Пептидомиметики с активностью антагонистов глюкагона и агонистов glp-1
Номер патента: 18000
Опубликовано: 30.04.2013
Авторы: Бахекар Раджеш Х., Джаин Мукул Р., Пател Панкадж Раманбхай











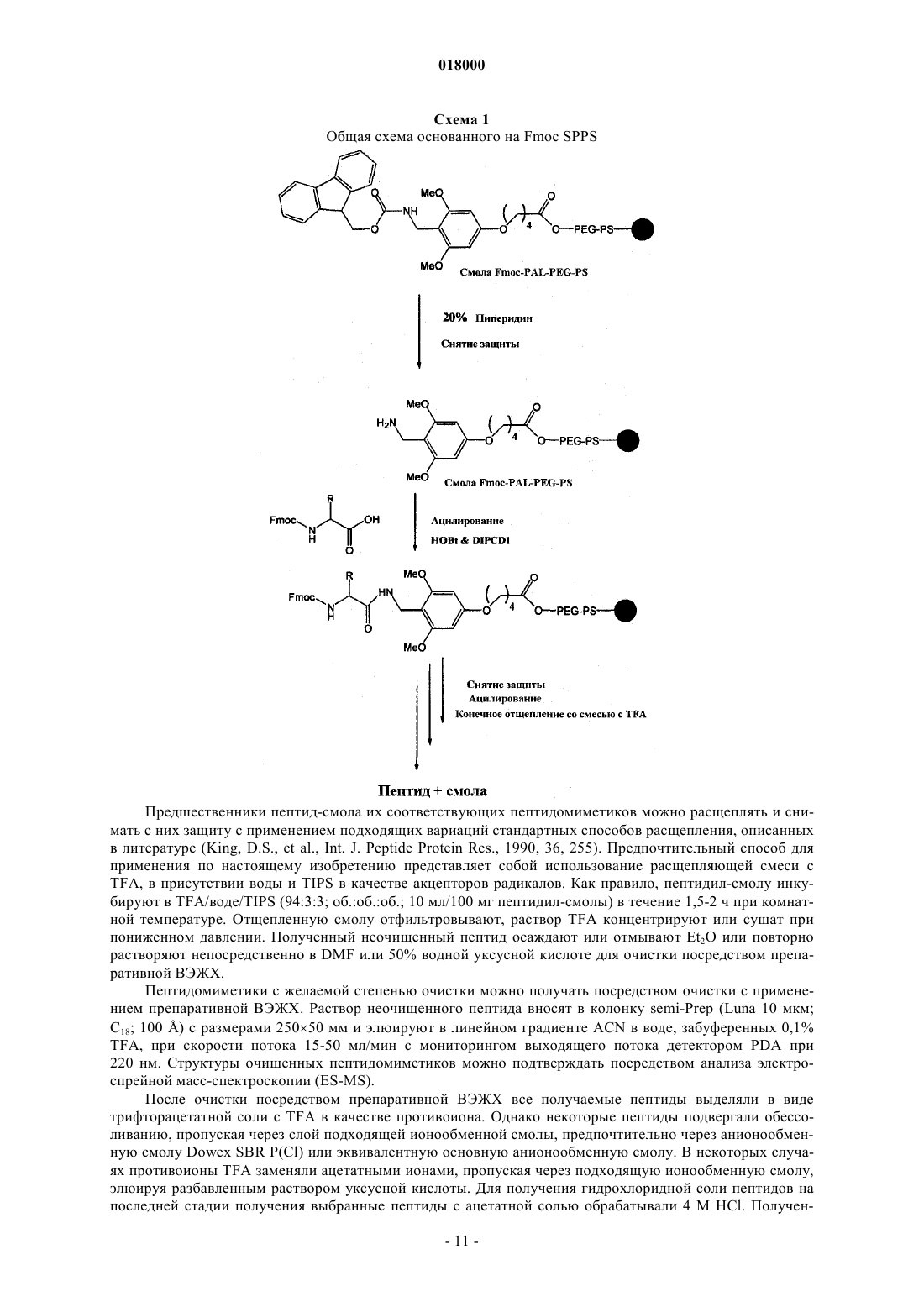











Формула / Реферат
1. Выделенные пептидомиметики с последовательностью формулы (I)

в которой A представляет собой группы -NH-R1, R3-CO-, R3-O-CO- или R3-SO2-, где R1 представляет собой водород или необязательно замещенную линейную или разветвленную (C1-С10)алкильную цепь; R3 выбран из линейных или разветвленных (C1-С10)алкильной, (С3-С6)циклоалкильной, арильной, гетероарильной или арилалкильной групп;
B представляет собой -COOR2, -CONHR2 или CH2OR2, где R2 представляет собой H, необязательно замещенные группы, выбранные из линейных или разветвленных (C1-С10)алкильной, арильной или аралкильной групп;
Z1-Z11 представляют собой природные или неприродные аминокислоты, связанные вместе амидной связью;
Z1 представляет собой L-гистидин (Н), D-гистидин (dH) или урокановую кислоту (UA):

Z2 представляет собой природные или неприродные аминокислоты, выбранные из группы, содержащей L-серин (S), D-серин (dS), L-аланин (А), D-аланин (dA), α-метилпролин (α-Me-Pro), α-аминоизомасляную кислоту (Aib), 1-аминоциклопропанкарбоновую кислоту (ACP) и 1-аминоциклопентанкарбоновую кислоту (APP):

Z3 представляет собой L-глутамин (Gln; Q), D-глутамин (dQ) или соединения формулы (II) (CNB или Hfl):

Z4 представляет собой глицин (G) или неприродные аминокислоты: 1-аминоциклопропанкарбоновую кислоту (АСР) или 1-аминоциклопентанкарбоновую кислоту (APP);
Z5 представляет собой природную или неприродную аминокислоту, содержащую гидроксильную боковую цепь; предпочтительно Z5 представляет собой L-треонин (Т), D-треонин (dT), L-аллотреонин (алло-Thr; алло-Т), D-аллотреонин (d-алло-Thr; d-алло-Т);
Z6 представляет собой природную или неприродную аминокислоту с двузамещенным альфа-атомом углерода с двумя боковыми цепями, где каждая из них может независимо быть необязательно замещена алкильной, или арильной, или аралкильной группой, где заместители могут быть выбраны из одной или нескольких алкильных групп или из одной или нескольких галогеновых групп;
Z7 и Z8 независимо представляют собой природную или неприродную аминокислоту, содержащую гидроксильную боковую цепь;
Z9 независимо представляет собой природную или неприродную аминокислоту с боковой аминокислотной цепью, содержащей кислую группу;
Z10 представляет собой L- или D-неприродную аминокислоту формул IIIa-IIIc:

Z11 представляет собой L- или D-неприродную аминокислоту формул IVa-IVl:

2. Соединение формулы (I) по п.1, где Z6 представляет собой группы Phe (F), альфа-метилфенилаланин (-α-Me-Phe-), альфа-метил-2-фторфенилаланин (-α-Me-2F-Phe-) или альфа-метил-2,6-дифторфенилаланин (-α-Ме-2,6-F-Phe-) или 2-фторфенилаланин (-2F-Phe-).
3. Соединение формулы (I) по п.1, где каждый из Z7 и Z8 выбран из треонина, серина, 1-аминоциклопропанкарбоновой кислоты.
4. Соединение формулы (I) по п.1, где Z9 выбран из L-аспарагиновой кислоты (D), D-аспарагиновой кислоты (dD) или соединений формулы (II).
5. Соединение формулы (I) по п.1, где арильная группа выбрана из фенильной, нафтильной, инданильной, флуоренильной или бифенильной групп; гетероарильная группа выбрана из пиридильной, тиенильной, фурильной, имидазолильной, бензофуранильной групп.
6. Соединение формулы (I) по п.1, где амидная связь между Z9 и Z10, или Z10 и Z11, или Z9-Z11 является дополнительно N-метилированной, представленной (NMe).
7. Соединение формулы (I) по п.1, где амидная связь между Z9 и Z10, или Z10 и Z11, или Z9-Z11 дополнительно является тиоамидной связью.
8. Соединение формулы (I) по п.7, где дополнительно тиоамидная связь между Z9 и Z10, или Z10 и Z11, или Z9-Z11 восстановлена до связи -CH2-.
9. Соединение формулы (I)
,
в которой A представляет собой группы -NH-R1, R3-CO-, R3-O-CO- или R3-SO2-, где R1 представляет собой водород или необязательно замещенную линейную или разветвленную (C1-С10)алкильную цепь; R3 выбран из линейных или разветвленных (C1-С10)алкильной, (С3-С6)циклоалкильной, арильной, гетероарильной или арилалкильной групп;
B представляет собой -COOR2, -CONHR2 или CH2OR2, где R2 представляет собой H, необязательно замещенные группы, выбранные из линейных или разветвленных (C1-С10)алкильной, арильной или аралкильной групп;
Z1 представляет собой L-гистидин (Н), D-гистидин (dH) или урокановую кислоту (UA);
Z2 выбран из L-серина, D-серина, L-аланина, D-аланина, α-аминоизомасляной кислоты, 1-аминоциклопропанкарбоновой кислоты (ACP) и 1-аминоциклопентанкарбоновой кислоты (APP):

Z3 представляет собой L-глутамин (Gln; Q), D-глутамин (dQ) или соединения формулы (II) (CNB или Hfl):

Z4 представляет собой глицин (G) или неприродные аминокислоты: 1-аминоциклопропанкарбоновую кислоту (ACP) или 1-аминоциклопентанкарбоновую кислоту (APP);
Z5 представляет собой L-треонин (T), D-треонин (dT), L-аллотреонин (алло-Thr; алло-Т), D-аллотреонин (d-алло-Thr; d-алло-T);
Z6 представляет собой фенилаланин (Phe; F), альфа-метилфенилаланин (-α-Me-Phe-), альфа-метил-2-фторфенилаланин (-α-Me-2F-Phe-), альфа-метил-2,6-дифторфенилаланин (-α-Me-2,6-F-Phe-) или 2-фторфенилаланин (-2F-Phe-):

Z7 и Z8 независимо выбраны из треонина, серина, 1-аминоциклопропанкарбоновой кислоты (АСР);
Z9 выбран из L-аспарагиновой кислоты (D), D-аспарагиновой кислоты (dD) или соединений формулы (II);
Z10 представляет собой L- или D-неприродную аминокислоту формул IIIa-IIIc:

Z11 представляет собой L- или D-неприродную аминокислоту формул IVa-IVl:

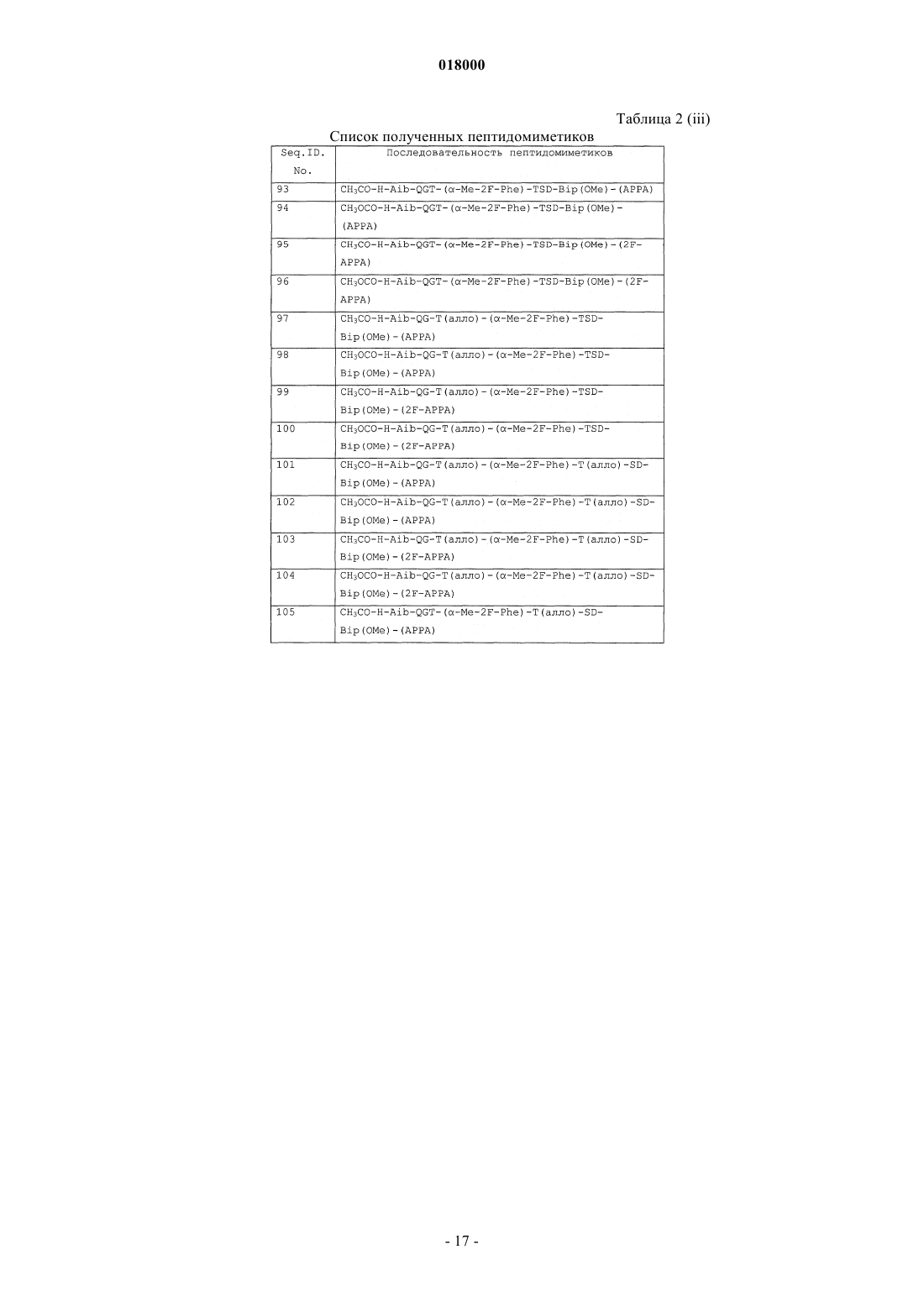
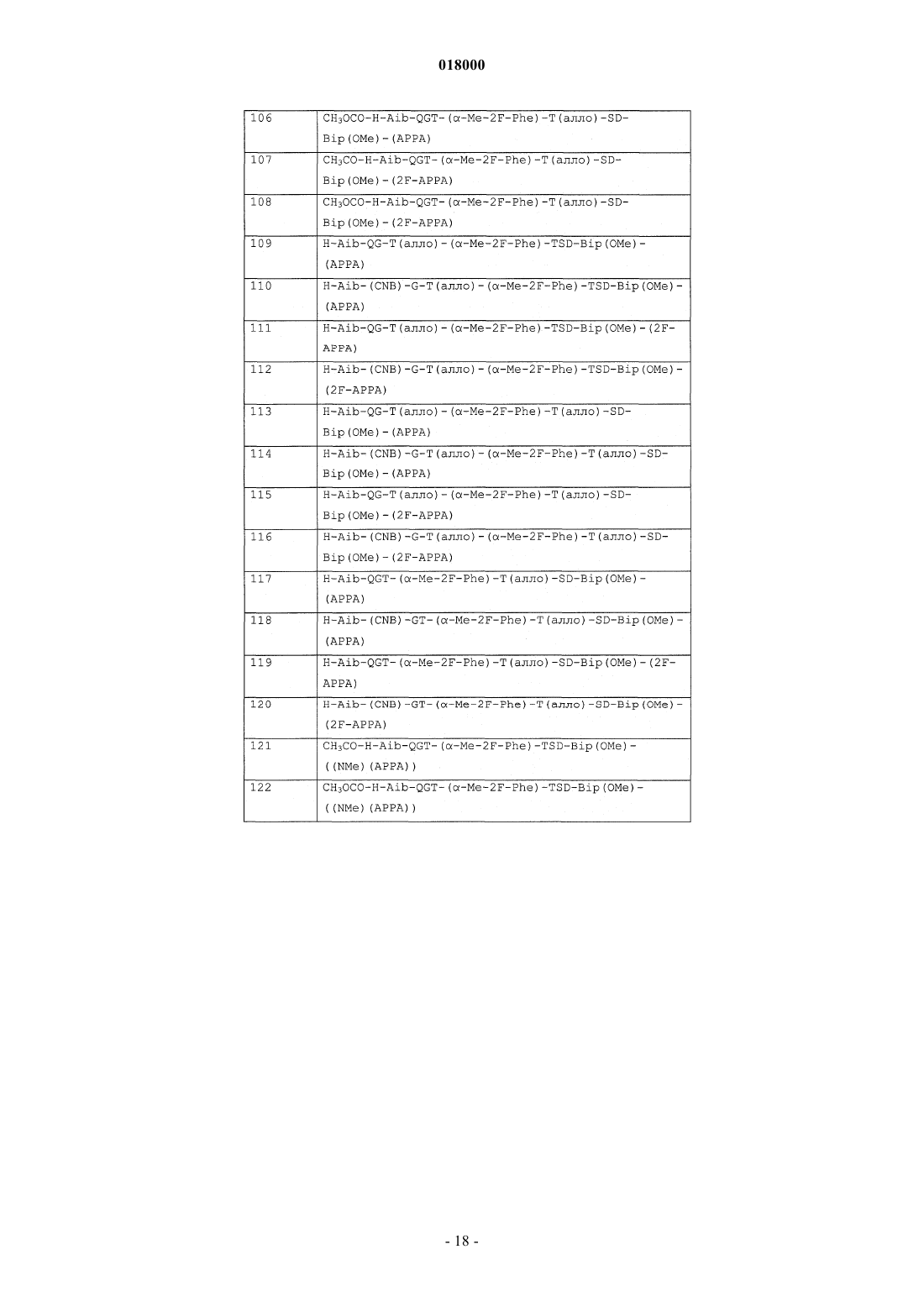
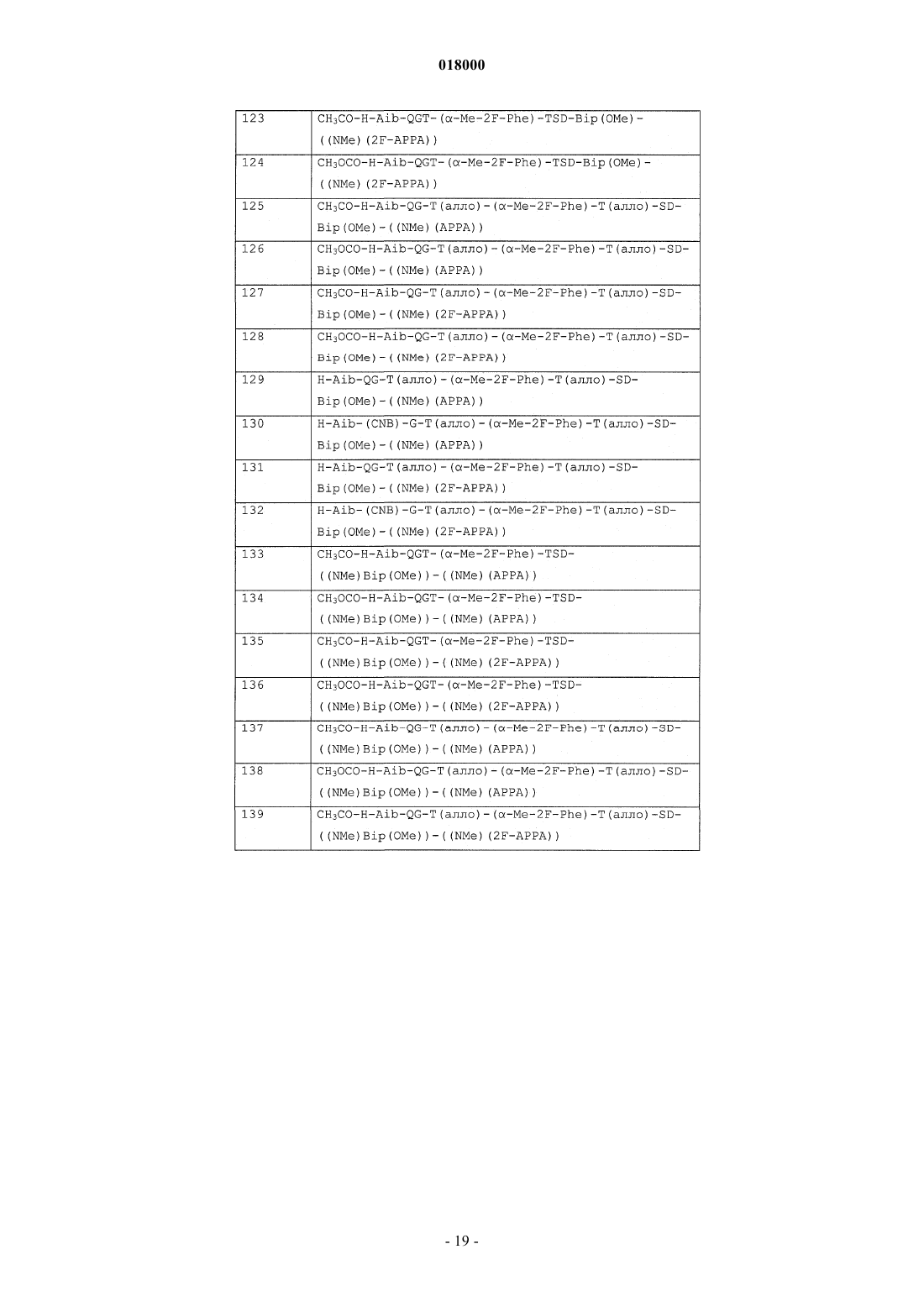
10. Соединение формулы (I), выбранное из:











11. Фармацевтическая композиция, предназначенная для снижения уровней циркулирующей глюкозы и для лечения диабета, содержащая соединения формулы (I) по любому из предшествующих пунктов и подходящий фармацевтически приемлемый носитель(и).
12. Способ профилактики или лечения заболеваний, вызываемых гиперлипидемией, гиперхолестеринемией, гипергликемией, гиперинсулинемией, повышенными уровнями свободных жирных кислот или глицерина в крови, гипертриглицеридемией; замедленным заживлением ран, нарушенной толерантностью к глюкозе, резистентностью к лептину, резистентностью к инсулину или другими осложнениями диабета, включающий введение нуждающемуся в этом пациенту эффективного нетоксического количества соединения формулы (I) по любому из пп.1-10.
13. Способ по п.12, где заболевание представляет собой диабет 2 типа, нарушенную толерантность к глюкозе, дислипидемию, гипертензию, атеросклероз, гиперлипидемию, ишемическую болезнь сердца, сердечно-сосудистые нарушения и другие заболевания, где лежащим в основе патофизиологическим механизмом является резистентность к инсулину.
14. Лекарственное средство для лечения/устранения любого из болезненных состояний, указанных в пп.12 и 13, включающее соединение формулы (I) по любому из пп.1-10 и фармацевтически приемлемый носитель, разбавитель, эксципиент или сольват.
15. Применение соединений формулы (I) по любому из пп.1-10 или их фармацевтической композиции по п.11 или содержащего их лекарственного средства по п.14 для лечения заболеваний, указанных в пп.12 и 13.

Текст
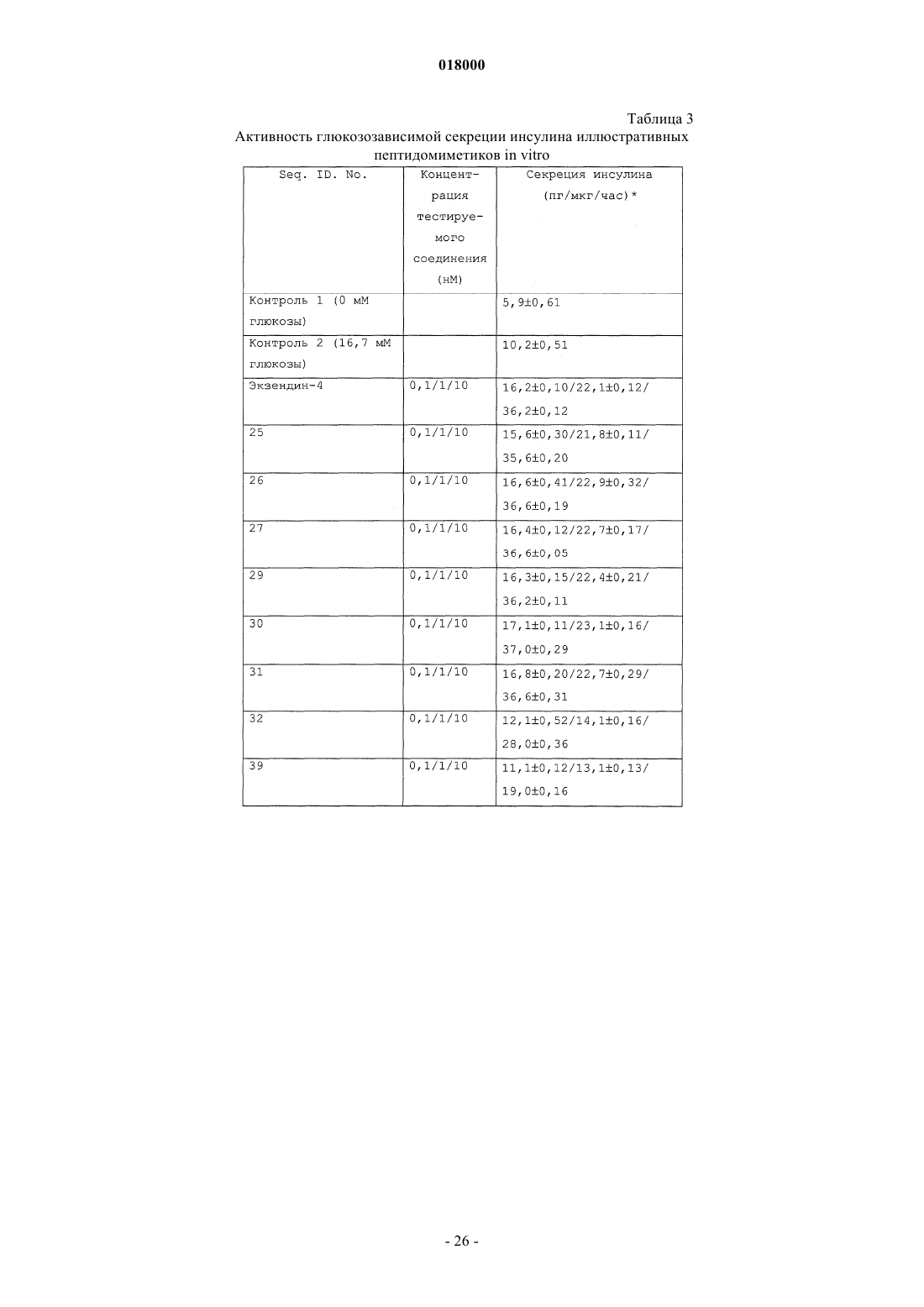
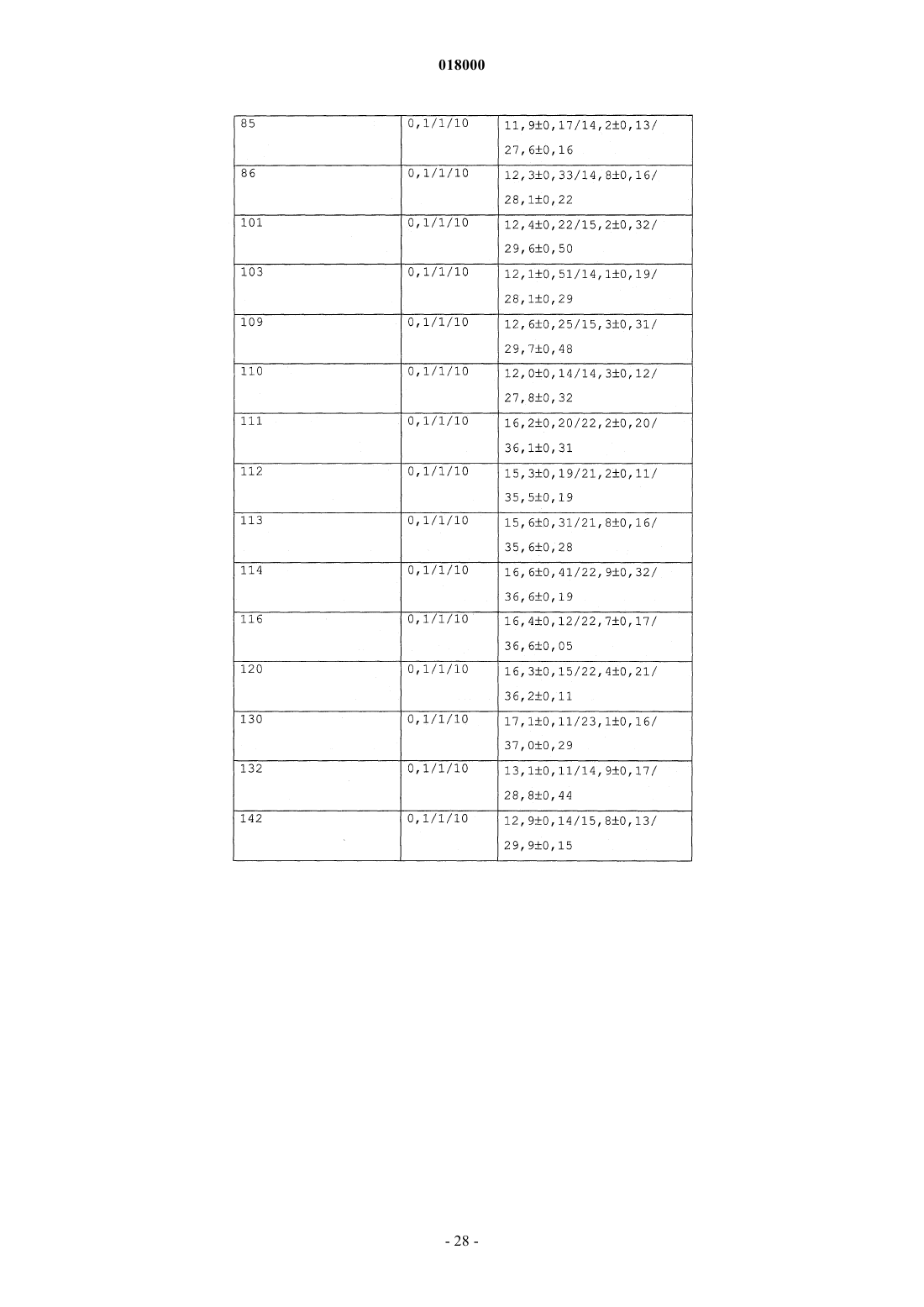
ПЕПТИДОМИМЕТИКИ С АКТИВНОСТЬЮ АНТАГОНИСТОВ ГЛЮКАГОНА И АГОНИСТОВ GLP-1 Изобретение относится к новым пептидомиметикам формулы (I), которые преимущественно действуют как стимуляторы глюкозозависимой секреции инсулина. Кроме того, выявлено, что эти пептидомиметики наряду с активностью агониста рецептора GLP-1 демонстрируют активность антагониста рецептора глюкагона.(71)(73) Заявитель и патентовладелец: КАДИЛА ХЕЛЗКЭР ЛИМИТЕД (IN) Область изобретения Настоящее изобретение относится к новым соединениям общей формулы (I), к их таутомерным формам, их фармацевтически приемлемым солям и к содержащим их фармацевтическим композициям Настоящее изобретение также относится к способу получения соединений общей формулы (I), их таутомерных форм, их фармацевтически приемлемых солей и содержащих их фармацевтических композиций. Предпосылки изобретения Диабет характеризуется нарушенной секрецией инсулина из -клеток поджелудочной железы, резистентностью к инсулину или и тем, и другим (Cavaghan, M.K., et al., J. Clin. Invest. 2000, 106, 329). Большинство пациентов с диабетом 2 типа можно лечить средствами, снижающими продукцию глюкозы печенью (антагонист глюкагона), снижающими всасывание глюкозы из ЖКТ, стимулирующими функцию -клеток (стимуляторы секреции инсулина), или средствами, увеличивающими чувствительность ткани пациентов к инсулину (сенсибилизаторы инсулина). Лекарственные средства, применяемые в настоящее время для лечения диабета 2 типа, включают ингибиторы -глюкозидазы, сенсибилизаторы инсулина, стимуляторы секреции инсулина и блокаторы KATP-каналов (Chehade, J.M., et al., Drugs, 2000, 60,95). Однако почти у половины индивидуумов с диабетом 2 типа через определенный промежуток времени исчезает ответ на эти средства, после чего им необходима инсулинотерапия. У лечения инсулином существует несколько недостатков, он является инъецируемым, вызывает гипогликемию и вызывает увеличение массы (Burge, M.R., Diabetes Obes. Metab., 1999, 1, 199). Недостатки современных способов лечения делают необходимыми новые средства лечения диабета 2 типа. В связи с этим признано, что терапевтически потенциальными являются агонист глюкагоноподобного пептида 1 (GLP-1), стимулирующий глюкозозависимую секрецию инсулина в поджелудочной железе, и антагонист рецептора глюкагона, ингибирующий продукцию глюкозы печенью, ингибируя гликогенолиз и глюконеогенез. Таким образом, выявлено, что агонист GLP-1 и антагонист глюкагона вместе снижают уровни циркулирующей глюкозы и представляют собой пригодные терапевтические средства для лечения и профилактики диабета 2 типа (Perry, Т.A., et al., Trends Pharmacol. Sci., 2003, 24,377). Глюкагон и GLP-1 являются представителями семейства структурно родственных пептидных гормонов (семейство секретина). Глюкагон и GLP-1 составляют набор высокогомологичных пептидов, так как эти два гормона происходят от общего предшественника, препроглюкагона, который после тканеспецифического процессинга приводит к продукции GLP-1, преимущественно в кишечнике, и глюкагона в поджелудочной железе (Jiang, G., et al., Am. J. Physiol. Endocrinol. Metab., 2003, 284, E671-678). Рецепторы этих двух пептидов являются гомологичными (идентичными на 58%) и принадлежат к классу В семейства связанных с G-белками рецепторов (GPCR). GPCR класса B также называют семейством секретиновых рецепторов, состоящем у людей из 15 связывающих пептиды рецепторов. РецепторыGPCR включают внеклеточный N-концевой домен из 100-160 остатков, соединенный с околомембранным доменом (J-домен) из семи трансмембранных -спиралей с промежуточными петлями, иC-концевой хвост (Brubaker, P.L., et al., Receptors Channels, 2002, 8, 179). GPCR класса B активируются эндогенными пептидными лигандами промежуточного размера, как правило, из 30-40 аминокислот(Hoare, S.R.J., Drug. Discovery Today, 2005, 10, 423; Gether, U., Endocrine Reviews, 2000, 21, 90). Глюкагон представляет собой пептидный гормон из 29 аминокислот, процессируемый РС 2 из проглюкагона в -клетках поджелудочной железы. Глюкагон действует через семь трансмембранных GPCR,состоящих из 485 аминокислот. Глюкагон высвобождается в кровоток при низком уровне циркулирующей глюкозы. Основной физиологической ролью глюкагона является стимуляция продукции глюкозы печенью, приводящая, таким образом, к увеличению гликемии (Tan, K., et al., Diabetologia, 1985, 28, 435). Глюкагон обеспечивает основной противодействующий инсулину механизм регуляции поддержания гомеостаза глюкозы in vivo. Глюкагон и его рецептор представляют собой потенциальные мишени для лечения диабета. Противодействие действию глюкагона посредством блокирования действия секретируемого глюкагона на рецептор глюкагона (антагонист глюкагона) или посредством ингибирования (супрессии) продукции самого глюкагона является новым направлением для воздействия на диабет и нарушения обмена веществ (Unson, С.G., et al., Peptides, 1989, 10, 1171; Parker, J.С., Diabetes, 2000, 49, 2079;Johnson, D.G., Science, 1982, 215, 1115; Ann, J.M., JMC, 2001, 44(9), 1372-1379). Амид GLP-1 (7-36) представляет собой продукт гена препроглюкагона, секретируемый L-клетками кишечника в ответ на потребление пищи. Физиологическое действие GLP-1 стало объектом значительного интереса. GLP-1 оказывает множественное действие, стимулируя секрецию инсулина из -клеток поджелудочной железы, независимым от глюкозы способом (инсулинотропное действие). GLP-1 снижает концентрацию циркулирующего в плазме глюкагона, ингибируя его секрецию (продукцию) из-клеток (Drucker D.J., Endocrinology, 2001, 142, 521-527). GLP-1 также проявляет свойства, подобные стимуляции роста -клеток, подавлению аппетита, задержке эвакуации желудочного содержимого и стимуляции чувствительности к инсулину (Nauck, M.A., Horm. Metab. Res., 2004, 36, 852). В настоящее вре-1 018000 мя на различных стадиях клинических исследований находятся различные аналоги GLP-1 и ЕХ-4, такие как лираглутид/NN2211 (Novo Nordisk; фаза III; WO 1998/008871), BIM 51077 (Ipsen; фаза II;WO 2001/004156) (Nauck M.A., Regulatory Peptides, 2004, 115, 13). Недавно на рынок США выпущена баета (BYETTA) (экзендин 4, АС 2933; US 5424286) (Amylin and Lilly). Однако все существующие агонисты GLP-1 вводят парентеральным способом введения так, что основной проблемой существующей терапии на основе GLP-1 является несоблюдение пациентом схемы лечения. Эффекторная система рецепторов глюкагона и GLP-1 представляет собой фермент аденилатциклазу(АС). Взаимодействие глюкагона или агониста GLP-1 с рецепторами глюкагона или GLP-1 (GLP-1R) соответственно вызывает активацию АС, преобразующую АТФ в цАМФ. Увеличение уровня внутриклеточного цАМФ увеличивает отношение АДФ/АТФ, таким образом вызывая деполяризацию клетки(вследствие закрытия KATP-каналов). Увеличение уровня внутриклеточного цАМФ также активирует протеинкиназу (PK-A и PK-C), увеличивающую концентрацию Са 2+ в цитозоле, открывая L-тип Са 2+ каналов. Увеличение внутриклеточного Са 2+ приводит к экзоцитозу инсулина в -клетках поджелудочной железы и пептида глюкагона в -клетках (Fehmann, H.C., Endocr. Rev., 1995, 16, 390). Приведенное ниже выравнивание последовательностей GLP-1 и глюкагона представляет соответствие первичных структур. Глюкагон:NH3(+)-1HAE(-)GTFTSD9(-)-CONH2 (Seq. ID. No. 3): суммарный заряд отрицательный. Первые N-концевые 1-9 остатки пептида глюкагона с C-концевым амидом:NH3(+)-1HSQGTFTSD9(-)-CONH2 (Seq. ID. No. 4): суммарный заряд нейтральный. Однобуквенные сокращения для аминокислот можно найти в Zubay, G., Biochemistry 2nd ed., 1988,MacMillan Publishing, New York, p. 33. Природные или синтетические пептиды GLP-1 быстро метаболизируются протеолитическими ферментами, такими как дипептидилпептидаза IV (DPP-IV), в неактивный метаболит, ограничивая, таким образом, применение GLP-1 в качестве лекарственного средства (Deacon, С.F., Regulatory Peptides, 2005,128, 117). Аналогично, в последние годы опубликовано несколько непептидных и пептидных антагонистов рецепторов глюкагона различной структуры, но ни один из них не находится в активной разработке или в клинических испытаниях (Kurukulasuriya, R., Expert Opinion Therapeutic Patents, 2005, 15, 1739; Lau,J., J. Med. Chem., 2007, 50, 113; Petersen, K.F. Diabetologia, 2001, 44, 2018; Cascieri, M.A., JBC, 1999, 274,8694). Полагают, что выявление непептидных лигандов (особенно агонистов) для GPCR класса B является основной трудностью при поиске лекарственных средств. HTS по-видимому дало несколько вариантов (US 2005/6927214; WO 2000/042026; US 2007/0043093), однако скрининг этих вариантов в отношении соответствующих рецепторов, особенно в условиях in vivo (модели на животных), имеет тенденцию к ложноотрицательным результатам (Murphy, K.G., PNAS, 2007, 104, 689). Глюкагон и GLP-1 играют большую роль в общем гомеостазе глюкозы (Drucker, D.J., J. Clin. Invest.,2007, 117, 24; Bollyky, J., J. Clin. Endocrinol. Metab., 2007, 92, 2879). Глюкагон увеличивает концентрации глюкозы в плазме, стимулируя глюконеогенез и гликогенолиз в печени, тогда как GLP-1 снижает концентрации глюкозы в плазме, опосредуемые глюкозозависимой секрецией инсулина (Mojsov, S., et al.,JBC, 1990, 265, 8001). С учетом важности пептида глюкагона и GLP-1 в поддержании нормальной концентрации глюкозы в крови в последние годы возник значительный интерес в идентификации одного лиганда, действующего в качестве антагонистов рецепторов глюкагона и агонистов рецепторов GLP-1(Claus, Т.Н., J. Endocrinology, 2007, 192, 371; Pan C.Q., JBC, 2006, 281, 12506). Хотя идентификация эффективного непептидного агониста GLP-1 может быть затруднительной(Chen, D., PNAS, 2007, 104, 943; Knudsen, L.В., PNAS, 2007, 104, 937), разработка гибридного пептидомиметика, действующего в качестве и антагониста глюкагона, и агониста рецептора GLP-1, вероятно,может предоставить новый подход для лечения диабета 2 типа (Claus, Т.Н., J. Endocrinology, 2007, 192,371). В последнее время опубликован ряд химерных пептидов, которые действуют и как агонист рецептора GLP-1, и как антагонист рецепторов глюкагона, сконструированных в основном посредством комбинирования N-концевых остатков пептида глюкагона (остатки 1-26) с последними 4 C-концевыми остатками пептида GLP-1 (VKGR) (Pan C.Q., et al., US 6864069 B2; Pan C.Q., JBC, 2006, 281, 12506). В литературе опубликованы исследования зависимости структура-активность (SAR) с определением роли индивидуальных аминокислот в последовательностях глюкагона и GLP-1 (Runge, S., JBC, 2003,278, 28005; Mann, R., Biochem. Soc. Trans., 2007, 35, 713). У глюкагона и GLP-1 в водном растворе нет определенной структуры, но в присутствии мицелл или в окружении мембраномиметиков они принимают в центральном участке альфа-спиральную структуру с подвижными N- и C-концевыми областями предположить, что спиральная структура необходима для связывания пептидных лигандов с их соответствующими рецепторами. Мутации или делеции аминокислот в N-концевой области у обоих пептидов приводят к антагонистам рецепторов или неактивным соединениям, что позволяет предположить важность N-конца для активации рецептора пептидами глюкагоном и GLP-1 (Hjorth, S.A., JBC, 1994, 269,30121; Green, В.D., J. Mol. Endocrinology, 2003, 31, 529). In vivo, GLP-1 быстро разрушается дипептидилпептидазой IV (DPP IV), протеазой, отвечающей за расщепление пептидов, содержащих остатки пролина и аланина в предпоследнем положении N-конца, что приводит к неактивным метаболитам. Замены чувствительных к DPP-IV участков, таких как замена Ala во 2 положении пептида GLP-1 на D-Ala, Aib илиHfl (гексафторлейцин) значительно увеличивают стабильность в плазме (Deacon, С.F., Diabetes, 1998, 47,764; Meng, H., J. Med. Chem., 2008, 51, 7303-7307). В настоящем исследовании авторы выявили, что связывание N-концевой последовательности пептида глюкагона (первые 1-9 остатки, Seq. ID. No. 4) с дипептидом из двух неприродных аминокислот,приводит к получению нового класса пептидомиметиков с активностью в качестве антагониста глюкагона и агониста GLP-1 с различной степенью селективности. Для увеличения продолжительности действия и стабильности в отношении фермента DPP-IV авторы сайт-специфически модифицировали гибридные пептидомиметики селективно в положении Z2 неприродными аминокислотами, такими как D-Ala, Aib,-метилпролин (-Me-Pro), 1-аминоциклопентанкарбоновая кислота (APP) и 1-аминоциклопропанкарбоновая кислота (АСР), и добились получения коротких пептидомиметиков. Некоторые из пептидомиметиков продемонстрировали эффективность даже при пероральном способе введения при сохранении активности антагониста глюкагона и агониста GLP-1. Предшествующий уровень техники и стратегия конструирования Опубликован ряд модифицированных по N-концу модуляторов GLP-1 с общей формулой Xaa1Xaa11, где Хаа 1-Хаа 9 представляет первые 1-9 остатки пептида GLP-1 (HAEGTFTSD; Seq. ID. No. 3), с некоторыми аналогами, где Хаа 2 или представляет собой Ala, или необязательно замещен Aib; Хаа 3 представляет собой аминокислоты с боковой цепью в виде карбоновой кислоты, такие как глутаминовая кислота и аспарагиновая кислота, но не Gln (Q), который консервативен в N-концевой последовательности пептида глюкагона (HSQGTFTSD, Seq. ID. No. 4); Хаа 6 представляет Phe или необязательно замещенMe-2F-Phe-; Хаа 9 представляет собой аминокислоты с боковыми цепями в виде карбоновой кислоты или амида, такие как аспарагиновая кислота, глутаминовая кислота, аспарагин и т.д.; Хаа 10 представляет собой производные замещенного или незамещенного бифенилаланина (Bip) и Хаа 11 представляет собой производные замещенного или незамещенного бифенилаланина (Bip) или производные замещенной или незамещенной 2-амино-5-фенилпентановой кислоты (APPA) WO 2003/033671 А 2; US 2004/0127423 A1;B2; US 2007/7238671 B2; US 2007/7238670 B2; US 2007/0287670 A1; US 2008/0045461 A1). Ранее опубликован ряд новых пептидомиметиков из 11 аминокислот, в основном состоящих изN-концевой последовательности пептида глюкагона (первые 1-9 остатки, Seq. ID. No. 4) в качестве активирующего компонента, ковалентно связанных с дипептидом из двух неприродных аминокислот (производные замещенного или незамещенного бифенилаланина (Bip в качестве связывающего компонента,которые преимущественно продемонстрировали глюкозозависимую секрецию инсулина. Кроме того, эти пептидомиметики, наряду с активностью агониста рецептора GLP-1, продемонстрировали активность антагониста рецептора глюкагона (WO 2008/062457 А 2). В настоящем изобретении предоставлены новые пептидомиметики формулы (I) (далее в настоящем документе обозначаемые как пептидомиметики). В настоящем изобретении вместо основанном на бифенилаланине (Bip) дипептидном связывающем компоненте авторы в качестве связывающих компонентов использовали производные на основе замещенного или незамещенного дипептида Bip(ОМе)-APPA. Неожиданно вместо первых 9 остатков N-концевой последовательности пептида GLP-1 (HAEGTFTSD; Seq.ID. No. 3), когда этот дипептид присоединили к первым 9 остаткам N-концевой последовательности пептида глюкагона (1HSQGTFTSD9; Seq. ID. No. 4), авторы обнаружили, что этот пептидомиметик (NH3+HSQGTFTSD-Bip(ОМе)-(APPA)-CONH2; Seq. ID. No. 5), наряду с активностью агониста рецептора GLP1, преимущественно демонстрировал активность антагониста рецептора глюкагона, фиг. 1. Новые пептидомиметики формулы (I) преимущественно действуют в качестве антагониста рецепторов глюкагона, а также демонстрируют эффекты агониста GLP-1R. Различные пептидомиметики, представленные в настоящем изобретении, продемонстрировали существенную глюкозозависимую секрецию инсулина (in vitro) и снижали уровни циркулирующей глюкозы (in vivo) с различным уровнем аффинности/селективности к рецепторам глюкагона и GLP-1. Кроме того, по сравнению с предыдущими опубликованными пептидомиметиками (например, как в WO 2008/062457 А 2), данные пептидомиметики с производными на основе замещенного или незамещенного дипептида Bip(ОМе)-APPA в качестве связывающих компонентов продемонстрировали увеличенную стабильность в отношении протеолитического расщепления, особенно в отношении DPP-IV, желудочных и кишечных ферментов. Таким образом, выявлено несколько стабильных в отношении ферментов ЖКТ и кислого рН желудка пептидомиметиков с улучшенной пероральной биодоступностью, что делает их подходящими кандидатами для лечения/облегчения/профилактики диабета 1 и 2 типа, нарушений обмена веществ и связанных нарушений. Сущность изобретения Настоящее изобретение относится к группе новых пептидомиметиков, функционирующих в качестве как антагониста рецептора глюкагона, так и агониста рецептора GLP-1 с различной степенью аффинности/селективности в отношении обоих рецепторов и пригодных для снижения уровней циркулирующей глюкозы и для лечения диабета. Эти пептидомиметики определены основной формулой (I), как приведено ниже. Пептидомиметики по настоящему изобретению пригодны для лечения человека или животного, регулируя действие инсулина и глюкагона. Вследствие встраивания нового и метаболически стабильного связывающего компонента пептидомиметики по настоящему изобретению продемонстрировали улучшенную пероральную биодоступность, и, таким образом, выявлено, что они подходят для лечения/облегчения/регуляции или профилактики диабета 1 и 2 типа и ассоциированных нарушений обмена веществ. Предпочтительные варианты осуществления В предпочтительном варианте осуществления настоящего изобретения предоставлены новые пептидомиметики общей формулы (I), их таутомерные формы, новые промежуточные соединения, участвующие в их синтезе, их фармацевтически приемлемые соли, их фармацевтически приемлемые сольваты и содержащие их или их смеси фармацевтические композиции, пригодные для лечения/облегчения/регуляции диабета. В другом предпочтительном варианте осуществления предоставлен способ получения новых пептидомиметиков общей формулы (I), их таутомерных форм, их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов и фармацевтических композиций, содержащих их. В дополнительном предпочтительном варианте осуществления предоставлены фармацевтические композиции, содержащие пептидомиметики общей формулы (I), их таутомерные формы, их фармацевтически приемлемые соли, сольваты и их смеси с фармацевтически приемлемыми носителями, растворителями, разбавителями, эксципиентами и другими средствами, как правило, применяемыми для их производства. В дополнительном предпочтительном варианте осуществления предоставлено применение новых пептидомиметиков по настоящему изобретению в качестве противодиабетических средств посредством введения терапевтически эффективного и нетоксичного количества пептидомиметиков формулы (I) или их фармацевтически приемлемых композиций млекопитающим, нуждающимся в таком лечении. Используемые сокращения В примерах и других разделах настоящего документа использовались следующие сокращения: г - грамм(ы),GLP-1R - рецептор 1 глюкагоноподобного пептида,глюкагон R - рецептор глюкагона,ч - час(ы),Hf1 - 5,5,5,5',5',5'-2S-гексафторлейцин,HOBt - гидроксибензотриазол,HOAt- 7-азагидроксибензотриазол,HBTU - гексафторфосфат 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметиламмония,ВЭЖХ - высокоэффективная жидкостная хроматография,в/б - внутрибрюшинно,л - литр,LC/MS - жидкостная хроматография/масс-спектрометрия,Me - метил,мин - минута(ы),мл - миллилитр,мкл - микролитр,мг - миллиграмм(ы),ммоль - миллимоль(и),MS - масс-спектрометрия,РуВОР - гексафторфосфат бензотриазол-1-илокси-трис-пирролидинофосфония,SPPS - твердофазный пептидный синтез,п/к - подкожно,TMS - триметилсилил,TIPS - триизопропилсилан,TFA - трифторуксусная кислота,TBTU - тетрафтороборат 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметиламмония,Trt - тритильная группа. Краткое описание чертежей На фиг. 1 проиллюстрирована активность антагониста рецептора глюкагона человека и активность агониста рецептора GLP-1 с Seq. ID. No. 5 in vitro. На фиг. 2 проиллюстрированы примеры ортогонально защищенных аминокислот, используемых в основанном на Fmoc твердофазном пептидном синтезе (SPPS) пептидомиметиков. На фиг. 3 проиллюстрировано определение DRC и ЕС 50 экзендина (фиг. А) и Seq. ID. No. 38(фиг. В) in vitro в анализе с HGLP-1R. На фиг. 4 проиллюстрировано снижение уровня глюкозы in vivo у мышей С 57, при примененииSeq. ID. No. 38, после внутрибрюшинного (в/б) введения. На фиг. 5 проиллюстрировано снижение уровня глюкозы in vivo у мышей С 57 при примененииSeq. ID. No. 38, после перорального (п/о) введения. На фиг. 6 проиллюстрировано снижение уровня глюкозы in vivo у мышей db/db при примененииSeq. ID. No. 38, после перорального (п/о) введения. На фиг. 7 проиллюстрированы уровни сывороточного инсулина после однократного перорального введения носителей/тестируемых пептидомиметиков (Seq. ID. No. 10, 20 и 25) мышам C57BL/6J (in vivo). Подробное описание изобретения По настоящему изобретению предоставлены синтетические пептидомиметики со структурной формулой (I), демонстрирующие глюкозозависимую секрецию инсулина. Кроме того, выявлено, что эти пептидомиметики, наряду с активностью агониста рецептора GLP-1, продемонстрировали активность антагониста рецептора глюкагона. Это двойное действие пептидомиметиков демонстрирует увеличенную стабильность в отношении протеолитического расщепления, особенно в отношении фермента DPP-IV(дипептидилпептидазы IV). Выявлено, что большинство пептидомиметиков стабильны в плазме крыс в пределах 24 ч (in vitro), демонстрируя увеличенную стабильность в отношении ферментов ЖКТ, таких как пепсин, и кислого рН желудка, а также в отношении микросом печени (in vitro). Вследствие увеличенной метаболической стабильности некоторые из этих пептидомиметиков для лечения или профилактики диабета и связанных нарушений обмена веществ также можно вводить пероральным способом в которой А представляет собой группы -NH-R1, R3-CO-, R3-O-CO- или R3-SO2-, где R1 представляет собой водород или необязательно замещенную линейную или разветвленную (C1-С 10)алкильную цепь;R3 выбран из линейных или разветвленных (C1-С 10)алкильной, (С 3-С 6)циклоалкильной, арильной, гетероарильной или арилалкильной групп; в предпочтительном варианте осуществления арильная группа выбрана из фенильной, дифенильной, нафтильной, инданильной, флуоренильной или бифенильной групп; гетероарильная группа выбрана из пиридильной, тиенильной, фурильной, имидазолильной, бензофуранильной групп;B представляет собой -COOR2, -CONHR2 или CH2OR2, где R2 представляет собой Н, необязательно замещенные группы, выбранные из линейных или разветвленных (C1-С 10)алкильной, арильной или аралкильной групп, как определено ранее;Z1-Z11 представляет собой природные или неприродные аминокислоты, связанные вместе амидной связью, если не указано иначе;Z1 представляет собой L-гистидин (Н), D-гистидин (dH) или урокановую кислоту (UA)Z2 представляет собой природную или неприродную аминокислоту, выбранную из группы, состоящей из L-серина (S), D-серина (dS), L-аланина (А), D-аланина (dA), -метилпролина (-Me-Pro),-аминоизомасляной кислоты (Aib), 1-аминоциклопропанкарбоновой кислоты (ACP), 1-аминоциклопентанкарбоновой кислоты (APP):Z3 представляет собой L-глутамин (Gln; Q), D-глутамин (dQ) или соединения формулы (II) (CNB или Hfl):Z4 представляет собой глицин (G) или неприродные аминокислоты 1-аминоциклопропанкарбоновую кислоту (АСР) или 1-аминоциклопентанкарбоновую кислоту (APP);Z5 представляет собой природную или неприродную аминокислоту, содержащую гидроксильную боковую цепь; предпочтительно Z5 представляет собой L-треонин (Т), D-треонин (dT), L-аллотреонинZ6 представляет собой природную или неприродную аминокислоту с двузамещенным альфа-атомом углерода с двумя боковыми цепями, где каждая из них независимо может необязательно быть замещена алкильной, или арильной, или аралкильной группой, где заместители можно выбирать из одной или нескольких алкильных групп или одной или нескольких галогеновых групп. Предпочтительно Z6 представляет собой фенилаланин (Phe; F), альфа-метилфенилаланин (Me-Phe-), альфа-метил-2 фторфенилаланин (Me-2F-Phe-) или альфа-метил-2,6-дифторфенилаланин (Me-2,6-F-Phe-) или 2 фторфенилаланин (-2F-Phe-):Z7 и Z8 независимо представляет собой природную или неприродную аминокислоту, содержащую гидроксильную боковую цепь, предпочтительные Z7 и Z8 независимо выбраны из треонина, серина,1-аминоциклопропанкарбоновой кислоты (АСР), как определено ранее;Z9 независимо представляет собой природную или неприродную аминокислоту с аминокислотной боковой цепью, содержащей кислотную группу. Предпочтительный Z9 выбран из L-аспарагиновой кислоты (D), D-аспарагиновой кислоты (dD) или соединений формулы (II), как определено ранее;Z10 представляет собой L или D неприродную аминокислоту формулы IIIa-IIIc, выбранную из замещенного или незамещенного 2'-этил-4'-метоксибифенилаланина (Bip(OMe и его производных:Z11 представляет собой L или D неприродную аминокислоту формулы IV (а-l), выбранную из замещенной или незамещенной 2-амино-5-фенилпентановой кислоты (APPA) и ее производных. В дополнительном варианте осуществления амидная связь между Z9 и Z10, или Z10 и Z11, или Z9-Z11 может являться N-метилированной, представленной аббревиатурой (NMe), такой как D-(NMe)(Bip(OMe; (Bip(OMe-(NMe)-APPA; D-(NMe)-(Bip(OMe-(NMe)-APPA, может представлять собой тиоамидную связь, представленную аббревиатурой C=S, такую как D-(C=S)-(Bip(OMe; Bip(OMe)(C=S)-(APPA); D-(C=S)-(Bip(OMe-(C=S)-(APPA), или тиоамидную связь между Z9 и Z10, или Z10 и Z11,или Z9-Z11 можно восстанавливать до группы -СН 2- (дезоксопептид), такой как D-(СН 2)-(Bip(ОМе; Определения Термин "природные аминокислоты" означает все 20 аминокислот, которые существуют в природе. Термин "неприродные аминокислоты" представляет или замещение L-аминокислот соответствующими D-аминокислотами, такое как замещение L-Ala на D-Ala и т.п., или подходящие модификации-алкилирования, таким как замещение Ala на -метил-Ala (Aib), замещение Phe на альфаметилфенилаланин (Me-Phe-), альфа-метил-2-фторфенилаланин (Me-2F-Phe-) или альфа-метил-2,6 дифторфенилаланин (Me-2,6-F-Phe-); замещения на боковой цепи аминокислоты, такого как замещение ароматической боковой цепи аминокислоты галогеном, (C1-С 3)алкильной, арильной группами, более конкретно, замещение Phe альфаметил-2-фторфенилаланином (Me-2F-Phe-) или альфа-метил-2,6-дифторфенилаланином (Me-2,6-FPhe-); замещения амидной связи тиоамидной связью, представленной аббревиатурой "C=S", где химически модификацию дипептида из амида в тиоамид можно осуществлять, обрабатывая защищенный дипептид, в фазе раствора или на твердой подложке реагентом Лавессона. Кроме того, тиоамидную связь в дипептиде можно конвертировать в группу "-СН 2-" (дезоксопептид) с использованием никель-боридного восстановления (Jr. Guziec, F.S., Tetrahedron Letters, 1990, 31(1), 23-26). В следующих абзацах описаны различные группы, радикалы и заместители, используемые по всему описанию. Термин "алкил", используемый в настоящем документе, отдельно или в сочетании с другими радикалами, означает линейный или разветвленный радикал, содержащий от 1 до 10 атомов углерода, такой как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, амил, трет-амил, н-пентил,н-гексил, изогексил, гептил, октил, децил и т.п. Термин "циклоалкил", используемый в настоящем документе, отдельно или в сочетании с другими радикалами, означает радикал, содержащий от 3 до 7 атомов углерода, такой как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и т.п. Термин "арил" или "ароматический", используемый в настоящем документе, отдельно или в сочетании с другими радикалами, означает ароматическую систему, содержащую один, два или три цикла,где такие циклы могут быть соединены вместе боковыми цепями или могут быть слитыми, такую как фенил, нафтил, тетрагидронафтил, индан, бифенил и т.п. Термин "арилалкил" означает алкильную группу, как определено выше, соединенную с арилом, таким как бензил, фенилэтил, нафтилметил и т.п. Термин "арилокси" означает арильный радикал, как определено выше, соединенный с алкоксигруппой, такой как фенокси, нафтилокси и т.п., которая может быть замещенной. Термин "аралкокси" означает арилалкильную группу, как определено выше, такую как бензилокси,фенэтилокси, нафтилметилокси, фенилпропилокси и т.п., которая может быть замещенной. Термин "гетероарил" или "гетероароматический", используемый в настоящем документе, отдельно или в сочетании с другими радикалами, означает ароматическую систему, содержащую один, два или три цикла, где такие циклы могут быть соединены вместе боковыми цепями или могут быть слитыми,содержащую один или несколько гетероатомов, выбранных из O, N или S. Термин "гетероаралкил", используемый в настоящем документе, отдельно или в сочетании с другими радикалами, означает гетероарильную группу, как определено выше, присоединенную к неразветвленной или разветвленной насыщенной углеродной цепи, содержащей от 1 до 6 атомов углерода. Термины "гетероарилокси", "гетероаралкокси", "гетероциклокси" означают гетероарильную, гетероарилалкильную группу соответственно, как определено выше, присоединенную к атому кислорода. Термин "ацил", используемый в настоящем документе, отдельно или в сочетании с другими радикалами, означает радикал, содержащий от 1 до 8 атомов углерода, такой как формил, ацетил, пропаноил,бутаноил, изобутаноил, пентаноил, гексаноил, гептаноил, бензоил и т.п., который может быть замещенным. Термин "карбоновая кислота", используемый в настоящем документе, отдельно или в сочетании с другими радикалами, означает группу -COOH и включает производные карбоновых кислот, такие как сложные эфиры и амиды. Термин "сложный эфир", используемый в настоящем документе, отдельно или в сочетании с другими радикалами, означает группу -COO- и включает производные карбоновых кислот,где остатки сложных эфиров представляют собой алкоксикарбонил, такой как метоксикарбонил, этоксикарбонил и т.п., который может быть замещенным. Если не указано иначе, термин "аминокислота", как применяют в настоящем документе отдельно или как часть другой группы, включает в качестве неограничивающих примеров аминогруппу и карбоксильную группу, связанные с одним и тем же атомом углерода, обозначаемым как -атом углерода. Абсолютную "S-конфигурацию у -атома углерода, как правило, обозначают как "L" или природную конфигурацию. "R"-конфигурацию у -атома углерода, как правило, обозначают как"D-аминокислоту. В случае, когда оба "-заместителя" являются одинаковыми, такими как водород или метил, аминокислоты представляют собой Gly или Aib и являются нехиральными. Строчная "d" означает хиральность D-аминокислот. В описании, когда бы ни была описана аминокислота, если не указано иначе, она включает и L-, и D-формы. Таким образом, в табл. 2 приведен список пептидомиметиков, получаемых по настоящему изобретению, в котором каждая из аминокислот может представлять собой или"L", или "D". Термин "антагонист рецептора" относится к соединениям, которые ингибируют активацию рецептора и образование вторичного мессенджера, такого как циклический АМФ, посредством конкурентного или неконкурентного связывания. Термин "антагонист рецептора глюкагона" относится к соединениям, которые ингибируют активацию рецептора глюкагона. Термин "модулятор или агонист рецептора GLP-1" относится к соединению, которое действует на рецептор GLP-1, изменяя его способность регулировать нисходящие сигнальные события, такие как про-8 018000 дукцию цАМФ и высвобождение инсулина. Примеры модуляторов рецепторов включают агонист, частичный агонист, обратный агонист и аллостерические потенцирующие средства. В соответствии с настоящим изобретением синтетически выделенные пептидомиметики, описываемые в настоящем документе, преимущественно действуют в качестве антагонистов рецепторов глюкагона. Кроме того, выявлено, что эти пептидомиметики также действуют в качестве агонистов рецепторов GLP-1. Эти синтетические пептидомиметики демонстрируют желаемые свойства антагонистов рецепторов глюкагона, а также активность агонистов рецепторов GLP-1 in vitro в клетках CHO, трансфицированных рецептором глюкагона или GLP-1 человека (Н глюкагон R или HGLP-1R), в диапазоне концентраций 1-100 нМ. Активность агониста HGLP-1R оценивают, рассчитывая количество высвобождаемого цАМФ, тогда как активность антагониста глюкагона оценивали, измеряя количество продуцируемого цАМФ, ингибируемого при тестировании пептидомиметиков, в присутствии пептида глюкагона. Новые пептидомиметики демонстрируют желаемую активность антагонистов рецепторов глюкагона invitro в клетках CHO, трансфицированных рецептором глюкагона человека, в диапазоне концентраций 1-100 нМ. Некоторые из полученных тестируемых пептидомиметиков, при тестировании in vivo в различных моделях диабета на животных, таких как гипергликемические мыши С 57, мыши ob/ob и db/db,продемонстрировали глюкозозависимое высвобождение инсулина и снижали гипергликемию при голодании, не вызывая гипогликемии, что, таким образом, делает их идеальными кандидатами в лекарственные средства для лечения и профилактики диабета 2 типа. Эти новые классы пептидомиметиков можно вводить пероральным или парентеральным способом введения. Настоящее изобретение относится к пептидомиметикам формулы (I), фармацевтическим композициям, где используют такие пептидомиметики отдельно или в сочетании, и к способам применения таких пептидомиметиков. В частности, настоящее изобретение относится к фармацевтической композиции,содержащей терапевтически эффективное количество пептидомиметиков формулы (I), отдельно или в сочетании(ях) с фармацевтически приемлемым носителем. Дополнительно предоставлен способ лечения или задержки развития или начала диабета, особенно диабета 2 типа, включая осложнения диабета, включая ретинопатию, нейропатию, нефропатию и замедленное заживление ран и связанные заболевания, такие как резистентность к инсулину (ухудшенный гомеостаз глюкозы), гипергликемия, гиперинсулинемия, повышенные уровни жирных кислот или глицерина в крови, гиперлипидемия, включая гипертриглицеридемию, синдром X, атеросклероз и гипертензия, где терапевтически эффективное количество пептидомиметиков формулы (I) или их сочетания(ий) вводят нуждающемуся в этом пациенту-млекопитающему, например человеку. Получение пептидомиметиков. Для получения пептидомиметиков по настоящему изобретению можно использовать несколько способов синтеза, хорошо известных специалисту в области пептидного синтеза. Пептидомиметики формулы (I), где все символы являются такими, как определено ранее, можно синтезировать описанными ниже способами совместно с общепринятыми способами, известными специалистам в области пептидного синтеза, или их вариаций, как очевидно специалистам в данной области. Указанные способы включают способы, но не ограничиваются ими, описанные ниже. Соответствующие пептидомиметики, описываемые в настоящем документе, можно получать химическим синтезом с использованием подходящих вариаций различных способов твердофазного синтеза,как правило, известных, таких как способы, описанные в G. Barany and R.В. Merrifield, "The peptides:peptide synthesis". 2nd Ed., Pierce chemical Co., Rockford, Il, 1984. Предпочтительная стратегия для получения пептидомиметиков по данному изобретению основана на применении основанного на Fmoc подхода SPPS, где группу Fmoc (9-флуоренилметилметилоксикарбонил) используют для временной защиты -аминогруппы, в сочетании с кислыми лабильными защитными группами, такими как трет-бутилоксикарбонильная (Boc), трет-бутильная (But), тритильная(Trt) группа (фиг. 2), для временной защиты боковых цепей аминокислот (см., например, Е. Atherton andMeienhofer, Eds., Academic Press, San Diego, 1987). Пептидомиметики можно синтезировать поэтапным способом на нерастворимой полимерной подложке (смоле), начиная с C-конца пептида. В одном из вариантов осуществления синтез начинают, присоединяя C-концевую аминокислоту пептида к смоле посредством образования амидной, сложноэфирной или простой эфирной связи. Это обеспечивает последующее высвобождение полученного пептида в виде C-концевого амида, карбоновой кислоты или спирта соответственно. В основанном на Fmoc SPPS необходимо, чтобы у C-концевой аминокислоты и всех остальных аминокислот, используемых в синтезе, их -аминогруппы и функциональные группы боковых цепей (если присутствуют) были защищены различным способом (ортогональная защита) так, чтобы в течение синтеза можно было селективно удалять -аминозащитную группу, используя подходящее основание,-9 018000 такое как 20% раствор пиперидина, без какого-либо предварительного отщепления пептида от смолы или удаления защитных групп боковых цепей, как правило, защищенных кислыми лабильными защитными группами. Связывание аминокислоты осуществляют, активируя ее карбоксильную группу в виде активного сложного эфира и проводя ее реакцию с разблокированной -аминогруппой N-концевой аминокислоты,добавляемой к смоле. После каждого связывания и снятия защиты пептидил-смолу отмывают избытком растворителей, таких как DMF, DCM и простой диэтиловый эфир. Последовательность снятия защиты с-аминогруппы и связывания повторяют до сборки желаемой пептидной последовательности (схема 1). Затем пептид отщепляют от смолы с одновременным снятием защиты функциональных групп боковых цепей, используя соответствующую смесь для отщепления, как правило, в присутствии соответствующих акцепторов радикалов для ограничения побочных реакций. В заключение полученный пептид очищают посредством ВЭЖХ с обратной фазой. В синтезе пептидил-смол, необходимых в качестве предшественников конечных пептидов, используют коммерчески доступные поперечно сшитые полистирольные полимерные смолы (Novabiochem, SanDiego, CA). Для применения по настоящему изобретению предпочтительны смола Fmoc-PAL-PEG-PS,4- (2',4'-диметоксифенил-Fmoc-аминометил)феноксиацетил-п-метилбензгидриламиновая смола (смолаFmoc-Rink амид MBHA), 2-хлортритилхлоридная смола или смола с п-бензилоксибензиловым спиртом(смола НМР), к которым уже может быть или не быть присоединена C-концевая аминокислота. ЕслиC-концевая аминокислота не присоединена, ее присоединение можно обеспечить посредством активного сложного эфира HOBt защищенной Fmoc аминокислоты, формируемого ее реакцией с DIPCDI. В случае 2-хлортритиловой смолы связывание первой защищенной Fmoc аминокислоты обеспечивают, применяяDIPEA. Для присоединения следующей аминокислоты N-концевую защиту пептидил-смолы селективно снимают, используя раствор 10-20% пиперидина. После каждого связывания и снятия защиты избыток аминокислот и связывающих реагентов удаляют посредством отмывки DMF, DCM и простым диэтиловым эфиром. Связывание последующих аминокислот можно осуществлять с применением активных сложных эфиров HOBt или HOAT, получаемых из DIPCDI/HOBt или DIPCDI/HOAT соответственно. В случае какого-либо сложного связывания, особенно связывания тех аминокислот, которые являются гидрофобными, или аминокислот с объемной защитой боковой цепи, полное связывание можно обеспечить,используя сочетание высокоэффективных средств связывания, таких как HBTU, PyBOP или TBTU, с добавками, такими как DIPEA. Синтез пептидомиметиков, описываемых в настоящем документе, можно осуществлять с использованием устройства для периодического пептидного синтеза или устройства для пептидного синтеза в непрерывном потоке, такого как пептидный синтезатор CS-Bio или AAPPTEC, с использованием технологии защиты Fmoc/трет-бутил. Неприродные некоммерческие аминокислоты, находящиеся в различных положениях, вводили в пептидную цепь одним или несколькими известными в данной области способами. В одном из подходов защищенные Fmoc неприродные аминокислоты получали в растворе соответствующими описанными в литературе способами. Например, защищенные Fmoc -метилированные аминокислоты, такие как Fmoc-Aib-OH, Fmoc-(-Me-2F-Phe)-ОН, Fmoc-(-Me-2,6-F-Phe)-OH, получали модифицированным описанным в литературе способом (Boesten, W.H.J., et al., Org. Lett., 2001, 3(8), 1121;(Fmoc-APPA) и ее производных, как приведено в формуле IV(a-l), модифицированным описанным в литературе способом (Betshrugge, J.V., Tetrahedron, 1998, 54, 1753-1762; WO 2003/087036). Fmoc-Bip(OMe)OH (2'-этил-4'-метоксибифенилаланин; 2-амино-3-(2'-этил-4'-метоксибифенил-4-ил)пропионовую кислоту) получали описанным в литературе способом (Kotha, S., et al., Tetrahedron. 2002, 58, 9633; US 2006/0004222 A1), a Fmoc-5,5,5,5',5',5'-2S-гексафторлейцин (Fmoc-(Hfl)-ОН) получали опубликованным способом (Chiu, H.P., Cheng, R.P., Org. Lett., 2007, 9(26), 5517-5520). Затем полученные производные использовали в поэтапном синтезе пептида. Альтернативно, необходимую неприродную аминокислоту получали непосредственно на смоле способами синтеза органической химии и достраивали линейную пептидную цепь. Схема 1 Общая схема основанного на Fmoc SPPS Предшественники пептид-смола их соответствующих пептидомиметиков можно расщеплять и снимать с них защиту с применением подходящих вариаций стандартных способов расщепления, описанных в литературе (King, D.S., et al., Int. J. Peptide Protein Res., 1990, 36, 255). Предпочтительный способ для применения по настоящему изобретению представляет собой использование расщепляющей смеси сTFA, в присутствии воды и TIPS в качестве акцепторов радикалов. Как правило, пептидил-смолу инкубируют в TFA/воде/TIPS (94:3:3; об.:об.:об.; 10 мл/100 мг пептидил-смолы) в течение 1,5-2 ч при комнатной температуре. Отщепленную смолу отфильтровывают, раствор TFA концентрируют или сушат при пониженном давлении. Полученный неочищенный пептид осаждают или отмывают Et2O или повторно растворяют непосредственно в DMF или 50% водной уксусной кислоте для очистки посредством препаративной ВЭЖХ. Пептидомиметики с желаемой степенью очистки можно получать посредством очистки с применением препаративной ВЭЖХ. Раствор неочищенного пептида вносят в колонку semi-Prep (Luna 10 мкм;TFA, при скорости потока 15-50 мл/мин с мониторингом выходящего потока детектором PDA при 220 нм. Структуры очищенных пептидомиметиков можно подтверждать посредством анализа электроспрейной масс-спектроскопии (ES-MS). После очистки посредством препаративной ВЭЖХ все получаемые пептиды выделяли в виде трифторацетатной соли с TFA в качестве противоиона. Однако некоторые пептиды подвергали обессоливанию, пропуская через слой подходящей ионообменной смолы, предпочтительно через анионообменную смолу Dowex SBR P(Cl) или эквивалентную основную анионообменную смолу. В некоторых случаях противоионы TFA заменяли ацетатными ионами, пропуская через подходящую ионообменную смолу,элюируя разбавленным раствором уксусной кислоты. Для получения гидрохлоридной соли пептидов на последней стадии получения выбранные пептиды с ацетатной солью обрабатывали 4 М HCl. Получен- 11018000 ный раствор фильтровали через мембранный фильтр (0,2 мкм) и затем высушивали в вакууме с выходом соли HCl с цветом от белого до кремового. В результате применения сходных способов и/или определенных подходящих модификаций, которые все хорошо известны специалистам в данной области, получали другие подходящие фармацевтически приемлемые соли пептидомиметиков по настоящему изобретению. Основной способ получения пептидомиметиков с применением подхода SPPS. Получение пептидомиметиков на смоле. Достаточному количеству (50-100 мг) смолы Fmoc-PAL-PEG-PS или смолы Fmoc-Rink амидMBHA, нагруженной 0,5-0,6 ммоль/г позволяли набухать в DMF (10-20 мл/100 мг смолы) в течение 2-10 мин. Затем группу Fmoc на смоле удаляли посредством инкубации смолы с 10-20% пиперидином вDMF (10-30 мл/100 мг смолы) в течение 10-30 мин. Смолу со снятой защитой фильтровали и отмывали избытком DMF, DCM и простого диэтилового эфира (450 мл). Отмытую смолу инкубировали в свежеперегнанном DMF (1 мл/100 мг смолы) в атмосфере азота в течение 5 мин. К смоле добавляли 0,5 М раствор первой защищенной Fmoc аминокислоты (1-3 экв.), предварительно активированной HOBt (1-3 экв.) и DIPCDI (1-2 экв.) в DMF и затем смолу перемешивали в течение 1-3 ч в атмосфере азота. Завершение связывания контролировали посредством качественной нингидриновой реакции. После связывания первой аминокислоты смолу отмывали DMF, DCM и простым диэтиловым эфиром (450 мл). Для присоединения следующей аминокислоты сначала снимали защиту Fmoc на первой аминокислоте, связанной со смолой с использованием 20% раствора пиперидина, с последующим присоединением защищенной Fmoc второй аминокислоты с использованием подходящих связывающих средств, как описано выше. Проводили повторяющиеся циклы снятия защиты, отмывки, присоединения и отмывки, как на основной схеме 1 выше, до получения желаемой пептидной цепи. В заключение, с защищенной Fmoc пептидил-смолы, полученной выше, снимали защиту посредством обработки 20% пиперидином, как описано выше, и пептидил-смолы отмывали DMF, DCM и простым диэтиловым эфиром (450 мл). Смолу, содержащую желаемый пептид, сушили в атмосфере азота в течение 10-15 мин и подвергали отщеплению/снятию защиты. Иллюстративный пример автоматизированного твердофазного синтеза последовательности пептидаID. No. 37 (H2N-H-Aib-QGT-(-Me-2F-Phe)-TSD-Bip (OMe)-(APPA)-CONH2). Линейную пептидную цепь H2N-H-Aib-QGT-(-Me-2F-Phe)-TSD-Bip(OMe)-(APPA)-PAL-PEG-PS получали на автоматизированном синтезаторе CS-Bio 536 PepSynthesiser способом твердофазного пептидного синтеза (SPPS) с Fmoc (схема 2). Fmoc-аминокислоты и 2-(1H-бензотриазол-1-ил)-1,1,3,3 тетраметилуронийтетрафтороборат (TBTU) помещали вместе в ампулы и располагали в модуле синтезатора для аминокислот. Исходный раствор диизопропилэтиламина (DIPEA; 0,9 М) и DMF хранили в бутылках для реагентов в атмосфере сухого азота. Смолу Fmoc-PAL-PEG-PS (0,38 ммоль/г; 1 г) сушили над Р 2 О 5, в вакууме (1 ч) и позволяли набухать в свежеперегнанном DMF (5 мл). Набухшую смолу в виде суспензии помещали в стеклянную колонку и размещали в синтезаторе. Все циклы синтеза проводили со скоростью потока 5 млмин-1, табл. 1. Смолу отмывали свежеперегнанным DMF в течение 10 мин. Снятие защиты группой Fmoc проводили 20% пиперидином в DMF в течение 10 мин, и снятие защиты контролировали посредством УФ-детектирования стока колонки при 304 нм. Избыток пиперидина удаляли тремя дополнительными циклами отмывки и циклом отмывки перегнанным DMF с длительностью каждого цикла 15 мин. Аминогруппу обрабатывали Fmocаминокислотой (4 экв.), предварительно активированной TBTU (3,9 экв.) в присутствии DIPEA (8 экв.) и повторяли цикл в течение 120 мин. Избыток аминокислоты и растворимые побочные продукты удаляли из колонки и контура четырьмя дополнительными циклами отмывки и циклами отмывки перегнаннымDMF с длительностью каждого цикла 10 мин. Далее циклы синтеза (снятие защиты, отмывка, ацилирование и отмывка) повторяли до полной сборки линейного пептида. Последний цикл снятия защиты проводили с 20% пиперидином в DMF в течение 15 мин с удалением концевой группы Fmoc с последующим циклом отмывки (104 мин). Полученный пептид-смолу фильтровали через полученный спеканием стеклянный фильтр, последовательно 3 раза отмывали DMF, DCM, метанолом, DMF и простым диэтиловым эфиром (100 мл каждого). Пептид-смолу сушили в вакууме над Р 2 О 5 (2 ч) и хранили при -20 С. Проводили реакцию смолы с нингидрином для проверки N-концевой свободной аминогруппы связанного со смолой пептида. Появление сине-фиолетового окрашивания раствора и гранул смолы указывает на наличие у связанного со смолой пептида свободной аминогруппы, и это рассматривали как положительный результат теста. Таблица 1 Автоматизированные циклы для твердофазного пептидного синтеза Проводили расщепление в малом масштабе для определения чистоты связанного со смолой пептида. Высушенную пептид-смолу (приблизительно 10 мг) обрабатывали смесью (1 мл) TFA, воды, триизопропилсилана (95: 2,5: 2,5 об./об.) в течение 90 мин при комнатной температуре со слабым периодиче- 13018000 ским помешиванием. Смолу отфильтровывали, тщательно отмывали чистым TFA (1 мл) и весь фильтрат выпаривали при пониженном давлении. Оставшийся TFA 3 раза подвергали азеотропному смешиванию с простым диэтиловым эфиром (2 мл). Полученный осадок суспендировали в дистиллированной воде(2 мл) и водный слой 3 раза экстрагировали простым диэтиловым эфиром (3 мл). Водный слой отделяли и лиофилизировали с выходом неочищенного пептида H2N-H-Aib-QGT-(-Me-2F-Phe)-TSD-Bip(OMe)(APPA)-CONH2. Лиофилизированный пептид H2N-H-Aib-QGT-(-Me-2F-Phe)-TSD-Bip(OMe)-(APPA)CONH2 растворяли в 0,1% водном TFA (приблизительно 1 мг/1 мл) и его чистоту анализировали посредством аналитической ОФ-ВЭЖХ и характеризовали масс-спектрометрией с электроспрейной ионизацией(ESI-MS). Процент чистоты: 90% (неочищенный пептид). ESI-MS; вычислено для H2N-H-Aib-QGT-(Me-2F-Phe)-TSD-Bip(OMe)-(APPA)-CONH2: 1465 (M+), 1487 (M+Na+) и 1503 (M+K+); выявлено (m/z): 1465 (M+), 1487 (M+Na+) и 1503 (M+K+). Используя указанный выше протокол и его подходящие вариации, которые известны специалистам в данной области, получали пептидомиметики, предусмотренные по настоящему изобретению, с применением подхода Fmoc-SPPS. Далее, связанные со смолой пептидомиметики отщепляли и снимали защиту, очищали и характеризовали с использованием приведенного ниже протокола. Отщепление и снятие защиты. Желаемые пептидомиметики отщепляли и снимали защиту из их соответствующих пептидил-смол посредством обработки смесью для отщепления с TFA, как указано ниже. К пептидил-смолам добавляли раствор TFA/воды/триизопропилсилана (95:2,5:2,5) (10 мл/100 мг пептидил-смолы) и смесь оставляли при комнатной температуре с периодическим перемешиванием. Смолу отфильтровывали, отмывали смесь для отщепления и объединенный фильтрат выпаривали до сухости. Полученный остаток растворяли в 10 мл воды и водный слой 3 раза экстрагировали простым диэтиловым эфиром (20 мл каждый) и в заключение водный слой лиофилизировали. Полученный после лиофилизации неочищенный пептид очищали препаративной ВЭЖХ, как указано ниже. Очистка неочищенных пептидомиметиков препаративной ВЭЖХ. Препаративную ВЭЖХ проводили на жидкостном хроматографе Shimadzu LC-8A. Раствор неочищенного пептида, растворенный в DMF или воде, наносили в колонку semi-Prep (Luna 10 мкм; C18; 100 ), с размерами 25050 мм и элюировали в линейном градиенте ACN в воде, забуференных 0,1%TFA, при скорости потока 15-50 мл/мин с мониторингом выходящего потока детектором PDA при 220 нм. Как правило, использовали градиент смеси вода-ACN от 20 до 70%, забуференных 0,1% TFA, в течение периода 50 мин с изменением градиента на 1%/мин. Элюируемый желаемый продукт собирали в одну фракцию 10-20 мл и чистые пептидомиметики получали в виде аморфных белых порошков посредством лиофилизации соответствующих фракций ВЭЖХ. Анализ ВЭЖХ очищенных пептидомиметиков. После очистки препаративной ВЭЖХ, как описано выше, каждый пептид анализировали посредством аналитической ОФ-ВЭЖХ на аналитической системе ВЭЖХ Shimadzu LC-10AD. Для анализа пептидомиметиков посредством аналитической ВЭЖХ использовали колонку Luna 5 мкм; C18; 100 , с размерами 2504,6 мм с линейным градиентом буфера 0,1% TFA и ACN, а оценку хроматограммы проводили при 220 нм с применением детектора PDA. Характеристика посредством масс-спектрометрии. Каждый пептид характеризовали посредством масс-спектрометрии с электроспрейной ионизацией(ESI-MS) в режиме впрыска в поток или LC/MS. Во всех анализах в режиме электроспрея положительных и отрицательных ионов использовали масс-спектрометры с тройным квадруполем (API-3000(MDS-SCIES, Canada). В диапазоне масс квадруполя, действующего при одиночном разрешении, получали данные полного сканирования. Во всех случаях экспериментально измеряемая молекулярная масса находилась в пределах 0,5 Да от рассчитанной моноизотопной молекулярной массы. Количественное определение массовой хроматограммы проводили с применением программного обеспечения Analyst 1.4.1. Используя способы синтеза, описываемые в настоящем документе, вместе с другими широкоизвестными способами и их подходящими вариациями, получали приведенные ниже новые пептидомиметики. Этот список указывает на различные группы пептидомиметиков, которые можно получать по настоящему изобретению, и подразумевают, что они включат, по меньшей мере, очевидные вариации этих пептидомиметиков. Однако такое описание не следует рассматривать как каким-либо образом ограничивающее объем изобретения. В табл. 2 (i-v) новые пептидомиметики по настоящему изобретению перечислены вместе с их соответствующими номерами Seq. ID. No. Исследования новых пептидомиметиков in vitro и in vivo. Пептидомиметики, полученные, как описано выше, тестировали на:a) глюкозозависимую секрецию инсулина in vitro (протокол скрининга анализа клеток RIN5F);c) активность антагониста глюкагона человека in vitro (определение циклического АМФ);e) демонстрацию эффективности тестируемых соединений (пептидомиметиков) in vivo у мышейC57BL/6J (in vivo), с использованием различных анализов in vitro и in vivo, как описано ниже. Исследования in vitro. Глюкозозависимая секреция инсулина in vitro (протокол скрининга анализа клеток RIN5F). Клетки RIN5F (инсулиномы крыс) культивировали в среде RPMI 1640, дополненной пируватом натрия (1 мМ) HEPES и глюкозой (4,5 г/л) во влажном инкубаторе (5% CO2) при 37 С. После трипсинизации клетки RIN5F высевали при концентрации 0,2106 клеток на лунку в 12-луночные планшеты. Клетки растили в течение ночи до 80% конфлюентности и проводили эксперименты с секрецией инсулина, как указано ниже (Montrose-Rafizadeh С., et al., Mol. Cell. Endo. 1997, 130, 109.; Wang, X., et al.,Endocrinology, 2001, 5, 1820). Клетки однократно отмывали раствором PBS с последующими 40 мин инкубации в свежем уравновешенном буфере Кребса-Рингера, содержащем NaCl (115 ммоль/л), KCl (4,7 ммоль/л), CaCl2(25 ммоль/л), содержащем глюкозу (1,1 мМ) и BSA (0,5%), рН 7,4. Через 40 мин буфер заменяли и клетки инкубировали (37 С) с тестируемыми пептидомиметиками при различных концентрациях в течение 30 мин в присутствии (16,7 мМ) и в отсутствие (0 мМ) глюкозной нагрузки. Супернатант собирали и количество инсулина измеряли ультрачувствительным набором ELISA для инсулина крыс (Crystal Chem,IL). Количество белка в супернатанте рассчитывали с применением набора Bicinchoninic Acid по протоколу производителя (Sigma Aldrich, МО). Общее содержание инсулина, полученное в пикограммах (пг),делили на общее содержание белка (мкг) для нормализации различий в плотности клеток между лунками. Активность глюкозозависимой секреции инсулина иллюстративных пептидомиметиков in vitro представлена в табл. 3. Таблица 3 Активность глюкозозависимой секреции инсулина иллюстративных пептидомиметиков in vitrovitro с различными концентрациями пептидомиметиков измеряли с использованием клеток инсулиномы крыс (RIN5F). Общее содержание инсулина(пг) делили на общее содержание белка (мкг) для нормализации различий в плотности клеток между лунками; n=3, значения представляют собой среднееS.D. При концентрации глюкозы 0 мМ для всех тестируемых соединений наблюдали базальную секрецию инсулина. Активность агониста GLP-1R человека in vitro (определение циклического АМФ). Новые пептидомиметики скринировали на активность агониста рецептора GLP-1 человека(HGLP-1R) (in vitro) с использованием анализа цАМФ в стабильно трансфицированных клетках(25 мМ), NaHCO3 (1,1 г/л) и дополненной сывороткой новорожденных телят (NBCS; 10%), пенициллином (50 Ед/мл (об./об. и стрептомицином (50 мкг/мл (об./об Клетки каждые 3 суток разделяли 1:8. Получение стабильных клеточных линий CHO, экспрессирующих рецептор GLP-1 человека. кДНК, кодирующую рецептор GLP-1 человека, выделяли посредством ОТ-ПЦР в соответствии со стандартным протоколом. Полноразмерную кДНК клонировали в pcDNA3.1(+). Для получения клеточных линий CHO, экспрессирующих рецептор GLP-1, клетки CHO трансфицировали 10 мкг экспрессирующей плазмиды pcDNA/hGLP-1R с использованием CaPO4 в соответствии со стандартным протоколом(Wheeler, M.B., et al., Endocrinology. 1993, 133, 57). Экспрессирующие рецептор клоны получали посредством селекции с G418 (800 мкг/мл активного, Sigma). Затем стабильные клоны поддерживали при 500 мкг/мл (G418). Выбранный клон для анализов цАМФ использовали между пересевами 9-25. Определение образования цАМФ. Клетки CHO, стабильно трансфицированные GLP-1R человека, поддерживали в среде Хама F12 + 10% NBCS + 500 мкг/мл G418 до конфлюентности 70-75%. Клетки трипсинизировали с использованием 2 мл TPVG (0,25% трипсин, 0,53 мМ ЭДТА, 1,38 мМ глюкоза). Трипсин инактивировали с использованием среды Хама F12, содержащей 10% NBCS, и клетки суспендировали в 2 мл полной среды. Затем 2105 клеток/лунку высевали в 12-луночный планшет и планшеты инкубировали во влажной атмосфере при 37 С в течение 16-18 ч (Fehmann, H.C., et al., Peptides
МПК / Метки
МПК: A61K 38/26, C07K 14/605
Метки: агонистов, glp-1, глюкагона, антагонистов, пептидомиметики, активностью
Код ссылки
<a href="https://eas.patents.su/30-18000-peptidomimetiki-s-aktivnostyu-antagonistov-glyukagona-i-agonistov-glp-1.html" rel="bookmark" title="База патентов Евразийского Союза">Пептидомиметики с активностью антагонистов глюкагона и агонистов glp-1</a>
Связанные с матрицей пептидомиметики β-шпилечной структуры с активностью антагонистов cxcr4

Номер патента: 10163
Опубликовано: 30.06.2008
Авторы: Меле Керстин, Димарко Стивен Дж., Фрийблуд Ян Вим, Лочиуро Серджио, Романьоли Барбара, Робинсон Джон Энтони, Хенце Хайко, Мукерджи Решми, Гомберт Франк, Лудин Кристиан, Обрехт Даниель, Цумбрунн Юрг
МПК: A61K 38/12, C07K 1/04, A61P 31/18...
Метки: связанные, антагонистов, cxcr4, матрицей, пептидомиметики, beta;-шпилечной, структуры, активностью
Формула / Реферат:
1. Соединения общей формулы где представляет собой группу, характеризуемую одной из формул A-CO означает DPro, DGln, 4-{2-[2-(метоксиэтокси)этокси]ацетиламино}-DPro или Gly; B-CO означает LPro, LGln, 4-{2-[2-(метоксиэтокси)этокси]ацетиламино}-LPro или Gly; но A-CO и B-CO не могут оба быть Gly; Z означает цепь из n a-аминокислотных остатков, причем n означает целое число 12, 14 или 18, а положения указанных аминокислотных остатков в цепи...
Связанные с матрицей бета-шпилечные пептидомиметики с ингибирующей активностью в отношении протеаз

Номер патента: 13814
Опубликовано: 30.08.2010
Авторы: Хенце Хайко, Демарко Стивен Дж., Лудин Кристиан, Гомберт Франк, Меле Керстин, Селлье Одиль, Юнг Франсуаза, Обрехт Даниель
МПК: C07K 7/06, A61K 38/04, C07K 7/08...
Метки: протеаз, отношении, матрицей, бета-шпилечные, пептидомиметики, ингибирующей, активностью, связанные
Формула / Реферат:
1. Соединение общей формулыгдеозначает группу одной из формулгдепредставляет собой Ala; Arg; Asn; Cys; Gln; Gly; His; Ile; Leu; Lys; Met; Phe; Pro; Pro(5RPhe), Ser; Thr; Trp; Tyr; Val; Cit; Orn; tBuA; Sar; t-BuG; 4AmPhe; 3AmPhe; 2AmPhe; Phe(mC(NH2)=NH); Phe(pC(NH2)=NH); Phe(mNHC(NH2)=NH); Phe(pNHC(NH2)=NH); Phg; Cha; C4al; C5al; Nle; 2-Nal; 1-Nal; 4Cl-Phe; 3Cl-Phe; 2Cl-Phe; 3,4Cl2Phe; 4F-Phe; 3F-Phe; 2F-Phe; Tic; Thi; Tza; Mso; AcLys; Dpr; A2Bu;...
Соединения пиперазинилпиразинов в качестве агонистов или антагонистов серотонин 5-ht2 рецептора

Номер патента: 6552
Опубликовано: 24.02.2006
Автор: Нильссон Бьерн М.
МПК: A61K 31/497, A61K 31/445, A61K 31/4427...
Метки: серотонин, качестве, рецептора, агонистов, антагонистов, пиперазинилпиразинов, соединения, 5-ht2
Формула / Реферат:
1. Соединение общей формулы (I) в которой: (i) X и Y оба являются азотом и Z является CH, образуя производное пиразина, или (ii) X и Z оба являются CH и Y является азотом, образуя производное пиридина, или (iii) X является C-CF3, Z является CH и Y является азотом, образуя производное 4-трифторметилпиридина, или (iv) Y и Z оба являются азотом и X является CH, образуя производное пиримидина, и где R1 и R2, каждый независимо, выбирают из группы A,...
Соединения мочевины, обладающие активностью антагонистов мускариновых рецепторов

Номер патента: 6437
Опубликовано: 29.12.2005
Авторы: Оуэр Дэвид, Маммен Матай
МПК: C07D 211/58, A61P 13/10, A61K 31/4468...
Метки: активностью, мочевины, мускариновых, соединения, рецепторов, обладающие, антагонистов
Формула / Реферат:
1. Соединение формулы где R46 означает C1-10алкил, C3-20циклоалкил или C1-40гетероцикл; R47 означает C1-10алкил, C6-20арил, HC(O)-, C1-10алкил-C(O)-, C3-20циклоалкил-C(O)-, C4-20циклоалкенил-C(O)-, C6-20арил-C(O)-, C1-15гетероарил-C(O)-, C1-40гетероциклил-C(O)-, C1-40гетероцикл или -COOR50, в котором R50 означает C1-10алкил; или R46 и R47 вместе с атомом азота, к которому они присоединены, образуют C1-40гетероцикл; X означает C3-20алкилен, где...
Пиперазинил-бензилиденил-лактамовые соединения в качестве агонистов и антагонистов 5-нт1а и 5-нт1d рецепторов, фармацевтическая композиция на их основе и способ лечения или профилактики нарушения илисостояния, связанных с недостаточной серотонергической нейротрансмиссией у млекопитающего

Номер патента: 2157
Опубликовано: 24.12.2001
Автор: Ховард Гэрри Ральф
МПК: A61K 31/4166, A61P 25/00, C07D 233/96...
Метки: основе, серотонергической, профилактики, илисостояния, способ, 5-нт1а, нейротрансмиссией, композиция, антагонистов, 5-нт1d, недостаточной, качестве, лечения, связанных, агонистов, рецепторов, пиперазинил-бензилиденил-лактамовые, нарушения, соединения, млекопитающего, фармацевтическая
Формула / Реферат:
1. Пиперазинил-бензилиденил лактамовые соединения формулы где R1 представляет пиперазин-1-ил или пиперазин-1-ил, замещенный в положении 4 (C1-C4) алкилом; R2 представляет водород; R представляет-(СН2)mВ, где m равно 0 или 1 и В представляет водород, фенил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, (C1-C6)алкила или трифторметила; Х представляет водород или галоген; Y представляет...