С-гликолипиды с улучшенным профилем th-1
Номер патента: 16042
Опубликовано: 30.01.2012
Авторы: Цудзи Мория, Фрэнк Ричард, Чэнь Гуану, Ян Гуанли









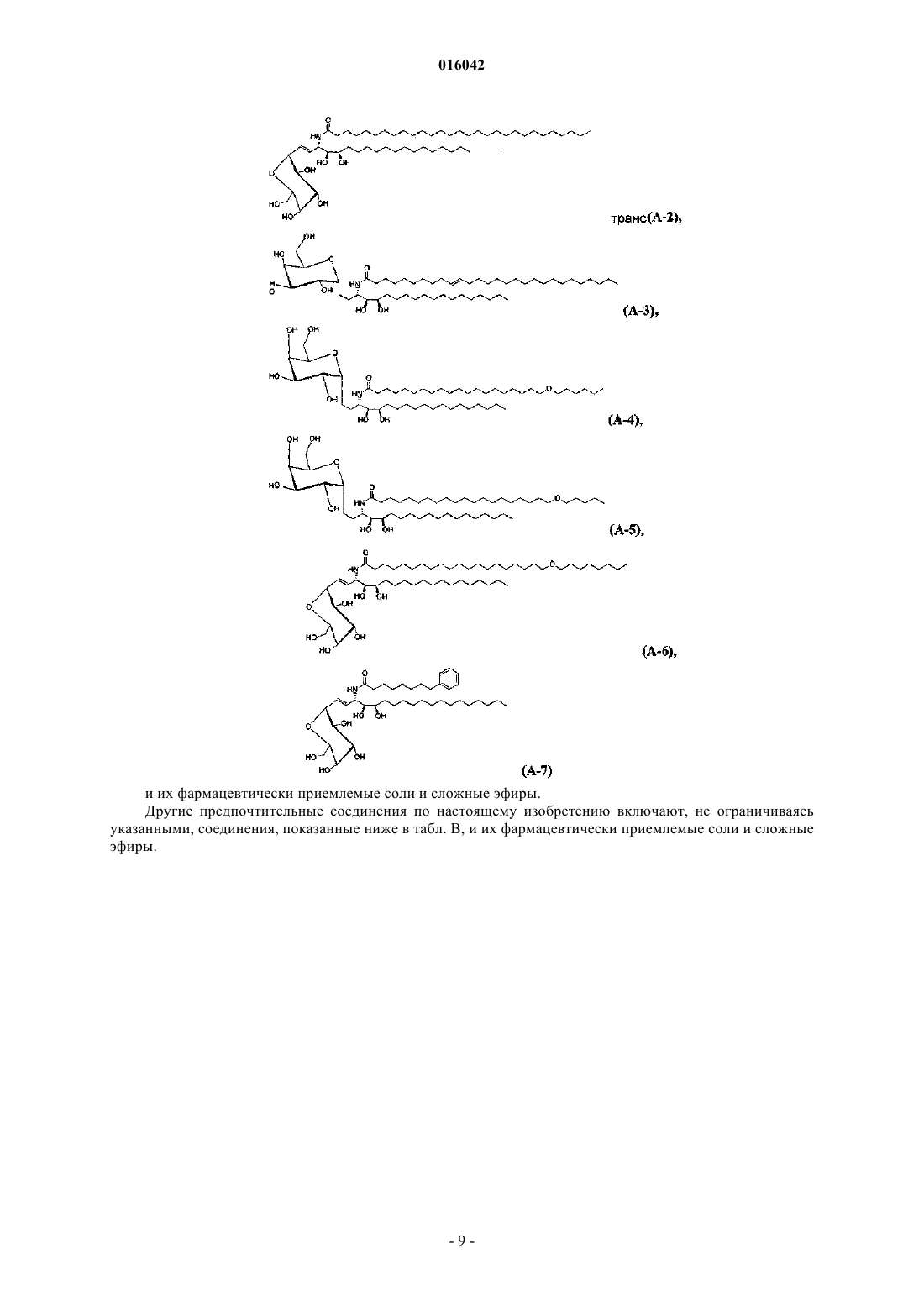
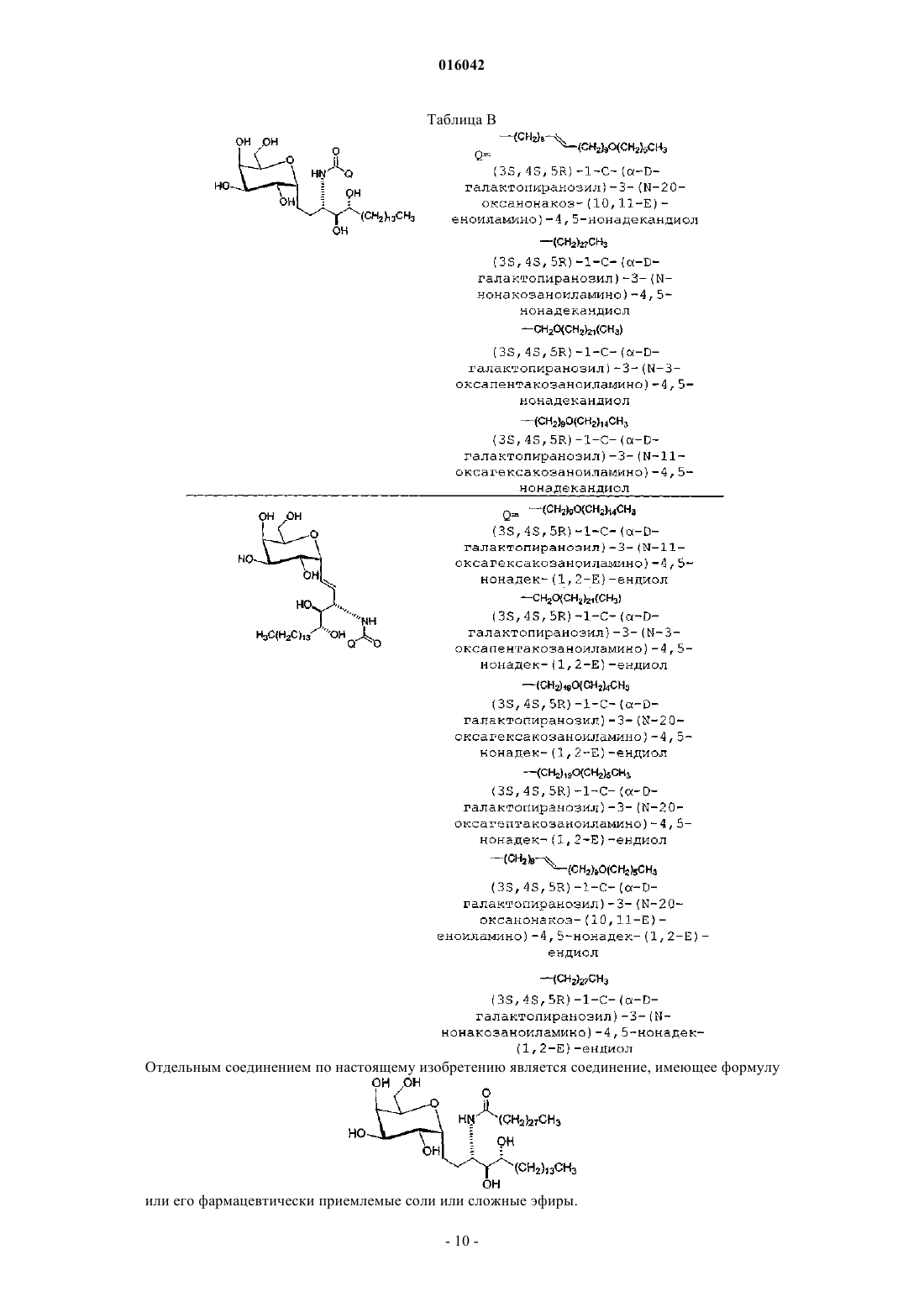











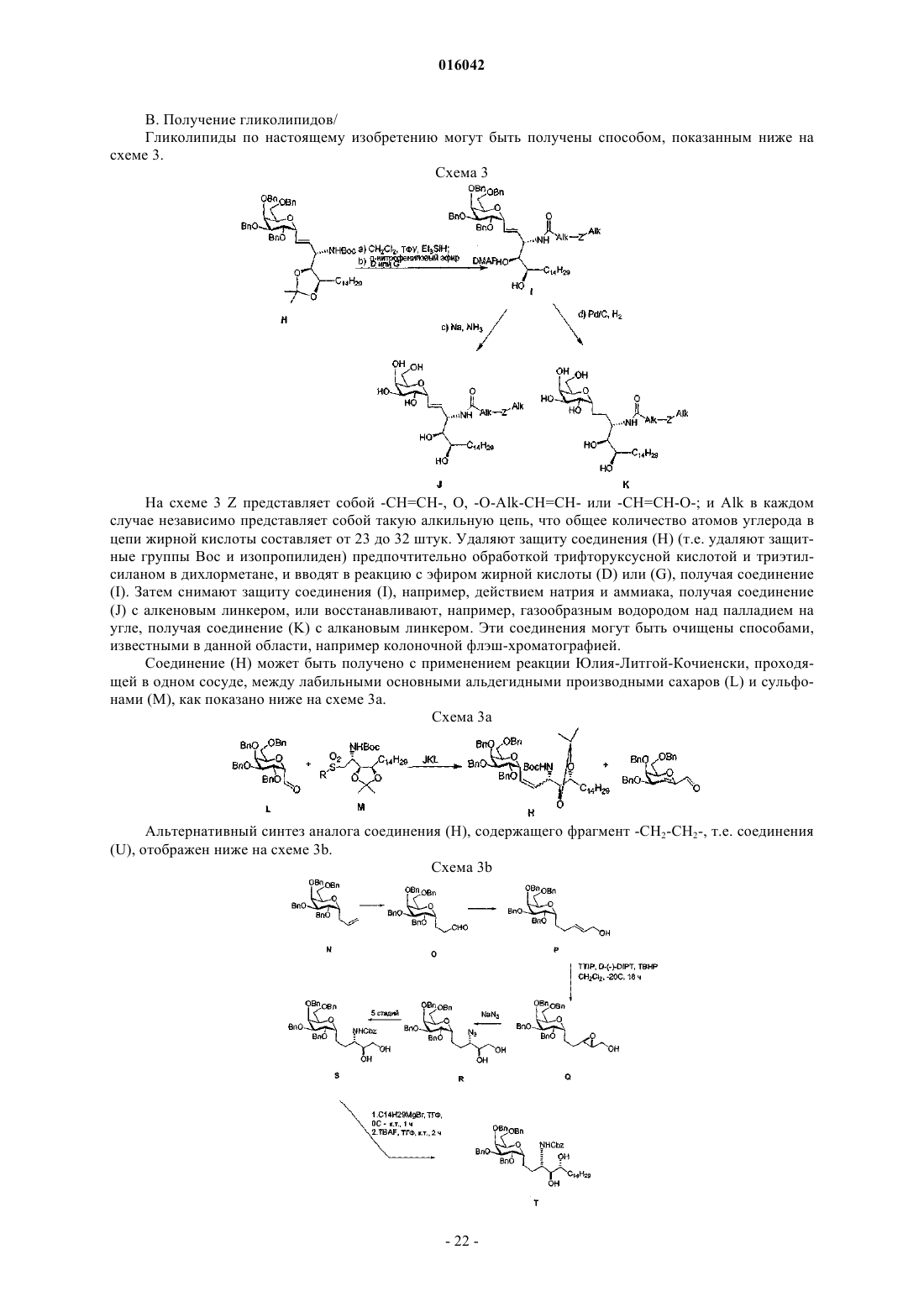



Формула / Реферат
1. Соединение формулы (I)

где X представляет собой О;
Y представляет собой -СН=СН-;
Q представляет собой С27-С32 алкил, С23-С32 алкенил, -R1-O-R2 или С6-C8 алкил, замещенный фенилом;
R1 и R2 представляют собой незамещенные алкильные или алкенильные группы, такие что R1 и R2 в совокупности включают от 23 до 32 атомов углерода;
R3 представляет собой -ОН или моносахарид и R4 представляет собой Н, или R3 представляет собой Н и R4 представляет собой -ОН или моносахарид; и
R5 представляет собой водород или моносахарид;
и его фармацевтически приемлемые соли.
2. Соединение по п.1, где X представляет собой О, R5 представляет собой Н, R3 представляет собой ОН, и R4 представляет собой Н.
3. Соединение по п.1 или 2, где Y представляет собой -СН=СН-, и соединение имеет транс-конфигурацию.
4. Соединение по п.3, где Q представляет собой -C8H16-СН=СН-С18Н37.
5. Соединение по п.1 или 2, где Y представляет собой -СН=СН-, и Q представляет собой С27-С28 алкил или С28-С32 алкенил.
6. Соединение по п.5, где Q представляет собой С28-С32 алкенил, имеющий одну, две или три двойных связи.
7. Соединение по п.6, где Q представляет собой С28-С32 алкенил, имеющий одну двойную связь.
8. Соединение по п.7, где двойная связь расположена между седьмым от карбонильной группы атомом углерода и двенадцатым от карбонильной группы атомом углерода.
9. Соединение по п.8, где двойная связь расположена между седьмым от карбонильной группы атомом углерода и десятым от карбонильной группы атомом углерода.
10. Соединение по п.9, где двойная связь расположена между девятым от карбонильной группы атомом углерода и десятым от карбонильной группы атомом углерода.
11. Соединение по п.1, имеющее формулу

и его фармацевтически приемлемые соли.
12. Соединение по п.1, 2 или 3, где Y представляет собой -СН=СН-, и Q представляет собой -R1-O-R2.
13. Соединение по п.12, где атом О во фрагменте -R1-O-R2 расположен между первым и двадцать пятым от карбонильной группы атомами углерода.
14. Соединение по п.13, где атом О во фрагменте -R1-O-R2 расположен между восемнадцатым и двадцать пятым от карбонильной группы атомами углерода.
15. Соединение по п.14, где Q представляет собой -C19H38-O-С6Н13.
16. Соединение по п.1, имеющее формулу

и его фармацевтически приемлемые соли.
17. Фармацевтическая композиция, содержащая соединение по любому из пп.1-16 в сочетании с фармацевтически приемлемым носителем.
18. Композиция по п.17, которая дополнительно содержит антиген.
19. Композиция по п.18, где антиген является вирусным или опухолевым антигеном.
20. Способ селективного индуцирования у млекопитающих иммунного ответа Th1-типа, где указанный способ включает введение млекопитающему эффективного количества соединения по любому из пп.1-16.
21. Способ по п.20, где указанное соединение индуцирует увеличение секреции IL-12.
22. Способ лечения заболевания, для контроля за которым требуется иммунный ответ Th1-типа, у млекопитающих, нуждающихся в этом, который включает введение млекопитающему эффективного количества соединения, определенного в любом из пп.1-16.
23. Способ по п.22, где заболевание выбрано из группы, состоящей из инфекции, рака, клеточного пролиферативного расстройства и аутоиммунного заболевания.
24. Способ по п.23, где заболевание представляет собой инфекционное невирусное заболевание.
25. Способ по п.23, где заболевание представляет собой инфекционное вирусное заболевание.
26. Способ по п.25, где заболевание является инфекцией, вызванной HIV.
27. Способ по п.25, где заболевание является инфекцией, вызванной HCV.
28. Способ по п.25, где заболевание является инфекцией, вызванной HBV.
29. Способ по п.25, где заболевание является малярийной инфекцией.
30. Способ по п.23, где заболевание представляет собой рак.
31. Способ по п.30, где заболевание представляет собой карциному простаты или меланому.
32. Способ по п.22, где заболевание является астмой или аллергией.
33. Способ повышения иммуногенности антигена у млекопитающих, где способ включает иммунизацию млекопитающего совместно антигеном и адъювантом, включающим соединение, определенное в любом из пп.1-16.
34. Способ по п.33, где антиген и адъювант вводят одновременно.
35. Способ по п.34, где антиген является HIV-специфичным.
36. Способ по п.34, где антиген является малярия-специфичным.
37. Способ получения соединения формулы (I)

где X представляет собой О;
Y представляет собой -СН=СН-;
Q представляет собой C27-C32 алкил, С23-С32 алкенил, -R1-O-R2 или С6-С8 алкил, замещенный фенилом;
R1 и R2 представляют собой незамещенные алкильные или алкенильные группы, такие что R1 и R2 в совокупности включают от 23 до 32 атомов углерода;
R3 представляет собой -ОН или моносахарид и R4 представляет собой Н, или R3 представляет собой Н и R4 представляет собой -ОН или моносахарид; и
R5 представляет собой водород или моносахарид;
и его фармацевтически приемлемой соли,
где способ включает стадии:
(а) удаление защитной группы Pg1 из соединения формулы (II)

с последующей его обработкой п-нитрофениловым эфиром, имеющим формулу (III):

где Q соответствует данным выше определениям; и
(b) последующего снятия защитных групп Pg2 и Pg3 и, необязательно, гидрирования двойной углерод-углеродной связи, расположенной по соседству с циклической группой, с образованием соединения формулы (I) и, необязательно, превращения соединения, полученного на стадии (b), в его фармацевтически приемлемую соль.
38. Способ по п.37, где соединение формулы (II) представляет собой

39. Соединение, имеющее формулу

и его фармацевтически приемлемые соли.
40. Соединение, имеющее формулу

и его фармацевтически приемлемые соли.
41. Соединение, имеющее формулу

и его фармацевтически приемлемые соли.
Текст
ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ЕВРАЗИЙСКОМУ ПАТЕНТУ Дата публикации и выдачи патента Изобретение направлено на новые синтетические С-гликолипиды формулы (I), которые селективно индуцируют иммунный ответ Th1-типа, характеризуемый повышением секрецииIL-12 и увеличением активации дендритных клеток. За счет этого соединения по настоящему изобретению применимы для лечения инфекций, раковых заболеваний, пролиферативных клеточных расстройств и аутоиммунных заболеваний, как непосредственно, так и в качестве адъювантов.(71)(73) Заявитель и патентовладелец: НЬЮ-ЙОРК ЮНИВЕРСИТИ; РИСЕРЧ ФАУНДЕЙШН ОФ ДЗЕ СИТИ ЮНИВЕРСИТИ ОФ НЬЮЙОРК; ДЗЕ ААРОН ДАЙМОНД ЭЙДС РИСЕРЧ СЕНТЕР ФОР ДЗЕ СИТИ ОФ НЬЮ-ЙОРК, ИНК. (US) 016042 Область техники, к которой относится изобретение Изобретение направлено на новые синтетические С-гликолипиды, которые селективно индуцируют иммунный ответ Th1-типа, характеризуемый улучшенной секрецией IL-12 и повышенной активацией антиген-презентирующих клеток (АРС), например дендритных клеток, и которые применимы при лечении инфекций, раковых заболеваний, пролиферативных клеточных расстройств и аутоиммунных заболеваний, как непосредственно, так и в качестве адъювантов. Предпосылки изобретения Иммунные ответы Th1-типа и Th2-типа первоначально определялись как иммунные ответы, опосредованные двумя различными подгруппами Т-клеток CD4+ (хелперных Т-клеток Th), которые секретируют две различные группы цитокинов. Для ознакомления с современным обзором см. Berkers andOvaa, Trends Pharmacol. Sci., 2005, 26(5):251-257 и цитированные в этом обзоре источники. Клетки Th1 секретируют цитокины Th1-типа, в число которых входят интерферон-гамма (IFN-) и интерлейкин 12 (IL-12). Основная функция цитокинов Th-1 типа заключается в поддержании клеточноопосредованного иммунитета, который служит для уничтожения опухолевых клеток, вирусов и других внеклеточных патогенов, за счет стимулирования опосредованной фагоцитами защиты и увеличения активности Т-клеток CD8+ (цитотоксичных Т-клеток) и натуральных клеток-киллеров (NK). Кроме того,цитокины Th1 ингибируют запуск синтеза иммуноглобулина В-клетками и подавляют выработку некоторых изотипов иммуноглобулина, таких как IgG1 и IgE, последний из которых является особенно важной причиной аллергии. IL-12 секретируется главным образом антиген-презентирующими клетками(АРС), в т.ч. дендритными клетками (DC) и макрофагами, и активирует Т-клетки CD8+ и NK-клетки. Клетки Th2 секретируют цитокины Th2-типа, включая IL-4, IL-5, IL-10 и IL-13. Основная функция цитокинов Th2-типа заключается в поддержке гуморального иммунитета (например, стимуляции IgE и иммунных реакций, опосредованных эозинофилами/мастоцитами) и понижающей регуляции иммунного ответа Th1 типа. Нарушение баланса между иммунными ответами Thl- и Th2-типов служит причиной заболеваний. Многие типы рака характеризуются преобладанием ответа Th2-типа и многие патогены отклоняют иммунную систему, вырабатывая цитокины, которые сдвигают баланс Thl-Th2 в сторону Th2 (Wilson andServet and Zitvogel, Curr Mol. Med., 2002, 2:739-756; Pinto, Pediatrics, 2006, Apr.17 [электронная публикация перед печатью]). Многие аутоиммунные заболевания, такие как астма, также характеризуются сдвигом баланса Th1-Th2 в сторону Th2. В противоположность этому такие аутоиммунные заболевания, как диабет 1 типа и рассеянный склероз, опосредуются аутопатогенными клетками Thl и характеризуются клетками Th2 с недостаточной чувствительностью, что ведет к Th1-подобному цитокиновому профилю(Hayakawa et al., 2004, Curr. Med. Chem., 11: 241-252; Wilson and Delovitch, 2003, Nat. Rev. Immunol.,3:211-222; Van Kaer, 2004, Immunol. Cell Biol., 82:315-322). Природные Т-клетки киллеры (NKT) играют решающую роль в регулировании иммунных ответовTh1- и Th2-типов. Клетки NKT представляют собой особую популяцию лимфоцитов, которые коэкспрессируют маркеры NK-клеток, наряду с полуинвариантным Т-клеточным рецептором (TCR). У мышейTCR большинства клеток NKT состоит из инвариантной цепи V, закодированной сегментами гена Vl4 и Jl8, спаренной с набором вариабельных цепей V, закодированных в основном сегментами генаV8.2, V7 или V2. Этот рецептор TCR дает возможность клеткам NKT распознавать молекулы CD1d,подобные классу I главного комплекса гистосовместимости (МНС), которые способны презентировать гидрофобные молекулы, например липиды и гидрофобные пептиды, клеткам NKT. До настоящего времени только в отношении нескольких молекул было показано, что они активируют клетки NKT. Из этих молекул лучше всего охарактеризован альфа-галактозилкерамид (-GalCer),т.е. гликолипид, первоначально выделенный из окинавских морских губок (Natori et al., Tetrahedron,50:2771-2784, 1994). Синтетический аналог -GalCer, именуемый KRN 7000 (2S,3S,4R)-1-O-(-Dгалактопиранозил)-2-(N-гексакозаноиламино)-1,3,4-октадекантриол, может быть приобретен у Pharmaceutical Research Laboratories, Kirin Brewery (Gumna, Japan) или синтезирован, как описано у Morita et al.,J. Med. Chem., 1995, 38:2176-2187. Другие производные -GalCer описаны в патенте США 5780441(Kirin). Следуя оригинальному раскрытию изобретения Kirin, была показана эффективность -GalCer в лечении нескольких заболеваний, включая первичные опухоли и их метастазы, инфекционные заболевания, такие как малярия и гепатит В, а также нескольких аутоиммунных заболеваний, таких как диабет и астма (см. Hayakawa et al, 2004, Curr. Med. Chem, 11:241-252; Wilson and Delovitch, 2003, Nat. Rev. Immunol., 3:211-222; Taniguchi et al, 2003, Annu. Rev. Immunol., 21:483-513; Van Kaer 2004, Immunol. Cell Biol. 82:315-322). Кроме того, было продемонстрировано, что -GalCer может применяться в качестве адъюванта, способного улучшить и/или увеличить продолжительность защитного иммунного ответа, вызванного другими антигенами (см. US 2003-0157135 и Gonzalez-Aseguinolaza et al., J Exp Med., 2002, 195:61724).-GalCer могут активировать клетки NKT как in vitro, так и in vivo (Kawano et al., 1997, Science,278:1626-1629; Burdin et al., 1998, J. Immunol., 161:3271-3281; Spada et al., 1998, J. Exp. Med., 188:1529-1 016042 1534; Brossay et al., 1998, J. Exp. Med. 188:1521-1528). Как показано на фиг. 1, в случае презентирования молекулы CD1d клетками АРС, такими как моноциты, образовавшиеся из моноцитов незрелые дендритные клетки и макрофаги, -GalCer взаимодействует с TCR клеток NKT, что впоследствии активирует как клетки NKT, так и АРС и приводит к выработке клетками NKT цитокина Th1-типа IFN- и цитокина Th2 типа IL-4. Затем на поверхности клеток NKT активируется рецептор IL-12, и одновременно IL-12 продуцируется активированными клетками АРС. Вырабатываемый АРС IL-12 продуцирует вторую волну выделения IFN- из клеток NKT и активирует также выработку IFN- клетками NK (Hayakawa et al, 2004,Curr. Med. Chem, 11:241-252; Kawano et al., 1997, Science, 278,1626-1629; Godfrey et al., 2000, Immunol.Today, 21:573-583; Wilson et al., 2002, Trends Mol. Med., 8:225-231; Matsuda et al., 2000, J. Exp. Med.,192:741-753). Таким образом, активация клеток NKT под действием -GalCer может приводить к вторичной активации клеток нескольких других типов, включая NK-клетки, В-клетки, Т-клетки CD8+, дендритные клетки и миелоидные клетки, а также к дифференцировке Т-клеток CD4+ на клетки Th1 илиTh2. Было показано, что введение -GalCer мышам приводит к быстрому проявлению значительной антималярийной активности, ингибирующей развитие стадий развития малярийных паразитов грызуновP.yoelii и P.berghei, протекающих внутри гепатоцитов (Gonzalez-Aseguinolaza et al., 2000, Proc. Natl. Acad.Sci. USA, 97:8461-8466). -GalCer был неспособен ингибировать развитие паразитов в печени мыши при недостатке либо IFN-, либо рецептора IFN-, и это свидетельствует о том, что антималярийная активность указанного гликолипида в первую очередь опосредуется IFN-. Стимулирование IL-4 под действием -GalCer позволяет облегчать течение ряда различных аутоиммунных заболеваний с применением этого гликолипида, включая аутоиммунный диабет типа 1 и аутоиммунный энцефаломиелит (Wilson etal. , 2002, Trends Mol. Med., 8:225-231). Важно, что помимо способности стимулировать иммунный ответ, -GalCer, как было показано, независимо от его дозировки, не проявляет токсического действия у грызунов и обезьян (Nakagawa et al.,1998, Cancer Res., 58:1202-1207). Однако эффективность терапевтического применения -GalCer в сильной степени ограничена одновременным стимулированием цитокинов Th1- и Th2-типов (т.е. IFN-, IL-12 и IL-4) (Pal et al., 2001, J.Immunol., 166:662-668; Berkers and Ovaa, Trends Pharmacol. Sci., 2005, 26:252-257). Действительно, при применении -GalCer у пациентов с солидными опухолями наблюдался незначительный эффект в исследовании фазы I (Giaccone et al., 2002, Clin. Cancer Res., 8:3702-3709). Было показано, что лечение GalCer становится более эффективным при смещении цитокинового профиля клеток NKT, например, в направлении Th1-типа, путем введения дендритных клеток, сенсибилизированных действием CD1d (Fujiiet al., 2002, Nat. Immunol., 3:867-874). Таким образом, аналог -GalCer, который мог бы селективно вызывать иммунный ответ Th1- илиTh2-типа, мог бы иметь более значительный терапевтический потенциал. В последнее время было разработано несколько аналогов -GalCer, в которых атом кислорода гликозидной связи заменен атомом углерода. См., например, патент США 6635622; Schmieg et al, 2003,J.Exp.Med, 198(11): 1631-1641; Chen et al. , Org Lett., 2004, 4077-80; Yang et al., Angew Chem Int Ed Engl.,2004, 43:3818-22, а также патентные заявки США 10/4 62 211 (US 2004-0127429); 11/193 852 (US 2006-0019246); 11/096 340 (US 2005-0222048). Такие аналоги устойчивы к дегликозилированию и, следовательно, имеют более продолжительный срок хранения (Bertozzi et al.,. Synthesis of C-glycosides: stable-C-GalCer CRONY 101 был первым примером С-гликозида, который обладал значительно лучшим терапевтическим потенциалом по сравнению с О-гликозидным аналогом. Как было показано в Schmieg etal. (2003, J.Exp.Med., 198:1631-1641) и Yang et al. (2004, Angew.Chem.Int.Ed.Engl., 43:3818-3822), in vivo введение CRONY 101 приводит к уменьшению выработки цитокина Th2-типа - IL-4 (по сравнению с GalCer) и улучшенной пролонгированной выработке цитокинов Th1-типа - IFN- и IL-12, что ведет к увеличению активности против метастазов меланомы и малярии в 100 и 1000 раз, соответственно. В последнее время цитокины Th1-типа IL-12 привлекли значительное внимание, благодаря их существенной роли во взаимодействии между врожденной и адаптивной ветвями иммунитета, за счет регулирования воспалительных реакций и врожденной сопротивляемости инфекциям и раку (обзор приведен в Colombo and Trinchieri, Cytokine Growth Factor Rev., 2002, 13:155-68; Watford, Cytokine Growth FactorRev., 2003, 14:361-368). Эндогенный IL-12 необходим для устойчивости ко многим патогенам и опухолям. Действительно, в экспериментальных моделях опухолей, применение рекомбинантного IL-12 приводило к существенному противоопухолевому воздействию на трансплантируемые опухоли, химически индуцированные опухоли и опухоли, самопроизвольно появляющиеся у генетически модифицированных мышей.-2 016042 Как было указано выше, интерлейкин IL-12 в основном секретируется различными АРС, такими как дендритные клетки и макрофаги, и содействует иммунному ответу Th1-типа (Roitt, Brostoff, Male, Immunology, Mosby ed., 6th ed.; Ma and Trinchieri, Adv Immunol., 2001, 79:55-92; Hilkens, Blood, 1997, 90:19201926; Szabo, Annu. Rev. Immunol., 2003, 21:713-58). IFN- и каскад других вторичных и третичных провоспалительных цитокинов, индуцируемый IL-12, оказывает прямое токсическое воздействие на инфицированные и опухолевые клетки и может активировать мощные анти-ангиогенные механизмы. Стимулирующее влияние IL-12 на антиген-специфичный иммунитет главным образом основывается на его способности определять или усиливать иммунные ответы Th1 и цитотоксичных Т-лимфоцитов. Благодаря этой способности IL-12 обладает мощной стимулирующей активностью при совместном действии с противораковыми и другими вакцинами. Многообещающие данные, полученные на доклинических моделях противоопухолевой иммунотерапии, породили надежду на то, что IL-12 мог бы стать эффективным терапевтическим средством против рака. Однако слишком высокая токсичность, наблюдаемая при клинических испытаниях IL-12, указывает на необходимость добиваться активации IL-12 локальным, а не системным путем. Сущность изобретения Как следует из предшествующего раздела "Предпосылки изобретения", в данной области существует значительная потребность в новых иммуностимулирующих соединениях, которые обладают низкой токсичностью in vivo, высокой стабильностью in vivo и способностью селективно инициировать иммунные ответы Th1-типа, в частности иммунные ответы Th1-типа, связанные с увеличенной местной выработкой IL-12. Настоящее изобретение отвечает на эти и другие имеющиеся в данной области потребности путем разработки новых синтетических С-гликолипидов. Соединения по настоящему изобретению могут применяться для лечения заболеваний, контроль за которыми требует иммунного ответа Th1-типа,включая, но не ограничиваясь перечисленными, различные инфекции, раковые заболевания, пролиферативные расстройства и аутоиммунные заболевания. Эти соединения способны также усиливать иммуногенность антигенов у млекопитающих. С-гликолипиды по настоящему изобретению включают соединения формулы (I)R1 и R2 представляют собой незамещенные алкильные или алкенильные группы, такие что R1 и R2 в совокупности включают от 23 до 32 атомов углерода;R5 представляет собой водород или моносахарид; и его фармацевтически приемлемые соли. В предпочтительном варианте осуществления Y представляет собой -СН=СН- в транс- или цисконфигурации. Более предпочтительно Y представляет собой -СН=СН- в транс-конфигурации. Предпочтительными являются те соединения, которые стимулируют повышенную секрецию IL-12. Такие соединения включают:(выше изображены транс- и цис-изомеры цепи жирной кислоты Q [именуемые также GCK109 иGCK151, соответственно]. В каждой формуле группа Y представляет собой транс-этилен. и их фармацевтически приемлемые соли. Эти соединения вызывают иммунный ответ Th1-типа с наибольшей специфичностью и обладают лучшим фармакокинетическим профилем по сравнению сCRONY. Не ограничиваясь какой-либо конкретной теорией, полагают, что эти соединения обеспечивают улучшенный баланс секреции IL-12 дендритными клетками и IFN- клетками NKT или NK, который дополнительно отражает специфичность иммунного ответа и улучшает безопасность соединений. Эти соединения не оказывают значительного стимулирующего действия на секрецию IL-4 клетками NKT илиNK. Другой вариант осуществления настоящего изобретения представляет собой фармацевтическую композицию, содержащую определенное выше соединение в сочетании с фармацевтически приемлемым носителем. Эта фармацевтическая композиция может дополнительно содержать антиген. Еще один вариант осуществления представляет собой способ селективного индуцирования у млекопитающих иммунного ответа Th1-типа, причем данный способ включает введение млекопитающему эффективного количества соединения по настоящему изобретению. Соединения по настоящему изобретению индуцируют иммунный ответ Th1-типа настолько селективно и эффективно, что они эффективны для лечения как при непосредственном применении, так и при применении в качестве адъюванта совместно со специфичными для болезни антигенами. Следовательно,еще один вариант осуществления настоящего изобретения представляет собой способ лечения заболеваний у млекопитающих, нуждающихся в этом, для контроля за которыми необходим иммунный ответTh1-типа, причем данный способ включает введение млекопитающему эффективного количества соединения формулы (I). Неограничивающие примеры заболеваний, для контроля за которыми необходим иммунный ответ Th1-типа, включают инфекции, раковые заболевания, клеточные пролиферативные расстройства и аутоиммунные заболевания Th2-типа. В предпочтительном варианте осуществления заболевание, для контроля над которым требуется иммунный ответ Th1-типа, представляет собой инфекционное вирусное заболевание, например вирусную инфекцию иммунодефицита человека (HIV), вирусную инфекцию гепатита С (HCV), вирусную инфекцию гепатита В (HBV), вирусную инфекцию герпеса или респираторно-синцитиальную вирусную инфекцию (RSV). В другом предпочтительном варианте осуществления заболевание представляет собой рак, например солидную опухоль, такую как карцинома простаты или молочной железы. В еще одном предпочтительном варианте осуществления заболевание представляет собой астму. Следующий вариант осуществления представляет собой способ повышения иммуногенности антигена у млекопитающих путем иммунизации млекопитающего антигеном совместно с адъювантом, включающим соединение формулы (I). В предпочтительном варианте осуществления антиген является HIVспецифичным, малярия-специфичным или специфичным в отношении рака простаты. Еще один вариант осуществления изобретения представляет собой способ получения соединений формулы (I), описанный в настоящей заявке. Отдельным вариантом осуществления является способ получения соединения формулы (I)Q представляет собой С 27-С 32 алкил, С 23-С 32 алкенил, -R1-O-R2 или C6-C8 алкил, замещенный фениR1 и R2 представляют собой незамещенные алкильные или алкенильные группы, такие что R1 и R2 в-4 016042 совокупности включают от 23 до 32 атомов углерода;R5 представляет собой водород или моносахарид; и его фармацевтически приемлемой соли,где указанный способ включает стадии:(а) удаление защитной группы Pg1 из соединения формулы (II) с последующей его обработкой п-нитрофениловым сложным эфиром, имеющим формулу (III): где заместитель Q соответствует данному выше определению; и(b) последующего снятия защитных групп Pg2 и Pg3, и необязательно гидрирования двойной углерод-углеродной связи,расположенной рядом с циклической группой, с получением соединения формулы (I). Согласно одному из предпочтительных вариантов осуществления соединение формулы (II) представляет собой Краткое описание иллюстрированного материала Фиг. 1 представляет собой схематическое изображение активации цитокинов и иммунных клеток под действием О- и С-гликолипидов. На фиг. 2 А-С отображены кинетические профили цитокина Th1-типа IFN-, который выделяется при введении соединений по настоящему изобретению или контрольных соединений мышам BALB/c(фиг. 2 А) и C57BL/6 (фиг. 2 В-С). Соединениями, представленными на данных графиках, являются А=соединение (транс-А-1), В=соединение (А-2), С=соединение (А-3), D=соединение (А-4),Е=соединение (А-5), Z=контроль (PBS без добавок), CRONY=-C-GalCer, KRN=-GalCer, GCK109 (А-1 транс-изомер), GCK151 (А-1 цис-изомер) и GCK152[A-7]. Уровни IFN- в сыворотке измеряли спустя 0,2, 6, 12, 24, 48 и 72 часа после введения соединения с помощью иммуноферментного твердофазного анализа (ELISA). Данные выражены в виде среднего значениястандартное отклонение (SD) для двух различных разбавлений смешанной сыворотки. На фиг. 3 А-С отображены кинетические профили цитокина Th1-типа IL-12, который выделяется при введении соединений по настоящему изобретению или контрольных соединений мышам BALB/c(фиг. 3 А) и C57BL/6 (фиг. 3 В-С). Соединениями, представленными на данных графиках, являются А=соединение(транс-А-1), GCK151 (цис-А-1) и GCK152[A-7]. Уровни IL-12 в сыворотке измеряли спустя 0, 2, 6, 12, 24,48 и 72 ч после введения соединения с помощью иммуноферментного твердофазного анализа (ELISA). Данные выражены в виде среднего значениястандартное отклонение (SD) для двух различных разбавлений смешанной сыворотки. На фиг. 4 А-С отображены кинетические профили цитокина Th2-типа IL-4, который выделяется при введении соединений по настоящему изобретению или контрольных соединений мышам BALB/c (фиг. 4 А) и C57BL/6 (фиг. 4 В-С). Соединениями, представленными на данных графиках, являются А=соединение (транс-А-1), В=соединение (А-2), С=соединение(А-3), D=соединение (А-4), Е=соединение(А-5), Z=контроль (PBS без добавок), CRONY=-C-GalCer, KRN=-GalCer, GCK109 (транс-А-1),GCK151 (цис-А-1) и GCK152[A-7]. Уровни IL-4 в сыворотке измеряли спустя 0, 2, 6, 12, 24, 48 и 72 ч после введения соединения с помощью иммуноферментного твердофазного анализа (ELISA). Данные выражены в виде среднего значениястандартное отклонение (SD) для двух различных разбавлений-5 016042 смешанной сыворотки. На фиг. 5 А и 5 В показаны уровни цитокинов, выделявшихся при действии соединений по настоящему изобретению, измеренные с помощью ELISA в экспериментальной системе человеческих клетокNKT in vitro. Концентрации IFN- или IL-4 определяли с помощью ELISA в супернатантах культур незрелых дендритных клеток (DC), которые культивировали в течение 18 часов совместно с сингенными моноядерными клетками периферической крови (РВМС) CD14- в присутствии различных гликолипидовCRONY=-C-GalCer, KRN=-GalCer). На фиг. 6 А и 6 В показано количество РВМС, секретирующих цитокины, при инкубировании с соединениями по настоящему изобретению, измеренное с помощью ELISPOT в экспериментальной системе человеческих клеток NKT in vitro. Количество РВМС, секретирующих IFN- или IL-4, определяли с помощью совместного культивирования незрелых DC с сингенными CD14-PBMC в планшете ELISPOT в течение 22-26 часов в присутствии различных гликолипидов (т.е. GCK142A [А-б], GCK109 [транс-А-1],GCK151 [цис-А-1], GCK127 [А-2], GCK152 [А-7] и контрольных соединений CRONY=-C-GalCer,KRN=-GalCer). Подробное описание изобретения Определения Термины "иммунный ответ Th1-типа" и "иммунный ответ Th2-типа" в настоящем описании относятся к иммунным ответам, опосредованным хелперными Т-клетками Th1 и Th2 CD4+, соответственно. Термины "цитокины Th1-типа" и "цитокины Th2-типа" относятся к цитокинам, продуцируемым Th1 и Th2 клетками, соответственно. Цитокины Th1-типа включают, не ограничиваясь указанными, IFN- иIL-12. Неограничивающим примером цитокинов Th2-типа является IL-4. Термин "заболевание, для контроля над которым требуется иммунный ответ Th1-типа" относится к заболеванию, характеризуемому преобладанием иммунного ответа Th2-типа и профилем цитокинов Th2 типа. Примеры таких заболеваний включают, не ограничиваясь перечисленными, инфекционные вирусные заболевания, например HIV-инфекцию, HCV-инфекцию, HBV-инфекцию, герпесную вирусную инфекцию и RSV-инфекцию; рак, например карциному простаты или карциному молочной железы; и аутоиммунные заболевания Th2-типа, например астму и аллергию. Термин "селективная индукция иммунного ответа Th1-типа" в настоящем описании относится к индукции и/или усилению, и/или увеличению продолжительности иммунного ответа Th1-типа, которые не вызывают одновременной индукции и/или усиления, и/или увеличения продолжительности иммунного ответа Th2-типа. Для соединений по настоящему изобретению селективная индукция иммунного ответа Th1-типа отражается в усилении секреции IL-12 и усилении активации дендритных клеток (по сравнению с соединениями известного уровня техники), которые имеют место без одновременного усиления секреции IL-4. Термин "моносахарид" относится к молекуле сахара, цепь которой состоит из 3-10 атомов углерода в форме альдегида (альдоза) или кетона (кетоза). Подходящие моносахариды, рассматриваемые для применения в настоящем изобретении, включают как природные, так и синтетические моносахариды. Неограничивающие примеры подходящих моносахаридов включают триозы, например глицерозу и дигидроксиацетон; тетрозы, например, эритрозу и эритрулозу; пентозы, например ксилозу, арабинозу, рибозу,ксилулозу, рибулозу; метилпентозы (6-дезоксигексозы), например рамнозу и фукозу; гексозы, например,глюкозу, маннозу, галактозу, фруктозу и сорбозу; и гептозы, например, глюкогептозу, галаманногептозу,седогептулозу и манногептулозу. Предпочтительные моносахариды включают, не ограничиваясь этим,гексозы."Эффективное количество" соединения для лечения заболевания, например рака, инфекционного заболевания или аутоиммунного заболевания, представляет собой количество, которое ведет к измеримому облегчению хотя бы одного симптома или параметра заболевания у млекопитающих, включая людей. В настоящем описании термин "их фармацевтически приемлемые соли или сложные эфиры" относится к тем солям (например, солям карбоновых кислот, аддитивным солям аминокислот) и сложным эфирам соединений по настоящему изобретению, которые в рамках обоснованного медицинского суждения подходят для применения в контакте с тканями пациентов, без неприемлемой токсичности, раздражения, аллергических реакций и т.п., в соответствии с разумным соотношением польза/риск и эффективны в намеченном для них применении, а также к цвиттер-ионным формам, в тех случаях, где они возможны, соединений по настоящему изобретению. Термин "лечить" применяется в настоящем описании для обозначения профилактики заболевания,или облегчения или улучшения как минимум одного симптома заболевания у субъекта. В смысле настоящего изобретения термин "лечить" может также означать продление инкубационного периода, т.е. периода между началом инфекции и клиническими проявлениями заболевания. Термин "терапевтически эффективный", использованный в отношении дозы или количества, относится к такому количеству соединения или фармацевтической композиции, которое достаточно для того,-6 016042 чтобы вызвать желаемую активность в случае введения млекопитающему при наличии такой необходимости. Термины "фармацевтически приемлемый" и "физиологически приемлемый" являются взаимозаменяемыми, и в случае их использования в связи с композициями по настоящему изобретению, относятся к молекулярным объектам и другим ингредиентам таких композиций, которые являются физиологически переносимыми и, как правило, не вызывают неблагоприятных реакций при введении людям. Предпочтительно в настоящем описании термин "фармацевтически приемлемый" означает одобренный регулирующим органом федеральной администрации или администрации штата или упомянутый в фармакопее США или других широко известных фармакопеях для применения у млекопитающих и более конкретно у людей. Термин "носитель" относится к разбавителю, эксципиенту или среде, вместе с которыми осуществляется введение соединения по настоящему изобретению. Эти фармацевтические носители могут представлять собой стерильные жидкости, например воду или масла, включая масла, полученные из нефти,животного, растительного или синтетического происхождения, например арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.п. Предпочтительно в качестве носителей применяются солевые растворы и водные растворы декстрозы и глицерина, в особенности с целью получения пригодных для инъекции растворов. Подходящие фармацевтические носители описаны E.W.Martin в "Remington'sPharmaceutical Sciences", 18 издание. Термины "адъювант" и "иммуноадъювант" в настоящем описании являются взаимозаменяемыми и относятся к соединению или смеси, которые могут быть неиммуногенными при введении субъекту в чистом виде, но усиливают иммунный ответ на другой антиген, при введении совместно с этим антигеном. В настоящем описании термины "совместное введение", "введенный совместно" и "совместно вводящийся" относятся к введению двух агентов, например иммуноадъюванта и антигена, одновременно в составе одной композиции или одновременно в составе разных композиций, или последовательно. Однако, для того, чтобы последовательное введение считалось "совместным", введение антигена и адъюванта должно быть разделено интервалом времени, который еще позволяет адъюванту усилить иммунный ответ на антиген. Например, если антиген представляет собой полипептид, антиген и адъювант вводят в один и тот же день, предпочтительно в течение часа друг за другом и наиболее предпочтительно одновременно. Однако, если субъекту вводят нуклеиновую кислоту и полипептидный антиген экспрессируется в клетках субъекта, адъювант предпочтительно вводят в течение 24 ч после введения нуклеиновой кислоты и более предпочтительно в течение 6 ч. Термин "субъект" в настоящем описании относится к животному, имеющему иммунную систему,предпочтительно млекопитающему. Субъекты, к которым применимо настоящее изобретение, включают, не ограничиваясь перечисленным, коров, лошадей, овец, свиней, домашнюю птицу (например, кур),коз, кошек, собак, грызунов (например, хомяков, мышей, крыс, кроликов), обезьян, приматов, а также людей. В предпочтительном варианте осуществления субъект является человеком. Термин "примерно" или "приблизительно" означает в пределах приемлемого диапазона ошибки для конкретной величины согласно мнению обычного специалиста в области техники, который частично будет зависеть от того, каким образом измерена или определена данная величина, т.е. ограничений измерительной системы. Например, "примерно" может означать в пределах одного или более чем одного стандартного отклонения, согласно практике, существующей в данной области. С другой стороны "примерно" может означать диапазон до 20%, предпочтительно до 10%, более предпочтительно до 5% и еще более предпочтительно до 1% от данной величины. В качестве альтернативы, в частности в отношении к биологическим системам или процессам, этот термин может означать в пределах порядка величины,предпочтительно в пределах 5-кратного значения величины и более предпочтительно в пределах 2 кратного значения величины. Если в заявке и формуле изобретения описаны конкретные величины и если не указано иначе, термин "примерно" следует понимать как означающий в диапазоне приемлемой ошибки для конкретной величины. В соответствии с настоящим изобретением могут применяться обычные методики молекулярной биологии, микробиологии и рекомбинантных ДНК, в пределах, известных специалисту в данной области техники. Такие методики хорошо известны и полностью объяснены в литературе. См., например, Sambrook, Fritsch and Maniatis, Molecular Cloning: A Laboratory Manual, Second Edition (1989) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York (в "Sambrook et ah, 1989"); DNA Cloning: A Practical[IRL Press, (1986)]; B. Perbal, A Practical Guide To Molecular Cloning (1984); F.M. Ausubel et a (eds.), Current Protocols in Molecular Biology, John WileySons, Inc. (1994). Соединения по настоящему изобретению Предпочтительными являются соединения формулы (I), где X представляет собой О, R5 представляет собой Н, R3 представляет собой ОН, и R4 представляет собой Н. Предпочтительно соединения по настоящему изобретению содержат как минимум одну нуклео-7 016042 фильную связь в боковой липидной цепи (т.е. группе Q). Каждая нуклеофильная связь предпочтительно представляет собой простую эфирную связь (т.е. Q представляет собой -R1-O-R2) или двойную связь (т.е.Q представляет собой алкенил). Нуклеофильная связь предпочтительно расположена на расстоянии минимум шести атомов углерода от концевого атома углерода боковой цепи Q. Например, двойная связь может находиться в любом положении от 6 до 22 атомов углерода от концевого атома углерода боковой цепи Q. Согласно одному из вариантов осуществления изобретения Q представляет собой С 23-С 32 алкенил,содержащий от 23 до 32 атомов углерода, предпочтительно от 25 до 32, более предпочтительно от 28 до 32 атомов углерода и имеющий одну, две или три двойные связи. Предпочтительно фрагмент Q имеет только одну двойную связь. Предпочтительно двойная связь расположена между атомами С 7 и C12 (от конца группы Q, связанного с карбонильной группой) и наиболее предпочтительно, между атомами С 7 и С 10 (например, между С 9 и С 10). Предпочтительно фрагмент Q представляет собой -C8H16-CH=CH-C18H37. Отдельное семейство образуют соединения, в которых Q представляет собой С 23-С 32 алкенил и Y представляет собой -СН=СН-, предпочтительно в транс-конфигурации. В другом варианте осуществления Q представляет собой -R1-O-R2. Атом кислорода может быть расположен в любом месте группы Q, но предпочтительно расположен между C1 и С 25 (считая от конца группы Q, связанного с карбонильной группой) (например, между C1 и С 2, или между C9 и С 10) и более предпочтительно между C18 и C25 (например, между С 19 и С 20). Например, Q может представлять собой-C19H38-O-C6H13. Отдельное семейство образуют соединения, в которых Q представляет собой -R1-O-R2, и У представляет собой -СН 2-СН 2-. В предпочтительном варианте осуществления Q представляет собой-R1-O-R2 и оба заместителя R1 и R2 являются алкильными группами. В еще одном варианте осуществления Q представляет собой -R1-O-R2 и как минимум один из заместителей R1 и R2 является алкенилом. Группа Q предпочтительно содержит от 1 до 3 двойных связей и более предпочтительно только одну двойную связь. Эта двойная связь и атом кислорода могут быть расположены в любом месте группы Q. Предпочтительно R1 представляет собой алкенил и более предпочтительно R1 представляет собой алкенил, и R2 представляет собой алкил. Если группа Q содержит одну двойную связь, она предпочтительно расположена между атомами С 7 и C12 (считая от конца группы Q, связанного с карбонильной группой),более предпочтительно между атомами С 7 и С 10 и еще более предпочтительно между атомами С 9 и С 10. Атом кислорода предпочтительно расположен между C1 и С 25 (считая от конца группы Q, связанного с карбонильной группой) (например, между C1 и С 2, или между C9 и Сю) и более предпочтительно между С 18 и С 25 (например, между C19 и С 20). Например, Q может представлять собой -C8H16-CH=CH-C9H18-OС 6 Н 13. Согласно другому предпочтительному варианту осуществленияY представляет собой -СН=СН- и Q представляет собой С 27-С 32 алкил, предпочтительно С 27 или С 28 алкил. Согласно другому предпочтительному варианту осуществленияY представляет собой -СН=СН- и Q представляет собой -R1-O-R2, где R1 и R2 соответствуют данным выше определениям. Предпочтительно Q содержит от 23 до 32 атомов углерода, более предпочтительно, от 25 до 32 атомов углерода и еще более предпочтительно от 28 до 32 атомов углерода. Например, Q может представлять собой -С 19 Н 38-O-С 6 Н 13. Предпочтительные соединения по настоящему изобретению включают, не ограничиваясь указанными: и их фармацевтически приемлемые соли и сложные эфиры. Другие предпочтительные соединения по настоящему изобретению включают, не ограничиваясь указанными, соединения, показанные ниже в табл. В, и их фармацевтически приемлемые соли и сложные эфиры. Отдельным соединением по настоящему изобретению является соединение, имеющее формулу или его фармацевтически приемлемые соли или сложные эфиры.- 10016042 Терапевтические применения Соединения по настоящему изобретению применимы для лечения любых заболеваний, при которых можно было бы получить благоприятный результат от селективного индуцирования иммунного ответаTh1-типа, особенно от иммунного ответа Th1-типа, связанного с увеличением секреции IL-12 и повышением активации антиген-презентирующих клеток (АРС), например дендритных клеток, которая происходит без значительного одновременного повышения секреции IL-4. Не ограничиваясь какой-либо конкретной теорией, авторы настоящего изобретения полагают, что соединения по настоящему изобретению опосредуют селективную индукцию иммунного ответа Th1 за счет связывания TCR клеток АРС, что активирует клетки NKT и приводит к секреции IFN- клеткамиNKT. Однако активированные таким образом клетки NKT не секретируют значительных количеств IL-4. Активированные клетки NKT далее связываются с антиген-презентирующими клетками (АРС), например дендритными клетками, через лиганд CD40, вызывающий улучшение местной секреции IL-12. Такое местное выделение IL-12 позволяет избежать негативных эффектов, связанных с токсичностью IL-12, и делает возможным генерацию очень эффективного и специфичного иммунного ответа Th1-типа, например против опухолей или патогенов. Авторы изобретения также полагают, что соединения по настоящему изобретению косвенно действуют на дендритные клетки и оказывают меньшее влияние на вторичную активацию клеток NK по сравнению с -GalCer или -C-GalCer (CRONY). Таким образом, неспецифическая активация клеток NK в значительной степени ограничена, что улучшает безопасность и переносимость соединений по настоящему изобретению по сравнению с соединениями известного уровня техники. Эти свойства делают соединения по настоящему изобретению особенно применимыми при лечении заболеваний, для контроля над которыми необходим иммунный ответ Th1-типа. В одном из вариантов осуществления соединения по настоящему изобретению применимы для лечения рака, например, в качестве противоопухолевых средств для ингибирования роста опухолей и для лечения пролиферативных клеточных расстройств. Соединения по настоящему изобретению могут применяться сами по себе или в комбинации с химиотерапией, радиотерапией или иммунотерапией. Более конкретно, соединения по настоящему изобретению применимы при лечении ряда раковых заболеваний, включая, но не ограничиваясь перечисленным, карциномы, например, мочевого пузыря,молочной железы, ободочной кишки, почек, печени, легких, включая мелкоклеточный рак легких, немелкоклеточный рак легких, пищевода, желчного пузыря, яичников, поджелудочной железы, яичек, желудка, почек, печени, шейки матки, щитовидной железы, простаты и кожи, включая карциному плоских клеток; гематопоэтические опухоли лимфоидной линии, включая лейкемию, острую лимфоцитарную лейкемию, острую лимфобластную лейкемию, В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, лимфому ворсинчатых клеток и лимфому Беркитта; гематопоэтические опухоли миелоидной линии, включая острые и хронические миелогенные лейкемии, миелодиспластический синдром и промиелоцитарную лейкемию; опухоли мезенхимального происхождения,включая фибросаркому и рабдомиосаркому; опухоли центральной или периферической нервной системы, включая астроцитому, нейробластому, глиому и шванномы; другие опухоли, включая меланому,семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоакантому, фолликулярный рак щитовидной железы и саркому Капоши. В предпочтительном варианте осуществления рак представляет собой солидную опухоль, например рак простаты или рак молочной железы. Клеточные пролиферативные расстройства, для лечения которых применимы соединения по настоящему изобретению, включают, не ограничиваясь перечисленными, доброкачественную гиперплазию простаты, семейный аденоматозный полипоз, нейрофиброматоз, псориаз, пролиферацию гладкомышечных клеток сосудов, связанную с атеросклерозом, легочный фиброз, артрит, гломерулонефрит и послеоперационные стенозы и рестенозы. В другом варианте осуществления соединения по настоящему изобретению применимы также для лечения инфекционных заболеваний, включая как вирусные, так и невирусные инфекции. Например, соединения по настоящему изобретению применимы для лечения вирусных инфекций,вызванных представителями семейства retroviridae (например, вирусами иммунодефицита человека, такими как HIV-1 (именуемым также HTLV-III, LAV или HTLV-III/LAV, или HIV-III; а также другими изолятами, например HIV-LP; Picornaviridae (например, полиовирусами, вирусом гепатита А, энтеровирусами, вирусами коксаки человека, риновирусами, эховирусами); Calciviridae (например, штаммами,которые вызывают гастроэнетрит); Togaviridae (например, вирусами энцефалита лошадей, вирусами краснухи); Flaviridae (например, вирусами лихорадки денге, вирусами энцефалита, вирусами желтой лихорадки); Coronaviridae (например, коронавирусами); Rhabdoviridae (например, вирусами везикулярного стоматита, вирусами бешенства); Filoviridae (например, вирусами лихорадки эбола); Paramyxoviridae (например, вирусами парагриппа, вирусом свинки, вирусом кори, респираторно-синцитиальным вирусом);Orthomyxoviridae (например, вирусами гриппа); Bungaviridae (например, вирусами Хантаан, вирусами бунья, флебовирусами и найровирусами); Arena viridae (вирусами геморрагической лихорадки); Reoviridae (например, реовирусами, орбивирусами и ротавирусами); Birnaviridae; Hepadnaviridae (вирусом гепа- 11016042 тита В); Parvoviridae (парвовирусами); Papovaviridae (вирусами папилломы, вирусами полиомы); Adenoviridae (большинством аденовирусов); Herpesviridae (вирусами простого герпеса (HSV) 1 и 2, вирусом ветряной оспы, цитомегаловирусом (CMV), вирусами герпеса); Poxviridae (вирусами оспы, вирусами вакцинии, вирусами чумы); и Iridoviridae (например, вирусом африканской лихорадки свиней); а также неклассифицированными вирусами (например, этиологическими агентами губчатых энцефалопатий,агентом дельта гепатита (который считается дефектным спутником вируса гепатита В), агентами гепатита, не являющегося гепатитом А и гепатитом В (класс 1=внутренне передаваемым; класс 2=парентерально передаваемым (т.е. гепатит С); Norwalk и родственными вирусами, а также астровирусами. Соединения по настоящему изобретению применимы также для лечения бактериальных инфекций,вызванных Helicobacter pylori, Borellia burgdorferi, Legionella pneumophilia, видами рода Mycobacteria(например, М.tuberculosis, M.avium, M.intracellulare, M.kansaii, M.gordonae), Staphylococcus aureus, Neisseria gonorrhoeae, Neisseria meningitidis, Listeria monocytogenes, Streptococcus pyogenes (стрептококк группы А), Streptococcus agalactiae (стрептококк группы В), Streptococcus (группы viridans), Streptococcus faecalis,Streptococcus bovis, Streptococcus (анаэробные виды), Streptococcus pneumoniae, патогенными видами рода Campylobacter, видами рода Enterococcus, видами рода Pseudomonas, видами рода Pneumococcus, видами рода Chlamidia, Haemophilus influenzae, Bacillus antracis, corynebacterium diphtheriae, видами родаcapsulatum, Coccidioides immitis, Blastomyces dermatitidis, Chlamydia trachomatis, Candidia albicans. Соединения по настоящему изобретению также применимы для лечения инфекций, вызванных другими инфекционными микроорганизмами, например протистами, включая виды рода Plasmodium, виды рода Leishmania, виды рода Schistosoma и виды рода Toxoplasma, а также дрожжами и другими грибками. В предпочтительном варианте осуществления соединения по настоящему изобретению применимы для лечения инфекций, вызванных вирусом иммунодефицита человека (HIV), вирусом гепатита С (HCV),вирусом гепатита В (HBV), вирусом герпеса, респираторно-синцитиальным вирусом (RSV) или малярии. В других вариантах осуществления соединения по настоящему изобретению применимы для лечения аутоиммунных заболеваний, контроль над которыми требует иммунного ответа Th1-типа, таких как астма или аллергия. Терапевтические и профилактические способы по настоящему изобретению могут применяться в сочетании с другими способами лечения. Например, лечение рака с применением соединений по настоящему изобретению может проводиться в комбинации с химиотерапией и/или радиотерапией. Лечение вирусных заболеваний с применением соединений по настоящему изобретению может проводиться в комбинации с введением IFN-. Помимо описанных выше способов, настоящее изобретение также относится к фармацевтическим и вакцинным композициям, включающим иммуногенно-эффективное количество соединения формулы (I) и, необязательно, дополнительное иммуностимулирующее средство, носитель или эксципиент (предпочтительно фармацевтически приемлемые). Способы введения Способы введения соединений и композиций по настоящему изобретению включают, не ограничиваясь перечисленным, пероральное, энтеральное, внутривенное, внутримышечное, внутриопухолевое,подкожное, трансдермальное, интраназальное, трансмукозальное (включая ректальное и буккальное) введение, а также ведение с помощью ингаляции. Предпочтительно применяются пероральный, трансдермальный, подкожный, ингаляционный или интраназальный способы введения (например, с применением твердых или жидких пероральных препаратов, кожных пластырей или назальных спреев, соответственно). В некоторых случаях действие соединений может быть усилено сингенными дендритными клетками с последующим внутривенным переносом в организм пациентов. Т.е. дендритные клетки могут быть инкубированы с соединениями по настоящему изобретению, чтобы дать возможность дендритным клеткам связать соединения с помощью их молекул CD1d. Эти дендритные клетки, связавшие соединения по настоящему изобретению, могут быть затем внутривенно перенесены в организм пациентов. Внутривенный перенос может быть либо местным, либо системным. Фармацевтические композиции Твердые дозированные формы для перорального введения соединений и композиций по настоящему изобретению включают капсулы, таблетки, пилюли, порошки, гранулы и суппозитории. В этих твердых дозированных формах действующее соединение по настоящему изобретению может быть смешано как минимум с одним обычным инертным эксципиентом (или носителем), таким как цитрат натрия или дикальцийфосфат; или с (а) наполнителями и средствами, увеличивающими объем, как, например, крахмалами, лактозой, сахарозой, глюкозой, маннитом и кремниевой кислотой; (b) связующими средствами,как, например, карбоксиметилцеллюлозой, альгинатами, желатином, поливинилпирролидоном, сахарозой и гуммиарабиком; (с) увлажняющими средствами, как, например, глицерином; (d) дезинтегрирую- 12016042 щими средствами, как, например, агар-агаром, карбонатом кальция, картофельным крахмалом или крахмалом маниоки, альгиновой кислотой, некоторыми комплексными силикатами, а также карбонатом натрия; (е) замедлителями растворения, например парафином; (f) ускорителями абсорбции, как, например,четвертичными аммониевыми соединениями; (g) смачивающими средствами, как, например, цетиловым спиртом и моностеаратом глицерина; (h) адсорбентами, как, например, каолином и бентонитом; и (i) лубрикантами, как, например, тальком, стеаратом кальция, стеаратом магния, твердыми полиэтиленгликолями, лаурилсульфатом натрия или их смесями. В случае капсул, таблеток и пилюль дозированные формы могут также включать буферные средства. Этими твердыми композициями, или твердыми композициями, подобными описанным выше, можно заполнять мягкие и твердые желатиновые капсулы с применением таких эксципиентов, как лактоза или молочный сахар, а также полиэтиленгликоли высокой молекулярной массы и т.п. Твердые дозированные формы, например таблетки, драже, капсулы, пилюли и гранулы, могут быть получены с покрытиями и оболочками, такими как кишечные покрытия или другие подходящие покрытия или оболочки. Несколько таких покрытий и/или оболочек хорошо известны в данной области, и они могут содержать средства, придающие непрозрачность, и могут также иметь такой состав, чтобы обеспечивать отсроченное высвобождение действующего соединения или соединений в определенных частях пищеварительного тракта. Примерами таких композиций, применимых по настоящему изобретению, в которых заключено действующее соединение, включают полимерные соединения и воски. Действующие соединения при необходимости могут также применяться в микроинкапсулированной форме, совместно с одним или несколькими из упомянутых выше наполнителей. Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Помимо действующих соединений эти жидкие дозированные формы могут содержать инертные разбавители, обычно применяемые в данной области, например воду или другие растворители, солюбилизирующие средства и эмульгаторы, как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла, в частности масло хлопчатника, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло, глицерин,тетрагидрофурфуриловый спирт, полиэтиленгликоли и эфиры жирных кислот и сорбитана, или смеси перечисленных соединений, и т.п. Если это желательно, композиция может также включать адъюванты,такие как смачивающие средства, эмульгирующие и суспендирующие средства, подсластители, вкусоароматические и/или ароматические средства. Композиция может включать носитель, определенный в настоящей заявке. Подходящие носители включают макромолекулы, которые растворимы в жидких средах организма и которые являются физиологически приемлемыми, согласно определению, данному в настоящей заявке. Носитель предпочтительно является относительно стабильным в кровеносной системе, имея приемлемое время полужизни в плазме для клиренса. Такие макромолекулы включают, не ограничиваясь перечисленным, соевый лецитин, олеиновую кислоту и триолеат сорбитана, причем триолеат сорбитана является предпочтительным. Суспензии, помимо действующих соединений, могут содержать суспендирующие средства, как, например, этоксилированные изостеариловые спирты, сложные эфиры сорбита, сорбитана и полиоксиэтилен сорбита, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар, трагакант и т.п. При желании можно применять смеси суспендирующих средств. Композиции для ректального введения предпочтительно являются суппозиториями, которые могут быть получены смешиванием соединений по настоящему изобретению с подходящими не раздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при обычной температуре, но становятся жидкими при температуре тела и, следовательно, плавятся в ректальной или вагинальной полости и высвобождают действующий компонент. Композиции, подходящие для парентеральных инъекций, могут включать физиологически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для восстановления в стерильные, пригодные для инъекций, растворы или дисперсии. Примеры подходящих водных и неводных носителей, разбавителей или сред включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и т.п.), их подходящие смеси, растительные масла (например, оливковое масло) и пригодные для инъекций органические сложные эфиры, например этилолеат. В случае дисперсий надлежащая текучесть может поддерживаться, например, путем применения покрытий, таких как лецитин,путем сохранения требуемого размера частиц, а также за счет применения ПАВ. Дозированные формы для местного введения соединений по настоящему изобретению включают мази, порошки, спреи и препараты для введения с помощью ингаляции. Действующий компонент может быть смешан в подходящих условиях (например, стерильных условиях) с физиологически приемлемым носителем и любыми консервантами, буферными компонентами, пропеллентами, которые могут оказаться необходимыми. Глазные составы, глазные мази, порошки и растворы также рассматриваются как составная часть настоящего изобретения. Соединения по настоящему изобретению могут быть преимущественно включены в композицию в- 13016042 форме, пригодной для введения с помощью ингаляции или интраназально, причем эти формы особенно применимы при лечении астмы или аллергических респираторных заболеваний. Выбор конкретных эксципиентов для таких препаратов зависит от желаемой дозированной формы, т.е. от того, предполагается ли вводить раствор в носовую полость в виде капель или спрея (аэрозоля) или суспензии, мази или геля. Для этой цели может применяться аэрозоль или препарат в упаковке под давлением. Такой аэрозольный препарат включает очень небольшие жидкие или твердые частицы, переносимые газообразным пропеллентом под давлением к участку терапевтического применения. Если в настоящем изобретении применяется фармацевтический аэрозоль, этот аэрозоль содержит терапевтически активное соединение,которое может быть растворено, суспендировано или эмульгировано в смеси жидкого носителя и пропеллента. Этот аэрозоль может иметь форму раствора, суспензии, эмульсии, порошка или полутвердого препарата. Применяемые по настоящему изобретению аэрозоли предназначены для введения в виде мелкодисперсных твердых частиц или капелек жидкости через дыхательные пути пациента. Могут применяться различные типы пропеллентов, известные специалисту в данной области техники. Примеры подходящих пропеллентов включают, не ограничиваясь перечисленным, углеводороды или другие подходящие газы. В случае аэрозоля в упаковке под давлением единица дозировки может быть определена путем задания величины для доставки отмеренного количества аэрозоля. Настоящее изобретение также может быть реализовано с применением распылителя, который представляет собой устройство, генерирующее очень мелкие жидкие частицы в основном однородного размера в потоке газа. Предпочтительно жидкость, содержащую соединения по настоящему изобретению, диспергируют в виде капелек. Эти маленькие капельки могут переноситься потоком газа через выходную трубку распылителя. Образующийся туман проникает в дыхательные пути пациента. В качестве альтернативы, млекопитающему при необходимости лечения можно вводить порошкообразную композицию, содержащую соединения по настоящему изобретению, а также содержащую или не содержащую лубрикант, носитель или пропеллент. Данный вариант осуществления изобретения может быть реализован с помощью обычного устройства для введения порошкообразных фармацевтических композиций с помощью ингаляции. Например, порошкообразная смесь соединения и подходящей порошковой основы, такой как лактоза или крахмал, может представлять собой единицу дозированной формы, например, в капсулах или картриджах, например, желатиновых, или в блистерных упаковках, из которых порошок можно вводить с помощью ингалятора. Для аэрозольного введения соединения по настоящему изобретению могут быть получены в форме тонкоизмельченных частиц размером от примерно 1 до примерно 20 мкм, предпочтительно от примерно 3 до примерно 10 мкм. В отдельном варианте осуществления соединения по настоящему изобретению доставляют с помощью липосом или мицеллярных частиц. При парентеральном введении внутривенным, подкожным или внутримышечным путем липосомы могут обеспечить регулируемое "депо" высвобождение инкапсулированного препарата на протяжении значительного периода времени и снизить побочные действия препарата за счет ограничения концентрации свободного лекарственного средства в кровотоке. Липосомы могут изменить картину распределения и поглощения препарата в тканях терапевтически предпочтительным путем и могут повысить удобство лечения, делая возможным менее частое введение лекарственных средств. При введении с помощью ингаляции липидная композиция липосом может быть подобрана таким образом, чтобы высвобождать находящееся внутри лекарственное средство с выбранной скоростью, которая может меняться, в терминах времени полужизни, от нескольких часов до нескольких дней. Кроме того, пока лекарственное средство заключено в липосомах, снижены побочные эффекты, связанные с быстрым всасыванием в дыхательные пути и кровоток. Дополнительное преимущество липосом для доставки лекарственных препаратов в ткани слизистых оболочек заключается в том, что поверхность липосом может быть модифицирована для увеличения адгезии к тканям, с целью улучшения времени пребывания липосом на целевом участке ткани. Известно несколько способов получения липосом с заключенными в них лекарственными средствами. В одном из способов липиды, образующие везикулы липосом, осаждают в виде тонкой пленки на боковых стенках колбы и медленно регидратируют добавлением водного буфера. Лекарственное средство, которое предполагается включить в липосомы, может быть введено либо в липидную пленку (в случае липофильного препарата) или в водную гидратирующую среду (в случае гидрофильного препарата). Липосомы, которые образуют многослойные везикулы (MLV), имеют гетерогенные размеры от примерно 0,05 до 10 микрон. MLV могут быть впоследствии обработаны, как правило, с помощью гомогенизации, ультразвука или экструзии через мембрану, для получения суспензии с частицами меньшего и более однородного размера. В основном предпочтительны липосомы, имеющие размер менее примерно 0,2-0,4 микрон. Липосомы этого размерного диапазона могут быть стерилизованы путем пропускания через фильтр с размером пор 0,45 мкм, имеют менее выраженную тенденцию к агрегации и могут также демонстрировать более предпочтительное распределение по органам в случае внутривенного введения (Gabizon). Как только липосомальный препарат получен, он может быть подвергнут лиофилизации, предпочтительно с применением криопротекторов, присутствующих как во внутренней, так и во внешней- 14016042 среде липосом. Эти криопротекторы могут быть выбраны из числа сахаров, таких как сахароза, трегалоза, лактоза,мальтоза, маннит, циклодекстрин и его производные. Упомянутые криопротекторы могут также представлять собой полимеры, такие как полиэтиленгликоль, декстран, поливинилпирролидон или гидроксиэтилкрахмал. Указанные криопротекторы могут применяться по отдельности или в комбинации. Кроме того, при лиофильной сушке липосомальных препаратов могут применяться аминокислоты. Криопротекторы вводят в водный слой, находящийся внутри липосом, во время получения незаполненных липосом, применяя эти криопротекторы, растворенные в гидратирующей среде. Введение криопротекторов во внешнюю среду производят во время диафильтрации, проводимой после завершения процесса введения препарата в липосомы. Желаемый криопротектор можно также вводить путем обмена внешнего буфера любой липосомальной суспензии с помощью диафильтрации. Полученной липосомальной суспензией наполняют пузырьки и лиофилизуют. Другие, нелипосомальные препараты, включающие соединения по настоящему изобретению, также могут быть подвергнуты лиофильной сушке. Применение в качестве адъювантов Помимо перечисленного выше, настоящее изобретение относится к способу повышения иммуногенности антигенов у млекопитающих, причем этот способ включает иммунизацию млекопитающих при совместном действии антигена и адъюванта, содержащего соединение формулы I. Согласно настоящему изобретению применение соединений формулы I в качестве адъювантов приводит к улучшению и/или увеличению продолжительности действия защитного иммунитета, вызванного антигеном. Например, как показано в настоящей заявке, совместное введение соединений формулы I и пептидов, соответствующих эпитопам Т-клеток или В-клеток, распознающим опухолевые или вирусные антигены, или конструктов ДНК, экспрессирующих эти антигены, улучшает антиген-специфичные иммунные ответы. Адъюванты формулы I можно вводить совместно с любыми антигенами, в частности с антигенами,полученными из инфекционных агентов или опухолей. Как указано выше, активность соединений по настоящему изобретению в качестве адъювантов относится хотя бы частично к их способности усиливать и/или продлевать опосредованный NKT и дендритными клетками антиген-специфичный Т-клеточный иммунный ответ Th1-типа и иммунные ответы Тклеток CD8+. По сравнению с соединениями известного уровня техники (включая KRN7000 и CRONY101), способность соединений по настоящему изобретению вызывать селективную индукцию иммунного ответаTh1-типа (т.е. вызывать эффективную, специфичную и локализованную активацию системы клеток NKT и, в частности, повышенную секрецию IL-12, а также эффективную и локализованную вторичную активацию дендритных клеток в отсутствие повышенной секреции IL-4) приводит к повышению локализованной цитотоксичности активированных клеток NKT в отсутствие неспецифичной цитотоксичности,что делает соединения по настоящему изобретению очень эффективными в увеличении иммуногенности различных опухолевых и инфекционных антигенов, характеризуемых низкой иммуногенностью. Кроме того, активация клеток NKT соединениями по настоящему изобретению зависит от молекулы CD1d, которая мономорфна у отдельных индивидуумов (Porcelli, Adv. Immunol., 59:1-98, 1995), поэтому адъюванты по настоящему изобретению могут применяться всеми пациентами, независимо от гаплотипа МНС. Согласно настоящему изобретению антиген и адъювант, содержащий соединение формулы (I), вводят совместно. Способы введения антигена и адъюванта включают, не ограничиваясь перечисленными,пероральное, энтеральное, внутривенное, внутримышечное, внутриопухолевое, подкожное, трансдермальное, интраназальное, трансмукозальное (включая ректальное и буккальное) и ингаляционное введение. Предпочтительно применяются пероральный, трансдермальный, подкожный, ингаляционный или интраназальный пути введения (например, с помощью твердых или жидких пероральных препаратов,кожных пластырей или назальных спреев, соответственно). Внутривенное вливание может быть местным или системным. Одновременное введение адъюванта, содержащего соединение по настоящему изобретению, и антигена является предпочтительным и, как правило, оно обеспечивает наиболее эффективную иммуностимуляцию. Если антиген и адъювант по настоящему изобретению входят в две различные композиции, их предпочтительно вводят в один и тот же участок организма и предпочтительно не далее чем в 1 см друг от друга. Поскольку адъювант по настоящему изобретению проявляет иммуностимулирующую активность в комбинации с большим числом различных антигенов, он применим для профилактических и терапевтических видов лечения. Соответственно, в еще одном аспекте настоящее изобретение относится к профилактическому и/или терапевтическому способу лечения заболеваний у млекопитающих, для контроля над которыми требуется иммунный ответ Th1-типа, включающему совместное введение указанному млекопитающему антигена и адъюванта, содержащего соединение формулы (I). Этот способ может быть применим, например, для защиты от и/или для лечения различных инфекций, а также для лечения различных неопластических заболеваний.- 15016042 Одним из вариантов осуществления является способ усиления иммунного ответа на вирусную инфекцию иммунодефицита человека (HIV), вирусную инфекцию гепатита С (HCV), вирусную инфекцию гепатита В (HBV), вирусную инфекцию герпеса или инфекцию респираторно-синцитиального вируса(RSV) у млекопитающих за счет совместного введения млекопитающему вирус-специфичного антигена и адъюванта, содержащего соединение формулы (I). Другим вариантом осуществления является способ усиления иммунного ответа на рак простаты или молочной железы у млекопитающих путем совместного введения млекопитающему антигена, специфичного для рака, и адъюванта, содержащего соединение формулы (I). Терапевтические и профилактические способы по настоящему изобретению могут применяться в сочетании с другими способами лечения. Например, лечение рака с применением опухоль-специфичного антигена и адъюванта по настоящему изобретению может применяться в комбинации с химиотерапией и/или радиационной терапией. Противовирусные вакцины, включающие адъюванты по настоящему изобретению, могут применяться в комбинации с лечением IFN-. Наряду со способами по настоящему изобретению в изобретении разработаны фармацевтические и вакцинальные композиции, включающие иммуногенно-эффективное количество антигена, а также иммуногенно-эффективное количество адъюванта, содержащего соединение формулы (I) и, необязательно,дополнительное иммуностимулирующее средство, носитель или эксципиент (предпочтительно все средства должны быть фармацевтически приемлемыми). Указанные антиген и адъювант могут либо входить в состав одной композиции, либо входить в состав двух отдельных композиций. Антигены, применяемые в иммуногенных (например, вакцинальных) композициях по настоящему изобретению, могут быть получены из эукариотических клеток (например, опухолей или паразитов, а также дрожжей или других грибков), бактериальных клеток, вирусных частиц или любых их частей. В случае если материал, на который должен быть направлен иммунный ответ, является слабым антигеном(например, синтетический антиген или фрагмент антигена), он может быть дополнительно конъюгирован с несущей молекулой, такой как альбумин или гаптен, с применением стандартной методики ковалентного связывания, например, с помощью одного из нескольких имеющихся в продаже наборов реагентов. Дополнительно он может включать определенные молекулы для индуцирования специфичного нацеливания на АРС, как, например, бета-субъединицу шига-токсина, пептиды со специфичной аффинностью к дендритным клеткам (Haicheur N, Bismuth E, Bosset S, Adotevi O, Warmer G, Lacabanne V, Regnault A,Desaymard C, Amigorena S, Ricciardi-Castagnoli P, Goud B, Fridman WH, Johannes L, Tartour E. The В subunit of Shiga toxin fused to a tumor antigen elicits CTL and targets dendritic cells to allow MHC class Irestricted presentation of peptides derived from exogenous antigens. J Immunol. 2000 Sep 15; 165 (6):3301-8),пептиды с аффинностью к рецептору Dec-205 (Sevilla et al., J.Exp.Med., 2000, 192:1249-1260), антитела против DC-SIGN (Tacken PJ, de Vries IJ, Gijzen K, Joosten B, Wu D, Rother RP, Faas SJ, Punt CJ, Torensmadendritic cells via a humanized anti-DC-SIGN antibody. Blood. 2005 Aug 15; 106(4):1278-85). Примеры предпочтительных антигенов по настоящему изобретению включают (i) малярияспецифичные антигены, как, например, подвергнутые облучению плазмодиальные спорозоиты или синтетические пептидные антигены, содержащие как минимум один Т-клеточный или В-клеточный эпитоп белка, окружающего малярийный спорозоит (CS) (см. ниже); (ii) вирусные белки или пептидные антигены, как, например, полученные из вируса гриппа (например, поверхностные гликопротеины гемагглютинин (НА) и нейраминидаза (NA) [такие как НА вируса гриппа индеек или НА вируса птичьего гриппа]); вируса иммунодефицита (например, антигена вируса иммунодефицита кошек (FIV), антигена вируса иммунодефицита обезьян (SIV) или антигена вируса иммунодефицита человека (HIV), таких как gp120,gp160, антигена р 18, Gag р 17/р 24, Tat, Pol, Nef и Env; вируса герпеса (например, гликопротеин, например, из вируса герпеса кошек, вируса герпеса лошадей, вируса герпеса коров, вируса псевдобешенства,вируса герпеса собак, вируса простого герпеса (HSV, например, HSV tk, gB, gD), вируса болезни Марека,вируса герпеса индеек (HVT), цитомегаловируса (CMV) или вируса Эпштейна-Барра); вируса гепатита(например, поверхностный антиген вируса гепатита В (HBsAg; (iii) бактериальные антигены, такие как липополисахариды, выделенные из стенок грамотрицательных бактериальных клеток и специфичные для стафилококков, стрептококков, пневмококков (например, PspA [см. публикацию РСТWO 92/14488]),Neisseria gonorrhea, Borrelia (например, антигены OspA, OspB, OspC видов Borrelia, связанных с болезнью Лайма, таких как Borrelia burgdorferi, Borrelia afzelli и Borrelia garinii [см., например, патент США 5523089; публикации РСТWO 90/04411, WO 91/09870, WO 93/04175, WO 96/06165, WO 93/08306;Borreliosis: Progress on the Development of Lyme Disease Vaccine, Vaccine 13: 133-135, 1995]); (iv) и опухоль-специфичные белки, такие как рецепторы ErbB, Melan A [MARTI], gp100, тирозиназа, TRP-1/gp 75 и TRP-2 (в меланоме); MAGE-1 и MAGE-3 (в раковых опухолях мочевого пузыря, головы и шеи и немелкоклеточной карциноме); белки HPV EG и Е 7 (в раковых опухолях шейки матки); Mucin [MUC-1] (в раковых опухолях молочной железы, поджелудочной железы, ободочной кишки и простаты); простатаспецифичный антиген [PSA] (в раковых опухолях простаты); карциноэмбриональный антиген [СЕА] (в- 16016042 раковых опухолях ободочной кишки, молочной железы и желудочно-кишечного тракта) и такие распространенные опухоль-специфичные антигены, как MAGE-2, MAGE-4, MAGE-6, MAGE-10, MAGE-12,BAGE-1, CAGE-1,2,8, CAGE-3-7, LAGE-1, NY-Eso-1/LAGE-2, NA-88, GnTV, TRP2-INT2 и WY-1 (ген опухоли Вильмса). Имеется в виду, что приведенный выше перечень антигенов является примерным, поскольку интересующий антиген может быть получен из любого животного или человеческого патогена или опухоли. Что касается ДНК, кодирующей антигены, относящиеся к интересующим патогенам, следует обратить внимание на, например, патенты США 4722848; 5174993; 5338683; 5494807; 5503834; 5505941; 5514375; 5529780; патент GB2269820 В; и публикации РСТWO 92/22641; WO 93/03145; WO 94/16716; WO 96/3941; PCT/US94/06652. Что касается антигенов, характерных для опухолевых вирусов, можно сослаться на Molecular Biology of Tumor Viruses, RNA Tumor Viruses, Second Edition, Edited by Weiss et al., Cold SpringHarbor Laboratory Press, 1982. Перечень дополнительных антигенов, применимых в композициях по настоящему изобретению, можно посмотреть также в Stedman's Medical Dictionary (24th edition 1982). В одном из вариантов осуществления композиции по настоящему изобретению обеспечивают защитный иммунитет против малярии, в частности против P.yoelii, и основных видов плазмодий человекаP.falciparum и P.vivax. Эти композиции включают один или несколько из следующих компонентов: (i) как минимум один из малярия-специфичных пептидов, включающих Т-клеточный эпитоп, способный вызывать антималярийный Т-клеточный ответ, предпочтительно у млекопитающих различного генетического происхождения (например, YNRNIVNRLLGDALNGKPEEK [SEQ ID NO:1] или SYVPSAEQIet al., Int. Immunol., 3:997, 1991; Moreno et al., J .Immunol., 151:489, 1993]); и/или (ii) как минимум один малярия-специфичный пептид, включающий В-клеточный эпитоп (например, (NANP)3 [SEQ ID No:5]. Вклеточный эпитоп, расположенный в повторяющейся области белка CS P.falciparum [Nardin et al., J. Exp.Med., 156:20, 1982; Nardin et al., Ann. Rev. Immunol., 11:687, 1993]), способный стимулировать выработку антималярийных (т.е. нейтрализующих) антител (например, направленных против стадии спорозоита малярийного микроорганизма). Предпочтительно иммуногенные композиции по настоящему изобретению включают как минимум один В-клеточный эпитоп и как минимум один Т-клеточный эпитоп. Вклеточные эпитопы предпочтительно вызывают выработку антител, которые специфично распознают и связываются с белком, окружающим малярийный спорозоит (CS). В качестве альтернативы или дополнения композиции по настоящему изобретению могут включать В-клеточные и/или Т-клеточные эпитопы, полученные из других малярийных компонентов и способные реагировать с ними, как, например,P.vivax, секретируемый эритроцитами белок-1 или -2 (PvESP-1 или PvESP-2) (см., например, патент США 5874527), поверхностный белок спорозоита P.falciparum, именуемый белком (анонимным), связанным с адгезией тромбоспондина (TRAP), a также именуемый поверхностным белком спорозоита 2(SSP2), Lsa-1, hsp70, SALSA, STARP, Help7, MSA, RAP-1 и RAP-2. В одном из вариантов осуществления компоненты, относящиеся к В-клеточным эпитопам и Т-клеточным эпитопам, включены в мультиантигенные пептиды (MAP), с образованием синтетических макромолекулярных полипептидов с высокой плотностью эпитопов. Способы синтеза MAP хорошо известны в данной области (см., например, Tam,Proc. Natl. Acad. Sci. USA, 85:5409, 1988; Tam, Meth. Enzymol., 168:7, 1989). Настоящее изобретение охватывает также В-клеточные и Т-клеточные эпитопы, полученные из других видов плазмодий, включая, но не ограничиваясь перечисленными, P.malariae, P.ovale,P.reichenowi, P.knowlesi, P.cynomolgi, P.brasilianum, P.berghei и P.chabaudi. Эти эпитопы, как правило,включают от 8 до 18 аминокислотных остатков, полученных из белков плазмодий. В другом отдельном варианте осуществления предпочтительный антиген по настоящему изобретению является HIV-специфичным (как, например, Т-клеточный эпитоп RGPGRAFVTI [SEQ ID NO:6]) белка р 18). В отдельном варианте осуществления антиген по настоящему изобретению может быть представлен рекомбинантным вирусом, экспрессирующим этот антиген. Предпочтительно указанный вирус выбран из группы, состоящей из рекомбинантного аденовируса, рекомбинантного вируса оспы и рекомбинантного вируса Sindbis. При применении в качестве адъювантов соединения по настоящему изобретению могут вводиться в виде составной части фармацевтической или вакцинальной композиции, включающей антиген, или в виде отдельного препарата, который вводят совместно со второй композицией, содержащей антиген. В любой из этих композиций соединения по настоящему изобретению могут быть объединены с другими адъювантами и/или эксципиентами/носителями. Упомянутые другие адъюванты включают, не ограничиваясь перечисленным, адъюванты на основе масляных эмульсий и адъюванты на основе эмульгаторов,такие как полный адъювант Фрейнда, неполный адъювант Фрейнда, MF59 или SAF; минеральные гели,такие как гидроксид алюминия (квасцы), фосфат алюминия или фосфат кальция; адъюванты, полученные из микробов, такие как токсин холеры (СТ), токсин коклюша, термолабильный токсин Escherichiabacterium parvum, фрагменты ДНК CpG, мурамил дипептид или монофосфорил липид А; адъюванты в форме микрочастиц, такие как иммуностимулирующие комплексы (ISCOM), липосомы, биоразрушаемые микросферы или сапонины (например, QS-21); цитокины, такие как IFN-, IL-2, IL-12 или GM-CSF; синтетические адъюванты, такие как неионные блок-сополимеры, аналоги мурамил пептида (например, Nацетилмурамил-L-треонил-D-изоглутамин [thr-MDP], N-ацетил-нор-мурамил-L-аланил-D-изоглутамин,N-ацетилмурамил-L-аланил-D-изоглутаминил-L-аланин-2-[1'-2'-дипальмитоил-sn-глицеро-3-гидроксифосфорилокси]этиламин), полифосфазены или синтетические полинуклеотиды, а также поверхностноактивные вещества, такие как лизолецитин, плуроновые полиолы, полианионы, пептиды, углеводородные эмульсии или гемоцианины лимфы улитки (KLH). Предпочтительно эти дополнительные адъюванты также фармацевтически приемлемы для применения у людей. Чистота композиций Соединения по настоящему изобретению предпочтительно очищают до степени чистоты по меньшей мере 75%, более предпочтительно по меньшей мере 85%, еще более предпочтительно по меньшей мере 90% и наиболее предпочтительно 90, 91, 92, 93, 94, 95, 96, 97, 98 или 99%. Эффективные дозировки Эффективные количества для лечения заболеваний могут быть легко определены с помощью эмпирических методик, известных специалистам в данной области техники, как, например, с помощью создания матрицы дозировок и частот введения и сравнения группы экспериментальных единиц или субъектов для каждого элемента матрицы. Точное количество препарата, которое следует вводить пациенту,должно меняться в зависимости конкретного заболевания, состояния и тяжести заболевания, а также физического состояния пациента. Измеримое облегчение любого симптома или параметра может быть определено врачом, являющимся специалистом в данной области, или на основании сообщений пациента врачу. Клинически значимое ослабление или улучшение означает ощутимое для пациента и/или для врача. Кроме того, следует понимать, что конкретная дозированная форма и уровень дозировки для любого конкретного пациента будет зависеть от ряда факторов, включающих активность конкретного применяемого соединения; возраст, массу тела, общее состояние здоровья и пол индивидуума, подвергаемого лечению; время и путь введения; скорость выведения; другие лекарственные препараты, которые водились ранее; а также тяжесть конкретного заболевания, подвергаемого лечению. Количество лекарственного средства, которое следует вводить пациенту, может находиться в пределах от примерно 0,1 до примерно 500 мкг/кг/за одно введение, предпочтительно от примерно 0,5 до примерно 100 мкг/кг/за одно введение и наиболее предпочтительно от примерно 1 до примерно 50 мкг/кг/день. Следует понимать, что фармацевтические композиции по настоящему изобретению не должны содержать полное количество агента, которое эффективно при лечении расстройства, поскольку введение этого эффективного количества может быть достигнуто введением нескольких доз такой фармацевтической композиции. Например, соединения по настоящему изобретению могут быть включены в состав капсул или таблеток,причем любая из этих форм предпочтительно содержит 0,06-3,00 мг соединений по настоящему изобретению. Токсичность и терапевтическая эффективность композиций, содержащих соединения по настоящему изобретению, может быть определена с помощью стандартных фармацевтических методик на экспериментальных животных, например, путем определения LD50 (доза, летальная для 50% экспериментальной группы) и ED50 (доза, терапевтически эффективная для 50% экспериментальной группы). Соотношение доз, проявляющих токсический и терапевтический эффект, представляет собой терапевтический индекс и его можно выразить соотношением LD50/ED50. Предпочтительны композиции, которые показывают более высокие терапевтические индексы. Если могут применяться терапевтические средства,которые демонстрируют побочные токсические действия (например, при лечении тяжелых форм рака или опасных для жизни инфекций), необходимо позаботиться о разработке системы доставки, которая направит такие иммуногенные композиции в конкретные участки организма (например, лимфоидные ткани, опосредующие иммунные ответ, опухоль или орган, поддерживающий репликацию агента инфекции) для минимизации потенциального вреда для других тканей и органов и, следовательно, уменьшения побочных эффектов. Как указано выше, данные, полученные в исследовании на животных, могут применяться при выборе диапазона доз для применения у людей. Терапевтически эффективная доза соединений по настоящему изобретению у людей предпочтительно находится в диапазоне концентраций в кровотоке, который включает ED50 при незначительной или отсутствующей токсичности. Доза может меняться в пределах этого диапазона, зависящего от примененной дозированной формы и выбранного пути введения. В идеальном случае должна применяться одна доза. Примеры Приведенные ниже примеры иллюстрируют настоящее изобретение, не ограничивая его объем. 1. Синтетические примеры. А. Получение промежуточных соединений. Промежуточные соединения для образования липидной цепи -C(O)-Q могут быть получены, как показано на схемах синтеза 1 А и 1 В ниже. На схемах 1 А и 1 В в каждом случае Alk независимо представляет собой алкильную цепь, такую,что общее количество атомов углерода в цепи жирной кислоты составляет от 23 до 32 атомов. На схеме 1 А карбоновую кислоту (А) превращают в сложный эфир паранитрофенола (В), используя паранитрофенол в присутствии DIC. Аналогично, алкенол (Е) превращают в алкен-эфир (F) обработкой гидридом натрия и подходящим галогенированным алканом. Наконец, сложный эфир паранитрофенола (В) превращают в конечную жирную кислоту (D) или (G) взаимодействием (В) с подходящим алкеном в присутствии катализатора Граббса второго поколения. Промежуточное соединение (D) или (G) может быть очищено известными в данной области способами, как, например, колоночной хроматографией на силикагеле. Синтез конкретных промежуточных соединений показан ниже на схемах 2 А-2 Е. Схема 2 А Соединение 2: к раствору исходной ундециленовой кислоты (4,1 г, 22,2 ммоль, 1,1 экв.) в безводномDCM (120 мл) добавляли паранитрофенол (2,9 г, 20,9 ммоль). В присутствии DIC (3,6 мл, 1,2 экв.) и 4DMAP (60 мг) реакционную смесь перемешивали при комнатной температуре в течение ночи, до тех пор,пока тонкослойная хроматография (РЕ-ЕА 6:1, Rf:0,52) не указала на завершение реакции. Суспензию разбавляли DCM и фильтровали через целит. Фильтрат концентрировали, получая остаток. Этот остаток очищали колоночной флэш-хроматографией (петролейный эфир CHCl3 2:1), получая сложный эфир 2(ушир. с, 1 Н), 3,87 (дд, J=9,8, 2,4 Гц, 1 Н), 3,51 (м, 2 Н), 2,50 (д, J=2,1 Гц, 1 Н). 13 С ЯМР (75 МГц, CDCl3): 171,1, 155,5, 145,2, 139,0, 125,1, 122,3, 114,2, 34,4, 33,8, 29,3, 29,2, 29,1,29,0, 24,9. Соединение 2 а. К раствору исходной ундециленовой кислоты в метаноле добавляли DCC и 4DMAP. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Соединение 4. К перемешиваемому раствору соединения 2 (197 мг, 0,646 ммоль) и 1-эйкозена (850 мг, 4,2 экв.) в сухом CH2Cl2 (15 мл) добавляли катализатор Граббса 2-ого поколения (65 мг, 12 мольн.%) и смесь кипятили с обратным холодильником в течение 1 дня, до тех пор, пока ТСХ (PE-DCM 1:1,Rf:0,40) не указала на исчезновение сложного эфира 2. Реакционную смесь концентрировали в вакууме и затем проводили колоночную хроматографию остатка с применением силикагеля (PE-DCM 2:1), получая указанное в заголовке соединение 4 (190 мг, 53%, транс/цис: 4:1). 1(т, J=7,5 Гц, 2 Н, Н-2), 2,02 (м, 4 Н, Н-9, Н-12), 1,76 (пент., J=7,5 Гц, 2 Н, Н-3), 1,42-1,25 (м, 42 Н, Н-4-8, Н 13-Н-28), 0,88 (т, J=6,7 Гц, 3 Н, Н-29). 13 С ЯМР (125 МГц, CDCl3): 171,3, 155,6, 145,3, 130,5, 130,2, 125,2, 122,4, 34,3, 32,6, 32,6, 31,9, 29,7,29,7, 29,6, 29,5, 29,4, 29,3, 29,2, 29,1, 29,0, 24,7, 22,7, 14,1. Соединение 4 а. К перемешиваемому раствору соединения 2 а и 1-эйкозена в сухом CH2Cl2 добавляли катализатор Граббса 2-ого поколения и смесь кипятили с обратным холодильником в течение 1 дня. Затем реакционную смесь концентрировали в вакууме и после этого очищали колоночной хроматографией с использованием силикагеля, получая указанное в заголовке соединение 4 а. Соединение 6. К раствору -ундециленилового спирта (2 мл, 98%, 9,74 ммоль) в безводном ТГФ(15 мл) и ДМФА (5 мл) добавляли гидрид натрия (60% в минеральном масле, 584 мг, 1,5 экв. ) при 0 С и затем через 10 минут добавляли TBAI (80 мг) и 1-бромпентан (1,82 мл, 1,5 экв.). После перемешивания в течение ночи при кипячении с обратным холодильником ТСХ (РЕ-ЕА 6:1) показала исчезновение спирта. Смесь разбавляли DCM и гасили водой. Водную фазу экстрагировали DCM (3), промывали насыщенным раствором бикарбоната натрия и затем насыщенным раствором соли. Органическую фазу высушивали (сульфатом натрия), концентрировали и остаток очищали колоночной флэш-хроматографией(петролейный эфир-DCM, 3:1), получая простой эфир 6 (2,21 г, 99%, Rf:0,12 с PE-DCM 3:1). Соединение 7 получали тем же способом, что и соединение 6 с выходом 94%. 6: 1H ЯМР (500 МГц, CDCl3) 5,81 (м, 1H, Н-10), 4,95 (м, 2H, Н-11), 3,38 (т, J=7,0 Гц, 4 Н, Н-1, Н-1'),2,03 (м, 2 Н, Н-9), 1,56 (м, 4 Н, Н-2, Н-2'), 1,37-1,27 (м, 16 Н, Н-3-8; Н-3'-4'), 0,88 (т, J=6,9 Гц, 3 Н, Н-5'). 13 С ЯМР (125 МГц, CDCl3): 138,9, 114,1, 71,0, 70,6, 33,8, 31,9, 29,8, 29,5, 29,5, 29,4, 29,1, 28,9, 26,2,- 20016042 19,4, 14,1. Соединение 8. К перемешиваемому раствору сложного эфира 2 (190 мг, 0,623 ммоль) и простого эфира 6 (200 мг, 0,833 ммоль, 1,3 экв.) в сухом хлороформе (20 мл) добавляли катализатор Граббса 2-го поколения (36 мг, 7 мольн.%) и кипятили смесь с обратным холодильником в течение 1 дня, после чего реакционную смесь концентрировали в вакууме и затем очищали колоночной хроматографией на силикагеле (PE-DCM 3:2), получая указанное в заголовке соединение 8 (174 мг, 84%, транс/цис: 4:1, Rf:0,16 при PE-DCM 1:1). Соединение 9 получали тем же способом, что и соединение 8 с выходом 81%. 8: 1H ЯМР (500 МГц, CDCl3) 8,26 (д, J=9,0 Гц, 2 Н), 7,27 (д, J=9,l Гц, 2 Н), 5,37 (м, 2 Н, Н-10, Н-11),3,38 (т, J=6,7 Гц, 4 Н, Н-20, Н-22), 2,58 (т, J=7,5 Гц, 2 Н, Н-2), 1,96 (м, 4 Н, Н-9, Н-12), 1,75 (пент., J=7,4 Гц,2 Н, Н-3), 1,56 (м, 4 Н, Н-19, Н-23), 1,39 (м, 2 Н, Н-4), 1,32-1,25 (м, 24 Н, Н-5-8, Н-13-18, Н-24-25), 0,88 (т,J=6,7 Гц, 3 Н, Н-26). 13 С ЯМР (125 МГц, CDCl3): 171,1, 155,6, 145,3, 130,5, 130,2, 125,2, 122,4, 71,0, 34,3, 32,6, 32,7, 29,8,29,7, 29,6, 29,5, 29,3, 29,2, 29,0, 26,2, 25,8, 24,7, 22,6, 14,1. Соединение 13. Соединение 4 а растворяли в DCM/MeOH (1:1) и к раствору добавляли 5% Pd на угле (30 мольн.%). Затем реакционную смесь перемешивали в атмосфере водорода до завершения реакции по данным ТСХ-анализа. Затем реакционную смесь концентрировали в вакууме и после этого очищали колоночной хроматографией на силикагеле, получая указанное в заголовке соединение 13. Соединение 14. Метиловый эфир 13 (147 мг, 0,32 ммоль) растворяли в 10 мл i-PrOH/ТГФ (1:1) и к полученному раствору добавляли 100 мг раствора NaOH в воде (2 мл). Смесь перемешивали при 60 С в течение 5 ч, после чего ТСХ (РЕ/ЕА 12:1) указала на завершение гидролиза. После нейтрализации 2 н.HCl, раствор непосредственно концентрировали в вакууме. К раствору полученного выше остатка жирной кислоты в DCM (10 мл) добавляли паранитрофенол (160 мг, 0,5 ммоль). В присутствии DCC (160 мг) и 4-DMAP (35 мг) реакционную смесь перемешивали в течение ночи при комнатной температуре, после чего ТСХ (РЕ/ЕА 10:1) показала завершение реакции. Полученную суспензию разбавляли DCM и добавляли к ней небольшое количество силикагеля. После упаривания остаток очищали колоночной флэшхроматографией (РЕ/ЕА 20:1), получая эфир 14 (154 мг, 86%). 1H ЯМР (500 МГц, CDCl3) 8,27 (д, J=9,1 Гц, 2H), 7,28 (д, J=9,1 Гц, 2H), 2,59 (т, J=7,5 Гц, 2H, Н-2),1,76 (пент., J=7,5 Гц, 2H, Н-3), 1,41 (пент., J=7,0 Гц, 2H, Н-4), 1,37-1,22 (м, 48 Н, H-5-H-28), 0,88 (т, J=7,0 Гц, 3 Н, Н-29). 13 С ЯМР (125 МГц, CDCl3): 171,3, 155,6, 145,3, 125,2, 122,4, 34,4, 33,7, 31,9, 30,2, 29,7, 29,7, 29,6,29,6, 29,5, 29,4, 29,4, 29,2, 29,2, 29,1, 26,7, 24,8, 22,7, 14,1. Альтернативный подход к синтезу отдельных промежуточных соединений с применением реакции сочетания по Юлия-Литгой-Кочиенски показан на схеме 2 Е. При использовании этого подхода коммерчески доступный алкилбромид 18 вводят во взаимодействие с коммерчески доступным бензотиазолтиолом 19 с получением карбоновой кислоты 20 после замещения атома брома. Затем тиоэфир карбоновой кислоты 20 окисляют действием mCPBA с получением сульфона 21. После этого сульфон 21 вводят во взаимодействие с альдегидом 23 (который получают из имеющегося в продаже спирта 22, используя,например, условия реакции Сверна) в присутствии гексаметилдисилазида лития в условиях реакции Юлия-Литгой-Кочиенски. Эта реакция приводит к образованию алкенкарбоновой кислоты 24. Карбоновую кислоту 24 затем превращают либо непосредственно в ненасыщенный п-нитрофениловый эфир 4 при обработке п-нитрофенолом в присутствии DCC и DMAP, или в насыщенный п-нитрофениловый эфир 15 через стадии (1) гидрирования с применением 5% палладия на угле и водорода и (2) обработки полученной насыщенной кислоты 25 п-нитрофенолом в присутствии DCC и DMAP. Схема 2E Альтернативный синтез соединений 4 и 14 с использованием реакции Юлия-Литгой-Кочиенски в качестве стадии образования связи С-СB. Получение гликолипидов/ Гликолипиды по настоящему изобретению могут быть получены способом, показанным ниже на схеме 3. Схема 3 На схеме 3 Z представляет собой -СН=СН-, О, -О-Alk-СН=СН- или -СН=СН-О-; и Alk в каждом случае независимо представляет собой такую алкильную цепь, что общее количество атомов углерода в цепи жирной кислоты составляет от 23 до 32 штук. Удаляют защиту соединения (Н) (т.е. удаляют защитные группы Boc и изопропилиден) предпочтительно обработкой трифторуксусной кислотой и триэтилсиланом в дихлорметане, и вводят в реакцию с эфиром жирной кислоты (D) или (G), получая соединение(I). Затем снимают защиту соединения (I), например, действием натрия и аммиака, получая соединение(J) с алкеновым линкером, или восстанавливают, например, газообразным водородом над палладием на угле, получая соединение (K) с алкановым линкером. Эти соединения могут быть очищены способами,известными в данной области, например колоночной флэш-хроматографией. Соединение (Н) может быть получено с применением реакции Юлия-Литгой-Кочиенски, проходящей в одном сосуде, между лабильными основными альдегидными производными сахаров (L) и сульфонами (М), как показано ниже на схеме 3 а. Схема 3 а Альтернативный синтез аналога соединения (Н), содержащего фрагмент -СН 2-СН 2-, т.е. соединения(U), отображен ниже на схеме 3b. Схема 3b- 22016042 На схеме 3b альдегид (О) получают из алкена (N). Удлинение цепи с помощью Реакции Виттига с последующим восстановлением до спирта приводит к получению аллилового спирта, который при обработке в условиях хорошо известной реакции эпоксидирования Шарплесса превращают в эпоксипроизводное (Q). Раскрытие цикла азидом натрия, происходящее с инверсией, приводит к получению защищенного аминогидрокси альдегида (S). Затем с помощью реакции Гриньяра получают спирт (Т). Синтез отдельных гликолипидов показан ниже на схеме 4. Схема 4 Типовая методика получения амидов: соединения 10 (53 мг, 0,054 ммоль) растворяли в DCM (4 мл) и добавляли к раствору соответствующие количества трифторуксусной кислоты (0,2 мл) и триэтилсилана(0,1 мл) при 0 С. После перемешивания при комнатной температуре в течение 2 ч смесь без дальнейшей обработки концентрировали в вакууме. К раствору остатка в ТГФ добавляли п-нитрофениловый эфир жирной кислоты 4 (31 мг, 1,03 экв.) и каталитическое количество DMAP. Затем смесь перемешивали при комнатной температуре в течение 2 ч, после чего ТСХ (петролейный эфир-EtOAc, 2:1) указала на завершение реакции амидирования. Полученную смесь упаривали и очищали колоночной флэшхроматографией (петролейный эфир-EtOAc-CHCl3, 2,5:1:0,5), получая соединения 11 (39 мг, выход 2 стадий 58%). Соединение 11 (Е-амидная цепь): 1(м, 56 Н (или 58 Н для А-4), Н-8-Н-18, Н-4"-Н-18", Н-24"-Н-25" (или -Н-26" для А-4, 0,85 (т, 6 Н, J=6,9 Гц, 3 Н-19, 3 Н-26"). 13 С ЯМР (125 МГц, пиридин-d5) 173,8, 78,8, 77,5, 74,1, 73,0, 72,5, 71,3, 71,0, 70,7, 63,1, 53,0, 37,3,34,8, 32,5, 32,3, 30,7, 30,6, 30,5, 30,4, 30,3, 30,2, 30,2, 30,0, 27,0, 26,9, 26,6, 23,3, 14,6. А-6. Соединение А-6 получали с применением той же методики, которая описана выше для соединения А-1, за исключением того, что соединение 10 вводили в реакцию с соединением 16 с образованием соединения А-6, защищенного бензилом. Бензильные группы затем удаляли действием натрия в аммиаке, получая соединение А-6. А-7. Соединение А-7 получали с применением той же методики, которая описана выше для соединения А-1, за исключением того, что соединение 10 вводили в реакцию с п-нитрофенил-фенилгептаноатом с образованием соединения А-7, защищенного бензилом. Бензильные группы затем удаляли действием натрия в аммиаке, получая соединение А-7. п-Нитрофенилфенилгептаноат получали взаимодействием зквимолярного количества пнитрофенола и фенилгептановой кислоты в дихлорметане в присутствии одного эквивалента DCC и 5 мольн.% DMAP. После завершения реакции осадок дициклогексилмочевины удаляли фильтрованием и удаляли дихлорметан в вакууме. С целью очистки соединения быстро проводили хроматографию на силикагеле.II. Биологические примеры. Пример 1. Определение кинетических профилей цитокинов Th1-и Th2-типов, которые выделяются при in vivo введении соединений по настоящему изобретению. Материалы и методики Протестированные соединения Другие протестированные синтетические С-гликолилиды были синтезированы, как описано выше. Введение соединений in vivo Самок мышей C57BL/6 и BALB/c в возрасте пяти-шести недель приобретали у Taconic. Каждый из гликолипидов хранили при концентрации 1 мг/мл в 100% ДМСО. Мышам осуществляли внутривенную инъекцию 1 мкг каждого из гликолипидов, разбавленного 200 мкл стерильного фосфатного буферного солевого раствора (PBS). В указанные моменты времени у каждого животного отбирали сыворотку и определяли концентрации IFN-, IL-4 и IL-12 в сыворотке с помощью иммуноферментного твердофазного анализа (ELISA). Определение цитокинов с помощью иммуноферментного твердофазного анализа (ELISA) Концентрации IFN-, IL-4 и IL-12 определяли с использованием набора для ELISA (eBioscience,SanDiego, CA). Вкратце, 96-луночные планшеты ELISA (NUNC) покрывали иммобилизованным антителом согласно рекомендациям производителя и инкубировали в течение ночи при 4 С. Планшеты промывали PBS + 0,05% Tween 20 (PBS-T), после чего инкубировали с предоставленным производителем блокирующим раствором в течение 1 часа при комнатной температуре для блокирования не специфически связывающихся сайтов. После промывания PBS-T сыворотку, разбавленную в соотношении 1:10, добавляли в лунки и инкубировали в течение 2 часов при комнатной температуре. Затем планшеты инкубировали при комнатной температуре в течение 1 часа с биотинилированным детектирующим антителом,промывали, инкубировали с авидином, конъюгированным с пероксидазой хрена (HRP), в течение 30 мин при комнатной температуре, промывали и на завершающем этапе инкубировали с раствором тетраметилбензидинового субстрата в темноте в течение 10 минут при комнатной температуре. Все компоненты,т.е. антитела, конъюгат авидин-HRP и раствор субстрата разбавляли согласно рекомендациям производителя. Реакции останавливали 2,0 н серной кислотой и определяли концентрацию цитокина в каждой лунке сравнением поглощения при 450 нм для каждой лунки с известными стандартами. Результаты Уровни цитокинов Th1- и Тп 2-типа определяли на двух генетически различных моделях, т.е. у мышей C57BL/6 и BALB/c. Вводили следующие соединения: А=соединение (А-1 транс-изомер),В=соединение (А-2), С=соединение (А-3), D=соединение (А-4), Е=соединение (А-5), Z=контроль (чистыйPBS), CRONY=-C-GalCer, KRN=-GalCer, GCK109 (А-1 транс-изомер), GCK151 (А-1 цис-изомер),GCK152 (А-7). Уровни цитокинов (пг/мл) измеряли через 0, 2, 6, 12, 24, 48 и 72 часа после введения соединений. Уровни цитокинов Th1-типа IFN- и IL-12 показаны на фиг. 2 А-С и 3 А-С, соответственно. Уровни цитокина Th2-тила IL-4 показаны на фиг. 4 А-С. Соответствующие числовые значения также приведены в табл. 1-6 ниже. В табл. 7 показаны соотношения IFN-/IL-4 и IL-12/IL-4 для различных соединений, причем эти соотношения можно использовать как индикаторы способности этих соединений селективно стимулировать иммунные ответы Th1-типа. Как следует из указанных графиков и таблиц, при сравнении с -GalCer (KRN), несколько протестированных синтетических С-гликолипидов по настоящему изобретению обеспечивали (i) сравнимые или более высокие уровни и увеличенное время секреции обоих цитокинов Th1-типа IFN- и IL-12 и (ii) значительно более низкие уровни цитокина Th2-типа IL-4. Этот эффект был выражен особенно явно в случае соединений А-1 (транс-изомера), А-2 и А-5. Способность соединений по настоящему изобретению селективно вызывать появление высоких уровней цитокина Th1-типа IL-12 при отсутствии индукции цитокина Th2-типа показывает, что описываемые соединения являются мощными селективными стимуляторами иммунного ответа Th1-типа, который развивается локально и связан со вторичной активацией дендритных клеток. Следовательно, соединения по настоящему изобретению применимы при лечении различных заболеваний, для контроля над которыми требуется иммунный ответ Th1-типа, как при непосредственном применении, так и в качестве адъювантов.- 25016042 Таблица 1. Уровни IFN- при введении гликолипидов мышам BALB/c- 26016042 Таблица 2. Уровни IFN- при введении гликолипидов мышам C57BL/6- 27016042 Таблица 3. Уровни IL-12 при введении гликолипидов мышам BALB/c- 28016042 Таблица 4. Уровни IL-12 при введении гликолипидов мышам C57BL/6
МПК / Метки
МПК: A61K 31/70, C07H 7/02
Метки: с-гликолипиды, улучшенным, профилем
Код ссылки
<a href="https://eas.patents.su/30-16042-s-glikolipidy-s-uluchshennym-profilem-th-1.html" rel="bookmark" title="База патентов Евразийского Союза">С-гликолипиды с улучшенным профилем th-1</a>
Буровой раствор с плоским реологическим профилем

Номер патента: 7842
Опубликовано: 27.02.2007
Авторы: Пейтел Арвинд Д., Фридхейм Джим, Хоксха Бернхэм, Ли Джон
МПК: C09K 8/32
Метки: реологическим, буровой, плоским, профилем, раствор
Формула / Реферат:
1. Буровой раствор, содержащий масляную жидкость, причем масляная жидкость является непрерывной фазой бурового раствора; немасляную жидкость, причем немасляная жидкость является дискретной фазой бурового раствора; эмульгатор, присутствующий в концентрации, достаточной для того, чтобы стабилизировать обратную эмульсию; модификатор вязкости, причем модификатор вязкости выбран из группы, состоящей из димера (С12-С22)-поликарбоновой жирной кислоты,...
Капсулы флуконазола с улучшенным высвобождением

Номер патента: 8585
Опубликовано: 29.06.2007
Авторы: Балогне Немеш Каталин, Клебович Имре, Фурдига Эва, Фекете Паль, Левентисне Хусар Магдольна, Виргула Юдит
МПК: A61K 9/16, A61K 31/4196, A61K 9/48...
Метки: флуконазола, капсулы, высвобождением, улучшенным
Формула / Реферат:
1. Капсулы, содержащие флуконазол, с быстрым и стабильным высвобождением активного ингредиента, включающие частицы флуконазола, представляющего собой [2,4-дифтор-(a),(a)-бис-(1Н-1,2,4-тиазол-1-илметил)бензиловый спирт], гранулированные с агентом, способным придавать поверхности указанных частиц гидрофильность. 2. Капсулы по п.1, гранулированные, кроме агента, способного придавать поверхности указанных частиц гидрофильность, с...
Многокомпонентный фильтр с улучшенным усилением аромата

Номер патента: 15119
Опубликовано: 30.06.2011
Авторы: Жордий Ив, Бессо Клемент, Кюрштайнер Шарль, Вайсс-Петерс Анн
МПК: A24D 3/04
Метки: усилением, фильтр, многокомпонентный, аромата, улучшенным
Формула / Реферат:
1. Многокомпонентный фильтр (4) для курительного изделия, содержащийпервый ароматовыделяющий сегмент (12) ближе к мундштучному концу фильтра ивторой ароматовыделяющий сегмент (14), расположенный непосредственно перед первым ароматовыделяющим сегментом (12),при этом первый ароматовыделяющий сегмент (12) содержит по меньшей мере одну ароматонесущую нить (16), а второй ароматовыделяющий сегмент (14) содержит ароматонесущие целлюлозные частицы...
Композиция с улучшенным терапевтическим спектром действия, содержащая ингибиторы синтеза нуклеотидов

Номер патента: 5136
Опубликовано: 30.12.2004
Авторы: Линднер Юрген, Хаазе Буркхард
МПК: A61K 31/74, A61P 37/00
Метки: нуклеотидов, содержащая, действия, синтеза, ингибиторы, композиция, спектром, улучшенным, терапевтическим
Формула / Реферат:
1. Композиция, включающая 1) по крайней мере одно соединение, выбранное из группы, включающей колестипол, колестирамин и активный углерод, которое в значительной мере препятствует энтерогепатической циркуляции ингибиторов синтеза нуклеотидов, или соединение, выбранное из группы, влючающей уридин, пурин, нуклеотиды пурина или нуклеотиды пиримидина, которое является антагонистом действия ингибиторов синтеза нуклеотидов со смещением во времени, и ...
Покрытая оболочкой разлагающаяся жевательная резинка с улучшенным сроком хранения и способ ее получения

Номер патента: 5626
Опубликовано: 28.04.2005
Авторы: Андерсен Лоне, Исаксен Анетте, Витторфф Хелле
МПК: A23G 3/30
Метки: получения, оболочкой, способ, резинка, покрытая, жевательная, разлагающаяся, улучшенным, сроком, хранения
Формула / Реферат:
1. Покрытый оболочкой элемент жевательной резинки, включающий примерно от 25 до 99,9 вес.% центральной части жевательной резинки, включающей по крайней мере один разлагаемый в окружающей среде эластомерный или смолистый полимер, и примерно от 0,1 до 75 вес.% внешнего покрытия. 2. Покрытый оболочкой элемент жевательной резинки по п.1, где внешнее покрытие до момента жевания жевательной резинки вызывает снижение степени разложения по крайней мере...
Предыдущий патент: Способы применения рецептора gpr119 для идентификации соединений, которые можно использовать для увеличения костной массы субъекта
Следующий патент: Косметические или дерматологические препараты, содержащие n-ацетилцистеин
Случайный патент: Применение антагонистов cgrp для устранения приливов в период менопаузы