Ингибиторы протеинкиназы, фармацевтический состав на их основе и применение в терапии
Номер патента: 18808
Опубликовано: 30.10.2013
Авторы: Санчес Мартинес Консепсьен, Перес Мартинес Карлос, Де Диос Магана Альфонсо, Мартин Ортега Фингер Мария Долорес, Де Прадо Гонсалес Ана, Мартинес Перес Хосе Антонио, Гельберт Лоуренс Марк, Матео -Эрранс Ана Исабель, Дель Прадо Каталина Мириам Филадельфа, Коутс Дэвид Эндрю, Гарсия Паредес Мария Кристина, Нобелок Джон Монте, Мартин Де Ла Нава Эва Мария







Формула / Реферат
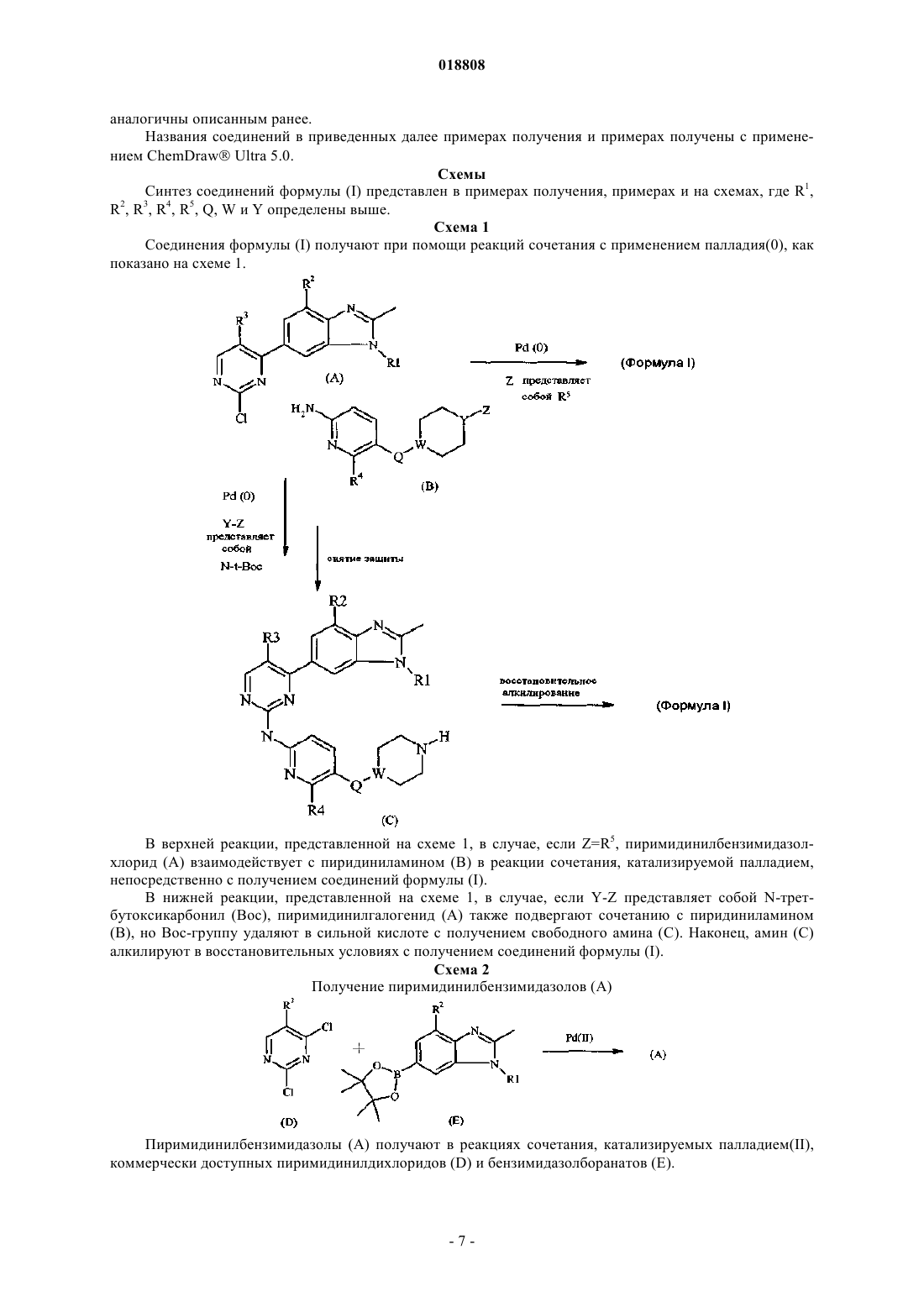
1. Соединение формулы

где R1 представляет собой C3-C5-алкил, C3-C5-циклоалкил или циклопропилметил;
R2 и R3 представляют собой Н или фтор, причем по меньшей мере один из R2 или R3 представляет собой фтор;
R4 представляет собой Н или СН3;
R5 представляет собой C1-C6-алкил или -NR6R7, причем R6 и R7 представляют собой C1-C3-алкил;
Q представляет собой СН2, О, S или прямую связь;
W и Y представляют собой С или N, причем по меньшей мере один из W или Y представляет собой N, при этом если Q представляет собой О или S, то W представляет собой С,
или его фармацевтически приемлемая соль.
2. Соединение по п.1 или его фармацевтически приемлемая соль, в котором R1 представляет собой изопропил, циклопропил, циклопентил или циклопропилметил.
3. Соединение по любому из предыдущих пунктов или его фармацевтически приемлемая соль, в котором каждый из R2 и R3 представляет собой фтор.
4. Соединение по любому из предыдущих пунктов или его фармацевтически приемлемая соль, в котором R4 представляет собой Н.
5. Соединение по любому из предыдущих пунктов или его фармацевтически приемлемая соль, в котором R5 представляет собой C1-C3-алкил.
6. Соединение по любому из предыдущих пунктов или его фармацевтически приемлемая соль, в котором Q представляет собой СН2.
7. Соединение по любому из предыдущих пунктов или его фармацевтически приемлемая соль, в котором W представляет собой N.
8. Соединение по любому из предыдущих пунктов или его фармацевтически приемлемая соль, выбранное из группы, состоящей из






9. Соединение по любому из предыдущих пунктов, которое представляет собой

или его фармацевтически приемлемая соль.
10. Соединение по любому из предыдущих пунктов, которое представляет собой мезилатную соль.
11. Кристаллическая форма III [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3Н-бензимидазол-5-ил)пиримидин-2-ил]амина, характеризующаяся порошковой дифракционной рентгенограммой (излучение CuKα, l=1,54056 Å), содержащей пик при 21,29 (2q±0,1°) и, необязательно, один или более пиков, выбранных из группы, содержащей 11,54, 10,91 и 12,13 (2q±0,1°).
12. Кристаллическая форма III [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3Н-бензимидазол-5-ил)пиримидин-2-ил]амина по п.11, которая дополнительно характеризуется спектром 13С ЯМР, содержащим пики с химическими сдвигами n(F1) [ppm] при 112,7, 127,3 и 129,4.
13. Фармацевтический состав, ингибирующий активность CDK4/6 или Pim-1 киназ, содержащий соединение по любому из пп.1-12 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель.
14. Фармацевтический состав, ингибирующий активность CDK4/6 или Pim-1 киназ, содержащий соединение по любому из пп.1-12 или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем и, при необходимости, в сочетании с другими терапевтическими ингредиентами.
15. Применение соединения по любому из пп.1-12 или его фармацевтически приемлемой соли для лечения заболевания или нарушения, характеризующегося аномальной пролиферацией клеток.
16. Применение соединения по любому из пп.1-12 или его фармацевтически приемлемой соли для лечения рака, выбранного из группы, состоящей из колоректального рака, рака груди, рака легких, рака простаты, глиобластомы, лимфомы из клеток мантийной зоны, хронической миелоидной лейкемии и острой миелоидной лейкемии.
Текст
ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ В изобретении предложено соединение формулы (I) Коутс Дэвид Эндрю, Де Диос Магана Альфонсо (US), Де Прадо Гонсалес Ана, Дель Прадо Каталина Мириам Филадельфа, Гарсия Паредес Мария Кристина (ES), Гельберт Лоуренс Марк, Нобелок Джон Монте (US),Мартин Де Ла Нава Эва Мария,Мартин Ортега Фингер Мария Долорес, Мартинес Перес Хосе Антонио, Матео Эрранс Ана Исабель,Перес Мартинес Карлос, Санчес Мартинес Консепсьен (ES) Медведев В.Н. (RU) или его фармацевтически приемлемая соль, фармацевтический состав на его основе,ингибирующий активность CDK4/6 или Pim-1 киназ, а также применение указанного соединения для лечения заболевания или нарушения, характеризующегося аномальной пролиферацией клеток.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Высокогомологичные циклинзависимые киназы (Cdk) CDK4 и CDK6 в комбинации с циклином D представляют собой ключевые регуляторы перехода через точку рестрикции R между фазами G1 (рост) иS (репликация ДНК) клеточного цикла. CDK4/6 осуществляют свое действие за счет фосфорилирования белка ретинобластомы (pRb). В результате фосфорилирования pRb теряет ингибирующее действие на транскрипцию генов, способствуя вхождению в фазу S. Напротив, специфическое ингибирование активности CDK4/6-киназы при помощи эндогенного белкового модулятора p16INK4 или низкомолекулярных ингибиторов приводит к получению гипофосфорилированного pRb и остановке развития клеток в точке рестрикции G1. Как основной механизм регулирования точки рестрикции G1 регулируемый указанными киназами путь подвергается изменению при широком спектре опухолей у человека, и, таким образом, ингибирование CDK4/CDK6 в указанных опухолях обладает терапевтическим преимуществом, заключающимся в предотвращении деления клеток.Pim-1 представляет собой серин/треонинкиназу, которая регулирует различные биологические функции,включая прохождение клеточного цикла, пути транскрипционной/сигнальной трансдукции и апоптоз, и функции, экспрессию которых связывают с различными раковыми заболеваниями, включая гематологические опухоли, опухоли простаты и полости рта (Bachmann, M. and Т. Moroy, Int. J. Biochem. Cell Biol.,2005, 37(4): p. 726-30). Ингибиторы киназ известны в данной области техники. В WO 98/11095 предложен ряд замещенных 2-пиридинаминов, которые охарактеризованы как ингибиторы киназы, в частностиp56lck, ZAP-70 киназ и протеинкиназы С. В WO 98/11095 не описано ингибирование Cdk. В WO 03/062236 предложен ряд 2-(пиридин-2-иламино)пиридо[2,3-d]пиримидин-7-онов, охарактеризованных как обладающие ингибирующей активностью в отношении CDK4/6. Указанные соединения охарактеризованы как подходящие для лечения нарушений клеточной пролиферации, таких как рак и рестеноз. Однако указанные соединения плохо растворяются в водном растворе и не проявляют заметной ингибирующей активности в отношении других (отличных от Cdk) мишеней-киназ. По-прежнему существует необходимость в создании ингибиторов CDK4/6, которые можно применять для лечения клеточных пролиферативных нарушений, таких как рак. В настоящем изобретении предложены ингибиторы CDK4/6. Конкретные соединения согласно настоящему изобретению являются более сильными ингибиторами CDK4/6 по сравнению с конкретными соединениями, известными в данной области техники. Дополнительно, существует необходимость в обеспечении ингибиторов CDK4/6, которые являются селективными для CDK4/6 по сравнению с другими Cdk и способны, таким образом, вызывать специфическую блокировку G1 при их присутствии в фармакологически значимых концентрациях. В настоящем изобретении предложены ингибиторы CDK4/6, которые способны вызывать специфическую блокировкуG1 при их присутствии в фармакологически значимых концентрациях. Также по-прежнему существует необходимость в обеспечении ингибиторов CDK4/6 с улучшенной растворимостью в водном растворе. Конкретные соединения согласно настоящему изобретению обладают улучшенной растворимостью в водном растворе по сравнению с конкретными соединениями, известными в данной области техники. Также существует необходимость в обеспечении ингибиторов CDK4/6, которые обладают улучшенным распространением в мозговую ткань и которые можно, таким образом, применять для лечения нарушений, протекающих в мозгу, например первичных или метастатических опухолей мозга. Конкретные соединения согласно настоящему изобретению обладают улучшенным распространением в мозговую ткань. Также существует необходимость в обеспечении ингибиторов CDK4/6 с хорошими фармакокинетическими свойствами, такими как пероральная доступность. Конкретные соединения согласно настоящему изобретению обладают улучшенной пероральной доступностью по сравнению с конкретными соединениями, известными в данной области техники. Дополнительно, существует необходимость в обеспечении ингибиторов киназы, которые обладают вторичной ингибирующей активностью в отношении других не-Cdks киназ, например Pim-1-киназы. Конкретные соединения согласно настоящему изобретению обладают двойной ингибирующей активностью в отношении CDK4/6 и Pim-1 киназ. В настоящем изобретении предложены соединения формулыR2 и R3 представляют собой Н или фтор, причем по меньшей мере один из R2 и R3 представляет собой фтор;Q представляет собой СН 2, О, S или прямую связь;W и Y представляют собой С или N, причем по меньшей мере один из W или Y представляет собойN и в случае если Q представляет собой О или S, то W представляет собой С; или их фармацевтически приемлемые соли. В настоящем изобретении предложен фармацевтический состав, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель. В настоящем изобретении предложено соединение согласно настоящему изобретению или его фармацевтически приемлемая соль для применения в терапии. В настоящем изобретении предложено соединение согласно настоящему изобретению или его фармацевтически приемлемая соль для применения при лечении рака, в частности, раковых заболеваний,выбранных из группы, состоящей из колоректального рака, рака груди, рака легких, в частности немелкоклеточного рака легких (НМРЛ), рака простаты, глиобластомы, лимфомы из клеток мантийной зоны(MCL), хронической миелоидной лейкемии (ХМЛ) и острой миелоидной лейкемии (ОМЛ). В настоящем изобретении также предложен способ лечения рака, выбранного из группы, состоящей из колоректального рака, рака груди, рака легких, в частности немелкоклеточного рака легких (НМРЛ),рака простаты, глиобластомы, лимфомы из клеток мантийной зоны, хронической миелоидной лейкемии и острой миелоидной лейкемии у млекопитающего, включающий введение млекопитающему, нуждающемуся в подобном лечении, эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Дополнительно, в настоящем изобретении предложено применение соединения согласно настоящему изобретению или его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака, в частности рака, выбранного из группы, состоящей из колоректального рака, рака груди, рака легких, в частности немелкоклеточного рака легких (НМРЛ), рака простаты, глиобластомы,лимфомы из клеток мантийной зоны, хронической миелоидной лейкемии и острой миелоидной лейкемии. Кроме того, в настоящем изобретении предложен фармацевтический состав для применения в терапии, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель. В изобретении также предложен фармацевтический состав для лечения колоректального рака, рака груди, рака легких, в частности немелкоклеточного рака легких (НМРЛ), рака простаты, глиобластомы, лимфомы из клеток мантийной зоны, хронической миелоидной лейкемии и острой миелоидной лейкемии, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель. Общие химические термины, используемые в формулах выше, имеют свои обычные значения. Например, термин "C3-C5-алкил" относится к линейной или разветвленной одновалентной насыщенной цепи, содержащей от 3 до 5 атомов углерода, и включает, но не ограничивается ими, н-пропил, изопропил,н-бутил, изобутил, втор-бутил и трет-бутил. Термин "C3-C5-циклоалкил" относится к насыщенной углеродной циклической системе, содержащей от 3 до 5 атомов углерода. Специалисту в данной области будет понятно, что большинство или все соединения согласно настоящему изобретению способны образовывать соли. Соединения согласно настоящему изобретению представляют собой амины и, соответственно, взаимодействуют с любыми неорганическими или органическими кислотами с образованием фармацевтически приемлемых солей присоединения кислот. Указанные фармацевтически приемлемые соли присоединения кислот и общие способы их получения хорошо известны в данной области техники. См., например, P. Stahl et al., Handbook of Pharmaceutical salts:Sciences, Vol. 66, No. l, January 1977. Предпочтительными являются гидрохлоридная и мезилатная соли. Мезилатная соль является особенно предпочтительной. Предпочтительно настоящее изобретение включает соединения формулы (I), где R1 представляет собой изопропил, циклопропил, циклопентил или циклопропилметил. Более предпочтительно R1 представляет собой изопропил. Предпочтительно настоящее изобретение включает соединения формулы (I), где R2 представляет собой фтор, a R3 представляет собой водород. Предпочтительно настоящее изобретение включает соединения формулы (I), где R2 представляет собой водород, a R3 представляет собой фтор. Более предпочтительно оба R2 и R3 представляют собой фтор. Предпочтительно настоящее изобретение включает соединения формулы (I), где R4 представляет собой водород. В качестве альтернативы, R4 предпочтительно представляет собой метил. Наиболее предпочтительно R4 представляет собой водород. Предпочтительно настоящее изобретение включает соединения формулы (I), где R5 представляет собой C1-C3-алкил или -NR6R7, где R6 и R7 представляют собой C1-C3-алкил. Более предпочтительно R6 иR7 представляют собой этил. Более предпочтительно R5 представляет собой C1-C3-алкил. Наиболее предпочтительно R5 представляет собой этил. Предпочтительно настоящее изобретение включает соединения формулы (I), где Q представляет собой СН 2 или прямую связь. Наиболее предпочтительно Q представляет собой СН 2. Предпочтительно настоящее изобретение включает соединения формулы (I), где Y представляет собой N. Предпочтительно настоящее изобретение включает соединения формулы (I), где W представляет собой N. Предпочтительно настоящее изобретение включает соединения формулы (I), где W и Y представляют собой N. Предпочтительные соединения согласно настоящему изобретению включают соединения формулыQ представляет собой СН 2, О или прямую связь;W представляет собой С или N, причем если Q представляет собой О, то W представляет собой С; или их фармацевтически приемлемые соли. Особенно предпочтительными являются соединения, примеры которых приведены в настоящем описании, или их фармацевтически приемлемые соли. Конкретно, более предпочтительным является соединение [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3Hбензимидазол-5-ил)пиримидин-2-ил]амин или его фармацевтически приемлемая соль.[5-(4-Этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3Hбензимидазол-5-ил)пиримидин-2-ил]амин, как альтернатива может быть назван как N-[5-[(4-этил-1 пиперазинил)метил]-2-пиридинил]-5-фтор-4-[4-фтор-2-метил-1-(1-метилэтил)-1H-бензимидазол-6-ил]-2 пиримидинамин. Особенно предпочтительной является кристаллическая форма III [5-(4-этилпиперазин-1 илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2 ил]амина, характеризующаяся порошковой дифракционной рентгенограммой (излучение CuK,=1,54056 ), содержащей пик при 21,29 (260,1) и, необязательно, один или более пиков, выбранных из группы, включающей 11,54, 10,91 и 12,13 (20,1). Кристаллическая форма III [5-(4-этилпиперазин-1 илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2 ил]амина может быть дополнительно охарактеризована спектром 13 С ЯМР, содержащим пики с химическими сдвигами (F1) [ppm] при 112,7, 127,3 и 129,4. Соединения согласно настоящему изобретению представляют собой специфические ингибиторыCDK4 и CDK6 и, таким образом, подходят для лечения заболевания или нарушения, характеризующегося аномальной пролиферацией клеток. В частности, соединения согласно настоящему изобретению подходят для лечения рака.CDK4 и CDK6 модулируют свое воздействие на клеточный цикл за счет фосфорилирования pRb. Полагают, что соединения согласно настоящему изобретению, которые являются сильными ингибиторами активности CDK4/6 и, таким образом, фосфорилирования pRb, ингибируют пролиферацию клеток(следовательно, и рост опухоли) при любом типе рака, при котором клетки являются пролиферирующими и содержат функциональный интактный ген Rb1 (который кодирует pRb). Соединения согласно настоящему изобретению, таким образом, подходят для лечения pRb+ раковых заболеваний, таких как ко-3 018808 лоректальный рак, рак груди, рак легких, рак простаты, хроническая миелоидная лейкемия, острая миелоидная лейкемия (Fry, D.W. et al. Mol. Cancer Ther. (2004), 3(11), 1427), лимфома из клеток мантийной зоны (Marzek, M. et al., Blood (2006), 108(5), 1744), рак яичников (Kim, T.M. et al., Cancer Research (1994),54, 605), рак поджелудочной железы (Schutte, M. et al., Cancer Research (1997), 57, 3126), злокачественная меланома и метастатическая злокачественная меланома (Maelandsmo, G.M. et al., British Journal of Cancer(1996), 73, 909) у млекопитающих. Также полагают, что соединения согласно настоящему изобретению подходят для лечения рабдомиосаркомы (Saab, R. et al., Mol. Cancer Ther. (2006), 5(5), 1299) и множественной миеломы (Baughn, L.B. et al., Cancer Res. (2006), 66(15), 7661) у млекопитающих. Предпочтительно млекопитающее, нуждающееся в лечении, представляет собой человека. Дополнительно, предпочтительные соединения согласно настоящему изобретению обладают преимущественным свойством, заключающимся в том, что указанные соединения обладают улучшенным распространением в мозговую ткань. Например, при введении крысам в модели заболевания на крысах отношение распределения в мозге:плазме соединения согласно примеру 16 (определенное с применением площади под кривой (AUC) или максимальных концентрациях в плазме и мозге (Cmax), см. табл. 6 с) составляет примерно 1, что показывает, что соединение согласно примеру 16 хорошо распространяется в мозг. С другой стороны, авторы настоящего изобретения определили, что предпочтительное соединение,представленное в WO 03/062236 (6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-илпиридин-2 иламино)-8H-пиридо[2,3-d]пиримидин-7-он), обладает отношением распределения в мозге:плазме, равным 0,17 (AUC) и 0,1 (Cmax), что показывает, что соединение сравнительно плохо распространяется в мозговую ткань в данной модели. Предпочтительные соединения согласно настоящему изобретению,таким образом, способны проникать в мозг и, таким образом, подходят для лечения первичных и метастатических опухолей мозга, в которых клетки являются пролиферирующими и содержат функциональный интактный ген Rb1. Примеры указанных pRb+ опухолей мозга включают глиобластому, а также медуллобластому и астроцитому (Lee, W.-H. et al., Science (1987), 235, 1394). Темозоломид представляет собой цитотоксический алкилирующий ДНК агент, применяемый для лечения опухолей мозга, включая глиобластому и астроцитому (Freedman, H.S., 2000, Clin. Cancer Res. 6(7): 2585-97), включая метастазы в мозг, вызванные меланомой, раком груди и НМРЛ (Siena, S. et al., 2009 Annals of Oncology,doi:10.1093/annonc/mdp343). Темозоломид взаимодействует с ДНК, вызывая химическую модификацию/повреждение (Marchesi et al., 2007, Pharmacol. Res. 56(4): 275-87). Соединения согласно настоящему изобретению можно применять в комбинации с темозоломидом для лечения первичных и метастатических pRb+ опухолей мозга, таких как глиобластома и астроцитома, например, в случае, если указанные метастазы вызваны меланомой, раком груди или НМРЛ. Гемцитабин HCl, аналог нуклеозида, который проявляет противоопухолевую активность, представляет собой моногидрохлорид 2'-дезокси-2',2'-дифторцитидина (-изомер), также известный как моногидрохлорид 2',2'-дифтор-2'-дезоксицитидина или как 1-(4-амино-2-оксо-1 Н-пиримидин-1-ил)-2-дезокси 2',2'-дифторрибоза. Гемцитабин HCl описан в патенте США 5464826. Структурная формула представлена ниже Гемцитабин HCl является эффективным для лечения немелкоклеточного рака легких (НМРЛ)(Sandler, A. and Ettinger, D.S., 1999, The Oncologist, 4, 241), рака поджелудочной железы (Pino, S.M. et al.,2004, Current Gastroenterology Reports, 6, 119), рака яичников (Pfisterer, J. et al., 2006, Journal of ClinicalOncology, 24(29, 4699 и метастатического рака груди (Chan, S. et al., 2009, Journal of Clinical Oncology,27(11, 1753. Соединения согласно настоящему изобретению можно применять в комбинации с гемцитабином HCl для лечения НМРЛ, рака поджелудочной железы, рака яичников и метастатического рака груди. Соединения согласно настоящему изобретению можно применять в способе лечения рака, в частности раковых заболеваний, описанных выше, у млекопитающего, включающем введение млекопитающему, нуждающемуся в подобном лечении, эффективного количества соединения согласно настоящему изобретению. Согласно предпочтительному варианту реализации соединения согласно настоящему изобретению можно применять в способе лечения рака, выбранного из группы, состоящей из колоректального рака, лимфомы из клеток мантийной зоны, рака груди, глиобластомы, острой миелоидной лейкемии и рака легких, в частности НМРЛ. Согласно другому предпочтительному варианту реализации соединения согласно настоящему изобретению можно применять в способе лечения рака, выбранного из группы,состоящей из колоректального рака, глиобластомы, острой миелоидной лейкемии и рака легких. Согласно другому предпочтительному варианту реализации соединение согласно настоящему изобретению можно применять в способе лечения глиобластомы или астроцитомы у млекопитающего, включающем введение млекопитающему, нуждающемуся в подобном лечении, терапевтически эффективной комбина-4 018808 ции соединения согласно настоящему изобретению и темозоломида. Согласно другому предпочтительному варианту реализации соединение согласно настоящему изобретению можно применять в способе лечения НМРЛ, рака поджелудочной железы, рака яичников или метастатического рака груди у млекопитающего, включающем введение млекопитающему, нуждающемуся в подобном лечении, терапевтически эффективной комбинации соединения согласно настоящему изобретению и гемцитабина HCl. Соединения согласно настоящему изобретению можно применять для лечения рака, в частности раковых заболеваний, описанных выше. Согласно одному из предпочтительных вариантов реализации соединения согласно настоящему изобретению можно применять для лечения рака, выбранного из группы,состоящей из колоректального рака, лимфомы из клеток мантийной зоны, рака груди, глиобластомы,острой миелоидной лейкемии и рака легких, в частности НМРЛ. Согласно другому предпочтительному варианту реализации соединения согласно настоящему изобретению можно применять для лечения рака,выбранного из группы, состоящей из колоректального рака, глиобластомы, острой миелоидной лейкемии и рака легких. Согласно другому предпочтительному варианту реализации в изобретении предложено соединение согласно настоящему изобретению для применения одновременно, раздельно или последовательно в комбинации с темозоломидом для лечения глиобластомы или астроцитомы. Согласно другому предпочтительному варианту реализации в изобретении предложено соединение согласно настоящему изобретению для применения одновременно, раздельно или последовательно в комбинации с гемцитабином HCl для лечения НМРЛ, рака поджелудочной железы, рака яичников или метастатического рака груди. Кроме того, соединения согласно настоящему изобретению можно применять для получения лекарственного средства для лечения рака, в частности раковых заболеваний, описанных выше. Согласно предпочтительному варианту реализации соединения согласно настоящему изобретению можно применять для получения лекарственных средств для лечения рака, выбранного из группы, состоящей из колоректального рака, лимфомы из клеток мантийной зоны, рака груди, глиобластомы, острой миелоидной лейкемии и рака легких, в частности НМРЛ. Согласно другому предпочтительному варианту реализации соединения согласно настоящему изобретению можно применять для получения лекарственного средства для лечения рака, выбранного из группы, состоящей из колоректального рака, глиобластомы, острой миелоидной лейкемии и рака легких. Согласно другому предпочтительному варианту реализации в изобретении предложено применение соединения согласно настоящему изобретению для получения лекарственного средства для лечения глиобластомы или астроцитомы, где лекарственное средство также содержит темозоломид или лекарственное средство следует вводить одновременно, раздельно или последовательно с темозоломидом. Согласно другому предпочтительному варианту реализации в настоящем изобретении предложено применение соединения согласно настоящему изобретению для получения лекарственного средства для лечения НМРЛ, рака поджелудочной железы, рака яичников или метастатического рака груди, где лекарственное средство также содержит гемцитабин HCl или лекарственное средство следует вводить одновременно, раздельно или последовательно с гемцитабином HCl. Также предложен фармацевтический состав для лечения рака, в частности раковых заболеваний,описанных выше, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем. Согласно предпочтительному варианту реализации также предложен фармацевтический состав для лечения рака, выбранного из группы, состоящей из колоректального рака, лимфомы из клеток мантийной зоны, рака груди, глиобластомы,острой миелоидной лейкемии и рака легких, в частности НМРЛ, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем. Согласно предпочтительному варианту реализации также предложен фармацевтический состав для лечения рака, выбранного из группы, состоящей из колоректального рака, глиобластомы, острой миелоидной лейкемии и рака легких, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем. Согласно другому варианту реализации в изобретении предложен фармацевтический состав для лечения глиобластомы или астроцитомы, содержащий соединение согласно настоящему изобретению и темозоломид совместно с фармацевтически приемлемым носителем. Согласно другому предпочтительному варианту реализации в изобретении предложен фармацевтический состав для лечения НМРЛ, рака поджелудочной железы, рака яичников или метастатического рака груди, содержащий соединение согласно настоящему изобретению и гемцитабин HCl совместно с фармацевтически приемлемым носителем. В изобретении также предложен фармацевтический состав, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и темозоломид совместно с фармацевтически приемлемым носителем, разбавителем или наполнителем. В изобретении также предложен фармацевтический состав, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и гемцитабин HCl совместно с фармацевтически приемлемым носителем, разбавителем или наполнителем. В изобретении также предложен фармацевтический состав, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль совместно с фармацевтически при-5 018808 емлемым носителем и, необязательно, другими терапевтическими ингредиентами. Далее, предпочтительны соединения, примеры которых приведены в настоящем описании, также являющиеся ингибиторами Pim-1. Как указано выше, Pim-1 представляет собой серин/треонин киназу,которая участвует в регулировании различных биологических функций, включающих прохождение клеточного цикла, пути транскрипционной/сигнальной трансдукции и апоптоз, и функций, экспрессию которых связывают с некоторыми раковыми заболеваниями. В частности, показано, что ингибированиеPim-1 низкомолекулярным ингибитором K00135 уменьшает выживаемость и клоногенный рост группы клеток острой лейкемии (Pogacic, V., et al., Cancer Res. (2007), 67(14), p. 6916-24). Дополнительно показано, что Pim-1 экспрессируется в неоинтимах у крыс с повреждением стенок сонных артерий балонным катетером и человеческих грудных аортах и коронарных артериях с утолщением интимы. Также специфическое ингибирование функции Pim-1 значительно подавляло образование неоинтимы после повреждения балонным катетером, а также пролиферацию культивированных клеток гладкой мускулатуры сосудов (VSMC), что позволяло сделать предположение о том, что Pim-1 играет важную роль в пролиферации указанных клеток. Пролиферация VSMC значительно осложнялась при патогенезе окклюзионных сосудистых заболеваний, таких как атеросклероз и рестеноз, и, таким образом, полагают, что ингибирование Pim-1 подавляет пролиферацию VSMC и, следовательно, подходит для лечения окклюзионных сосудистых заболеваний (Katakami N., et al., JBC (2004), 279(52), 54742-54749). Соответственно, предпочтительные соединения согласно настоящему изобретению или их фармацевтически приемлемые соли можно применять в способе лечения окклюзионных сосудистых заболеваний, таких как атеросклероз или рестеноз, у млекопитающего, включающем введение млекопитающему,нуждающемуся в подобном лечении, эффективного количества соединения согласно настоящему изобретению. Предпочтительные соединения согласно настоящему изобретению или их фармацевтически приемлемые соли можно применять для лечения окклюзионного сосудистого заболевания, такого как атеросклероз или рестеноз. Кроме того, предпочтительные соединения согласно настоящему изобретению или их фармацевтически приемлемые соли можно применять для получения лекарственного средства для лечения окклюзионного сосудистого заболевания, такого как атеросклероз или рестеноз. Также предложен фармацевтический состав для лечения окклюзионного сосудистого заболевания, такого как атеросклероз или рестеноз, содержащий предпочтительное соединение согласно настоящему изобретению или его фармацевтически приемлемую соль. Используемые в настоящем описании термины обозначают следующее:"ч" - час или часы,"мин" - минута или минуты,"CDK" - циклинзависимая киназа,"pRb" - белок ретинобластомы,"MCL" - лимфома из клеток мантийной зоны,"ОМЛ" - острая миелоидная лейкемия,"Boc" - N-трет-бутоксикарбонил,"ЭА" - этилацетат,"ДХМ" - дихлорметан,"ДМСО" - диметилсульфоксид,"ДМА" - диметилацетамид,"ТГФ" - тетрагидрофуран,"МТБЭ" - метил-трет-бутиловый эфир,"ТЭА" - триэтиламин,"ЭБС" - эмбриональная бычья сыворотка,"PBS" - фосфатный буферный солевой раствор,"БСА" - альбумин бычьей сыворотки,"КТ" - комнатная температура,"мг/кг" - миллиграммам на килограмм,"ро" - пероральный,"qd" - один раз в день,"ВЭЖХ" - высокоэффективная жидкостная хроматография,"q2d" - введение одной дозы каждые 2 дня,"q2dx10" - введение одной дозы каждые 2 дня в течение 10 дней,"VSMC" - клетка гладкой мускулатуры сосудов и"XRD" - дифракционная рентгенограмма. Соединения формулы (I) могут быть получены специалистом в данной области техники при помощи известных в данной области техники способов и методик. Более конкретно, соединения формулы (I) могут быть получены в соответствии с представленными схемами, способами и примерами, приведенными далее. Для специалиста в данной области техники очевидно, что для получения соединений формулы (I) индивидуальные стадии представленных далее схем могут отличаться. Реагенты и исходные вещества легко доступны специалисту в данной области техники. Все заместители, если не указано иное,-6 018808 аналогичны описанным ранее. Названия соединений в приведенных далее примерах получения и примерах получены с применением ChemDraw Ultra 5.0. Схемы Синтез соединений формулы (I) представлен в примерах получения, примерах и на схемах, где R1,R2, R3, R4, R5, Q, W и Y определены выше. Схема 1 Соединения формулы (I) получают при помощи реакций сочетания с применением палладия(0), как показано на схеме 1. В верхней реакции, представленной на схеме 1, в случае, если Z=R5, пиримидинилбензимидазолхлорид (А) взаимодействует с пиридиниламином (В) в реакции сочетания, катализируемой палладием,непосредственно с получением соединений формулы (I). В нижней реакции, представленной на схеме 1, в случае, если Y-Z представляет собой N-третбутоксикарбонил (Boc), пиримидинилгалогенид (А) также подвергают сочетанию с пиридиниламином(В), но Вос-группу удаляют в сильной кислоте с получением свободного амина (С). Наконец, амин (С) алкилируют в восстановительных условиях с получением соединений формулы (I). Схема 2 Получение пиримидинилбензимидазолов (А) Бензимидазолборанаты (Е) получают в результате катализируемого Pd(II) боранилирования бромида бензимидазолов (Н) при помощи бис-(пинаколато)диборана. Бензимидазолы (Н), в свою очередь, получают при помощи циклизации амидинов (F) с т-бутоксидом калия или конденсации бензолдиаминов(G) с триэтилортоацетатом/уксусной кислотой. Амидины (F) получают при помощи известной специалисту в области органического синтеза конденсации 4-бром-2,6-дифторфениламина с моноацетамидным производным аминов R1-NH2 в присутствии хлорокиси фосфора. Бензолдиамины (G) получают в две стадии при помощи известного специалисту в области органического синтеза замещения брома в положении 2 2,4-дибромнитробензола аминомR1-NH2 с последующим восстановлением нитрогруппы до аминогруппы. Схема 4 Получение пиридиниламинов (В), где Q представляет собой S или О и W представляет собой С Синтез пиридиниламинов (В), где Q представляет собой S или О и W представляет собой С, проводят при помощи замещения 5-галогенида в пиридине (I) при помощи коммерчески доступного тиола или спирта (J). В случае, если необходимо применение нитропиридина (I), продукт замещения дополнительно подвергают стадии восстановления нитрогруппы с получением (В). Следует отметить, что соединения(I) являются универсальными реагентами в указанных схемах, но только некоторые из них коммерчески доступны как пиридиламины, а некоторые как нитропиридины. Коммерчески доступные соединения (I),тем не менее, способны подвергаться химическому превращению в результате реакций окисления амина или восстановления нитрогруппы, известных в данной области техники, для применения в представленных здесь и ниже схемах. Схема 5 Получение пиридиниламинов (В), где Q представляет собой СН 2 Синтез пиридиниламинов (В), где Q представляет собой СН 2, осуществляют двумя способами: 1) коммерчески доступные карбальдегиды (K) подвергают восстановительному аминированию со свободным амином (L) с последующим замещением пиридинбромида в результате катализируемогоPd(0) аминирования 1,1,1,3,3,3-гексаметилдисилазаном лития или жидким аммиаком и оскидом меди; 2) коммерчески доступный 1,1-диметилэтиловый эфир 4-метиленпиперидин-1-карбоновой кислотыPd(II). Схема 6 Получение пиридиниламинов (В), где Q представляет собой прямую связь Синтез пиридиниламинов (В), где Q представляет собой прямую связь, осуществляют двумя способами: 1) коммерчески доступный 1,1-диметилэтиловый эфир 3,6-дигидро-4-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)пиридин-1(2H)-карбоновой кислоты (N) подвергают сочетанию с нитропиридином(I) в присутствии палладия(II) с последующим восстановлением нитрогруппы и двойной связи; 2) бромид нитропиридина (I) замещают свободным амином (L) с последующим восстановлением нитрогруппы. Пример получения 1. трет-Бутиловый эфир 4-(6-аминопиридин-3-илсульфанил)пиперидин-1-карбоновой кислоты. К смеси 2,9-диметил-1,10-фенантролина (76,52 мг), йодида меди(I) (69,27 мг), трет-бутоксида натрия (475,59 мг), трет-бутилового эфира 4-меркаптопиперидин-1-карбоновой кислоты (583,5 мг), магния(49,10 мг) и 2-амино-5-йодпиридина (550 мг) добавляли осушенный толуол (6,06 мл). Через смесь барботировали азот с применением ультразвука и перемешивали суспензию при 110C в запаянной трубке в течение 24 ч. Охлаждали и фильтровали через целит. Промывали толуолом и удаляли растворитель в вакууме. Добавляли гексан/ЭА (1/1) и фильтровали через подушку целит/силикагель, дважды промывали гексаном/ЭА (1/1), затем ЭА. Удаляли растворитель в вакууме. Очищали при помощи колоночной хроматографии на силикагеле, элюируя смесями гексан/ЭА (50-75%), с получением 630 мг указанного в заголовке соединения. МС (ES+): m/z=310 (M+H)+. Пример получения 2. 5-Фтор-2-нитропиридин. К серной кислоте (46 мл) при 0C на открытом воздухе добавляли 25% пероксид водорода(26,98 мл). Через 5 мин через капельную воронку по каплям добавляли холодный раствор 2-амино-5-фторпиридина (9 г) в концентрированной серной кислоте (46 мл). Перемешивали полученный темный раствор при температуре от 0C до КТ на бане в течение ночи. Выливали в 200 мл смеси ледвода и экстрагировали ДХМ. Промывали объединенные органические слои 5% водным раствором бисульфита натрия и сушили над безводным сульфатом натрия. Удаляли растворитель в вакууме и очищали при помощи колоночной хроматографии на силикагеле, элюируя ДХМ, с получением 7,5 г указанного в заголовке соединения. МС (ES+): m/z=143 (M+H)+. Представленное далее соединение получали, по существу, в соответствии с описанием получения 5-фтор-2-нитропиридина с применением соответствующего амина. Пример получения 4. 1-Изопропил-4-(2-метил-6-нитропиридин-3-ил)пиперазин. Перемешивали 3-бром-2-метил-6-нитропиридин (2,46 г), 1-изопропилпиперазин (2,74 г), йодид тетра-н-бутиламмония (418,69 мг) и карбонат калия (1,72 г) в диметилсульфоксиде (ДМСО, 20 мл) при 65C в течение ночи. Добавляли ЭА и воду, разделяли фазы и сушили органический слой над сульфатом магния и удаляли растворитель в вакууме. Очищали при помощи сильного катионита, элюируя метанолом,затем 2 н. смесью метанол-NH3 с получением 2,58 г указанного в заголовке соединения. МС (ES+): m/z=265 (М+Н)+. Следующие промежуточные соединения получали, по существу, в соответствии с описанием получения 1-изопропил-4-(2-метил-6-нитропиридин-3-ил)пиперазина с применением соответствующего бромпроизводного. Пример получения 7. 5-(4-Изопропилпиперазин-1-ил)-6-метилпиридин-2-иламин. Перемешивали 1-изопропил-4-(2-метил-6-нитропиридин-3-ил)пиперазин (2,52 г) и 10% палладий на угле (600 мг) в метаноле (38 мл) и ЭА (38 мл) в атмосфере H2 (баллон) в течение ночи. Фильтровали через подушку целита и удаляли растворитель в вакууме. Очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью ДХМ/метанол (0-10%) с получением 2,23 г указанного в заголовке соединения. МС (ES+): m/z=143 (M+Н)+. Следующие промежуточные соединения получали, по существу, в соответствии с описанием получения 5-(4-изопропилпиперазин-1-ил)-6-метилпиридин-2-иламина с применением соответствующего нитропроизводного. Пример получения 10. трет-Бутиловый эфир 4-(6-нитропиридин-3-илокси)пиперидин-1-карбоновой кислоты. К раствору трет-бутил 4-гидрокси-1-пиперидинкарбоксилата (8,76 г) и диметилацетамида (ДМА,39 мл) при 0C в атмосфере азота добавляли трет-бутоксид калия (4,84 г). Перемешивали в течение 1 ч и по каплям добавляли раствор 5-фтор-2-нитропиридина (5 г) в ДМА (78 мл). Оставляли реакционную смесь перемешиваться при КТ на ночь. Добавляли воду и выстаивали в течение 1 ч. Фильтровали, промывали водой. Очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью ДХМ/ЭА (0-15%) с получением 5,65 г указанного в заголовке соединения. МС (ES+): m/z=324 (M+H)+.(5,65 г) в смеси тетрагидрофуран (ТГФ)/метанол (30/30 мл/мл) добавляли 10% палладий на угле (0,6 г). Гидрировали в аппарате Парра при 2 атм в течение ночи. Фильтровали через подушку целита, промывали ДХМ и метанолом. Очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью ДХМ/метанол (10%)/аммиак (1%) с получением 5 г указанного в заголовке соединения. МС (ES+): m/z=294 (M+H)+. Пример получения 12. трет-Бутиловый эфир 6-амино-2-метил-3',6'-дигидро-2'H-[3,4']бипиридинил-1'-карбоновой кислоты Барботировали азот через смесь трет-бутилового эфира 4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан 2-ил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты (2,46 г) и 5-бром-6-метилпиридин-2-иламина(1,49 г) в 1,4-диоксане (31,82 мл) в течение 5 мин, затем добавляли N-гидрат трехосновного фосфата калия (5,07 г), ацетат палладия (35,72 мг), дициклогексил-(2',6'-диметоксибифенил-2-ил)фосфан(134,69 мг), воду (7,96 мл) и перемешивали при 90C в течение 3 ч. Разбавляли ДХМ и промывали водой. Сушили над сульфатом натрия и удаляли растворитель в вакууме. Очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью ДХМ/этанола 5%/NH3 0,1%, затем при помощи сильного катионита (SCX), элюируя метанолом, затем 2 М смесью метанол-NH3 с получением 2,12 г указанного в заголовке соединения. МС (ES+): m/z=292 (M+H)+. Пример получения 13. трет-Бутиловый эфир 6-амино-2-метил-3',4',5',6'-тетрагидро-2'H-[3,4']бипиридинил-1'-карбоновой кислоты. Перемешивали смесь трет-бутилового эфира 6-амино-2-метил-3',6'-дигидро-2'H-[3,4']бипиридинил 1'-карбоновой кислоты (2,12 г) и 10% влажного палладия на угле (330 мг) в метаноле (29,30 мл) в атмосфере Н 2 (45 psi) в течение 48 ч. Фильтровали через подушку целита и удаляли растворитель в вакууме с получением 2,07 г указанного в заголовке соединения. МС (ES+): m/z=292 (M+H)+. Пример получения 14. трет-Бутиловый эфир 6-нитро-3',6'-дигидро-2'H-[3,4']бипиридинил-1-карбоновой кислоты. Барботировали азот через смесь трет-бутилового эфира 4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан 2-ил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты (19,6 г), 5-бром-2-нитропиридина (12,87 г), 2 М раствора карбоната натрия в воде (63,39 мл) и хлорида бис-(трифенилфосфино)палладия(II) (4,45 г) в 1,4-диоксане (316,94 мл) в течение 5 мин и перемешивали при 80C в течение 5 ч. Разбавляли ДХМ и промывали водой. Сушили над сульфатом магния и удаляли растворитель в вакууме. Очищали при помощи хроматографии на силикагеле, элюируя смесью ДХМ/ЭА (0-40%) с получением 8,72 г указанного в заголовке соединения. МС (ES+): m/z=306 (M+H)+. Пример получения 15. трет-Бутиловый эфир 6-амино-3',4',5',6'-тетрагидро-2'H-[3,4']бипиридинил-1'-карбоновой кислоты. Растворяли трет-бутиловый эфир 6-нитро-3',6'-дигидро-2'H-[3,4']бипиридинил-1-карбоновой кислоты (1,89 г) в этаноле (123,80 мл). Гидрировали при помощи палладия на угле (установка H-Cube, 70 бар,50C, 1 мл/мин) с получением 1,72 г указанного в заголовке соединения. МС (ES+): m/z=278 (M+H)+. Пример получения 16. трет-Бутиловый эфир 4-(6-аминопиридин-3-илметил)пиперидин-1-карбоновой кислоты. В течение 5 мин перемешивали трет-бутиловый эфир 4-метиленпиперидин-1-карбоновой кислоты(3,8 г), карбонат калия (3,87 г) и хлорид 1,1'-(бис-(дифенилфосфино)ферроцен)палладия(II) (538,10 мг) и дегазированную смесь ДМФ (47,83 мл) и воды (4,78 мл). Перемешивали при 60C в течение 4 ч, затем при КТ в течение выходных. Добавляли воду и ЭА. Разделяли и экстрагировали водный слой ЭА. Объединяли органические слои и сушили над сульфатом натрия и удаляли растворить в вакууме. Очищали при помощи колоночной хроматографии на силикагеле, элюируя смесями от ДХМ/метанол (1%)/аммиак(0,1%) до ДХМ/метанол (3%)/аммиак (0,3%). Растирали остаток с ЭА с получением 1,85 г указанного в заголовке соединения. МС (ES+): m/z=292 (M+H)+. Следующее соединение получали, по существу, в соответствии с описанием получения третбутилового эфира 4-(6-аминопиридин-3-илметил)пиперидин-1-карбоновой кислоты с применением соот- 11018808 Пример получения 18. 1-(6-Бромпиридин-3-илметил)-4-этилпиперазин. К смеси 6-бромпиридин-3-карбальдегида (300 г) и ДХМ (5000 мл) добавляли чистый 1-этилпиперазин (221,44 мл). Затем по частям добавляли триацетоксиборгидрид натрия (372,09 г) и перемешивали при КТ в течение 12 ч. Добавляли ДХМ (1000 мл) и 2 н. водный раствор гидроксида натрия(1500 мл). Разделяли слои и дважды экстрагировали водный слой ДХМ (600 мл). Объединяли органические слои и удаляли растворитель в вакууме, добавляли ЭА и выпаривали с получением 451,3 г указанного в заголовке соединения. МС (ES+): m/z=285 (M+H)+. Следующее соединение получали, по существу, в соответствии с описанием получения 1-(6-бромпиридин-3-илметил)-4-этилпиперазина с применением соответствующего амина. Пример получения 20. 5-(4-Этилпиперазин-1-илметил)пиридин-2-иламин. К дегазированной смеси 1-(6-бромпиридин-3-илметил)-4-этилпиперазина (250 г), (дициклогексилфосфино)бифенила (18,50 г), трис-(дибензилиденацетон)дипалладия (24,17 г) и ТГФ (250 мл) при 50C медленно добавляли 1,1,1,3,3,3-гексаметилдисилазан (1055 мл) лития. Нагревали смесь при 65C в течение ночи. Охлаждали до 37C и добавляли воду (500 мл). Удаляли половину растворителя в вакууме и добавляли ДХМ (2,5 л). Фильтровали через подушку целита и удаляли часть растворителя. К смеси добавляли метанол (300 мл) и метил-трет-бутиловый эфир (МТБЭ, 600 мл) и охлаждали на ледяной бане. Затем добавляли 2 М раствор хлороводородной кислоты в диэтиловом эфире (800 мл) и 32% водный раствор хлороводородной кислоты (100 мл). Удаляли органический слой и добавляли 2 M водный раствор гидроксида натрия (2500 мл). Трижды экстрагировали водную фазу ДХМ и удаляли растворитель в вакууме. Растворяли в 90 мл толуола при 50C до полного растворения и затем добавляли 80 мл МТБЭ. Перемешивали в течение ночи при КТ. Дополнительно добавляли МТБЭ (100 мл) для полного осаждения. Отфильтровывали твердое вещество и сушили с получением 108,24 г указанного в заголовке соединения. МС (ES+): m/z=221 (M+H)+. Следующее соединение получали,по существу,в соответствии с описанием получения 5-(4-этилпиперазин-1-илметил)пиридин-2-иламина с применением соответствующего 2-бромпиридинового производного. Пример получения 22. 2,4-Дибром-1-нитробензол. К раствору 1,3-дибромбензола (102,51 мл) в концентрированной серной кислоте (322,79 мл) и воде(62,39 мл) при 0C по каплям добавляли дымящую азотную кислоту (101,40 мл). Нагревали до КТ и перемешивали в течение 12 ч. Выливали реакционную смесь в смесь лед-вода (1500 мл). Отфильтровывали полученное желтое твердое вещество в вакууме с получением 178,46 г указанного в заголовке соединения. МС (ES+): m/z=281 (M+H)+.(32 мл). Нагревали смесь при 100C в течение ночи. Удаляли растворитель в вакууме, добавляли воду и экстрагировали ЭА. Последовательно промывали органический слой водным раствором бикарбоната натрия, а затем водой. Сушили над сульфатом магния и удаляли растворитель в вакууме с получением 22 г указанного в заголовке соединения. МС (ES+): m/z=286 (M+H)+. Пример получения 24. 4-Бром-N2-циклопентилбензол-1,2-диамин. К раствору (5-бром-2-нитрофенил)циклопентиламина (22 г), ТГФ (150 мл), воды (150 мл) и гидроксида аммония (30 мл) добавляли дитионит натрия (107,47 г). Перемешивали смесь при КТ в течение ночи. Дважды экстрагировали ЭА, сушили над сульфатом магния и удаляли растворитель в вакууме с получением 14,80 г указанного в заголовке соединения. МС (ES+): m/z=256 (M+H)+. Пример получения 25. 6-Бром-1-циклопентил-2-метил-1H-бензимидазол. Нагревали смесь 4-бром-N2-циклопентилбензол-1,2-диамина (10,6 г), триэтилортоацетата (9,5 мл) и уксусной кислоты (6,3 мл) при 100C в течение 2,5 ч. Разбавляли ДХМ и выливали в насыщенный водный раствор бикарбоната натрия. Сушили над сульфатом натрия и удаляли растворитель в вакууме. Очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью ДХМ/этанол-10%N-Изопропилацетамид. К раствору 2-пропанамина (10 г) в ДХМ (100 мл) при 0C добавляли ТЭА (23,58 мл). Затем осторожно по каплям добавляли ангидрид уксусной кислоты (16,15 мл). Перемешивали при КТ в течение ночи. Удаляли растворитель в вакууме, разбавляли диэтиловым эфиром (эфиром) и отфильтровывали твердое вещество. Удаляли растворитель в вакууме. Разбавляли масло эфиром, добавляли карбонат калия и перемешивали в течение ночи при КТ. Отфильтровывали твердое вещество и удаляли растворитель в вакууме с получением 15,62 г указанного в заголовке соединения. ЯМР (CDCl3): 4,06 (м, 1H), 1,94 (с, 3 Н), 1,14 (д, 6H). Следующие амиды получали, по существу, в соответствии с описанием получения(6,70 мл) в толуоле (150 мл) добавляли ТЭА (10,05 мл). Кипятили смесь с обратным холодильником в течение 3 ч. Охлаждали смесь и удаляли растворитель в вакууме. Растворяли неочищенное вещество в ДХМ, несколько раз промывали насыщенным водным раствором бикарбоната натрия для удаления всех следов кислоты. Сушили над сульфатом натрия и удаляли растворитель в вакууме с получением 14 г указанного в заголовке соединения. МС (ES+): m/z=292 (М+Н)+. Следующие промежуточные соединения получали, по существу, в соответствии с описанием получения N-(4-бром-2,6-дифторфенил)-N'-изопропилацетамида с применением соответствующего ацетамида.(20 мл) добавляли трет-бутоксид калия (811,43 мг). Нагревали смесь при 100C в течение 2 ч. Охлаждали до КТ, добавляли ДХМ (150 мл), трижды промывали насыщенным водным раствором хлорида натрия(солевой раствор, 300 мл), сушили над сульфатом магния и удаляли растворитель в вакууме. Добавляли гексан и встряхивали с применением ультразвука в течение нескольких минут. Отфильтровывали твердое вещество, дважды повторяли добавление гексана/фильтрование с получением 1,86 г указанного в заголовке соединения. МС (ES+): m/z=272 (M+H)+. Следующие промежуточные соединения получали, по существу, в соответствии с описанием получения 6-бром-4-фтор-1-изопропил-2-метил-1H-бензимидазола с применением соответствующего ацетамидина. Пример получения 38. 4-Фтор-1-изопропил-2-метил-6-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1H-бензимидазол. Барботировали азот через смесь 6-бром-4-фтор-1-изопропил-2-метил-1H-бензимидазола (30,0 г),бис-(пинаколато)диборана (42,15 г), трициклогексилфосфина (5,43 г), ацетата калия (32,58 г) и ДМСО(200 мл). Добавляли ацетат палладия (2,8 г) и нагревали на предварительно нагретой масляной бане при 90C в течение 1 ч. Разбавляли ЭА (200 мл) и фильтровали через подушку целита. Промывали смесь солевым раствором (100 мл), сушили над сульфатом натрия и удаляли растворитель в вакууме. Растирали с гексаном и отфильтровывали твердое вещество с получением 27 г указанного в заголовке соединения. МС (ES+): m/z=319 (M+H)+. Следующие промежуточные соединения получали, по существу, в соответствии с описанием получения 4-фтор-1-изопропил-2-метил-6-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1H-бензимидазола с применением соответствующих производных 6-бромбензимидазола. Пример получения 43. 6-(2-Хлор-5-фторпиримидин-4-ил)-4-фтор-1-изопропил-2-метил-1H-бензимидазол. Барботировали азот через смесь 2,4-дихлор-5-фторпиримидина (12,7 г), хлорида бис-(трифенилфосфино)палладия(II) (4,9 г), 2 М раствора карбоната натрия в воде (103,7 мл) и 1,2-диметоксиэтана (120 мл). Нагревали на предварительно нагретой масляной бане при 80C и по каплям добавляли раствор 4-фтор-1-изопропил-2-метил-6-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1Hбензимидазола (22 г) в 1,2-диметоксиэтане (200 мл). Перемешивали смесь при 84C в течение 1 ч. Охлаждали до КТ, добавляли ЭА (800 мл) и дважды промывали солевым раствором (100 мл). Сушили над сульфатом магния и удаляли растворитель в вакууме. Растирали с ацетонитрилом с получением 14,4 г указанного в заголовке соединения. МС (ES+): m/z=323 (M+H)+. Следующие промежуточные соединения получали, по существу, в соответствии с описанием получения 6-(2-хлор-5-фторпиримидин-4-ил)-4-фтор-1-изопропил-2-метил-1H-бензимидазола с применением соответствующих дихлорпиримидиновых и боранатных производных. Следующие промежуточные соединения получали, по существу, в соответствии с описанием получения[5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3Hбензимидазол-5-ил)пиримидин-2-ил]амина с применением соответствующих аминовых и хлорпиримидиновых производных. К смеси трет-бутилового эфира 4-6-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3H-бензимидазол-5 ил)пиримидин-2-иламино]пиридин-3-илметилпиперазин-1-карбоновой кислоты (150 мг) в ДХМ (10 мл) и метаноле (10 мл) добавляли 4 М раствор хлороводорода в диоксане (194 мкл). Перемешивали в течение 10 мин и удаляли растворитель в вакууме. Очищали при помощи сильного катионита (SCX), элюируя метанолом, затем 2 М смесью метанол-NH3, затем при помощи колоночной хроматографии на силикагеле, элюируя 2 М смесью ДХМ/метанол-NH3 (3%) с получением 120 мг указанного в заголовке соединения. МС (ES+): m/z=479 (М+Н)+. Следующие промежуточные соединения получали, по существу, в соответствии с описанием получения [5-фтор-4-(7-фтор-3-изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]-(5-пиперазин-1 илметилпиридин-2-ил)амина. Барботировали азот через смесь 6-(2-хлор-5-фторпиримидин-4-ил)-4-фтор-1-изопропил-2-метил- 19018808(1,82 г),4,5-бис-(дифенилфосфино)-9,9 диметилксантена (2,35 г) в 1,4-диоксане (197,06 мл). Нагревали смесь на предварительно нагретой масляной бане при 110C в течение 2 ч. Охлаждали до КТ, разбавляли ДХМ и фильтровали через подушку целита. Удаляли растворитель в вакууме и очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью ДХМ/метанол (2%), затем смесью ДХМ/метанол-NH3 2 M 2% с получением 22,11 г указанного в заголовке соединения. МС (ES+): m/z=507 (M+H)+. Следующие примеры получали, по существу, в соответствии с описанием получения[5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3Hбензимидазол-5-ил)пиримидин-2-ил]амина с применением соответствующих аминовых и хлорпиримидиновых производных.[5-фтор-4-(7-фтор-3-изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]-(5 пиперазин-1-илметилпиридин-2-ил)амина (130 мг), ацетона (31,6 мкл), 1,2-дихлорэтана (9 мл) и уксусной кислоты (16,3 мкл) добавляли триацетоксиборгидрид натрия (299,9 мг). Нагревали при 60C в течение 1 ч. Удаляли растворитель в вакууме. Очищали с применением сильного катионита (SCX), элюируя метанолом, затем смесью метанол-NH3 2 М, затем при помощи колоночной хроматографии на силикагеле,элюируя смесью ДХМ/метанол-NH3 2 М (3%) с получением 115 мг указанного в заголовке соединения. МС (ES+): m/z=521 (M+H)+. Следующие примеры получали, по существу, в соответствии с описанием получения [5-фтор-4-(7 фтор-3-изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]-[5-(4-изопропилпиперазин-1 илметил)пиридин-2-ил]амина с применением соответствующих аминов.(2709 мкл) добавляли триацетоксиборгидрид натрия (720 мг). Нагревали при 60C в течение 1 ч. Удаляли растворитель в вакууме. Очищали с применением сильного катионита (SCX), элюируя метанолом, затем смесью метанол-NH3 2 M, а затем при помощи колоночной хроматографии на силикагеле, элюируя смесью ДХМ/метанол-NH3 2 М (3%) с получением 80 мг указанного в заголовке соединения. МС (ES+): m/z=504 (M+H)+. Следующие примеры получали, по существу, в соответствии с описанием получения [4-(3 циклопропил-7-фтор-2-метил-3H-бензимидазол-5-ил)-5-фторпиримидин-2-ил]-[5-(1-этилпиперидин-4 илметил)пиридин-2-ил]амина с применением соответствующих аминов. Пример 16. Метансульфонат [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2 метил-3H-бензимидазол-5-ил)пиримидин-2-ил]амина. К раствору [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил 3H-бензимидазол-5-ил)пиримидин-2-ил]амина (17,3 г) в смеси ДХМ (100 мл) и метанола (100 мл) добавляли метансульфокислоту (63,59 мл). Перемешивали раствор в течение 1 ч и удаляли растворитель в вакууме. Растирали с МТБЭ и отфильтровывали твердое вещество с получением 20,4 г указанного в заголовке соединения. МС (ES+): m/z=507 (M+H)+. Следующие примеры получали, по существу, в соответствии с описанием получения метансульфоната[5-(4-Этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3Hбензимидазол-5-ил)пиримидин-2-ил]амин. Кристаллическая форма I. Смешивали 102,1 мг аморфного [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор 3-изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]амина с 2 мл ацетона. Выделяли осажденное твердое вещество при помощи вакуумного фильтрования с получением светло-желтого осадка и сушили непосредственно в устройстве для фильтрования в течение 30 мин с получением 72,1 мг твердого вещества. Помещали твердое вещество в вакуумный сушильный шкаф при 100C на 3 ч. Типичные пики XRD формы I представлены в табл. 1. Пример 32.[5-(4-Этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3Hбензимидазол-5-ил)пиримидин-2-ил]амин. Кристаллическая форма III. Смешивали 208 мг аморфного [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3 изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]амина с 4 мл ацетона. Суспендировали в течение 2 ч при 60C, перемешивая со скоростью 1000 об/мин, затем выделяли твердое вещество при помощи вакуумного фильтрования с получением светло-желтого осадка. Сушили непосредственно в устройстве для фильтрования в течение 30 мин с получением 112 мг твердого вещества (выход 54%). Помещали в вакуумный сушильный шкаф при 80C на 3 ч. Типичные пики XRD формы III представлены в табл. 2. Положения пиков проверяли с применением внешнего стандарта. Порошковая дифракционная рентгенограмма. Рентгенограммы XRD кристаллов получали на порошковом дифрактометре Bruker D8 Advance,оборудованном источником излучения CuK (=1,54056 ) и детектором Vantec, с рабочими характеристиками 50 кВ и 40 мА. Каждый образец сканировали в диапазоне от 4 до 402 с шагом 0,022 и скоростью сканирования 9,0 с на шаг, с 1 мм дивергенцией и щелями приемника и 0,1 мм щелью детектора. Сухой порошок помещали в углубленный держатель образца с верхней загрузкой, ровную поверхность получали с применением предметного стекла. Дифракционные рентгенограммы кристаллических форм получали при температуре и относительной влажности окружающей среды. Перед снятием пиков для кристаллов формы III убирали фон, тогда как для формы I фон не убирали. В области кристаллографии хорошо известно, что для каждой заданной кристаллической формы относительные интенсивности дифракционных пиков могут изменяться в зависимости от предпочтительной ориентации, зависящей от факторов, таких как морфология и форма кристалла. В случае, если присутствуют эффекты предпочтительной ориентации, интенсивности пиков изменяются, но положения характеристических пиков полиморфа остаются неизменными. См., например, Фармакопея США 23,- 26018808 Национальный формуляр 18, стр. 1843-1844, 1995. Кроме того, в области кристаллографии хорошо известно, что для каждой заданной кристаллической формы положение угловых пиков может незначительно изменяться. Например, положения пиков могут сместиться в результате изменения температуры или влажности, при которых анализируют образец, замены образца или наличия или отсутствия внутреннего стандарта. В настоящем случае погрешность 0,12 положения пиков учитывает указанные потенциальные изменения без помех для точного определения исследуемой формы кристалла. Подтверждение формы кристалла может быть проведено на основании любой уникальной комбинации различных пиков (в единицах 2), как правило, более крупных пиков. Таким образом, полученный образец кристаллической формы I [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор 3-изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]амина характеризуется рентгенограммойXRD, полученной с применением излучения CuK, содержащим пики дифракции (в единицах 2-тета),представленные в табл. 1, в частности содержащим пики при 4,51 в комбинации с одним или более пиком, выбранным из группы, состоящей из 13,09, 16,31 и 18,82; с погрешностью углов дифракции, равной 0,1. Таблица 1 Пики порошковой дифракционной рентгенограммы кристаллической формы I [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3 изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]амина Полученный образец кристаллической формы III [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5 фтор-4-(7-фтор-3-изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]амина характеризуется рентгенограммой XRD, полученной с применением излучения CuK, содержащей дифракционные пики(в единицах 2-тета), описанные в табл. 2, и, в частности, содержащей пики при 21,29 в комбинации с одним или более пиком при 11,54, 10,91 и 12,13; с погрешностью углов дифракции, равной 0,1. Таблица 2 Пики порошковой дифракционной рентгенограммы кристаллической формы III [5-(4-этилпиперазин-1-илметил)пиридин-2-ил]-[5-фтор-4-(7-фтор-3 изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]амина Спектры ЯМР кросс-поляризации/вращения под магическим углом (CP/MAS) (твердофазного ЯМР или SSNMR) получали на ЯМР-спектрометре широких линий Bruker Advance III 400 с рабочими частотами для 1H и 13 С, равными 400,131 и 100,623 МГц соответственно, с применением 4 мм датчиков двойного резонанса Bruker. Скорость MAS устанавливали на 5 или 10 кГц с применением контролирующего устройства Bruker-MAS-II; скорости вращения поддерживали внутри 2 Гц диапазона от установленного значения. Развязку SPINAL64 при частоте нутации протонов, равной 100 кГц, применяли для гетероядерной развязки. Боковые полосы от вращения удаляли при помощи пятиимпульсной последовательности для полного удаления боковых полос (TOSS). Контактное время СР для переноса намагниченности с протонов на углерод устанавливали равным 4 мс для увеличения эффективности СР в канале 1 Н применяли постепенное изменение частоты от 93,5 до 46,9 кГц. Устанавливали экспозицию, равную 34 мс, и получали спектры со спектральной шириной, равной 30 кГц, с задержкой перед повторным снятием спектра, равной 5 с. Поддерживали температуру образца, равную 2971 К для минимизации нагревания за счет трения, вызванного вращением образца. Химические сдвиги 13 С соотносили с развязанным от протонов пиком 13 С внешнего стандарта (0,05 ppm) чистого (жидкого) тетраметилсилана с применением сильнопольного резонанса адамантина (=29,5 ppm). Список химических сдвигов кристаллической формы III [5-(4-этилпиперазин-1-илметил)пиридин-2 ил]-[5-фтор-4-(7-фтор-3-изопропил-2-метил-3H-бензимидазол-5-ил)пиримидин-2-ил]амина представлен далее: 13 С-ЯМР:(F1) (ppm) 11,7; 12,9; 20,5; 48,6; 52,5; 59,4; 108,9; 110,0; 112,7; 127,3; 129,4; 135,5; 136,4; 148,8; 150,1; 152,2; 154,5; 156,3.a) 1-(6-Бромпиридин-3-илметил)-4-этилпиперазин. К смеси 6-бромпиридин-3-карбальдегида (8,3 кг) и ДХМ (186 кг) добавляли чистый 1-этилпиперазин (5,6 кг). Затем по частям добавляли триацетоксиборгидрид натрия (10,9 кг) и перемешивали при 20-30C в течение 12 ч. Реакцию гасили, выливая реакционную массу в смесь ДХМ (36 кг) и 2 н. водного раствора гидроксида натрия (46 кг). Отделяли слои и дважды экстрагировали водный слой ДХМ (242 кг). Объединяли органические слои, промывали солевым раствором (502 кг) и удаляли растворитель в вакууме с получением 11,5 кг указанного в заголовке соединения. МС (ES+): m/z=285 (M+H)+.(200 г) и МеОН (57 кг) при Т 40C добавляли жидкий аммиак (50,0 кг). Нагревали смесь при 65-75C в течение ночи. Охлаждали до 20-30C и фильтровали через подушку целита. Концентрировали фильтрат и добавляли ДХМ (113 кг) и доводили рН до 12-14 2 н. гидроксидом натрия (23 кг), разделяли фазы и промывали органическую фазу ДХМ (582 кг) и объединяли органические слои. Фильтровали смесь через целит и концентрировали. Растворяли остаток в толуоле (9,7 кг) и кристаллизовали при помощи добавления МТБЭ (8,3 кг) с получением 6,0 кг указанного в заголовке соединения. Проводили дополнительную очистку перекристаллизацией из толуола. МС (ES+): m/z=221 (M+H)+.(28 кг). Охлаждали смесь примерно до 5-0 C и добавляли ацетилхлорид (16,7 кг) со скоростью, равной примерно 2-3 кг/ч. Перемешивали до завершения реакции, определяемой с помощью газовой хроматографии. Гасили реакционную смесь водой (0,8 кг) и фильтровали реакционную смесь и концентрировали с получением 13,4 кг указанного в заголовке соединения. ЯМР (CDCl3) 4,06 (м, 1 Н), 1,94 (с, 3 Н), 1,14 (д, 6H).d) N-(4-Бром-2,6-дифторфенил)-N'-изопропилацетамид. К смеси 4-бром-2,6-дифторфениламина (14,5 кг), N-изопропилацетамида (8,5 кг), ТЭА (10,6 кг) в толуоле (115 кг) при 20C добавляли хлорокись фосфора (16,0 кг). Перемешивали при 10-20C до завершения реакции, определяемой с помощью ВЭЖХ. Удаляли растворитель в вакууме и добавляли МТБЭ (64 кг). Регулировали рН смеси 10% водным карбонатом натрия (250 кг). Фильтровали смесь и промывали осадок МТБЭ (112 кг). Разделяли фазы и промывали водный слой МТБЭ (222 кг). Объединяли органические слои и концентрировали, фильтровали и промывали циклогексаном (0,6 кг) и сушили с получением 17,2 кг указанного в заголовке соединения. МС (ES+): m/z=292 (M+H)+.(76 кг) добавляли по частям трет-бутоксид калия (6,9 кг), поддерживая температуру 30C. Нагревали смесь при 70-75C до завершения реакции, определяемой с помощью ВЭЖХ. Охлаждали до 20-30C и гасили при помощи добавления в воду (227 кг), затем экстрагировали МТБЭ (374 кг). Промывали объединенные органические фазы солевым раствором (492 кг) и концентрировали до объема, равного 25-30 л, добавляли н-гексан (64 кг) и фильтровали суспензию с получением 11 кг указанного в заголовке соединения. МС (ES+): m/z=272 (M+H)+. Дополнительную очистку проводили при помощи растворения неочищеного соединения в ДХМ и фильтрования через подушку силикагеля и целита с последующим выделением из смеси МТБЭ/гексан.f) 4-Фтор-1-изопропил-2-метил-6-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1H-бензимидазол. Барботировали азот через смесь 6-бром-4-фтор-1-изопропил-2-метил-1H-бензимидазола (600 г),бис-(пинаколато)диборана (843 г), трициклогексилфосфина (106 г), ацетата калия (652 г) и ДМСО (3,6 л). Добавляли ацетат палладия (49 г) и нагревали при 100C до завершения реакции, определяемой с помощью ВЭЖХ. Охлаждали реакционную смесь и разбавляли водой (18 л), затем фильтровали для выделе- 29
МПК / Метки
МПК: C07D 401/14, A61P 35/00, A61K 31/517
Метки: протеинкиназы, основе, состав, применение, фармацевтический, ингибиторы, терапии
Код ссылки
<a href="https://eas.patents.su/30-18808-ingibitory-proteinkinazy-farmacevticheskijj-sostav-na-ih-osnove-i-primenenie-v-terapii.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы протеинкиназы, фармацевтический состав на их основе и применение в терапии</a>
Производные оксаиндена, способ их получения (варианты), фармацевтический состав на их основе, их применение и способ лечения с их использованием

Номер патента: 25
Опубликовано: 26.02.1998
Авторы: Новак Лайош, Ковач Габор, Семереди Каталин, Гиглер Габор, Драбант Шандор, Сантаи Чаба, Божинг Даниэль, Эдьед Андраш, Шимиг Дьюла, Блашко Габор, Чёргё Маргит, Ковач Петер, Пирок Дьордь, Гадо Клара, Такач Габорне
МПК: A61K 31/34, C07D 307/77
Метки: применение, получения, состав, производные, фармацевтический, варианты, использованием, оксаиндена, основе, лечения, способ
Формула / Реферат:
1. Производные оксаиндена общей формулы (I), где R1 представляет собой низший С1-4 алкил, низшую С1-4 алкоксигруппу или низший С3-6 циклоалкил, R2 представляет собой водород, низший С1-4 алкил или низший С3-6 циклоалкил, R3 представляет собой водород, низший С1-4 алкил, низшую С1-4 алкоксигруппу или бензилоксигруппу, Х представляет собой водород, алифатический С1-4 ацил или бензоиловую или нафтоиловую группу, возможно замещенные одной или...
7α, 17α-замещённые 11β-галогеностероиды, способы их получения, применение и фармацевтический препарат на их основе

Номер патента: 10572
Опубликовано: 30.10.2008
Авторы: Больманн Рольф, Нуббемейер Райнхард, Галлус Норберт, Цорн Людвиг, Мун Ханс-Петер, Кюнцер Германн
МПК: C07J 11/00, A61K 31/56, C07J 1/00...
Метки: препарат, основе, применение, 7&alpha, фармацевтический, способы, получения, 17α-замещённые, 11β-галогеностероиды
Формула / Реферат:
1. 7a,17a-замещенные 11b-галогенстероиды общей формулы 8,10,12 в которой U-V-W-X-Y-Z представляет собой одну из циклических структур С1-С2-С3-С4=С5-С10, С1-С2-C3-С4-С5=С10 или С1-С2-С3-C4-С5-С10, при этом в данном случае оксогруппа (=O) связана с W (=С3), или представляет собой циклическую структуру С1=С2-С3=С4-С5=С6, при этом в данном случае остаток OR3 связан с W (=С3), R3 представляет собой Н, С1-С4алкил, С1-С4алканоил, R7 представляет собой...
Фармацевтическая композиция на основе 3,6-ангидрогалактопиранозы и/или ее производных, и/или растворимого сахарида, содержащего данное соединение, применение композиции и входящих в ее состав ингредиентов, пищевой продукт, содержащий указанную композицию, и применение пищевого продукта

Номер патента: 4148
Опубликовано: 26.02.2004
Авторы: Икаи Катсусиге, Сакаи Такеси, Томинага Таканари, Като Икуносин, Нисийама Еидзи, Йу Фу-Гонг, Сагава Хироаки, Еноки Татсудзи, Койама Нобуто
МПК: A23L 1/30, A61K 31/70, A01N 1/00...
Метки: пищевого, содержащего, ингредиентов, основе, данное, 3,6-ангидрогалактопиранозы, производных, растворимого, композицию, применение, композиции, пищевой, фармацевтическая, содержащий, продукта, входящих, состав, указанную, соединение, сахарида, композиция, продукт
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента по меньшей мере одно соединение, выбранное из группы, состоящей из 3,6-ангидрогалактопиранозы, представленной формулой I ее альдегида и гидрата и 2-O-метилированных производных 3,6-ангидрогалактопиранозы, ее альдегида и гидрата; и растворимого сахарида, содержащего данное соединение на восстанавливающем конце. 2. Фармацевтическая композиция по п.1, где сахарид...
Ингибиторы akt (протеинкиназы в)

Номер патента: 15712
Опубликовано: 31.10.2011
Авторы: Ли Жэньхуа, Шмид Кристофер Рэндалл, Майерс Майкл Рэй, Джозеф Саджан, Абуруб Актхам, Дай Дженни Пингкви
МПК: A61K 31/435, A61P 35/00, A61P 31/18...
Метки: протеинкиназы, ингибиторы
Формула / Реферат:
1. Соединение, которое представляет собой 4-[5-(2-аминоэтансульфонил)изохинолин-7-ил]фенол или его фармацевтически приемлемую соль либо гидрат указанного соединения или его фармацевтически приемлемой соли.2. Соединение по п.1, которое представляет собой дигидрохлорид 4-[5-(2-аминоэтансульфонил)изохинолин-7-ил]фенола.3. Соединение по п.1, которое представляет собой полугидрат моногидрохлорида 4-[5-(2-аминоэтансульфонил)изохинолин-7-ил]фенола.4....
Ингибиторы протеинкиназы с.

Номер патента: 598
Опубликовано: 29.12.1999
Авторы: Джироусек Майкл Р., Макдональд Джон Х., Хит Уильям Ф., Рито Кристофер Дж.
МПК: A61K 47/22, C07D 471/22
Метки: ингибиторы, протеинкиназы
Формула / Реферат:
1. Соединение формулы где W является О, -S- или NH, R1 представляет собой независимо водород, галоген, С1-С4-алкил, гидрокси, С1-С4-алкокси, галогеналкил, нитро, -NH(С1-С4-алкил), N(С1-С4-алкил)2 или -NНСО(С1-С4-алкил), R2 является водородом, СН3СО-, NН2 или гидроксигруппой, Z является -(СН2)р- или (СН2)р-O-(СН2)p-, R6 является -NH(CF3) или -N(CF3)(СН3), m независимо является 0, 1, 2 или 3, р независимо является 0, 1 или 2 или...
Предыдущий патент: Тиазолилпиразолопиримидины в качестве синтетических промежуточных соединений и связанные с ними способы синтеза
Следующий патент: Способ извлечения редкоземельных элементов из твердой смеси, содержащей галофосфат и соединение одного или нескольких редкоземельных элементов
Случайный патент: Новая полиморфная форма кристаллической гемикальциевой соли аторвастатина