Антагонисты неосновного рецептора-1 меланинконцентрирующего гормона
Номер патента: 16126
Опубликовано: 28.02.2012
Авторы: Мэнфреди Марк С., Уошберн Уильям Н., Робл Джеффри А., Хернандес Андрес С.






Формула / Реферат
1. Соединение, или его фармацевтически приемлемая соль, или стереоизомер, или пролекарство, или сольват формулы I

где R1 независимо выбран из группы, состоящей из водорода, галогена, С1-8алкила, С3-10циклоалкила, фенила, нафтила, CF3, CN, NR7R7, OR6 и SR6;
R2 выбран из группы, состоящей из водорода и С1-8алкила;
R3 независимо выбран из группы, состоящей из водорода и С1-8алкила;
R4 представляет собой G-D2-Zn;
R5 выбран из группы, состоящей из водорода, галогена, С1-8алкила, CF3, SR6, С1-8алкокси, CN, SOR6, SO2R6, CO2R6 и COR6;
m представляет собой целое число от 0 до 1;
n представляет собой целое число от 1 до 3;
G выбран из группы, состоящей из О и S;
D2 выбран из группы, состоящей из прямой связи, С1-8алкила и С3-10циклоалкила;
Z выбран из группы, состоящей из гидроксила, С3-10циклоалкила, С3-10циклоалкокси, OCOR6, CN, OSO2R6, SR6, SOR6, SO2R6, CO2R7, OPO(OR6)2 и COR6;
R6 независимо выбран из группы, состоящей из С1-8алкила и С3-10циклоалкила;
R7 независимо выбран из группы, состоящей из водорода, С1-8алкила и С3-10циклоалкила;
причем пролекарства представляют собой ацетаты, пивалаты, метилкарбонаты, бензоаты, образованные аминокислотами сложные эфиры, фосфаты, фосфоацетали или О-глюкозиды, образованные по гидроксильной связи одной или более гидроксильных групп соединения формулы I.
2. Соединение по п.1, где R5 выбран из группы, состоящей из водорода, С1-8алкила и С1-8алкокси.
3. Соединение по п.1, где R1 выбран из группы, состоящей из галогена и С1-8алкила.
4. Соединение по п.3, где галоген представляет собой хлор.
5. Соединение по п.4, где n представляет собой целое число от 1 до 2.
6. Соединение по п.5, где G представляет собой О, D2 выбран из группы, состоящей из С1-8алкила и С3-10циклоалкила.
7. Соединение по п.6, где R2 представляет собой водород и по крайней мере один R3 представляет собой водород.
8. Фармацевтическая композиция, содержащая
по крайней мере одно соединение по п.1 и
по крайней мере один фармацевтически приемлемый разбавитель или носитель.
9. Фармацевтическая композиция по п.8, дополнительно содержащая по крайней мере один дополнительный терапевтический агент.
10. Фармацевтическая комбинация, содержащая
по крайней мере одно соединение по п.1 и
по крайней мере один дополнительный терапевтический агент, где дополнительный терапевтический агент может быть введен пациенту перед соединением по п.1, одновременно с соединением по п.1 или после соединения по п.1.
11. Способ лечения ожирения, включающий введение пациенту терапевтически эффективного количества по крайней мере одного соединения по п.1 отдельно или в комбинации с одним или более терапевтическим агентом.
12. Способ лечения диабетов, включающий введение пациенту терапевтически эффективного количества по крайней мере одного соединения по п.1 отдельно или в комбинации с одним или более терапевтическим агентом.
13. Способ по п.12, где диабет является диабетом типа II.
14. Способ лечения депрессии, включающий введение пациенту терапевтически эффективного количества по крайней мере одного соединения по п.1 отдельно или в комбинации с одним или более терапевтическим агентом.
15. Способ лечения анексии, включающий введение пациенту терапевтически эффективного количества по крайней мере одного соединения по п.1 отдельно или в комбинации с одним или более терапевтическим агентом.
16. Соединение, выбранное из группы, состоящей из













или его фармацевтически приемлемая соль либо стереоизомер.
17. Соединение по п.1, которое имеет формулу

представленное его пролекарством, где
R1 независимо выбран из группы, состоящей из галогена, C1-8алкила, С3-10циклоалкила, фенила, нафтила, CF3, CN, OR6 и SR6;
R2 представляет собой водород;
R4 представляет собой G-D2-Zn;
n представляет собой целое число от 1 до 3;
R5 выбран из группы, состоящей из водорода, галогена, С1-8алкила, CF3, SR6, С1-8алкокси, CN, SOR6, SO2R6, CO2R6 и COR6;
G представляет собой О;
D2 выбран из группы, состоящей из С1-8алкила и С3-10циклоалкила;
Z выбран из группы, состоящей из гидроксила, С3-10алкокси, С3-10циклоалкокси, OCOR6, CN, OSO2R6, SR6, SOR6, SO2R6, CO2R7, OPO(OR6)2 и COR6;
R6 независимо выбран из группы, состоящей из С1-8алкила и С3-10циклоалкила;
R7 независимо выбран из группы, состоящей из водорода, С1-8алкила и С3-10циклоалкила;
причем пролекарства представляют собой ацетаты, пивалаты, метилкарбонаты, бензоаты, образованные аминокислотами сложные эфиры, фосфаты, фосфоацетали или О-глюкозиды, образованные по гидроксильной связи одной или более гидроксильных групп соединения формулы I.
18. Соединение по п.17, где R1 выбран из группы, состоящей из галогена, С1-8алкила, CF3, CN, OR6 и SR6.
19. Соединение по п.3, где R5 выбран из группы, состоящей из водорода, С1-8алкила и С1-8алкокси.
20. Соединение по п.17, где n представляет собой целое число от 1 до 2.
21. Соединение по п.17, где
R1 выбран из группы, состоящей из галогена, С1-8алкила, CF3, CN, OR6 и SR6;
R5 выбран из группы, состоящей из водорода, С1-8алкила и С1-8алкокси;
n представляет собой целое число от 1 до 2.
Текст
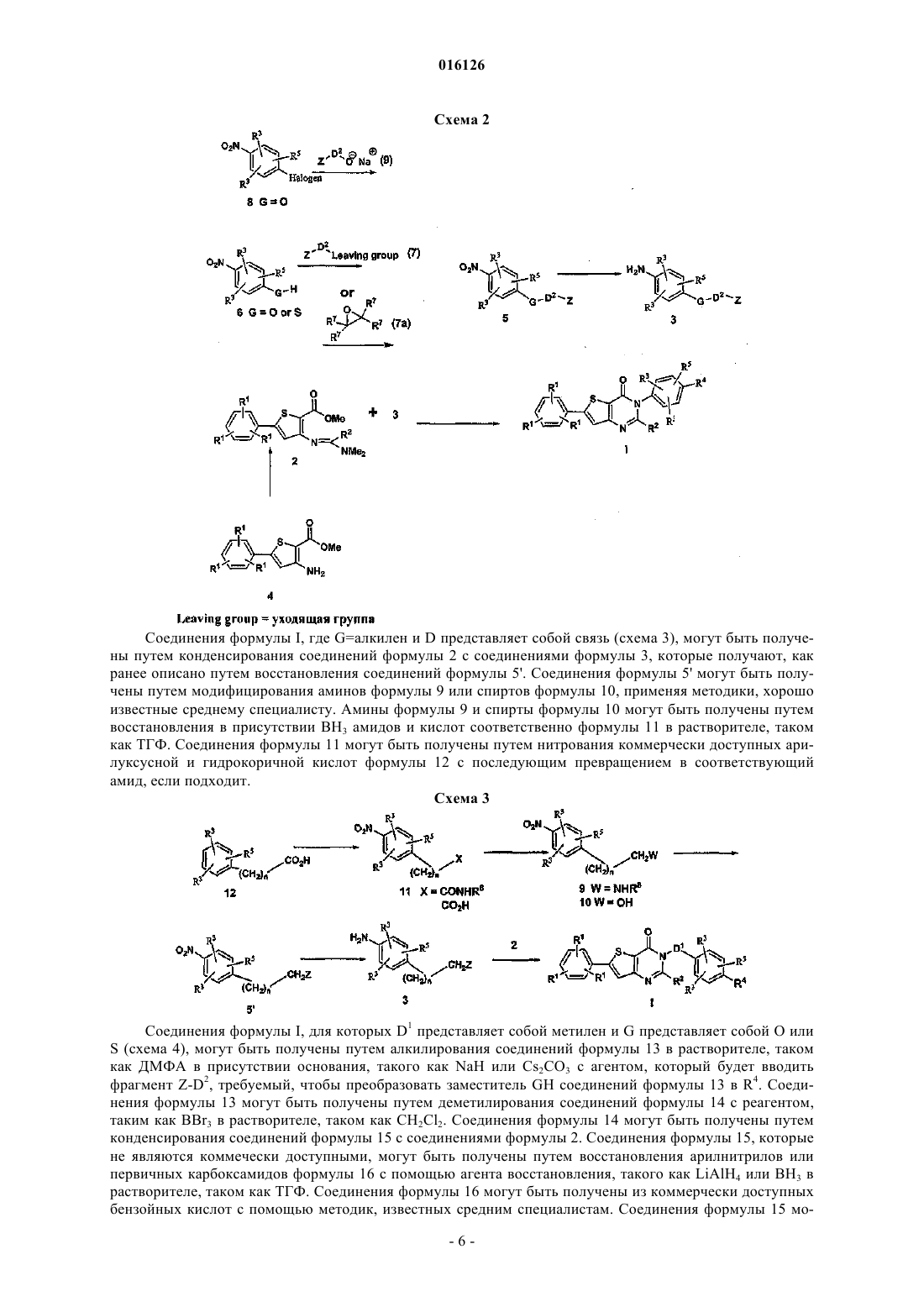
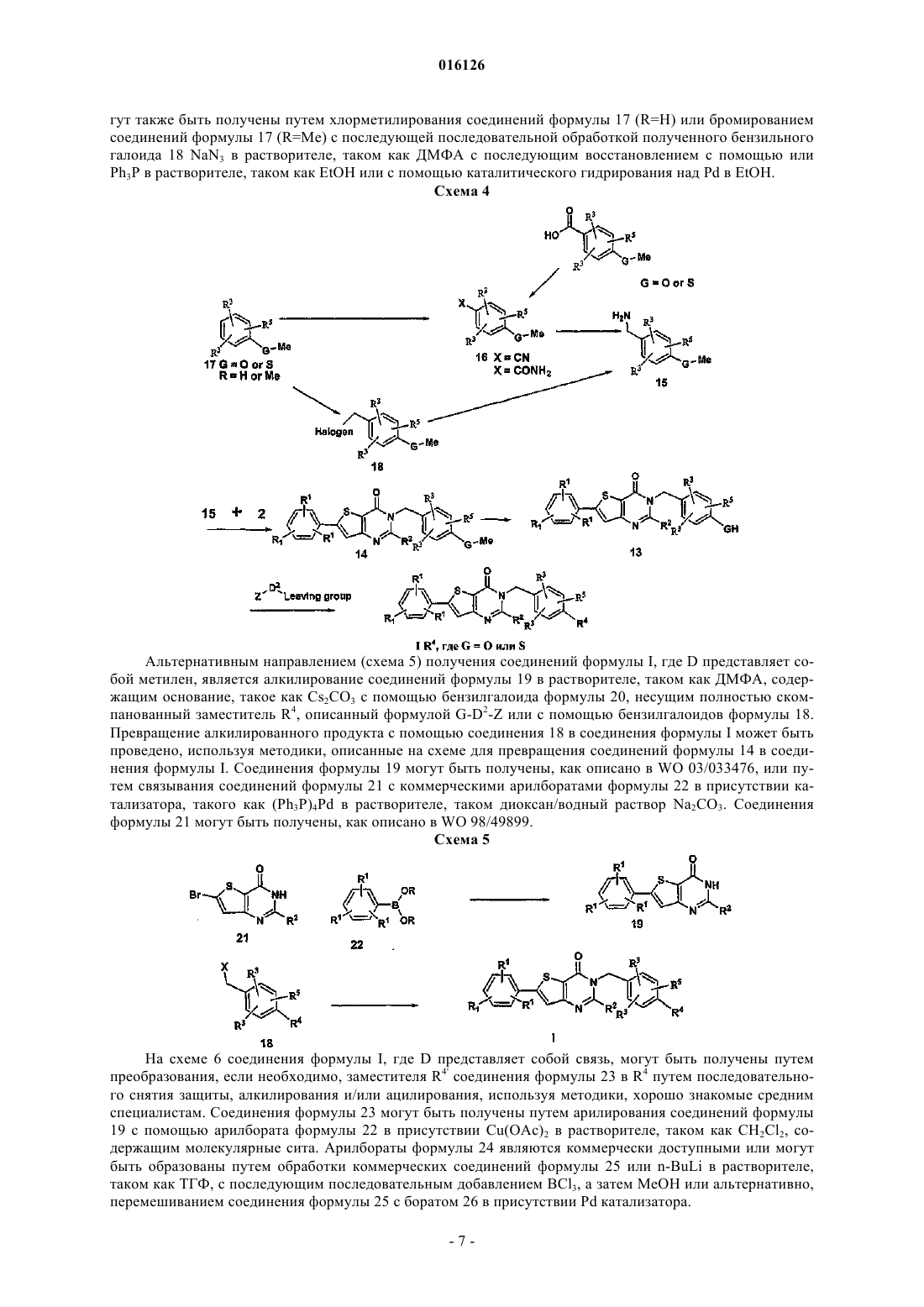
Настоящее изобретение обеспечивает соединения, включая все стереоизомеры, сольваты,пролекарства и их фармацевтически приемлемые формы в соответствии с формулой I. Кроме того, настоящее изобретение обеспечивает фармацевтические композиции, содержащие по крайней мере одно соединение в соответствии с формулой I и необязательно по крайней мере один дополнительный терапевтический агент. Наконец, настоящее изобретение обеспечивает способы лечения пациента, страдающего от MCHR-I модулированного заболевания или расстройства,такого как, например, ожирение, диабеты, депрессия или аннексия, путем введения терапевтически эффективной дозы соединения в соответствии с формулой I 016126 Уровень техники Некоторые цепочки фармакологических и генетических доказательств указывают на роль рецептора меланинконцентрирующего гормона 1 типа (далее "MCHR1") в качестве модулятора потребления пищи и массы тела. Центральное введение МСН увеличивает потребление пищи и массу тела у крыс и у мышей. Длительная ICV инфузия МСН вызывает повышенное потребление пищи и исключительное ожирение у мышей, тогда как инфузия пептидного антагониста МСН блокирует вызванное МСН потребление пищи и приводит к потере веса и пониженному питанию у мышей с ожирением, вызванным диетой. Экспрессия и МСН пептида и рецептора модулируется состоянием питания. МСН мРНК является нерегулируемой и у чрезмерно питающихся мышей с ожирением (ob/ob) и у голодающих животных. Направленное разрушение гена, отвечающего за МСН пептид, приводит к гипофагии и истощению. Разрушение гена MCHR1 вызывает истощение, нарушенный метаболизм и повышенную двигательную активность в сочетании с умеренной гиперфагией. Наоборот, сверхэкспрессия МСН пептида приводит к гиперфагии, ожирению и диабету. Низкомолекулярные антагонисты MCHR1, как было продемонстрировано, вызывают потерю веса и образа питания у грызунов как после перорального, так и после интраперитонеального введения; Eur. J. Pharmacol., 438,129-135, 2002, Nat. Med., 8, 825-830, 2002, Eur. J. Pharmacol,497, 41-47, 2004. Раскрыто множество непептидных антагонистов MCHR1. Рамки рода для каждого отражают общее понимание в отношении критериев, необходимых для распознавания лиганда, как MCHR1 агонистов. Недавний патентный обзор MCHR1 раскрывает подчеркнутую общность этих структур следующим описанием: "вездесущими в патентной литературе, посвященной МСН, являются молекулы, состоящие из центрального скелета, к которому прикрепляются связующие агенты с арильной или гетероарильной группой и основная аминная функциональная группа" (Т.J. Kowalski and М.D. MacBriar, Expert Opin. Investig. Drags 13, 1113-1122, 2004). Модели фармакофоров этих семейств соответственно предусматривают предполагаемое необходимое электростатическое взаимодействие между основным аминным центром лиганда антагониста и аспарагиновой кислотой 123 рецептора, который, по-видимому, предназначен имитировать вынужденное взаимодействие между аргинином 14 агонистов МСН пептида и аспарагиновой кислотой 123 MCHR1 рецептора (Т. Ulven, J. Med. Chem. 2005, 48, 5684-5697). Тем не менее,введение этой основной аминной группы в структуру антагониста MCHR1 существенно повышает вероятность связывания нецелевых ионных каналов и рецепторов биогенных аминов. Предлагается ряд новых высокоаффинных селективных MCHR1 антагонистов, которые были получены путем замены основной аминной функциональной группы, описанной в WO 03/033476 на неосновные полярные функциональные группы. Более того, эта структурная модификация приводит к неожидаемому прекращению связывания с другими рецепторами биогенных аминов, также как и связывания с рецептором HERG в сердце. Уменьшение/ликвидация сродства к рецептору HERG особенно важно, поскольку занятость лиганда связана с инициацией фатальных аритмий. Подробное описание изобретения Изобретение раскрывает соединения, включая все стереоизомеры, сольваты, пролекарства и их фармацевтически приемлемые формы в соответствии с формулой I. Кроме того, оно охватывает фармацевтические композиции, содержащие по крайней мере одно соединение в соответствии с формулой I и необязательно по крайней мере один дополнительный терапевтический агент. Наконец, изобретение включает в себя способы лечения пациента, страдающего от MCHR-I модулированного заболевания или расстройства, такого как, например, ожирение, диабеты, депрессия или аннексия, путем введения терапевтически эффективной дозы соединения в соответствии с формулой I где R1 независимо выбран из группы, состоящей из водорода, галогена, С 1-8 алкила, С 3-10 циклоалкила,фенила, нафтила, CF3, CN, NR7R7, OR6 и SR6;R2 выбран из группы, состоящей из водорода и С 1-8 алкила;R3 независимо выбран из группы, состоящей из водорода, галогена, С 1-8 алкила;m представляет собой целое число от 0 до 1;n представляет собой целое число от 1 до 3;D2 выбран из группы, состоящей из прямой связи, С 1-8 алкила и С 3-10 циклоалкила;R6 независимо выбран из группы, состоящей из С 1-8 алкила и С 3-10 циклоалкила;R7 независимо выбран из группы, состоящей из водорода, С 1-8 алкила и С 3-10 циклоалкила; причем пролекарства представляют собой ацетаты, пивалаты, метилкарбонаты, бензоаты, образованные аминокислотами сложные эфиры, фосфаты, фосфоацетали или О-глюкозиды, образованные по гидроксильной связи одной или более гидроксильных групп соединения формулы I. Определения Если иное не указано, термин "низший алкил", как используют в настоящем описании сам по себе или как часть другой группы, включает, обе, прямую и разветвленную углеводородную цепь, содержащую от 1 до 8 атомов углерода, и термины "алкил" и "алк", как используют в настоящем описании сами по себе или как часть другой группы, включают обе прямую и разветвленную углеводородную цепь, содержащую от 1 до 20 атомов углерода, предпочтительно от 1 до 10 атомов углерода, более предпочтительно от 1 до 8 атомов углерода в нормальной цепи, такой как метил, этил, пропил, изопропил, бутил,трет-бутил, изобутил, пентил, гексил, изогексил, гептил, 4,4-диметилпентил, октил, 2,2,4-триметилпентил, нонил, децил, ундецил, додецил, различные их изомеры с разветвленной цепью и им подобные,так же как такие группы, включая от 1 до 4 заместителей, таких как галоген, например F, Br, Cl или I илиCF3, алкил, алкокси, арил, арилокси, арил(арил) или диарил, арилалкил, арилалкилокси, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкилалкил, циклоалкилалкилокси, гидрокси, гидроксиалкил,ацил, алканоил, гетероарил, гетероарилокси, циклогетероалкил, арилгетероарил, арилалкоксикарбонил,гетероарилалкил, гетероарилалкокси, арилоксиалкил, арилоксиарил, алкиламидо, алканоиламино, арилкарбониламино, нитро, циано, тиол, галогеналкил, тригалогеналкил и/или алкилтио. Если иное не указано, термин "циклоалкил", как используют в настоящем описании сам по себе или как часть другой группы, включает насыщенные или частично ненасыщенные (содержащие 1 или 2 двойных связей) циклические углеводородные группы, содержащие от 1 до 3 колец, любое одно из которых может необязательно быть спирозамещенным циклоалкилом, включая моноциклический алкил, бициклический алкил и трициклический алкил, содержащий общее число атомов углерода от 3 до 20, образующих кольцо, предпочтительно от 3 до 10 атомов углерода, образующих кольцо и которые могут быть конденсированы с 1 или 2 ароматическими кольцами, как описано для арила, которые включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил и циклододецил,циклогексенил, любая из указанных групп может быть необязательно замещена от 1 до 4 заместителями, такими как галоген, алкил, алкокси, гидрокси, арил, арилокси, арилалкил, циклоалкил, алкиламидо, алканоиламино,оксо, ацил, арилкарбониламино, нитро, циано, тиол и/или алкилтио и/или любым из алкильных заместителей. Термин "гетероцикло", "гетероцикл", "гетероциклил" или "гетероциклическое кольцо", как используют в настоящем описании, представляет собой незамещенную или замещенную стабильную от 4- до 7 членную моноциклическую кольцевую систему, которая может быть насыщенной или ненасыщенной и которая состоит из атомов углерода, от одного до четырех гетероатомов, выбранных из азота, кислорода или серы, и где гетероатомы азота и серы могут необязательно быть окислены и гетероатом азота может необязательно быть кватернизован. Гетероциклическое кольцо может быть присоединено по любому гетероатому или атому углерода, что приводит к созданию стабильной структуры. Примеры таких гетероциклических групп включают, но без ограничения, пиперидинил, пиперазинил, оксопиперазинил, оксопиперидинил, оксопирролидинил, оксоазепинил, азепинил, пирролил, пирролидинил, фуранил, тиенил,пиразолил, пиразолидинил, имидазолил, имидазолинил, имидазолидинил, пиридил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изооксазолил, изоксазолидинил, морфолинил, тиазолил,тиазолидинил, изотазолил, тиадиазолил, тетрагидропиранил, тиаморфолинил, тиаморфолинил сульфоксид, тиаморфолинил сульфон, оксадиазолил и другие гетероциклы, описанные у Katritzky, A.R. and Rees,С.W., eds. Comprehensive Heterocyclic Chemistry: The Structure, Reactions, Synthesis and Uses of Heterocyclic Compounds 1984, Pergamon Press, New York, NY; and Katritzky, A.R., Rees, С.W., Scriven, E.F., eds.Comprehensive Heterocyclic Chemistry II: A Review of the Literature 1982-1995 1996, Elsevier Science, Inc.,Tarrytown, NY; и являющимися ссылками. Термин "алканоил", как используют в настоящем описании сам по себе или как часть другой группы, относится к алкилу, связанному с карбонильной группой. Термин "галоген" или "гало", как используют в настоящем описании сам по себе или как часть другой группы, относится к хлору, брому, фтору и йоду, с хлором или фтором, которые являются предпочтительными. Термин "ион металла" относится к ионам щелочных металлов, таких как натрий, калий или литий и-2 016126 ионам щелочно-земельных металлов, таких как магний и кальций, так же как цинк и алюминий. Если иное не указано, термин "арил" или "Арил", как используют в настоящем описании сам по себе или как часть другой группы, относится к моноциклическим и бициклическим ароматическим группам, содержащим от 6 до 10 атомов углерода в части кольца (такого как фенил или нафтил, включая 1 нафтил и 2-нафтил) и могут необязательно включать от одного до трех дополнительных колец, конденсированных с карбоциклическим кольцом или гетероциклическим кольцом, таким как арильные, циклоалкильные, гетероарильные или циклогетероалкильные кольца, например и могут быть необязательно замещены по доступным атомам углерода с помощью 1, 2 или 3 групп, выбранных из водорода, галогена, галогеналкила, алкила, галогеналкила, алкокси, галогеналкокси, алкенила, трифторметила, трифторметокси, алкинила, циклоалкилалкила, циклогетероалкила, циклогетероалкилалкила, арила, гетероарила, арилалкила, арилокси, арилоксиалкила, арилалкокси, алкоксикарбонила,арилкарбонила, арилалкенила, аминокарбониларила, арилтио, арилсульфинила, арилазо, гетероарилалкила, гетероарилалкенила, гетероарилгетероарила, гетероарилокси, гидрокси, нитро, циано, тиола, алкилтио, арилтио, гетероарилтио, арилтиоалкила, алкоксиарилтио, алкилкарбонила, арилкарбонила, алкиламинокарбонила, ариламинокарбонила, алкоксикарбонила, аминокарбонила, алкилкарбонилокси, арилкарбонилокси, алкилкарбониламино, арилкарбониламино, арилсульфинила, арилсульфинилалкила, арилсульфониламино и арилсульфонаминокарбонила и/или любым из алкильных заместителей, представленных в настоящем описании. Если иное не указано, термин "гетероарил", как используют в настоящем описании сам по себе или как часть другой группы, относится к 5- или 6-членному ароматическому кольцу, которое включает 1, 2,3 или 4 гетероатома, таких как азот, кислород или сера. Такие кольца могут быть конденсированы с арилом, циклоалкилом, гетероарилом или гетероциклилом и включают возможные N-оксиды, как описано уKlatritzky, A.R. and Rees, С.W., eds. Comprehensive Heterocyclic Chemistry: The Structure, Reactions, Synthesis and Uses of Heterocyclic Compounds, 1984, Pergamon Press, New York, NY; and Katritzky, A.R., Rees,С.W., Scriven, E.F., eds. Comprehensive Heterocyclic Chemistry II: A Review of the Literature, 1982-1995,1996, Elsevier Science, Inc., Tarrytown, NY; и являющимися ссылками. Кроме того, "гетероарил", как определено в настоящем описании, могут необязательно быть замещен одним или большим количеством заместителей, таких как заместители, охваченные выше в определении терминов "замещенный алкил" и"замещенный арил". Примеры гетероарильных групп включают следующие: и им подобные. Если иное не указано, термин "низший алкокси", "алкокси", "арилокси" или "аралкокси", как используют в настоящем описании сам по себе или как часть другой группы, включает любую из указан-3 016126 ных выше алкильных, аралкильных или арильных групп, связанных с атомом кислорода. Если иное не указано, термин "низший алкилтио", алкилтио", "арилтио" или "аралкилтио", как используют в настоящем описании сам по себе или как часть другой группы, включает любую из указанных выше алкильных, аралкильных или арильных групп, связанных с атомом серы. Термин "полигалогеналкил", как используют в настоящем описании, относится к "алкильной" группе, как определено выше, которая включает от 2 до 9, предпочтительно от 2 до 5, галогеновых заместителей, таких как F или Cl, предпочтительно F, таких как CF3CH2, CF3 или CF3CF2CH2. Термин "полигалогеналкилокси", как используют в настоящем описании, относится к "алкокси" или"алкилокси" группе, как определено выше, которая включает от 2 до 9, предпочтительно от 2 до 5, галогеновых заместителей, таких как F или Cl, предпочтительно F, таких как CF3CH2O, CF3O илиCF3CF2CH2O. Соединения формулы I по настоящему изобретению могут быть получены, как показано на следующих реакционных схемах и в их описании, так же как согласно релевантным процессам из уровня техники, которые могут быть использованы средним специалистом. Типичные реагенты и методики для указанных реакций представлены в дальнейшем и в рабочих примерах. Сокращения. Следующие сокращения применены в настоящем описании:i-PrOH - изопропанол; НОАс или АсОН - уксусная кислота;-4 016126 Способы получения соединений Как суммировано на схеме 1, соединения формулы I могут быть получены или путем конденсирования соединений формулы 2 с соединениями формулы 3, чтобы получить тиофенопиримидоновый центральный бициклический остаток in situ, или через алкилирование/арилирование соединений формулы 18 агентами анкетирования формулы 19 или агентами арилирования, таким как бораты формулы 24. В зависимости от полученной конкретной молекулы формулы I R4 может или быть полностью сформирован, или преобразован после формирования ядра структуры формулы I. Схема 1 В частности, соединения формулы I могут быть получены путем конденсирования соединений формулы 2 с соединениями формулы 3 в растворителе, таком как горячий EtOH (схема 2). Соединения формулы 2 могут быть получены, как описано в WO 2003/033476, путем нагревания соединения формулы 4 с диметилформамид диметилацеталем. Получение соединения формулы 4 описано вWO 1998/49899. Амины формулы 3, для которых D представляет собой связь, могут быть получены путем восстановления нитроароматиков формулы 5 или путем каталитического гидрирования, используя катализатор, такой как Pd/C в растворителе, таком как EtOH или путем восстановления с помощью SnCl2 в растворителе, таком EtOAc. Соединения формулы 5, где G=О или S и D представляет собой связь, могут быть получены путем алкилирования соответствующего фенола или тиофенола формулы 6 с подходящим агентом алкилирования формулы 7 в присутствии основания, такого как Cs2CO3 или K2CO3 в растворителе, таком как ДМФА, используя методики, хорошо известные среднему специалисту. Альтернативно, соединения формулы 5 могут быть получены путем нагревания соли щелочного металла соединений формулы 6 с эпоксидами формулы 7 а термически или предпочтительно в микроволновой печи в растворителе, таком как 15% H2O/MeCN, содержащим NaH2PO4. Соединения формулы 5 могут быть также получены путем нагревания соединений формулы 8 с предварительно полученными натриевыми солями соединений формулы 9 в растворителе, таком как ДМФА. Соединения формулы 6 и 8 являются или коммерчески доступными, или могут быть легко получены в случае соединения формулы 6 из соответствующего ненитрованного фенола путем последовательного ацилирования, нитрирования или сульфонилирования, используя способы, известные средним специалистам. Соединения формулы I, где G=алкилен и D представляет собой связь (схема 3), могут быть получены путем конденсирования соединений формулы 2 с соединениями формулы 3, которые получают, как ранее описано путем восстановления соединений формулы 5'. Соединения формулы 5' могут быть получены путем модифицирования аминов формулы 9 или спиртов формулы 10, применяя методики, хорошо известные среднему специалисту. Амины формулы 9 и спирты формулы 10 могут быть получены путем восстановления в присутствии ВН 3 амидов и кислот соответственно формулы 11 в растворителе, таком как ТГФ. Соединения формулы 11 могут быть получены путем нитрования коммерчески доступных арилуксусной и гидрокоричной кислот формулы 12 с последующим превращением в соответствующий амид, если подходит. Схема 3 Соединения формулы I, для которых D1 представляет собой метилен и G представляет собой О илиS (схема 4), могут быть получены путем алкилирования соединений формулы 13 в растворителе, таком как ДМФА в присутствии основания, такого как NaH или Cs2CO3 с агентом, который будет вводить фрагмент Z-D2, требуемый, чтобы преобразовать заместитель GH соединений формулы 13 в R4. Соединения формулы 13 могут быть получены путем деметилирования соединений формулы 14 с реагентом,таким как BBr3 в растворителе, таком как CH2Cl2. Соединения формулы 14 могут быть получены путем конденсирования соединений формулы 15 с соединениями формулы 2. Соединения формулы 15, которые не являются коммечески доступными, могут быть получены путем восстановления арилнитрилов или первичных карбоксамидов формулы 16 с помощью агента восстановления, такого как LiAlH4 или ВН 3 в растворителе, таком как ТГФ. Соединения формулы 16 могут быть получены из коммерчески доступных бензойных кислот с помощью методик, известных средним специалистам. Соединения формулы 15 мо-6 016126 гут также быть получены путем хлорметилирования соединений формулы 17 (R=Н) или бромированием соединений формулы 17 (R=Me) с последующей последовательной обработкой полученного бензильного галоида 18 NaN3 в растворителе, таком как ДМФА с последующим восстановлением с помощью илиPh3P в растворителе, таком как EtOH или с помощью каталитического гидрирования над Pd в EtOH. Схема 4 Альтернативным направлением (схема 5) получения соединений формулы I, где D представляет собой метилен, является алкилирование соединений формулы 19 в растворителе, таком как ДМФА, содержащим основание, такое как Cs2CO3 с помощью бензилгалоида формулы 20, несущим полностью скомпанованный заместитель R4, описанный формулой G-D2-Z или с помощью бензилгалоидов формулы 18. Превращение алкилированного продукта с помощью соединения 18 в соединения формулы I может быть проведено, используя методики, описанные на схеме для превращения соединений формулы 14 в соединения формулы I. Соединения формулы 19 могут быть получены, как описано в WO 03/033476, или путем связывания соединений формулы 21 с коммерческими арилборатами формулы 22 в присутствии катализатора, такого как (Ph3P)4Pd в растворителе, таком диоксан/водный раствор Na2CO3. Соединения формулы 21 могут быть получены, как описано в WO 98/49899. Схема 5 На схеме 6 соединения формулы I, где D представляет собой связь, могут быть получены путем преобразования, если необходимо, заместителя R4' соединения формулы 23 в R4 путем последовательного снятия защиты, алкилирования и/или ацилирования, используя методики, хорошо знакомые средним специалистам. Соединения формулы 23 могут быть получены путем арилирования соединений формулы 19 с помощью арилбората формулы 22 в присутствии Cu(OAc)2 в растворителе, таком как CH2Cl2, содержащим молекулярные сита. Арилбораты формулы 24 являются коммерчески доступными или могут быть образованы путем обработки коммерческих соединений формулы 25 или n-BuLi в растворителе,таком как ТГФ, с последующим последовательным добавлением BCl3, а затем МеОН или альтернативно,перемешиванием соединения формулы 25 с боратом 26 в присутствии Pd катализатора. На схеме 7 соединения формулы I, где G=CH2 и D представляет собой связь, могут быть получены путем конденсации соответствующего бензальдегида формулы 27 с подходящим кетоном формулы 28 или сложным эфиром формулы 29 в присутствии основания, такого как NaOH или NaOR6 в растворителе, таком как ДМФА или EtOH, с помощью методики, хорошо известные среднему специалисту. Получение соединения формулы 3, где G=СН 2 и D представляет собой связь, может быть завершено путем восстановления кетонкарбонильных соединений формулы 30 агентом восстановления, таким как NaBH4 в растворителе, таком как EtOH с последующим каталитическим гидрированием, используя Н 2 и Pd/C вEtOH или смесь EtOH/EtOAc. Альтернативно, соединения формулы 31 могут быть преобразованы в соединения формулы 3 путем каталитического гидрирования, используя Н 2 и Pd/C в EtOH или смесях Термин "пролекарство" включает оба термина "пролекарственные сложные эфиры" и термин "пролекарственные простые эфиры". Термин "пролекарственные сложные эфиры", как используют в настоящем описании, включает сложные эфиры и карбонаты, образованные путем взаимодействия одного или большего количества гидроксильных соединений формулы I с или алкил, алкокси, или арилзамещенными агентами ацилирования, или агентом фосфорилирования, применяя методики, известные среднему специалисту, чтобы получить ацетаты, пивалаты, метилкарбонаты, бензоаты, сложные эфиры аминокислот, фосфаты и им подобные. Примеры таких пролекарственных сложных эфиров включают Термин "пролекарственные простые эфиры" включает оба фосфатацетали и О-глюкозиды. Характерные примеры таких пролекарственных эфиров включают Соединения формулы I могут присутствовать в виде солей, которые также находятся в границах на-8 016126 стоящего изобретения. Фармацевтически приемлемые (т.е. нетоксичные, физиологически приемлемые) соли являются предпочтительными. Если соединения формулы I имеют, например, по крайней мере один основной центр, они могут образовывать кислотно-аддитивные соли. Они образуются, например, с сильными неорганическими кислотами, такими как неорганические кислоты, например серная кислота, фосфорная кислота или галоидводородная кислота, с органическими карбоновыми кислотами, такими как алканкарбоновые кислоты, содержащими от 1 до 4 атомов углерода, например уксусная кислота, которая является незамещенной или замещенной, например, с помощью галогена, как хлоруксусная кислота, такая как насыщенные или ненасыщенные дикарбоновые кислоты, например щавелевая, малоновая, янтарная, малеиновая, фумаровая, фталевая или терефталевая кислота, такие как гидроксикарбоновые кислоты, например аскорбиновая, гликолевая, молочная, яблочная, винная или лимонная кислота, такие как аминокислоты (например, аспарагиновая или глутаминовая кислота, или лизин, или аргинин) или бензойная кислота, или с органическими сульфоновыми кислотами, такими как (C1-С 4)алкильные или арилсульфоновые кислоты, которые являются незамещенными или замещенными, например, с помощью галогена, например метил- или п-толуол-сульфоновая кислота. Соответствующие кислотно-аддитивные соли могут также быть образованы в случае наличия, если необходимо, дополнительного основного центра. Соединения формулы I, имеющие по крайней мере одну кислотную группу (например, СООН), могут также образовывать соли с основаниями. Подходящие соли с основаниями являются, например, соли металла, такие как соли щелочных металлов или соли щелочно-земельных металлов, например соли натрия, калия или магния, или аммонийные соли, или соли органическим амином, таким как морфолин,тиоморфолин, пиперидин, пирролидин, моно-, ди- или тринизший алкиламин, например этил, третбутил, диэтил, диизопропил, триэтил, трибутил или диметилпропиламин или моно-, ди- или тригидрокси низший алкиламин, например моно-, ди- или триэтаноламин. Кроме того, могут быть образованы соответствующие внутренние соли. Соли, которые являются неподходящими для фармацевтических применений, но которые могут быть использованы, например, для выделения или очистки свободных соединений формулы I или их фармацевтически приемлемых солей, являются также включенными. Предпочтительные соли соединений формулы I, которые содержат основную группу, включают моногидрохлорид, гидросульфат, метансульфонат, фосфат, нитрат или ацетат. Предпочтительные соли соединений формулы I, которые содержат кислотную группу, включают соли натрия, калия и магния и фармацевтически приемлемые органические амины. Рассмотрены все стереоизомеры соединений по настоящему изобретению или в примесной, или в чистой, или в основном чистой форме. Соединение по настоящему изобретению могут имеют ассиметричные центры на любом из атомов углерода, включая любой один из R заместителей. Следовательно,соединение формулы I может находиться в энантиомерных или диастереомерных формах или в их смесях. Способы получения могут использовать рацематы, энантиомеры или диастереомеры в виде исходных продуктов. Когда получены энантиомерные или диастереомерные продукты, они могут быть отделены с помощью обычных способов, например, хроматографически или фракционной кристаллизацией. Примеры Следующие примеры служат для лучшей иллюстрации, но не для ограничения, некоторых из предпочтительных воплощений патента. Где возможно, модульный конвергентный способ используют для получения подходящего анилина с помощью последующих примеров осуществления синтеза, конденсации с формамидином, чтобы получить ядро бициклического тиенопиримидона с последующей разработкой, если необходимо, боковой цепи. Используют два условия для создания тиенопиримидонового бицикла: анилин и формамидин нагревают при температуре кипения с обратным холодильником в EtOH в течение 18 ч. При охлаждении продукт высаживают и выделяют фильтрованием. Выходы составляют, как правило, 20-40% и редко превышают 40%. Альтернативно, анилин и формамидин в феноле нагревают до температуры 130 С в течение 10-30 мин; затем разбавляют МеОН, продукт выделяют фильтрованием и выходы достигают приблизительно 80%. Следует отметить, если продукт не очищают, выделение становится длительным и выходы могут резко упасть. Значительно большая часть пара-алкоксианилинов являются или коммерчески доступными, или синтезированы по одной из трех методик: А, В или С. Методика А (нуклеофильное ароматическое замещение). Требуется предварительное формирование алкоксида натрия с помощью NaH в ДМФА с последующим добавлением 2-хлор-5-нитроанизола. Как правило, реакционную смесь нагревают в течение 1 ч при температуре 90 С. После очистки с помощью хроматографии на силикагеле нитрованный ариловый эфир восстанавливают до желаемого анилина с помощью 10% Pd/C катализатора гидрирования (давление 50 пси (3.45 бар) Н 2) в EtOH. Методика В (промотированное основанием фенольное алкилирование). Осуществляют нагревание смеси калийной или натриевой соли нитрофенола с алкилгалоидом в ДМФА в течение 2-4 ч при температуре 90 С. После выделения и очистки с помощью хроматографии на силикагеле продукт восстанавливают до желаемого анилина, как описано ранее. Методика С (промотированное основанием фенольное алкилирование). Методику используют, когда методики А или В не подходят. Концентрированную суспензию калийной или натриевой соли фено-9 016126 ла, NaH2PO4 и подходящго эпоксида в смеси 9:1 MeCN/H2O нагревают до температуры 120-180 С в течение 30-90 мин в микроволновой печи или в течение 1-8 ч в стальном автоклаве (буферизация с помощью NaH2PO4 является необходимой для предупреждения обращения продукта в исходный фенол при увеличении значения рН во время реакции. Следует подчеркнуть, для небольшого масштаба реакции следует избегать значений температур более чем 180 С, чтобы минимизировать возможное последующее взрывоопасное разложение калийной соли 2-хлор-4-нитрофенола, быстро разлагающейся при температуре 210 С, с выделением газообразных продуктов). Последующее выделение и очистка с помощью хроматографии на силикагеле восстанавливает продукт до желаемого анилина, как описано ранее. Используют несколько аналитических HPLC способов; все контролируют УФ-абсорбцией при 220 нМ. Способ 1. Phenomenex Luna C18 S5 колонка 4.650 мм, 4 мин градиент при 4 мл/мин,10% МеОН/90% Н 2 О/0.2% Н 3 РО от 4 до 90% МеОН/10% Н 2 О/0.2% Н 3 РО 4 с 1 мин удержания в конце градиента. Способ 2. YMC S5 С 18 4.650 мм колонка, 4 мин градиент при 4 мл/мин, 10% МеОН/90% Н 2 О/0.2% Н 3 РО от 4 до 90% МеОН/10% Н 2 О/0.2% Н 3 РО 4 с 1 мин удержания в конце градиента. Способ 3. Phenomenex S5 С 18 4.630 мм колонка, 2 мин градиент при 4 мл/мин, 10% МеОН/90% Н 2 ОА (от 1% TFA до 90% МеОН/10% Н 2 ОА). 1% TFC 1 мин удержания в конце градиента. Способ 4. Phenomenex S5 С 18 4.630 мм колонка, 2 мин градиент при 4 мл/мин, 10% MeCN/90%H2OA (от 1% TFA до 90% MeCN/10% H2OA). 1% TFC 1 мин удержания в конце градиента. Способ 5. Phenomenex Luna C18 S5 колонка 4.650 мм, 4 мин градиент при 4 мл/мин, 10%MeCN/90% H2OA (1% TFA и 90% MeCN/10% H2OA). 1% TFC 1 мин удержания в конце градиента. Способ 6. Zorbax SB C18 S5 колонка 4.675 мм, 8 мин градиент от 50% растворитель В до 100% растворитель В при 2.5 мл/мин; растворитель А=10% МеОН/90% Н 2 О/0.2% Н 3 РО 4; растворитель В=90% МеОН/10% Н 2 О/0.2% Н 3 РО 4 с 2 мин удержания при 100% растворителе В. Способ 7. YMC A300-ODS S-5, 4.650 мм; 4 мин градиент при 4 мл/мин.; А=90:10 вода:метанол+0.2% фосфорная кислота, В=10:90 вода:метанол+0.2% фосфорная кислота; 0% В до 100% В 4 мин с 1 мин удержания в конце градиента. Препаративные HPLC условия использованы YMC C18 колонки, используя применяемый градиент элюирования с подходящей смесью от 10% МеОН/90% Н 2 ОА/0.1% TFA до 90% МеОН/10% Н 2 О/0.1%TFA. Иногда используют смеси от 10% MeCN/90% H2O/0.1% TFA и 90% MeCN/10% H2O/0.1% TFA. Если молекула содержит кислотный сенситивный компонент, TFA отсутствует. Масс-спектральные данные получают, используя Waters ZMD простой квадропольный массспектрометр. Типичными условиями являются Phenomenex C18 колонка 4.650 мм с обращенной фазой, 4 минутный градиент, от 10% МеОН/90% Н 2 О/0.1% TFA до 90% МеОН/10% Н 2 О/0.1% TFA, 1 мин удержания; 4 мл/мин, УФ определение при 220 нм. Пример 1. N-(2-(4-(6-(4-Хлорфенил)-4-оксотиено[3,2-d]пиримидин-3-(4 Н)-ил)-2-метоксифенокси)этил)пиваламид Часть А. N-(2-Гидроксиэтил)пиваламид. К перемешиваемому при температуре 4 С водному раствору, содержащему этаноламин (610 мг,10 ммоль), KOH (840 мг, 15 ммоль) и H2O (15 мл), добавляют пивалоилхлорид (1.44 г, 12 ммоль), после чего реакционному раствору дают возможность медленно нагреться до температуры 20 С. После перемешивания в течение 18 ч раствор экстрагируют 4EtOAc. Объединенные органические слои промывают рассолом, сушат над Na2SO4 до концентрации, используя роторный испаритель, чтобы получить на выходе 737 мг прозрачного масла. Часть В. N-(2-(4-Амино-2-метоксифенокси)этил)пиваламид К перемешиваемому раствору ДМФА (2 мл), содержащему N-(2-гидроксиэтил)пиваламид (360 мг,2.5 ммоль), под N2 добавляют 60% NaH/парафин (100 мг, 2.5 ммоль). Когда прекратится выделение газа,добавляют 2-хлор-5-нитроанилин (285 мг, 1.55 ммоль) и реакционную смесь перемешивают в течение 18 ч при температуре 20 С. Реакционную смесь разбавляют Н 2 О и экстрагируют EtOAc 3. Органические слои промывают дважды водным раствором Na2CO3, рассолом и сушат над Na2SO4 до концентра- 10016126 ции. Хроматография на силикагеле, используя 25% смесь EtOAc/CH2Cl2 в качестве элюента, дает 360 мгN-(2-(4-нитро-2-метоксифенокси)этил)пиваламида в виде твердого вещества желтого цвета. После растворения N-(2-(4-нитро-2-метоксифенокси)этил)пиваламида (360 мг, 1.2 ммоль) в EtOH(30 мл), содержащем 30 мг 10% Pd/C, смесь перемешивают под давлением 50 пси Н 2 в течение 1 ч. Реакционную смесь фильтруют через плотный слой целита и концентрируют перед хроматографированием остатка на силикагеле, используя EtOAc, чтобы элюировать N-(2-(4-амино-2-метоксифенокси)этил)пиваламид (310 мг). Часть С. Метил-5-(4-хлорфенил)-3-диметиламино)метиленамино)тиофен-2-карбоксилат Указанное в заголовке соединение получают, следуя методике, описанной в WO 2003033476. Конденсация 2.00 г метил-3-амино-5-(4-хлорфенил)тиофен-2-карбоксилата с диметилформамид диметилацеталем в течение 3 ч с нагреванием при температуре кипения с обратным холодильником EtOH дает на выходе 2.52 г (100%) после удаления летучих веществ в вакууме. 1(316 мг, 1.2 ммоль) конденсируют с метил-5-(4-хлорфенил)-3-диметиламино)метиленамино)тиофен-2 карбоксилатом (355 мг, 1.1 ммоль) путем нагревания раствора EtOH (3 мл) из двух компонентов до температуры кипения с обратным холодильником в течение 15 ч. После охлаждения и фильтрации, указанное в заголовке соединение выделяют в виде твердого вещества белого цвета. 1 Н ЯМР (CDCl3)1.22 (с, 9 Н), 3.71 (м, 2 Н), 3.90 (с, 3 Н), 4.176 (т, 2 Н, J=5 Гц), 6.35 (м, 1H), 6.94 (дд,1H, J=8.2 Гц, J=2.1 Гц), 6.99 (д, 1 Н, J=2.1 Гц), 7.05 (д, 1 Н, J=8.2 Гц), 7.45 (д, 2 Н, JAB=8.9 Гц), 7.54 (с, 1H),7.67 (д, 2 Н, JAB=8.9 Гц), 8.41 (с, 1 Н); Аналогичную методику, описанную для получения соединений по примеру 1, применяют, чтобы превратить N-этилэтаноламин в указанное в заголовке соединение. 1 Н ЯМР (CDCl3)1.16 (т, 0.75 Н, J=7 Гц), 1.24 (т, 2.25 Н, J=7 Гц), 2.13 (с, 2.25 Н), 2.24 (с, 0.75 Н), 2.25 Суспензию K2CO3 (280 мг, 2 ммоль), NaI (700 мг, 5 ммоль), калийной соли 2-метокси-4 нитрофенола (1.37 г, 6.6 ммоль), и трет-бутил-2-хлорэтилкарбамата (1.4 г, 8 ммоль) в ДМФА (8 мл) нагревают при температуре 90 С в течение 6 ч. После разбавления Н 2 О, смесь экстрагируют 4 CH2Cl2. Объединенные органические слои промывают 2 водным раствором K2CO3, затем рассолом до высушивания над Na2SO4. После удаления растворителя в вакууме, остаток хроматографируют на силикагеле,используя 5% смесь EtOAc/CH2Cl2, чтобы элюировать 870 мг нитрофенилового эфира. Восстановление на катализаторе Pd в этаноле, как описано в примере 1, дает на выходе трет-бутил-2-(4-амино-2 метоксифенокси)этилкарбамат (700 мг) в виде твердого вещества грязно-белого цвета, которое переносят на следующую стадию без дополнительной очистки. Часть В. Указанное в заголовке соединение (560 мг) получают фильтрацией твердого вещества, которое образуется при стоянии при температуре 20 С после нагревания смеси трет-бутил-2-(4-амино-2 метоксифенокси)этилкарбамата (700 мг) и метил-5-(4-хлорфенил)-3-диметиламино)метиленамино)тиофен-2-карбоксилата (800 мг, 2.5 ммоль) в EtOH (3 мл) при температуре кипения с обратным холодильником в течение 15 ч. 1 Н ЯМР (CDCl3)1.46 (с, 9 Н), 3.59 (м, 2 Н), 3.90 (с, 3 Н), 4.14 (т, 2 Н, J=4.8 Гц), 5.14 (м, 1H), 6.94 (дд,1 Н, J=8.4 Гц, J=2.2 Гц), 6.97 (д, 1 Н, J=2.2 Гц), 7.02 (д, 1H, J=8.4 Гц), 7.45 (д, 2 Н, JAB=8.9 Гц), 7.53 (с, 1H),7.65 (д, 2 Н, JAB=8.9 Гц), 8.13 (с, 1 Н); 10%-ный Раствор TFA/CH2Cl2 (0.5 мл), содержащий трет-бутил-2-(4-(6-(4-хлорфенил)-4 оксотиено[3,2-d]пиримидин-3-(4 Н)-ил)-2-метоксифенокси)этилкарбамата (15 мг, 0.03 ммоль) (пример 3),перемешивают в течение 2.5 ч, после чего летучие вещества удаляют в вакууме. К остатку, содержащему 3-(4-(2-аминоэтокси)-3-метоксифенил)-6-(4-хлорфенил)тиено[3,2-d]пиримидин-4-(3 Н)-он,последовательно добавляют CH2Cl2 (0.25 мл), Ас 2 О (20 мг, 0.2 ммоль) и Et3N (74 мг, 0.7 ммоль). После стояния в течение ночи реакционную смесь разбавляют водным раствором Na2CO3 и экстрагируют 4 CH2Cl2. Объединенные органические слои промывают рассолом и сушат над Na2SO4. После удаления растворителя остаток хроматографируют на силикагеле, используя 5-10% смесь МеОН/EtOAc, чтобы элюировать желаемый ацетамид (12 мг). 1- 12016126 Примеры 5-11. Указанные производные получают в соответствии с методикой, аналогичной методике, описанной в примере 4, или согласно близкого аналога путем ацилирования или сульфонилирования первичного амина Нитроариловый эфир из части А превращают в соединение по примеру 12, следуя методике, описанной в примере 1 и выделяют в виде твердого вещества белого цвета. 1 Н ЯМР (CDCl3)2.02-2.08 (м, 2H), 2.39-2.42 (м, 2H), 3.65-3.68 (м, 2H), 3.74-3.76 (м, 2 Н), 3.89 (с,3 Н), 4.21-4.23 (м, 2H), 6.93 (дд, J=8.25 Гц, 2.20 Гц, 1H), 6.97 (д, J=2.20 Гц, 1H), 6.99 (д, J=8.25 Гц, 1H),7.44 (д, J=8.24 Гц, 2 Н), 7.53 (с, 1H), 7.66 (д, J=8.25 Гц, 2H), 8.14 (с, 1H); 13 С ЯМР (CDCl3)18.20, 30.77, 42.22, 48.99, 56.11, 67.92, 111.03, 113.22, 119.18, 120.84, 123.17,127.63, 129.43, 130.28, 131.50, 135.66, 148.12, 148.72, 149.90, 151.66, 156.79, 157.38, 175.36; К раствору пирролидина при температуре 0 С (1.23 мл, 14.8 ммоль) в CH2Cl2 (мл) добавляют раствор хлорметилсульфонилхлорида (1.0 г, 65.71 ммоль) в течение более 30 мин. Баню удаляют и реакционную смесь перемешивают в течение 3 ч при температуре 20 С, после чего ее промывают 1 N HCl(20 мл). После высушивания над MgSO4, концентрации, получают на выходе твердое вещество желтого цвета (899 мг), которое в дальнейшем не очищают перед использованием для алкилирования калийной соли 2-метокси-4-нитрофенола, чтобы в конечном счете получить указанное в заголовке соединение,следуя методике, описанной в примере 3. 1- 14016126 Раствор 4-нитрогваяколкалийной соли (613 мг; 2.96 ммоль) и трет-бутилбромацетата (0.65 мл; 4.43 ммоль) в ДМФА (7 мл) перемешивают при комнатной температуре в течение 0.5 ч до разбавленияH2O и экстрагирования EtOAc. Органический слой промывают Н 2 О, сушат над MgSO4, фильтруют и фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью флэшхроматографии (силикагель, смесь гексан/EtOAc, градиент от 100:0 до 1:1), чтобы получить указанное в заголовке соединение (738 мг; 88%) в виде твердого вещества бежевого цвета. 1 Н ЯМР (CDCl3)1.46 (с, 9 Н), 3.96 (с, 3 Н), 4.67 (с, 2 Н), 6.77 (д, J=8.79 Гц, 1H), 7.76 (д, J=2.74 Гц,1H), 7.85 (дд, J=8.80 Гц, 2.75 Гц, 1H); 13 С ЯМР (CDCl3)27.98, 56.37, 66.18, 83.09, 107.07, 111.52, 117.28, 142.17, 149.19, 152.69, 166.66; Восстановление 718 мг трет-бутил-2-(2-метокси-4-нитрофенокси)ацетата из части А, как описано в примере 1, дает на выходе 601 мг (94%). 1 Н ЯМР (CDCl3)1.45 (с, 9H), 3.48 (уширенный с, 2H), 3.80 (с, 3 Н), 4.46 (с, 2 Н), 6.16 (дд, J=8.80 Гц,2.75 Гц, 1 Н), 6.28 (д, J=2.19 Гц, 1H), 6.70 (д, J=8.80 Гц, 1H); 13 С ЯМР (CDCl3)28.02, 55.71, 67.98, 81.77, 100.78, 106.36, 116.92, 140.41, 141.97, 150.69, 168.60;(740 мг) и трет-бутил-2-(4-амино-2-метокси-фенокси)ацетата из части С (581 мг), как описано в примере 1, дает на выходе указанное в заголовке соединение 388 мг (34%). 1 Раствор трет-бутил-2-(4-(6-(4-хлорфенил)-4-оксотиено[3,2-d]пиримидин-3-(4 Н)-ил)-2-метоксифенокси)ацетата из примера 14 (370 мг; 0.741 ммоль) в смеси 1:1 TFA/CH2Cl2 (6 мл) перемешивают при комнатной температуре в течение 1 ч. После концентрации при пониженном давлении и азеотропной отгонки с толуолом остаток растирают в порошок с МеОН, чтобы получить указанное в заголовке соединение (290 мг; 88%) в виде твердого вещества белого цвета. 1 Раствор 2-(4-(6-(4-хлорфенил)-4-оксотиено[3,2-d]пиримидин-3-(4 Н)-ил)-2-метоксифенокси)уксусной кислоты из примера 15 (20 мг; 0.045 ммоль), пиперидина (7 мкл; 0.068 ммоль), гидрохлорида EDC (10 мг; 0.054 ммоль) и гидрата гидроксибензотриазола (8 мг; 0.054 ммоль) в 70 мкл ДМФА и 415 мкл CH2Cl2 охлаждают до температуры 0 С. После завершения добавления N-метилморфолина (6 мкл; 0.054 ммоль),раствору дают возможность нагреться до комнатной температуры, после чего перемешивают в течение 23 ч. После концентрации при пониженном давлении и разбавления водой, смесь экстрагируют EtOAc. Органический слой промывают водой и рассолом, сушат над MgSO4, фильтруют и фильтрат концентрируют при пониженном давлении. Остаток растирают в порошок в МеОН, чтобы получить указанное в заголовке соединение (20 мг; 87%) в виде твердого вещества белого цвета. 1MS (ES): м/е 510 [M+H]+. Примеры 19-29. Следующие амиды синтезированы из соединения по примеру 15 и коммерчески доступных аминов в соответствии с методикой, аналогичной методике получения соединения, описанной в примере 18.- 17016126 Примеры 30-75. Указанные амиды получают в виде библиотеки, используя следующую методику. Следующие растворы последовательно добавляют в реакционный сосуд: 2-(4-(6-(4-хлорфенил)-4 оксотиено[3,2-d]пиримидин-3-(4 Н)-ил)-2-метоксифенокси)уксусную кислоту (получение описано в примере 15) (13.29 мг, 30 мкмоль) в 300 мкл ДМФА, HOBt (5.07 мг, 38 мкмоль) в 150 мкл ДМФА и 150 мкл ДМФА, содержащего EDC (7.19 мг, 38 мкмоль) и NEt(iPr)2 (26.1 мг, 150 мкмоль). Затем объединенные растворы перемешивают в течение 10 мин, добавляют 150 мкл ДМФА, содержащего амин (38 мкмоль) для осуществления ацилирования. Полученную смесь перемешивают в течение ночи при температуре 65 С, после чего реакционную смесь охлаждают до комнатной температуры и переносят в пластиковые микротрубки, используя ДМФА в виде промывочного растворителя. Для очистки сырые реакционные смеси вводят непосредственно из пластиковых микротрубок на препаративную LCMS, которая инициирует CMS на желаемую массу иона, чтобы собрать подходящие фракции. Детали очистки включают градиент элюирования, используя 20-100% растворитель В от Waters Sunfire 19100 5 мкм колонка с силикагелем в течение более 10 мин с 5-минутной задержкой. Растворитель А: 10/90 ацетонитрил/вода, содержащий 0.1% TFA. Растворитель В: 90/10 ацетонитрил/вода, содержащий 0.1% TFA. УФ 220 нМ; MSESI+. Фракции сушат в течение ночи, разбавляют 1 мл ДМФА и переносят в чистые, тарные пластиковые микротрубки. Небольшую аликвоту отбирают для анализа с помощью аналитической LCMS, разбавляют ДМФА для растворения. Микротрубки сушат в течение ночи и затем взвешивают. Данные аналитических хроматографии содержат оценку чистоты с помощью градиента элюирования от Waters Sunfire 4.650, 5 мкм колонка с силикагелем, используя 0-100% В в течение более 4 мин, 1-минутной задержкой,общее время выполнения 5 мин, контролирование УФ 220 нМ и MSESI+. Растворитель А: 10/90 метанол/вода, содержащий 0.1% TFA. Растворитель В: 90/10 метанол/вода, содержащий 0.1% TFA. Примеры 76-81. Следуя методике, описанной в примере 1, соединения по примерам 76-81 получают из соответственно замещенного 4-нитроанизола. Стальной автоклав, оснащенный внутренней термопарой, манометром и предохранительным выпускным клапаном, настроенный на давление 3000 пси, с объемом 55 мл, загружают калийной солью 2 метокси-4-нитрофенола (6 г, 29 ммоль), NaH2PO4 (3.3 г, 27.7 ммоль), изобутиленоксидом (2.8 г, 35 ммоль) и 30 мл 15% Н 2 О/MeCN. Герметизированный автоклав нагревают при температуре 170 С в течение трех ч. После охлаждения, HPLC выявляет, что весь исходный фенол превращен в продукт. Двухфазный раствор концентрируют, используя роторный испаритель перед распределением между CH2Cl2 и Н 2 О. Водную фазу экстрагируют 3 CH2Cl2; объединенные CH2Cl2 фракции промывают 3 водным растворомKHCO3/K2CO3 и один раз Н 2 О. После высушивания над Na2SO4, концентрация в вакууме дает на выходе 6.9 г желаемого продукта в виде твердого вещества желто-коричневого цвета.(Буферизация с помощью NaH2PO4 является необходимой для предупреждения обращения продукта в исходный фенол при увеличении значения рН в течение реакции. Следует подчеркнуть, для небольшого маштаба реакции следует избегать значений температур более чем 180 С, чтобы снизить возможное последующее взрывоопасное разложение калийной соли 2-хлор-4-нитрофенола, быстро разлагающейся при температуре 210 С, с выделением газообразных продуктов). 1 Н ЯМР (CDCl3)1.42 (с, 6H), 2.5 (с, 1 Н), 3.91 (с, 3 Н), 3.94 (с, 3 Н), 6.90 (д, J=9 Гц, 1H), 7.74 (д, J=2 Гц, 1H), 7.87 (дд, J=2 и 9 Гц, 1H); 13 С ЯМР (CDCl3)26.02, 56.18, 70.00, 77.42, 106.77, 111.74, 117.56, 141.73, 149.37, 153.95;HPLC (способ 1): 3.26 мин время задержки (99% API); Перемешиваемую EtOH суспензию (320 мл), содержащую 1-(2-метокси-4-нитрофенокси)-2 метилпропан-2-ол, из части А (13.6 г, 56.4 ммоль) и 10% Pd/C (0.25 г) гидрируют при давлении 60 пси H2 в течение 6 ч. HPLC анализ выявляет отсутствие исходного нитрокатехольного эфира. Полученную черного цвета суспензию фильтруют через стекловолокнистую фильтровальную бумагу под газообразнымN2. Полученный прозрачный, окрашенный в темно-красный цвет раствор сразу концентрируют, используя роторный испаритель в вакууме, чтобы получить на выходе 12.4 желаемого продукта в виде масла темно-оранжевого цвета, которое используют на следующей стадии без дополнительной очистки. 1 Превращения 1-(2-метокси-4-аминофенокси)-2-метилпропан-2-ола в указанное в заголовке соединение достигается с помощью методики, описанной в примере 1. 1 Н ЯМР (CDCl3)1.38 (с, 6 Н), 2.74, (с, 1H), 3.87 (с, 3 Н), 3.89 (с, 2 Н), 6.93 (дд, J=8.25 Гц, 2.20 Гц,1H), 6.96 (д, J=2.20 Гц, 1H), 7.01 (д, J=8.25 Гц, 1 Н), 7.44 (д, J=8.79 Гц, 2 Н), 7.52 (с, 1H), 7.65 (д, J=8.80 Гц,2 Н), 8.14 (с, 1H);LCMS (ES): м/е 457 [М+Н]. Примеры 83-88. Указанные соединения получают в соответствии с методикой, аналогичной методике получения соединения по примеру 82. К перемешиваемому при температуре 4 С раствору NaSMe (710 мг, 10. 1 ммоль) в Н 2 О (20 мл) последовательно добавляют порошок Cu (30 мг, ммоль), а затем водный раствор BF4 соли 2-гидрокси-5 нитробензолдиазония (298 мг, 1.8 ммоль), который получают, как описано у Can J. Chem, 1972, 50, 20252030). После перемешивания в течение 30 мин, реакционную смесь нагревают до температуры 20 С и перемешивают еще 2 ч перед фильтрованием. Значение рН фильтрата доводят до 5 с помощью 1 N HCl до экстрагирования 3 EtOAc. После высушивания над Na2SO4 и концентрации, твердое вещество черного цвета очищают с помощью хроматографии на силикагеле, используя 20-30% смесь EtOAc/гексан, чтобы элюировать (190 мг) в виде твердого вещества черного цвета.K2CO3 (134 мг, 0.97 ммоль) в 10% смеси H2O/MeCN (3 мл) нагревают при микроволновом излучении до температуры 125 С в течение 1 ч. После охлаждения и разбавления Н 2 О, смесь экстрагируют 3 EtOAc. После высушивания объединенной органической фракции над Na2SO4 и концентрации остаток хроматографируют на силикагеле, используя 30-50% смесь Смесь EtOAc/гексан, чтобы элюировать нитроариловый эфирный продукт (70 мг). Часть С. Указанный продукт из части В в МеОН (10 мл), содержащем 10% Pd/C (20 мг) перемешивают при давлении 60 пси H2 в течение 1 ч. Когда подтверждается завершение восстановления, с помощью LCMS анализа, реакционную смесь фильтруют. После концентрации остаток вместе с формамидином из примера 1, часть С (119 мг, 0.36 ммоль) и фенолом (0.5 г) нагревают до температуры 130 С в течение 30 мин. После охлаждения и разбавления МеОН, полученное твердое вещество собирают фильтрованием. Частичное окисление до соответствующего сульфоксида происходит во время концентрации при температуре 50 С, фракции собирают во время последней очистки с помощью препаративной HPLC, используя градиент водного раствора МеОН, содержащий 0.1% TFA. При повторении хроматографического отделении, чистый сульфид и сульфоксид получают, если фракции концентрируют при температуре 20 С. 1 Н ЯМР (CDCl3)141 (с, 6 Н), 2.45 (с, 3 Н), 3.93 (с, 2 Н), 6.45 (д, 1 Н), 7.13 (с, 2 Н), 7.45 (ABq, J=8.3 Гц,2 Н), 7.55 (с, 1H), 7.67 (ABq, J=8.3 Гц, 2 Н), 8.16 (с, 1 Н); К 0.5 М 9-BBN/ТГФ раствору (26.0 мл, 13.0 ммоль) добавляют пропаргилбромид (0.71 мл,6.37 ммоль, 80% в толуоле) и смесь нагревают при температуре 65 С в течение 5 ч. После охлаждения до температуры 20 С добавляют предварительно дегазированный раствор NaOH (749 мг, 18.7 ммоль) в воде(6.3 мл) и перемешивание продолжают в течение 1,5 ч. Смесь переносят во вторую колбу, содержащую 2-бром-4-нитроанизол (1.29 г, 5.45 ммоль) и тетракис(трифенилфосфин)палладия(0) (187 мг,0.16 ммоль) в ТГФ (8.0 мл). После нагревания при температуре 60 С в течение 14 ч, реакцию гасят водой(15 мл) и экстрагируют эфиром (340 мл). Объединенные органические экстракты непродолжительно промывают 2 М NaOH (320 мл) и водой (320 мл), сушат (Na2SO4) и концентрируют. Очистка с помощью двухсекционной хроматографической колонки (SiO2, первое элюирование со смесью 9/1 гек- 22016126 сан/EtOAc, второе элюирование со смесью 7/3 гексан/CH2Cl2) дает возможность выделить указанное в заголовке соединение (113 мг, 11% выход) в виде масла желтоватого цвета. К раствору соединения из части А (113 мг, 0.59 ммоль) в CH2Cl2 (1.0 мл) добавляют бортрифториддиметилсульфидный комплекс (0.37 мл, 3.52 ммоль) и смесь перемешивают при температуре 20 С в течение 7.5 ч. После завершения добавления МеОН (6.0 мл), смесь перемешивают в течение 20 мин и упаривают в вакууме. Хроматография (SiO2 230-400 ячеек, смесь 4/1 гексан/EtOAc) остатка дает указанное в заголовке соединение (44.3 мг, 42% выход) в виде твердого вещества красноватого цвета. Смесь соединения из части В (84.0 мг, 0.47 ммоль), карбонат калия (195 мг, 1.41 ммоль), CH3CN(4.0 мл), воду (0.4 мл) и изобутиленоксид (0,26 мл, 2.84 ммоль) нагревают при температуре 130 С в течение 3 ч в микроволновом реакторе. Конечную смесь упаривают и остаток распределяют между EtOAc(50 мл) и водой (8.0 мл). Органический слой промывают рассолом (8.0 мл), сушат (Na2SO4) и концентрируют. Хроматография (SiO2 230-400 ячеек, смесь от 7/3 до 3/2 гексан/EtOAc) сырого продукта дает указанное в заголовке соединение (73.0 мг, 62% выход) в виде твердого вещества белого цвета. К раствору соединения из части С (43.5 мг, 0.17 ммоль) в EtOAc (2.5 мл) добавляют 5% Pd-C(9.0 мг) и суспензию гидрируют (1 атм) в течение 40 мин. Суспензию фильтруют через плотный слой целита и фильтровальную лепешку промывают МеОН (20 мл). Объединенные фильтраты упаривают,чтобы получить на выходе указанное в заголовке соединение (38.0 мг, квант, но содержит 6% от его н-пропильного производного) в виде бесцветного масла. Продукт из части D (62.2 мг, 0.27 ммоль) и формамидин, описанный в части С примера 1 (185 мг,0.57 ммоль) смешивают с фенолом (430 мг) и нагревают при температуре 130 С в течение 35 мин. После охлаждения до температуры 20 С, смесь растворяют в эфире (8.0 мл) и дают возможность отстояться в течение 1 ч. Эфирный раствор декантируют и образованное твердое вещество кристаллизуют из смесиCH2Cl2/эфир, чтобы получить желаемое соединение (65.0 мг, 50% выход, содержит 5% от его нпропильного производного) в виде твердого вещества грязно-белого цвета. 1 Н ЯМР(CD2Cl2, част./млн) 8.09 (с, 1H), 7.69 (д, J=8.5 Гц, 2H), 7.56 (с, 1H), 7.46 (д, J=8.5 Гц, 2 Н),7.18 (дд, J=8.5, 2.4 Гц, 1 Н), 6.97 (д, J=8.5 Гц, 1 Н), 6.92 (д, J=2.4 Гц, 1H), 3.90 (с, 2 Н), 2.21 (м, 1H), 1.38 (с,6 Н), 0.98 (м, 2 Н), 0.67 (м, 2 Н); К раствору метил-2-метокси-5-нитробензоата (389 мг, 1.84 ммоль) в ТГФ (3.0 мл) добавляют 33% раствор MeNH2/EtOH (2.0 мл, 16.0 ммоль) и смесь перемешивают при температуре 20 С в течение ночи. Раствор упаривают и остаток хроматографируют (SiO2 230-400 ячеек), элюируя со смесью 7:3 гексан/EtOAc, чтобы удалить 2-(метиламино)-5-нитробензоатные производные. Дальнейшее элюированиеEtOAc обеспечивает указанное в заголовке соединение (268.5 мг, 70% выход) в виде твердого вещества желтоватого цвета. К раствору соединения из части А (268.5 мг, 1.28 ммоль), Ph3P (370 мг, 1.41 ммоль) и азидотриметилсилана (0.19 мл, 1.44) в ТГФ (12.6 мл) добавляют диэтилазодикарбоксилат (0.23 мл, 1.42 ммоль) и смесь перемешивают при температуре 20 С в течение 15.5 ч. Добавляют дополнительные количестваPPh3 (370 мг), TMSN3 (0.19 мл) и DEAD (0.23 мл) и смесь нагревают при температуре 50 С в течение 14 ч. Добавляют третью часть каждого из PPh3 (370 мг), TMSN3 (0.19 мл) и DEAD (0.23 мл) и нагревание при температуре 50 С продолжают в течение 15 ч. Большую часть растворителя удаляют путем продувания азота через реакционную смесь. Остаток хроматографируют (SiO2 230-400 ячеек, смесь от 1/1 до 7/3 гексан/EtOAc), что дает желаемый тетразол, загрязненный Ph3PO. Очистка с помощью хроматографии(SiO2 230-400 ячеек, смесь от 9/1 до 4/1 CH2Cl2/эфир) дает указанное в заголовке соединение (150.8 мг,50% выход) в виде твердого вещества желтоватого цвета. К раствору соединения из части В (150.8 мг, 0.64 ммоль) в CH2Cl2 (1.0 мл) добавляют бортрифторид-диметилсульфидный комплекс (0.41 мл, 3.90 ммоль) и смесь перемешивают при температуре 20 С в течение 28 ч. После завершения добавления МеОН (6.0 мл), смесь перемешивают в течение 1 ч и упаривают в вакууме. Хроматография (SiO2 230-400 ячеек, смесь 4:1 CH2Cl2/эфир) остатка обеспечивает указанное в заголовке соединение (135 мг, 96% выход) в виде твердого вещества красноватого цвета. К раствору соединения из части С (64.0 мг, 0.29 ммоль) в EtOAc (5.0 мл) и МеОН (0.3 мл) добавляют 5% Pd-C (15.0 мг) и суспензию гидрируют (1 атм) в течение 4.5 ч. Суспензию фильтруют через плотный слой целита и фильтровальную лепешку промывают МеОН (20 мл). Объединенные фильтраты упаривают, чтобы получить указанное в заголовке соединение (52.2 мг, 93% выход) в виде масла красноватого цвета: MS (электроспрей, + ионы) м/е 192 (М+Н). Часть Е. 6-(4-Хлорфенил)-3-(4-гидрокси-3-(1-метил-1H-тетразол-5-ил)фенил)тиено[3,2d]пиримидин-4-(3 Н)-он Продукт из части D (52.2 мг, 0.27 ммоль) и формамидин, описанный в части С Примера 1 (175 мг,0.54 ммоль) смешивают с фенолом (295 мг) и нагревают при температуре 125 С в течение 46 мин. После охлаждения до температуры 20 С, смесь растворяют в эфире (8.0 мл) и дают возможность отстояться в течение 1 ч. Эфирный раствор декантируют и образованное твердое вещество кристаллизуют из смесиCH2Cl2/эфир, чтобы получить указанное в заголовке соединение (64.5 мг, 55% выход) в виде твердого вещества грязно-белого цвета: MS (электроспрей, + ионы) м/е 437 (М+Н). Смесь продукта из части Е (30.3 мг, 0.07 ммоль), K2CO3 (11.0 мг, 0.08 ммоль), CH3CN (2.0 мл), воды(0.2 мл) и изобутиленоксида (45 мкл, 0.51 ммоль) нагревают при температуре 125 С в течение 2.5 ч в микроволновом реакторе. Конечную смесь разбавляют CH2Cl2 (40 мл) и промывают водой (5.0 мл). Водный раствор промывают, снова экстрагируют CH2Cl2 (25 мл) и объединенные органические экстракты сушат (Na2SO4) и концентрируют. Хроматография (SiO2 230-400 ячеек, смесь от 1/4 до 1/9 CH2Cl2/EtOAc) сырого продукта дает указанное в заголовке соединение (17.0 мг, 48% выход) в виде твердого вещества желтоватого цвета. 1 Н ЯМР (CD2Cl2+CD3OD капли, 35 С, част./млн)8.23 (с, 1 Н), 7.70 (д, J=8.4 Гц, 2 Н), 7.66 (дд, J=8.8,2.6 Гц, 1H), 7.58 (с, 1H), 7.56 (д, J=2.6 Гц, 1 Н), 7.45 (д, J=8.4 Гц, 2 Н), 7.31 (д, J=8.8 Гц, 1H), 4.09 (с, 3 Н),3.95 (с, 2 Н), 1.15 (с, 6 Н);(5.0 мл) добавляют гидрохлорид гидроксиламина (1.75 г, 25.2 ммоль). После перемешивания при температуре 20 С в течение 10 ч, смесь разбавляют CH2Cl2 (125 мл) и промывают водой (470 мл) и рассолом(50 мл). Органический слой сушат (Na2SO4) и упаривают. Остаток растирают в порошок с EtOH и фильтруют. Полученное твердое вещество промывают эфиром и сушат в вакууме, чтобы получить указанное в заголовке соединение (1.86 г, 81% выход) в виде твердого вещества желтоватого цвета: MS (электроспрей, + ионы) м/е 183 (М+Н). Часть В. Этил-3-(2-гидрокси-5-нитрофенил)изоксазол-4-карбоксилат К раствору соединения из части А (365 мг, 1.96 ммоль) и пиридина (35 мкл, 0.43 ммоль) в 1,2 дихлорэтан (6.0 мл) добавляют N-хлорсукцинимид (296 мг, 2.17 ммоль) порциями в течение более 15 мин. После завершения добавления смесь перемешивают при температуре 20 С в течение 25 мин и при температуре 50 С в течение 20 мин. Добавляют дополнительное количество NCS (150 мг, 1.10 ммоль) и нагревание при температуре 50 С продолжают в течение 20 мин. В это время добавляют этил(3 диметиламино)акрилат (0.56 мл, 3.91 ммоль) с последующим добавлением раствора Et3N (0.30 мл,2.15 ммоль) в 1,2-DCE (3.0 мл) в течение более 40 мин. Перемешивание продолжают затем при температуре 50 С в течение 1.5 ч и при температуре 20 С в течение ночи. Смесь разбавляют EtOAc (100 мл) и промывают 0.5 М KH2PO (30 мл) и рассолом (30 мл). Органический слой сушат (Na2SO4 и концентрируют. Сырой продукт очищают с помощью двух последовательных хроматографий (SiO2 230-400 ячеек,первый элюент: смесь 7:3 гексан/EtOAc, второй элюент: смесь 95/5 CH2Cl2/эфир), чтобы получить указанное в заголовке соединение (134.6 мг, 25% выход) в виде твердого вещества желтоватого цвета: MS Применяют аналогичные методики, описанные для получения соединений в частях D, Е и F примера 92, чтобы превратить продукт из части В в соответствующую карбоновую кислоту указанного в заголовке соединения. Обработка указанной кислоты смесью 3 M HCl/CH2Cl2, МеОН, MeOAc (получают путем добавления AcCl в раствор 3:2 CH2Cl2/МеОН при температуре 0 С и последующее перемешивание при температуре 20 С в течение 30 мин) дает на выходе желаемый метилэфирное соединение. 1 Н ЯМР (CD2Cl2 + CD3OD капли, част./млн)8.36 (с, 1H), 8.21 (с, 1H), 8.05 (д, J=2.6 Гц, 1 Н), 7.72 (д,J=8.5 Гц, 2 Н), 7.61 (с, 1H), 7.57 (дд, J=8.8, 2.6 Гц, 1H), 7.48 (д, J=8.5 Гц, 2 Н);MeCN (130 мл) при температуре 0 С добавляют по каплям TFAA (12 мл, 83 ммоль) с такой скоростью,чтобы поддержать значение внутренней температуры ниже 7 С. После перемешивания в течение 5 мин реакционному раствору дают возможность нагреться до комнатной температуры. Через 30 мин при этой температуре реакционную смесь выливают в воду со льдом (1.5 л). Смесь перемешивают в течение 40 мин. Твердое вещество собирают фильтрованием, промывают водой (250 мл) и частично сушат на фильтре. Осадок соместно упаривают трижды с EtOH (200-мл порции), чтобы обеспечить метил-3(трифторацетиламино)-2-тиофенкарбоксилат в виде твердого вещества желтого цвета (15 г, 96%). Часть В. Метил-3-амино-5-бромтиофен-2-карбоксилат К перемешиваемому раствору изо-Pr2NH (6.2 мл, 3.2 ммоль) в ТГФ (100 мл) на бане сухой лед-изоPrOH добавляют по каплям н-бутиллития (2.5 N в гексане, 17 мл, 43 ммоль). Через 5 мин смесь помещают на баню воды со льдом. Через еще 5 мин смесь охлаждают на бане сухой лед-изо-PrOH. К указанному перемешиваемому раствору добавляют по каплям в течение более 2 мин раствор метил-3(трифторацетиламино)-2-тиофенкарбоксилата (3.5 г, 14 ммоль) в ТГФ (15 мл). Через 5 мин добавляют одной порцией 1,2-диброметан (4 мл). Через 5 мин последовательно добавляют МеОН (3.5 мл) и насыщенный раствор NaHCO3 (150 мл), содержащий тиосульфат натрия (5 г). Смесь удаляют из охлаждающей бани и дают возможность нагреться до комнатной температуры, после чего добавляют EtOAc(250 мл). После разделения водный слой экстрагируют EtOAc (2100 мл). Три объединенных органических слоя промывают рассолом (2100 мл), сушат (MgSO4 и концентрируют в вакууме. Остаток абсорбируют на Целите 545 и хроматографируют (12 г силикагеля, от 10% смесь EtOAc-гексан до 25% смесьEtOAc/гексан, 50 мл/мин). Фракции, содержащие продукт объединяют и повторно хроматографируют(12 г силикагеля, 10% смесь EtOAc-гексан, 50 мл/мин), чтобы обеспечить метил-5-бром-3(трифторацетиламино)-2-тиофенкарбоксилат в виде твердого вещества бледно-желтого цвета 0.98 г(21%). Метил-5-бром-3-(трифторацетиламино)-2-тиофенкарбоксилат (2.0 г, 6.2 ммоль) перемешивают в 120 мл 0.25 N K2CO3 в смеси 7:3 МеОН/вода. Через 2.5 ч основную часть МеОН удаляют в вакууме. Остаток распределяют между рассолом (75 мл) и CH2Cl2 (100 мл). Органический слой сушат (MgSO4) и- 26016126 концентрируют в вакууме, чтобы обеспечить метил-5-бром-3-амино-2-тиофенкарбоксилат в виде твердого вещества желтого цвета (1.4 г, 100%). Часть С. Раствор этил-5-бром-3-амино-2-тиофенкарбоксилата (1.0 г, 4.2 ммоль) и диметилформамид диметилацеталя (1.1 мл, 8.5 ммоль) в EtOH (30 мл), нагревают до температуры кипения с обратным холодильником в течение 50 мин. После охлаждения растворитель удаляют в вакууме. Остаток азеотропно упаривают с толуолом (220 мл), чтобы получить на выходе метил-5-бром-3-диметиламино)метилен)амино)-2-тиофенкарбоксилат. Остаток и 1-4-амино-2-метокси)фенокси)-2-метил-2-пропанол(0.90 мг, 4.2 ммоль) (описано в части В примера 82) смешивают с фенолом (4 г) до нагревания при температуре 130 С в течение 40 мин. После охлаждения смесь хроматографируют (120 г силикагеля, отCH2Cl2 до 50% смеси EtOAc-CH2Cl2), чтобы обеспечить 1-(4-(6-бром-4-оксо-тиено[3,2-d]пиримидин-3(4 Н)-ил)-2-метоксифенокси)-2-метил-2-пропанол в виде твердого вещества желто-коричневого цвета(1 мл) и N2-дегазированную воду (0.5 мл) нагревают на бане при температуре 110 С в течение 30 мин. Смесь после охлаждения выливают в полунасыщенный раствор NaHCO3 (3 мл). Смесь экстрагируютCH2Cl2 (25 мл). Объединенные органические слои сушат (MgSO4) и концентрируют в вакууме. Остаток хроматографируют (4 г силикагеля, от CH2Cl2 до 50% смеси EtOAc-CH2Cl2), чтобы обеспечить 1-(4-(6(2,4-дихлорфенил)-4-оксо-тиено[3,2-d]пиримидин-3-(4 Н)-ил)-2-метоксифенокси)-2-метил-2-пропанол в виде твердого вещества грязно-белого цвета (41 мг, 71%). 1 Н ЯМР (CDCl3)8.09 (с, 1H), 7.54 (с, 1H), 7.49 (м, 2 Н), 7.29 (дд, J=8,2 Гц, 1H), 6.84-6.96 (м, 3 Н),3.81 (с, 5 Н), 1.30 (с, 6 Н);LCMS (ES) м/е 491 (М+Н). Примеры 95-103. Следующие соединения получают из 1-(4-(6-бром-4-оксо-тиено[3,2-d]-пиримидин-3-(4 Н)-ил)-2 метоксифенокси)-2-метил-2-пропанола, используя методики, аналогичные тем, которые описаны в примере 94, часть Е. Время реакции меняется от 20 мин до 17 ч и температура меняется от комнатной температуры до 100 С. В некоторых случаях используют большой избыток (до 3 эквивалентов) подходящей бороновой кислоты. Используют растирание в порошок хроматографированных продуктов с метанолом,чтобы удалить следы 1-(4-оксотиено[3,2-d]пиримидин-3-(4 Н)-ил)-2-метоксифенокси)-2-метил-2 пропанола. Для хроматографии смеси МеОН/CH2Cl2, EtOAc/CH2Cl2 или Смесь EtOAc/гексан используют в виде растворителей. Смесь калийной соли 4-нитрогваякола (2.00 г, 9.65 ммоль) и хлорацетона (1.15 мл, 14.5 ммоль) в 20 мл ДМФА нагревают при температуре 80 С в течение 3 ч. Суспензию охлаждают до комнатной температуры и разбавляют водой. Образуется осадок, который фильтруют, промывают водой и сушат в вакууме, чтобы получить указанное в заголовке соединение (1.63 г) в виде твердого вещества грязнобелого цвета. 1 Н ЯМР (CDCl3)2.29 (с, 3 Н), 3.96 (с, 3 Н), 4.71 (с, 2 Н), 6.74 (д, J=8.80 Гц, 1H), 7.76 (д, J=2.20 Гц,1 Н), 7.84 (дд, J=8.79 Гц, 2.20 Гц, 1 Н); 13 С ЯМР (CDCl3)26.37, 56.33, 73.62, 107.11, 111.88, 117.36, 142.33, 149.23, 152.49, 203.40; К смеси 1-(2-метокси-4-нитрофенокси)пропан-2-она (300 мг, 1.33 ммоль) в 1.3 мл МеОН и 1.3 мл Н 2 О при комнатной температуре добавляют по каплям в течение более 5 мин раствор NaBH4 (53 мг, 1.40 ммоль) в 0.5 мл Н 2 О. Суспензию перемешивают при комнатной температуре в течение 1 ч и разбавляют 0.2 мл НОАс, а затем 2 мл Н 2 О. Осадок фильтруют, промывают Н 2 О и сушат в вакууме, чтобы получить указанное в заголовке соединение (270 мг) в виде твердого вещества желтого цвета. 1 1-(2-Метокси-4-нитрофенокси)пропан-2-ол превращают в указанное в заголовке соединение соответствии с методикой, аналогичной методике, ранее описанной в примере 1. 1 Н ЯМР (DMSO-d6)1.16 (д, J=6.05 Гц, 3 Н), 3.77-3.82, (м, 4 Н), 3.88-3.91 (м, 1H), 3.94-4.02 (м, 1H),4.89 (д, J=4.39 Гц, 1H), 7.03 (дд, J=8.79 Гц, 1.64 Гц, 1H), 7.10 (д, J=8.25 Гц, 1H), 7.18 (д, J=1.65 Гц, 1H),7.57 (д, J=8.25 Гц, 2 Н), 7.92 (д, J=8.25 Гц, 2 Н), 7.97 (с, 1 Н), 8.39 (с, 1 Н); 13 С ЯМР (DMSO-d6)20.03, 55.58, 64.21, 73.88, 111.63, 112.68, 119.43, 121.56, 121.84, 127.66,129.10, 129.57, 131.02, 134.09, 148.27, 148.80, 149.32, 149.60, 155.91, 157.23; Часть А. (R)-2-2-Метокси-4-нитрофенокси)метил)оксиран. К раствору Ph3P (23.3 г; 88.7 ммоль) в 450 мл ТГФ, охлажденному до температуры 0 С, добавляют раствор ди-трет-бутилазодикарбоксилата (20.4 г; 88.7 ммоль) в 50 мл ТГФ в течение более 15 мин. После перемешивания при температуре 0 С в течение 10 мин добавляют 4-нитрогваякол (10.0 г; 59.1 ммоль), а затем (S)-глицидный спирт (6.3 мл; 94.6 ммоль) в течение более 10 мин, смеси дают возможность нагреться до комнатной температуры и перемешивают в течение 2 ч. Раствор концентрируют и остаток растворяют в EtOAc, промывают Н 2 О и рассолом, сушат над безводным MgSO4, фильтруют и фильтрат концентрируют при пониженном давлении. Остаток очищают с помощью флэш-хроматографии (силикагель, смесь гексан/EtOAc; градиент от 100:0 до 1:3). Фракции с примесями концентрируют при пониженном давлении и остаток затем очищают с помощью флэш-хроматографии (силикагель, смесь гексан/EtOAc; градиент от 100:0 до 0:100). Чистые фракции после обеих очисток объединяют и концентрируют при пониженном давлении, чтобы получить 9.58 г (72%) указанного в заголовке соединения в виде твердого вещества желтого цвета. 1 Н ЯМР (CDCl3)2.78-2.80 (м, 1H), 2.94-2.96 (м, 1H), 3.40-3.43 (м, 1H), 3.96 (с, 3 Н), 4.06-4.11 (м,1H), 4.41-4.44 (м, 1H), 6.98 (д, J=8.79 Гц, 1 Н), 7.75 (д, J=2.20 Гц, 1H), 7.89 (дд, J=8.79 Гц, 2.19 Гц, 1 Н); 13 С ЯМР (CDCl3)28.20, 44.57, 49.75, 56.28, 70.17, 106.80, 111.71, 117.50, 141.92, 149.20, 153.38; К 160 мл Et2O, охлажденному до температуры 0 С, добавляют порциями LiClO4 (80 г; 752 ммоль) в течение более 20 мин. Смеси дают возможность нагреться до комнатной температуры и добавляют (R)-22-метокси-4-нитрофенокси)метил)оксиран (9.55 г; 42.5 ммоль). Суспензию перемешивают в течение 10 мин, добавляют диметиламиноборан (3.40 г; 46.6 ммоль) и суспензию перемешивают при комнатной температуре в течение 2.5 ч. Суспензию разбавляют CH2Cl2 и перемешивают в химическом стакане с Н 2 О, пока не прекратится выделение газа. Органический слой промывают Н 2 О, сушат над безводнымMgSO4, фильтруют и фильтрат концентрируют при пониженном давлении. Остаток растирают в порошок в CH2Cl2, фильтруют и фильтрат очищают с помощью флэш-хроматографии (силикагель, смесь гексан/EtOAc; градиент от 100:0 до 100:0), чтобы получить 6.83 г (70%) указанного в заголовке соединения в виде твердого вещества желтого цвета.
МПК / Метки
МПК: C07D 495/04, A61K 31/522, A61P 5/00, A61P 25/00
Метки: меланинконцентрирующего, антагонисты, рецептора-1, гормона, неосновного
Код ссылки
<a href="https://eas.patents.su/30-16126-antagonisty-neosnovnogo-receptora-1-melaninkoncentriruyushhego-gormona.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты неосновного рецептора-1 меланинконцентрирующего гормона</a>
Антагонисты гормона, высвобождающего гонадотропин.

Номер патента: 992
Опубликовано: 28.08.2000
Авторы: Эштон Уоллес Т., Чу Лин, Виврэтт Мэтью Дж., Лин Питер, Гаулет Марк, Фишер Майкл Х., Джиротра Нариндар Н.
МПК: C07D 401/12, A61P 13/08, A61K 31/4045...
Метки: гормона, антагонисты, гонадотропин, высвобождающего
Формула / Реферат:
1. Соединение формулы где А представляет C1-C6алкил, замещенный C1-C6алкил, С3-С7 циклоалкил, замещенный С3-С7циклоалкил, С3-С6алкенил, замещенный С3-С6алкенил, С3-С6алкинил, замещенный С3-С6 алкинил, C1-C6 алкокси или (C0-C5алкил)-S(О)n-(С0-С5алкил), (C0-C5алкил)-O-(C0-C5алкил), (C0-C5алкил)-NR18-(C0-C5алкил), где R18 и (C0-C5 алкил) могут быть соединены, образуя кольцо, или одинарную связь; R0 представляет водород, C1-C6алкил,...
Производные пиримидин-2,4-диона в качестве антагонистов рецептора гонадотропин-высвобождающего гормона

Номер патента: 10370
Опубликовано: 29.08.2008
Авторы: Го Чжицян, Чень Юншэн, Хуанг Чарльз К., Чен Чен, Туччи Фабио К., Уэйд Уоррен, У Дунпэй, Дуайт Уэсли Дж.
МПК: A61K 31/513, C07D 239/54, A61P 5/02...
Метки: антагонистов, гормона, рецептора, качестве, производные, гонадотропин-высвобождающего, пиримидин-2,4-диона
Формула / Реферат:
Соединение, имеющее следующую структуру: или его стереоизомер или фармацевтически приемлемая соль, где R1a, R1b, R1c являются одинаковыми или разными и независимо представляют собой водород, галоген, С1-4алкил, гидрокси или алкокси, или R1a и R1b, взятые вместе, образуют -OCH2O- или -ОСН2СН2-; R2a и R2b являются одинаковыми или разными и независимо представляют собой водород, галоген, трифторметил, циано или -SO2CH3; R3 представляет собой...
Peg – илированный паратиреоидный гормон в качестве модулятора рецептора паратиреоидного гормона и его применение

Номер патента: 14696
Опубликовано: 30.12.2010
Авторы: Браун-Аугсбургер Патриция Ли, Кон Вэйн Дэвид
МПК: A61P 19/10, A61K 47/48, A61K 38/29...
Метки: гормона, рецептора, паратиреоидный, паратиреоидного, илированный, гормон, модулятора, качестве, применение
Формула / Реферат:
1. Соединение, имеющее последовательностьгде Хаа8 и Xaa18 представляют собой Met или Хаа8 и Xaa18 представляют собой Nle;mPEG представляет собой монометоксиполиэтиленгликоль со средней молекулярной массой от 1500 до 5500 Да;или его фармацевтически приемлемая соль.2. Соединение по п.1, имеющее последовательность аминокислот3. Соединение по п.1, имеющее последовательность аминокислот4. Соединение, выбранное из группы, включающейгде mPEG...
Антагонисты рецептора il-8

Номер патента: 1436
Опубликовано: 26.02.2001
Авторы: Юревич Энтони Джозеф, Ратледж Мельвин Кларенс Мл., Виддаусон Кетрин Луиза, Херцберг Роберт Филип, Вебер Дэниел Франк
МПК: A61P 11/06, A61K 31/17
Метки: антагонисты, рецептора
Формула / Реферат:
1. Способ лечения болезненного состояния, опосредованного хемокином, где хемокин связывается у млекопитающих, нуждающихся в таком лечении, с IL-8 а- или b-рецептором, включающий введение млекопитающему эффективного количества соединения формулы где Х является кислородом или серой; R является любой функциональной группой, имеющей ионизируемый водород и рКа, равный 10 или менее; R1 независимо выбирают из водорода; галогена; нитро; циано;...
Бензоксазиноновые антагонисты рецептора допамина d4.

Номер патента: 1486
Опубликовано: 23.04.2001
Авторы: Вайс Лауренс Дейвид, Вустров Дейвид Юрген, Беллиотти Томес
МПК: A61K 31/535, C07D 265/36
Метки: антагонисты, рецептора, допамина, бензоксазиноновые
Формула / Реферат:
1. Соединение формулы (I) где R1 и R2 означают независимо водород или алкил с числом углеродных атомов от 1 до 6, X означает С, N или СН, при этом в случае X=С штрихованная линия означает связь, а в других случаях отсутствует; R3 означает фенил, нафтил, гетероарил, замещенный фенил, замещенный нафтил или замещенный гетероарил, при этом каждый заместитель независимо выбран из группы, включающей галоген, алкоксигруппу с числом углеродных...
Предыдущий патент: Обессеривание дизельного топлива с использованием окисления и экстракции
Следующий патент: Применение производных триазина для производства лекарственного средства, обладающего заживляющим или ангиогенным эффектом
Случайный патент: Производные n1-замещенного 5-фтор-2-оксопиримидинон-1(2h)-карбоксамида и их применение в качестве фунгицидов