем обрабатывался гидрохлоридом палладия на угле; полученная смесь перемешивалась в атмосфере Н 2 в течение ночи. Неочищенный материал очищался с помощью препаративной ВЭЖХ с получением (2R,3R,4R,5S,6S)-6-(2-3-[2-третбутилсульфанилметил-4-(2,2-диметилпропиониламино)фенокси]-4-метоксифенилацетокси)-3,4,5 тригидрокситетрагидропиран-2-карбоновой кислоты. Данные масс-спектроскопии (М+Н) соединений приведены в табл. 1. Пример 48. Определение CRTH2. Пример 48 а. Анализ на связывание с DP2/CRTH2. Способность соединения связываться с человеческим рецептором DP2 оценивалась с помощью анализа на связывание радиолиганда (меченого лиганда) с использованием [3 Н]PGD2. Клетки линииHEK293, стабильно экспрессирующие рекомбинантный человеческий DP2, ресуспендировались в среде с ГЭПЭС (10 ммоль/л), 7,4, содержащей 1 ммоль/л ДТТ, лизировались и центрифугировались при 75000 г для осаждения мембран. Мембраны ресуспендировались в среде ГЭПЭС (10 ммоль/л), 7,4, содержащей 1 ммоль/л ДТТ и 10% глицерола, до концентрации около 5 мг белка/мл. Мембраны (2-10 мкг на лунку) инкубировались на 96-луночных пластинах в присутствии 1 нмоль/л [3H]PGD2 и исследуемого соединения в аналитическом буфере (50 ммоль/л ГЭПЭС, 10 ммоль/л MnCl2, 1 ммоль/л ЭДТУК, 0,2% человеческого сывороточного альбумина (не обязательно), pH 7,4) в течение 60 мин при комнатной температуре. Реакции останавливались путем быстрой фильтрации через фильтровальные пластины из стекловолокнаGF/C. Фильтровальные пластины предварительно замачивались в 0,33% полиэтиленимине на 30 мин при комнатной температуре, а затем промывались промывочным буфером (50 ммоль/л ГЭПЭС, 0,5 моль/лNaCl, pH 7,4). После сбора фильтровальные пластины промывались 3 раза 1 мл холодного промывочного буфера, а затем высушивались. На пластины наносился сцинтиллянт; радиоактивность вещества, оставшегося на фильтрах, определялась с помощью аппарата Packard TopCount (Perkin Elmer). Специфическое связывание определялось как общая связанная радиоактивность минус неспецифическое связывание в присутствии 10 мкмоль/л PGD2. Значения IC50 определялись с помощью анализа кривых титрования препарата (GraphPad Prism). Исследованные с помощью данного анализа соединения характеризовались значениями IC50 менее 20 мкмоль/л. Пример 48b. Анализ GTPS. Способность соединения ингибировать связывание ГТФ с DP2 оценивалась с помощью мембранного анализа GTPS. Клетки линии CHO (яичников китайских хомяков), стабильно экспрессирующие рекомбинантный человеческий рецептор CRTH2, ресуспендировались в среде, содержащей 10 ммоль/л ГЭПЭС, 7,4, 1 ммоль/л ДТТ, лизировались и центрифугировались при 75000 г для осаждения мембран. Мембраны ресуспендировались в среде, содержащей ГЭПЭС (10 ммоль/л), pH 7,4, 1 ммоль/л ДТТ и 10% глицерола. Мембраны (12,5 мкг на лунку) инкубировались на 96-луночной пластине с 0,05 нмоль/л(50 ммоль/л ГЭПЭС, pH 7,4, 100 ммоль/л NaCl, 5 ммоль/л MgCl2 и 0,2% человеческого сывороточного альбумина) в течение 60 мин при 30 С. Реакции завершались путем быстрой фильтрации через фильтровальные пластины из стекловолокна GF/B. Фильтровальные пластины трижды промывались 1 мл холодного аналитического буфера, а затем высушивались. На пластины наносился сцинтиллянт; радиоактивность вещества, оставшегося на фильтрах, определялась с помощью аппарата Packard TopCount (PerkinElmer). Специфическое связывание определялось как общее связывание радиоактивности минус неспецифическое связывание в отсутствие лиганда (80 нмоль/л PGD2). Значения IC50 определялись с помощью анализа кривых титрования препаратов (GraphPad Prism). Пример 48 с. Анализ изменения формы эозинофилов цельной крови. Кровь забиралась у добровольцев в вакутайнер с ЭДТУК; анализ проводился в течение 1 ч от момента забора. Аликвоты крови по 98 мкл смешивались с 2 мкл исследуемого соединения (в 50% ДМСО) в полипропиленовых пробирках на 1,2 мл. Кровь перемешивалась круговыми движениями и инкубировалась при 37 С в течение 15 мин. В пробирки добавлялось по 5 мкл раствора PGD2 в ФСБ (1 мкмоль/л) до конечной концентрации 50 нмоль/л; содержимое пробирок размешивалось круговыми движениями в течение непродолжительного времени. Реакционная смесь инкубировалась в течение 5 мин (точно) при 37 С, а затем реакция останавливалась путем помещения пробирок на лед и немедленного добавления 250 мкл ледяного раствора Cytofix (BD Biosciences), разведенного в соотношении 1:4. Реакционная смесь переносилась в полистиреновые круглодонные пробирки 1275 ммоль/л, эритроциты лизировались путем добавления 3 мл лизирующего раствора хлорида аммония (150 ммоль/л NH4Cl, 10 ммоль/л KHCO3,0,1 ммоль/л динатриевой соли ЭДТУК) и пробирки инкубировались при комнатной температуре в течение 15 мин. Клетки осаждались путем центрифугирования при 1300 об/мин в течение 5 мин при 4 С и однократно промывались 3 мл ледяного ФСБ. Клетки ресуспендировались в 0,2 мл ледяного раствораCytofix (BD Biosciences), разведенного 1:4, и анализировались на приборе FACSCalibur (BD Biosciences) в течение 2 ч. Эозинофилы отбирались на основании аутофлюоресценции в канале FL2, анализировалось изменение формы клеток на 500 эозинофилах путем определения прямого и бокового рассеяния. Специфическое изменение формы, индуцированное PGD2, рассчитывалось как разница между процентным содержанием эозинофилов с высоким прямым рассеянием в присутствии и в отсутствие PGD2. ЗначенияIC50 определялись с помощью анализа кривых титрования препаратов (Graphpad Prism). Пример 48d. Анализ на связывание с DP1. Способность соединения связываться с человеческим рецептором DP1 оценивалась с помощью мембранного анализа на связывание с радиолигандом с использованием DP1-селективного синтетического лиганда [3H]BWA868C. Упакованные тромбоциты человека (Biological Specialty Corporation) ресуспендировались в 6 объемах буфера ГЭПЭС/HBSS (10 ммоль/л ГЭПЭС, 1 ммоль/л ДТТ в сбалансированном солевом растворе Хэнкса (ССРХ), лизировались и центрифугировались при 75000 г для осаждения мембран. Мембраны ресуспендировались в буфере с ГЭПЭС/ССРХ до концентрации белка около 12 мг/мл. Мембраны (20 мкг белка на лунку) инкубировались на 96-луночных пластинах в присутствии 2 нмоль/л [3H]BWA868C и исследуемого соединения в аналитическом буфере (50 ммоль/л ГЭПЭС,10 ммоль/л MnCl2, 1 ммоль/л ЭДТУК, 0,2% человеческого сывороточного альбумина (не обязательно),pH 7,4) в течение 60 мин при комнатной температуре. Реакции останавливались путем быстрой фильтрации через фильтровальные пластины из стекловолокна GF/C. Фильтровальные пластины предварительно замачивались в 0,33% полиэтиленимине на 30 мин при комнатной температуре, а затем промывались промывочным буфером (50 ммоль/л ГЭПЭС, 0,5 моль/л NaCl, pH 7,4). После сбора фильтровальные пластины промывались 3 раза 1 мл холодного промывочного буфера, а затем высушивались. На пластины наносился сцинтиллянт; радиоактивность вещества, оставшегося на фильтрах, определялась с помощью аппарата Packard TopCount (Perkin Elmer). Специфическое связывание определялось как общее связывание радиоактивности минус неспецифическое связывание в присутствии 10 мкмоль/л BW A868C. Значения IC50 определялись с помощью анализа кривых титрования препаратов (GraphPad Prism). Пример 49. Модель аллергического ринита у мышей. Способность соединения подавлять аллерген-индуцированное чихание и потирание носа оценивалась с помощью модели аллергического ринита у мышей. Приводится описание адаптированного метода,разработанного на основании оригинального метода Nakaya, M., et al. (Laboratory Investigation, 86:917926, 2006). Мыши линии BALB/c (самки массой тела 20-25 г) иммунизировались путем внутрибрюшинной инъекции 2 мкл овальбумина (OVA) в комплексе с квасцами (общий объем инокулята 0,2 мл) на ну- 28019351 левой и 14 дни. Через 7 дней (на 21 день) проводилась интраназальная провокация 20 мкл раствора OVA(10 мг/мл). Период ежедневных провокаций длился с 21 по 25 день. Мыши (по 5-7 мышей в группе) рандомизировались к введению исследуемого соединения или носителя и получали пищу через рот за 1-2 ч до каждого введения OVA. Число чиханий и потираний носа подсчитывалось независимым наблюдателем в течение 8 мин после провокации OVA на 21, 23 и 25 дни. Значительное повышение частоты аллерген-индуцированного чихания и потирания носа наблюдалось по прошествии 5-дневной провокации. Подавление этого эффекта исследуемыми соединениями определялось статистически с использованиемGraphPad Prism. Пример 50. Повышение лейкоцитоза в периферической крови у морских свинок, индуцированное внутривенным ДК-PGD2. Способность соединений ингибировать миграцию лейкоцитов in vivo оценивалась с помощью внутривенной инъекции 13,14-дигидро-15-кетопростагландина D2 (ДК-PGD2). Приводится описание адаптированного метода, разработанного на основании оригинальной методики Shichijo et al. (Journal ofPharmacology and Experimental Therapeutics, 307:518-525, 2003). Самцы морских свинок Хартли [Hartley] иммунизировались овальбумином (OVA) в нулевой день путем внутрибрюшинного введения 1 мл раствора (100 мкг/мл в Imject Alum). Затем эти особи использовались для проведения анализа (14-21 дни). Субъекты рандомизировались к введению носителя (0,5% метилцеллюлозы, 4 мл/кг перорально) или одной из трех-четырех доз исследуемого соединения. Через 2-18 ч после введения дозы животные под кетаминовым наркозом провоцировались DK-PGD2 (1 мг/кг внутривенно). Через 30 мин после внутривенного введения препарата у животных забиралась кровь из краевой ушной вены в пробирку с ЭДТУК для клеточного анализа. 10 мкл крови лизировалось в 190 мкл воды, затем раствор разводился 1:20 ФСБ. Фракция раствора (10 мкл) смешивалась с равным объемом трипана синего и загружалась в гемоцитометр. Клетки визуализировались при 40-кратном увеличении с помощью светового микроскопа LabPro; число клеток фиксировалось. Число клеток выражалось в n108 на 1 мл крови. Подавление этого эффекта исследуемыми соединениями определялось статистически с использованием GraphPad Prism. В табл. 2 указаны исследуемые соединения, значение IC50 для которых в анализе на связывание
МПК / Метки
МПК: A61K 31/167, A61K 31/10, A61P 11/00, A61P 37/08, C07C 321/20, C07C 323/57
Метки: простагландина, рецепторов, антагонисты
Код ссылки
<a href="https://eas.patents.su/30-19351-antagonisty-receptorov-prostaglandina-d2.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты рецепторов простагландина d2</a>
N,n-дизамещенные аминоалкилбифенилы в качестве антагонистов рецепторов простагландина d2

Номер патента: 18901
Опубликовано: 29.11.2013
Авторы: Волкотс Дебора, Труонг Ен Фам, Ванг Бовей, Арруда Дженни М., Хатчинсон Джон Говард, Стирнс Брайан Эндрю, Сейдерс Томас Ён, Ропп Джефри Роджер, Сток Николас Саймон, Скотт Джилл Мелисса
МПК: C07C 235/34, C07C 313/06, C07C 237/10...
Метки: рецепторов, простагландина, аминоалкилбифенилы, антагонистов, n,n-дизамещенные, качестве
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая сольгде Q представляет собой тетразолил или -C(=O)-Q1;Q1 представляет собой -ОН, -О(C1-C4-алкил), -NHSO2R12, -N(R13)2, -NH-OH или -NH-CN;каждый R1 независимо выбран из Н, F и -СН3;каждый из R2, R3, R4, R5, R6, R7 и R9 независимо представляет собой Н, галоген, -CN, -NO2, -ОН, -OR13, -SR12, -S(=O)R12, -S(=O)2R12, -NHS(=O)2R12, -C(=O)R12, -OC(=O)R12, -CO2R13, -OCO2R13, -CH(R13)2,...
Антагонисты рецепторов эндотелина

Номер патента: 952
Опубликовано: 28.08.2000
Авторы: Эллиотт Джон Дункан, Луэнго Хуан Игнасио
МПК: C07D 405/02, A61K 31/415, A61P 9/02...
Метки: рецепторов, эндотелина, антагонисты
Формула / Реферат:
1. Соединение общей формулы I в которой Z представляет собой радикал в котором R1 и R3 являются водородом, R4 является хлором или н.алкоксигруппой, R5 является группой -О-СН2-Аr, в которой Аr является фенилом, замещенным одним или двумя заместителями, выбранными из СООН, хлора, или Аr является пиридилом, замещенным СООН, R15 является низшим алкилом; Р представляет СООН; R2 представляет группу в которой Z1 и Z2 является...
Новые антагонисты мсн рецепторов

Номер патента: 15500
Опубликовано: 31.08.2011
Авторы: Грин Стивен Джеймс, Хембр Эрик Джеймс, Амегадзи Алберт Кудзови, Гардинир Кевин Мэттью, Люй Цзяньлян, Спинадзе Патрик Джанпьетро, Дао Ен, Гармен Дэвид Джозеф
МПК: C07D 495/04, A61P 3/04, A61K 31/4365...
Метки: антагонисты, рецепторов, новые, мсн
Формула / Реферат:
1. Соединение формулыгде ---- необязательно представляет собой связь, образующую двойную связь;R1 независимо выбирают из группы, включающей водород, С1-С4 алкил, галоген, С1-С4галоалкил, С1-С4 алкокси, С1-С4галоалкокси и -О-С3-С4 циклоалкил;Ra и Rb независимо представляют собой водород, фтор, хлор или метокси;R2 представляет собой водород или метил;L1 выбирают из группы, включающей связь, -ОСН2СН2-, -ОСН2СН2СН2-, -C(O)NHCH2CH2-, -C(O)CH2CH2-,...
Антагонисты рецепторов возбудительной аминокислоты

Номер патента: 1769
Опубликовано: 27.08.2001
Авторы: Вэлли Мэттью Дж., Домингес-Фернандес Кармен, Монн Джеймс А.
МПК: A61K 31/196, A61P 25/00
Метки: рецепторов, аминокислоты, антагонисты, возбудительной
Формула / Реферат:
1. Соединение формулы где Х представляет связь, S, SO или SO2; и R представляет С1-6-алкильную группу; фенильную группу, которая не замещена или замещена одним или двумя заместителями, выбранными независимо из галогена, C1-4-алкильной и C1-4-алкоксигруппы; или фенил (C1-4)-алкильную или дифенил (C1-4)-алкильную группу, в которой фенил может быть не замещен или замещен одним или двумя заместителями, выбранными независимо из атома галогена,...
Агонисты и антагонисты рецепторов 5-ht2c

Номер патента: 5820
Опубликовано: 30.06.2005
Авторы: Тор Маркус, Нильссон Бьерн, Пелькман Беньямин, Йенссон Маттиас, Тейбрант Ян, Рингберг Эрик, Нильссон Йонас
МПК: C07D 241/18, A61P 25/00, A61K 31/497...
Метки: рецепторов, агонисты, 5-ht2c, антагонисты
Формула / Реферат:
1. Соединение общей формулы (Ib) где X1 представляет -O-, -S- или -N(R27)-; Y1 представляет -O-, -S-, -N(R27)- или -CH2-; Ra представляет C3-8-циклоалкил, арил или гетероарил, каждый из которых может быть, необязательно, замещен в одном или в нескольких положениях независимо друг от друга C1-6-алкилом, C1-6-алкокси, фтор-C1-6-алкокси, 2,2,2-трифторэтокси, C3-5-алкинилокси, C3-5-алкенилокси, диметиламино-C1-6-алкокси, метиламино-C1-6-алкокси,...
Предыдущий патент: Производные хиназолина как модуляторы raf-киназы и способы их применения
Следующий патент: Способ модификации некрахмального углеводного материала
Случайный патент: Ингаляционное устройство для транспульмонального введения