Новые бензальдоксимы, способ получения бензальдоксимов и его применение
Номер патента: 10633
Опубликовано: 30.10.2008
Авторы: Лохтман Рене, Бауманн Эрнст, Мисслитц Ульф, Фон Дейн Вольфганг, Гебхардт Йоахим, Хаген Хельмут, Гётц Норберт, Витшель Маттиас, Ракк Михаель, Кейль Михаэль, Райнхаймер Йоахим







Формула / Реферат
1. Способ получения соединений формулы XV

где остатки имеют следующие значения:
X - NO2, S(O)nRy;
Rx, Ry независимо друг от друга означают галоген, карбоксил, карбоксамид, N-алкилкарбоксамид, N,N-диалкилкарбоксамид, фенил, С1-С6алкил, C1-C6алкокси, C1-C6алкилтио;
m равно 0, 1, 2, 3 или 4;
n равно 0, 1 или 2;
включающий взаимодействие соединений формулы XVI

где заместители имеют вышеприведенные значения, с органическим нитритом общей формулы R-O-NO, где R представляет собой алифатический или ароматический остаток, при воздействии основания, при этом взаимодействие проводят при температуре от -50 до -20шС в присутствии диполярного апротонного растворителя, и необязательно включающий превращение оксимовой группы -CH=NOH в соединении формулы XV с получением соответствующих альдегидов -СНО, нитрилов (-CN) или нитрилоксидов (-CNO).
2. Способ по п.1, при котором в качестве растворителя применяют диметилформамид.
3. Соединения формулы XV

где остатки имеют следующие значения:
X - S(O)nRy;
R1 - водород, C1-C6алкил, галоген, C1-C6алкокси, C1-C6алкилтио;
Rx означает галоген, карбоксил, карбоксамид, N-алкилкарбоксамид, N,N-диалкилкарбоксамид, фенил, C1-C6алкил, C1-C6алкокси, C1-C6алкилтио;
Ry - C1-C6алкил;
m равно 1;
n равно 0, 1 или 2.
4. Применение способа получения по п.1 для осуществления способа получения соединения формулы X

где заместители имеют следующие значения:
R1 - водород, C1-C6алкил;
R2 - С1-С6алкил;
R3, R4, R5 - водород, C1-C6алкил или R4 и R5 образуют вместе связь;
n равно 0, 1 или 2,
характеризующегося:
а) циклизацией оксима формулы XV, когда X означает NO2, m означает 0 или 1, а, если m равно 1, Rx означает C1-C6алкил, расположенный в орто-положении относительно группы CH=NOH, алкеном формулы IV

где остатки R3 до R5 имеют приведенные выше значения, в присутствии основания с получением 4,5-дигидроизоксазола формулы V

где остатки R1, R3 до R5 имеют приведенные выше значения;
б) восстановлением нитрогруппы в присутствии катализатора с получением анилина формулы VI

где остатки R1, R3 до R5 имеют приведенные выше значения;
в) взаимодействием анилина формулы VI с диалкилдисульфидом формулы VII
![]()
в присутствии огранического нитрита и необязательно катализатора с получением простого тиоэфира формулы VIII

где остатки R1 до R5 имеют приведенные выше значения;
г) бромированием простого тиоэфира формулы VIII средством бромирования с получением соединения формулы X, где n равно 0; и необязательно
д) окислением соединения формулы X, где n равно 0, окислительным агентом с получением соединения формулы X, где n имеет значения 1 или 2.
Текст
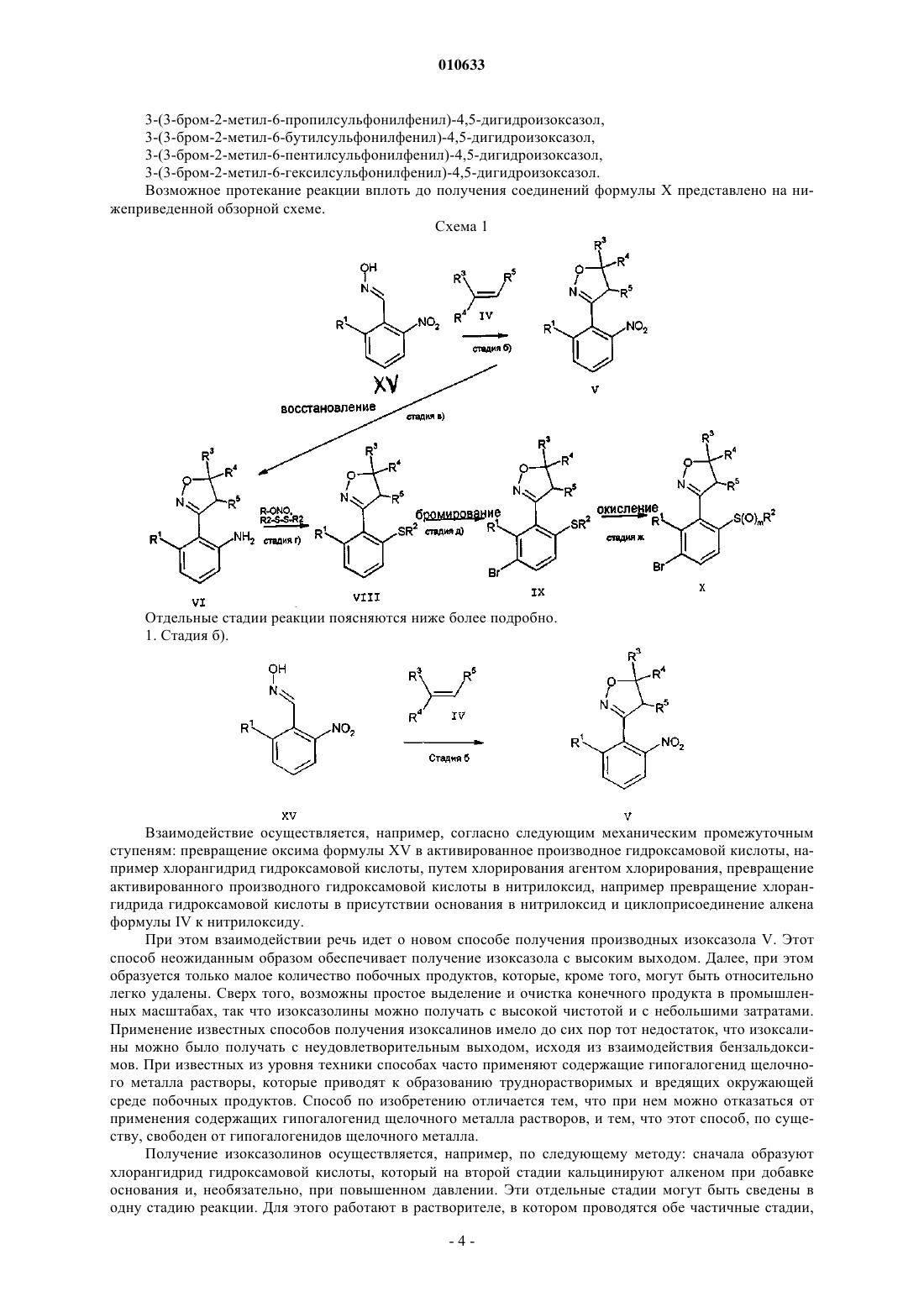
010633 Настоящее изобретение относится к способу получения бензальдоксимов, к новым бензальдоксимам, а также к применению способа получения бензальдоксимов для синтеза промежуточных продуктов для получения изоксазолин-3-ил-ацилбензолов. Изоксазолин-3-ил-ацилбензолы являются ценными соединениями, которые могут применяться в области защиты растений. Например, в заявке WO 98/31681 2-алкил-3-(4,5-дигидроизоксазол-3-ил)ацилбензолы описываются как гербицидные действующие вещества. Задачей изобретения является разработка промежуточных соединений и способов их получения, предназначенных для альтернативного способа получения замещенных 3-гетероциклилом производных бензоила. Описанный в заявке WO 98/31681 способ получения 2-алкил-3-(4,5-дигидроизоксазол-3-ил)ацилбензолов, соответственно, их промежуточных продуктов (производных 2-алкил-3-(4,5-дигидроизоксазол-3-ил) бромбензола) плохо пригоден для производства таких соединений в крупных промышленных масштабах,так как синтез осуществляется в несколько стадий и выход соответствующего конечного продукта в пересчете на использованный на первой стадии исходный продукт является относительно низким. Получение соединений, соответственно, промежуточных соединений, которые по структуре схожи с соединениями формулы I, известно из различных публикаций. Из WO 96/26206 известен способ получения 4-[3-(4,5-дигидроизоксазол-3-ил)бензоил]-5-гидроксипиразолов, при котором на последней стадии 5-гидроксипиразол подвергают взаимодействию с производным 3-(4,5-дигидроизоксазол-3-ил)бензойной кислоты. Необходимое для этого способа производное 3-(4,5 дигидроизоксазол-3-ил)бензойной кислоты можно получать сложным путем с использованием множества стадий. Поэтому такой способ является относительно дорогим и не оптимальным по своей экономичности. В DE 19709118 описывается способ получения 3-(4,5-дигидроизоксазол-3-ил)бензойных кислот, исходя из 3-бром-(4,5-дигидроизоксазол-3-ил)бензола, соединений Гриньяра и диоксида углерода. Неожиданным образом было установлено, что число стадий способа получения замещенных 3-гетероциклилом производных бензоила по сравнению с описанным в WO 98/31681 способом можно снизить,если синтез осуществлять по отобранным промежуточным соединениям. Кроме того, предлагаемый способ имеет то преимущество, что общий выход конечного продукта формулы I, соответственно, промежуточных продуктов X, в пересчете на использованные исходные вещества, является выше, чем выход по описанному в WO 98/31681 способу. Сверх того, соответствующие промежуточные продукты отдельных стадий способа могут быть получены с хорошим выходом продукта. Кроме того, отдельные стадии способа пригодны для технического (промышленного) получения промежуточных продуктов, так как они позволяют экономичное и недорогое изготовление. Далее, преимуществом является то, что применяемыми исходными веществами являются простые в изготовлении химические вещества, которые можно покупать у многих независимых поставщиков химреагентов в большом количестве. В общем, способ по изобретению представляет собой недорогой, экономичный и надежный способ получения гербицидноактивных действующих веществ формулы I в промышленных масштабах. Объектом настоящего изобретения является способ, пригодный для получения бензальдоксимов формулы XV где остатки имеют следующие значения:Rx, Ry независимо друг от друга означают галоген, карбоксил, карбоксамид, N-алкилкарбоксамид,N,N-диалкилкарбоксамид, фенил, C1-C6 алкил, C1-C6 алкокси, C1-C6 алкилтио;Rx, Ry представляют собой органические остатки, которые могут быть одинаковыми или различными и при выбранных условиях реакции химически инертными. Например, остаток Rx может быть галогеном, таким как хлор, бром или йод; карбоксилом; карбоксиамидом; N-алкилкарбоксимидом и N,Nдиалкилкарбоксамидом; фенилом; C1-C6 алкилом, таким как метил, этил; C1-C6 алкокси; C1-C6 алкилтио. В том случае, если m1, Rx может быть одинаковым или различным. Предпочтительно Rx имеет такое же значение, что и R1, и находится в орто-положении относительно оксимовой группы -CH=NOH. В частности, m означает 2, причем один из заместителей Rx имеет такое же значение, что и R1, и второй заместитель Rx представляет собой атом галогена, который предпочтительно находится в мета-положении к оксимовой группе. Ry означает C1-C6 алкил, например метил, этил, пропил. Способ включает взаимодействие соединений формулы XVI-1 010633 где заместители имеют вышеприведенное значение, с органическим нитритом общей формулы R-O-NO,где R представляет собой алифатический или ароматический остаток, при воздействии основания, при этом взаимодействие проводят при температуре от -50 до -20 С в присутствии диполярного апротонного растворителя, и необязательно включающий превращение оксимовой группы -CH=NOH в соединении формулы XV с получением соответствующих альдегидов -СНО, нитрилов (-CN) или нитрилоксидов (-CNO). В качестве растворителя применяют диметилформамид. Объектом настоящего изобретения являются также новые соединения формулы XV где остатки имеют следующее значение:n равно 0, 1 или 2. Предпочтительными соединениями формулы XV являются такие соединения, при которых X означает группу SO2-Ry, m равно 2. В этом случае один из остатков Rx предпочтительно представляет собой галоген (например, бром или хлор) и находится в мета-положении к оксимовой группе. Второй остатокRx предпочтительно является C1-C6 алкилом (например, метилом, этилом) и находится в орто-положении по отношению к оксимовой группе. В рамках настоящей заявки описан способ получения соединениий формулы XV для осуществления способа получения соединения формулы X где заместители имеют следующее значение:n равно 0, 1 или 2,характеризующегося: а) циклизацией оксима формулы XV, когда X означает NO2, m означает 0 или 1, а, если m равно 1,Rx означает С 1-C6 алкил, расположенный в орто-положении относительно группы CH=NOH, алкеном формулы IV где остатки R3 до R5 имеют приведенные выше значения, в присутствии основания с получением 4,5 дигидроизоксазола формулы V где остатки R1, R3 до R5 имеют приведенные выше значения; б) восстановлением нитрогруппы в присутствии катализатора с получением анилина формулы VI где остатки R1, R3 до R5 имеют приведенные выше значения; в) взаимодействием анилина формулы VI с диалкилдисульфидом формулы VII в присутствии огранического нитрита и необязательно катализатора с получением простого тиоэфира формулы VIII где остатки R1 до R5 имеют приведенные выше значения; г) бромированием простого тиоэфира формулы VIII средством бромирования с получением соединения формулы X, где n равно 0; и необязательно д) окислением соединения формулы X, где n равно 0, окислительным агентом с получением соединения формулы X, где n имеет значения 1 или 2.C1-C6 Алкил, соответственно, C1-С 4 алкил во всех случаях означает разветленную или неразветвленную алкильную группу с 1-6, соответственно, 1-4 атомами углерода, такую как метил, этил, н-пропил,изопропил, н-бутил, изобутил, н-пентил или н-гексил. Аналогичное действительно для C1-C6 алкоксигруппы.R1 предпочтительно означает алкильную группу, в частности метил, этил, изопропил, н-пропил или н-бутильную группу.R3, R4 или R5 означают предпочительно водород. R4 и R5 могут вместе представлять собой связь, так что они образуют соответствующие производные изоксазола. В этом случае R3 предпочтительно означает водород. Соединения формулы X (3-(4,5-дигидроизоксазол-3-ил)бромбензолы) представляют собой ценные промежуточные соединения для получения действующих веществ формулы где заместители имеют следующее значение:n равно 0, 1 или 2. Соединения I пригодны, например, как описано в WO 96/26206 и WO 97/35850, в качестве средств защиты растений, в частности гербицидов. Предпочтительными соединениями формулы X являются следующие соединения: 3-(3-бром-2-метил-6-метилсульфонилфенил)-4,5-дигидроизоксазол,3-(3-хлор-2-метил-6-метилсульфонилфенил)-4,5-дигидроизоксазол,3-(3-бром-6-метилсульфонилфенил)-4,5-дигидроизоксазол,3-(3-бром-2-этил-6-метилсульфонилфенил)-4,5-дигидроизоксазол,3-(3-бром-2-изопропил-6-метилсульфонилфенил)-4,5-дигидроизоксазол,3-(3-бром-2-метил-6-этилсульфонилфенил)-4,5-дигидроизоксазол,-3 010633 3-(3-бром-2-метил-6-пропилсульфонилфенил)-4,5-дигидроизоксазол,3-(3-бром-2-метил-6-бутилсульфонилфенил)-4,5-дигидроизоксазол,3-(3-бром-2-метил-6-пентилсульфонилфенил)-4,5-дигидроизоксазол,3-(3-бром-2-метил-6-гексилсульфонилфенил)-4,5-дигидроизоксазол. Возможное протекание реакции вплоть до получения соединений формулы X представлено на нижеприведенной обзорной схеме. Схема 1 Отдельные стадии реакции поясняются ниже более подробно. 1. Стадия б). Взаимодействие осуществляется, например, согласно следующим механическим промежуточным ступеням: превращение оксима формулы XV в активированное производное гидроксамовой кислоты, например хлорангидрид гидроксамовой кислоты, путем хлорирования агентом хлорирования, превращение активированного производного гидроксамовой кислоты в нитрилоксид, например превращение хлорангидрида гидроксамовой кислоты в присутствии основания в нитрилоксид и циклоприсоединение алкена формулы IV к нитрилоксиду. При этом взаимодействии речь идет о новом способе получения производных изоксазола V. Этот способ неожиданным образом обеспечивает получение изоксазола с высоким выходом. Далее, при этом образуется только малое количество побочных продуктов, которые, кроме того, могут быть относительно легко удалены. Сверх того, возможны простое выделение и очистка конечного продукта в промышленных масштабах, так что изоксазолины можно получать с высокой чистотой и с небольшими затратами. Применение известных способов получения изоксалинов имело до сих пор тот недостаток, что изоксалины можно было получать с неудовлетворительным выходом, исходя из взаимодействия бензальдоксимов. При известных из уровня техники способах часто применяют содержащие гипогалогенид щелочного металла растворы, которые приводят к образованию труднорастворимых и вредящих окружающей среде побочных продуктов. Способ по изобретению отличается тем, что при нем можно отказаться от применения содержащих гипогалогенид щелочного металла растворов, и тем, что этот способ, по существу, свободен от гипогалогенидов щелочного металла. Получение изоксазолинов осуществляется, например, по следующему методу: сначала образуют хлорангидрид гидроксамовой кислоты, который на второй стадии кальцинируют алкеном при добавке основания и, необязательно, при повышенном давлении. Эти отдельные стадии могут быть сведены в одну стадию реакции. Для этого работают в растворителе, в котором проводятся обе частичные стадии,-4 010633 например в сложном эфире карбоновой кислоты, таком как сложный этиловый эфир уксусной кислоты,хлорбензол или ацетонитрил. Получение хлорангидридов гидроксамовой кислоты посредством N-хлорсукцинимида в диметилформамиде известно из литературных источников (Лиу и др., J. Org. Chem. 1980, 45: стр. 3916-3918). Однако здесь можно найти указание на то, что о-нитробензальдоксимы могут переводиться хлорированием в хлорангидриды гидроксамовой кислоты с плохим выходом (Chiang, J. Org. Chem. 1971, 36: стр. 21462155). В качестве побочной реакции следует при этом ожидать образование бензалхлорида. По вышеописанному способу были неожиданным образом предложены условия, которые позволяют получение хлорангидридов гидроксамовой кислоты с высоким выходом. В частности, особенно преимущественным является то, что при этом можно применять недорогой хлор. Реакция взаимодействия осуществляется, например, при следующих условиях: растворитель - галогеналканы, такие как 1,2-дихлорэтан или метиленхлорид; ароматы, такие как бензол, толуол, холрбензол,нитробензол или ксилол; полярные апротонные растворители, например N,N-диалкилформамиды, -ацетамиды, N-метилпирролидон, диметилпропиленмочевина, тетраметилмочевина, ацетонтитрил, пропионитрил; спирты, такие как метанол, этанол, н-пропанол или изопропанол; карбоновые кислоты, такие как уксусная кислота или пропионовая кислота, сложные эфиры карбоновой кислоты, такие как сложный этиловый эфир уксусной кислоты. Предпочтение отдается следующим растворителям: уксусной кислоте,метанолу, этанолу, 1,2-дихлорэтану, метиленхлориду или хлорбензолу или сложному этиловому эфиру уксусной кислоты. Взаимодействие осуществляют при температуре от -40 до 100 С; предпочтительно от-10 до 40 С, соответственно, от 0 до 30 С. В качестве средства галогенирования могут применяться Nхлорсукцинимид, элементарный хлор, предпочтительно хлор. Стехиометрическое соотношение составляет, например, 1-3 экв. средства галогенирования, предпочтительно 1-1,5 экв. Дозировка производится в случае хлора посредством инициирования, при N-хлорсукцинимиде в качестве твердого вещества или, в случае необходимости, в подходящем растворителе. Переработка осуществляется, например, по следующей схеме: а) без очистки. Раствор применяют далее; б) замена растворителя путем отгонки растворителя; в) подача воды и экстракция хлорангидрида гидроксамовой кислоты подходящим растворителем. При добавке основания из хлорангидрида гидроксамовой кислоты образуются нитрилоксиды. Так как они нестабильны, проблема состояла в нахождении таких условий, которые позволяют стабилизировать нитрилоксиды и преобразовывать их в желаемые продукты. Эта задача неожиданным образом была решена за счет того, что были выбраны следующие условия реакции: в качестве растворителя применяли галогеналканы, такие как 1,2-дихлорэтан или метиленхлорид; в качестве ароматов такие, как бензол, толуол, хлорбензол, нитробензол или ксилол; полярные апротонные растворители, например N,Nдиалкилформамиды, -ацетамиды, N-метилпирролидон, диметилпропиленмочевину, тетраметилмочевину,ацетонитрил, пропионитрил; сложные эфиры карбоновой кислоты, такие как сложный этиловый эфир уксусной кислоты. Предпочтительно применяют 1,2-дихлорэтан, метиленхлорид, толуол, ксилол, сложный этиловый эфир уксусной кислоты или хлорбензол. Температура реакции взаимодействия составляет от 0 до 100 С, предпочтительно от 0 до 50 С или от 0 до 30 С. В качестве основания применяют третичные амины, например триэтиламин, цикличные амины, такие как N-метилпиперидин или N,N'-диметилпиперазин, пиридин, карбонаты щелочных металлов, например карбонат натрия или карбонат калия, гидрокарбонаты щелочных металлов, например гидрокарбонат натрия или гидрокарбонат калия, карбонаты щелочно-земельных металлов, например карбонат кальция, гидроокиси щелочных металлов, например гидроокись натрия или гидроокись калия. Предпочтительно применяют триэтиламин, карбонат натрия, гидрокарбонат натрия или гидроокись натрия. Стехиометрическое соотношение составляет, например, 1-3 экв. основания, предпочтительно 1-1,5 экв.; 1-5 экв. алкена, предпочтительно 1-2 экв. Дозировка осуществляется предпочтительно при повышенном давлении алкена посредством медленной подачи основания. Взаимодействие происходит при нормальном давлении до 10 атм, предпочтительно 1-6 атм. 2. Стадия в). При этой реакции речь идет о новом хемоселективном гидрировании нитрогруппы наряду с изоксазолином, которое до сих пор не было известным. Неожиданным образом было установлено, что при вы-5 010633 бранных условиях реакции расщепляется связь N-O изоксалинового цикла. Каталитическое гидрирование ароматических нитросоединений с получением анилинов уже давно известно (см. Хубен-Вейл, BdIV/1c, стр. 506 и след.). С другой стороны, также известно, что N-O-связь изоксалинов может расщепляться каталитическим гидрированием, например со скелетным никелем (Ранея) (Curran и др., Synthesis 1986, стр. 12-315) или палладием (Auricchio и др., Tetrahedron, 43, стр. 3983-3986, 1987) в качестве катализатора. Превращение происходит, например, при следующих условиях: в качестве растворителя пригодны ароматы, такие как бензол, толуол, ксилол; полярный апротонный растворитель, например N,N-диалкилформамиды, -ацетамиды, N-метилпирролидон, диметилпропиленмочевина, тетраметилмочевина; сложный эфир карбоновой кислоты, такой как сложный этиловый эфир уксусной кислоты, простой эфир, такой как диэтиловый эфир или метил-трет.-бутиловый эфир, циклический эфир, такой как тетрагидрофуран или диоксан; спирты, такие как метанол, этанол, н-пропанол или изопропанол; карбоновые кислоты,такие как уксусная кислота или пропионовая кислота. Предпочтительно применяют следующие растворители: этилацетат, толуол, ксилол, метанол. Взаимодействие осуществляют при температуре от -20 до 100 С; предпочтительно от 0 до 50 С, особенно предпочтительно от 0 до 30 С. В качестве катализатора применяют платиновый или палладиевый катализатор на носителе из активного угля при содержании от 0,1 до 15 мас.% в пересчете на активный уголь носителя. При применении палладиевого катализатора он может быть легирован примесью серы или селена, чтобы получить лучшую селективность. Предпочтительно применяют платину/активный уголь или палладий/активный уголь с содержанием платины или палладия 0,5-10 мас.%. Для реакции взаимодействия имеются, например, следующие стехиометрические соотношения: от 0,001 до 1 мас.% платины или палладия в пересчете на нитросоединение; предпочтительно от 0,01 до 1 мас.% платины. Дозировку воды проводят непрерывно или прерывно, предпочтительно прерывно при нормальном давлении до 50 атм, предпочтительно до 10 атм. Переработка реакционной смеси осуществляется отделением катализатора посредством фильтрации. Катализатор, в случае необходимости, может снова применяться. Растворитель отгоняется. Для последующего превращения на следующей стадии способа продукт может применяться непосредственно,без очистки. При потребности продукт может также далее очищаться. Очистку продукта осуществляют,например, по следующей схеме: если это требуется, анилин может очищаться посредством загрузки в разбавленную минеральную кислоту, например водную соляную кислоту или разбавленную серную кислоту, экстрагирования подходящим органическим агентом, например галогеналканами, такими как 1,2 дихлорэтан или метиленхлорид, ароматами, такими как бензол, толуол, хлорбензол или ксилол, простыми эфирами, такими как диэтиловый эфир или метил-трет.-бутиловый эфир, сложными эфирами карбоновой кислоты, такими как сложный этиловый эфир уксусной кислоты, и с помощью основания снова высвобождаться. 3. Стадия г). Реакция взаимодействия осуществляется при следующих условиях: в качестве растворителя применяют, например, галогеналканы, такие как 1,2-дихлорэтан или метиленхлорид, ароматы, такие как бензол, толуол, хлорбензол, нитробензол или в избытке диалкилдисульфид как растворитель. Предпочтительно применяют избыток диалкилсульфида в качестве растворителя. Температура реакции взаимодействия составляет от 40 до 150, предпочтительно от 50 до 100, особенно предпочтительно от 60 до 90 С. В качестве реагентов применяют нитриты (R-ONO), как, например, алкилнитриты, предпочтительно нбутилнитрит, (изо)амилнитрит или трет.-бутилнитрит. При этом R означает любой органический или химически инертный остаток, который не оказывает влияния на реакцию. Остаток R является, например,C1-C6 алкилом или C2-C6 алкенильной группой. При взаимодействии соединений имеется следующее стехиометрическое соотношение: 1-3 экв. алкилнитрита, предпочтительно 1-1,5 экв. алкилнитрита. В качестве катализатора могут применяться медный порошок, элементарная медь другой формы, как, например, стружка, проволока, гранулят, дробь,стержни; соли меди(I), например хлорид меди(I), бромид меди(I) или йодид меди(I), соли меди(II), или элементарный йод, в частности предпочтительно медный порошок. При проведении реакции в растворителе применяют 1-3 экв. диалкилдисульфида, предпочтительно 1-2 экв. При одной предпочтительной-6 010633 форме проведения способа диалкилдисульфид применяют в качестве растворителя в избытке и после этого регенерируют отгонкой. На дальнейших стадиях продукт может применяться без очистки. В случае необходимости, перед этим можно проводить очистку продукта путем дистилляции или кристаллизации с помощью подходящего растворителя, например простого диизопропилового эфира. 4. Стадия д). Бромирование происходит аналогично описанному в WO 98/31676 методу. В качестве растворителя предпочтительной являтся уксусная кислота. 5. Стадия ж). Окисление осуществляется анаолгично описанному в WO 98/31676 методу. С помощью приведенных ниже примеров выполнения изобретение поясняется более подробно. Пример 1. Получение 2-метил-6-нитробензальдоксима (вариант А). Раствор из 274 г (2,6 моль) н-бутилнитрита (97%) и 300 г (2,0 моль) 3-нитро-о-ксилола (97%) в 750 мл диметилформамида охлаждают до температуры -55 - -60 С и при этой температуре добавляют по каплям раствор из 522 г (4,56 моль) трет.-бутилата калия в 750 мл диметилформамида в течение 2,5 ч. Окраска раствора изменяется от желтой до темно-красной, и консистенция становится более вязкой. За реакцией следят с помощью ЖХВД. Для обработки сначала примешивают 300 мл воды и после этого приблизительно 300 мл ледяного уксуса до достижения значения рН 5-6. Температура повышается при этом до -10 С, и образуется желтая суспензия. Реакционную смесь выливают потом на 6 кг ледяной воды и образовавшийся осадок отсасывают, промывают 5 л воды и сушат в сушильном шкафу при 30 С на протяжении ночи. Получают 339 г светло-бежевого сырого продукта, который освобождается от загрязнений приблизительно в 3 л толуола при температуре от 80 до 90 С в течение 2 ч. После охлаждения продукт отсасывают и сушат. Получают 276 г 2-нитро-6-метилбензальдоксима. Выход: 77%. Тпл.: 190-192 С. Чистота (по ЖХВД) составляет 98%. Пример 2. Получение 2-метил-6-нитробензальдоксима (вариант В). 1200 мл безводного диметилформамида подают в четырехлитровую реакционную колбу и охлаждают до -40 С. При перемешивании при этой температуре подают 336,5 г (4,56 моль) метилата калия (95%) и реакционную смесь суспендируют. После этого подают по каплям смесь из 300 г (1,92 моль) 3-нитро-оксилола (97%) и 274 г (2,52 моль) н-бутилнитрита (95%) в течение 7 ч при -40 С (при соответствущей мощности охлаждения продолжительность подачи может быть сокращена, продление еще не проверялось; колебания температуры между -35 и -45 С являются допустимыми). Полное превращение исходного материала контролируется с помощью ЖХВД. Потом реакционную смесь подают в смесь из 300 мл воды и 300 мл ледяного уксуса при -5 С до 0 С при перемешивании. Затем реакционную смесь выливают на 6 кг ледяной воды, твердое вещество отделяют фильтрацией (без проблем, сопротивление фильтра еще не определено) и 2 раза промывают каждый раз в 500 мл воды (внимание: сырой продукт сильно пахнет). Очистку сырого продукта (ЖХВД: 96%) осуществляют суспендированием влажного твердого вещества в 800 мл толуола в течение более 1,5 ч. Твердое вещество отфильтровывают (без проблем, сопротивление фильтра еще не определено) и сушат в вакуумном сушильном шкафу при 50 С. Выход: 306 г (ЖХВД: 99,4% продукта; E/Z-смесь), соответствует 85% от теории. Пример 3. Получение 3-(2-метил-6-нитрофенил)-4,5-дигидроизоксазола. а) К раствору из 5 г (28 ммоль) 2-метил-6-нитробензальдоксима в 50 мл ацетонитрила подают при 60 С небольшое количество раствора из 3,71 г (28 ммоль) N-хлорсукцинимида в 30 мл ацетонитрила. По-7 010633 сле того, как реакция запустилась, медленно по каплям подают при 40-50 С остальной раствор. Реакционную смесь перемешивают в течение 29 мин и контролируют с помощью ЖХВД полноту превращения. Получается раствор оранжевого цвета, который осторожно концентрируют. Остаток суспендируют приблизительно 1,5 ч в 50 мл толуола и раствор отделяют от сукцинимида. Фильтрат имеет все еще оранжевый цвет. Раствор заполняют в миниавтоклав и запресовывают при 30 бар этилен. Потом дозируют в течение 5 ч раствор из 4,7 г гидрокарбоната натрия в 50 мл воды и еще 5 ч при 30 бар давления этилена перемешивают. Для очистки отделяют фазы, фазу толуола промывают 2 раза раствором NaHCO3 и 1 раз водой, сушат и концентрируют. Выход составляет 4,9 г (86%) коричневатых кристаллов. Тпл.: 100-105 С. 1 Н-ЯМР (CDCl3): =8,00 (d, 1H); 7,57 (d, 1H); 7,49 (t, 1H); 4,60 (t, 2H); 3,32 (t, 2H); 2,41 (s, 3H). б) 100 г 2-метил-6-нитробензальдоксима растворяют в 750 мл ледяного уксуса, после чего в течение 2 ч вводят хлор. Избыточный хлор выводят азотом. Потом ледяной уксус отгоняют и остаток суспендируют в 1000 мл толуола. Реакционную смесь заполняют в автоклав и впресовывают при 6 бар этилен. В течение 1 ч дозируют 55,6 г триэтиламина (1 экв.) в 300 мл толуола и перемешивают 10 ч при комнатной температуре и при 6 бар давления этилена. Реакционную смеь промывают 1 раз водным раствором NaHCO3 и 1 раз водой. Органическую фазу сушат над сульфатом натрия, отфильтровывают и перемешивают. Выход составляет 96,3 г (87% от теории). Пример 4. Получение 2-(4,5-дигидроизоксазол-3-ил)-3-метиланилина. а) В автоклав гидрирования подают раствор из 117 г (0,57 моль) 3-(2-метил-6-нитрофенил)-4,5-дигидроизоксазола в 1,2 л этилацетата и 11,7 г катализатора, который содержит 5 мас.% платины на угле. Потом автоклав продувается 2 раза азотом. После этого при 20 бар давления водорода и при интенсивном перемешивании в течение 48 ч при 25-30 С производят гидрирование. Реакционную смесь отсасывают над силикагелем и растворитель отводят под вакуумом. Получают 94 г коричневого твердого твещества, который загружают в метил-трет.-бутиловый эфир, и воду экстрагируют посредством 1 М соляной кислоты. Водную фазу устанавливают на значение рН 10-11 и экстрагируют метиленхлоридом. Метиленхлоридную фазу сушат над сульфатом магния и отводят растворитель. Выход составляет 87 г (87%) твердого вещества оранжевого цвета. Тпл.: 86-88 С. Чистота по ЖХВД составляет 97%. Посредством перемешивания с обратным холодильником продукт подвергают дальнейшей очистке. Тпл. составляет 90-91 С. Чистота составляет по ЖХВД 100%. б) В автоклав гидрирования подают раствор из 1000 г (4,85 моль) 3-(2-метил-6-нитрофенил)-4,5-дигидроизоксазола в 5,5 л метанола и 4,6 г катализатора, который содержит 10 мас.% палладия на угле. Потом автоклав продувают 2 раза азотом. После этого при 2,5 бар давления водорода при интенсивном перемешивании гидрируют в течение 17 ч при 25-30 С. Реакционную смесь отсасывают над силикагелем и растворитель отгоняют в вакууме. Получают 781,7 г светло-коричневого твердого вещества. Выход: 781,7 г (85%) (содержание по ЖХВД: 93%). Пример 5. Получение 3-(2-метил-6-метилтиофенил)-4,5-дигидроизоксазола. 19,5 г (170 ммоль) трт.-бутилнитрита и 20 г медного порошка подают в 30 мл диметилдисульфида и при температуре от 50 до 55 С прибавляют по каплям раствор из 20 г (114 ммоль) 2-(4,5-дигидроизоксазол-3-ил)-3-метиланилина в 100 мл диметилдисульфида. После этого перемешивают в течение 1,5 ч при 60 С. Для переработки отсасывают твердое вещество, разбавляют метиленхлоридом и экстрагируют разбавленной соляной кислотой. Органическую фазу промывают насыщенным водным раствором NaHCO3,сушат над сульфатом натрия, отгоняют и концентрируют. Избыточный диметилсульфид удаляют в вакууме мясляного насоса. Получают 23,4 г (99%) темного масла, которое через некоторое время застывает (содержание по ЖХВД 100%). Продукт может подвергаться дальнейшей очистке перемешиванием в метил-трет.-бутиловом эфире. Тпл.: 66-67 С. Пример 6. Получение 3-(3-бром-2-метил-6-метилтиофенил)-4,5-дигидроизоксазола. К 120 мл конц. серной кислоты по порциям добавляют при 0 С 10 г (48 ммоль) 3-(2-метил-6-метилтиофенил)-4,5-дигидроизоксазола и перемешивают в течение приблизительно 30 мин. Потом подают по каплям 3,7 г (23 ммоль) брома и перемешивают в течение 2,5 ч при 0 С. Затем реакционную смесь нагревают приблизительно 45 мин до комнатной температуры. При этом образуется гомогенный раствор. Для переработки реакционную смесь выливают на ледяную воду и 3 раза экстрагируют метиленхлоридом. Органическую фазу промывают гидрокарбонатом натрия, сушат над сульфатом магния и концентрируют. Получают 11,4 г сырого продукта, который без дальнейшей очистки может быть использован на следующей стадии. Пример 7. Получение 3-(3-бром-2-метил-6-метилсульфонилфенил)-4,5-дигидроизоксазола. К раствору из 11,4 г (40 ммоль) 3-(3-бром-2-метил-6-метилтиофенил)-4,5-дигидроизоксазола и 400 мг вольфрамгидрата натрия в 100 мл ледяного уксуса при 45 С подают по каплям 11,3 г (100 ммоль) 30% перикиси водорода. Реакционную смесь перемешивют в течение ночи при комнатной температуре. Далее-8 010633 смесь выливют на ледяную воду, экстрагируют метиленхлоридом, органическую фазу промывают водным раствором сульфита натрия, сушат над сульфатом магния и концентрируют. Выход составляет 9,6 г. Для очистки продукт выкристаллизовывают из 65 мл изопропанола. Выход: 7,7 г (50% по двум стадиям). Тпл.: 137-139 С. В нижеследующих примерах выполнения более подробно поясняется получение бензальдоксимовXV (стадия способа а). Пример 8. Получение 2-метил-6-нитробензальдоксима (вариант А). Раствор из 274 г (2,6 моль) н-бутилнитрита (97%) и 300 г (2,0 моль) 3-нитро-о-ксилола (97%) в 750 мл диметилформамида охлаждают до температуры от -55 до -60 С и при этой температуре подают по каплям раствор из 522 г (4,56 моль) трет.-бутилата калия в 750 мл диметилформамида в течение 2,5 ч. Окраска раствора изменяется при этом от желтой до темно-красной, и консистенция раствора сгущается. Реакцию контроллируют с помощью ЖХВД. Затем к реакционной смеси подают сначала 300 мл воды и потом приблизительно 300 мл ледяного уксуса до достижения значения рН 5-6. При этом температура повышается до значения -10 С и образуется желтая суспензия. После этого реакционную смесь выливают на 6 кг ледяной воды и отсасывают образовавшийся осадок, промывают посредством 5 л воды и сушат в сушильном шкафу при 30 С на протяжении ночи. Получают 339 г светло-бежевого сырого продукта, который освобождают суспендированием в приблизительно 3 л толуола при 80-90 С в течение 2 ч от загрязнений. После охлаждения продукт отсасывают и сушат. Получают 276 г 2-нитро-6-метилбензальдоксима. Выход: 77%. Тпл.: 190-192 С. Чистота составляет (по ЖХВД) 98%. Пример 9. Получение 2-метил-6-нитробензальдоксима (вариант В). 1200 мл безводного диметилформамида подают в реакционную колбу вместимостью 4 л и охлаждают до -40 С. При перемешивании подают при этой температуре 336,5 г (4,56 моль) метилата калия(95%) и суспендируют. После этого подают по каплям смесь из 300 г (1,92 моль) 3-нитро-о-ксилола(97%) и 274 г (2,52 моль) н-бутилнитрита (95%) в течение 7 ч при -40 С (при соответствующей интенсивности охлаждения время подачи можно сократить). Полное превращение исходного вещества контролируют с помощью ЖХВД. После этого реакционную смесь подают в смесь из 300 мл воды и 300 мл ледяного уксуса при температуре от -5 до 0 С при перемешивании. Затем реакционную смесь выливают на 6 кг ледяной воды, твердое вещество отделяют фильтрацией и 2 раза промывают каждый раз в 500 мл воды. Очистку сырого продукта (ЖХВД: 96 жид.%) осуществляют суспендированием влажного твердого вещества в 800 мл толуола в течение 1,5 ч. Твердое вещество отфильтровывают и сушат в сушильном вакуумном шкафу при 50 С. Выход: 306 г (ЖХВД 99,4 жид.% продукт; E/Z-смесь), соответствует 85% от теории. Пример 10. Получение 2-хлор-6-нитробензальдоксима. Раствор из 4,1 г (40 ммоль) н-бутилнитрита (97%) и 5 г (29 ммоль) 2-хлор-6-нитротолуола в 50 мл диметилформамида охлаждают до температуры от -55 до -60 С и при этой температуре подают по каплям раствор из 3,3 г (29,5 ммоль) трет.-бутилата калия в 30 мл диметилформамида в течение 20 мин. Реакцию контролируют с помощью ЖХВД. Затем к реакционной смеси примешивают воду и потом устанавливают ледяным уксусом на значение рН 5-6. Продукт выделяют экстракцией этилацетатом. Получают 5,7 г 2-хлор-6-нитробензальдоксима. 1 Н-ЯМР (CDCl3): =8,00 (d, 1H); 7,84 (s, 1H); 7,76 (d, 1H); 7,52 (t, 1H). Пример 11. Получение 3-хлор-2-метил-6-метилсульфонилбензальдоксима. Раствор из 12,7 г (119 ммоль) н-бутилнитрита (97%) и 20 г (92 ммоль) 2,3-диметил-4-метилсульфонилхлорбензола в 100 мл диметилформамида охлаждают до температуры от -55 до 60 С и при этой температуре подают к нему по каплям раствор из 16,8 г (147 ммоль) трет.-бутилата калия в 70 мл диметилформамида в течение 30 мин. Реакцию контролируют с помощью ЖХВД. Реакционную смесь сначала смешивают с 50 мл воды и потом устанавливают с помощью приблизительно 30 мл ледяного уксуса на значение рН 5-6. Затем реакционную смесь выливают на 0,7 кг ледяной воды и водную фазу экстрагируют метиленхлоридом. Органическую фазу промывают раствором гидрокарбоната натрия, сушат над сульфатом магния и концентрируют. Получают 18,4 г светло-бежевого сырого продукта, который подвергают очистке перекристаллизацией из приблизительно 30 мл толуола. Выход: 6,15 г (27%) белых кристаллов. Тпл.: 164-168 С. Чистота составляет (по ЖХВД) 100%. Пример 12. Получение 3-бром-2-метил-6-метилсульфонилбензальдоксима. Раствор из 2,1 г (20 ммоль) н-бутилнитрита (97%) и 4 г (15 ммоль) 2,3-диметил-4-метилсульфонилбромбензола в 50 мл диметилформамида охлаждают до температуры от -55 до -60 С и при этой температуре добавляют по каплям раствор из 2,8 г (25 ммоль) трет.-бутилата калия в 35 мл диметилформамида в течение 20 мин. Реакцию контролируют с помощью ЖХВД. Реакционную смесь сначала смешивают с 10 мл воды и затем посредством приблизительно 9 мл ледяного уксуса устанавливают на значение рН 5-6. После этого реакционную смесь выливают на 100 мл ледяной воды и водную фазу экстрагируют метиленхлоридом. Органическую фазу промывают гидрокарбонатом натрия, сушат над сульфатом магния и концентрируют.-9 010633 Получают 3,6 г масляного сырого продукта (по ЖХВД 90%), который можно очищать перекристаллизацией из толуола. Выход: 1,22 г (27%). Тпл.: 192-194 С. Чистота составляет (по ЖХВД) 99%. Пример 13. Получение дифениламида 3-гидроксиимино-2-метил-4-метилсульфонилбензойной кислоты. а) Получение предварительного продукта. 5 г (3 ммоль) 2,3-диметилтиоанизола и 7,6 г (33 ммоль) дифенилкарбамоилхлорида растворяют в 50 мл 1,2-дихлорэтана и при комнатной температуре примешивают 4,8 г (36 ммоль) безводного хлорида алюминия. Реакционную смесь кипятят с обратным холодильником в течение 3 ч, выливают на смесь изо льда и концентрированной соляной кислоты и водную фазу 2 раза экстрагируют метиленхлоридом. Органическую фазу промывают раствором гидрокарбоната натрия, сушат над сульфатом магния и концентрируют. Получают 10,8 г сырого продукта, который может очищться хроматографией на силикагеле с растворителем из толуола и этилацетата. Выход: 7,8 г дифениламида 2,3-диметил-4-метилтиобензойной кислоты. К раствору из 7 г (20 ммоль) дифениламида 2,3-диметил-4-метилтиобензойной кислоты и 200 млг гидрата вольфрамата натрия в 50 мл ледяного уксуса подают по каплям при макс. 45 С 5,7 г (50 ммоль) 30% перекиси водорода. Реакционную смесь перемешивают в течение ночи при комнатной температуре. Для переработки реакционную смесь выливют на ледяную воду, экстрагируют метиленхлоридом, органическую фазу промывают водным раствором сульфита натрия, сушат над сульфатом магния и концентрируют. Выход: 7,4 г дифениламида 2,3-диметил-4-метилсульфонилбензойной кислоты. Тпл.: 155-165 С. б) Получение дифениамида 3-гидроксимино-2-метил-4-метилсульфонилбензойной кислоты. Раствор из 0,7 г (6,9 ммоль) н-бутилнитрита (97%) и 2 г (5,3 ммоль) дифениламида 2,3-диметил-4 метилсульфонилбензойной кислоты в 30 мл диметилформамида охлаждают до -55 - -60 С и при этой температуре по каплям подают раствор 1,4 г (12 ммоль) трет.-бутилата калия в 10 мл диметилформамида в течение 20 мин. За реакцией наблюдают с помощью ЖХВД. Затем к реакционной смеси подмешивают 10 мл воды и после этого ледяным уксусом устанавливают на значение рН 5-6. Реакционную смесь выливают на 100 мл ледяной воды и водную фазу экстрагируют этилацетатом. Органическую фазу промывают раствором гидрокарбоната натрия, сушат над сульфатом магния и концентрируют. Получают 3,0 г частично кристаллического сырого продукта, который подвергают очистке хроматографией на силикагеле с толуолом и ацетоном в качестве растворителя. Выход: 1,0 г (46%). Тпл.: 208-211 С. Пример 14. Получение 3-бром-2-метил-6-метилсульфонилбензальдегида. 7,1 г 3-бром-2-метил-6-метилсульфонилбензальдоксима (23 ммоль) перемешивают в смеси из 17 г 5% соляной кислоты, 2 г 37% раствора формальдегида, 15 мл воды и 30 мл тетрагидрофурана в течение 32 ч при 65 С. При этом подают по порциям в 0,5 г еще 3,5 г 37% раствора формальдегида. После этого реакционную смесь охлаждают до комнатной температуры и продукт отсасывают. Получают 5,1 г (79%), чистота составляет 94% (по ГХ). Пример 15. Получение 2-метил-6-нитробензальдегида. 14 г 2-метил-6-нитробензальдоксима (80 ммоль) перемешивают в смеси из 55 мл 5% соляной кислоты, 37 г 37% раствора формальдегида, 50 мл воды и 100 мл тетрагидрофурана в течение 24 ч при 65 С. Затем разделяют фазы и темную фазу экстрагируют метиленхлоридом и водой. Органическую фазу сушат сульфатом натрия и концентрируют. Получают 10,1 г сырого продукта, который подвергают очистке посредством фильтрирования над силикагелем с толуолом в качестве растворителя. Выход: 7,2 г (54%). Пример 16. Получение 2-метил-6-нитробензонитрила. Раствор из 16 г (150 ммоль) н-бутилнитрита (97%) и 7,7 г (50 ммоль) 3-нитро-о-ксилола (97%) в 50 мл диметилформамида охлаждают до температуры от -5 до -10 С и при этой температуре к нему подают по каплям раствор из 11 г (100 ммоль) трет.-бутилата калия в 50 мл диметилформамида в течение 1,5 ч. Реакционную смесь перемешивают еще 6 дней при комнатной температуре. Далее реакционную смесь выливают на ледяную воду, соляной кислотой устанавливают значение рН на 1 и водную фазу экстрагируют этилацетатом. Органическую фазу промывают водой, сушат над сульфатом магния и концентрируют.- 10010633 Получают 8,2 г продукта. Хроматографией на силикагеле с толуолом в качестве растворителя можно очищать 2-метил-6-нитробензонитрил. Тпл.: 101-103 С. В нижеследующих примерах выполнения более подробно поясняется получение тиоэфиров формулы VIIIa (стадия способа г). Пример 17. а) Сравнительный пример. При взаимодействии 2-(4,5-дигидроизоксазол-3-ил)-3-метиланилина с диметилдисульфидом и трет.-бутилнитритом без катализатора образуются побочные продукты. Получают смесь из соединений А и Б в соотношении 2:1 по ЖХВД. б) Способ по изобретению. Аналогично описанному для стадии а) методу осуществляют взаимодействие в присутствии медного порошка. В этом случае не был обнаружен побочный продукт А. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединений формулы XV где остатки имеют следующие значения:Rx, Ry независимо друг от друга означают галоген, карбоксил, карбоксамид, N-алкилкарбоксамид,N,N-диалкилкарбоксамид, фенил, С 1-С 6 алкил, C1-C6 алкокси, C1-C6 алкилтио;n равно 0, 1 или 2; включающий взаимодействие соединений формулы XVI где заместители имеют вышеприведенные значения, с органическим нитритом общей формулы R-O-NO,где R представляет собой алифатический или ароматический остаток, при воздействии основания, при этом взаимодействие проводят при температуре от -50 до -20 С в присутствии диполярного апротонного растворителя, и необязательно включающий превращение оксимовой группы -CH=NOH в соединении формулы XV с получением соответствующих альдегидов -СНО, нитрилов (-CN) или нитрилоксидов (-CNO). 2. Способ по п.1, при котором в качестве растворителя применяют диметилформамид. 3. Соединения формулы XV где остатки имеют следующие значения:n равно 0, 1 или 2. 4. Применение способа получения по п.1 для осуществления способа получения соединения формулы X где заместители имеют следующие значения:n равно 0, 1 или 2,характеризующегося: а) циклизацией оксима формулы XV, когда X означает NO2, m означает 0 или 1, а, если m равно 1, Rx означает C1-C6 алкил, расположенный в орто-положении относительно группы CH=NOH, алкеном формулы IV где остатки R3 до R5 имеют приведенные выше значения, в присутствии основания с получением 4,5-дигидроизоксазола формулы V где остатки R1, R3 до R5 имеют приведенные выше значения; б) восстановлением нитрогруппы в присутствии катализатора с получением анилина формулы VI где остатки R1, R3 до R5 имеют приведенные выше значения; в) взаимодействием анилина формулы VI с диалкилдисульфидом формулы VII в присутствии огранического нитрита и необязательно катализатора с получением простого тиоэфира формулы VIII где остатки R1 до R5 имеют приведенные выше значения; г) бромированием простого тиоэфира формулы VIII средством бромирования с получением соединения формулы X, где n равно 0; и необязательно д) окислением соединения формулы X, где n равно 0, окислительным агентом с получением соединения формулы X, где n имеет значения 1 или 2. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6
МПК / Метки
МПК: C07D 251/40, C07D 319/14, C07D 413/10, C07D 261/04
Метки: получения, способ, новые, бензальдоксимов, бензальдоксимы, применение
Код ссылки
<a href="https://eas.patents.su/13-10633-novye-benzaldoksimy-sposob-polucheniya-benzaldoksimov-i-ego-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Новые бензальдоксимы, способ получения бензальдоксимов и его применение</a>
Новые производные эритромицина, способ их получения и их применение в качестве медикаментов.

Номер патента: 1376
Опубликовано: 26.02.2001
Авторы: Шанто Жан-Франсуа, Агуридас Константэн
МПК: A61P 31/00, A61K 31/70, C07H 17/08...
Метки: способ, медикаментов, производные, получения, эритромицина, новые, применение, качестве
Формула / Реферат:
1. Производные эритромицина формулы (I): в которых Х представляет собой радикал СН2 или SO2 или атом кислорода, Y представляет собой радикал (СН2)m-(СН=СН)n((CH2)o, где m+n+o<= 8, n=0 или 1, Аr представляет собой арил, возможно замещенный, W представляет собой атом водорода или остаток карбаматной функции где R" представляет собой алкил, включающий до 8 атомов углерода, или арил, возможно замещенный, Z представляет собой атом...
Новые 2, 3, 3а, 4, 9, 9а – гексагидро – 8 – гидрокси – 1н – бенз[f]индолы, способ их получения и их применение в качестве лекарственных средств

Номер патента: 3370
Опубликовано: 24.04.2003
Авторы: Паллук Райнер, Грауэрт Маттиас, Бехтель Вольф-Дитрих, Картер Адриан, Вайзер Томас, Пшорн Уве, Хёнке Кристоф
МПК: A61P 9/06, A61K 31/40, C07D 209/60...
Метки: средств, получения, применение, новые, бенз[f]индолы, способ, гидрокси, лекарственных, гексагидро, качестве
Формула / Реферат:
1. Производные индола общей формулы 1 где X обозначает простую связь, -O-, C1-C4алкил, C1-C3алкоксигруппу, -O-CH2-CH2-O- или -O-CH2-CH2-NH-, R1 обозначает водород, метил, этил или фенил, R2 обозначает водород или метил, R3 обозначает водород, F, Cl, Br, гидрокси- или метоксигруппу, R4 обозначает водород, метил или этил, R5 обозначает водород, метил или этил, R6 обозначает водород, метил или этил, R7 обозначает трет-бутил, циклогексил, ...
Новые ароматические амиды, замещенные рибозой, способ их получения и их применение в качестве лекарств

Номер патента: 4304
Опубликовано: 26.02.2004
Авторы: Мусицки Бранислав, Клиш Мишель, Демассе Жак
МПК: A61P 31/04, A61K 31/70, C07H 17/075...
Метки: применение, лекарств, амиды, получения, качестве, ароматические, способ, рибозой, новые, замещенные
Формула / Реферат:
1. Соединения формулы (I) в которой R1 обозначает алкил, алкенил или алкинил, O-алкил, O-алкенил или O-алкинил, линейный, разветвленный или циклический, содержащий до 8 атомов углерода, возможно замещенный одним или несколькими атомами галогена, возможно прерванный атомом кислорода, серы или азота, арил или аралкил, содержащий до 18 атомов углерода, возможно замещенный, ароматический или неароматический гетероциклический радикал, моно- или...
Новые производные эритромицина, способ их получения и их применение в качестве медикаментов

Номер патента: 2168
Опубликовано: 28.02.2002
Авторы: Пежак Жан-Мари, Дени Алексис, Шанто Жан-Франсуа, Агуридас Константэн
МПК: A61P 31/04, A61K 31/7048, C07H 17/08...
Метки: получения, производные, медикаментов, эритромицина, новые, способ, применение, качестве
Формула / Реферат:
1. Производные эритромицина формулы I в которой R означает атом водорода; алкильный радикал, содержащий до 12 атомов углерода, который может быть замещен галогеном или радикалами (СН2)mАr или в которых m - целое число от 1 до 8, n и р, одинаковые или различные, означают целые числа от 0 до 6; А и В, одинаковые или различные, означают атом водорода или галогена или алкил, содержащий до 8 атомов углерода; Аr - фенил или нафтил или...
Применение фенэтилакриламидов, новые фенэтилакриламиды, способ их получения, а также содержащие их средства

Номер патента: 5764
Опубликовано: 30.06.2005
Авторы: Гевер Маркус, Мюллер Бернд, Штирль Райнхард, Фольк Торстен, Грамменос Василиос, Штратман Зигфрид, Гётц Норберт, Заутер Хуберт, Аммерманн Эберхард, Тормо И Бласко Хорди, Гулльманн Оливер, Лоренц Гизела
МПК: C07C 233/69, A01N 37/18
Метки: также, фенэтилакриламиды, способ, средства, фенэтилакриламидов, новые, содержащие, получения, применение
Формула / Реферат:
1. Применение фенэтилакриламидов формулы I где заместители имеют следующие значения: X означает галоген, C1-C4алкил, C1-C4галогеналкил, C1-C8алкокси, C1-C4галогеналкокси и -O-C(Ra,Rb)-Cу C-Rc; Ra, Rb означают независимо друг от друга водород или C1-C6алкил; Rc означает водород, C1-C8алкил, C3-C8циклоалкил и фенил, который может быть замещен галогеном, циано, нитро, CF3, C1-C4алкилом и/или C1-C4алкокси; m равно от 1 до 4, причем остатки X могут...