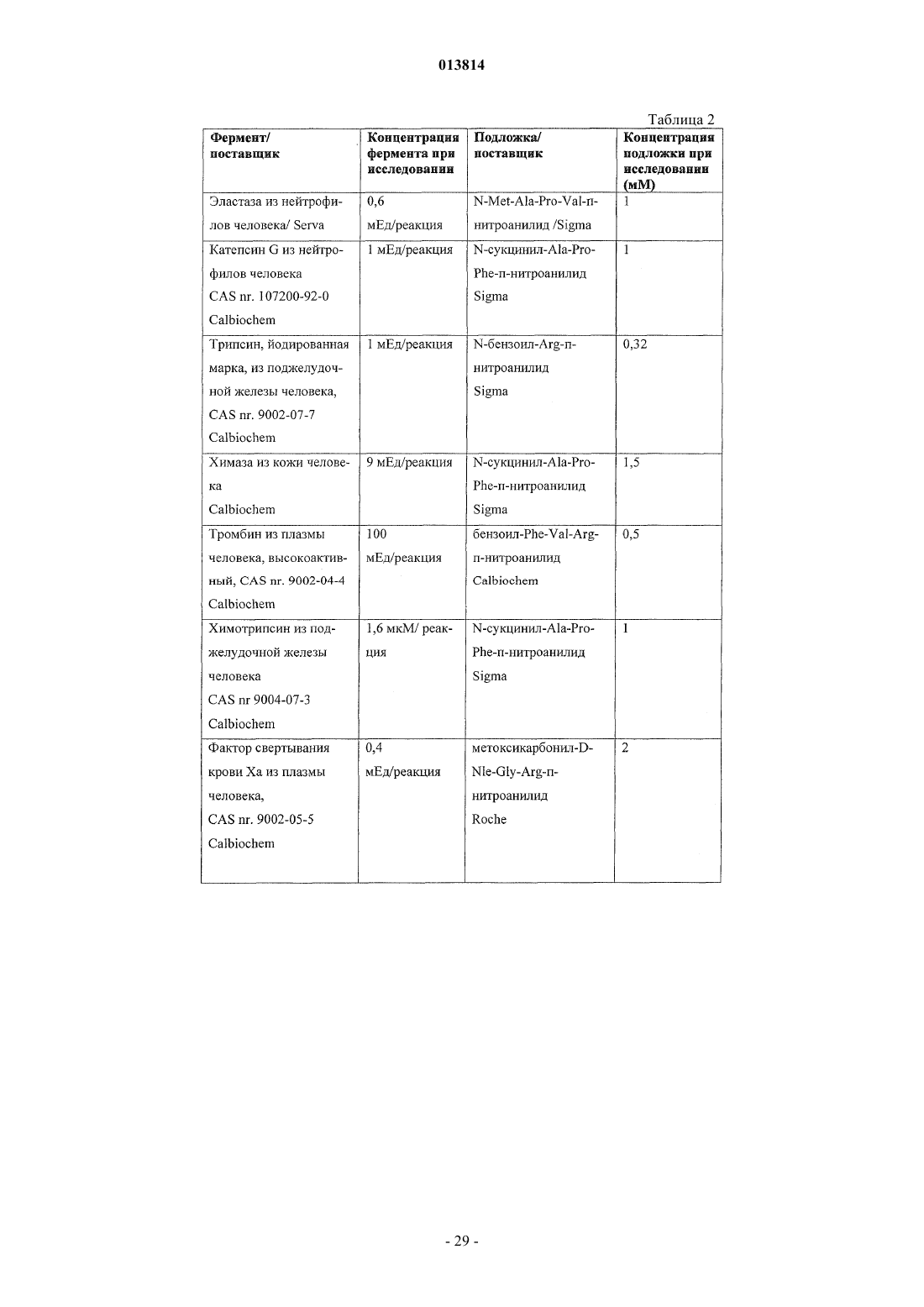
Связанные с матрицей бета-шпилечные пептидомиметики с ингибирующей активностью в отношении протеаз
Номер патента: 13814
Опубликовано: 30.08.2010
Авторы: Меле Керстин, Демарко Стивен Дж., Хенце Хайко, Лудин Кристиан, Обрехт Даниель, Гомберт Франк, Селлье Одиль, Юнг Франсуаза





























Формула / Реферат
1. Соединение общей формулы

где

означает группу одной из формул

где

представляет собой Ala; Arg; Asn; Cys; Gln; Gly; His; Ile; Leu; Lys; Met; Phe; Pro; Pro(5RPhe), Ser; Thr; Trp; Tyr; Val; Cit; Orn; tBuA; Sar; t-BuG; 4AmPhe; 3AmPhe; 2AmPhe; Phe(mC(NH2)=NH); Phe(pC(NH2)=NH); Phe(mNHC(NH2)=NH); Phe(pNHC(NH2)=NH); Phg; Cha; C4al; C5al; Nle; 2-Nal; 1-Nal; 4Cl-Phe; 3Cl-Phe; 2Cl-Phe; 3,4Cl2Phe; 4F-Phe; 3F-Phe; 2F-Phe; Tic; Thi; Tza; Mso; AcLys; Dpr; A2Bu; Dbu; Abu; Aha; Aib; Y(Bzl); Bip; S(Bzl); T(Bzl); hCha; hCys; hSer, hArg; hPhe; Bpa; Pip; OctG; MePhe; MeNle; MeAla; MeIle; MeVal или MeLeu
![]()
означает группу формулы

Z означает цепь из 11 a-аминокислотных остатков, положения указанных аминокислотных остатков в боковой цепи отсчитываются от N-концевой аминокислоты, причем указанные аминокислотные остатки в положениях 1-11 цепи Z представляют собой

![]()
при дополнительном условии, что, если матрица представляет собой DProLPro, то аминокислотные остатки в положениях Р1-Р6 и Р8-Р11 не означают

и их фармацевтически приемлемые соли.
2. Соединение по п.1, в котором a-аминокислотные остатки в положениях 1-11 представляют собой

![]()
3. Соединение по п.1, в котором a-аминокислотные остатки в положениях 1-11 представляют собой

4. Соединение по п.1, в котором a-аминокислотные остатки в положениях 1-11 цепи Z представляют собой

5. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

6. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

7. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой


8. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

9. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой


10. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

11. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

![]()
12. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

13. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

14. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

15. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

16. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой


17. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

18. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

![]()
19. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой:

20. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

21. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

22. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

23. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой
![]()

24. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

25. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой


26. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

27. Соединение формулы (I) по п.1, где матрица представляет собой DPro-LPro и аминокислотные остатки в положениях 1-11 представляют собой

28. Энантиомеры соединения формулы (I) по п.1.
29. Соединения по любому из пп.1-28 для применения в качестве терапевтически активных веществ.
30. Соединения по п.29, обладающие селективной ингибирующей активностью по отношению к протеазе, в частности против катепсина G, или эластазы, или триптазы, и/или противораковой активностью, и/или противовоспалительной активностью, и/или противоинфекционной активностью, и/или лечебной активностью при сердечно-сосудистых заболеваниях, и/или антииммунологической активностью, и/или антинейродегенеративной активностью.
31. Фармацевтическая композиция, содержащая соединение по любому из пп.1-28 и фармацевтически инертный носитель.
32. Композиции по п.31 в форме, пригодной для перорального, местного, чрескожного, трансбуккального, проводимого через слизистую оболочку, или легочного введения, или введения с помощью инъекции или ингаляции.
33. Композиции по п.31 или 32 в форме таблеток, драже, капсул, растворов, жидкостей, гелей, пластыря, кремов, мазей, сиропа, взвесей, суспензий, спрея, распыляемого средства или суппозиториев.
34. Применение соединений по любому из пп.1-28 для приготовления лекарственного средства, предназначенного для применения в качестве ингибитора протеазных ферментов.
35. Применение по п.34, в котором указанное ингибирующее протеазу лекарственное средство предназначено для применения с целью предупреждения инфекций у здоровых индивидуумов или для замедления инфекций у инфицированных пациентов; или когда рак опосредуется активностью протеазы или обусловлен ее активностью; или когда иммунологические заболевания опосредуются активностью протеазы или обусловлены ее активностью, или когда воспаление опосредуется активностью протеазы или обусловлено ее активностью; или когда иммунологическая реакция опосредуется активностью протеазы или обусловлена ее активностью.
36. Применение соединений по любому из пп.2 и 5-11 для приготовления лекарственного средства, предназначенного для применения в качестве ингибитора катепсина G.
37. Применение соединений по любому из пп.3 и 12-22 для приготовления лекарственного средства, предназначенного для применения в качестве ингибитора эластазы.
38. Применение соединений по любому из пп.4 и 23-24 для приготовления лекарственного средства, предназначенного для применения в качестве ингибитора триптазы.
39. Способ получения соединений по любому из пп.1-27, который предусматривает:
(a) проведение реакции сочетания соответствующим образом функционализированной твердой подложки с подходящим N-защищенным производным аминокислоты, которая в искомом конечном продукте находится в положении 5, 6 или 7, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;
(b) удаление N-защитной группы из полученного таким образом продукта;
(c) проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится на одно положение ближе к N-концевому аминокислотному остатку, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;
(d) удаление N-защитной группы из полученного таким образом продукта;
(e) повторение стадий (с) и (d), пока не будет введен N-концевой аминокислотный остаток;
(f) проведение реакции сочетания полученного таким образом продукта с соединением общей формулы

где

является такой, как определено в п.1, и X означает N-защитную группу, или, альтернативно, если

является группой (a1) или (а2), как определено в п.1,
(fa) проведение реакции сочетания продукта, полученного на стадии (е), с соответствующим образом N-защищенным производным аминокислоты общей формулы
![]()
или
![]()
в которой В и А являются такими, как определено в п.1, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;
(fb) удаление N-защитной группы из полученного таким образом продукта и
(fc) проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты указанной выше общей формулы IV и, соответственно, III, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;
(g) удаление N-защитной группы из продукта, полученного на стадии (f) или (fc);
(h) проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится в положении 11, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;
(i) удаление N-защитной группы из полученного таким образом продукта;
(j) проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится на одно положение дальше от положения 11, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;
(k) удаление N-защитной группы из полученного таким образом продукта;
(l) повторение стадий (j) и (k), пока не будут введены все аминокислотные остатки;
(m) при желании селективное снятие защиты у одной или нескольких защищенных функциональных групп, содержащихся в молекуле, и соответствующее замещение реакционноспособной группы (групп), высвободившихся при этом;
(n) при желании образование внутри цепи связи между боковыми цепями соответсвующих аминокислотных остатков в положениях 2 и 10;
(о) отделение полученного таким образом продукта от твердой подложки;
(р) циклизацию продукта, отделенного от твердой подложки;
(q) удаление любых защитных групп, находящихся на функциональных группах любых элементов цепи аминокислотных остатков, и при желании любой защитной группы (групп), которые дополнительно могут находиться в молекуле; и
(r) при желании превращение полученного таким образом продукта в фармацевтически приемлемую соль или превращение полученной таким образом фармацевтически приемлемой или неприемлемой соли в соответствующее свободное соединение формулы (I) или в другую фармацевтически приемлемую соль.
40. Способ получения соединений по любому из пп.1-27, который предусматривает:
(а') проведение реакции сочетания соответствующим образом функционализированной твердой подложки с соединением общей формулы

в которой

является такой, как определено в п.1, и X означает N-защитную группу, или, альтернативно, если

является группой (a1) или (а2), как определено в п.1,
(а'а) проведение реакции сочетания указанной соответствующим образом функционализированной твердой подложки с соответствующим образом N-защищенным производным аминокислоты общей формулы
![]()
или
![]()
в которой В и А являются такими, как определено в п.1, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена; (а'b) удаление N-защитной группы из полученного таким образом продукта; и (а'с) проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты указанной выше общей формулы IV и, соответственно, III, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена; (b') удаление N-защитной группы из продукта, полученного на стадии (а') или (а'с); (с') проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится в положении 11, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена; (d') удаление N-защитной группы из полученного таким образом продукта; (е') проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится на одно положение дальше от положения 11, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;
(f') удаление N-защитной группы из полученного таким образом продукта;
(g') повторение стадий (е') и (f'), пока не будут введены все аминокислотные остатки;
(h') при желании селективное снятие защиты у одной или нескольких защищенных функциональных групп, содержащихся в молекуле, и соответствующее замещение реакционноспособной группы (групп), высвободившихся при этом;
(i') при желании образование внутри цепи связи между боковыми цепями соответствующих аминокислотных остатков в положениях 2 и 10;
(j') отделение полученного таким образом продукта от твердой подложки;
(k') циклизацию продукта, отделенного от твердой подложки;
(l') удаление любых защитных групп, находящихся на функциональных группах любых элементов цепи аминокислотных остатков, и при желании любой защитной группы (групп), которые дополнительно могут находиться в молекуле; и
(m') при желании превращение полученного таким образом продукта в фармацевтически приемлемую соль или превращение полученной таким образом фармацевтически приемлемой или неприемлемой соли в соответствующее свободное соединение формулы (I) или в другую фармацевтически приемлемую соль.
41. Модификация способов по п.39 или 40 для получения соединений по п.28, в которой используются энантиомеры всех хиральных исходных веществ.
Текст
где Z означает цепь из 11 -аминокислотных остатков, которые в зависимости от их положений в цепи (при отсчете начиная с N-концевой аминокислоты) представляют собой Gly, или Pro, илиPro(4NHCOPhe), или остатки определенных типов, которые, как и другие обозначения в приведенной выше формуле, определены в описании и формуле изобретения, и их соли, обладают способностью ингибировать протеазы, в частности сериновые протеазы, в особенности катепсин G,или эластазу, или триптазу. Эти -шпилечные пептидомиметики можно приготовить способами,которые основаны на смешанной стратегии твердофазного и жидкофазного синтеза. 013814 Настоящее изобретение относится к связанным с матрицей -шпилечным пептидомиметикам,включающим связанную с матрицей цепь из 11 -аминокислотных остатков, которые в зависимости от их положения в цепи представляют собой Gly, или Pro, или Pro(4NHCOPhe), или являются остатками определенных типов, определенных ниже в настоящем изобретении. Эти связанные с матрицей шпилечные пептидомиметики применимы в качестве ингибиторов протеазных ферментов. Они являются особенно полезными в качестве ингибиторов различных сериновых протеаз, таких как катепсин G, эластаза или триптаза человека. Кроме того, настоящее изобретение относится к эффективному способу, с помощью которого эти соединения при желании можно получить в виде библиотеки.-Шпилечные пептидомиметики, соответствующие настоящему изобретению, обнаруживают улучшенную эффективность, биологическую доступность при пероральном введении, увеличенный период полувыведения и, что наиболее важно, большое отношение селективности по отношению к различным сериновым протеазам, которое зависит от надлежащего выбора некоторых типов -аминокислотных остатков и их положения в указанной цепи. Кроме того, эти -шпилечные пептидомиметики характеризуются небольшим гемолизом на эритроцитах и низкой цитотоксичностью. Установлено, что ингибиторы протеаз перспективны для применения в медицине с целью лечения таких заболеваний, как раковые заболевания (R.P. Beckett, A. Davidson, A.H. Drummond, M. Whittaker,Drug Disc. Today 1996, 1, 16-26; L.L. Johnson, R. Dyer, D.J. Hupe, Curr. Opin. Chem. Biol., 1998, 2, 466-71;Stubbs, W.A. Bode, Thromb. Res., 1993, 69, 1-58; H. Fukami et al., Current Pharmaceutical Design, 1998, 4,439-453) и нейродегенеративные нарушения, включая болезнь Альцгеймера (R. Vassar, В.D. Bennett, S.Babu-Kahn, S. Kahn, E.A. Mendiaz, Science, 1999, 286, 735-41), ангиогенез (Kaatinen M. et al., Atherosklerosis, 1996, 123 1-2, 123-131) и рассеянный склероз (Ibrahim M.Z. et al., J. Neuroimmunol, 1996, 70, 131-138). Поскольку большинство протеаз связываются со своими субстратами в растянутой конформации или конформациях -цепи, эффективные ингибиторы должны быть способны имитировать такую конформацию. Поэтому -шпилечные миметики идеально подходят для фиксации последовательностей пептидов в растянутой конформации. Из числа протеаз сериновые протеазы являются важными объектами терапевтического воздействия. Сериновые протеазы классифицируют по их специфичности по отношению к субстрату, в особенности по типам остатков, находящихся у Р 1, как трипсиноподобные (положительно заряженные остаткиAla/Val у P1) или химотрипсиноподобные (большие гидрофобные остатки Phe/Tyr/Leu у Р 1). Сериновые протеазы, для которых в базе данных PDB (PDB: www.rcsb.org/pdb) имеются рентгеноструктурные данные для кристаллов ингибиторов протеазы, включают трипсин, -химотрипсин, -химотрипсин, эластазу нейтрофилов человека, тромбин, субтилизин, цитомегаловирус человека, протеиназу А, ахромобактер,катепсин G человека, специфичную по отношению к глутаминовой кислоте протеазу, карбопептидазу D,фактор свертывания крови VIIa, фактор свиней 1 ХА, мезентерикопептидазу, HCV протеазу и термитазу. Другие сериновые протеазы, которые представляют терапевтический интерес, включают триптазу, комплементарную конвертазу, протеазу гепатита C-NS3. Ингибиторы тромбина (например, J.L. Metha, L.Y.Respir. Dis., 1991, 144, 875-83) включены в клинические исследования по лечению эмфиземы и других заболеваний легких, а ингибиторы триптазы находятся в фазе II клинических исследований по лечению астмы (С. Seife, Science, 1997, 277, 1602-3), ингибиторы урокиназы - по лечению рака молочной железы и ингибиторы химазы - по лечению заболеваний, связанных с сердцем. Наконец, катепсин G и эластаза непосредственно участвуют в модуляции активности цитокинов и их рецепторов. В частности, на участках воспаления большие концентрации катепсина G, эластазы и протеиназы 3, высвобождающиеся из инфильтрованных полиморфноядерных клеток, находятся в хорошей временной корреляции с повышен-1 013814 ными содержаниями воспалительных цитокинов, что убедительно указывает на то, что эти протеазы участвуют в регулировании биологической активности и доступности цитокинов (U. Bank, S. Ansorge, J.Leukoc. Biol., 2001, 69, 177-90). Таким образом, ингибиторы эластазы и катепсина G являются важными объектами воздействия новых предлагаемых лекарственных средств, в частности, для лечения хронического обструктивного заболевания легких (Ohbayashi H., Epert Opin. Investig. Drugs., 2002, 11, 965-980). Из числа многих появившихся белковых ингибиторов сериновой протеазы один представляет собой содержащий 14 аминокислот циклический пептид, выделенный из семян подсолнечника, названный подсолнечниковым ингибитором трипсина (SFTI-1) (S. Luckett, R. Santiago Garcia, J.J. Barker, A.V. Konarev,P.R. Shewry, A.R. Clarke, R.L. Brady, J. Mol. Biol., 1999, 290, 525-533; Y.-Q. Long, S.-L. Lee, C.-Y. Lin, I.J.Enyedy, S. Wang, P. Li, R.B. Dickson, P.P. Roller, Biorg.Med. Chem. Lett., 2001, 11, 2515-2519), который обладает последовательностью и конформацией, сходными с характеристиками трипсиновой реакционноспособной петли ингибиторов семейства Баумана-Бирк (Bowman-Birk) сериновых протеаз. Ингибитор принимает -шпилечную конформацию при связывании с активным участком бычьего -трипсина.(Ki7,4 мкМ) и тромбин (Ki136 мМ). В настоящем изобретении иллюстрируется подход к разработке ингибитора, который включает трансплантацию -шпилечной петли из природного пептида в индуцированную шпилькой матрицу. На основании хорошо известной трехмерной структуры -шпилечных миметиков можно получить библиотеки соединений, которые в конечном счете могут привести к новым ингибиторам, обладающим разными профилями специфичности по отношению к нескольким классам протеаз. Связанные с матрицей шпилечные миметические пептиды описаны в литературе (D. Obrecht, M. Altorfer, J.A. Robinson, Adv. Med. Chem., 1999, 4, 1-68; J.A. Robinson, Syn. Lett., 2000, 4, 429-441) и ингибирующие сериновую протеиназу связанные с матрицей пептидомиметики и методики их синтеза описаны в международной патентной заявке WO 2003/054000 А 1 и в публикации Descours A., Moehle K., RenardA., Robinson J. ChemBioChem., 2002, 3, 318-323, но описанные ранее молекулы не обладают высокой селективностью и, в особенности, высокой активностью. Однако недавно установлена возможность получения -шпилечных пептидомиметиков с помощью комбинаторного метода и метода параллельного синтеза (L. Jiang, K. Moehle, В. Dhanapal, D. Obrecht, J.A. Robinson, Helv. Chim. Acta., 2000, 83, 3097-3112). Эти методы позволяют синтезировать и выполнить скрининг больших библиотек шпилечных миметиков, что, в свою очередь, облегчает проведение исследований зависимостей структура-активность и,следовательно, обнаружение новых молекул, обладающих очень эффективной и селективной ингибирующей активностью по отношению к сериновой протеазе, пероральной биологической доступностью,низкой гемолитической активностью по отношению к эритроцитам человека и низкой цитотоксичностью.-Шпилечные пептидомиметики, соответствующие настоящему изобретению, являются соединениями общей формулы означает группу одной из формулZ означает цепь из 11-аминокислотных остатков, положения указанных аминокислотных остатков в боковой цепи отсчитывается от N-концевой аминокислоты, причем указанные аминокислотные остатки в положениях 1-11 цепи Z представляют собой при дополнительном условии, что, если матрица представляет собой DProLPro, то аминокислотные остатки в положениях Р 1-Р 6 и Р 8-Р 11 не означают и их фармацевтически приемлемые соли. В соответствии с настоящим изобретением эти -шпилечные пептидомиметики можно получить способом, который предусматривает:(a) проведение реакции сочетания соответствующим образом функционализированной твердой-3 013814 подложки с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится в положении 5, 6 или 7, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;(b) удаление N-защитной группы из полученного таким образом продукта;(c) проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится на одно положение ближе к N-концевому аминокислотному остатку, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;(d) удаление N-защитной группы из полученного таким образом продукта;(e) повторение стадий (с) и (d), пока не будет введен N-концевой аминокислотный остаток;(f) проведение реакции сочетания полученного таким образом продукта с соединением общей формулы означает указанную выше группу (a1) или (а 2),(fa) проведение реакции сочетания продукта, полученного на стадии (е), с соответствующим образом N-защищенным производным аминокислоты общей формулы или в которой В и А являются такими, как определено выше, причем любая функциональная группа,которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;(fb) удаление N-защитной группы из полученного таким образом продукта и(fc) проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты указанной выше общей формулы IV и, соответственно,III, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;(g) удаление N-защитной группы из продукта, полученного на стадии (f) или (fc);(h) проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится в положении 11, причем любая функциональная группа, которая может содержаться в указанномN-защищенном производном аминокислоты, также соответствующим образом защищена;(i) удаление N-защитной группы из полученного таким образом продукта;(j) проведение реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится на одно положение дальше от положения 11, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;(k) удаление N-защитной группы из полученного таким образом продукта;(l) повторение стадий (j) и (k), пока не будут введены все аминокислотные остатки;(m) при желании селективное снятие защиты у одной или нескольких защищенных функциональ-4 013814 ных групп, содержащихся в молекуле, и соответствующее замещение реакционноспособной группы(n) при желании образование внутри цепи связи между боковыми цепями соответсвующих аминокислотных остатков в положениях 2 и 10;(о) отделение полученного таким образом продукта от твердой подложки;(р) циклизацию продукта, отделенного от твердой подложки;(q) удаление любых защитных групп, находящихся на функциональных группах любых элементов цепи аминокислотных остатков, и при желании любой защитной группы (групп), которые дополнительно могут находиться в молекуле; и(r) при желании превращение полученного таким образом продукта в фармацевтически приемлемую соль или превращение полученной таким образом фармацевтически приемлемой или неприемлемой соли в соответствующее свободное соединение формулы (I) или в другую фармацевтически приемлемую соль. Альтернативно, пептидомиметики, соответствующие настоящему изобретению, можно получить с помощью:(а') проведения реакции сочетания соответствующим образом функционализированной твердой подложки с соединением общей формулы означает указанную выше группу (a1) или (а 2),(а'а) проведения реакции сочетания указанной соответствующим образом функционализированной твердой подложки с соответствующим образом N-защищенным производным аминокислоты общей формулы или в которой В и А являются такими, как определено выше, причем любая функциональная группа,которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;(а'b) удаления N-защитной группы из полученного таким образом продукта и(а'с) проведения реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты указанной выше общей формулы IV и, соответственно,III, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;(b') удаления N-защитной группы из продукта, полученного на стадии (а') или (а'с);(с') проведения реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится в положении 11, причем любая функциональная группа, которая может содержаться в указанномN-защищенном производном аминокислоты, также соответствующим образом защищена;(d') удаления N-защитной группы из полученного таким образом продукта;(е') проведения реакции сочетания полученного таким образом продукта с соответствующим образом N-защищенным производным аминокислоты, которая в искомом конечном продукте находится на одно положение дальше от положения 11, причем любая функциональная группа, которая может содержаться в указанном N-защищенном производном аминокислоты, также соответствующим образом защищена;(f) удаления N-защитной группы из полученного таким образом продукта;(g') повторения стадий (е') и (f), пока не будут введены все аминокислотные остатки;(h') при желании селективного снятия защиты у одной или нескольких защищенных функциональных групп, содержащихся в молекуле, и соответствующего замещения реакционноспособной группы(i') при желании образования внутри цепи связи между боковыми цепями соответствующих аминокислотных остатков в положениях 2 и 10;(j') отделения полученного таким образом продукта от твердой подложки;(k') циклизации продукта, отделенного от твердой подложки;(l') удаления любых защитных групп, находящихся на функциональных группах любых элементов цепи аминокислотных остатков, и при желании любой защитной группы (групп), которые дополнительно могут находиться в молекуле; и(m') при желании превращения полученного таким образом продукта в фармацевтически приемлемую соль или превращение полученной таким образом фармацевтически приемлемой или неприемлемой соли в соответствующее свободное соединение формулы (I) или в другую фармацевтически приемлемую соль. Пептидомиметики, соответствующие настоящему изобретению, также могут представлять собой энантиомеры соединений формулы (I). Эти энантиомеры можно получить путем модификации указанных выше способов, в которых используются энантиомеры всех хиральных исходных веществ. Структурный элемент -А-СО- означает аминокислотные структурные фрагменты, которые в комбинации со структурным элементом -В-СО- образуют матрицы (a1) и (а 2). Матрицы (a1) и (а 2) образуют структурные фрагменты, которые содержат N-конец и С-конец, ориентированные в пространстве таким образом, что расстояние между этими двумя группами может составлять 4,0-5,5 А. Пептидная цепь Z связана с С-концом и N-концом матриц (a1) и (а 2) через соответствующие N- и С-концы, так что матрица и цепь образуют циклическую структуру, такую как представлена в формуле (I). В таком случае, как описанный в настоящем изобретении, когда расстояние между N- и С-концами матрицы составляет 4,0-5,5 А, матрица вызовет образование сетки Н-связей, необходимой для образования -шпилечной конформации пептидной цепи Z. Эта матрица и пептидная цепь образуют -шпилечный миметик.-Шпилечная конформация тесно связана со способностью -шпилечных миметиков, соответствующих настоящему изобретению, ингибировать активность сериновой протеазы. Свойства матриц (a1) и (а 2) стабилизировать конформацию -шпильки играют ключевую роль не только для селективной ингибирующей способности, но и для описанного выше синтеза, поскольку включение матриц в начале или вблизи от середины, содержащих защитные группы линейных предшественников пептидов, значительно повышает выход реакции циклизации. Так, например, DPro-LPro образует прототип матриц (a1) и LPro-DPro образует прототип матрицы(а 2). Следует понимать, что структурный фрагмент -А-СО-, в котором А имеет (D)-конфигурацию, несет водород (Н) в -положении к N-концу. Специалисты в данной области техники должны понимать, что А приведена в (D)-конфигурации, которая соответствует (R)-конфигурации. Боковые цепи, состоящие преимущественно из (L)-аминокислот в противолежащих положениях региона -цепи, могут образовывать межцепочечную связь. Наиболее известной связью является дисульфидный мостик, образованный цистеинами и гомоцистеинами, расположенными в противолежащих положениях -цепи. Известны различные методики образования дисульфидных связей, включая описанные в публикациях: J.P. Tarn et al. Synthesis, 1979, 955-957; Stewart et al. Solid Phase Peptide Synthesis, 2d Ed.,Pierce Chemical Company, III., 1984; Ahmed et al. J. Biol. Chem., 1975, 250, 8477-8482 и Pennington et al.Peptides, p. 164-166, Giralt and Andreu, Eds., ESCOM Leiden, The Netherlands, 1990. Для объема настоящего изобретения дисульфидные связи предпочтительнее всего можно получить с использованием ацетамидометильных (Acm)-защитных групп для цистеина. Положениями для межцепочечной связи являются положения Р 2 и Р 10, взятые совместно. Известно, что такие межцепочечные связи стабилизируют -шпилечные конформации и тем самым образуют структурный элемент, важный для образования -шпилечных миметиков. Наиболее предпочтительным аминокислотными остатками в цепи Z являются образованные из природных -аминокислот. Ниже приведен перечень аминокислот, которые или остатки которых пригодны для задач настоящего изобретения, и использованы общепринятые аббревиатуры: Другие -аминокислоты, которые или остатки которых пригодны для задач настоящего изобретения, включают: Обычно пептидная цепь Z в -шпилечных миметиках, соответствующих настоящему изобретению,включает 11 аминокислотных остатков. Положения Р 1-Р 11 каждого аминокислотного остатка в цепи Z однозначно определяются следующим образом: Р 1 означает первую аминокислоту в цепи Z, которая своим N-концом связана с С-концом группы -В-СО- в матрице (a1), или группы -А-СО- в матрице (а 2); и Р 11 означает последнюю аминокислоту в цепи Z, которая своим С-концом связана с N-концом группы-Аминокислотные остатки в положениях 1-11 в цепи Z особенно предпочтительно представляют собой при дополнительном условии, что, если матрица представляет собой DProLPro, то аминокислотные остатки в положениях Р 1-Р 6 и Р 8-Р 11 не означают Для ингибиторов катепсина G -аминокислотные остатки в положениях 1-11 наиболее предпочтительно представляют собой- 11013814 Для ингибиторов эластазы -аминокислотные остатки в положениях 1-11 наиболее предпочтительно представляют собой Для ингибиторов триптазы -аминокислотные остатки в положениях 1-11 в цепи Z наиболее предпочтительно представляют собой: остатки Cys, содержащиеся в Р 2 и Р 10, могут образовывать дисульфидный мостик. Особенно предпочтительные -пептидомиметики, соответствующие настоящему изобретению,включают описанные в примерах 5, 19, 20, 22, 23, 38, 39, 40 и 75 в качестве ингибиторов катепсина G; в примерах 91, 121, 153, 154, 155, 156, 157, 158, 159, 160, 161, 177 и 178 в качестве ингибиторов эластазы и в примерах 193, 194 и 195 в качестве ингибиторов триптазы. Способы, соответствующие настоящему изобретению, с успехом могут быть осуществлены как параллельный матричный синтез (parallel array synthesis) с получением библиотек связанных с матрицей-шпилечных пептидомиметиков указанной выше общей формулы (I). Такой параллельный синтез позволяет получить группы, содержащие множество (обычно от 24 до 192, в типичном случае 96) соединений общей формулы (I) с высокими выходами и определенной чистотой со сведением к минимуму образования димерных и полимерных побочных продуктов. Поэтому ключевую роль играет надлежащий выбор функционализированной твердой подложки (т.е. твердой подложки с молекулой-линкером), матрицы и положения циклизации. Функционализированную твердую подложку обычно получают из полистирола, предпочтительно сшитого с 1-5% дивинилбензола; полистирола с покрытием из полиэтиленгликолевых спейсеровSeries, vol. 17, Pergamon, Elsevier Science, 1998). Твердую подложку обычно функционализируют с помощью линкера, т.е. бифункциональной спейсерной молекулы, которая на одном конце содержит якорную группу для присоединения к твердой подложке, а на другом конце содержит селективно отщепляемую функциональную группу, использующуюся для проведения последующих химических превращений и отщепления. Для задач настоящего изобретения используют два типа линкеров. Линкеры типа 1 предназначены для высвобождения амидной группы в кислой среде (Rink H., Tetrahedron Lett., 1987, 28, 3783-3790). Линкеры этого типа образуют амиды карбоксигрупп аминокислот; примеры смол, функционализированных такими линкерными структурами, включают смолу 4-[2,4 диметоксифенил)Fmoc-аминометил)феноксиацетамидо)аминометил] ПС (полистирол), смолу 4-[2,4 диметоксифенил)Fmoc-аминометил)феноксиацетамидо)аминометил]-4-метилбензгидриламин ПС (амидная смола Rink MBHA ПС) и смолу 4-[2,4-диметоксифенил)Fmoc-аминометил)феноксиацетами- 12013814 до)аминометил]бензгидриламин ПС (амидная смола Rink BHA ПС). Предпочтительно, если подложка образована из полистирола, предпочтительно сшитого с 1-5% дивинилбензола, и функционализирована с помощью 4-2,4-диметоксифенил)Fmoc-аминометил)феноксиацетамидного) линкера. Линкеры типа 2 предназначены для высвобождения в конечном счете карбоксильной группы в кислой среде. Линкеры этого типа образуют лабильные в кислой среде эфиры с карбоксигруппами аминокислот, обычно лабильные в кислой среде бензиловые, бензгидриловые и тритиловые эфиры; примеры таких линкерных структур включают 2-метокси-4-гидроксиметилфеноксигруппу (линкер SasrinR), 4-(2,4 диметоксифенилгидроксиметил)феноксигруппу (линкер Rink), 4-(4-гидроксиметил-3-метоксифеноксигруппу)масляную кислоту (линкер НМРВ), тритил и 2-хлортритин. Предпочтительно, если подложка получена из полистирола, наиболее предпочтительно сшитого с 1-5% дивинилбензола и функционализированного с помощью 2-хлортритильного линкера. При проведении параллельного матричного синтеза способы, соответствующие настоящему изобретению, можно с успехом применять так, как описано ниже в настоящем изобретении, но для специалистов в данной области техники должно быть совершенно очевидно, как эти методики следует модифицировать в случае, когда необходимо синтезировать одно конкретное соединение приведенной выше формулы (I). В количество реакционных сосудов (обычно от 24 до 192, в типичном случае 96), равное общему количеству соединений, синтезируемых по параллельной методике, помещают от 25 до 1000 мг, предпочтительно 100 мг соответствующим образом функционализированной подложки, которая предпочтительно получена из полистирола, сшитого с 1-3% дивинилбензола, или из смолы Tentagel. Использующиеся растворители должны обеспечивать набухание смолы и они включают, но не ограничиваются только ими, дихлорметан (ДХМ), диметилформамид (ДМФ), N-метилпирролидон (NMP),диоксан, толуол, тетрагидрофуран (ТГФ), этанол (EtOH), трифторэтанол (ТФЭ), изопропиловый спирт и т.п. Смеси растворителей, содержащие в качестве по меньшей мере одного компонента полярный растворитель (например, 20% ТФЭ/ДХМ, 35% ТГФ/NMP), прекрасно подходят для обеспечения высокой реакционной способности и сольватации связанных со смолой пептидных цепей (Fields, G.В., Fields,С.G., J. Am. Chem. Soc., 1991, 113, 4202-4207). После разработки различных линкеров, которые высвобождают С-концевую карбоксигруппу в слабокислой среде, без воздействия на лабильные в кислой среде защитные группы функциональных групп в боковой цепи (цепях), достигнут значительный прогресс в синтезе защищенных пептидных фрагментов. Полученный из 2-метокси-4-гидроксибензилового спирта линкер (линкер SasrinR, Mergler et al., Tetrahedron Lett., 1988, 29 4005-4008) отщепляется с помощью разбавленной трифторуксусной кислоты(0,5-1% ТФК в ДХМ) и стабилен при условиях, использующихся для удаления защитной группы Fmoc при синтезе пептидов, и дополнительные защитные группы на основе Boc/tBu совместимы с этой схемой защиты. Другие линкеры, которые являются подходящими для способов, соответствующих настоящему изобретению, включают лабильный в суперкислоте 4-(2,4-диметоксифенилгидроксиметил)феноксильный линкер (линкер Rink, Rink, H. Tetrahedron Lett., 1987, 28, 3787-3790), когда для удаления пептида требуется 10% уксусная кислота в ДХМ или 0,2% трифторуксусная кислота в ДХМ; полученный из 4-(4-гидроксиметил-3-метоксифенокси)масляной кислоты линкер (линкер НМРВ, FlorsheimerRiniker,Peptides, 1991, 1990, 131) который также отщепляется с помощью 1% ТФК/ДХМ с получением пептидного фрагмента, содержащего все лабильные в кислой среде защитные группы боковой цепи; и, в дополнение, 2-хлортритилхлоридный линкер (Barlos et al., Tetrahedron Lett., 1989, 30, 3943-3946), который позволяет отщепить пептид с использованием смеси ледяная уксусная кислота/трифторэтанол/ДХМ (1:2:7) в течение 30 мин. Защитными группами, подходящими для аминокислот и, соответственно, для их остатков, являются, например, для аминогруппы (содержащейся, например, также в боковой цепи лизина) для карбоксигруппы (содержащейся, например, также в боковой цепи аспарагиновой или глутаминовой кислоты) путем превращения в эфиры со спиртами для гуанидиновой группы (содержащейся, например, в боковой цепи аргинина) для гидроксигруппы (содержащейся, например, в боковой цепи треонина и серина) Защищенные с помощью 9-флуоренилметоксикарбонила (Fmoc) производные аминокислот предпочтительно используют в качестве структурных фрагментов для получения связанных с матрицей-шпилечных петлевых миметиков формулы (I). Для удаления защитной группы, т.е. отщепления группыFmoc, можно использовать 20% пиперидина в ДМФ или 2% DBU/2% пиперидина в ДМФ. Количество реагента, т.е. производного аминокислоты, обычно составляет 1-20 экв. в расчете на количество миллиэквивалентов на 1 г (мэкв./г) функционализированной твердой подложки (обычно 0,1-2,85 мэкв./г для полистирольных смол), в исходном состоянии отвешенных в пробирку для проведения реакций. При необходимости для доведения реакции до конца за разумное время можно использовать дополнительное количество эквивалентов реагентов. Пробирки для проведения реакций совместно с блоком держателя и коллектором повторно вставляют в блок резервуара и аппаратуру скрепляют друг с другом. Через коллектор подают поток газа для образования регулируемой среды, например азота, аргона, воздуха и т.п. До подачи в коллектор поток газа также можно нагреть или охладить. Нагрев или охлаждение ячеек для проведения реакций с целью проведения необходимых реакций синтеза проводят путем нагревания или охлаждения реакторного блока снаружи смесью изопропанол/твердый диоксид углерода и т.п. Перемешивание проводят путем встряхивания или с помощью магнитной мешалки (в пробирке для проведения реакций). Предпочтительными установками (без наложения ограничений) являются установка Labsource's Combi-chem и синтезатор MultiSyn Tech's-Syro. Для образования амидной связи необходима активация -карбоксигруппы с целью проведения стадии ацилирования. Если активацию проводят с помощью обычно применяющихся карбодиимидов, таких как дициклогексилкарбодиимид (ДЦК, SheehanHess, J. Am. Chem. Soc., 1955, 77, 1067-1068) или диизопропилкарбодиимид (ДИК, Sarantakis et al. Biochem. Biophys. Res. Commun., 1976, 73, 336-342), полученная дициклогексилмочевина и диизопропилмочевина нерастворима и, соответственно, растворима в обычно применяющихся растворителях. В варианте методики с использованием карбодиимида в качестве добавки к смеси для сочетания включают 1-гидроксибензотриазол (HOBt, KnigGeiger, Chem. Ber.,1970, 103, 788-798). HOBt предотвращает дегидратацию, подавляет рацемизацию активированных аминокислот и выступает в качестве катализатора для улучшения протекания медленных реакций сочетания. В качестве реагентов для прямого сочетания используют некоторые фосфониевые реагенты, такие как бензотриазол-1-илокси-трис-(диметиламино)фосфонийгексафторфосфат (ВОР, Castro et al., TetrahedronLett., 1975, 14, 1219-1222; Synthesis, 1976, 751-752) или бензотриазол-1-илокси-триспирролидинофосфонийгексафторфосфат (Ру-ВОР, Coste et al., Tetrahedron Lett. 1990, 31, 205-208), или 2(1 Н-бензотриазол-1-ил-)1,1,3,3-тетраметилуронийтетрафторборат (TBTU) или -гексафторфосфат (HBTU,Knorr et al., Tetrahedron Lett., 1989, 30, 1927-1930); эти фосфониевые реагенты также пригодны для образования in situ эфиров HOBt с защищенными производными аминокислоты. С недавних пор в качестве реагентов для реакции сочетания используют дифеноксифосфорилазид (ДФФА) или O-(7-азабензотриа- 14013814 зол-1-ил)-N,N,N',N'-тетраметилуронийтетрафторборат (TATU) или O-(7-азабензотриазол-1-ил)-N,N,N',N'тетраметилуронийгексафторфосфат (HATU)/7-аза-1-гидроксибензотриазол (HOAt, Carpino et al., Tetrahedron Lett., 1994, 35, 2279-2281). Поскольку важно протекание реакций сочетания с выходом, близким к количественному, желательно получать экспериментальные доказательства завершения реакций. После каждой стадии сочетания можно легко и быстро провести нингидриновую реакцию (Kaiser et al., Anal. Biochemistry, 1970, 34,595), при которой положительная колориметрическая реакция аликвоты связанного со смолой пептида является качественным подтверждением наличия первичного амина. Использование группы Fmoc позволяет спектрофотометрически определить хромофор Fmoc, если он высвобождается с основанием (Meienhofer et al., Int. J. Peptide Protein Res., 1979, 13, 35-42). В каждой пробирке для проведения реакций связанный со смолой промежуточный продукт промывают для удаления избытка сохранившихся реагентов, растворителей и побочных продуктов путем многократной обработки чистым растворителем (растворителями) по одной из двух описанных ниже методик: 1) ячейки проведения реакций заполняют растворителем (предпочтительно 5 мл), пробирки для проведения реакций совместно с блоком держателя и коллектором погружают и перемешивают в течение от 5 до 300 мин, предпочтительно 15 мин, и сливают под действием силы тяжести, а затем через входное отверстие коллектора подают газ под давлением (закрыв выходное отверстие) для удаления растворителя; 2) коллектор удаляют из блока держателя, аликвоты растворителя (предпочтительно 5 мл) подают через верхнюю часть пробирок для проведения реакций и через фильтр сливают под действием силы тяжести в приемный сосуд, такой как пробирка или флакон. Обе указанные выше процедуры промывки повторяют до около 50 раз (предпочтительно примерно 10 раз), контролируя удаление реагента, растворителя и побочного продукта по таким методикам, как ТСХ (тонкослойная хроматография), ГХ (газовая хроматография), или путем исследования промывочных растворов. Описанную выше методику реакции связанного со смолой соединения с реагентом в ячейках для проведения реакций с последующим удалением избытка реагентов, побочных продуктов и растворителей повторяют для каждого последовательного превращения, пока не будет получен конечный связанный со смолой полностью защищенный линейный пептид. До отделения этого связанного со смолой полностью защищенного линейного пептида от твердой подложки при необходимости можно селективно удалить защитные группы от одной или нескольких функциональных групп, содержащихся в молекуле, и соответствующим образом заместить высвобожденную таким образом реакционноспособную группу (группы). Для этого рассматриваемую функциональную группу (группы) необходимо сначала защитить защитной группой, которую можно селективно удалить без воздействия на остальные защитные группы. Alloc (аллилоксикарбонил) является примером такой защитной группы аминогруппы, которую можно селективно удалить, например, с помощью Pd0 и фенилсилана в CH2Cl2 без воздействия на остальные защитные группы, такие как Fmoc, содержащиеся в молекуле. Высвободившуюся таким образом реакционноспособную группу затем можно обработать реагентом, пригодным для введения необходимого заместителя. Так, например, аминогруппу можно ацилировать с помощью ацилирующего реагента, соответствующего вводимому ацильному заместителю. До отделения этого связанного со смолой полностью защищенного линейного пептида от твердой подложки при необходимости также можно образовывать межцепочечные связи между боковыми цепями подходящих аминокислотных остатков в положениях 2 и 10. Межцепочечные связи и их образование обсуждены выше в связи с пояснениями выше, которые,например, могут представлять собой дисульфидные мостики, образованные цистеиновым и гомоцистеиновыми остатками в противолежащих положениях -цепи; или лактамные мостики, образованные остатками глутаминовой и аспарагиновой кислот, связывающими орнитиновые и, соответственно, лизиновые остатки, или остатками глутаминовой кислоты, связывающими остатки 2,4-диаминомасляной кислоты,расположенными в противолежащих положениях -цепи, путем образования амидной связи. Образование таких межцепочечных связей можно провести по методикам, хорошо известным в данной области техники. Для образования дисульфидных мостиков предпочтительно используют раствор 10 экв. раствора йода в ДМФ или в смеси CH2Cl2/МеОН в течение 1,5 ч и после отфильтровывания раствора йода обработку повторяют в течение еще 3 ч с использованием свежего раствора йода, или в смеси ДМСО (диметилсульфоксид) и уксусной кислоты, значение рН которого доведено до 5-6 с помощью 5% NaHCO3, в течение 4 ч, или в воде, значение рН которой доведено до 8 раствором гидроксида аммония, при перемешивании в течение 24 ч, или в аммонийацетатном буфере, значение рН которого доведено до 8, в присутствии воздуха, или в растворе NMP и три-н-бутилфосфина (предпочтительно 50 экв.). Отделение связанного со смолой полностью защищенного линейного пептида от твердой подложки проводят путем погружения пробирок для проведения реакций совместно с блоком держателя и коллек- 15013814 тором в ячейки для проведения реакций, содержащие раствор отщепляющего реагента (предпочтительно от 3 до 5 мл). Поток газа, регулирование температуры, перемешивание и мониторинг протекания реакции проводят так, как это описано выше и необходимо для проведения реакции отделения. Пробирки для проведения реакций совместно с блоком держателя и коллектором отделяют от блока резервуара и поднимают выше уровня раствора, но ниже верхнего края ячеек для проведения реакций и через входное отверстие коллектора подают газ под давлением (закрыв выходное отверстие) для полного слива раствора конечного продукта в ячейки резервуара. Затем смолу, оставшуюся в пробирках для проведения реакций, от 2 до 5 раз промывают так, как выше, с помощью от 3 до 5 мл подходящего растворителя для экстракции (вымывания) как можно большего количества отделенного продукта. Полученные таким образом растворы продукта объединяют, соблюдая осторожность, чтобы не допустить перекрестного смешивания. Затем отдельные растворы/экстракты обрабатывают так, как это необходимо для выделения конечных соединений. Типичная обработка включает, но не ограничивается только ими, выпаривание,концентрирование, жидкостно/жидкостную экстракцию, подкисление, подщелачивание, нейтрализацию или дополнительные реакции в растворе. Растворы, содержащие производные полностью защищенного линейного пептида, которые отделены от твердой подложки и нейтрализованы основанием, выпаривают. Затем циклизацию проводят в растворах с использованием таких растворителей, как ДХМ, ДМФ, диоксан, ТГФ и т.п. Для циклизации можно использовать различные реагенты реакции сочетания, которые указаны выше. Длительность циклизации составляет примерно 6-48 ч, предпочтительно примерно 16 ч. За протеканием реакции следят,например, с помощью ОФ-ВЭЖХ (высокоэффективная жидкостная хроматография с обращенной фазой). Затем растворитель удаляют выпариванием, производное полностью защищенного линейного пептида растворяют в растворителе, который не смешивается с водой, таком как ДХМ, и для удаления избытка реагента сочетания раствор экстрагируют водой или смесью смешивающихся с водой растворителей. В заключение производное полностью защищенного линейного пептида обрабатывают с помощью 95% ТФК, 2,5% H2O, 2,5% ТИС или другой комбинации поглотителей для проведения отщепления защитных групп. Длительность реакции отщепления обычно составляет от 30 мин до 12 ч, предпочтительно примерно 2,5 ч. Летучие вещества выпаривают досуха и неочищенный пептид и неочищенный пептид растворяют в 20% АсОН в воде и экстрагируют водой и изопропиловым эфиром или другими подходящими для этого растворителями. Водный слой собирают и выпаривают досуха и в качестве конечного продукта получают производное циклического пептида с полностью удаленными защитными группами формулы (I). Альтернативно, отделение, циклизацию и полное отделение полностью защищенного пептида от твердой подложки можно провести вручную в стеклянных сосудах. В зависимости от его чистоты это производное пептида можно использовать непосредственно для биологических исследований или его необходимо дополнительно очистить, например, с помощью препаративной ВЭЖХ. Как отмечено выше, при необходимости после этого можно превратить полученный таким образом продукт с полностью удаленными защитными группами формулы (I) в фармацевтически приемлемую соль или превратить полученную таким образом фармацевтически приемлемую или неприемлемую соль в соответствующее свободное соединение формулы (I) или в другую фармацевтически приемлемую соль. Любую из этих операций можно провести по методикам, хорошо известным в данной области техники. Исходные вещества матриц формулы II, использующиеся в способах, соответствующих настоящему изобретению, вещества для получения исходных веществ и получение исходных веществ и веществ для получения исходных веществ описаны в заявке на международный патент РСТ/ЕР 02/01711 тех же заявителей, опубликованной, как WO 02/070547 А 1.-Шпилечные пептидомиметики, соответствующие настоящему изобретению, можно использовать в самых различных случаях, когда воспалительные заболевания или заболевания легких, или инфекционные или иммунологические заболевания, или сердечно-сосудистые заболевания, или нейродегенеративные заболевания опосредуются активностью сериновой протеазы или обусловлены ее активностью,или когда рак опосредуется активностью сериновой протеазы или обусловлен ее активностью. Для лечения или предупреждения данного нарушения или заболевания, которое поддается лечению ингибиторами протеазы, -шпилечные пептидомиметики можно вводить по отдельности или можно применять в виде подходящего состава совместно с носителями, разбавителями или инертными наполнителями, хорошо известными в данной области техники. При использовании для лечения или предупреждения таких заболеваний, как эмфизема легких,ревматоидный артрит, остеоартрит, атеросклероз, псориаз, кистозный фиброз, рассеянный склероз, респираторный дистресс-синдром взрослых, панкреатит, астма, аллергический ринит, воспалительные дерматозы, рестеноз после ангиопластики, гипертрофия сердца, сердечная недостаточность или рак, такой как, но не ограничиваясь только ими, рак молочной железы или рак, связанный с ангиогенезом или метастазированием, -шпилечные пептидомиметики можно вводить по отдельности, в виде смесей нескольких -шпилечных пептидомиметиков, в комбинации с другими противовоспалительными средствами- 16013814 или противомикробными средствами, или противораковыми средствами и/или комбинации с другими фармацевтически активными средствами. -Шпилечные пептидомиметики можно вводить по отдельности или в виде фармацевтических композиций. Фармацевтические композиции, содержащие -шпилечные пептидомиметики, соответствующие настоящему изобретению, можно приготовить с помощью обычных технологий смешивания, растворения, гранулирования, изготовления таблеток с покрытием, растирания в порошок, эмульгирования, капсулирования, включения или лиофилизации. Фармацевтические композиции можно приготовить обычным образом с использованием одного или большего количества физиологически приемлемых носителей, разбавителей, инертных наполнителей или вспомогательных веществ, которые облегчают переработку активных -шпилечных пептидомиметиков в препараты, которые можно использовать фармацевтически. То, какой препарат является подходящим, зависит от выбранного пути введения. Для местного введения -шпилечные пептидомиметики, соответствующие настоящему изобретению, можно приготовить в виде растворов, гелей, мазей, кремов, суспензий и т.п., хорошо известных в данной области техники. Системные препараты включают предназначенные для введения путем инъекции, например подкожной, внутривенной, внутримышечной, внутриоболочечной или внутрибрюшинной инъекции, а также предназначенные для чрескожного, проводимого через слизистую оболочку, перорального или легочного введения. Для инъекции -шпилечные пептидомиметики, соответствующие настоящему изобретению, можно приготовить в виде надлежащих растворов предпочтительно в физиологически совместимых буферах,таких как раствор Хенка (Hink's solution), раствор Рингера или физиологический солевой раствор. Растворы могут содержать использующиеся для приготовления вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие вещества. Альтернативно, -шпилечные пептидомиметики, соответствующие настоящему изобретению, могут находиться в виде порошка, предназначенного для проводимого перед использованием комбинирования с подходящим разбавителем, например стерильной апирогенной водой. Для введения через слизистые оболочки в препаратах, известных в данной области техники, используют обеспечивающие проницаемость вещества, соответствующие барьеру, проникновение через который необходимо обеспечить. Для перорального введения -шпилечные пептидомиметики, соответствующие настоящему изобретению, можно легко приготовить путем их объединения с фармацевтически приемлемыми носителями,хорошо известными в данной области техники. Такие носители позволяют приготовить -шпилечные пептидомиметики, соответствующие настоящему изобретению, в виде таблеток, пилюль, драже, капсул,жидкостей, гелей, сиропов, взвесей, суспензий и т.п., предназначенных для проглатывания подвергающимися лечению пациентами. Подходящие для пероральных препаратов, таких как, например, порошки,капсулы и таблетки, инертные наполнители включают наполнители, такие как сахара, например лактоза,сахароза, маннит и сорбит; препараты целлюлозы, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовую камедь, метилцеллюлозу, гидроксипропилметилцеллюлозу, натриевую соль карбоксиметилцеллюлозы и/или поливинилпирролидон (ПВП); гранулирующие средства и связывающие вещества. При необходимости можно прибавить вещества, обеспечивающие распадаемость, такие как сшитые поливинилпирролидоны, агар или альгиновую кислоту или ее соль, такую как альгинат натрия. При необходимости с помощью стандартных технологий на твердые дозированные формы можно нанести покрытие из сахара или энтеросолюбильное покрытие. Для приготовления пероральных жидких препаратов, таких как, например, суспензии, эликсиры и растворы, подходящие носители, разбавители или инертные наполнители включат воду, гликоли, масла,спирты и т.п. Кроме того, можно прибавить ароматизаторы, консерванты, красители и т.п. Для трансбуккального введения композиция может находится в форме таблеток, пастилок и т.п.,приготовленных обычным образом. Для введения путем ингаляции -шпилечные пептидомиметики, соответствующие настоящему изобретению, обычно готовят в виде аэрозолей, распыляемых из находящихся под давлением упаковок или распылителя с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением разовую дозу можно подавать с помощью клапана, подающего отмеренное количество. Можно изготовить капсулы и катриджи, например, изготовленные из желатина, предназначенные для использования в ингаляторе или аппарате для вдувания порошка, содержащие порошкообразную смесь -шпилечных пептидомиметиков, соответствующих настоящему изобретению, и подходящей порошкообразной основы,такой как лактоза или крахмал. Соединения также можно приготовить в виде композиций для ректального или вагинального введения, таких как суппозитории, совместно с подходящей основой для суппозиториев, такой как масло какао или другие глицериды. В дополнение к описанным выше композициям -шпилечные пептидомиметики, соответствующие- 17013814 настоящему изобретению, также можно приготовить в виде препаратов-депо (depot preparations). Такие препараты длительного действия можно вводить путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Для приготовления таких препаратов-депо-шпилечные пептидомиметики, соответствующие настоящему изобретению, можно приготовить вместе с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в подходящем масле) или ионообменными смолами, или в виде умеренно растворимых солей. Кроме того, можно использовать другие фармацевтические системы доставки, такие как липосомы и эмульсии, хорошо известные в данной области техники. Также можно использовать некоторые органические растворители, такие как диметилсульфоксид. Кроме того, -шпилечные пептидомиметики, соответствующие настоящему изобретению, можно вводить с использованием систем пролонгированного действия, таких как полупроницаемые матрицы из твердых полимеров, содержащие лекарственное средство. Имеются различные материалы пролонгированного действия, которые хорошо известны специалистам в данной области техники. В зависимости от своей химической природы капсулы пролонгированного действия выделяют соединения в течение от нескольких недель до более 100 дней. В зависимости от химической природы и биологической стабильности лекарственного средства можно использовать дополнительные стратегии или стабилизацию белка. Поскольку -шпилечные пептидомиметики, соответствующие настоящему изобретению, могут содержать заряженные остатки, их можно включать в любые описанные выше препараты сами по себе или в виде фармацевтически приемлемых солей. Фармацевтически приемлемые соли обычно лучше растворимы в водных и других протонных растворителях, чем соответствующие свободные формы.-Шпилечные пептидомиметики, соответствующие настоящему изобретению, или их композиции обычно используют в количествах, эффективных для достижения поставленной цели. Следует понимать,что использующееся количество зависит от конкретного случая применения. Для местного введения с целью лечения или предупреждения заболеваний, которые поддаются лечению -шпилечными пептидомиметиками, терапевтически эффективную дозу можно определить с использованием, например, исследований in vitro, описанных в примерах. Лечение можно проводить, когда заболевание проявляется или даже когда оно не проявляется. Специалист с общей подготовкой в данной области техники будет способен без проведения чрезмерно большого количества экспериментов определить терапевтически эффективные количества, необходимые для лечения местных заболеваний. Для системного введения терапевтически эффективную дозу можно определить заранее по данным исследований in vitro. Например, дозу можно приготовить для подопытных животных для определения диапазона концентраций циркулирующего в кровотоке -шпилечного пептидомиметика с использованием значения IC50, определенного для клеточной культуры. Такую информацию можно использовать для более точного определения доз, подходящих для людей. Начальные дозы также можно определить по данным исследований in vivo, например, на подопытных животных, по методикам, которые хорошо известны в данной области техники. Специалист с общей подготовкой в данной области техники может легко оптимизировать введение людям на основании данных, полученных для животных. Дозы для использования в качестве агентов, ингибирующих сериновую протеазу, можно подобрать индивидуально с обеспечением в плазме содержания -шпилечных пептидомиметиков, соответствующих настоящему изобретению, которое достаточно для поддержания терапевтического эффекта. Терапевтически эффективные содержания в сыворотке можно обеспечить путем введения нескольких доз ежедневно. В случае местного введения или селективного потребления эффективная локальная концентрация-шпилечных пептидомиметиков, соответствующих настоящему изобретению, может не быть связана с концентрацией в плазме. Специалист с общей подготовкой в данной области техники должен быть способен без проведения чрезмерно большого количества экспериментов оптимизировать терапевтически эффективные локальные дозы. Количество вводимых -шпилечных пептидомиметиков, разумеется, зависит от подвергающегося лечению субъекта, массы субъекта, тяжести заболевания, пути введения и решения лечащего врача. Обычно терапевтически эффективная доза -шпилечных пептидомиметиков, описанных в настоящем изобретении, обеспечит благоприятное терапевтическое воздействие и не приведет к существенной токсичности. Токсичность -шпилечных пептидомиметиков, соответствующих настоящему изобретению, можно определить с помощью стандартных фармацевтических процедур на клеточных культурах или подопытных животных, например, путем определения LD50 (дозы, летальной для 50% популяции) илиLD100(дозы, летальной для 100% популяции). Отношение доз, приводящих к токсическому и терапевтическому воздействию, называется терапевтическим индексом. Предпочтительными являются соединения, обладающие большим терапевтическим индексом. Данные, полученные с помощью клеточных культур или подопытных животных, можно использовать для определения диапазона доз, которые не являются токсичными при введении людям. Дозы -шпилечных пептидомиметиков, соответствующих- 18013814 настоящему изобретению, предпочтительно находятся в диапазоне находящихся в кровотоке концентраций, которые включают эффективную дозу при слабой токсичности или при отсутствии токсичности. Дозы могут меняться в определенном диапазоне, зависящем от использующихся дозированной формы и пути введения. Точный состав препарата, путь введения и дозу может подобрать каждый врач с учетом состояния пациента (см., например, Fingl et al., 1975, In: The Pharmacological Basis of Therapeutics, Ch.l, p.l). Приведенные ниже примеры более подробно иллюстрируют настоящее изобретение, но никоим образом не ограничивают его объем. В примерах использованы следующие аббревиатуры:HATU: O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (Carpino et al. Tetrahedron Lett., 1994, 35, 2279-2281). Примеры 1. Синтез пептидов. Примеры 1-45, 48, 51-63, 65-67, 70, 71, 74-114, 122-125, 129, 131-162, 164, 166, 167, 169, 170, 172,173, 175, 177-182, 184-189 и 191-196 иллюстрируют изобретение и примеры 46, 47, 49, 50, 64, 68, 69, 72,73, 115-121, 126-128, 130, 163, 165, 168, 171, 174, 183 и 190 представлены ради полноты описания. Связывание первого защищенного аминокислотного остатка со смолой. 0,5 г 2-хлортритилхлоридной смолы (Barlos et al. Tetrahedron Lett., 1989, 30, 3943-3946)(0,83 ммоль/г, 0,415 ммоль) помещают в высушенную колбу. Смолу суспендируют в CH2Cl2 (2,5 мл) и ей дают набухать при комнатной температуре при непрерывном перемешивании в течение 30 мин. Смолу обрабатывают с помощью 0,415 ммоль (1 экв.) первого соответствующим образом защищенного аминокислотного остатка (см. ниже) и 284 мкмоль (4 экв.) диизопропилэтиламина (ДИЭА) в CH2Cl2 (2,5 мл),смесь встряхивают при 25 С в течение 4 ч. Цвет смолы переходит в пурпурный и раствор остается желтоватым. Смолу встряхивают (CH2Cl2 /МеОН/ДИЭА:17/2/1), 30 мл в течение 30 мин; затем промывают в следующем порядке с помощью СH2Cl2 (1), ДМФ (1), CH2Cl2 (1), МеОН (1), CH2Cl2 (1), МеОН (1),CH2Cl2 (2), Et2O (2) и сушат в вакууме в течение 6 ч. Содержание аминокислоты обычно составляет 0,6-0,7 ммоль/г. Получают следующие смолы, содержащие аминокислоты: Fmoc-Pro-2-хлортритиловая смола,Fmoc-Asp (OtBu)-2-хлортритиловая смола, Fmoc-Pro(5RPhe)-2-хлортритиловая смола, Fmoc-Leu-2 хлортритиловая смола, Fmoc-Glu(OtBu)-2-хлортритиловая смола, Fmoc-Asp(OtBu)-2-хлортритиловая смола, Fmoc-Phe-2-хлортритиловая смола, Fmoc-Gln(Trt)-2-хлортритиловая смола, Fmoc-Ser(OtBu)-2 хлортритиловая смола, Fmoc-Val-2-хлортритиловая смола, Fmoc-Thr(OtBu)-2-хлортритиловая смола иFmoc-Ile-2-хлортритиловая смола. Синтез полностью защищенного пептидного фрагмента. Синтез проводят с помощью синтезатора Syro-peptide (Multisyntech) с использованием от 24 до 96 сосудов для проведения реакций. В каждый сосуд помещают 60 мг (масса смолы до введения аминокислоты) указанной выше смолы. Программируют и выполняют следующие циклы реакций: Для добавления каждой аминокислоты повторяют стадии 3-6. После завершения синтеза полностью защищенного пептидного фрагмента выполняют процедуру А или процедуру В, описанные ниже, в зависимости от того, необходимо ли формировать межцепочечные связи (т.е. дисульфидные связи в -цепи).- 19013814 Процедура А. Циклизация и обработка основной цепи циклизованных пептидов. Отщепление полностью защищенного пептидного фрагмента. После завершения синтеза смолу суспендируют в 1 мл (0,39 ммоль) 1% ТФК в CH2Cl2 (об./об.) в течение 3 мин, фильтруют и фильтрат нейтрализуют с помощью 1 мл (1,17 ммоль, 3 экв.) 20% ДИЭА вCH2Cl2 (об./об.). Эту процедуру повторяют дважды, чтобы обеспечить завершение отщепления. Из аликвоты (200 мкл) фильтрата полностью удаляют защитные группы с помощью 0,5 мл смеси для отщепления, содержащей 95% трифторуксусной кислоты (ТФК), 2,5% воды и 2,5% триизопропилсилана (ТИС) и анализируют с помощью ЖХ-МС с обращенной фазой (жидкостная хроматография-масс-спектроскопия) для мониторинга эффективности синтеза линейного пептида. Циклизация линейного пептида. Полностью защищенный линейный пептид растворяют в ДМФ (8 мл, концентрация 10 мг/мл). Прибавляют 2 экв. HATU (0,72 ммоль) в 1 мл ДМФ и 4 экв. ДИЭА (1,44 ммоль) в 1 мл ДМФ и смесь перемешивают при комнатной температуре в течение 16 ч. Летучие вещества выпаривают досуха. Неочищенный циклизованный пептид растворяют в 7 мл CH2Cl2 и трижды экстрагируют с помощью 10% ацетонитрила в воде (4,5 мл). Содержащий CH2Cl2 слой выпаривают досуха. Удаление защитных групп и очистка циклического пептида. Полученный циклический пептид растворяют в 3 мл смеси для отщепления, содержащей 95% трифторуксусной кислоты (ТФК), 2,5% воды и 2,5% триизопропилсилана (ТИС). Смесь выдерживают при 20 С в течение 2,5 ч и затем концентрируют в вакууме. Неочищенный пептид растворяют в 20% АсОН в воде (7 мл) и трижды экстрагируют диизопропиловым эфиром (4 мл). Водный слой собирают, выпаривают досуха и остаток очищают с помощью препаративной ЖХ-МС с обращенной фазой. После лиофилизации получают продукты в виде белых порошков и их анализируют с помощью ЖХ-МС. Данные анализа, включающие чистоту после препаративной ВЭЖХ и ИЭР-МС (масс-спектроскопия с ионизацией электрораспылением), приведены в табл. 1. Методика анализа. Времена удерживания по данным анализа с помощью ВЭЖХ (ВУ, в мин) определяют с помощьюJupiter Proteo 90A, 1502,0 мм, (cod. 00F4396-B0 - Phenomenex) с использованием следующих растворителей А (H2O + 0,1% ТФК), В (CH3CN + 0,1% ТФК) и градиентного режима: 0 мин: 95% А, 5% В; 20 мин: 40% А, 60% В; 21-23 мин: 0% А, 100% В; 23,1-30 мин: 95% А, 5% В. Процедура В. Циклизация и обработка основной цепи циклизованных пептидов, содержащих дисульфидные связи в -цепи. Образование дисульфидной связи в -цепи. После завершения синтеза смоле дают набухать в 3 мл сухого ДМФ в течение 1 ч. Затем в реактор прибавляют 10 экв. раствора йода в ДМФ (6 мл), затем перемешивают в течение 1,5 ч. Смолу отфильтровывают и прибавляют свежий раствор йода (10 экв.) в ДМФ (6 мл), затем перемешивают в течение еще 3 ч. Смолу отфильтровывают и промывают с помощью ДМФ (3) и CH2Cl2 (3). Циклизация основной цепи, отщепление и очистка пептида. После образования дисульфидной связи в -цепи смолу суспендируют в 1 мл (0,39 ммоль) 1% ТФК в CH2Cl2 (об./об.) в течение 3 мин и фильтруют и фильтрат нейтрализуют с помощью 1 мл (1,17 ммоль,3 экв.) 20% ДИЭА в CH2Cl2 (об./об.). Эту процедуру повторяют дважды, чтобы обеспечить завершение отщепления. Смолу промывают с помощью 2 мл CH2Cl2. Содержащий СН 2 С 12 слой выпаривают досуха. Полностью защищенный линейный пептид солюбилизируют в 8 мл сухого ДМФ. Затем к пептиду прибавляют 2 экв. HATU в сухом ДМФ (1 мл) и 4 экв. ДИПЭА в сухом ДМФ (1 мл), затем перемешивают в течение 16 ч. Летучие вещества выпаривают досуха. Неочищенный циклизованный пептид растворяют в 7 мл CH2Cl2 и трижды экстрагируют с помощью 10% ацетонитрила в воде (4,5 мл). СодержащийCH2Cl2 слой выпаривают досуха. Для полного удаления защитных групп из пептида прибавляют 3 мл смеси для отщепления ТФК:ТИС:H2O (95:2,5:2,5) и смесь выдерживают в течение 2,5 ч. Летучие вещества выпаривают досуха и неочищенный пептид растворяют в 20% АсОН в воде (7 мл) и трижды экстрагируют диизопропиловым эфиром (4 мл). Водный слой собирают и выпаривают досуха и остаток очищают с помощью препаративной ЖХ-МС с обращенной фазой. После лиофилизации получают продукты в виде белых порошков и их анализируют с помощью аналитической методики ИЭР-МС, описанной выше. Данные анализа, включающие чистоту после препаративной ВЭЖХ и ИЭР-МС, приведены в табл. 1. Соединения примеров 1-45, 52-63, 65-67, 70-71, 75-114, 129, 131-162 и 179-196 приведены в табл. 1. Пептиды синтезируют с использованием в качестве исходной аминокислоты Pro, которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейные пептиды синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DPro-P11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Соединения примеров 1-6, 9-45, 52-63, 65-67, 70-71, 75-103, 112-114, 129, 131, 133, 136-138, 140-141, 143-146, 148-153, 155,157-162 и 179-196 отщепляют от смолы, вводят в реакцию образования дисульфидных мостиков, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Соединения примеров 82,- 20013814 123, 149, 159, 161 и 178 отщепляют от смолы так, как указано в процедуре В. Дисульфидные мостики образуют по следующей процедуре: Неочищенный продукт солюбилизируют в аммонийацетатном буфере 0,1 М (рН доводят до 8)(концентрация: 1 мг неочищенного продукта в 1 мл). Смесь перемешивают при комнатной температуре в присутствии воздуха. За протеканием реакции следят с помощью ЖХ-МС с обращенной фазой. После завершения реакции раствор выпаривают досуха и остаток очищают с помощью препаративной ЖХ-МС с обращенной фазой. Циклизацию основной цепи проводят так, как указано в процедуре А. Удаление защитных групп проводят по следующей методике: Для полного удаления защитных групп из пептида прибавляют 5 мл смеси для отщепления ТФК:H2O:фенол:тиоанизол:этандитиол (82,5:5:5:5:2,5) и смесь выдерживают в течение 5 ч при комнатной температуре. Петид осаждают путем прибавления холодного диэтилового эфира (10 мл). После центрифугирования надосадочную фазу удаляют. Осадок трижды промывают с помощью 5 мл диэтилового эфира и очищают с помощью препаративной ЖХ-МС с обращенной фазой. После лиофилизации получают продукты в виде белых порошков и их анализируют с помощью аналитической методики ИЭР-МС, описанной выше. Соединения примеров 7, 8, 104-111, 132, 134, 135, 139, 142, 147, 154 и 156 отщепляют от смолы,циклизуют, удаляют защитные группы и очищают так, как указано в процедуре А. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике (далее аббревиатура прим. означает "соединение примера") прим. 1 (15,37), прим. 2 (11,54), прим. 3 (7,82), прим. 4 (8,62), прим. 5 (16,51), прим. 6 (13,67), прим. 7 (3,61), прим. 8 (4,11), прим. 9 (5,82), прим. 10 (7,98), прим. 11 (8,38), прим. 12 (6,80), прим. 13 (7,41),прим. 14 (6,20), прим. 15 (8,68), прим. 16 (9,82), прим. 17 (5,59), прим. 20 (7,32), прим. 21 (8,66), прим. 22(10,74), прим. 196 (9,94). Соединение примера 46 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro, которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DAsp(OtBu)-P11-P10-P9-P8-P7-P6-P5-P4-P3-P2P1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 46 (8,94). Соединение примера 47 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Asp,которую прививают к смоле. Исходной смолой является Fmoc-Asp(OtBu)-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Asp(OtBu)-DPro-P11-P10-P9-P8-P7-P6-P5P4-P3-P2-P1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 47 (7,29).Pro(5RPhe), которую прививают к смоле. Исходной смолой является Fmoc-Pro(5RPhe)-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro(5RPhe)-DPro-Р 11-Р 10-Р 9-Р 8 Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют,удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 48 (10,07). Соединение примера 49 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro, которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DAla-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 49 (8,09). Соединение примера 50 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro, которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DIle-P11-P10-P9-P8-P7-P6-P5-P4-P3-P2-P1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 50 (9,78). Соединение примера 51 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Leu,которую прививают к смоле. Исходной смолой является Fmoc-Leu-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Leu-DPro-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 51 (8,94). Соединение примера 64 приведено в табл. 7. Пептид синтезируют, начиная с аминокислоты Glu, которую прививают к смоле. Исходной смолой является Fmoc-Glu(OtBut)-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Glu(OtBu)-DPro-P11-P10-P9-P8-P7-P6-P5P4-P3-P2-P1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 64 (13,17). Соединение примера 68 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Asp,которую прививают к смоле. Исходной смолой является Fmoc-Asp(OtBu)-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Asp(OtBu)-DAla-P11-P10-P9-P8-P7-P6-P5P4-P3-P2-P1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 68 (12,44). Соединение примера 69 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro, которую прививают к смоле. Исходной смолой является Frnoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола Pro-DAsn(Trt)-P11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 69 (12,97). Соединение примера 72 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro, которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DThr(OtBu)-P11-P10-P9-P8-P7-P6-P5-P4-P3-P2-P1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В.- 22013814 Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 72 (13,34). Соединение примера 73 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro, которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DIle-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 73 (9,78). Соединение примера 74 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Leu,которую прививают к смоле. Исходной смолой является Fmoc-Leu-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Leu-DPro-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 74 (8,94). Соединение примера 115 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro,которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DAsp(OtBu)-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4 Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 115 (4,82). Соединение примера 116 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro,которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DPhe-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 116 (5,98). Соединение примера 117 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro,которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DArg(Trt)-P11-P10-P9-P8-P7-P6-P5-P4-P3P2-P1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 117 (4,48). Соединение примера 118 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro,которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DSer(OtBu)-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3 Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 118 (4,73). Соединение примера 119 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro,которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DVal-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 119 (5,47). Соединение примера 120 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro,которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DPip-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1.- 23013814 Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше градиентной методике 1: прим. 120 (5,48). Соединение примера 121 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Asp,которую прививают к смоле. Исходной смолой является Fmoc-Asp(OtBu)-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Asp(OtBu)-DPro-Р 11-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6 Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 121 (4,56). Соединения примеров 122 и 167 приведены в табл. 1. Пептиды синтезируют, начиная с аминокислоты Phe, которую прививают к смоле. Исходной смолой является Fmoc-Phe-2-хлортритильная смола,которую получают так, как описано выше. Линейные пептиды синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Phe-DPro-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5 Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 122 (5,75); 167 (5,75). Соединения примеров 123, 164, 169, 170, 172, 173, 175, 177 и 178 приведены в табл. 1. Пептиды синтезируют, начиная с аминокислоты Gln, которую прививают к смоле. Исходной смолой являетсяFmoc-Gln(Trt)-2-хлортритильная смола, которую получают так, как описано выше. Линейные пептиды синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Gln(Trt)-DPro-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 123 (4,35), 164 (13,20), 169 (16,81), 170 (14,57), 172 (16,78), 173 (13,57), 175 (15,94), 177 (16,78), 178(17,45). Соединение примера 124 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Ser,которую прививают к смоле. Исходной смолой является Fmoc-Ser(OtBu)-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Ser(OtBu)-DPro-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5 Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 124 (4,46). Соединение примера 125 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Val,которую прививают к смоле. Исходной смолой является Fmoc-Val-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Val-DPro-Р 11-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3 Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 125 (18,42). Соединение примера 126 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Thr,которую прививают к смоле. Исходной смолой является Fmoc-Thr(OtBu)-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Thr(OtBu)-DThr(OtBu)-P11-P10-P9-P8-P7P6-P5-P4-P3-P2-P1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют,удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 126 (4,35). Соединения примеров 127, 163, 165 и 174 приведены в табл. 1. Пептиды синтезируют, начиная с аминокислоты Glu, которую прививают к смоле. Исходной смолой является Fmoc-Glu(OtBu)-2 хлортритильная смола, которую получают так, как описано выше. Линейные пептиды синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Glu(OtBu)DLys(Вос)-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 127 (4,11), 163 (14,93), 165 (14,40), 174 (12,73). Соединение примера 128 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Thr,- 24013814 которую прививают к смоле. Исходной смолой является Fmoc-Thr(OtBu)-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Thr(OtBu)-DPhe-Р 11-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6 Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше градиентной методике 1: прим. 128 (5,26). Соединение примера 130 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Pro,которую прививают к смоле. Исходной смолой является Fmoc-Pro-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Pro-DAla-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 130 (14,79). Соединение примера 166 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Не,которую прививают к смоле. Исходной смолой является Fmoc-Ile-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Ile-DPhe-Р 11-Р 10-Р 9-Р 8-Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 166 (16,80). Соединение примера 168 приведено в табл. 1. Пептид синтезируют, начиная с аминокислоты Asp,которую прививают к смоле. Исходной смолой является Fmoc-Asp(OtBu)-2-хлортритильная смола, которую получают так, как описано выше. Линейный пептид синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Asp(OtBu)-DPro-P11-P10-P9-P8-P7-P6-P5P4-P3-P2-P1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют, удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 168 (4,56). Соединения примеров 171 и 176 приведены в табл. 1. Пептиды синтезируют, начиная с аминокислоты Gln, которую прививают к смоле. Исходной смолой является Fmoc-Gln(Trt)-2-хлортритильная смола, которую получают так, как описано выше. Линейные пептиды синтезируют на твердой подложке по описанной выше методике в следующей последовательности: Смола-Gln(Trt)-DGln(Trt)-Р 11-Р 10-Р 9-Р 8 Р 7-Р 6-Р 5-Р 4-Р 3-Р 2-Р 1. Затем образуют дисульфидный мостик и пептид отщепляют от смолы, циклизуют,удаляют защитные группы и очищают так, как указано в процедуре В. Время удерживания для ВЭЖХ (мин) определяют по описанной выше аналитической методике: прим. 171 (15,40), 176 (13,67). Таблица 1 Примеры а) Чистота %-чистота соединения после препаративной ВЭЖХ Cys в положениях 2 и 10 в соединениях примеров 1-6, 9-103,112-131, 133, 136-138, 140-141, 143-146, 148-153, 155, 157-196 образует дисульфидный мостик. 2. Биологические методика. 2.1. Приготовление образцов пептидов. Лиофилизированные пептиды отвешивают на микровесах (Mettler MT5) и растворяют в стерильной воде до конечной концентрации, равной 1 мМ, если не указано иное. Исходные растворы держат при+4 С в защищенном от воздействия света месте. 2.2. Исследования ферментов. Характеристики ферментов и субстратов приведены в табл. 2. Кинетические исследования проводят при полном объеме реакционной смеси, равном 100 мкл, в- 27013814 плоскодонных 96-луночных планшетах (Greiner) на устройстве считывания планшетов Genios (Tecan). Фермент объединяют с пептидами (ингибиторами) в буфере, содержащем 100 мМ HEPES (N-2 гидроксиэтилпиперазин-N-2-этансульфоновая кислота) (рН 7,5), 50 мМ CaCl2, 0,025% Tween-20,5% ДМСО и 1 мМ субстрата. Скорость гидролиза субстрата измеряют путем мониторинга изменения поглощения при 405 нм в течение 30 мин для проверки линейности кривой реакции. Для всех расчетов используют среднюю скорость в промежутке от минуты 1 до минуты 10. Начальные значения вычитаемого фона, средней скорости, двойного усреднения и выраженного в % ингибирования проводят с использованием программного обеспечения Magellan (version 5), выпускающегося компанией Tecan. Расчеты IC50% проводят с использованием программного обеспечения Grafit (version 5.0.10), выпускающегося компанией Erithacus Software, путем аппроксимации данных для 6 разных концентраций ингибитора с помощью 4-параметрического уравнения: В этом уравнении s означает коэффициент наклона, х означает концентрации ингибитора и у означает выраженное в % ингибирование при данной концентрации ингибитора. Определение Km/Ki. Значения Km для субстрата сериновой протеазы определяют по зависимости Lineweaver-Burke(Grafit v5). Значения Ki для ингибиторов рассчитывают по формуле Ki=IC50%/(1+([субстрат]/Km. При реакции с ферментом используют увеличивающиеся концентрации субстрата и строят зависимость скорости каждой реакции (ABS/mSec) от концентрации субстрата. Для получения Km и Vmax(вставка) (см. ниже ссылку 1) также строят обратную зависимость (Lineweaver-Burke).
МПК / Метки
МПК: C07K 7/08, C07K 7/64, A61K 38/04, C07K 7/06
Метки: бета-шпилечные, отношении, активностью, ингибирующей, пептидомиметики, связанные, матрицей, протеаз
Код ссылки
<a href="https://eas.patents.su/30-13814-svyazannye-s-matricejj-beta-shpilechnye-peptidomimetiki-s-ingibiruyushhejj-aktivnostyu-v-otnoshenii-proteaz.html" rel="bookmark" title="База патентов Евразийского Союза">Связанные с матрицей бета-шпилечные пептидомиметики с ингибирующей активностью в отношении протеаз</a>
Связанные с матрицей пептидомиметики β-шпилечной структуры с активностью антагонистов cxcr4

Номер патента: 10163
Опубликовано: 30.06.2008
Авторы: Меле Керстин, Димарко Стивен Дж., Обрехт Даниель, Робинсон Джон Энтони, Гомберт Франк, Лудин Кристиан, Хенце Хайко, Романьоли Барбара, Мукерджи Решми, Лочиуро Серджио, Цумбрунн Юрг, Фрийблуд Ян Вим
МПК: C07K 1/04, A61P 31/18, A61K 38/12...
Метки: структуры, активностью, beta;-шпилечной, антагонистов, матрицей, пептидомиметики, связанные, cxcr4
Формула / Реферат:
1. Соединения общей формулы где представляет собой группу, характеризуемую одной из формул A-CO означает DPro, DGln, 4-{2-[2-(метоксиэтокси)этокси]ацетиламино}-DPro или Gly; B-CO означает LPro, LGln, 4-{2-[2-(метоксиэтокси)этокси]ацетиламино}-LPro или Gly; но A-CO и B-CO не могут оба быть Gly; Z означает цепь из n a-аминокислотных остатков, причем n означает целое число 12, 14 или 18, а положения указанных аминокислотных остатков в цепи...
Тиольные производные, обладающие ингибирующей активностью в отношении металлопептидаз.

Номер патента: 991
Опубликовано: 28.08.2000
Авторы: Романьяно Стефано, Пеллачини Франко, Норчини Габриеле, Семераро Клаудио, Фантуччи Марио, Сантанджело Франческо
МПК: A61K 31/425, A61P 9/00, C07D 277/30...
Метки: ингибирующей, тиольные, отношении, активностью, металлопептидаз, обладающие, производные
Формула / Реферат:
1. Соединение формулы где R обозначает меркаптогруппу или группу R4COS, которая превращается в организме в меркаптогруппу; R1 обозначает С2-С4алкильную группу с прямой или разветвленной цепью или арильную или арилалкильную группу, имеющую от 1 до 6 атомов углерода в алкильном фрагменте, где арил обозначает фенил или 5- или 6-членный ароматический гетероцикл с одним или двумя гетероатомами, выбранными из группы, включающей азот, кислород и...
Производные фосфоновой кислоты, обладающие ингибирующей активностью в отношении металлопептидаз

Номер патента: 743
Опубликовано: 28.02.2000
Авторы: Норчини Габриеле, Сантанджело Франческо, Ботта Даниела
МПК: C07K 5/062, A61K 38/05
Метки: металлопептидаз, отношении, производные, фосфоновой, ингибирующей, кислоты, активностью, обладающие
Формула / Реферат:
1. Соединение формулы где R обозначает С1-С6алкильную группу с прямой или с разветвленной цепью, необязательно замещенную одним или несколькими атомами фтора, арильную или арилалкильную группу с 1-6 атомами углерода в алкильном фрагменте, где арил обозначает фенильную, 1-нафтильную или 2-нафтильную группу или 5- или 6-членный ароматический гетероцикл с 1 или 2 гетероатомами, выбранными из группы, включающей азот, кислород и серу,...
Новые соединения, обладающие ингибирующей активностью в отношении натрийзависимого транспортёра

Номер патента: 10299
Опубликовано: 29.08.2008
Авторы: Уита Киитиро, Номура Сумихиро, Каваниси Ийдзи
МПК: C07H 19/06
Метки: соединения, отношении, активностью, натрийзависимого, ингибирующей, обладающие, транспортёра, новые
Формула / Реферат:
1. Соединение формулы где кольцо А представляет собой где R1a, R2a, R3a, R1b, R2b и R3b, каждый независимо, представляет собой атом водорода, атом галогена, гидроксигруппу, алкоксигруппу, алкильную группу, галогеналкильную группу, галогеналкоксигруппу, гидроксиалкильную группу, алкоксиалкильную группу, алкоксиалкоксигруппу, алкенильную группу, алкинильную группу, циклоалкильную группу, циклоалкилиденметильную группу, циклоалкенильную группу,...
Фенил- и пиридил-тетрагидропиридины, обладающие ингибирующей активностью в отношении фактора некроза опухоли (фно)

Номер патента: 4508
Опубликовано: 29.04.2004
Авторы: Кардамоне Розанна, Бурри Бернар, Барони Марко, Гуцци Умберто, Казелла Пьер
МПК: A61K 31/4709, C07D 401/06, A61P 9/00...
Метки: активностью, отношении, фно, ингибирующей, фактора, фенил, опухоли, пиридил-тетрагидропиридины, некроза, обладающие
Формула / Реферат:
1.Соединение формулы (I) где X представляет собой N или CH; R1 представляет собой атом водорода или галогена или группу CF3; R2 и R3 независимо представляют собой атом водорода или метильную группу; n равно 0 или 1; A представляет собой группу формулы (a) или (b) где R4 представляет собой атом водорода или галогена, (C1-C4)алкильную группу, группу CF3, аминогруппу, моно(C1-C4)алкиламиногруппу или ди(C1-C4)алкиламиногруппу; R5 представляет...
Предыдущий патент: Применение иматиниба для лечения гепатита с
Следующий патент: Дисперсия порошка меламина, способ ее получения и ее применение
Случайный патент: Датчик