Замещённые индолильные алкиламинопроизводные в качестве новых ингибиторов гистондеацетилазы
Номер патента: 10652
Опубликовано: 30.10.2008
Авторы: Вердонк Марк Густаф Селин, Тен Холтэ Петер, Пилатт Изабелль Ноэлль Констанс, Артс Янине, Ру Брюно, Ван Эмелен Кристоф, Анжибо Патрик Рене







Формула / Реферат
1. Соединение формулы (I)

его N-оксидные формы, фармацевтически приемлемые аддитивные соли и их стереохимически изомерные формы, где
каждый из n представляет собой целое число со значением 0, 1 или 2 и, когда n равно 0, тогда подразумевается прямая связь;
каждый из m представляет собой целое число со значением 1 или 2;
каждый из X независимо представляет собой N или СН;
каждый из Y независимо представляет собой О, S или NR4; где
каждый из R4 представляет собой водород, С1-6алкил, С1-6алкилоксиС1-6алкил, С3-6циклоалкил, С3-6циклоалкилметил, фенилС1-6алкил, -С(=O)-CHR5R6 или -S(=O)2-N(CH3)2; где
каждый из R5 и R6 независимо представляет собой водород, амино, C1-6алкил или аминоС1-6алкил и,
когда Y представляет собой NR4 и R2 находится в 7-положении индолила, тогда R2 и R4 вместе могут образовывать двухвалентный радикал
-(СН2)2- (а-1) или
-(СН2)3- (а-2);
R1 представляет собой водород, C1-6алкил, гидроксиС1-6алкил, C1-6алкилсульфонил, C1-6алкилкарбонил или моно- или ди(C1-6алкил)аминосульфонил;
R2 представляет собой водород, гидрокси, амино, галоген, C1-6алкил, циано, С2-6алкенил, полигалогенС1-6алкил, нитро, фенил, C1-6алкилкарбонил, гидроксикарбонил, C1-6алкилкарбониламино, C1-6алкилокси или моно- или ди(C1-6алкил)амино;
R3 представляет собой водород, C1-6алкил или C1-6алкилокси и,
когда R2 и R3 представляют собой соседние атомы углерода, они могут образовывать двухвалентный радикал -О-СН2-О-.
2. Соединение по п.1, в котором
каждый из n представляет собой целое число со значением 0 или 1;
каждый из R4 представляет собой водород, C1-6алкил, С1-6алкилоксиС1-6алкил, С3-6циклоалкил или фенилС1-6алкил;
R1 представляет собой водород, С1-6алкил, гидроксиС1-6алкил, C1-6алкилкарбонил или C1-6алкилсульфонил и
R2 представляет собой водород, галоген, C1-6алкил, циано, нитро, полигалогенС1-6алкил или C1-6алкилокси.
3. Соединение по пп.1 и 2, в котором
каждый из n представляет собой целое число со значением 1;
каждый из m представляет собой целое число со значением 1;
каждый из X независимо представляет собой N;
каждый из Y независимо представляет собой NR4;
каждый из R4 представляет собой C1-6алкил;
R1 представляет собой водород;
R2 представляет собой водород или галоген и
R3 представляет собой водород.
4. Соединение по пп.1, 2 и 3, в котором указанное соединение представляет собой соединение ь 1а, соединение ь 30, соединение ь 39 и соединение ь 50.

5. Фармацевтическая композиция, содержащая фармацевтически приемлемые носители и, в качестве активного ингредиента, терапевтически эффективное количество соединения по пп.1-4.
6. Способ получения фармацевтической композиции по п.5, в котором фармацевтически приемлемые носители и соединение по пп.1-4 тщательно смешиваются.
7. Применение соединения по любому из пп.1-4 для производства лекарственного средства для лечения пролиферативных заболеваний.
8. Сочетание противоракового агента и соединения по любому из пп.1-4 в качестве ингибитора HDAC.
9. Способ получения соединения по п.1, включающий взаимодействие промежуточного соединения формулы (II), где Q представляет собой тетрагидропиранилоксиаминокарбонил, обозначенного как промежуточное соединение формулы (II-а), с соответствующей кислотой, такой, например, как трифторуксусная кислота, с получением гидроксамовой кислоты формулы (I)

10. Способ получения соединения по п.1, включающий взаимодействие промежуточного соединения формулы (II), где Q представляет собой C1-6алкилоксикарбонил, обозначенного как промежуточное соединение формулы (II-с), с гидроксиламином в присутствии основания и в соответствующем растворителе с получением гидроксамовой кислоты формулы (I)

11. Соединение формулы (II)

его N-оксидные формы, фармацевтически приемлемые аддитивные соли и их стереохимически изомерные формы, где
каждый из n представляет собой целое число со значением 0, 1 или 2 и, когда n равно 0, тогда подразумевается прямая связь;
каждый из m представляет собой целое число со значением 1 или 2;
каждый из X независимо представляет собой N или СН;
каждый из Y независимо представляет собой О, S или NR4; где
каждый из R4 представляет собой водород, C1-6алкил, С1-6алкилоксиС1-6алкил, С3-6циклоалкил, С3-6циклоалкилметил, фенилС1-6алкил, -С(=O)-CHR5R6 или -S(=O)2-N(CH3)2; где
каждый из R5 и R6 независимо представляет собой водород, амино, C1-6алкил или аминоС1-6алкил и,
когда Y представляет собой NR4 и R2 находится в 7-положении индолила, тогда R2 и R4 вместе могут образовывать двухвалентный радикал
-(СН2)2- (а-1) или
-(СН2)3- (а-2);
R1 представляет собой водород, C1-6алкил, гидроксиС1-6алкил, C1-6алкилсульфонил, C1-6алкилкарбонил или моно- или ди(C1-6алкил)аминосульфонил;
R2 представляет собой водород, гидрокси, амино, галоген, C1-6алкил, циано, С2-6алкенил, полигалогенС1-6алкил, нитро, фенил, C1-6алкилкарбонил, гидроксикарбонил, C1-6алкилкарбониламино, C1-6алкилокси или моно- или ди(C1-6алкил)амино;
R3 представляет собой водород, C1-6алкил или C1-6алкилокси;
когда R2 и R3 представляют собой соседние атомы углерода, они могут образовывать двухвалентный радикал -О-СН2-О- и
Q представляет собой С1-2алкилоксикарбонил, гидроксикарбонил или тетрагидропиранилоксиаминокарбонил.
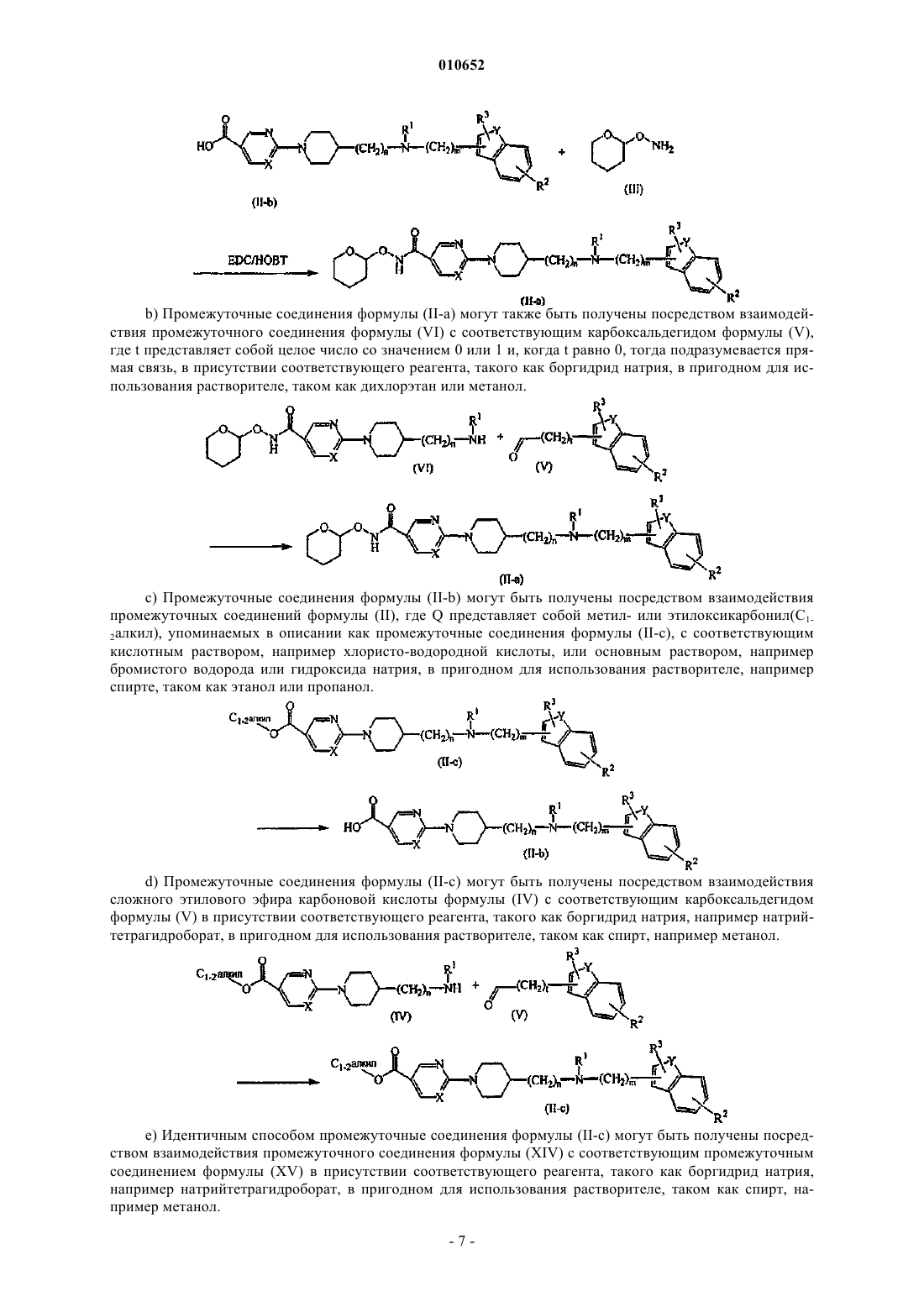
12. Способ получения соединения по п.11, включающий взаимодействие соединения формулы (II), где Q представляет собой гидроксикарбонил, обозначенный как соединение формулы (II-b), с промежуточным соединением формулы (III) в присутствии соответствующих реагентов, таких как моногидрохлорид N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина (EDC) и 1-гидрокси-1H-бензотриазол (НОВТ), с образованием соединения формулы (II-а)

13. Способ получения соединения по п.11, включающий взаимодействие промежуточного соединения формулы (VI) с соответствующим карбоксальдегидом формулы (V), где t представляет собой целое число со значением 0 или 1 и, когда t равно 0, тогда подразумевается прямая связь, в присутствии соответствующего реагента с образованием соединения формулы (II-а)

14. Способ получения соединения по п.11, включающий взаимодействие соединения формулы (II), где Q представляет собой метил- шыш этилоксикарбонил (C1-2алкил), обозначенный как соединение формулы (II-с), с соответствующим кислотным раствором или основным раствором с образованием соединения формулы (II-b)

15. Способ получения соединения по п.11, включающий взаимодействие сложного этилового эфира карбоновой кислоты формулы (IV) с соответствующим карбоксальдегидом формулы (V) в присутствии соответствующего реагента с образованием соединения формулы (II-c)

16. Способ получения соединения по п.11, включающий взаимодействие промежуточного соединения формулы (XIV) с соответствующим промежуточным соединением формулы (XV) в присутствии соответствующего реагента в пригодном для использования растворителе с образованием соединения формулы (II-с)

17. Способ получения соединения по п.11, включающий взаимодействие промежуточного соединения формулы (X) с промежуточным соединением формулы (XI), где W представляет собой соответствующую уходящую группу, такую, например, как галоген, например фтор, хлор, бром или йод, или сульфонилоксирадикал, такой как метилсульфонилокси, с образованием соединения формулы (II-c)

18. Способ получения соединения по п.11, включающий взаимодействие промежуточного соединения формулы (XII) с промежуточным соединением формулы (XIII), где W представляет собой соответствующую уходящую группу, как описано выше, с образованием соединения формулы (II-с)

Текст
010652 Настоящее изобретение относится к соединениям, имеющим ферментативную активность ингибирования гистондеацетилазы (HDAC). Кроме того, оно относится к способам их получения, к композициям, содержащим их, а также к их применению как in vitro, так и in vivo для ингибирования HDAC, а также в качестве лекарственного средства, например в качестве лекарственного средства для ингибирования пролиферативных состояний, таких как рак и псориаз. Нуклеарные гистоны известны в качестве интегральных и динамических компонентов механизма,ответственного за регуляцию транскрипции гена и другие процессы, кодируемые ДНК, такие как репликация, репарация, рекомбинация и разделение хромосом. Они являются субъектом посттрансляционных модификаций, включая ацетилирование, фосфорилирование, метилирование, убиквитинирование и АТФ-рибозилирование. Гистондеацетилаза(ы), упоминаемые в описании как "HDAC", представляют собой ферменты, которые катализируют удаление ацетильной модификации на лизиновых остатках белков, включая ядерные нуклеосомальные гистоны Н 2 А, Н 2 В, Н 3 и Н 4. Вместе с гистонацетилтрансферазой(ами), упоминаемой как "HAT", HDAC регулируют уровень ацетилирования гистонов. Баланс ацетилирования нуклеосомальных гистонов играет важную роль в транскрипции многих генов. Гипоацетилирование гистонов ассоциируется с конденсированной структурой хроматина, приводящей к подавлению транскрипции гена, в то время как ацетилированные гистоны ассоциируются с более открытой структурой хроматина и активированием транскрипции. Описаны одиннадцать структурно родственных HDAC, и они попадают в два класса. Класс I HDAC состоит из HDAC 1, 2, 3, 8 и 11, в то время как класс II HDAC состоит из HDAC 4, 5, 6, 7, 9 и 10. Элементы третьего класса HDAC являются структурно несвязанными с HDAC класса I и класса II. HDAC классаI/II работают посредством цинкзависимых механизмов, в то время как HDAC класса III являются NADзависимыми. В дополнение к гистонам, другие белки также представляют собой субстрат для ацетилирования в конкретных факторах транскрипции, таких как р 53, GATA-1 и E2F; ядерных рецепторах, таких как рецептор глюкокортикоидов, тироидные рецепторы, рецепторы эстрогенов; и в белках, регулирующих клеточный цикл, таких как pRb. Ацетилирование белков связано со стабилизацией белка, такой как стабилизация р 53, рекрутингом кофакторов и увеличением связывания ДНК. р 53 представляет собой супрессор опухоли, который может индуцировать приостановку клеточного цикла или апоптоз в ответ на различные сигналы стресса, такие как повреждение ДНК. Главной мишенью для индуцированной р 53 приостановки клеточного цикла, видимо, является ген р 21. После его активирования посредством р 53 р 21 идентифицируется посредством его ассоциации с комплексами циклин/циклинзависимая киназа, приводящей к приостановке клеточного цикла в фазах как G1, так и G2, его положительной регуляции во время сенесценции и к его взаимодействию с ядерным антигеном пролиферирующих клеток. Исследование ингибиторов HDAC показывает, что они играют важную роль в приостановке клеточного цикла, клеточной дифференциации, апоптозе и обращении трансформированных фенотипов. Ингибитор трихостатин A (TSA), например, вызывает приостановку клеточного цикла в фазах какG1, так и в G2, обращает трансформированный фенотип различных линий клеток и индуцирует дифференциацию клеток лейкемии Френда и других. TSA (и субероиланилид гидроксамовой кислоты SAHA),как сообщается, ингибируют рост клеток, индуцируют конечную дифференциацию и предотвращают образование опухолей у мышей (Finnin et al., Nature, 401: 188-193, 1999). Трихостатин А также, как сообщается, является пригодным для лечения фиброза, например фиброза печени и цирроза печени (Geerts et al., заявка на европейский патент ЕР 0827742, опубликованная 11 марта 1998 года). Фармакофор для ингибиторов HDAC состоит из домена связывания металла, который взаимодействует с цинксодержащим активным сайтом HDAC, домена линкера и поверхностного распознающего домена или кэпирующего участка, который взаимодействует с остатками на вспомогательном участке активного сайта. Ингибиторы HDAC также, как сообщается, индуцируют экспрессию гена р 21. Транскрипционное активирование гена р 21 посредством этих ингибиторов промотируется модификацией хроматина после ацетилирования гистонов Н 3 и Н 4 в промоторной области р 21. Это активирование р 21 осуществляется способом, не зависимым от р 53, и таким образом ингибиторы HDAC работают в клетках с мутировавшими генами р 53, отличительным признаком многочисленных опухолей. В дополнение к этому, ингибиторы HDAC могут иметь опосредованные активности, такие как аугментация иммунных реакций хозяина и ингибирование ангиогенеза опухолей, и таким образом могут подавлять рост первичных опухолей и блокировать метастазы (Mai et al., Medicinal Research Reviews, 25: 261-309). С учетом всего изложенного выше, ингибиторы HDAC могут иметь большой потенциал при лечении пролиферативных заболеваний или состояний клеток, включая опухоли с мутировавшими генами р 53. Заявка на европейский патент ЕР 1472216, опубликованная 14 августа 2003 года, описывает бициклические гидроксаматы в качестве ингибиторов гистондеацетилазы. Заявки на европейский патент ЕР 1485099, ЕР 1485348, ЕР 1485353, ЕР 1485354, ЕР 1485364,-1 010652 ЕР 1485365, ЕР 1485370, ЕР 1485378, опубликованные 18 сентября 2003 года, среди прочего, описывают замещенные пиперазинилпиримидинилгидроксамовые кислоты в качестве ингибиторов гистондеацетилаз, кроме того, ЕР 1485365 описывает R306465. Заявка на европейский патент ЕР 1492534, опубликованная 9 октября 2003 года, описывает соединения карбаминовой кислоты, содержащие пиперазиновую связь, в качестве ингибиторов HDAC. Заявка на европейский патент ЕР 1495002, опубликованная 23 октября 2003 года, описывает замещенные пиперазинилфенилбензамидные соединения в качестве ингибиторов гистондеацетилазы. Заявка на международный патент WO04/009536, опубликованная 29 января 2004 года, описывает производные, содержащие алкильный линкер между арильной группой и гидроксаматом, в качестве ингибиторов гистондеацетилазы. Заявка на европейский патент ЕР 1525199, опубликованная 12 февраля 2004 года, описывает (гетеро)арилалкенилзамещенные бициклические гидроксаматы в качестве ингибиторов гистондеацетилазы. Заявка на международный патент WO04/063146, опубликованная 29 июля 2004 года, описывает производные N-гидроксибензамидных производных с противовоспалительной и противоопухолевой активностью. Заявка на международный патент WO04/063169, опубликованная 29 июля 2004 года, описывает замещенные арилгидроксаматные производные в качестве ингибиторов гистондеацетилазы. Заявка на международный патент WO04/072047, опубликованная 26 августа 2004 года, описывает индолы, бензимидазолы и нафтимидазолы в качестве ингибиторов гистондеацетилазы. Заявка на международный патент WO04/082638, опубликованная 30 сентября 2004 года, описывает гидроксаматы, связанные с неароматическими гетероциклическими кольцевыми системами, в качестве ингибиторов гистондеацетилазы. Заявка на международный патент WO04/092115, опубликованная на 28 октября 2004 года, описывает гидроксаматные производные в качестве ингибиторов гистондеацетилазы. Заявка на международный патент WO05/028447, опубликованная 31 марта 2005 года, описывает бензимидазолы в качестве ингибиторов гистондеацетилазы. Заявки на международный патент WO05/030704 и WO05/030705, опубликованные 7 апреля 2005 года, описывает бензамиды в качестве ингибиторов гистондеацетилазы. Заявка на международный патент WO05/040101, опубликованная 6 мая 2005 года, описывает связанные с ацилмочевиной и связанные с сульфонилмочевиной гидроксаматы в качестве ингибиторов гистондеацетилазы. Заявка на международный патент WO05/040161 также опубликованная 6 мая 2005 года, описывает биарилсвязанные гидроксаматы в качестве ингибиторов гистондеацетилазы. Соединения по настоящему изобретению отличаются от соединений, известных из литературы,структурой, их фармакологической активностью и/или силой фармакологического действия. Проблема, которая должна решаться, представляет собой создание ингибиторов гистондеацетилазы с высокой ферментативной и клеточной активностью, которые имеют повышенную биологическую доступность и/или сильное биологическое действие in vivo. Новые соединения по настоящему изобретению решают описанную выше проблему. Соединения по настоящему изобретению показывают превосходную ферментативную и клеточную активность ингибирования гистондеацетилазы. Они имеют высокую способность к активированию гена р 21 как на клеточном уровне, так и на уровне in vivo. Они имеют желаемый фармакокинетический профиль и низкое сродство к ферментам Р 450, что понижает риск вредного взаимодействия лекарственных средств, также делая возможным более широкие пределы безопасности. Преимущественными особенностями настоящих соединений являются метаболическая стабильность, растворимость и/или способность индуцирования р 21. Более конкретно, соединения по настоящему изобретению имеют увеличенное время полураспада в гепатоцитах крыс, имеют повышенную растворимость/стабильность в водных растворах и/или имеют повышенные способности индуцирования промотора р 21 in vivo. Настоящее изобретение относится к соединениям формулы (I) к их N-оксидным формам, фармацевтически приемлемым аддитивным солям и их стереохимически изомерным формам, где каждый из n представляет собой целое число со значением 0, 1 или 2 и, когда n равно 0, тогда подразумевается прямая связь; каждый из m представляет собой целое число со значением 1 или 2; каждый из X независимо представляет собой N или СН;-2 010652 каждый из Y независимо представляет собой О, S или NR4; где каждый из R4 представляет собой водород, C1-6 алкил, С 1-6 алкилоксиС 1-6 алкил, С 3-6 циклоалкил, С 3-6 циклоалкилметил, фенил С 1-6 алкил, -C(=O)-CHR5R6 или -S(=O)2-N(CH3)2; где каждый из R5 и R6 независимо представляет собой водород, амино, С 1-6 алкил или аминоС 1-6 алкил и,когда Y представляет собой NR4 и R2 находится в 7-положении индолила, тогда R2 и R4 вместе могут образовывать двухвалентный радикалR3 представляет собой водород, C1-6 алкил или C1-6 алкилокси и,когда R2 и R3 представляют собой соседние атомы углерода, они могут образовывать двухвалентный радикал -О-СН 2-О-. Линии, нарисованные в бициклических кольцевых системах от заместителей, показывают, что связи могут присоединяться к любому из соответствующих кольцевых атомов бициклической кольцевой системы. Термин "ингибитор гистондеацетилазы" или "ингибитор деацетилазы гистона" используется для идентификации соединения, которое способно взаимодействовать с деацетилазой гистона и ингибировать ее активность, более конкретно ее ферментативную активность. Ингибирование ферментативной активности гистондеацетилазы означает уменьшение способности гистондеацетилазы к удалению ацетильной группы из гистона. Предпочтительно такое ингибирование является специфичным, т.е. ингибитор гистондеацетилазы уменьшает способность гистондеацетилазы к удалению ацетильной группы с гистона при концентрации, которая ниже, чем концентрация ингибитора, которая требуется для получения какого-либо другого биологического эффекта, не связанного с данным. Как используется в приведенных выше определениях и далее, галоген представляет собой фтор,хлор, бром и йод; С 1-2 алкил означает насыщенные углеводородные радикалы с прямой цепью, имеющие 1 или 2 атома углерода, такие, например, как метил или этил; С 1-6 алкил определяет С 1-2 алкил и насыщенные углеводородные радикалы с прямой и разветвленной цепью, имеющие от 3 до 6 атомов углерода, таких, например, как пропил, бутил, 1-метилэтил, 2-метилпропил, пентил, 2-метилбутил, гексил, 2-метилпентил и т.п.; и полигалогенС 1-6 алкил определяет С 1-6 алкил, содержащий три идентичных или различных галогеновых заместителя, например трифторметил; С 2-6 алкенил определяет углеводородные радикалы с прямой и разветвленной цепью, содержащие одну двойную связь и имеющие от 2 до 6 атомов углерода,такие, например, как этенил, 2-пропенил, 3-бутенил, 2-пентенил, 3-пентенил, 3-метил-2-бутенил и т.п.; С 3-6 циклоалкил включает циклические углеводородные группы, имеющие от 3 до 6 атомов углерода, такие как циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексил и т.п. Фармацевтически приемлемые аддитивные соли охватывают фармацевтически приемлемые аддитивные соли кислот и фармацевтически приемлемые аддитивные соли оснований. Фармацевтически приемлемые аддитивные соли кислот, как рассмотрено выше, как предполагается, включают терапевтически активные нетоксичные формы аддитивных солей кислот, которые способны формировать соединения формулы (I). Соединения формулы (I), которые имеют основные свойства, могут преобразовываться в их фармацевтически приемлемые аддитивные соли кислот посредством обработки указанной основной формы соответствующей кислотой. Соответствующие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористо-водородная или бромистоводородная кислота; серная; азотная; фосфорная и т.п. кислоты; или органические кислоты, такие, например, как уксусная, трифторуксусная, пропановая, гидроксиуксусная, молочная, пировиноградная кислота, щавелевая, малоновая, янтарная (т.е. бутанкарбоновая кислота), малеиновая, фумаровая, яблочная,винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и т.п. кислоты. Соединения формулы (I), которые имеют кислотные свойства, могут преобразовываться в их фармацевтически приемлемые аддитивные соли оснований посредством обработки указанной кислотной формы соответствующим органическим или неорганическим основанием. Соответствующие формы основных солей включают, например, соли аммония, соли щелочных и щелочно-земельных металлов, например соли лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, например соли бензатина, N-метил-D-глюкамина, гидрабамина, и соли с аминокислотами, такими, например, как аргинин, лизин и т.п. Термин "аддитивные соли кислот или оснований" также включает гидраты и формы добавления растворителя, которые способны образовывать соединения формулы (I). Примеры таких форм представляют собой, например, гидраты, алкоголяты и т.п. Термин "стереохимические изомерные формы соединений формулы (I)", как используется в описа-3 010652 нии, определяет все возможные соединения, состоящие из одних и тех же атомов, соединенных посредством одной и той же последовательности связей, но имеющие различные трехмерные структуры, которые не являются взаимозаменяемыми, которые могут иметь соединения формулы (I). Если только не рассматривается или не указывается иного, химическое обозначение соединения охватывает смесь всех возможных стереохимически изомерных форм, которые может иметь указанное соединение. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Все стереохимические изомерные формы соединений формулы (I) как в чистой форме, так и в смеси друг с другом, как предполагается, находятся в объеме настоящего изобретения.N-Оксидные формы соединений формулы (I), как предполагается, включают те соединения формулы (I), где 1 или несколько атомов азота окисляются до так называемого N-оксида, в частности те N-оксиды, где N-окисленными являются 1 или несколько атомов азота пиперидина, пиперазина или пиридазинила. Некоторые из соединений формулы (I) могут также существовать в их таутомерных формах. Такие формы, хотя не указываются в явном виде в указанной выше формуле, как предполагается, являются включенными в объем настоящего изобретения. Везде, где он используется далее, термин "соединения формулы (I)", как предполагается, включает также фармацевтически приемлемые аддитивные соли и все стереоизомерные формы. Как используется в описании, термины "гистондеацетилаза" и "HDAC", как предполагается, относятся к любому из семейства ферментов, которые удаляют ацетильные группы из -аминогрупп лизиновых остатков на N-окончании гистона. Если по контексту не указывается иного, термин "гистон", как предполагается, относится к любому белку гистона, включая H1, Н 2 А, Н 2 В, Н 3, Н 4 и Н 5, из любых видов. Белки или генные продукты HDAC человека включают, но не ограничиваясь этим, HDAC-1, HDAC2, HDAC-3, HDAC-4, HDAC-5, HDAC-6, HDAC-7, HDAC-8, HDAC-9, HDAC-10 и HDAC-11. Гистондеацетилаза также может быть получена из источника протозоала или грибков. Первая группа представляющих интерес соединений состоит из тех соединений формулы (I), к которым применимо одно или несколько из следующих ограничений:c) R3 представляет собой водород. Вторая группа представляющих интерес соединений состоит из таких соединений формулы (I), к которым применимо одно или несколько из следующих ограничений:a) каждый из n представляет собой целое число со значением 0 или 1;b) каждый из X независимо представляет собой N;c) каждый из R4 представляет собой водород или С 1-6 алкил;d) R1 представляет собой водород, С 1-6 алкил или гидроксиС 1-6 алкил илиe) R2 представляет собой водород, галоген, C1-6 алкил или C1-6 алкилокси. Третья группа представляющих интерес соединений состоит из таких соединений формулы (I), к которым применимо одно или несколько из следующих ограничений:a) каждый из n представляет собой целое число со значением 0 или 1;b) каждый из R4 представляет собой водород, С 1-6 алкил, С 1-6 алкилоксиС 1-6 алкил, С 3-6 циклоалкил или фенилС 1-6 алкил;d) R2 представляет собой водород, галоген, С 1-6 алкил, циано, нитро или С 1-6 алкилокси. Четвертая группа представляющих интерес соединений состоит из таких соединений формулы (I), к которым применимо одно или несколько из следующих ограничений: а) каждый из n представляет собой целое число со значением 0 или 1;b) каждый из m представляет собой целое число со значением 1;c) каждый из R4 представляет собой водород, С 1-6 алкилоксиС 1-6 алкил, С 3-6 циклоалкил или фенилС 1-6 алкил;f) R3 представляет собой C1-6 алкилокси; или,g) когда R2 и R3 представляют собой соседние атомы углерода, они могут образовывать двухвалентный радикал -О-СН 2-O-. Пятая группа представляющих интерес соединений состоит из таких соединений формулы (I), к которым применимо одно или несколько из следующих ограничений:a) каждый из n представляет собой целое число со значением 1;b) каждый из m представляет собой целое число со значением 1;c) каждый из X независимо представляет собой N;d) каждый из Y независимо представляет собой NR4;g) R2 представляет собой водород или галоген/ илиh) R3 представляет собой водород. Группа предпочтительных соединений состоит из тех соединений формулы (I), где каждый из n представляет собой целое число со значением 0 или 1; каждый из R4 представляет собой водород, C1-6 алкил, С 1-6 алкилоксиС 1-6 алкил, С 3-6 циклоалкил или фенилС 1-6 алкил; R1 представляет собой водород, С 1-6 алкил, гидроксиС 1-6 алкил, C1-6 алкилкарбонил или С 1-6 алкилсульфонил и R2 представляет собой водород,галоген, С 1-6 алкил, циано, нитро, полигалогенС 1-6 алкил или С 1-6 алкилокси. Группа более предпочтительных соединений состоит из тех соединений формулы (I), где каждый изn представляет собой целое число со значением 1; каждый из m представляет собой целое число со значением 1; каждый из X независимо представляет собой N; каждый из Y независимо представляет собойNR4; каждый из R4 представляет собой C1-6 алкил; R1 представляет собой водород; R2 представляет собой водород или галоген и R3 представляет собой водород. Наиболее предпочтительные соединения представляют собой соединение 1 а, соединение 30,соединение 39 и соединение 50. Соединения формул (I) и (II), их фармацевтически приемлемые соли и N-оксиды и их стереохимически изомерные формы могут быть получены обычным способом. Исходные материалы и некоторые промежуточные соединения представляют собой известные соединения и являются коммерчески доступными или могут быть получены в соответствии с обычными процедурами реакций, как, в целом, известно в данной области или как описано в заявках на европейский патент ЕР 1485099, ЕР 1485348,ЕР 1485353, ЕР 1485354, ЕР 1485364, ЕР 1485365, ЕР 1485370 и ЕР 1485378. Некоторые способы получения будут описаны далее более подробно. Другие способы получения конечных соединений формулы (I) описываются в примерах. а) Гидроксамовые кислоты формулы (I) могут быть получены посредством взаимодействия промежуточных соединений формулы (II), где Q представляет собой тетрагидропиранилоксиаминокарбонил,упоминаемых как промежуточные соединения формулы (II-а), с соответствующей кислотой, такой, например, как трифторуксусная кислота. Указанную реакцию осуществляют в соответствующем растворителе, таком, например, как метанол или дихлорметан.b) Альтернативно, гидроксамовые кислоты формулы (I) могут быть получены посредством взаимодействия промежуточных соединений формулы (II), где Q представляет собой С 1-6 алкилоксикарбонил,упоминаемых как промежуточные соединения формулы (II-с), с гидроксиламином в присутствии основания, такого, например, как гидроксид натрия. Указанную реакцию осуществляют в соответствующем растворителе, таком, например, как метанол. Соединения формулы (I) могут также преобразовываться в любое другое посредством известных в данной области реакций или преобразований, функциональных групп. Ряд таких преобразований уже описан выше. Другие примеры представляют собой гидролиз сложных эфиров карбоновых кислот до соответствующей карбоновой кислоты или спирта; гидролиз амидов до соответствующих карбоновых кислот или аминов; гидролиз нитрилов до соответствующих амидов; аминогруппы на имидазоле или фениле могут заменяться водородом посредством известных в данной области реакций диазотирования и последующей замены диазогруппы водородом; спирты могут преобразовываться в сложные эфиры и простые эфиры; первичные амины могут преобразовываться во вторичные или третичные амины; двойные связи могут гидрироваться до соответствующей одинарной связи; йодорадикал на фенильной группе может преобразовываться в сложноэфирную группу посредством включения монооксида углерода в присутствии соответствующего палладиевого катализатора. Настоящее изобретение также относится к промежуточным соединениям формулы (II) их N-оксидным формам, фармацевтически приемлемым аддитивным солям и стереохимически изомерным формам, где каждый из n представляет собой целое число со значением 0, 1 или 2 и, когда n равно 0, тогда подразумевается прямая связь; каждый из m представляет собой целое число со значением 1 или 2; каждый из X независимо представляет собой N или СН; каждый из Y независимо представляет собой О, S или NR4; где каждый из R4 представляет собой водород, С 1-6 алкил, С 1-6 алкилоксиС 1-6 алкил, С 3-6 циклоалкил, С 35 6 6 циклоалкилметил, фенилС 1-6 алкил, -С(=O)-CHR R или -S(=O)2-N(CH3)2; где 5 6 каждый из R и R независимо представляет собой водород, амино, C1-6 алкил или аминоС 1-6 алкил и,когда Y представляет собой NR4 и R2 находится в 7-положении индолила, тогда R2 и R4 вместе могут образовывать двухвалентный радикалR3 представляет собой водород, С 1-6 алкил или С 1-6 алкилокси; когда R2 и R3 представляют собой соседние атомы углерода, они могут образовывать двухвалентный радикал -О-СН 2-О иQ представляет собой С 1-2 алкилоксикарбонил, гидроксикарбонил или тетрагидропиранилоксиаминокарбонил. Группы представляющих интерес предпочтительных, более предпочтительных и наиболее предпочтительных соединений могут определяться для соединений формулы (II) в соответствии с группами,определенными для соединений формулы (I). а) Промежуточные соединения формулы (II-а) могут быть получены посредством взаимодействия промежуточного соединения формулы (III) с промежуточным соединением формулы (II), где Q представляет собой гидроксикарбонил, упоминаемых в описании как промежуточные соединения формулы(II-b), в присутствии соответствующих реагентов, таких как N'-(этилкарбонимидоил)-N,N-диметил-1,3 пропандиамин, моногидрохлорид (EDC) и 1-гидрокси-1 Н-бензотриазол (НОВТ). Реакция может осуществляться в присутствии основания, такого как триэтиламин, в пригодном для использования растворителе, таком как смесь дихлорметана и тетрагидрофурана.b) Промежуточные соединения формулы (II-а) могут также быть получены посредством взаимодействия промежуточного соединения формулы (VI) с соответствующим карбоксальдегидом формулы (V),где t представляет собой целое число со значением 0 или 1 и, когда t равно 0, тогда подразумевается прямая связь, в присутствии соответствующего реагента, такого как боргидрид натрия, в пригодном для использования растворителе, таком как дихлорэтан или метанол. с) Промежуточные соединения формулы (II-b) могут быть получены посредством взаимодействия промежуточных соединений формулы (II), где Q представляет собой метил- или этилоксикарбонил(С 12 алкил), упоминаемых в описании как промежуточные соединения формулы (II-с), с соответствующим кислотным раствором, например хлористо-водородной кислоты, или основным раствором, например бромистого водорода или гидроксида натрия, в пригодном для использования растворителе, например спирте, таком как этанол или пропанол.d) Промежуточные соединения формулы (II-с) могут быть получены посредством взаимодействия сложного этилового эфира карбоновой кислоты формулы (IV) с соответствующим карбоксальдегидом формулы (V) в присутствии соответствующего реагента, такого как боргидрид натрия, например натрийтетрагидроборат, в пригодном для использования растворителе, таком как спирт, например метанол. е) Идентичным способом промежуточные соединения формулы (II-с) могут быть получены посредством взаимодействия промежуточного соединения формулы (XIV) с соответствующим промежуточным соединением формулы (XV) в присутствии соответствующего реагента, такого как боргидрид натрия,например натрийтетрагидроборат, в пригодном для использования растворителе, таком как спирт, например метанол.f) Промежуточные соединения формулы (II-с) могут также быть получены посредством взаимодействия промежуточного соединения формулы (X) с промежуточным соединением формулы (XI), где W представляет собой соответствующую уходящую группу, такую, например, как галоген, например фтор,хлор, бром или йод, или сульфонилоксирадикал, такой как метилсульфонилокси, 4-метилфенилсульфонилокси и т.п. Реакция может осуществляться в растворителе, инертном для реакции, таком, например,как спирт, например метанол, этанол, 2-метоксиэтанол, пропанол, бутанол и т.п.; простой эфир, например 4,4-диоксан, 1,1'-оксибиспропан и т.п.; кетон, например 4-метил-2-пентанон; или N,N-диметилформамид, нитробензол, ацетонитрил и т.п. Добавление соответствующего основания, такого, например,как карбонат или бикарбонат щелочного или щелочно-земельного металла, например триэтиламина или карбоната натрия, может использоваться для захвата кислоты, которая высвобождается в ходе реакции. Малое количество соответствующего йодида металла, например йодида натрия или калия, может быть добавляться для ускорения реакции. Перемешивание может увеличить скорость реакции. Реакцию может быть удобным осуществлять при температуре, находящейся в пределах между комнатной температурой и температурой кипения с обратным холодильником реакционной смеси, и, если это желательно, реакция может осуществляться при повышенном давлении.g) Идентичным способом промежуточные соединения формулы (II-с) могут быть получены посредством взаимодействия промежуточного соединения формулы (XII) с промежуточным соединением формулы (XIII), где W представляет собой соответствующую уходящую группу, как описано выше. Промежуточные соединения формулы (VI) могут быть получены посредством взаимодействия промежуточного соединения формулы (VII) с пиперидином в пригодном для использования растворителе, например дихлорметане.-8 010652 Промежуточные соединения формулы (VII) могут быть получены посредством взаимодействия промежуточного соединения формулы (VIII) с промежуточным соединением формулы (III) в присутствии соответствующих реагентов, таких как моногидрохлорид N'-(этилкарбонимидоил)-N,N-диметил-1,3 пропандиамина, (EDC) и 1-гидрокси-1 Н-бензотриазол (НОВТ). Реакция может осуществляться в присутствии основания, такого как триэтиламин, в пригодном для использования растворителе, таком как смесь дихлорметана и тетрагидрофурана. Промежуточные соединения формулы (VIII) могут быть получены посредством взаимодействия промежуточного соединения формулы (IX) с промежуточным соединением формулы (IV), где R1 представляет собой водород, упоминаемых в описании как промежуточные соединения формулы (IV-а), в присутствии гидроксида натрия, в пригодном для использования растворителе, таком как тетрагидрофуран, с последующей нейтрализацией хлористо-водородной кислотой и с добавлением карбоната натрия. Соединения формулы (I) и некоторые из промежуточных соединений могут иметь в своей структуре по меньшей мере один стереогенный центр. Этот стереогенный центр может присутствовать в R- илиS-конфигурациях. Соединения формулы (I), как получают в описанных выше способах, как правило, представляют собой рацемические смеси энантиомеров, которые могут отделяться друг от друга, следуя известным в данной области процедурам разрешения. Рацемические соединения формулы (I) могут преобразовываться в соответствующие формы диастереомерных солей посредством взаимодействия с соответствующей хиральной кислотой. Указанные формы диастереомерных солей впоследствии разделяют, например, посредством селективной или фракционной кристаллизации, и энантиомеры высвобождаются из них посредством щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включают жидкостную хроматографию с использованием хиральной стационарной фазы. Указанные чистые стереохимические изомерные формы могут также быть получены из соответствующих чистых стереохимических изомерных форм соответствующих исходных материалов при условии, что реакция осуществляется стереоспецифично. Предпочтительно, если желательным является конкретный стереоизомер, указанное соединение может синтезироваться посредством стереоспецифичных способов получения. Эти способы будут преимущественно использоваться энантиомерно чистые исходные материалы. Соединения формулы (I), их фармацевтически приемлемые аддитивные соли кислот и их стереоизомерные формы имеют ценные фармакологические свойства, заключающиеся в том, что они имеют эффект ингибирования гистондеацетилазы (HDAC). Настоящее изобретение предусматривает способ ингибирования аномального роста клеток, включая трансформированные клетки, посредством введения эффективного количества соединения по настоящему изобретению. Аномальный рост клеток относится к росту клеток, не зависимому от нормальных регуляторных механизмов (например, к потере контактного ингибирования). Это включает ингибирование роста опухоли, как непосредственно, вызывая приостановку роста, конечную дифференциацию и/или апоптоз раковых клеток, так и опосредованно, посредством ингибирования неоваскуляризации опухолей.-9 010652 Настоящее изобретение также предусматривает способ ингибирования роста опухоли посредством введения эффективного количества соединения по настоящему изобретению субъекту, например млекопитающему (а более конкретно, человеку), нуждающемуся в таком лечении. В частности, настоящее изобретение предусматривает способ ингибирования роста опухолей посредством введения эффективного количества соединений по настоящему изобретению. Примеры опухолей, которые могут ингибироваться, но не ограничиваясь этим, включают рак легких (например, аденокарциному, и включая немелкоклеточный рак легких), раковые заболевания поджелудочной железы (например, карциному поджелудочной железы, такую, например, как экзокринная карцинома поджелудочной железы), раковые заболевания толстой кишки (например, карциномы толстой и прямой кишки, такие, например, как аденокарцинома толстой кишки и аденома толстой кишки), рак простаты, включая запущенное заболевание, гематопоэтические опухоли лимфоидной ткани слизистой оболочки (например, острую лимфоцитную лейкемию,лимфому В-клетки, лимфому Burkitt), миелоидные лейкемии (например, острую миелогенную лейкемию(AML, фолликулярный рак щитовидной железы, миелодиспластический синдром (MDS), опухоли мезенхимального происхождения (например, фибросаркомы и рабдомиосаркомы), меланомы, тератокарциномы, нейробластомы, глиомы, злокачественные опухоли кожи (например, кератоакантомы), карциному груди (например, запущенный рак груди), карциному почек, карциному яичников, карциному мочевого пузыря и эпидермальную карциному. Соединение в соответствии с настоящим изобретением может использоваться для других терапевтических целей, например:a) сенсибилизации опухолей для радиационной терапии посредством введения соединения по настоящему изобретению до, во время или после облучения опухоли для лечения рака;b) лечения артропатий и остеопатологических состояний, таких как ревматоидный артрит, остеоартрит, ювенильный артрит, подагра, полиартрит, псориатический артрит, анкилозный спондилит и системная красная волчанка;c) ингибирования пролиферации клеток гладких мышц, включая пролиферативные расстройства сосудов, атеросклероз и рестеноз;d) лечения воспалительных состояний и состояний кожи, таких как язвенный колит, болезнь Крона,аллергический ринит, заболевание отторжения трансплантатов, конъюнктивит, астма, ОРДС, болезнь Бехчета, отторжение трансплантов, уртикария (аллергическая сыпь), аллергический дерматит, гнездная плешивость, склеродерма, экзантема, экзема, дерматомиозит, прыщи, диабет, системная красная волчанка,болезнь Кавасаки, множественный склероз, эмфизема, фиброз мочевого пузыря и хронический бронхит;e) лечения эндометриоза, фиброидов матки, дисфункционального маточного кровотечения и эндометриальной гиперплазии;f) лечения васкуляризации глаз, включая васкулопатию, воздействующую на ретинальные и хороидальные сосуды;h) ингибирования иммунодепрессивных состояний, такого как лечение инфекции ВИЧ;l) лечения нейропатологии, например болезни Паркинсона, или нейропатологии, которая приводит к когнитивному расстройству, например болезни Альцгеймера или нейрональным заболеваниям, связанным с полиглютамином;n) ингибирования нервномышечной патологии, например амилотропного латерального склероза;o) лечения спинальной мышечной атрофии; р) лечения других патологических состояний, поддающихся лечению посредством усиления экспрессии гена;s) лечения паразитоза, такого как малярия. Следовательно, настоящее изобретение описывает соединения формулы (I) для использования в качестве лекарства, а также использования этих соединений формулы (I) для производства медикамента для лечения одного или нескольких из указанных выше состояний. Соединения формулы (I), их фармацевтически приемлемые аддитивные соли кислот и стереоизомерные формы могут иметь ценные диагностические свойства, заключающиеся в том, что они могут использоваться для детектирования или идентификации HDAC в биологическом образце, включая детектирование или измерение образования комплекса между меченым соединением и HDAC. Способы детектирования или идентификации могут использовать соединения, которые метятся метящими агентами, такими как радиоактивные изотопы, ферменты, флуоресцентные вещества, светящиеся вещества и т.п. Примеры радиоактивных изотопов включают 125I, 131I, 3 Н и 14 С. Ферменты обычно де- 10010652 лаются детектируемыми посредством конъюгирования с соответствующим субстратом, который, в свою очередь, катализирует детектируемую реакцию. Их примеры включают, например, бета-галактозидазу,бета-глюкозидазу, щелочную фосфатазу, пероксидазу и малатдегидрогеназу, предпочтительно пероксидазу хрена. Светящиеся вещества включают, например, люминол, производные люминола, люциферин,аэкворин и люциферазу. Биологические образцы могут определяться как телесные ткани или телесные жидкости. Примеры телесных жидкостей представляют себой спинно-мозговую жидкость, кровь, плазму, сыворотку, мочу,мокроту, слюну и т.п. С учетом их полезных фармакологических свойств, рассматриваемые соединения могут приготавливаться в различных фармацевтических формах для целей введения. Для получения фармацевтических композиций по настоящему изобретению эффективное количество конкретного соединения в форме аддитивной соли основания или кислоты в качестве активного ингредиента объединяется в тесную смесь с фармацевтически приемлемым носителем, этот носитель может принимать разнообразные формы в зависимости от формы препарата, желаемой для введения. Эти фармацевтические композиции являются желательными в виде стандартной лекарственной формы, пригодной, предпочтительно, для введения перорально, ректально, подкожно или посредством парентеральной инъекции. Например, при получении композиций в форме пероральной дозировки может использоваться любая из обычных фармацевтических сред, такая, например, как вода, гликоли, масла, спирты и т.п, в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие материалы,разрыхляющие агенты и т.п, в случае порошка, пилюль, капсул и таблеток. Благодаря простоте их введения таблетки и капсулы представляют собой наиболее преимущественную форму пероральной единичной дозировки, в этом случае, очевидно, используются твердые фармацевтические носители. Для парентеральных композиций носитель будет обычно содержать стерильную воду по меньшей мере в большей части, хотя могут включаться и другие ингредиенты, например, для увеличения растворимости. Например, могут приготавливаться растворы для инъекций, в которых носитель содержит солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Также могут приготавливаться суспензии для инъекций, в этом случае могут использоваться соответствующие жидкие носители, суспендирующие агенты и т.п. В композициях, пригодных для подкожного введения, носитель необязательно содержит агент для увеличения проницаемости и/или соответствующий смачивающий агент, необязательно объединенный с соответствующими добавками любой природы в малых пропорциях, эти добавки не вызывают значительного отрицательного воздействия на кожу. Указанные добавки могут облегчить введение в кожу и/или могут быть полезны для получения желаемых композиций. Эти композиции могут вводиться различными путями, например как трансдермальный пластырь, как наклейка или как мазь. Особенно преимущественным является приготовление указанных фармацевтических композиций в стандартной лекарственной форме для простоты введения и однородности дозировки. Стандартная лекарственная форма, как используется в описании и формуле изобретения, относится к физически отдельным дозированным формам, пригодным в качестве отдельных доз, каждая дозированная форма содержит заданное количество активного ингредиента, рассчитанное дляоказания желаемого терапевтического воздействия, в ассоциации с требуемым фармацевтическим носителем. Примеры таких стандартных лекарственных форм представляют собой таблетки (включая разламываемые таблетки или таблетки с покрытием), капсулы, пилюли, пакетики с порошком, пластинки, растворы или суспензии для инъекций,дозы для заполнения чайной ложки, дозы для заполнения столовой ложки и т.п. и их разделенные наборы. Специалист в данной области может легко определить эффективное количество по результатам исследований, представленных далее. Как правило, предполагается, что терапевтически эффективное количество должно составлять от 0,005 до 100 мг/кг массы тела, и в частности от 0,005 до 10 мг/кг массы тела. Может быть соответствующим введение необходимой дозы в виде 2, 3, 4 или более субдоз через соответствующие интервалы в течение дня. Указанные субдозы могут приготавливаться в виде стандартных лекарственных форм, например, содержащих от 0,5 до 500 мг, и в частности от 10 до 500 мг,активного ингредиента на стандартную лекарственную форму. В качестве другого аспекта настоящего изобретения рассматривается сочетание ингибитора HDAC с другим противораковым агентом, в частности, для использования в качестве лекарственного средства,более конкретно, при лечении рака или связанных с ним заболеваний. Для лечения указанных выше состояний соединения по настоящему изобретению преимущественно могут использоваться в сочетании с одним или несколькими другими лекарственными агентами, более конкретно с другими противораковыми агентами. Примеры противораковых агентов представляют собой координационные соединения платины, например цисплатин, карбоплатин или оксалиплатин; таксановые соединения, например паклитаксель или доцетаксель; ингибиторы топоизомеразы I, такие как соединения камптотецина, например иринотекан или топотекан; ингибиторы топоизомеразы II, такие как противоопухолевые подофиллотоксиновые производные,- 11010652 например этопозид или тенипозид; противоопухолевые алкалоиды винка, например винбластин, винкристин или винорельбин; противоопухолевые нуклеозидные производные, например 5-флуороурацил, гемцитабин или капецитабин; алкилирующие агенты, такие как азотная горчица или нитрозомочевина, например циклофосфамид,хлорамбуцил, кармустин или ломустин; противоопухолевые антрациклиновые производные, например даунорубицин, доксорубицин, идарубицин или митоксантрон; антитела HER2, например трастузумаб; антагонисты рецепторов эстрогенов или селективные модуляторы рецепторов эстрогенов, например тамоксифен, торемифен, дролоксифен, фаслодекс или ралоксифен; ингибиторы ароматазы, такие как эксеместан, анастрозол, летразол и ворозол; дифференцирующие агенты, такие как ретиноиды, витамин D и агенты, блокирующие метаболизм ретиноевой кислоты (RAMBA), например аккутан; ингибиторы метилтрансферазы ДНК, например азацитидин; ингибиторы киназы, например флавоперидол, иматиниб мезилат или гефитиниб; ингибиторы фарнезилтрансферазы; другие ингибиторы HDAC; ингибиторы убиквитин-протеасомного пути, например Velcade; илиYondelis. Термин "координационное соединение платины" используется в описании для обозначения любого координационного соединения платины, ингибирующего рост клеток опухоли, которое поставляет платину в форме иона. Термин "таксановые соединения" указывает класс соединений, имеющих таксановую кольцевую систему, и получаемый из экстрактов от определенных видов тисовых деревьев (Taxus) или связанный с ними. Термин "ингибиторы топизомеразы" используется для указания ферментов, которые способны изменять топологию ДНК в эукариотических клетках. Они являются критичными для важных клеточных функций и пролиферации клеток. Имеются два класса топоизомераз в эукариотических клетках, а именно тип I и тип II. Топоизомераза I представляет собой мономерный фермент с молекулярной массой приблизительно 100000. Фермент связывается с ДНК и производит временный разрыв одной цепочки, развивает двойную спираль (или позволяет ей развиться), впоследствии соединяет разрыв перед диссоциацией с цепочки ДНК. Топизомераза II имеет сходный механизм действия, который включает индуцирование разрывов цепочки ДНК или образование свободных радикалов. Термин "соединения камптотецина" используется для указания соединений, которые получаются из исходного соединения камптотецина, который представляет собой водонерастворимый алкалоид, полученный из аралии китайской Camptothecin acuminata и фикуса Nothapodytes foetida, или связаны с ним. Термин "соединения подофиллотоксина" используется для указания соединений, которые связаны c или получаются из исходного подофиллотоксина, который экстрагируют из растения мандрагора (Mandrake). Термин "противоопухолевые алкалоиды винка" используется для указания соединений, которые связаны или получаются из экстрактов растения барвинок (Vinca rosea). Термин "алкилирующие агенты" охватывает группу разнообразных соединений, которые имеют общий признак - способность к введению, при физиологических условиях, алкильных групп в биологически важные макромолекулы, такие как ДНК. Вместе с большинством более важных агентов, таких как азотистый иприт и нитрозомочевины, активные алкилирующие остатки генерируются in vivo после сложных деградационных реакций, некоторые из которых являются ферментативными. Наиболее важными фармакологическими действиями алкилирующих агентов являются те, которые нарушают фундаментальные механизмы, связанные с пролиферацией клеток, при конкретном синтезе ДНК и делении клеток. Способность алкилирующих агентов вмешиваться в функционирование и целостность ДНК в быстро пролиферирующих тканях обеспечивает основу для их терапевтических применений и для многих их токсических свойств. Термин "противоопухолевые производные антрациклина" включает антибиотики, полученные от грибков Strep. peuticus var. Caesius, и их производные, отличающиеся тем, что они имеют тетрациклиновую кольцевую структуру с необычным сахаром, даунозамином, присоединенным посредством гликозидной связи. Амплификация белка рецептора 2 фактора эпидермального роста человека (HER2) в первичных карциномах груди, как показано, коррелирует с плохим клиническим прогнозом для определенных пациентов. Трастузумаб представляет собой каппа-антитело к очеловеченному моноклональному IgG1, полученному с помощью рекомбинантной ДНК высокой очистки, которое связывается с высоким сродством и специфичностью с внеклеточным доменом рецептора HER2. Многие виды рака груди имеют рецепторы эстрогенов, и рост этих опухолей может стимулироваться эстрогенами. Термины "антагонисты рецепторов эстрогенов" и "селективные модуляторы рецепторов эстрогенов" используются для указания конкурентных ингибиторов связывания эстрадиола с рецептором эстрогенов (ER). Селективные модуляторы рецепторов эстрогенов, когда связываются с ER, вызывают изменение трехмерной формы рецептора, модулируя его связывание с эстрогенчувствительным элементом (ERE) на ДНК.- 12010652 У женщин после менопаузы главный источник циркулирующего эстрогена возникает из-за преобразования андрогенов надпочечников и яичников (андростендиона и тестостерона) в эндогены (эстрон и эстрадиол) посредством фермента ароматазы в периферийных тканях. Устранение влияния эстрогена посредством ингибирования или инактивирования ароматазы представляет собой эффективное и селективное лечение для некоторых пациентов после менопаузы с гормонозависимым раком груди. Термин "антиэстрогенный агент" используется в описании не только для антагонистов рецепторов эстрогенов и селективных модуляторов рецепторов эстрогенов, но также и для ингибиторов ароматазы,как обсуждается выше. Термин "дифференцирующие агенты" охватывает соединения, которые могут различными путями ингибировать пролиферацию клеток и индуцировать дифференциацию. Витамин D и ретиноиды, как известно, играют главную роль в регуляции роста и дифференциации разнообразных нормальных и злокачественных типов клеток. Агенты, блокирующие метаболизм ретиноевой кислоты (RAMBA), увеличивают уровни эндогенных ретиноевых кислот посредством ингибирования катаболизма ретиноевых кислот, опосредуемого цитохромом Р 450. Изменения метилирования ДНК находятся среди наиболее распространенных аномалий неоплазии человека. Гиперметилирование в промоторах избранных генов обычно связывается с инактивированием рассматриваемых генов. Термин "ингибиторы метилтрансферазы ДНК" используется для указания соединений, которые действуют посредством фармакологического ингибирования метилтрансферазы ДНК и реактивирования экспрессии гена, подавляющего опухоль. Термин "ингибиторы киназы" включает мощные ингибиторы киназ, которые вовлечены в развитие клеточного цикла и программируемую гибель клетки (апоптоз). Термин "ингибиторы фарнезилтрансферазы" используются для указания соединений, которые разработаны для предотвращения фарнезилирования Ras и других внутриклеточных белков. Они, как показано, имеют воздействие на пролиферацию и выживание злокачественных клеток. Термин "другие ингибиторы HDAC" включает, но не ограничиваясь этим,карбоксилаты, например бутират, коричную кислоту, 4-фенилбутират или вальпроиновую кислоту; гидроксамовые кислоты, например субероиланилид гидроксамовую кислоту (SAHA), содержащие пиперазиновые аналоги SAHA, биарилгидроксамат А-161906 и его карбозилэфирные, тетрагидропиридиновые и тетралоновые аналоги, бициклические арил-N-гидроксикарбоксамиды, пироксамид, CG-1521,PXD-101, сульфонамидгидроксамовую кислоту, LAQ-824, LBH-589, трихостатин A (TSA), оксамфлатин,скриптаид, родственные скриптаиду трициклические молекулы, бис-гидроксамат м-карбоксикоричной кислоты (СВНА), СВНА-подобные гидроксамовые кислоты, трапоксиновый аналог гидроксамовой кислоты,R306465 и родственные бензоил- и гетероарилгидроксамовые кислоты, аминосубераты и малонилдиамиды; циклические тетрапептиды, например трапоксин, апидицин, депсипептид, соединения, родственные спирухостатину, RedFK-228, сульфгидрилсодержащие циклические тетрапептиды (SCOP), циклические тетрапептиды, содержащие гидроксамовые кислоты, (CHAP), TAN-174S и азумамиды; бензамиды, например MS-275 или CI-994; или депудецин. Термин "ингибиторы убиквитин-протеасомного пути" используется для идентификации соединений, которые ингибируют целевое разрушение клеточных белков в протеасоме, включая регуляторные белки клеточного цикла. Для лечения рака соединения в соответствии с настоящим изобретением могут вводиться пациенту,как описано выше, в сочетании с облучением. Облучение означает ионизирующее излучение, и в частности гамма-излучение, в частности испускаемое линейными ускорителями или радионуклидами, которые в настоящее время находятся в повсеместном использовании. Облучение опухоли радионуклидами может быть наружным или внутренним. Настоящее изобретение также относится к комбинации в соответствии с настоящим изобретением из противоракового агента и ингибитора HDAC в соответствии с настоящим изобретением. Настоящее изобретение также относится к комбинации в соответствии с настоящим изобретением для использования в лекарственной терапии, например для ингибирования роста опухолевых клеток. Настоящее изобретение также относится к комбинациям в соответствии с настоящим изобретением для ингибирования роста опухолевых клеток. Настоящее изобретение также относится к способу ингибирования роста опухолевых клеток у субъекта-человека, который включает введение субъекту эффективного количества комбинации в соответствии с настоящим изобретением. Настоящее изобретение, кроме того, предусматривает способ ингибирования аномального роста клеток, включая трансформированные клетки, посредством введения эффективного количества комбинации в соответствии с настоящим изобретением. Другой лекарственный агент и ингибитор HDAC может вводиться одновременно (например, в виде отдельных или объединенных композиций) или последовательно, в любом порядке. В последнем случае два соединения будут вводиться в пределах периода, и в количестве, и способом, которые являются дос- 13010652 таточными, чтобы обеспечить достижения преимущественного или синергического воздействия. Будет понятно, что предпочтительные способ, и порядок введения, и соответствующие величины и режимы дозировки для каждого компонента комбинации будут зависеть от конкретного другого лекарственного агента и ингибитора HDAC, которые вводятся, их способа введения, конкретной опухоли, которая лечится, и конкретного субъекта, которого лечат. Оптимальные способ, и порядок введения, и величины, и режим дозировки легко могут быть определены специалистом в данной области с использованием обычных способов и с учетом информации, приведенной в описании. Координационное соединение платины преимущественно вводится при дозировке от 1 до 500 мг на квадратный метр (мг/м 2) площади поверхности тела, например 50-400 мг/м 2, в частности для цисплатина при дозировке примерно 75 мг/м 2 и для карбоплатина примерно при 300 мг/м 2 на курс лечения. Соединение таксана преимущественно вводится при дозировке от 50 до 400 мг на квадратный метр(мг/м 2) площади поверхности тела, например 75-250 мг/м 2, в частности для паклитаксела при дозировке от примерно 175 до 250 мг/м 2 и для доцетаксела примерно при 75-150 мг/м 2 на курс лечения. Соединение камптотецина преимущественно вводится при дозировке от 0,1 до 400 мг на квадратный метр (мг/м 2) площади поверхности тела, например 1-300 мг/м 2, в частности для иринотекана при дозировке от примерно 100 до 350 мг/м 2 и для топотекана примерно при 1-2 мг/м 2 на курс лечения. Противоопухолевое производное подофиллотоксина преимущественно вводится при дозировке от 30 до 300 мг на квадратный метр (мг/м 2) площади поверхности тела, например 50-250 мг/м 2, в частности для этопозида при дозировке от примерно 35 до 100 мг/м 2 и для тенипозида примерно при 50-250 мг/м 2 на курс лечения. Противоопухолевый алкалоид винка преимущественно вводится при дозировке от 2 до 30 мг на квадратный метр (мг/м 2) площади поверхности тела, в частности для винбластина при дозировке от примерно 3 до 12 мг/м 2, для винкристина при дозировке от примерно 1 до 2 мг/м 2 и для винорельбина при дозировке от примерно 10 до 30 мг/м 2 на курс лечения. Противоопухолевое нуклеозидное производное преимущественно вводится при дозировке от 200 до 2500 мг на квадратный метр (мг/м 2) площади поверхности тела, например 700-1500 мг/м 2, в частности для 5-FU при дозировке от 200 до 500 мг/м 2, для гемцитабина при дозировке от примерно 800 до 1200 мг/м 2 и для капецитабина от примерно 1000 до 2500 мг/м 2 на курс лечения. Алкилирующий агент, такой как азотная горчица или нитрозомочевина, преимущественно вводится при дозировке от 100 до 500 мг на квадратный метр (мг/м 2) площади поверхности тела, например 120-200 мг/м 2,в частности для циклофосфамида при дозировке от примерно 100 до 500 мг/м 2, для хлорамбуцила при дозировке от примерно 0,1 до 0,2 мг/кг, для кармустина при дозировке от примерно 150 до 200 мг/м 2 и для ломустина при дозировке от примерно 100 до 150 мг/м 2 на курс лечения. Противоопухолевое антрациклиновое производное преимущественно вводится при дозировке от 10 до 75 мг на квадратный метр (мг/м 2) площади поверхности тела, например 15-60 мг/м 2, в частности для доксорубицина при дозировке от примерно 40 до 75 мг/м 2, для даунорубицина при дозировке от примерно 25 до 45 мг/м 2 и для идарубицина при дозировке от примерно 10 до 15 мг/м 2 на курс лечения. Трастузумаб преимущественно вводится при дозировке от 1 до 5 мг на квадратный метр (мг/м 2) площади поверхности тела, в частности от 2 до 4 мг/м 2 на курс лечения. Антиэстрогенный агент преимущественно вводится при дозировке от примерно 1 до 100 мг в день,в зависимости от конкретного агента и от состояния, которое лечится. Тамоксифен преимущественно вводится перорально при дозировке от 5 до 50 мг, предпочтительно от 10 до 20 мг дважды в день, продолжая терапию в течение времени, достаточного для достижения и поддержания терапевтического воздействия. Торемифен преимущественно вводится перорально при дозировке от примерно 60 мг 1 раз в день, продолжая терапию в течение времени, достаточного для достижения и поддержания терапевтического воздействия. Анастрозол преимущественно вводится перорально при дозировке примерно 1 мг 1 раз в день. Дролоксифен преимущественно вводится перорально при дозировке примерно 20-100 мг 1 раз в день. Ралоксифен преимущественно вводится перорально при дозировке примерно 60 мг 1 раз в день. Эксеместан преимущественно вводится перорально при дозировке от примерно 25 мг 1 раз в день. Эти дозировки могут вводиться, например, 1, 2 или более раз на курс лечения, который может повторяться, например, каждые 7, 14, 21 или 28 дней. С учетом их полезных фармакологических свойств, компоненты комбинаций в соответствии с настоящим изобретением, т.е. другой лекарственный агент и ингибитор HDAC, могут приготавливаться в различных фармацевтических формах, в зависимости от введения. Компоненты могут приготавливаться по отдельности в виде отдельных фармацевтических композиций или в виде объединенной фармацевтической композиции, содержащей оба компонента. Следовательно, настоящее изобретение также относится к фармацевтической композиции, содержащей другой лекарственный агент и ингибитор HDAC вместе с одним или несколькими фармацевтическими носителями. Настоящее изобретение также относится к комбинации в соответствии с настоящим изобретением в форме фармацевтической композиции, содержащей противораковый агент и ингибитор HDAC в соответствии с настоящим изобретением, вместе с одним или несколькими фармацевтическими носителями.- 14010652 Кроме того, настоящее изобретение относится к применению комбинации в соответствии с настоящим изобретением при производстве фармацевтической композиции для ингибирования роста опухолевых клеток. Кроме того, настоящее изобретение относится к продукту, содержащему в качестве первого активного ингредиента ингибитор HDAC в соответствии с настоящим изобретением, а в качестве второго активного ингредиента противораковый агент в качестве объединенного препарата для одновременного,раздельного или последовательного использования при лечении пациентов, страдающих от рака. Экспериментальная часть Следующие далее примеры приводятся для целей иллюстрации. Далее "DCM" определяется как дихлорметан, "DIPE" определяется как простой диизопропиловый эфир, "DMA" определяется как N,N-диметилацетамид, "DMSO" определяется как диметилсульфоксид, "EDC" определяется как моногидрохлоридN'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина, "EtOAc" определяется как этилацетат, "EtOH" определяется как этанол, "HOBt" определяется как 1-гидрокси-1H-бензотриазол, "МеОН" определяется как метанол, "TFA" определяется как трифторуксусная кислота и "ТГФ" определяется как тетрагидрофуран. А. Получение промежуточных соединений. Пример А 1. а) Получение промежуточного соединения 1. Смесь сложного этилового эфира 2-[4-(аминометил)-1-пиперидинил]-5-пиримидинкарбоновой кислоты (0,0114 моль), 1-метил-1H-индол-3-карбоксальдегида (0,017 моль) и MgSO4 (0,5 г) в МеОН (80 мл) перемешивают и нагревают с обратным холодильником в течение 15 ч, затем охлаждают до комнатной температуры. Натрийтетрагидроборат (0,018 моль) добавляют по частям. Смесь перемешивают при комнатной температуре 5 ч, выливают в воду и экстрагируют EtOAc. Органический слой отделяют, сушат(MgSO4), фильтруют и растворитель выпаривают. Остаток (6,6 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 94/6/0,5). Чистые фракции собирают и растворитель выпаривают с получением 4,3 г (90%) промежуточного соединения 1.b) Получение промежуточного соединения 2. Смесь промежуточного соединения 1 (0,0037 моль) и гидроксида натрия (0,0074 моль) в EtOH (60 мл) перемешивают при 50 С в течение 15 ч, затем охлаждают до комнатной температуры и растворитель выпаривают досуха с получением 1,5 г (100%) промежуточного соединения 2. с) Получение промежуточного соединения 3.(0,015 моль) добавляют при комнатной температуре к смеси промежуточного соединения 2 (0,005 моль) вDCM (100 мл) и ТГФ (100 мл) в потоке N2. Смесь перемешивают при 40 С в течение 4 ч, выливают в воду со льдом и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (4 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм)(элюент: DCM/MeOH/NH4OH 96/4/0,5). Чистые фракции собирают и растворитель выпаривают с получением 1 г (42%) промежуточного соединения 3. Фракцию (0,051 г) кристаллизуют из DIPE. Осадок отфильтровывают и сушат с получением 0,03 г промежуточного соединения 3, температура плавления 70 С. Пример А 2. а) Получение промежуточного соединения 4.- 15010652 Смесь сложного этилового эфира 2-[4-(аминометил)-1-пиперидинил]-5-пиримидинкарбоновой кислоты (0,0072 моль) в ТГФ (40 мл) и гидроксида натрия 1 н. (40 мл), перемешивают при комнатной температуре в течение ночи. Добавляют хлористо-водородную кислоту 1 н. (40 мл). Смесь перемешивают в течение 10 мин. Добавляют карбонат натрия (0,0216 моль). Смесь перемешивают в течение 10 мин. 1(9H-Флуорен-9-илметокси)карбонил]окси]-2,5-пирролидиндион (0,0072 моль) добавляют по частям. Смесь перемешивают при комнатной температуре в течение 6 ч, затем охлаждают до 0 С и подкисляют хлористо-водородной кислотой. Осадок фильтруют, промывают простым диэтиловым эфиром и сушат с получением 4,1 г (100%) промежуточного соединения 4.b) Получение промежуточного соединения 5. Триэтиламин (0,02 моль), EDC (0,0082 моль) и HOBt (0,0082 моль) добавляют при комнатной температуре к смеси промежуточного соединения 4 (0,0068 моль) в DCM/ТГФ (200 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 15 мин. Добавляют О-(тетрагидро-2H-пиран-2-ил) гидроксиламин (0,0082 моль). Смесь перемешивают при комнатной температуре в течение 48 ч, выливают в воду и экстрагируют DCM. Органический слой промывают NaHCO3 10%, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (4 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 98/2/0,1). Чистые фракции собирают и растворитель выпаривают с получением 3,4 г (89%) промежуточного соединения 5. с) Получение промежуточного соединения 6. Смесь промежуточного соединения 5 (0,0355 моль) и пиперидина (0,089 моль) в DCM (400 мл) перемешивают при 35 С в течение 72 ч. Растворитель выпаривают. Остаток очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 80/20/2). Чистые фракции собирают и растворитель выпаривают с получением 6,7 г (56%). Часть остатка (0,79 г) кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат с получением 0,62 г промежуточного соединения 6, температура плавления 129 С.d) Получение промежуточного соединения 7. Смесь промежуточного соединения 6 (0,0009 моль) и 5-хлор-1H-индол-3-карбоксальдегида (0,0012 моль) в 1,2-дихлорэтане (30 мл) перемешивают при комнатной температуре в течение ночи. Натрий трис(ацето-О)гидроборат (0,0013 моль) добавляют по частям. Смесь перемешивают при комнатной температуре в течение 4 ч, выливают в воду/NaOH 3 н. и экстрагируют DCM. Органический слой отделяют, сушат(MgSO4), фильтруют и растворитель выпаривают. Остаток (0,5 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент DCM/MeOH/NH4OH 93/7/0,5). Две фракции собирают и растворитель выпаривают с получением 0,07 г (16%) промежуточного соединения 7. Пример A3. а) Получение промежуточного соединения 8. Раствор сложного этилового эфира 2-(метилсульфонил)-5-пиримидинкарбоновой кислоты (0,094 моль) в ацетонитриле (40 мл) добавляют при 10 С к суспензии 4-пиперидинметанола (0,086 моль) и карбоната калия (0,172 моль) в ацетонитриле (200 мл) в потоке N2. Смесь доводят до комнатной температуры, затем перемешивают в течение 4 ч, выливают в воду и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (23 г) кристаллизуют из ацетонитрила/простого диэтилового эфира. Осадок отфильтровывают и сушат с получением 7,8 г (34%) промежуточного соединения 8. Слой маточного раствора выпаривают. Остаток (17 г) очищают с помощью коло- 16010652 ночной хроматографии на силикагеле (20-45 мкм) (элюент: DCM/MeOH/NH4OH 97/3/0,1). Чистые фракции собирают и растворитель выпаривают с получением 4,6 г (20%) промежуточного соединения 8, температура плавления 129 С.b) Получение промежуточного соединения 9. Триэтиламин (0,038 моль), затем метансульфонилхлорид (0,025 моль) добавляют при 0 С к раствору промежуточного соединения 8 (0,0189 моль) в DCM (80 мл) в потоке N2. Смесь перемешивают при 0 С в течение 2 ч и выливают в воду со льдом. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают с получением 6,5 г (100%) промежуточного соединения 9. с) Получение промежуточного соединения 10. Смесь промежуточного соединения 9 (0,0189 моль), N-метил-1H-индол-3-этанамина (0,0172 моль) и карбоната калия (0,0344 моль) в ацетонитриле (180 мл) перемешивают и нагревают с обратным холодильником в течение 24 ч, выливают в воду и экстрагируют EtOAc. Органический слой отделяют, сушат(MgSO4), фильтруют и растворитель выпаривают. Остаток (8,5 г) очищают с помощью колоночной хроматографии на силикагеле (70-200 мкм) (элюент: DCM/MeOH/NH4OH 98/2/0-97/3/0,1). Чистые фракции собирают и растворитель выпаривают с получением 1,25 г (20%) промежуточного соединения 10.d) Получение промежуточного соединения 11. Смесь промежуточного соединения 10 (0,003 моль) и гидроксида натрия (0,006 моль) в EtOH (80 мл) перемешивают и нагревают с обратным холодильником в течение ночи, затем охлаждают до комнатной температуры и растворитель выпаривают досуха с получением 1,3 г (100%) промежуточного соединения 11, температура плавления 260 С. с) Получение промежуточного соединения 12.EDC (0,0045 моль), затем HOBt (0,0045 моль) добавляют при комнатной температуре к смеси промежуточного соединения 11 (0,003 моль) в ТГФ (100 мл) и DCM (100 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 15 мин. Добавляют О-(тетрагидро-2H-пиран-2-ил)гидроксиламин (0,012 моль). Смесь перемешивают при комнатной температуре в течение 72 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (3 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент:DCM/MeOH/NH4OH 94/6/0,1). Чистые фракции собирают и растворитель выпаривают. Остаток (0,82 г) извлекают в простом диэтиловом эфире. Осадок отфильтровывают и сушат с получением 0,78 г промежуточного соединения 12, температура плавления 154 С.- 17010652 Пример А 4. а) Получение промежуточного соединения 13. Смесь сложного этилового эфира 2-[4-(аминометил)-1-пиперидинил]-5-пиримидинкарбоновой кислоты (0,0049 моль), метансульфоната 1H-индол-3-этанола (сложный эфир) (0,0054 моль) и карбоната калия (0,01 моль) в ацетонитриле (20 мл) перемешивают и нагревают с обратным холодильником в течение ночи, затем охлаждают, выливают в воду со льдом и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (2,2 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 96/4/0,2). Чистые фракции собирают и растворитель выпаривают с получением 0,442 г (22%) промежуточного соединения 13, температура плавления 238 С.b) Получение промежуточного соединения 14. Смесь промежуточного соединения 13 (0,0025 моль), (2-бромэтокси)(1,1-диметилэтил)диметилсилана (0,0034 моль) и N-этил-N-(1-метилэтил)-2-пропанамина (0,0038 моль) в DMSO (20 мл) перемешивают при 50 С в течение 15 ч, затем охлаждают до комнатной температуры, выливают в воду со льдом и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (1,7 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 98/2/0,1). Чистые фракции собирают и растворитель выпаривают с получением 0,76 г (54%) промежуточного соединения 14. с) Получение промежуточного соединения 15. Смесь промежуточного соединения 14 (0,0013 моль) и гидроксида натрия (0,0027 моль) в EtOH (40 мл) перемешивают при 80 С в течение ночи, затем охлаждают до комнатной температуры и растворитель выпаривают с получением 0,75 г (100%) промежуточного соединения 15.d) Получение промежуточного соединения 16.EDC (0,002 моль), затем HOBt (0,002 моль) добавляют при комнатной температуре к смеси промежуточного соединения 15 (0,0013 моль) в ТГФ (80 мл) и DCM (80 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 15 мин. Добавляют О-(тетрагидро-2H-пиран-2-ил)гидроксиламин(0,0068 моль). Смесь перемешивают при комнатной температуре в течение 72 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (1,3 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент:DCM/MeOH/NH4OH 95/5/0,1). Чистые фракции собирают и растворитель выпаривают с получением 0,38 г промежуточного соединения 16.- 18010652 е) Получение промежуточного соединения 17. Смесь промежуточного соединения 16 (0,0011 моль) и тетрабутиламмонийфторида (0,0032 моль) в ТГФ (10 мл) перемешивают при комнатной температуре в течение 72 ч, выливают в воду и экстрагируютEtOAc. Органический слой промывают водой, сушат (MgSO4), фильтруют и растворитель выпаривают с получением 0,5 г (88%) промежуточного соединения 17. Пример А 5. а) Получение промежуточного соединения 45. Раствор сложного метилового эфира 2-хлор-5-пиримидинкарбоновой кислоты (0,058 моль) в DMA(80 мл) добавляют по каплям к раствору 4-пиперидинметанамина (0,116 моль) и N-этилдиизопропиламина (0,145 моль) в DMA (150 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 1 ч и 30 мин, выливают в воду со льдом и экстрагируют EtOAc, затем DCM. Органический слой промывают водой, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток кристаллизуют из DIPE. Осадок отфильтровывают и сушат с получением 10 г (65%) промежуточного соединения 45.b) Получение промежуточного соединения 46. Смесь промежуточного соединения 45 (0,0024 моль), 1-метил-5-нитро-1H-индол-3-карбоксальдегида (0,0036 моль) и MgSO4 (0,25 г) в МеОН (80 мл) перемешивают при 60 С в течение ночи, затем охлаждают до комнатной температуры. Натрийтетрагидроборат (0,0041 моль) добавляют по частям. Смесь перемешивают при комнатной температуре в течение 18 ч, выливают в воду и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (1,1 г) кристаллизуют из ацетонитрила. Осадок отфильтровывают и сушат с получением 0,9 г (86%) промежуточного соединения 46, температура плавления 150 С. с) Получение промежуточного соединения 47. Смесь промежуточного соединения 46 (0,002 моль) и гидроксида натрия (0,008 моль) в EtOH (60 мл) перемешивают при 60 С в течение ночи, затем охлаждают до комнатной температуры и выпаривают. Остаток извлекают в простом диэтиловом эфире. Осадок отфильтровывают и сушат с получением 0,6 гd) Получение промежуточного соединения 48.EDC (0,0019 моль) и HOBt (0,0019 моль) добавляют при комнатной температуре к раствору промежуточного соединения 47 (0,0013 моль) и триэтиламина (0,0039 моль) в DCM/ТГФ (50/50) (100 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 15 мин. Добавляют О-(тетрагидро 2 Н-пиран-2-ил)гидроксиламин (0,0026 моль). Смесь перемешивают при комнатной температуре в течение 72 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (1 г) очищают с помощью колоночной хроматографии на кромасиле (5 мкм) (элюент: DCM/MeOH/NH4OH 98/2/0,2-92/8/0,2). Чистые фракции собирают и растворитель выпаривают с получением 0,101 г (15%) промежуточного соединения 48. Пример А 6. а) Получение промежуточного соединения 49. Раствор сложного метилового эфира 2-хлор-5-пиримидинкарбоновой кислоты (0,033 моль) в DCM(80 мл) добавляют при комнатной температуре к раствору 4-пиперидинметанола (0,066 моль) и N-этилдиизопропиламина (0,083 моль) в DCM (100 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 3 ч и 30 мин, выливают в воду со льдом и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток извлекают в пентане. Осадок отфильтровывают и сушат с получением 7,88 г (95%) промежуточного соединения 49.b) Получение промежуточного соединения 50.DMSO (0,058 моль) добавляют по каплям при -78 С к раствору этандиоилдихлорида (0,0278 моль) вDCM (50 мл) в потоке N2. Смесь перемешивают в течение 15 мин. Раствор промежуточного соединения 49(0,023 моль) в DCM (200 мл) добавляют по каплям. Смесь перемешивают при -78 С в течение 1 ч и 30 мин. Триэтиламин (0,118 моль) добавляют по каплям. Смесь перемешивают при -78 С в течение 1 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат с получением 3,06 г (54%) промежуточного соединения 50. с) Получение промежуточного соединения 51. Промежуточное соединение 50 (0,0122 моль) добавляют при 5 С к раствору 1-метил-1H-индол-3 этанамина (0,0122 моль) в МеОН (270 мл) в потоке N2. Смесь перемешивают несколько минут. Добавляют натрийцианоборгидрид (0,0183 моль) и уксусную кислоту (0,0183 моль). Смесь перемешивают при комнатной температуре в течение 48 ч, выливают в карбонат калия 10% и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (4,9 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 97/3/0,1). Чистые фракции собирают и растворитель выпаривают с получением 1,2 г (24%) промежуточного соединения 51.d) Получение промежуточного соединения 52.- 20010652 Смесь промежуточного соединения 51 (0,0009 моль) и гидроксида натрия (0,0039 моль) в EtOH (60 мл) перемешивают и нагревают с обратным холодильником в течение 15 ч, затем выпаривают досуха с получением промежуточного соединения 52. Это промежуточное соединение используют непосредственно на следующей стадии реакции. е) Получение промежуточного соединения 53.HOBt (0,0019 моль), затем EDC (0,0019 моль) добавляют при комнатной температуре к раствору промежуточного соединения 52 (0,0009 моль) и О-(тетрагидро-2H-пиран-2-ил)гидроксиламина (0,0019 моль) в DCM/ТГФ (130 мл). Смесь перемешивают при комнатной температуре в течение 48 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (0,93 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 97/3/0,1). Чистые фракции собирают и растворитель выпаривают с получением 0,155 г (33%) промежуточного соединения 53. Пример А 7. а) Получение промежуточного соединения 54. Смесь сложного этилового эфира 2-[4-(аминометил)-1-пиперидинил]-5-пиримидинкарбоновой кислоты (0,0038 моль), 1-этил-1H-индол-3-карбоксальдегида (0,0049 моль) и Pd/C 10% (0,5 г) в МеОН (20 мл),содержащем 1 мл 10% раствор тиофена в EtOH, гидрируют при комнатной температуре в течение 24 ч при давлении 3 бара, затем фильтруют через целит. Растворитель выпаривают досуха. Остаток (1,8 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 95/5/0,2-93/7/0,5). Чистые фракции собирают и растворитель выпаривают с получением 0,7 г (44%) промежуточного соединения 54.b) Получение промежуточного соединения 55. Гидрид натрия 60% (0,009 моль) добавляют при 0 С к раствору промежуточного соединения 54(0,0045 моль) в ТГФ (50 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 1 ч. Раствор йодэтана (0,0062 моль) в ТГФ (10 мл) добавляют по каплям. Смесь перемешивают при комнатной температуре в течение ночи, выливают в воду и экстрагируют EtOAc. Органический слой отделяют,сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (0,6 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 95/5/0,1). Чистые фракции собирают и растворитель выпаривают с получением 0,16 г (8%) промежуточного соединения 55. с) Получение промежуточного соединения 56.- 21010652 Смесь промежуточного соединения 55 (0,0003 моль) и гидроксида натрия (0,03 г) в EtOH (15 мл) перемешивают при 80 С в течение 6 ч, затем выпаривают досуха с получением 0,16 г (100%) промежуточного соединения 56.d) Получение промежуточного соединения 57.EDC (0,0005 моль) и HOBt (0,0005 моль) добавляют при комнатной температуре к смеси промежуточного соединения 56 (0,0003 моль) в DCM (20 мл) и ТГФ (20 мл) в потоке N2. Смесь перемешивают в течение 15 мин. Добавляют О-(тетрагидро-2 Н-пиран-2-ил)гидроксиламин (0,0007 моль). Смесь перемешивают при комнатной температуре в течение 72 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (0,3 г) очищают с помощью колоночной хроматографии на кромасиле (5 мкм) (элюент: DCM/MeOH/NH4OH 93/7/0,35). Чистые фракции собирают и растворитель выпаривают с получением 0,03 г (16%) промежуточного соединения 57. Пример А 8. а) Получение промежуточного соединения 58. Гидрид натрия (0,011 моль) добавляют при 5 С к раствору промежуточного соединения 13 (0,0037 моль) в ТГФ (30 мл) в потоке N2. Смесь перемешивают в течение 30 мин. Раствор йодметана (0,0081 моль) в ТГФ (10 мл) добавляют по каплям. Смесь перемешивают при 10 С в течение 2 ч, затем доводят до комнатной температуры в течение 1 ч и 30 мин, выливают в воду со льдом и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (1,7 г) очищают с помощью колоночной хроматографии на кромасиле (15-40 мкм) (элюент: DCM/MeOH/NH3OH 98/2/0,1). Две фракции собирают и растворитель выпаривают с получением 0,265 г промежуточного соединения 58 и 0,57 г (17%) промежуточного соединения 10.b) Получение промежуточного соединения 59. Смесь промежуточного соединения 58 (0,0006 моль) и гидроксида натрия (0,0012 моль) в EtOH (30 мл) перемешивают при 80 С в течение ночи, затем охлаждают до комнатной температуры и растворитель выпаривают с получением 0,26 г (100%) промежуточного соединения 59. с) Получение промежуточного соединения 60.EDC (0,0009 моль) и HOBt (0,0009 моль) добавляют при комнатной температуре к раствору промежуточного соединения 59 (0,0006 моль) в ТГФ (30 мл) и DCM (30 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 15 мин. Добавляют О-(тетрагидро-2H-пиран-2-ил)гидроксиламин(0,0012 моль). Смесь перемешивают при комнатной температуре в течение 24 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (0,6 г) очищают с помощью колоночной хроматографии на кромасиле (5 мкм) (элюент:DCM/MeOH/NH4OH 99/1/0,05 и 94/6/0,3). Чистые фракции собирают и растворитель выпаривают с получением 0,1 г (33%) промежуточного соединения 60. Пример А 9. а) Получение промежуточного соединения 61.DMSO (0,127 моль) добавляют при -78 С к раствору этандиоилдихлорида (0,061 моль) в DCM (110 мл) в потоке N2. Смесь перемешивают в течение 15 мин. Добавляют раствор промежуточного соединения 8(0,051 моль) в DCM (200 мл). Смесь перемешивают при -78 С в течение 1 ч и 30 мин. Триэтиламин (0,26 моль) добавляют по каплям. Смесь перемешивают при -78 С в течение 15 мин, затем доводят до комнатной температуры в течение 2 ч и 30 мин. Добавляют воду. Смесь экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (14 г) очищают с помощью колоночной хроматографии на силикагеле (20-45 мкм) (элюент: циклогексан/EtOAc 70/30). Чистые фракции собирают и растворитель выпаривают с получением 7,6 г (57%) промежуточного соединения 61.b) Получение промежуточного соединения 62. Натрийцианоборгидрид (0,049 моль) и уксусную кислоту (0,034 мл) добавляют при комнатной температуре к раствору 5-хлор-1-метил-1H-индол-3-этанамина (0,031 моль) и промежуточного соединения 61 (0,034 моль) в МеОН (700 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 24 ч, выливают в карбонат калия 10% и экстрагируют DCM. Органический слой отделяют, сушат(MgSO4), фильтруют и растворитель выпаривают. Остаток (14,8 г) очищают с помощью колоночной хроматографии на силикагеле (20-45 мкм) (элюент: DCM/MeOH/NH4OH 95/5/0,2). Чистые фракции собирают и растворитель выпаривают с получением 4,52 г (32%) промежуточного соединения 62. с) Получение промежуточного соединения 63. Метансульфонилхлорид (0,0049 моль) добавляют при 5 С к раствору промежуточного соединения 62(0,004 моль) и триэтиламина (0,008 моль) в DCM (150 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 24 ч, выливают в воду со льдом и экстрагируют DCM. Органический слой отделяют,сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (2,39 г) извлекают в DIPE. Осадок отфильтровывают и сушат с получением 1,78 г (84%) промежуточного соединения 63, температура плавления 162 С.d) Получение промежуточного соединения 64. Смесь промежуточного соединения 63 (0,0032 моль) и гидроксида натрия (0,0128 моль) в EtOH (150 мл) перемешивают и нагревают с обратным холодильником в течение 5 ч, затем охлаждают до комнатной температуры и извлекают в простом диэтиловом эфире. Осадок отфильтровывают и сушат с получением- 23010652 1,57 г (99%) промежуточного соединения 64, температура плавления 260 С. е) Получение промежуточного соединения 65.EDC (0,0064 моль) и HOBt (0,0064 моль) добавляют при комнатной температуре к раствору промежуточного соединения 64 (0,0032 моль) в ТГФ (160 мл) и DCM (160 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют О-(тетрагидро-2H-пиран-2-ил)гидроксиламин (0,0064 моль). Смесь перемешивают при комнатной температуре в течение 3 дней, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (2,77 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 97/3/0,1). Чистые фракции собирают и растворитель выпаривают. Остаток(0,385 г) кристаллизуют из CH3CN/простого диэтилового эфира. Осадок отфильтровывают и сушат с получением 0,084 г промежуточного соединения 65, температура плавления 179 С. Пример А 10. а) Получение промежуточного соединения 66. Раствор 2-(метилсульфонил)-5-пиримидинкарбоновой кислоты, сложного этилового эфира (0,094 моль) в ацетонитриле (240 мл) добавляют при комнатной температуре к раствору сложного 1,1-диметилэтилового эфира 4-пиперидинилкарбаминовой кислоты (0,078 моль) и карбоната калия (0,156 моль) в ацетонитриле(120 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение ночи, выливают в воду со льдом и экстрагируют EtOAc. Органический слой промывают водой, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток кристаллизуют из простого диэтилового эфира. Осадок отфильтровывают и сушат с получением 14,4 г (53%) промежуточного соединения 66, температура плавления 160 С.b) Получение промежуточного соединения 67.TFA (20 мл) добавляют при комнатной температуре к раствору промежуточного соединения 66(0,0225 моль) в DCM (110 мл). Смесь перемешивают при комнатной температуре в течение 15 ч, выливают в воду и подщелачивают карбонатом калия. Смесь экстрагируют DCM. Органический слой отделяют, сушат(MgSO4), фильтруют и растворитель выпаривают с получением 5,5 г (98%) промежуточного соединения 67. Пример A11. а) Получение промежуточного соединения 68. Гидрид натрия 60% в масле (0,0069 моль) добавляют при 0 С к раствору сложного этилового эфира 2-метил-1H-индол-3-уксусной кислоты (0,0046 моль) в ТГФ (10 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 1 ч. Добавляют йодэтан (0,006 моль). Смесь перемешивают при комнатной температуре в течение 18 ч и выливают в EtOAc и насыщенный раствор NaCl. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают досуха. Остаток (1,1 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: циклогексан/EtOAc 80/20). Чистые фракции собирают и растворитель выпаривают с получением 0,73 г (65%) промежуточного соединения 68.b) Получение промежуточного соединения 69. Раствор диизобутилалюминийгидрида в толуоле (0,0045 моль) добавляют по каплям при -78 С к раствору промежуточного соединения 68 (0,003 моль) в DCM (15 мл) и 1,2-диметоксиэтана (15 мл) (молекулярные сита: 3 ) в потоке N2. Смесь перемешивают при -78 С в течение 3 ч, затем гасят HCl 3 н. и экстрагируют DCM. Органический слой промывают водой, сушат (MgSO4), фильтруют и растворитель выпаривают досуха с получением 0,7 г (100%) промежуточного соединения 69. с) Получение промежуточного соединения 70. Титан(IV) этоксид (0,0023 моль) добавляют к смеси промежуточного соединения 67 (0,0021 моль) и промежуточного соединения 69 (0,0021 моль) в 1,2-дихлорэтане (25 мл). Смесь перемешивают при комнатной температуре в течение 30 мин. Трис(ацетоО)гидроборат натрий (0,0023 моль) добавляют по частям. Смесь перемешивают при комнатной температуре в течение 18 ч, затем гасят с NaHCO3 и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают досуха. Остаток (1,2 г) очищают с помощью колоночной хроматографии на силикагеле (5 мкм) (элюент:DCM/MeOH/NH4OH 95/5/0,1). Чистые фракции собирают и растворитель выпаривают с получением 0,2 гd) Получение промежуточного соединения 71. Смесь промежуточного соединения 70 (0,0004 моль) и гидроксида натрия (0,0009 моль) в EtOH (30 мл) перемешивают при 60 С в течение ночи, затем охлаждают до комнатной температуры и выпаривают с получением 0,2 г (100%) промежуточного соединения 71. е) Получение промежуточного соединения 72.EDC (0,0007 моль) и HOBt (0,1 г) добавляют при комнатной температуре к раствору промежуточного соединения 71 (0,0004 моль) и триэтиламина (0,0009 моль) в DCM/ТТФ (40 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 15 мин. Добавляют О-(тетрагидро-2H-пиран-2-ил) гидроксиламин (0,0009 моль). Смесь перемешивают при комнатной температуре в течение 72 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (0,4 г) очищают с помощью колоночной хроматографии на кромасиле (5 мкм)(элюент: DCM/MeOH/NH4OH 98/2/0,1-90/10/1). Чистые фракции собирают и растворитель выпаривают с получением 0,087 г (37%) промежуточного соединения 72. Пример А 12. а) Получение промежуточного соединения 73. Промежуточное соединение 50 (0,0046 моль) добавляют при 5 С к раствору 6-метокси-1-метил-1Hиндол-3-этанамина (0,0046 моль) в МеОН (100 мл) в потоке N2. Смесь перемешивают в течение 30 мин. Добавляют натрийцианоборгидрид (0,0068 моль), затем уксусную кислоту (0,0046 моль) по частям. Смесь перемешивают при комнатной температуре в течение 48 ч, выливают в карбонат калия 10% и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (4 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент:DCM/MeOH/NH4OH 96/4/0,2). Чистые фракции собирают и растворитель выпаривают. Часть (0,7 г) остатка (2,2 г) кристаллизуют из ацетонитрила. Осадок отфильтровывают и сушат с получением 0,43 гb) Получение промежуточного соединения 74. Смесь промежуточного соединения 73 (0,0015 моль) и гидроксида натрия (0,006 моль) в EtOH (90 мл) перемешивают и нагревают с обратным холодильником в течение 8 ч, затем выпаривают досуха с получением промежуточного соединения 74. с) Получение промежуточного соединения 75.HOBt (0,003 моль), затем EDC (0,003 моль) добавляют при комнатной температуре к раствору промежуточного соединения 74 (0,0015 моль) и О-(тетрагидро-2H-пиран-2-ил)гидроксиламина (0,003 моль) в ТГФ/DCM (200 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 48 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (1,1 г) очищают с помощью колоночной хроматографии на силикагеле (10 мкм) (элюент: DCM/MeOH/NH4OH 95/5/0,5). Чистые фракции собирают и растворитель выпаривают. Остаток (0,24 г) очищают с помощью колоночной хроматографии на силикагеле (10 мкм) (элюент: DCM/MeOH/NH4OH 95/5/0,5). Чистые фракции собирают и растворитель выпаривают с получением 0,17 г (21%) промежуточного соединения 75. Пример А 13. а) Получение промежуточного соединения 76.b) Получение промежуточного соединения 77. Раствор промежуточного соединения 76 (0,017 моль) в DMF (19 мл) добавляют по каплям при комнатной температуре к суспензии гидрида натрия (0,034 моль) в DMF (11 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 1 ч 30 мин. Добавляют 1-йодпропан (0,034 моль). Смесь перемешивают при комнатной температуре в течение 1 ч и 15 мин. Добавляют насыщенный растворNaCl. Смесь экстрагируют EtOAc. Органический слой промывают водой, сушат (MgSO4), фильтруют и растворитель выпаривают с получением 4,9 г промежуточного соединения 77. Этот продукт используют непосредственно на следующей стадии реакции. с) Получение промежуточного соединения 78. Смесь промежуточного соединения 77 (0,068 моль) и гидразина, моногидрата (0,068 моль) в EtOH(60 мл) перемешивают и нагревают с обратным холодильником в течение 1 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают с получением 3,53 г промежуточного соединения 78. Этот продукт используют непосредственно на следующей стадии реакции.d) Получение промежуточного соединения 79. Натрийцианоборгидрид (0,024 моль) и уксусную кислоту (0,0167 моль) добавляют при комнатной температуре к раствору промежуточного соединения 61 (0,0167 моль) и промежуточного соединения 78(0,015 моль) в МеОН (380 мл) в потоке N2. Смесь перемешивают в течение 30 мин, выливают в карбонат калия 10% и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (8,84 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм)(элюент: DCM/MeOH/NH4OH 96/4/0,2). Чистые фракции собирают и растворитель выпаривают с получением 2,85 г (40%) промежуточного соединения 79. е) Получение промежуточного соединения 80. Смесь промежуточного соединения 79 (0,0028 моль), йодэтана (0,0056 моль) и триэтиламина(0,0085 моль) в DMF (60 мл) перемешивают при 50 С в течение 7 ч, выливают в воду со льдом и экстрагируют EtOAc. Органический слой промывают водой, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (1,8 г) очищают с помощью колоночной хроматографии на кромасиле (5 мкм) (элюент:DCM/MeOH 100/0-95/5). Чистые фракции собирают и растворитель выпаривают с получением 1,1 гf) Получение промежуточного соединения 81. Смесь промежуточного соединения 80 (0,0022 моль) и гидроксида натрия (0,0088 моль) в EtOH (100 мл) перемешивают и нагревают с обратным холодильником в течение 6 ч, затем перемешивают при комнатной температуре в течение ночи и выпаривают досуха. Остаток извлекают в простом диэтиловом эфире. Осадок отфильтровывают и сушат с получением 0,965 г (88%) промежуточного соединения 81, температура плавления 260 С.g) Получение промежуточного соединения 82.EDC (0,0038 моль) и HOBt (0,0038 моль) добавляют при комнатной температуре к раствору промежуточного соединения 81 (0,0019 моль) в ТГФ (100 мл) и DCM (100 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют О-(тетрагидро-2H-пиран-2-ил)гидроксиламин (0,0038 моль). Смесь перемешивают при комнатной температуре в течение 48 ч, выливают в воду и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (1,2 г) очищают с помощью колоночной хроматографии на кромасиле (5 мкм) (элюент:DCM/MeOH/NH4OH 99/1/0,05-93/7/0,35). Чистые фракции собирают и растворитель выпаривают с получением 0,114 г промежуточного соединения 82. Пример А 14. а) Получение промежуточного соединения 83. Смесь промежуточного соединения 62 (0,0034 моль) и гидроксида натрия (0,0134 моль) в EtOH (150 мл) перемешивают при 80 С в течение 3 ч, затем охлаждают до комнатной температуры и выпаривают досуха. Остаток извлекают в простом диэтиловом эфире. Осадок отфильтровывают и сушат с получением 1,18 г (78%) промежуточного соединения 83, температура плавления 260 С.b) Получение промежуточного соединения 84.EDC (0,0052 моль) и HOBt (0,0052 моль) добавляют при комнатной температуре к раствору промежуточного соединения 83 (0,0026 моль) в ТГФ (120 мл) и DCM (120 мл) в потоке N2. Смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют О-(тетрагидро-2H-пиран-2-ил)гидроксиламин (0,0052 моль). Смесь перемешивают при комнатной температуре в течение 6 дней, выливают в воду со льдом и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (2 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм)(элюент: DCM/MeOH/NH4OH 96/4/0,5). Чистые фракции собирают и растворитель выпаривают. Остаток(0,75 г, 55%) очищают с помощью колоночной хроматографии на кромасиле (10 мкм) (элюент:DCM/MeOH/NH4OH 96/4/0,5). Чистые фракции собирают и растворитель выпаривают с получением 0,625 г промежуточного соединения 84. Этот продукт используют непосредственно на следующей стадии реакции.- 28010652 Пример А 15. а) Получение промежуточного соединения 85. 4-Пиперидинметанамин (0,65 моль) и карбонат калия (96 г) перемешивают в ацетонитриле (1000 мл),а затем добавляют раствор сложного этилового эфира 2-(метилсульфонил)-5-пиримидинкарбоновой кислоты (0,37 моль) в ацетонитриле (500 мл) по каплям при комнатной температуре в течение 1 ч. Реакционную смесь перемешивают в течение ночи при комнатной температуре и растворитель выпаривают. Остаток перемешивают в воде и смесь экстрагируют DCM (2500 мл). Органический слой отделяют,промывают водой, сушат (MgSO4), отфильтровывают и растворитель выпаривают. Остаток очищают с помощью колоночной хроматографии на силикагеле (элюент 1: EtOAc/гексан 1/1; элюент 2: MeOH+малое количество NH4OH). Фракции продукта собирают, перемешивают с суспензией карбоната калия и смесь экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), отфильтровывают и растворитель выпаривают с получением 31 г (32%) промежуточного соединения 85. Пример А 16. а) Получение промежуточного соединения 86.(0,0056 моль) в ТГФ (52 мл) в потоке N2. Смесь перемешивают при 0 С в течение 1 ч. Добавляют 1-йодпропан (0,0067 моль). Смесь перемешивают при комнатной температуре в течение 2 дней, выливают в воду и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают с получением 1,5 г промежуточного соединения 86. Этот продукт используют непосредственно на следующей стадии реакции.b) Получение промежуточного соединения 87. Натрийцианоборгидрид (0,0068 моль) и уксусную кислоту (0,0046 моль) добавляют при комнатной температуре к раствору промежуточного соединения 85 (0,0042 моль) и промежуточного соединения 86(0,0051 моль) в MeOH (120 мл) в потоке N2. Смесь перемешивают и нагревают с обратным холодильником в течение 2 дней, затем охлаждают до комнатной температуры, выливают в карбонат калия 10% и экстрагируют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (2,42 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 95/5/0,2). Чистые фракции собирают и растворитель выпаривают с получением 1,2 г (60%) промежуточного соединения 87. с) Получение промежуточного соединения 88. Смесь промежуточного соединения 87 (0,0025 моль) и гидроксида натрия (0,01 моль) в EtOH (120 мл) перемешивают и нагревают с обратным холодильником в течение 4 ч, затем выпаривают досуха. Остаток извлекают в простом диэтиловом эфире. Осадок отфильтровывают и сушат с получением 0,845 г (72%) промежуточного соединения 88, температура плавления 260 С.d) Получение промежуточного соединения 89.EDC (0,0036 моль) и HOBt (0,0036 моль) добавляют при комнатной температуре к раствору промежуточного соединения 88 (0,0018 моль) в ТГФ (90 мл) и DCM (90 мл) в потоке N2. Смесь перемешивают в течение 30 мин. Добавляют О-(тетрагидро-2H-пиран-2-ил)гидроксиламин (0,0036 моль). Смесь перемешивают при комнатной температуре в течение 3 дней, выливают в воду со льдом и экстрагируют(1,3 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент:DCM/MeOH/NH4OH 92/8/0,5). Чистые фракции собирают и растворитель выпаривают с получением 0,54 г(56%) промежуточного соединения 89. Пример А 17. а) Получение промежуточного соединения 90. Метансульфонилхлорид (0,004 моль) добавляют при 10 С к раствору 1,2-диметил-1H-индол-3-этанола (0,0026 моль) и триэтиламина (0,008 моль) в DCM (10 мл) в потоке N2. Смесь перемешивают при 10 С в течение 4 ч. Растворитель выпаривают досуха с получением промежуточного соединения 90. Этот продукт используют непосредственно на следующей стадии реакции.b) Получение промежуточного соединения 91. Смесь промежуточного соединения 85 (0,0054 моль), промежуточного соединения 90 (0,0075 моль) и карбоната калия (0,021 моль) в ацетонитриле (150 мл) перемешивают и нагревают с обратным холодильником в течение 2 дней, затем охлаждают до комнатной температуры, выливают в воду со льдом и экстрагируют EtOAc. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток (1,88 г) очищают с помощью колоночной хроматографии на силикагеле (15-40 мкм) (элюент: DCM/MeOH/NH4OH 97/3/0,1). Чистые фракции собирают и растворитель выпаривают с получением 0,15 г (7%) промежуточного соединения 91. с) Получение промежуточного соединения 92. Смесь промежуточного соединения 91 (0,0003 моль) в гидроксиде натрия (0,0014 моль) и EtOH (20 мл) перемешивают и нагревают с обратным холодильником в течение 1 дня, затем охлаждают до комнатной температуры и выпаривают досуха. Остаток извлекают в простом диэтиловом эфире. Осадок отфильтровывают и сушат с получением 0,12 г (82%) промежуточного соединения 92, температура плавления 260 С.d) Получение промежуточного соединения 93.
МПК / Метки
МПК: A61P 35/00, C07D 409/14, A61K 31/506, C07D 405/14, C07D 401/14
Метки: качестве, новых, индолильные, ингибиторов, алкиламинопроизводные, гистондеацетилазы, замещённые
Код ссылки
<a href="https://eas.patents.su/30-10652-zameshhyonnye-indolilnye-alkilaminoproizvodnye-v-kachestve-novyh-ingibitorov-gistondeacetilazy.html" rel="bookmark" title="База патентов Евразийского Союза">Замещённые индолильные алкиламинопроизводные в качестве новых ингибиторов гистондеацетилазы</a>
Сульфонилпроизводные в качестве новых ингибиторов гистон-деацетилазы

Номер патента: 6707
Опубликовано: 24.02.2006
Авторы: Дяткин Алексей Борисович, Ван Брандт Свен Францискус Анна, Де Винтер Ханс Луи Йос, Вердонк Марк Густаф Селин, Ван Эмелен Кристоф, Пилатт Изабелль Ноэлль Констанс, Ван Хьюсден Джимми Арнольд Вивиан, Мерпул Ливен, Артс Янине, Бакс Лео Якобус Йозеф, Понселе Виржини Софи
МПК: C07D 213/82, C07D 207/28, C07D 215/36...
Метки: ингибиторов, сульфонилпроизводные, гистон-деацетилазы, качестве, новых
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, его фармацевтически приемлемые соли добавления кислот и его стереохимически изомерные формы, где n равно 0, 1, 2 или 3 и, если n равно 0, то имеется в виду прямая связь; t равно 0, 1, 2, 3 или 4, и если t равно 0, то имеется в виду прямая связь; каждый Q представляет собой азот или каждый X представляет собой азот или каждый Y представляет собой азот или каждый Z представляет собой азот или ...
6-замещённые циклогексилалкил 2-замещённые хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли (adp-рибоза) полимеразы

Номер патента: 10592
Опубликовано: 30.10.2008
Авторы: Ван Дюн Якобус Альфонсус Йозефус, Мабир Доминик Жан-Пьер, Ваутерс Вальтер Баудевейн Леопольд, Сомерс Мария Викторина Франциска
МПК: C07D 401/06, C07D 241/44
Метки: ингибиторов, adp-рибоза, циклогексилалкил, полимеразы, хинолиноны, поли, 2-замещённые, 6-замещённые, 2-хиноксалиноны, качестве
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, аддитивные соли и стереохимические изомерные формы, в котором n представляет собой 0 или 1; s представляет собой 0 или 1; X представляет собой -N= или -СН=; Y представляет собой -N< или -СН<; Q представляет собой -NH-, -O-, -С(О)-, -СН2-СН2- или -CHR5-, где R5 является водородом, гидрокси, C1-6алкилом, арилС1-6алкилом, C1-6алкилоксикарбонилом, C1-6 алкилоксиC1-6алкиламино или...
2-амино-6-(2-замещенные-4-фенокси)-замещенные-пиридины в качестве ингибиторов синтазы окиси азота

Номер патента: 3834
Опубликовано: 30.10.2003
Авторы: Волкманн Роберт Альфред, Новаковски Йоланта, Лауэ Джон Адамс III
МПК: A61K 31/4418, C07D 213/73, A61P 25/00...
Метки: ингибиторов, 2-амино-6-(2-замещенные-4-фенокси)-замещенные-пиридины, азота, синтазы, качестве, окиси
Формула / Реферат:
1. Соединение формулы где R1 и R2 выбраны независимо из группы, включающей водород, галоид, гидрокси, (C1-C6)алкокси, (C1-C7)алкил, (C2-C6)алкенил, (C2-C10)алкоксиалкил; G выбирают из группы, включающей водород, (C1-C6)алкил, (C1-C6)алкокси(C1-C3)алкил, аминокарбонил-(C1-C3)алкил-, (C1-C3)алкиламинокарбонил-(C1-C3)алкил-, ди-[(C1-C3)алкил]аминокарбонил-(C1-C3)алкил и N(R3)(R4)(C0-C4)алкил-, где R3 и R4 независимо выбирают из группы, включающей...
Триамид-замещённые индолы, бензофураны и бензотиофены в качестве ингибиторов микросомального белка, переносящего триглицериды, (мтр) и/или ингибиторов секреции аполипопротеина в (аро в)

Номер патента: 7008
Опубликовано: 30.06.2006
Авторы: Бронк Брайан Скотт, Хуатан Хип, Блайз Алан Элвуд, Бертинато Питер, Ли Дзин, Мейсон Клайв Филип, Ченг Хенгмиао
МПК: A61K 31/4439, A61K 31/404, A61K 31/343...
Метки: качестве, секреции, триглицериды, белка, бензофураны, переносящего, ингибиторов, мтр, триамид-замещённые, аполипопротеина, бензотиофены, аро, микросомального, индолы
Формула / Реферат:
1. Соединение формулы 1b или его фармацевтически приемлемая соль, где R1 представляет собой заместитель в 5 или 6 положении формулы 1b и имеет структуру или, когда R7 представляет собой фенил, пиридил, фенил-Z1-или пиридил-Z1-, необязательно замещенные от одного до пяти, независимо, выбранными R12, то R1 представляет собой (C1-С6)алкил, (С3-С8)циклоалкил, (С5-С10)бициклоалкил, -(CRaRb)tO(C1-C6 алкил), -(СRaRb)tS(C1-C6 алкил), -(CRaRb)rC(O)R15,...
Замещённые производные пиррола и их применение в качестве ингибиторов hmg-cоa

Номер патента: 9646
Опубликовано: 28.02.2008
Авторы: Саттигери Джитендра, Чугх Анита, Кумар Ятендра, Салман Мохаммад, Ариян Рам Чандер, Раманатан Викрам Кришна
МПК: C07D 207/34, A61K 31/366, A61K 31/40...
Метки: производные, ингибиторов, замещённые, пиррола, hmg-cоa, применение, качестве
Формула / Реферат:
1. Соединения, имеющие структурную формулу I их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, полиморфы, таутомеры, или N-оксиды, где или R1 может быть С1-С6алкил, С3-С6 циклоалкил или необязательно замещенный фенил (где до трех заместителей независимо выбраны из галоидов, C1-С6алкила, циано или С1-С3перфторалкила); R2 может быть необязательно замещенный фенил (где до трех заместителей независимо выбраны из циано,...
Предыдущий патент: Применение замещенных хинолиновых производных для лечения заболеваний, вызываемых микобактериями, резистентными к лекарственным средствам
Следующий патент: Нацеливание на опухолевую сосудистую сеть при использовании меченного радиоактивным изотопом антитела l 19 к ed – b фибронектину