Соединения 2-алкилиндазола для лечения некоторых расстройств цнс
Номер патента: 19217
Опубликовано: 28.02.2014
Авторы: Омбрато Розелла, Ализи Мария Алессандра, Мауджери Катерина, Фурлотти Гвидо, Поленцани Лоренцо, Каццолла Никола







Формула / Реферат
1. Соединение 2-алкилиндазола общей формулы (I)
где X обозначает -С(О)N(R5)-;
Y обозначает СН или N;
W обозначает СН или N;
при условии, что по меньшей мере один из Y и W является атомом азота;
n обозначает целое число, выбранное из 1, 2 и 3;
m обозначает целое число, выбранное из 0, 1, 2 и 3;
р обозначает целое число, выбранное из 1 и 2;
R1 обозначает Н, метил или метоксигруппу;
R2 обозначает метил, этил, изопропил или метоксиэтил;
R3 обозначает Н, 4-гидрокси, 4-метокси или 4-фтор;
R4 обозначает Н или 2-фтор;
R5 обозначает Н, метил, метоксигруппу или вместе с R6 образует 6-членное насыщенное кольцо, выбранное из группы, включающей пиперидин и пиперазин;
R6 вместе с R5 образует 6-членное насыщенное кольцо, выбранное из группы, включающей пиперидин и пиперазин; или вместе с R7 образует 5- или 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолин, пиперидин и пиперазин; и
R7 обозначает Н, этил или вместе с R6 образует 5- или 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолин, пиперидин и пиперазин.
2. Соединение по п.1, отличающееся тем, что
X обозначает -C(O)N(R5)-;
Y обозначает СН;
W обозначает N;
n обозначает целое число, выбранное из 1 и 2;
m обозначает 2;
р обозначает целое число, выбранное из 1 и 2;
R1 обозначает Н или метоксигруппу;
R2 обозначает метил или метоксиэтил;
R3 обозначает Н, 4-гидрокси, 4-метокси или 4-фтор;
R4 обозначает Н или 2-фтор;
R5 обозначает Н, метил или метокси, или вместе с R6 образует 6-членное насыщенное кольцо, выбранное из группы, включающей пиперидин и пиперазин;
R6 вместе с R5 образует 6-членное насыщенное кольцо, выбранное из группы, включающей пиперидин и пиперазин; или вместе с R7 образует 5- или 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолин, пиперидин и пиперазин; и
R7 обозначает Н, этил или вместе с R6 образует 5- или 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолин, пиперидин и пиперазин.
3. Способ получения (i) соединения 2-алкилиндазола общей формулы (I), в котором R1, R2, R3, R4, R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1 или (ii) его соли с фармацевтически приемлемой органической или неорганической кислотой, отличающийся тем, что включает в себя:
1а) конденсацию амина формулы (II)
где Y, W, R3, R4, R5, R6, R7, n, m и р имеют значения, установленные ранее в п.1, с производным индазолкарбоновой кислоты формулы (III)
где R1 и R2 имеют значения, установленные ранее в п.1, и
Z выбирается из группы, включающей атом галогена, OR и OC(O)R группу, где R обозначает неразветвленную или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода,
для получения соединения 2-алкилиндазола общей формулы (I), в которой R1, R2, R3, R4, R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1; причем указанная конденсация выполняется в присутствии подходящего апротонного, полярного или неполярного растворителя при температуре от 0 до 140°С, в течение от 0,5 до 24 ч.
4. Способ получения (i) соединения 2-алкилиндазола общей формулы (I) по п.3, отличающийся тем, что Z выбирается из группы, включающей Cl или Br.
5. Способ получения (i) соединения 2-алкилиндазола общей формулы (I), в котором Rl, R2, R3, R4, R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1 или (ii) его соли с фармацевтически приемлемой органической или неорганической кислотой, отличающийся тем, что включает в себя:
1b) взаимодействие амина общей формулы (IV)
где Rl, R2, R5, R6, R7, Y, n и р имеют значения, установленные ранее в п.1,
с соединением общей формулы (V)
где R3, R4 и m имеют значения, установленные ранее в п.1, и
Q обозначает уходящую группу, выбранную из группы, включающей атом галогена, мезилатную группу (MeSO3-) и тозилатную группу (p-MePhSO3-),
для получения соединения 2-алкилиндазола общей формулы (I), в которой R1, R2, R3, R4, R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1;
причем указанная реакция выполняется в присутствии подходящего апротонного, полярного или неполярного растворителя при температуре, находящейся в пределах от комнатной до 100°С, при этом время реакции составляет от 6 до 24 ч.
6. Способ получения (i) соединения 2-алкилиндазола общей формулы (I) по п.5, отличающийся тем, что Q обозначает уходящую группу, выбранную из группы, включающей Cl или Br.
7. Способ получения (i) соединения 2-алкилиндазола общей формулы (I), в котором R1, R2, R3, R4, R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1 или (ii) его соли с фармацевтически приемлемой органической или неорганической кислотой, отличающийся тем, что включает в себя:
1с) конденсацию амина общей формулы (VI)
где R1, R2 и R5 имеют значения, установленные ранее в п.1,
с производным карбоновой кислоты общей формулы (VII)
где R3, R4, R6, R7, Y, W, n, m и р имеют значения, установленные ранее в п.1, и
Z имеет значения, установленные ранее в отношении соединения формулы (III),
для получения соединения 2-алкилиндазола общей формулы (I), в которой R1, R2, R3, R4, R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1; и
2) возможное образование соли кислотного присоединения соединения 2-алкилиндазола общей формулы (I), полученного таким образом, с фармацевтически приемлемой органической или неорганической кислотой, причем указанная конденсация выполняется в присутствии подходящего апротонного, полярного или неполярного растворителя при температуре от 0 до 140°С, при этом время реакции составляет от 0,5 до 24 ч.
8. Промежуточное соединение формулы (II)
где R3, R4 и n имеют значения, установленные ранее в п.1,
R5 обозначает C1-3алкил,
R6 и R7 вместе образуют 5- или 6-членное насыщенное кольцо, выбранное из группы, состоящей из пирролидина, имидазолина, пиразолидина, пиперидина и пиперазина,
Y обозначает СН,
W обозначает N,
р является целым числом, выбранным из 0 и 1,
m является 2.
9. Промежуточное соединение формулы (IV)
в которой R1, R2, R5, R6, R7, Y, n, p имеют значения, установленные ранее в п.1.
10. Промежуточное соединение формулы (VI)
R1 и R2 имеют значения, установленные ранее в п.1; и
R5 обозначает Н, C1-3алкил или C1-3алкоксигруппу.
11. Промежуточное соединение общей формулы (XX)
в которой T1 обозначает Н, C1-3алкил или C1-3алкоксигруппу, или R-СО, в которой R обозначает Н или C1-3алкил,
R3, R4, n и p имеют значения, установленные ранее в п.1,
R6 и R7 вместе образуют 5- или 6-членное насыщенное кольцо, выбранное из группы, состоящей из пирролидина, имидазолина, пиразолидина, пиперидина и пиперазина,
Y обозначает СН,
W обозначает N и
m является 2.
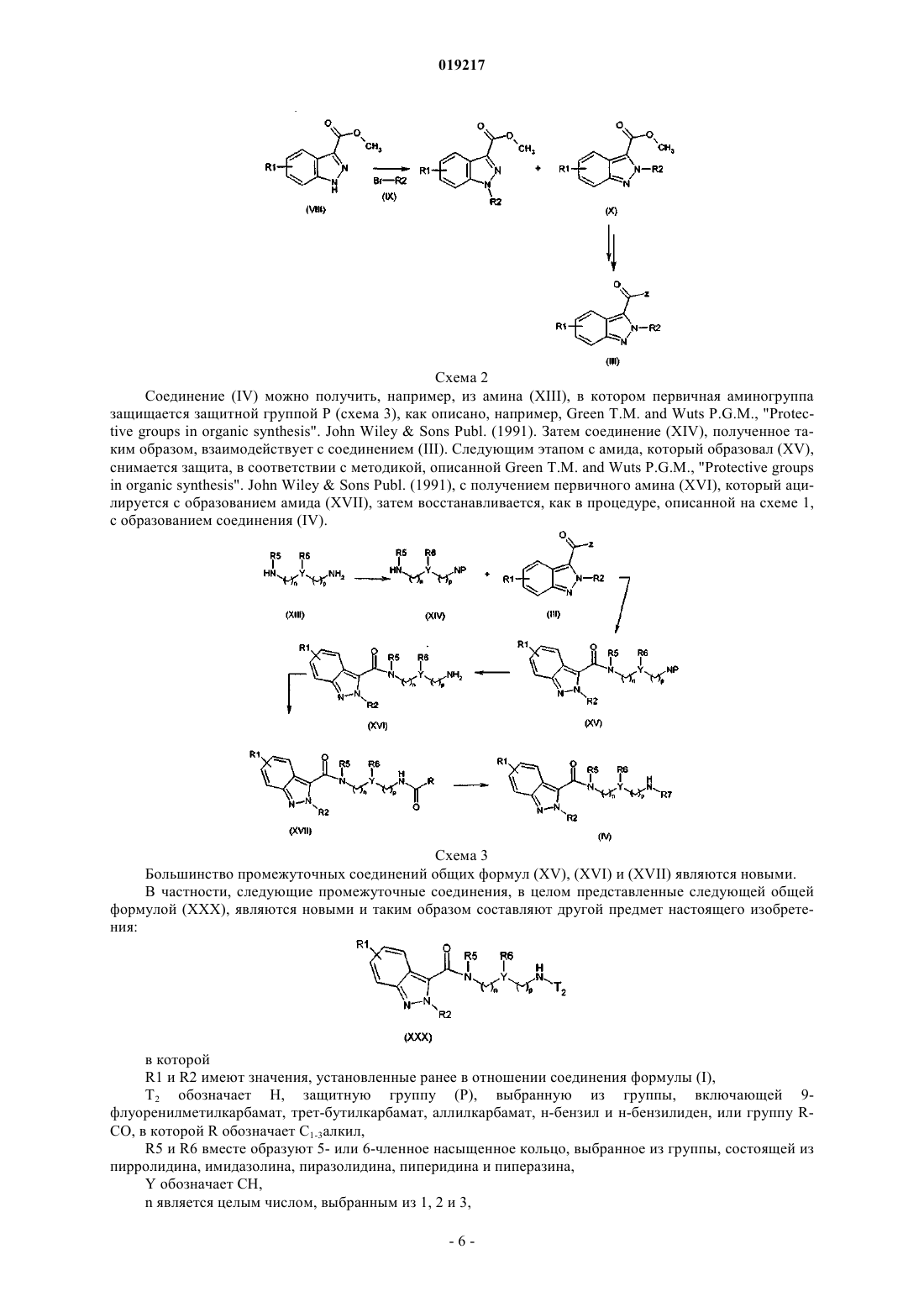
12. Промежуточное соединение общей формулы (XXX)
в которой R1, R2, n и p имеют значения, установленные ранее в п.1,
Т2 обозначает Н, защитную группу (Р), выбранную из группы, включающей 9-флуоренилметилкарбамат, трет-бутилкарбамат, аллилкарбамат, N-бензил и N-бензилиден или группу R-CO, в которой R обозначает C1-3алкил,
R5 и R6 вместе образуют 5- или 6-членное насыщенное кольцо, выбранное из группы, состоящей из пирролидина, имидазолина, пиразолидина, пиперидина и пиперазина, и
Y обозначает СН.
13. Фармацевтическая композиция, содержащая эффективную дозировку по меньшей мере одного соединения 2-алкилиндазола формулы (I), в которой R1, R2, R3, R4, R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1, или его соль с фармацевтически приемлемой органической или неорганической кислотой и по меньшей мере один фармацевтически приемлемый наполнитель.
14. Фармацевтическая композиция по п.13, отличающаяся тем, что содержит такое количество соединения 2-алкилиндазола формулы (I), или его фармацевтически приемлемую соль присоединения кислоты, чтобы обеспечить уровень введения от 0,0001 до 100 мг/кг/день.
15. Фармацевтическая композиция по п.13, отличающаяся тем, что содержит такое количество соединения 2-алкилиндазола формулы (I), или его фармацевтически приемлемую соль присоединения кислоты, чтобы обеспечить уровень введения от 0,001 до 50 мг/кг/день.
16. Фармацевтическая композиция по п.14, отличающаяся тем, что содержит такое количество соединения 2-алкилиндазола формулы (I), или его фармацевтически приемлемую соль присоединения кислоты, чтобы обеспечить уровень введения от 0,01 до 10 мг/кг/день.
17. Применение фармацевтической композиции по любому из пп.13-16 для лечения патологических состояний, выбранных из группы, включающей расстройства сна, шизофрению, беспокойство, нарушения в гладких мышцах как желудочно-кишечного тракта, так и сердечно-сосудистой системы.
Текст
СОЕДИНЕНИЯ 2-АЛКИЛИНДАЗОЛА ДЛЯ ЛЕЧЕНИЯ НЕКОТОРЫХ РАССТРОЙСТВ ЦНС Соединение 2-алкилиндазола и его фармацевтически приемлемые соли присоединения кислоты,способ и промежуточные соединения для их получения, фармацевтическая композиция,содержащая их, и дальнейшее применение. Соединение 2-алкилиндазола имеет следующую общую формулу (I), в которой R1, R2, R3, R4, R6, R7, X, Y, W, n, p и m имеют значения, установленные в описании.(71)(73) Заявитель и патентовладелец: АЦЬЕНДЕ КИМИКЕ РЬЮНИТЕ АНДЖЕЛИНИ ФРАНЧЕСКО Настоящее изобретение относится к соединениям 2-алкилиндазола, способу и промежуточным соединениям для его получения, фармацевтической композиции, содержащей его, и дальнейшему применению. В частности, настоящее изобретение относится к соединению 2-алкилиндазола, обладающему селективным сродством к 5-НТ 2 рецептору или к другим серотониновым рецепторам, таким как 5-HT1, 5 НТ 3, 5-НТ 4, 5-НТ 5, 5-НТ 6 и 5-НТ 7 рецепторы. Что касается различных субтипов 5-НТ 2 рецептора, то соединения настоящего изобретения обладают селективным сродством к 5-HT2A рецептору или к 5-HT2B рецептору и, в общем случае, показывают преференциальное сродство к 5-HT2A рецептору по сравнению с 5-HT2C рецептором. В еще более частном случае, настоящее изобретение относится к соединению, которое может использоваться при лечении некоторых патологий, которые затрагивают 5-HT2A рецептор, например некоторые расстройства центральной нервной системы, такие как расстройства сна, шизофрения и беспокойство, а также нарушения в гладких мышцах или желудочно-кишечного тракта, или сердечно-сосудистой системы. Серотонин 2 А рецептор (5-HT2A) является рецептором, связанным с G-белком, который присутствует у многих видов и широко распространен в теле человека. Способ трансмиссии сигнала, возникающего при активации рецептора, полностью не ясен. Известно, что активация этого рецептора через связанный с ним G-белок приводит к активации различных энзимов, таких как фосфолипаза С (с последующим гидролизом фосфатидилинозитол дифосфата и получением инозитол трифосфата) или фосфолипаза А 2 (которая приводит к высвобождению арахидоновой кислоты). Более того, рецепторный отклик вызывает поток ионов кальция (Са 2+) в клетке и активацию функциональных белков, таких как протеинкиназа С. Рецептор присутствует в центральной нервной системе, в клетках сосудов и гладких мышцах желудочно-кишечного тракта, и в тромбоцитах. В настоящее время обнаружено соединение 2-алкилиндазоламида, имеющее преференциальное сродство к 5-HT2A рецептору. Биологическая активность заметно отличается от активности известных соединений 2 алкилиндазола, описанных как пестициды (арил или эфир-арил 2-алкилиндазоламиды, WO 94/05642), как инсектициды (пиридиновые соединения 2-метилиндазоламида, ЕР-А-0726266) или как антагонисты CRF1 рецептора (7-арил или 7-эфир-арил-2-алкилиндазоламид, US 2004/0110815). Эта биологическая активность также отличается от активности соединений 1-алкилиндазола, обладающих анальгетической активностью (WO 04/101548). Более того, соединения настоящего изобретения имеют структуру, которая заметно отличается от известных соединений с преференциальным сродством к 5-HT2A рецептору, которая достигла клинических фаз развития, например, EMD-281014 (7-4-[2-(4-фторфенил)этил]пиперазин-1-карбонил-1 Ниндол-3-карбонитрил гидрохлорид) в фазе I для бессонницы и MDL-100907 R)-(+)-(2,3 диметоксифенил)-1-[2-(4-фторфенил)этил]пиперидин-4-илметанол) в фазе II для бессонницы. Первый аспект настоящего изобретения относится к соединению 2-алкилиндазола общей формулы (I)Y обозначает СН или N;W обозначает СН или N; при условии, что по меньшей мере один из Y и W является атомом азота;R2 обозначает неразветвленный или разветвленный С 1-6 алкил, С 1-3 алкил-С 1-3 алкокси, арил-С 1-3 алкил, где арильная группа может замещаться 1 или 2 заместителями, выбранными из галогена, C1-3 алкила и C1-3 алкокси;R3 и R4, которые могут быть одинаковыми или разными, обозначают Н, гидрокси, C1-3 алкил, C1-3 алкокси, R'RN-, нитро, галоген, трифторметил или R'CON(R)-;R5 обозначает Н, C1-3 алкил, C1-3 алкокси или вместе с R6 образует 5- или 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолидин, пиперидин и пиперазин;R7 обозначает Н, неразветвленный или разветвленный C1-6 алкил, арил-С 1-3 алкил, где арильная группа может быть замещена 1 или 2 заместителями, выбранными из галогена, C1-3 алкила, гидрокси, C1-3 алкокси, нитрогруппы, трифторметила; иR' и R, которые могут быть одинаковыми или разными, обозначают Н или C1-3 алкил; и их соли присоединения фармацевтически приемлемых органических и неорганических кислот. Типичными примерами фармацевтически приемлемых кислот являются щавелевая, малеиновая, метансульфоновая, паратолуолсульфоновая, янтарная, лимонная, винная, молочная, хлористо-водородная,фосфорная и серная. Предпочтительно, когда X обозначает -C(O)N(R5)-;Y обозначает СН или N;W обозначает СН или N; при условии, что по меньшей мере один из Y и W является атомом азота;R5 обозначает Н, метил, метоксигруппу или вместе с R6 образует 6-членное насыщенное кольцо,выбранное из группы, включающей пирролидин, имидазолин, пиразолидин, пиперидин и пиперазин;R7 вместе с R6 образует 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолидин, пиперидин и пиперазин. Еще более предпочтительно X обозначает -С(О)N(R5)-; Y обозначает CH; W обозначает N; n обозначает целое число, выбранное из 1 и 2; m обозначает 2; р обозначает целое число, выбранное из 1 и 2;R1 обозначает Н или метоксигруппу; R2 обозначает метил или метоксиэтил; R3 обозначает Н, 4 гидрокси, 4-метокси или 4-фтор; R4 обозначает Н или 2-фтор; R5 обозначает Н, или вместе с R6 образует 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолидин, пиперидин и пиперазин; R6 вместе с R5 или R7 образует 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолидин, пиперидин и пиперазин; R7 обозначает Н, этил или вместе с R6 образует 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолидин, пиперидин и пиперазин. Второй аспект настоящего изобретения относится к способу получения соединения 2 алкилиндазола общей формулы (I) и его солям присоединения фармацевтически приемлемых органических и неорганических кислот, отличающийся тем, что включает в себя: 1 а) конденсацию амина формулы (II)Y, W, R3, R4, R5, R6, R7, n, m и р имеют значения, установленные ранее в отношении соединения формулы (I), с производным индазолкарбоновой кислоты формулы (III)R1 и R2 имеют значения, установленные ранее в отношении соединения формулы (I), иZ выбирается из группы, включающей атом галогена, предпочтительно Cl или Br, OR и OC(O)R группу, где R обозначает неразветвленную или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода,для получения соединения 2-алкилиндазола общей формулы (I); или 1b) реакцию амина общей формулы (IV)R1, R2, R5, R6, R7, Y, n и р имеют значения, установленные ранее в отношении соединения формулы (I), с соединением общей формулы (V)R3, R4 и m имеют значения, установленные ранее в отношении соединения формулы (I), иQ обозначает уходящую группу, выбранную из группы, включающей атом галогена, предпочтительно Cl или Br, мезилатную группу (MeSO3-) и тозилатную группу (p-MePhSO3-),для получения соединения 2-алкилиндазола общей формулы (I); или 1 с) конденсацию амина общей формулы (VI)R1, R2 и R5 имеют значения, установленные ранее в отношении соединения формулы (I), с производным карбоновой кислоты общей формулы (VII)R3, R4, R6, R7, Y, W, n, m и р имеют значения, установленные ранее в отношении соединения формулы (I), иZ имеет значения, установленные ранее в отношении соединения формулы (III),для получения соединения 2-алкилиндазола общей формулы (I); и 2) возможное образование соли присоединения кислоты соединения 2-алкилиндазола общей формулы (I), полученной таким образом с фармацевтически приемлемой органической или неорганической кислотой. Стадия 1(а) может выполняться с помощью традиционных методик. Например, амин формулы (II) взаимодействует с ацилгалогенидом формулы (III), в которой Z обозначает предпочтительно атом хлора,необязательно в присутствии подходящего основания. Типичными примерами приемлемых оснований являются триэтиламин и диизопропилэтиламин. Преимущественно, реакция выполняется в присутствии подходящего растворителя при температуре от 0 до 140 С в течение от 0,5 до 24 ч. Предпочтительно температура реакции в интервале от 10 до 60 С, тогда как время реакции составляет от 1 до 12 ч. Обычно растворитель, который используют, является апротонным, полярным или неполярным. Примерами подходящих неполярных апротонных растворителей являются ароматические углеводороды, такие как толуол и ксилол. Примерами подходящих полярных апротонных растворителей являются дихлорметан и N,N-диметилформамид. Предпочтительно используется неполярный апротонный растворитель. Полученное на стадии 1(а) соединение можно также очистить с помощью таких традиционных методик, как флэш-хроматография и кристаллизация. Стадия 1(b) также может выполняться с помощью традиционных методик. Например, амин формулы (IV) взаимодействует с соединением формулы (V), в котором Q обозначает предпочтительно атом брома или метансульфоновую группу, необязательно в присутствии подходящего основания. Типичными примерами подходящих оснований являются карбонат калия и карбонат натрия. Предпочтительно реакция выполняется в присутствии подходящего растворителя при температуре от комнатной до 160 С и за время от 1 до 48 ч. Предпочтительно температура реакции находится в пределах от комнатной до 100 С,принимая во внимание, что время реакции составляет от 6 до 24 ч. Обычно используемый растворитель является полярным, протонным или апротонным. Примерами подходящих полярных протонных растворителей являются спирты, такие как этанол, тогда как примером подходящего полярного апротонного растворителя является ацетон. Соединение, полученное на стадии 1(b), может также очищаться с помощью таких традиционных методик, как флэш-хроматография и кристаллизация. Стадия 1(с) может также выполняться с помощью традиционных методик. Например, амин формулы (VI) взаимодействует с ацилгалогенидом формулы (VII), в которой Z обозначает предпочтительно атом хлора, необязательно в присутствии походящего основания. Типичными примерами приемлемых оснований являются триэтиламин и диизопропилэтиламин. Преимущественно реакция выполняется в присутствии подходящего растворителя при температуре от 0 до 140 С, за промежуток времени от 0,5 до 24 ч. Предпочтительно температура реакции от 10 до 60 С, тогда как время реакции от 1 до 12 ч. Обычно используемый растворитель является апротонным, полярным или неполярным. Примерами подходящих неполярных апротонных растворителей являются ароматические углеводороды, такие как толуол и ксилол. Примерами подходящих полярных апротонных растворителей являются дихлорметан и N,Nдиметилформамид. Предпочтительно используется неполярный апротонный растворитель. Соединение, полученное на стадии 1(с), также может очищаться с помощью таких традиционных методик, как флэш-хроматография и кристаллизация. Образование соли присоединения кислоты соединения 2-алкилиндазола общей формулы (I) с фармацевтически приемлемой органической или неорганической кислотой также может выполняться с помощью традиционных методик. Например, может выполняться с помощью растворения соединения формулы (I) в подходящем растворителе и при обработке раствора, полученного таким образом, органическим или водным раствором требуемой кислоты. Типичными примерами подходящих растворителей являются этанол, изопропанол, этилацетат и диэтиловый эфир. Соль, которая образуется, можно затем отделить с помощью традиционных методик и, при необходимости, очистить кристаллизацией. Некоторые промежуточные соединения формулы (II), (IV) и (VI) являются новыми, и, следовательно, они составляют другой аспект настоящего изобретения. В частности, следующие соединения являются новыми:R3 и R4, которые могут быть одинаковыми или разными, обозначают Н, гидроксигруппу, С 1-3 алкил,С 1-3 алкокси, R'RN-, нитро, галоген, трифторметил или R'CON(R)-, однако при условии, что R3 и R4 не являются оба атомом Н,R5 обозначает С 1-3 алкил,R6 и R7 вместе образуют 5- или 6-членное насыщенное кольцо, выбранное из группы, состоящей из пирролидина, имидазолина, пиразолидина, пиперидина и пиперазина,Y обозначает СН,W обозначает N,n является целым числом, выбранным из 1, 2 и 3,р является целым числом, выбранным из 0 и 1,m является 2,R' и R, которые могут быть одинаковыми или разными, обозначают Н или С 1-3 алкил;R1, R2, R5, R6, R7, Y, n и р имеют значения, установленные ранее в отношении соединения формулы (I); в которой оба R1 и R2 имеют значения, установленные ранее в отношении соединения формулы (I), иR5 обозначает C1-3 алкил или C1-3 алкоксигруппу. Амин (II) можно получить с помощью традиционных методик, например, как описано в заявленной авторами патентной заявке WO 04/101548. Когда R5 отличается от Н, амин (II) преимущественно получают (схема 1) путем амидирования амина (XI) для получения соединения (XII) и затем полным восстановлением соединения (XII) с образованием амина (II). Схема 1 Большинство промежуточных соединений общей формулы (XI) и (XII) являются новыми. В частности, следующие промежуточные соединения, в целом, представленные следующей общей формулой (XX), являются новыми и таким образом составляют другой предмет настоящего изобретения:T1 обозначает Н, C1-3 алкил или C1-3 алкоксигруппу, или R-СО, в которой R обозначает Н или C1-3 алкил,R3 и R4, которые могут быть одинаковыми или разными, обозначают Н, гидроксигруппу, C1-3 алкил,С 1-3 алкокси, R'RN-, нитро, галоген, трифторметил или R'CON(R)-, где R' и R, которые могут быть одинаковыми или разными, обозначают Н или C1-3 алкил, однако при условии, что R3 и R4 не являются оба атомом Н,R6 и R7 вместе образуют 5- или 6-членное насыщенное кольцо, выбранное из группы, состоящей из пирролидина, имидазолина, пиразолидина, пиперидина и пиперазина,Y обозначает СН,W обозначает N,n является целым числом, выбранным из 1, 2 и 3,р является целым числом, выбранным из 0, 1 и 2, иm является 2. Индазолкарбоновую кислоту (III) можно получить традиционными способами. Например, при алкилировании сложного эфира индазол-3-карбоновой кислоты (VIII) с алкилгалогенидом (IX), в соответствии с Chem. Pharm. Bull. (1995) 43(11), 1912-1930, с последующим отделением 2-алкилированного соединения (X) от реакционной смеси хроматографией, затем подвергая 2-алкилированное соединение (X) основному гидролизу и, в итоге, преобразуя полученное таким образом соединение до требуемого карбонового соединения (III), как показано на следующей схеме 2: Схема 2 Соединение (IV) можно получить, например, из амина (XIII), в котором первичная аминогруппа защищается защитной группой Р (схема 3), как описано, например, Green T.M. and Wuts P.G.M., "Protective groups in organic synthesis". John WileySons Publ. (1991). Затем соединение (XIV), полученное таким образом, взаимодействует с соединением (III). Следующим этапом с амида, который образовал (XV),снимается защита, в соответствии с методикой, описанной Green T.M. and Wuts P.G.M., "Protective groupsin organic synthesis". John WileySons Publ. (1991), с получением первичного амина (XVI), который ацилируется с образованием амида (XVII), затем восстанавливается, как в процедуре, описанной на схеме 1,с образованием соединения (IV). Схема 3 Большинство промежуточных соединений общих формул (XV), (XVI) и (XVII) являются новыми. В частности, следующие промежуточные соединения, в целом представленные следующей общей формулой (XXX), являются новыми и таким образом составляют другой предмет настоящего изобретения:R1 и R2 имеют значения, установленные ранее в отношении соединения формулы (I),Т 2 обозначает Н, защитную группу (P), выбранную из группы, включающей 9 флуоренилметилкарбамат, трет-бутилкарбамат, аллилкарбамат, н-бензил и н-бензилиден, или группу RCO, в которой R обозначает C1-3 алкил,R5 и R6 вместе образуют 5- или 6-членное насыщенное кольцо, выбранное из группы, состоящей из пирролидина, имидазолина, пиразолидина, пиперидина и пиперазина,Y обозначает СН,n является целым числом, выбранным из 1, 2 и 3,-6 019217m является 2. Однако по процедуре (1 с) амин (VI) можно получить в соответствии с традиционными способами,например теми, что описаны Fusco R., "The chemistry of heterocyclic compounds, Pyrazoles, Pyrazolines,Indazoles and condensed rings" (1967) Publ. Wiley N.Y., или Katritsky A. and Rees C.W., "Comprehensiveheterocyclic chemistry", vol. 5 (1984) Publ. Pergamon Press. Соединение (VII) можно также получить традиционными способами, например, теми, что описаны в патентной заявке WO 04/101548. Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество по меньшей мере одного соединения 2-алкилиндазола формулы (I) или его соль с фармацевтически приемлемой органической или неорганической кислотой и по меньшей мере один фармацевтически приемлемый наполнитель. Предпочтительно фармацевтическая композиция настоящего изобретения получается в подходящих дозированных формах, содержащих эффективное количество по меньшей мере одного соединения 2-алкилиндазола формулы (I) или его соль с фармацевтически приемлемой органической или неорганической кислотой и по меньшей мере один фармацевтически приемлемый наполнитель. Типичными примерами патологических состояний, которым может помочь лечение фармацевтической композицией, соответствующей настоящему изобретению, являются некоторые расстройства центральной нервной системы, такие как нарушения сна, шизофрения и беспокойство, а также нарушения деятельности гладких мышц, как желудочно-кишечного тракта, так и сердечно-сосудистой системы. Следовательно, другой аспект настоящего изобретения относится к применению вышеупомянутой фармацевтической композиции при лечении патологических состояний, выбранных из группы, включающей нарушения сна, шизофрению, беспокойство, нарушения в гладких мышцах, как желудочнокишечного тракта, так и сердечно-сосудистой системы. Примерами подходящих дозировочных форм являются таблетки, капсулы, таблетки в оболочке,гранулы, растворы и сиропы для перорального приема; медицинские пластыри для трансдермального назначения; суппозитории для ректального введения и инъекционные стерильные растворы. Другими подходящими дозировочными формами являются формы с замедленным высвобождением и формы на основе липосом для перорального, инъекционного или трансдермального назначения. Дозировочные формы могут также содержать другие традиционные ингредиенты, такие как консерванты, стабилизаторы, поверхностно-активные вещества, буферы, соли для регулирования осмотического давления, эмульгаторы, подсластители, красители, вкусовые добавки и тому подобное. Если предъявляются специфические терапевтические требования, то фармацевтическая композиция может содержать другие фармацевтически активные ингредиенты, совместная комбинация которых будет полезна при назначении. Количество соединения 2-алкилиндазола формулы (I) или его фармацевтически приемлемой соли присоединения кислоты в фармацевтической композиции настоящего изобретения может варьироваться в широком диапазоне в зависимости от известных факторов, например типа патологии, тяжести заболевания, веса пациента, дозировочной формы, выбранного способа введения, частоты приемов препарата в день и эффективности выбранного соединения 2-алкилиндазола формулы (I). Однако оптимальное количество может быть легко и просто определено специалистом в этой области. Обычно количество соединения 2-алкилиндазола формулы (I) или его фармацевтически приемлемой соли присоединения кислоты в фармацевтической композиции настоящего изобретения будет таким,чтобы гарантировать уровень введения от 0,0001 до 100 мг/кг/день. Предпочтительно уровень введения составляет от 0,001 до 50 мг/кг/день, еще более предпочтительно от 0,01 до 10 мг/кг/день. Дозировочные формы фармацевтической композиции настоящего изобретения можно получать методиками, которые хорошо известны фармацевтам и включают перемешивание, гранулирование, сдавливание, растворение, стерилизацию и тому подобное. Фармакологические свойства соединений формулы (I), соответствующих настоящему изобретению,оценили с помощью способов, описанных позже в тексте патентной заявки, в разделах Тесты А, В и С. Кратко, сродство 5-HT2A рецептора крысы демонстрировали стандартной методикой в Leysen J.E.,Niemegeers C.J., Van Nueten J.M., Laduron P.M., (1982) "[3H-Ketanserin, a selective 3H-ligand for serotonin 2receptor binding sites. Binding properties, brain distribution and functional role", Molecular Pharmacology 21: 301-314. (Тест А) Значения сродства некоторых соединений формулы (I), соответствующих настоящему изобретению, для 5-HT2A рецептора показаны в табл. 1, в которой сродство больше для более высокого значения Селективное сродство соединений формулы (I), соответствующих настоящему изобретению, для других рецепторов или систем для транспортировки и введения серотонина также определили, используя стандартную методику (Тест В). Эти тесты позволили измерить ингибирование, выраженное в %, вызванное соединениями формулы(I), соответствующими настоящему изобретению, при концентрации 1 мкМ для рецепторов: 5-HT1A, 5HT1B, 5-HT1D, 5-HT2b, 5-HT3, 5-HT4, 5-НТ 4(b), 5-HT5(A), 5-НТ 6 и 5-НТ 7. Кроме этого, определи процентное ингибирование, вызванное соединениями формулы (I), соответствующими настоящему изобретению,при 1 мкМ на транспортную систему серотонина и при 10 мкМ на клеточную систему для введения серотонина. Во всех классах соединения формулы (I), соответствующие настоящему изобретению, вызывали процентное ингибирование, при использовании концентрации меньше чем 50%. Преференциальное сродство соединений формулы (I), соответствующих настоящему изобретению,для 5-HT2A рецептора по сравнению с 5-HT2C рецептором, также определили, используя стандартную методику (Тест В). В соответствии с этими тестами измерили процентное ингибирование, вызванное соединениями формулы (I), соответствующими настоящему изобретению, при шести различных концентрациях, из которых получили значения pKi для соединений по отношению к индивидуальным рецепторам. Значения сродства некоторых соединений формулы (I), соответствующих настоящему изобретению, для 5-HT2A и 5-HT2C рецепторов рекомбинантных клеток человека показаны в табл. 2, в которой сродство больше для более высоких значений pKi. Таблица 2 Более того, соединения формулы (I), соответствующие настоящему изобретению, испытали in vivo в организмах живых мышей на модели, описывающей "судорожные движение головой" (Тест С). Этот стандартный тест для определения любого взаимодействия с серотонинергической системой, как описано в Sztanke К., Fidecka S., Kedzierska E., Karczmarzyk Z., Pihlaja K., Matosiuk D. (2005) "Antionociceptiveactivity of new imidazole carbonyl derivatives. Part 4. Synthesis and pharmacological activity of 8-aryl-3,4dioxo-2H,8H-6,7-dihydrimidazo[2,1c][1,2,4]triazine", Eur. J. Med. Chem. 40: 127-134, и в Corne S.J.,Pickering R.W., Warner B.T., (1963) "A method for assessing the effects of drugs on the central actions of 5hydroxytryptamine", Br. J. Pharmacol. Chemother. 20: 106-120. В этом тесте исследуемые соединения вызвали уменьшение числа "судорожных движений головой" у животных, обработанных только метилцеллюлозой (МТС), таким образом демонстрируя способность для антагонизма серотонинергических эффектов, вызванных введением 5-гидрокситриптофана (5-НТР). Значения ингибирования, полученные в тесте "Судорожные движения головой" с тестируемыми соединениями, показаны на фиг. 1. Описание, которое представлено далее в тексте патентной заявки, предназначено для дополнительной иллюстрации настоящего изобретения, без ограничений. Пример 1. 2-Метил-N-[1-(2-фенилэтил)пиперидин-4-ил]метил-2 Н-индазол-3-карбоксамид гидрохлорид гидрат (соединение 1)R6+R7=-CH2-CH2-) 1a) Метиловый эфир 2-метил-2 Н-индазол-3-карбоновой кислоты (Соединение X: R1=H, R2=CH3) Карбонат калия (207,1 г; 1,498 моль) и по каплям метилиодид (31 мл; 0,50 моль) добавляли к суспензии метилового эфира 1 Н(2 Н)-индазол-3-карбоновой кислоты (74,0 г; 0,499 моль) в ацетоне (750 мл). Реакционную смесь нагревали с обратным холодильником в течение 4 ч, охлаждали и фильтровали. Растворитель удаляли выпариванием при пониженном давлении и остаток дважды промывали в этилацетате. Приблизительно 80 г сырого соединения получили таким образом. Твердое вещество кристаллизовали дважды из смеси н-гексан/этилацетат, получая метиловый эфир 2-метил-2 Н-индазол-3-карбоновой кислоты (50,0 г) . 1H-ЯМР (, ДМСО-d6): 3,93 (с, 3 Н), 4,17 (с, 3 Н), 7,3-7,4 (м, 1 Н), 7,5-7,6 (м, 1 Н), 7,7-7,8 (м, 1 Н), 8,08,1 (м, 1 Н). 1b) Хлорид 2-метил-2 Н-индазол-3-карбоновой кислоты (Соединение III; R1=H, R2=CH3, Z=Cl) Гидролиз метилового эфира 2-метил-2 Н-индазол-3-карбоновой кислоты (1 а) (50,0 г; 0,260 моль) выполняли в воде (300 мл), содержащей гидроксид натрия (20,9 г; 0,520 моль). Реакционную смесь кипятили с обратным холодильником в течение 2 ч, охлаждали до комнатной температуры и подкисляли 2 нHCl до тех пор, пока не выпадет белый осадок. После фильтрования и сушки в печи под вакуумом, получили 45,2 г 2-метил-2 Н-индазол-3-карбоновой кислоты и ее использовали без дальнейшей очистки в последующих реакциях. Тионилхлорид (39 мл; 0,54 моль) добавляли к суспензии 2-метил-2 Н-индазол-3-карбоновой кислоты (45,2 г; 0,270 моль) в толуоле (350 мл) и реакционную смесь нагревали с обратным холодильником 24 ч. Растворитель удаляли выпариванием при пониженном давлении. Остаток дважды промывали в толуоле. Таким образом получили 47,0 г хлорида 2-метил-2 Н-индазол-3-карбоновой кислоты. 1 Н-ЯМР (, ДМСО-d6): 3,97 (с, 3 Н); 7,3-7,4 (м, 1 Н), 7,5-7,6 (м, 1 Н); 7,7-7,8 (м, 1 Н); 8,0-8,1 (м, 1 Н). 1c) 1-[1-(2-Фенилэтил)-4-пипериденил]метанамин (Соединение XI: R3=R4=H, R6+R7 = -CH2-CH2-,n=1, р=2, m=2) Бензальдегид (28,0 г; 0,264 моль) добавляли по каплям к раствору 4-аминометилпиперидина (30,2 г; 0,264 моль) в толуоле (100 мл). Полученный таким образом раствор продолжали перемешивать при комнатной температуре. Через 3 ч растворитель удалили выпариванием при пониженном давлении и остаток дважды промывали в толуоле. 53,4 г N-гексагидро-4-пиридинилметил-N-фенилметилиденамина получили таким образом и использовали в последующих реакциях без дополнительной очистки.N-Гексагидро-4-пиридинилметил-N-фенилметилиденамин (53,4 г; 0,264 моль) растворили в абсолютном этаноле (500 мл), содержащем безводный карбонат калия (73,0 г; 0,528 моль) и фенэтилбромид(39,5 мл; 0,290 моль). Полученную таким образом реакционную смесь кипятили с обратным холодильником 24 ч. После охлаждения до комнатной температуры суспензию фильтровали и растворитель выпаривали при пониженном давлении. Полученный таким образом остаток суспендировали в 3 н HCl (200 мл) и перемешивали при комнатной температуре 2 ч. Кислую водную фазу промывали 4 раза этилацетатом и затем подщелачивали до рН приблизительно 13 6 н NaOH и экстрагировали дихлорметаном. Органическую фазу, полученную таким образом, сушили над безводным Na2SO4 и растворитель удалили выпариванием при пониженном давлении. Таким образом получили 50,0 г 1-[1-(2-фенилэтил)-4-пипериденил]метанамина. 1-CH2-CH2-) 1-[1-(2-Фенилэтил)-4-пипериденил]метанамина (1 с) (20,3 г; 0,0930 моль) добавляли к раствору хлорида 2-метил-2 Н-индазол-3-карбоновой кислоты (1b) (18,0 г; 0,0930 моль) в толуоле (100 мл). Реакционную смесь продолжали перемешивать при комнатной температуре в течение ночи и затем фильтровали. Твердое вещество промывали в 1 н NaOH и экстрагировали дихлорметаном (DCM). Органические фазы объединили и затем сушили над обезвоженным Na2SO4, и растворитель удаляли выпариванием при пониженном давлении. Неочищенное соединение, полученное таким образом, преобразовывали до соответствующего гидрохлорида растворением в этилацетате, обработкой избытком 5 н хлористо-водородным этанолом при комнатной температуре 3 ч, удалением растворителя выпариванием при пониженном давлении и кристаллизацией твердого вещества, полученного из этилацетата/абсолютного этанола. Получили 7,2 г 2-метил-N-[1-(2-фенилэтил)пиперидин-4-ил]метил-2 Н-индазол-3-карбоксамид гидрохлорид гидрата. Т.пл.=130-140 С. Элементный анализ для C23H31C1N4O2(Соединение I: R1=H, R2=CH3, Х=-C(О)N(R5)-, R3=R4=H, R5=CH3, n=1, p=2, m=2, Y=CH, W=N,R6+R7=-CH2-CH2-) 2a) N,2-Диметил-N-[1-(2-фенилэтил)пиперидин-4-ил]метил-2 Н-индазол-3-карбоксамид гидрохлорид хемигидрат После перемешивания в течение 1 ч при комнатной температуре добавляли метилиодид (0,83 мл; 0,013 моль) к суспензии 2-метил-N-[1-(2-фенилэтил)пиперидин-4-ил]метил-2 Н-индазол-3 карбоксамида (1d) (5,0 г; 0,013 моль) и 60% гидрида натрия (0,51 г; 0,013 моль) в тетрагидрофуране(ТГФ) (50 мл). Полученную таким образом смесь перемешивали при комнатной температуре 48 ч. Затем добавляли дистиллированную воду, чтобы удалить любой избыток гидрида. Органическую фазу отделяли и концентрировали выпариванием при пониженном давлении. Полученный остаток очищали, применяя флэш-хроматографию (элюенты CHCl3/CH3OH/NH3 в пропорции 85/14/1) и преобразовывали до гидрохлорида растворением в этилацетате, обработкой избытком 5 н хлористо-водородного этанола при комнатной температуре в течение 3 ч и удалением растворителя выпариванием при пониженном давлении. Соединение, полученное таким образом, кристаллизовали из смеси этилацетата/абсолютного спирта. 5,2 г N,2-диметил-N-[1-(2-фенилэтил)пиперидин-4-ил]метил-2 Н-индазол-3-карбоксамид гидрохлорид хемигидрат получили таким образом. Т.пл.=206-207 С. Элементный анализ для C24H33ClN4O2(Соединение I: R1=H, R2=CH3, Х=-С(О)N(R5)-, R3=4-OH, R4=H, R5=H, n=1, p=2, m=2, Y=CH,W=N, R6+R7 = -CH2-CH2-) 3a) 1-(2-(4-Гидроксифенил)этил)-4-пиридинил метанамин (Соединение XI: R3=4-OH, R4=H,R6+R7=-CH2-CH2-, n=1, p=2, m=2) Получение было таким же, как описано для получения (1 с), начиная реакцию с N-гексагидро-4 пиридинилметил-N-фенилметилиденамина (7,5 г, 0,037 моль) и 2-(4-гидроксифенил)этил бромида (7,5 г; 0,037 моль) [полученного, как описано в Acta Chemica Scandinava (1967), 21 (1), 53-62]. Таким образом получили 9,3 г 1-(2-(4-гидроксифенил)этил)-4-пиридинил метанамина. 1H-ЯМР (, CDCl3 + D2O): 1,15-1,41 (м, 3 Н); 1,74 (д, J=9 Гц, 2 Н); 1,9-2,1 (м, 2 Н), 2,4-2,6 (м, 4 Н); 2,65-2,75 (м, 2 Н); 3,01 (д, J=12 Гц, 2 Н); 6,75 (д, J=9 Гц, 2 Н); 7,00 (д, J=9 Гц, 2 Н). 3b) N3-(1-[2-(4-Гидроксифенил)этил]гексагидро-4-пиридинилметил)-2-метил-2 Н-3-индазолкарбоксамид гидрохлорид гидрат 1-(2-(4-гидроксифенил)этил)-4-пиридинил метанамин (3 а) (5,6 г, 0,024 моль) добавляли небольшими порциями к раствору хлорида 2-метил-2 Н-индазол-3-карбоновой кислоты (1b) (4,7 г, 0,024 моль) в толуоле (100 мл). Смесь, полученную таким образом, перемешивали 18 ч при комнатной температуре. Затем фильтровали. Осадок очищали несколько раз кристаллизацией из смеси этилацетата/абсолютного этанола и преобразовывали до гидрохлорида растворением в этилацетате, обработкой избытком 5 н хлористоводородного этанола при комнатной температуре в течение 3 ч и удалением растворителя выпариванием при пониженном давлении. После кристаллизации из этилацетата/абсолютного этанола получили 5,0 г N3-(1-[2-(4 гидроксифенил)этил]гексагидро-4-пиридинилметил)-2-метил-2 Н-3-индазолкарбоксамид гидрохлорид гидрата. Т.пл.=236-238 С. Элементный анализ для C23H29ClN4O2 3/4 Н 2 О(Соединение X: R1=5-CH3O, R2=CH3) Смесь 5-метоксииндазол-3-карбоновой кислоты [полученная в соответствии с Gazzetta Chimica Italiana (1963) 93, 3-14] (11,8 г, 0,0610 моль), метанола (200 мл) и серной кислоты (2 мл) перемешивали при комнатной температуре 4 ч. Затем смесь разводили дистиллированной водой. Твердое вещество, которое образовалось, отделяли фильтрованием, сушили в печи (9,6 г) и использовали без дальнейшей очистки для последующей реакции. Гидроксид калия (3,6 г; 0,064 моль) добавляли небольшими порциями к суспензии, содержащей метиловый эфир 5-метоксииндазол-3-карбоновой кислоты (9,6 г; 0,047 моль) и метилиодид (3,4 мл; 0,054 моль) в диметоксиэтане (DME) (50 мл). Реакционную смесь нагревали с обратным холодильником 18 ч и затем охлаждали. Растворитель удаляли выпариванием при пониженном давлении. Твердое вещество обрабатывали толуолом и промывали несколько раз водой и 6 н NaOH. Затем растворитель выпаривали при пониженном давлении и полученный осадок очищали флэш-хроматографией (нгексан/этилацетат=7/3). Таким образом получали 5,0 г метиловый эфир 5-метокси-2-метил-2 Н-индазол-3 карбоновой кислоты. 1(Соединение III: R1=5-CH3O, R2=CH3, Z=Cl) Гидроксид натрия (1,0 г; 0,025 моль) добавляли к суспензии метилового эфира 5-метокси-2-метил 2 Н-индазол-3-карбоновой кислоты (полученного, как описано в предшествующем примере 4 а) (2,8 г; 0,013 моль) в дистиллированной воде (40 мл). Смесь, полученную таким образом, оставляли взаимодействовать с обратным холодильником в течение 4 ч, охлаждали и подкисляли 2 н HCl до тех пор, пока осадок 5-метокси-2-метил-2 Н-индазол-3-карбоновой кислоты полностью не выпадет, и после фильтрования его использовали без дополнительной очистки в последующих реакциях. Тионилхлорид (0,8 мл; 0,01 моль) добавляли в колбу, содержащую 5-метокси-2-метил-2 Н-индазол 3-карбоновую кислоту (1,1 г; 0,0050 моль) и толуол (30 мл). Реакция в смеси проходила с обратным холодильником 5 ч и после выпаривания растворителя при пониженном давлении остаток промывали несколько раз в толуоле. Таким образом получили 1,2 г хлорида 5-метокси-2-метил-2 Н-индазол-3 карбоновой кислоты. 1N-(1-[2-(4-Гидроксифенил)этил]пиперидин-4-ил)метил-5-метокси-2-метил-2 Н-индазол-3 карбоксамид 1-(2-(4-Гидроксифенил)этил)-4-пиперидинил метанамин (3 а) (1,25 г; 0,0050 моль) добавляли небольшими порциями к суспензии хлорида 5-метокси-2-метил-2 Н-индазол-3-карбоновой кислоты (4b) (1,2 г; 0,0050 моль) в толуоле (30 мл). Смесь, полученную таким образом, оставляли взаимодействовать 4 ч при комнатной температуре и кипятили с обратным холодильником дополнительно 4 ч. Затем суспензию охлаждали и фильтровали. Твердое вещество кристаллизовали из н-гексан/этилацетата. Таким образом получили 1,5 г N-(1-[2-(4-гидроксифенил)этил]пиперидин-4-илметил)-5-метокси 2-метил-2 Н-индазол-3-карбоксамида. Т.пл.=218-219 С (разл.) 1(Соединение I: R1=H, R2=CH3, Х=-С(О)N(R5)-, R3=4-CH3O, R4=H, R5=H, n=1, p=2, m=2, Y=CH,W=N, R6+R7=-CH2-CH2-) 5a) 1-[2-(4-Метоксифенил)этил]-4-пиперидинилметанамин (Соединение XI: R3=4-CH3O, R4=H,R6+R7=-CH2-CH2-, n=l, p=2, m=2) Соединение получали таким же способом, как описан для получения 1 с), начиная с N-гексагидро-4 пиридинилметил-N-фенилметилиденамина (12,9 г; 0,0631 моль) и 2-(4-метоксифенил)этилбромида (15,0 г; 0,0698 моль) [полученного, как описано в Acta Chemica Scandinava (1967), 21 (1), 53-62]. Получили 5,0 г 1-[2-(4-метоксифенил)этил]-4-пиперидинилметанамина. 1 Н-ЯМР (, CDCl3): 1,1-1,4 (м, 3 Н); 1,6-1,9 (м, 4 Н); 1,9-2,1 (м, 2 Н); 2,4-2,6 (м, 4 Н); 2,7-2,8 (м, 2 Н); 2,9-3,1 (м, 2 Н); 3,77 (с, 3 Н); 6,7-6,9 (м, 2 Н); 7,0-7,2 (м, 2 Н). 5b)N-(1-[2-(4-Метоксифенил)этил]пиперидин-4-илметил)-2-метил-2H-индазол-3-карбоксамид гидрохлорид 1-[2-(4-Метоксифенил)этил]-4-пиперидинилметанамин (5 а) (6,8 г; 0,027 моль) добавляли небольшими порциями к суспензии хлорида 2-метил-2 Н-индазол-3-карбоновой кислоты (1b) (5,3 г; 0,027 моль) в толуоле (200 мл). Смесь, полученную таким образом, перемешивали 48 ч при комнатной температуре и затем фильтровали. Твердое соединение суспендировали в 1 н NaOH и экстрагировали 3 раза с дихлорметаном. Растворитель удаляли выпариванием при пониженном давлении и остаток кристаллизовали сначала из смеси изопропиловый эфир/изопропанол и затем из смеси этилацетат/абсолютный этанол. Затем соединение преобразовывали до его гидрохлорида путем растворения в этаноле с последующей обработкой 5 н хлористо-водородным этанолом в течение 3 ч при комнатной температуре и затем удалили растворитель выпариванием при пониженном давлении. После перекристаллизации из смеси этилацетат/абсолютный этанол получили 5,0 г N-(1-[2-(4 метоксифенил)этил]пиперидин-4-илметил)-2-метил-2 Н-индазол-3-карбоксамид гидрохлорида. Т.пл.=80 С (разл.) 1(Соединение I: R1=H, R2=CH3, Х=-С(О)N(R5)-, R3=4-CH3O, R4=H, R5=CH3, n=1, p=2, m=2, Y=CH,W=N, R6+R7 = -CH2-CH2-) 6a) (1-[2-(4-Метоксифенил)этил]-4-пиперидинилметил)формамид (Соединение XII: R3=4-CH3O,R4=H, R6+R7 = -CH2-CH2-, n=1, p=2, m=2, R=H) Муравьиную кислоту (0,60 мл, 0,016 моль) добавляли по каплям, в атмосфере азота, при температуре около 0 С, в колбу, содержащую уксусный ангидрид (1,25 мл; 0,0112 моль). Полученную таким образом смесь нагревали с обратным холодильником 2 ч и после охлаждения до комнатной температуры добавляли раствор 1-[2-(4-метоксифенил)этил]-4-пиперидинил-метанамина (5 а) (1,24 г; 502 ммоль) в ТГФ(10 мл). Смесь реагировала в течение 18 ч. Затем суспензию концентрировали выпариванием при пониженном давлении. Таким образом получили 1,0 г(Соединение II: R3=4-CH3O, R4=H, R5=CH3, R6+R7=-CH2-CH2-, n=l, p=2, m=2) Раствор комплекса боран-диметилсульфида 2 н в н-гексане (6 мл) добавляли по каплям к суспензии(1-[2-(4-метоксифенил)этил]-4-пиперидинилметил)формамида (1,0 г; 0,0041 моль (6 а) в 20 мл ТГФ при 0 С. Смесь нагревали с обратным холодильником 5 ч и затем охлаждали до 0 С. При этой температуре затем добавляли 2 мл метанола и реакционную смесь энергично перемешивали 1 ч. Следующим этапом барботировали газообразный HCl в реакционную смесь до тех пор, пока подкисление не было завершено. Затем смесь нагревали с обратным холодильником в течение 1 ч. После охлаждения смеси добавляли метанол (10 мл) и растворитель выпаривали при пониженном давлении. Остаток переносили в воду и промывали 3 раза этилацетатом. Водную фазу подщелачивали бн NaOH и экстрагировали 3 раза дихлорметаном. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали при пониженном давлении. Таким образом получили 0,6 г 1-1-[2-(4-метоксифенил)этил]-4 пиперидинил-N-метилметанамина.N3-2-Диметил-N3-[(1-2-[4-(метилокси)фенил]этилпиперидин-4-ил)метил]-2 Н-индазол-3 карбоксамид гидрохлорид гидрат 1-1-[2-(4-Метоксифенил)этил]-4-пиперидинил-N-метилметанамин (полученный, как описано в предшествующем примере 6b) (1,4 г; 0,0052 моль) добавляли небольшими порциями к раствору хлорида 2-метил-2 Н-индазол-3-карбоновой кислоты (1b) (1,1 г; 0,0061 моль) в толуоле (20 мл). Смесь, полученную таким образом, перемешивали 18 ч при комнатной температуре и затем фильтровали. Твердое соединение промывали в 1 н NaOH и экстрагировали дихлорметаном. Объединенные органические фазы сушили над Na2SO4 и растворитель удаляли выпариванием при пониженном давлении. Твердый остаток преобразовывали до гидрохлорида растворением в этаноле, добавлением избытка 5 н хлористо-водородного этанола, перемешивая 3 ч при комнатной температуре, и затем удаляли растворитель выпариванием при пониженном давлении. После перекристаллизации из смеси этилацетат/абсолютный этанол получили 0,8 г N3-2-диметилN3-[(1-2-[4-(метилокси)фенил]этилпиперидин-4-ил)метил]-2 Н-индазол-3-карбоксамид гидрохлорид гидрата. Т.пл.=132,5-136,5 С. Элементный анализ для C25HT35ClN4O3(Соединение I: R1=H, R2=CH2CH2OCH3, Х=-С(О)N(R5)-, R3=4-F, R4=H, R5=H, n=1, p=2, m=2,Y=CH, W=N, R6+R7=-CH2-CH2-) 7a) 2-(2-Метоксиэтил)-2 Н-индазол-3-этилкарбоксилат (Соединение III: R1=H, R2=CH3OCH2CH2,Z=CH3CH2O) Карбонат калия (33,6 г; 0,243 моль) и по каплям, 2-бромэтилметиловый эфир (46 мл; 0,49 моль) добавляли к раствору этилового эфира 1H(2 Н)-индазол-3-карбоновой кислоты (15,4 г; 0,0811 моль) в абсолютном этаноле (200 мл). Реакционную смесь перемешивали 48 ч. Затем растворитель выпаривали при пониженном давлении. Остаток переносили в хлороформ и промывали водой 3 раза. Органическую фазу сушили над Na2SO4 и после фильтрования и выпаривания растворителя получили приблизительно 10 г сырого продукта. Его очищали флэш-хроматографией, используя нгексан/этилацетат 7:3 смесь в качестве элюента. Таким образом, получили 4,5 г 2-(2-метоксиэтил)-2 Ниндазол-3-этилкарбоксилата. 1(т, J=6 Гц, 2 Н); 7,2-7,4 (м, 2 Н); 7,79 (д, J=9 Гц, 1 Н); 8,02 (д, J=9 Гц, 1 Н). 7b) 2-(2-Метоксиэтил)-2 Н-индазол-3-карбонилхлорид (Соединение III: R1=H, R2=CH3OCH2CH2,Z=Cl) Сложный эфир 2-(2-метоксиэтил)-2 Н-индазол-3-этилкарбоксилата (полученный, как описано в предшествующем примере 7 а) (4,26 г; 0,0171 моль) гидролизовали в водном растворе (30 мл) гидроксида натрия (1,16 г; 0,0291 моль). Реакционную смесь нагревали с обратным холодильником 2 ч, охлаждали до комнатной температуры и подкисляли 6 н HCl до тех пор, пока полностью не выпадала в осадок 2-(2 метоксиэтил)-2 Н-индазол-3-карбоновая кислота, которая после отделения фильтрованием и сушки в печи под вакуумом была в виде твердого вещества белого цвета (3,6 г), который использовали без дальнейшей очистки в следующей реакции. Тиенилхлорид (2,6 г; 0,036 моль) добавляли к суспензии 2-(2-метоксиэтил)-2 Н-индазол-3 карбоновой кислоты (3,6 г; 0,016 моль) в толуоле (100 мл) и реакционную смесь нагревали с обратным холодильником 24 ч. Растворитель удаляли выпариванием при пониженном давлении и остаток промывали дважды в н-гексане. Таким образом получали 3,9 г 2-(2-метоксиэтил)-2 Н-индазол-3 карбонилхлорида. 1H-ЯМР (, CDCl3): 3,29 (с, 3 Н); 3,84 (т, J=6 Гц, 2 Н); 5,11 (т, J=6 Гц, 2 Н); 7,1-7,4 (м, 2 Н); 7,74 (д, J=9 Гц, 1 Н); 7,99 (д, J=9 Гц, 1 Н). 7 с) 1-(2-Бромэтил)-4-фторбензол (Соединение V: R3=4-F, R4=H, m=2, Q=Br) 60,8 г 47% бромисто-водородной кислоты (0,353 моль) медленно добавляли к раствору 2-(4 фторфенил)этанола (20 г; 0,143 моль) в концентрированной серной кислоте (8,5 мл; 0,16 моль). Смесь нагревали с обратным холодильником 3 ч, затем охлаждали до комнатной температуры и добавляли 50 мл дихлорметана. Органическую фазу отделяли и успешно промывали 1 н NaOH и водой и затем сушили над Na2SO4. После фильтрования и выпаривания растворителя при пониженном давлении получили приблизительно 13 г сырого соединения, которое очищали флэш-хроматографией (н-гексан/этилацетат=9/1). 10,6 г 1-(2 бромэтил)-4-фторбензола получили таким образом. 1N-гексагидро-4-пиридинилметил-N-фенилметилиденамин (17,9 г; 0,0879 моль) растворили в абсолютном этаноле (100 мл), содержащем безводный карбонат калия (24,3 г; 0,176 моль) и 1-(2-бромэтил)-4 фторбензол (полученный, как описано в предшествующем примере 7 с) (18,0 г; 0,088 моль). Реакционную смесь нагревали с обратным холодильником 24 ч. После охлаждения до комнатной температуры суспензию фильтровали и растворитель выпаривали при пониженном давлении. Полученный остаток суспендировали в 3 н HCl (120 мл) и перемешивали при комнатной температуре 3 ч. Кислую водную фазу промывали 4 раза этилацетатом и затем подщелачивали до рН приблизительно 13 6 н NaOH и окончательно экстрагировали 3 раза этилацетатом. Полученную таким образом органическую фазу сушили над безводным Na2SO4 и после фильтрования растворитель выпаривали при пониженном давлении, чтобы получить 17,2 г 1-[2-(4-фторфенил)этил]-4-пиперидин метанамина. 1 Н-ЯМР (, CDCl3): 1,2-1,5 (м, 5H); 1,6-1,8 (м, 2 Н); 1,9-2,1 (м, 2 Н); 2,4-2,6 (м, 4 Н); 2,7-2,8 (м, 2 Н); 2,9-3,1 (м, 2 Н); 6,9-7,2 (м, 4 Н).N-(1-[2-(4-Фторфенил)этил]пиперидин-4-илметил)-2-(2-метоксиэтил)-2 Н-индазол-3 карбоксамид гидрохлорид 1-[2-(4-Фторфенил)этил]-4-пиперидинметанамин (полученный, как описано в предшествующем примере 7d) (3,23 г; 0,0131 моль) и триэтиламин (5,4 мл; 0,039 моль) добавляли к раствору 2-(2 метоксиэтил)-2 Н-индазол-3-карбонилхлорида (полученному, как описано в предшествующем примере 7b) (3,26 г; 0,0131 моль) в толуоле (50 мл). Реакционную смесь нагревали с обратным холодильником 5 ч и затем растворитель выпаривали при пониженном давлении. Остаток обрабатывали в этилацетате и успешно промывали 0,1 н NaOH и водой. Органическую фазу сушили над безводным Na2SO4 и после фильтрования растворитель выпаривали при пониженном давлении. Получили 4,7 г сырого продукта, который очищали флэш-хроматографией(CHCl3/CH3OH/NH3=98/2/0,2). Полученное соединение (1,5 г) преобразовывали до соответствующего гидрохлорида растворением в этилацетате, обработкой избытком 5 н хлористо-водородного этанола при комнатной температуре в течение 3 ч, удалили растворитель выпариванием при пониженном давлении и окончательно кристаллизовали из абсолютного этанола. Таким образом получили 0,6 г N-(1-[2-(4-фторфенил)этил]пиперидин-4-илметил)-2-(2 метоксиэтил)-2H-индазол-3-карбоксамид гидрохлорида. Т.пл.=193,0-194,0 С. Элементный анализ для C25H31N4O2FHCl(CHCl3/CH3OH/NH3=97/3/0,3). Полученное таким образом соединение (4,1 г) преобразовали до соответствующего гидрохлорида растворением в этаноле, обработкой избытком 5 н хлористо-водородным этанолом при комнатной температуре 3 ч, удалением растворителя выпариванием при пониженном давлении и в конце кристаллизацией из абсолютного этанола/этилацетата. 4,3 г N-(1-[2-(4-фторфенил)этил]пиперидин-4-илметил)-2-метил-2 Н-индазол-3-карбоксамид гидрохлорида получили таким образом. Т.пл.=208,0-209,0 С. Элементный анализ для C23H27N4OFHClN-Гексагидро-4-пиридинилметил-N-фенилметилиденамин (11,3 г; 0,0558 моль) и триэтиламин (9,3 мл; 0,066 моль) добавляли к раствору хлорида 2-метил-2 Н-индазол-3-карбоновой кислоты (полученной,как описано в предшествующем примере 1b) (10,8 г; 0,0558 моль) в толуоле (150 мл). Смесь нагревали с обратным холодильником 5 ч. Затем реакционную смесь охлаждали до комнатной температуры и фильтровали. Растворитель выпаривали при пониженном давлении. Остаток перемешивали при комнатной температуре с 3 н HCl (80 мл) 5 ч. Кислый раствор промывали 3 раза этилацетатом. Затем подщелачивали 12 н NaOH. Щелочную водную фазу экстрагировали дихлорметаном 3 раза. Объединенные органические фазы сушили над безводным Na2SO4. Затем их фильтровали и растворитель выпаривали при пониженном давлении. Таким образом получили приблизительно 16 г сырого продукта и очищали флэш-хроматографией (CHCl3/СН 3 ОН/NH3=95/5/0,5). Получили 13,4 г 1-1-[(2-метил-2 Н-индазол-3-ил)карбонил]пиперидин-4-илметанамина.(Соединение V: R3=2-F, R4=4-F, m=2, Q=CH3OSO2) Раствор (2,4-дифторфенил)уксусной кислоты (15,0 г; 0,0871 моль) в этиловом эфире (100 мл) добавляли медленно к суспензии гидрида литий-алюминия (6,58 г; 0,174 моль) в этиловом эфире (100 мл). Смесь нагревали с обратным холодильником 5 ч и затем довели до комнатной температуры. Избыток гидрида удаляли добавлением 1 н HCl (150 мл). Кислотную фазу отделяли и затем экстрагировали 3 раза этиловым эфиром. Объединенные органические фазы промыли дважды 1 н NaOH, сушили над безводнымNa2SO4 и после фильтрования растворитель выпарили при пониженном давлении. Таким образом получили 6,4 г требуемого соединения и использовали без дальнейшей очистки для последующей реакции. Метансульфонилхлорид (3,3 мл; 0,043 моль) и триэтиламин (5,9 мл; 0,043 моль) добавляли к раствору 2-(2,4-дифторфенил)этанола (6,4 г; 0,0426 моль) в дихлорметане (100 мл). Растворитель охлаждали в течение 1 ч до 0 С и затем довели до комнатной температуры за 1 ч. Затем реакционную смесь обрабатывали, успешно промывая 5 н серной кислотой (20 мл), водой (50 мл), 5% раствором NaHCO3 (50 мл) и водой (50 мл). Затем органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток промывали дважды в CCl4, таким образом получили 6,42 г 2-(2,4 дифторфенил)этилметансульфоната. 1(полученный, как описано в предшествующем примере 9 а) (6,73 г, 0,0247 моль) добавляли к раствору 2(2,4-дифторфенил)этилметансульфоната (9b) (6,42 г; 0,0271 моль) в абсолютном спирте (150 мл). Смесь нагревали с обратным холодильником 30 ч и затем растворитель выпаривали при пониженном давлении. Остаток обрабатывали этилацетатом и промывали 3 раза водой. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Таким образом получили приблизительно 15 г сырого продукта,который очищали флэш-хроматографией(CHCl3/CH3OH/NH3=99/1/0,1). Полученное таким образом соединение (2,5 г) преобразовали до гидрохлорида растворением в абсолютном этаноле и обработкой избытком 5 н хлористо-водородным этанолом 3 ч. После выпаривания растворителя при пониженном давлении твердый остаток кристаллизовали из этанола/этилацетата, получая 0,7 г 2-(2,4-дифторфенил)-N-(1-[(2-метил-2 Н-индазол-3-ил)карбонил]пиперидин-4-илметил)этанамин гидрохлорид. Т.пл.=108,0-110,0 С. Элементный анализ для C23H26N4OF2HCl(Соединение I: R1=H, R2=CH3, Х=-С(О)N(R5)-, R3=4-F, R4=H, R5+R6=-CH2-CH2-, n=2, p=1, m=2,Y=CH, W=N, R7=H) 10a) 2-(4-Фторфенил)-N-(1-[(2-метил-2H-индазол-3-ил)карбонил]пиперидин-4-илметил)этанамин гидрохлорид Требуемое соединение получили, как описано в примере 9 с), используя 4,10 г 1-(2-бромэтил)-4 фторбензол (полученный, как описано в предшествующем примере 7 с) (0,0201 моль), 5,0 г 1-1-[(2 метил-2 Н-индазол-3-ил)карбонил]пиперидин-4-илметанамин (полученный, как описано в предшествующем примере 9 а) (0,018 моль), 2,77 г K2CO3 (0,0201 моль) и 100 мл абсолютного этанола. Таким образом получили приблизительно 7 г сырого продукта, который очищали флэшхроматографией (CHCl3/СН 3 ОН/NH3=98/2/0,2). Полученное таким образом соединение (3,5 г) преобразовали до гидрохлорида растворением в абсолютном этаноле и обработкой избытком 5 н хлористоводородным этанолом 3 ч. После выпаривания растворителя при пониженном давлении твердый остаток кристаллизовали из этанола/диизопропилового эфира, получая 2,5 г 2-(4-фторфенил)-N-(1-[(2-метил 2 Н-индазол-3-ил)карбонил]пиперидин-4-илметил)этанамин гидрохлорид. Т.пл.=198,0-199,0 С. Элементный анализ для C23H27N4OFHClK2CO3 (0,71 г; 0,0052 моль), иодид калия (10 мг; 0,062 ммоль) и этилбромид (0,54 мл; 0,0074 моль) добавляли к раствору 2-(4-фторфенил)-N-(1-[(2-метил-2 Н-индазол-3-ил)карбонил]пиперидин-4 илметил)этанамина (10 а) (1,7 г; 0,0043 моль) в абсолютном этаноле (20 мл). Смесь нагревали с обратным холодильником 48 ч и затем растворитель выпаривали при пониженном давлении. Остаток обрабатывали хлороформом и промывали дважды водой. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный в результате (1,5 г) сырой продукт очищали флэш-хроматографией (этилацетат/н-гексан/аммоний=96/4/0,2). Полученное соединение (0,5 г) преобразовали до гидрохлорида растворением в абсолютном этаноле и обработкой избытком 5 н хлористоводородным этанолом 3 ч. После выпаривания растворителя при пониженном давлении, твердый остаток кристаллизовали из этанола/этилацетата, получая 0, 15 г N-этил-2-(4-фторфенил)-N-(1-[(2-метил-2 Ниндазол-3-ил)карбонил]пиперидин-4-илметил)этанамин гидрохлорид. 1 Н-ЯМР (, CDCl3): 0,6-2,4 (м, 5 Н); 1,44 (т, J=9 Гц, 3 Н); 2,7-3,7 (м, 12 Н); 4,28 (с, 3 Н), 6,9-7,4 (м, 6 Н); 7,46 (д, J=9 Гц, 1 Н); 7,72 (д, J=9 Гц, 1 Н); 12,0-12,7 (ушир.с, 2 Н). фторфенил)этил)-4-пиперидин метанамин (пример 12 а) (1,06 г; 4,5 ммоль). Таким образом получили 350 мг N-(1-[2-(2-фторфенил)этил)пиперидин-4-илметил)-2-(2 метоксиэтил)-2 Н-индазол-3-карбоксамид. т.пл.=128-130 С. Элементный анализ для C23H27N4OF(Соединение X: R1=H, R2=CH2CH3) Соединение получали в соответствии с методикой, описанной в примере 1 а), используя в качестве реагентов метиловый эфир 1 Н (2 Н)-индазол-3-карбоновой кислоты (7,4 г; 49,9 ммоль) и этилбромид(Соединение III: R1=H, R2=CH2CH3, Z=Cl) Соединение получали в соответствии с методикой, описанной в примере 1b), используя в качестве реагента метиловый эфир 2-этил-2 Н-индазол-3-карбоновой кислоты (пример 13 а) (4,0 г; 19,6 ммоль). Получили 3,5 г 2-этил-2 Н-индазол-3-карбонилхлорида. 1H-ЯМР (, ДМСО-d6): 1,51 (т, J=7,3 Гц, 3 Н); 4,59 (кв, J=7,3 Гц, 2 Н); 7,3-7,4 (м, 1 Н); 7,5-7,6 (м, 1 Н); 7,7-7,8 (м, 1 Н); 7, 9-8, 1 (м, 1H). 13 с) N-(1-[2-(4-фторфенил)этил]пиперидин-4-илметил)-2-этил-2H-индазол-3-карбоксамид гидрохлорид Соединение получали в соответствии с методикой, описанной в примере 7 е), используя 2-этил-2 Ниндазол-3-карбонилхлорид (пример 13b) (1,05 г; 5,4 ммоль) и 1-[2-(4-фторфенил)этил]-4-пиперидин метанамин (пример 7d) (1,1 г; 5,4 ммоль). Получили 190 мг N-(1-[2-(4-фторфенил)этил]пиперидин-4-илметил)-2-этил-2 Н-индазол-3 карбоксамид гидрохлорида. Т.пл.=182-186 С. Элементный анализ для C24H29N4OFHCl(Соединение XI: R3=4-F, R4=H, R6+R7=-CH2-CH2-, Y=CH, W=N, n=0, p=2, m=2) Бензальдегид (21,2 г; 0,2 моль) добавляли по каплям к раствору 4-аминопиперидина (20 г; 0,2 моль) в толуоле (80 мл). Полученный таким образом раствор перемешивали при комнатной температуре. Через 3 ч растворитель удалили выпариванием при пониженном давлении и остаток промывали дважды в толуоле. Таким образом получили 31 г N-(фенилметилен)пиперидин-4-амина и использовали в последующих реакциях без дополнительной очистки. Аликвоту этого продукта (1,88 г; 10 ммоль) использовали в соответствии с методикой, описанной в примере 7d), вместе с 1-(2-бромэтил)-4-фторбензолом (пример 7 с) (2,03 г; 10 моль). Таким образом получили 1,3 г 1-[2-(4-фторфенил)этил]пиперидин-4-амина и использовали в следующей реакции без дополнительной очистки.(пример 14 а) (1,1 г; 5,8 ммоль). Таким образом получили 305 мг N-l-[2-(4-фторфенил)этил]пиперидин-4-ил-2-метил-2 Н-индазол 3-карбоксамид гидрохлорид. Т.пл.=224-228 С. Элементный анализ для C22H25N4OFHCl(Соединение I: R1=H, R2=CH3, Х=-С(О)N(R5)-, R3=4-F, R4=H, R5=H, R6+R7=-CH2-CH2-, n=0, p=1,m=2, Y=CH, W=N) 15a) 1-[2-(4-фторфенил)этил]пирролидин-3-амин (Соединение XI: R3=4-F, R4=H, R6+R7=-CH2-CH2-,Y=CH, W=N, n=0, p=1, m=2) Бензальдегид (21,2 г; 0,20 моль) добавляли по каплям к раствору 3-аминопирролидина (17,2 г; 0,20 моль) в толуоле (80 мл). Полученный таким образом раствор перемешивали при комнатной температуре. Через 3 ч растворитель удалили выпариванием при пониженном давлении и остаток промывали дважды в толуоле. Таким образом получили 21 г N-(фенилметилен)пирролидин-3-амина и использовали в последующих реакциях без дополнительной очистки. Аликвоту этого продукта (1,7 г; 10 ммоль) использовали в соответствии с методикой, описанной в примере 7d), вместе с 1-(2-бромэтил)-4-фторбензолом (пример 7 с) (2,0 г; 10 ммоль). Таким образом получили 1,4 г 1-[2-(4-фторфенил)этил]пирролидин-3-амина и использовали в следующей реакции без дополнительной очистки. 15b) N-1-[2-(4-фторфенил)этил]пирролидин-3-ил-2-метил-2H-индазол-3-карбоксамид Соединение получили, как описано в примере 7 е), используя в качестве реагентов 2-метил-2 Ниндазол-3-карбонилхлорид (пример 1b) (1,3 г; 6,7 ммоль) и 1-[2-(4-фторфенил)этил]пирролидин-3-амин(пример 15 а) (1,4 г; 6,7 ммоль). Таким образом получили 235 мг N-1-[2-(4-фторфенил)этил]пирролидин-3-ил-2-метил-2 Ниндазол-3-карбоксамид. Т.пл.=117-119 С. Элементный анализ для C21H23N4OF(Соединение XI: R3=4-F, R4=H, R6+R7=-CH2-CH2-CH2-, Y=CH, W=N, n=0, p=1, m=2) Бензальдегид (21,2 г; 0,2 моль) добавляли по каплям к раствору 3-аминопиперидина (20 г; 0,2 моль) в толуоле (80 мл). Полученный таким образом раствор перемешивали при комнатной температуре. Через 3 ч растворитель удалили выпариванием при пониженном давлении и остаток промывали дважды в толуоле. Таким образом получили 29 г N-(фенилметилен)пиперидин-3-амина и использовали в последующих реакциях без дополнительной очистки. Аликвоту этого продукта (1,88 г; 10 ммоль) использовали, в соответствии с методикой, описанной в примере 7d), вместе с 1-(2-бромэтил)-4-фторбензолом (пример 7 с) (2,0 г; 10 ммоль). Таким образом получили 1,5 г 1-[2-(4-фторфенил)этил]пиперидин-3-амина и использовали в следующей реакции без дополнительной очистки.(пример 16 а) (1,3 г; 5,8 ммоль). Таким образом получили 335 мг N-l-[2-(4-фторфенил)этил]пирролидин-3-ил-2-метил-2 Ниндазол-3-карбоксамид. Т.пл.=194-197 С. Элементный анализ для C22H25N4OFl-(1-2-[4-(трифторметил)фенил]этилпиперидин-4-ил)метанамин получили в соответствии с методикой, описанной в примере 7d), используя в качестве реагентов N-гексагидро-4-пиридинилметил-Nфенилметилиденамин (2,36 г; 10 ммоль) и 1-(2-бромэтил)-4-(трифторметил)бензол (2,5 г; 10 ммоль). Получили 1,8 г 1-(1-2-[4-(трифторметил)фенил]этилпиперидин-4-ил)метанамин и использовали в следующей реакции без дополнительной очистки. 17b) 2-Метил-N-[(1-2-[4-(трифторметил)фенил]этилпиперидин-4-ил)метил]-2 Н-индазол-3-карбоксамид Соединение получили, как описано в примере 7 е), используя в качестве реагентов 2-метил-2 Ниндазол-3-карбонилхлорид (пример 1b) (1,28 г; 6,6 ммоль) и 1-(1-2-[4-(трифторметил)фенил]этилпиперидин-4-ил)метанамин (пример 17 а) (1,8 г; 6,6 ммоль). Таким образом получили 415 мг 2-метил-N-[(1-2-[4-(трифторметил)фенил]этилпиперидин-4 ил)метил]-2 Н-индазол-3-карбоксамид. Т.пл.=174-176 С. Элементный анализ для C24H27N4OF3N-(фенилметилен)пиперидин-4-амин (пример 14 а) (1,8 г; 10 ммоль) и 1-(2-бромэтил)-2-фторбензол (2,5 г; 10 ммоль). Таким образом получили 1,3 г 1-[2-(2-фторфенил)этил]пиперидин-4-амина и использовали в следующей реакции без дополнительной очистки. 18b) N-1-[2-(2-фторфенил)этил]пиперидин-4-ил-2-метил-2 Н-индазол-3-карбоксамид Соединение получили, как описано в примере 7 е), используя в качестве реагентов 2-метил-2 Ниндазол-3-карбонилхлорид (пример 1b) (1,14 г; 5,8 ммоль) и 1-[2-(2-фторфенил)этил]пиперидин-4-амин(пример 18 а) (1,3 г; 5,8 ммоль). Таким образом получили 188 мг N-l-[2-(2-фторфенил)этил]пиперидин-4-ил-2-метил-2H-индазол 3-карбоксамид. Т.пл.=164-166 С. Элементный анализ для C22H25N4OFK2CO3 (1,61 г; 11,7 ммоль) добавляли небольшими порциями к раствору, содержащему 4-(4 фторфенил)пиперидин (1,05 г; 5,85 ммоль) в безводном ацетоне (25 мл). Затем раствор (2 бромэтил)фталимида (164 г; 6,44 ммоль) в безводном ацетоне (25 мл) добавляли медленно к смеси при перемешивании при комнатной температуре. В конце добавления (30 мин) смесь нагревали до температуры флегмы в течение 24 ч. В конце реакции смесь охлаждали и фильтровали. Остаточный раствор затем разводили этилацетатом (50 мл) и экстрагировали 1,5 н HCl (320 мл). Объединенные кислотные фазы промывали этилацетатом (210 мл), затем сильно подщелачивали 3 н NaOH и экстрагировали дихлорметаном (520 мл). Объединенные органические фазы сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Полученный остаток (607 мг) затем растворяли в метаноле (11 мл) и обрабатывали гидразин моногидратом (2 мл; 41 ммоль) при 90 С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли дихлорметаном (50 мл) и экстрагировали несколько раз 3 н NaON (515 мл). Органическую фазу затем обрабатывали безводным Na2SO4 и выпаривали при пониженном давлении. Таким образом получили 400 мг N-(2-аминоэтил)-4-(4-фторфенил)пиперидин и использовали в следующей реакции без дополнительной очистки. 19b) N-2-[4-(4-фторфенил)пиперидин-1-ил]этил-2-метил-2 Н-индазол-3-карбоксамид Соединение получили в соответствии с методикой, описанной в примере 7 е), используя в качестве реагентов 2-метил-2 Н-индазол-3-карбонилхлорид (пример 1b) (240 мг; 1,22 моль) и N-(2-аминоэтил)-4(4-фторфенил)пиперидин (пример 19 а) (400 мг). Таким образом получили 190 мг N-2-[4-(4-фторфенил)пиперидин-1-ил]этил-2-метил-2 Н-индазол 3-карбоксамид. Т.пл.=105-107 С. Элементный анализ для C22H25FN4O(Соединение II: R5 = H, R6+R7 = -CH2-CH2-, R3 = 4-F, R4 = H, Y = N, W = CH, n = 3, p = 2, m = 0) Соединение получили в соответствии с методикой, описанной в примере 19 а), используя в качестве реагентов 4-(4-фторфенил)пиперидин (1,10 г; 6,14 моль) и N-(3-бромпропил)фталимида (1,82 г; 6,78 ммоль). Таким образом получили 1,6 г N-(3-аминопропил)-4-(4-фторфенил)пиперидин и использовали в следующей реакции без дополнительной очистки. 20b) N-3-[4-(4-фторфенил)пиперидин-1-ил]пропил-2-метил-2 Н-индазол-3-карбоксамид Соединение получили в соответствии с методикой, описанной в примере 7 е), используя в качестве реагентов 2-метил-2 Н-индазол-3-карбонилхлорид (пример 1b) (236 мг; 1,21 моль) и N-(3-аминопропил)4-(4-фторфенил)пиперидин (пример 20 а) (550 мг). Таким образом получили 120 мг N-3-[4-(4-фторфенил)пиперидин-1-ил]пропил-2-метил-2 Н- 20019217 индазол-3-карбоксамид. Т.пл.=136-140 С. Элементный анализ для C23H27FN4O(Соединение XI: R3 = H, R4 = H, R6+R7 = -CH2-CH2-, Y = CH, W = N, n = 0, p = 2, m = 1) Соединение получили в соответствии с методикой, описанной в примере 7d), используя в качестве реагентов N-[фенилметилен]пиперидин-4-амин (пример 14 а) (1,88 г; 10 ммоль) и бензилбромид (1,7 г; 10 моль). Таким образом получили 1,7 г 1-бензилпиперидин-4-амина и использовали в следующей реакции без дополнительной очистки. 21b) N-(1-бензилпиперидин-4-ил)-2-метил-2 Н-индазол-3-карбоксамид Соединение получили в соответствии с методикой, описанной в примере 7 е), используя в качестве реагентов 2-метил-2 Н-индазол-3-карбонилхлорид (пример 1b) (1,74 г; 8,9 ммоль) и 1-бензилпиперидин-4 амин (пример 21 а) (1,7 г; 8,9 ммоль). Таким образом получили 460 мг N-(1-бензилпиперидин-4-ил)-2-метил-2 Н-индазол-3-карбоксамида. Т.пл.=167-169 С. Элементный анализ для C21H24N4O(Соединение XI: R3 = 4-F, R4 = H, R6+R7 = -CH2-CH2-, Y = CH, W = N, n = 0, p = 2, m = 3) Соединение получили в соответствии с методикой, описанной в примере 7d), используя в качестве реагентов N-(фенилметилен)пиперидин-4-амин (пример 14 а) (1,9 г; 10 моль) и 1-(3-бромпропил)-4 фторбензол (2,2 г; 10 ммоль). Таким образом получили 1,8 г 1-[3-(4-фторфенил)пропил]пиперидин-4-амина и использовали в следующей реакции без дополнительной очистки. 22b) N-1-[3-(4-фторфенил)пропил]пиперидин-4-ил-2-метил-2 Н-индазол-3-карбоксамид Соединение получили в соответствии с методикой, описанной в примере 7 е), используя в качестве реагентов 2-метил-2 Н-индазол-3-карбонилхлорид (пример 1b) (1,49 г; 7,6 ммоль) и 1-[3-(4 фторфенил)пропил]пиперидин-4-амин (пример 22 а) (1,8 г; 7,6 ммоль). Таким образом получили 165 мг N-1-[3-(4-фторфенил)пропил]пиперидин-4-ил-2-метил-2 Ниндазол-3-карбоксамид. Т.пл.=162-165 С. Элементный анализ для C23H27N4OF(Соединение XI: R3 = 4-F, R4 = 2-F, R6+R7 = -CH2-CH2-, Y= CH, W = N, n = 1, p = 2, m = 2) 1-[2-(2,4-Дифторфенил)этил]-4-пиперидин)метанамин получили в соответствии с методикой, описанной в примере 7d), используя в качестве реагентов N-гексагидро-4-пиридинил-N-фенилметилиден амин (2,0 г; 10 ммоль) и 1-(2-бромэтил)-2,4-дифторбензол (2,2 г; 10 моль). Таким образом получили 1,9 г 1-[2-(2,4-дифторфенил)этил]-4-пиперидинметанамина и использовали в следующей реакции без дополнительной очистки. 23b) N-(1-[2-(2,4-дифторфенил)этил]пиперидин-4-илметил)-2-метил-2 Н-индазол-3-карбоксамид Соединение получили в соответствии с методикой, описанной в примере 7 е), используя в качестве реагентов 2-метил-2 Н-индазол-3-карбонилхлорид (пример 1b) (1,45 г; 7,4 ммоль) и 1-[2-(2,4 дифторфенил)этил]-4-пиперидинметанамина (пример 23 а) (1,9 г; 7,4 ммоль). Таким образом получили 350 мг N-(1-[2-(2,4-дифторфенил)этил]пиперидин-4-илметил)-2-метил 2 Н-индазол-3-карбоксамида. Т.пл.=122-126 С. Элементный анализ для C23H26N4OF2(м, 1H); 7,09-7,25 (м, 2 Н); 7,27-7,42 (м, 2 Н); 7,67 (д, J=8,77 Гц, 1 Н); 7,77 (д, J=8,18 Гц, 1 Н); 8,54 (т, J=5,70 Гц, 1 Н). Фармакология Тест А. Связывание с серотониновым рецептором 5-HT2A крыс выполняли, используя, в качестве исходного материала, получение мембран из гомогенизата коры головного мозга крыс, предварительно инкубированного в течение 15 мин при 37 С и предварительно обработанного 100 нМ празозином и 100 нМ пириламином для предотвращения связывания [3H]-кетансерина с 1 адренергическим и H1 гистаминергическим рецепторами. Исследования перемещения проводили, используя 1 нМ [3 Н]-кетансерин в качестве радиолиганда. Эту концентрацию выбрали на основе исследований сатурации, используя разнообразные концентрации радиолиганда (от 0,04 до 10 нМ), который сделал возможным получение Вmax 709,9 нМ и Kd 1,7 нМ. Неспецифическое связывание измерили в присутствии 10 мкМ метисергида. Анализ провели в TrisHCl 50 мМ буфере (рН 7,4 при 37 С), с инкубированием в течение 2 ч при 37 С. Тестируемые соединения растворили в ДМСО, затем развели в буфере (конечная концентрация ДМСО 0,01%) и поместили в 96-луночный планшет. Метисергид и/или кетансерин использовали в качестве сравнительных соединений. Связывание инициируется добавлением 200 мкл гомогенизата (450 мкг/мл содержание белка); после инкубирования мембраны фильтровали через стекловолоконные фильтры (GF/B) (Unifilter, Packard),обработанные 0,3% полиэтиленимина. Затем фильтры промывали буферным раствором и сушили в печи 30 мин при 45 С. Сцинтилляционную жидкость добавляли в каждую лунку и через 10 ч измеряли радиоактивность в течение 1 мин, используя TopCount (Packard). Тестируемые соединения анализировали дважды при 8 концентрациях (от 10-12 до 10-5 M).IC50 значения для каждого соединения вычисляли, используя анализ нелинейной регрессииclassification and nomenclature", Neuropharmacol. 33: 261-273 и в May J.A., McLaughlin M.A., Sharif N.A.,Hellberg M.R. and Dean T.R. (2003) "Evaluation of the ocular hypotensive response of serotonin 5-HT1A and 5HT2 receptor ligands in conscious ocular hypertensive cynomolgus monkeys", J. Pharmacol. Exp. Ther. 306(1): 301-309. Рекомбинантные СНО клетки для экспрессии энзима человека и метерголин, в качестве сравнительного соединения, использовали как исходный материал. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Связывание с серотониновым 5-HT1B рецептором выполняли, используя стандартные способы, описанные в Hoyer D., Engel G. and Kalkman H.O. (1985) "Characterization of the 5-HT1B recognition site in ratbrain: binding studies with (-)[125]iodocyanopindolol", Eur. J. Pharmacol. 18: 1-12. Гомогенат из коры головного мозга крыс Wistar использовали в качестве исходного материала и серотонин как сравнительное соединение. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Связывание с серотониновым 5-HT1D рецептором выполняли, используя стандартные способы, описанные в Heuring R.E. and Peroutka S.J. (1987) "Characterization of a novel 3H-5-hydroxytryptamine bindingsite subtype in bovine brain membrance", J. Neurosci. 7(3): 894-903. Гомогенат из бычьего церебрального каудатума использовали в качестве исходного материала, а серотонин - как сравнительное соединение. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Связывание с серотониновым 5-HT2A рецептором человека выполняли с помощью стандартной методики, описанной в Bonhaus D.W., Bach С, De Souza A., Salazar F.H., Matsuoka B.D., Zuppan P., Chancells: agonist-induced down-regulation involves decreases in receptor RNA and number", J. Neurochem. 68(5): 1998-2011. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ и затем при шести концентрациях, чтобы обнаружить pKi значения отдельных соединений. Связывание с серотониновым 5-HT2B рецептором выполняли, используя стандартную методику,описанную в Bounhaus D.W., Bach С, De Souza A., Rich Salazar F.H., Matsuoka B.D., Zuppan P., Chan H.W.and Eglen R.M. (1995) "The pharmacology and distribution of human 5-hydroxytryptamine 2B (5-HT2B) receptor gene products: comparison with 5-HT2A and 5-HT2C receptors", Br. J. Pharmacol. 115: 622-628. CHO-K1 рекомбинантные клетки для экспрессии энзима человека использовали в качестве исходного материала, а кетансерин - в качестве сравнительного соединения. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Связывание с серотониновым 5-HT2C рецептором выполняли, используя стандартную методику,описанную в Wolf W.A. and Schultz J.S. (1997) "The serotonin 5-HT2C receptor is a prominent serotonin inbasal ganglia: evidence from functional studies on serotonin-mediated phosphoinositide hydrolysis", J. Neurochem. 69: 1449-1458. CHO-K1 рекомбинантные клетки для экспрессии энзима человека использовали в качестве исходного материала, a SB242084 - качестве сравнительного соединения. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ и затем при шести концентрациях,чтобы определить pKi значения отдельных соединений. Связывание с серотониновым 5-НТ 3 рецептором выполняли, используя стандартную методику,описанную в Boess F.G., Steward L.J., Steele J.A., Liu D., Reid J., Glencorse T.A. and Martin I.L. (1997)rat cortex and the radioligand [3H]GR65630, [3H]BRL43694 and [3H]LY278584", Synapse 11: 58-66. HEK293 рекомбинантные клетки для экспрессии энзима человека использовали в качестве исходного материала иMDL-72222, в качестве сравнительного соединения. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Связывание с серотониновым 5-НТ 4 рецептором выполняли, используя стандартные способы, описанные в Grossman C.J., Kilpatrick G.J. and Bunce K.Т. (1993) "Development of a radioligand binding assayfor 5-HT4 receptors in guinea-pig and rat brain" Br. J. Pharmacol. 109: 618-624. Гомогенат из церебрального стриатума (полосатого тела) морской свинки использовали в качестве исходного материала, a RS23597190 как сравнительное соединение. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Селективность тестируемых соединений для 5-НТ 4(b) рецептора рекомбинантных клеток человека определяли, используя методику, выведенную из стандартной методики, описанной в Mialet J., BerqueBestel I., Eftekhari P., Gastineau M., Giner M., Dahmoune Y., Donzeau-Gouge P., Hoebeke J., Langlois M.,Sicsic S., Fischmeister R. and Lezoualc'h F. (2000) "Isolation of the serotoninergic 5-HT4(e) receptor from humanheart and comparative analysis of its pharmacological profile in C6-glial and CHO cell lines" Br. J. Pharmacol. 129: 771-781. Связывание с 5-HT4(b) рецептором выполняли, используя получение НЕК 293 рекомбинантных клеток для 5-НТ 4(b) рецептора человека в качестве сходного материала. В день эксперимента клеточный пеллет, который хранили при -80 С, размораживали и хранили во льду. Затем пеллет гомогенезировали в буфере (25 мМ Tris-HCl pH 7,4; 0,1 мМ EDTA) и суспензию центрифугировали при 30000 г в течение 20 мин. Пеллет промывали три раза в промывочном буфере и окончательно ресуспендировали в буфере для инкубации (25 мМ Tris-HCl pH 7,4; 0,5 мМ EDTA; 10 мМ MgSO4) при конечной концентрации приблизительно 6105 клеток/мл. Исследования перемещения проводили, используя 0,2 нМ [3 Н]-GR113808 как радиолиганд. Эту концентрацию выбрали, основываясь на исследованиях сатурации, используя разные концентрации радиолиганда (от 0,02 нМ до 2,5 нМ). Неспецифическое связывание измеряли в присутствии 10 мкМ Piboserod. Анализ выполняли в общем объеме 250 мкл буфера 25 мМ Tris-HCl pH 7,4; 0,5 мМEDTA; 10 мМ MgSO4 с инкубацией в течение 2 ч при 27 С. Тестируемые соединения растворили в ДМСО и затем развели в буфере (конечная концентрация ДМСО равна 0,01%) и поместили на 96-луночный планшет. Пибосерод использовали в качестве сравнительного соединения. Связывание инициировали добавлением 200 мкл клеточного гомогената (35-92 мкг/мл содержание белка); после инкубирования мембраны фильтровали через стекловолоконные фильтры (GF/B) (Unifilter, Packard), обработанных 0,1% полиэтилимина. Затем фильтры промывали буферным раствором и сушили в печи 30 мин при 45 С. Сцинтилляционную жидкость добавляли в каждую лунку и через 16 ч измеряли радиоактивность в течение 1 мин, используя TopCount (Packard). Концентрацию белка определяли, используя ВСА способ(Pierce) с BSA в качестве стандарта. Тестируемые соединения анализировали дважды при 8 концентрациях (от 10-12 до 10-5 M), IC50 значения для каждого соединения вычисляли, используя анализ нелинейной регрессии (GraphPad PRISM software) и константы ингибирования Ki определяли, используя выравнивание, описанное Cheng Y. and Prussof W.H. (1973) "Relationship between the inhibition constant (Ki)and characterization of the human 5-HT5A serotonin receptor. FEBS Lett. 355: 242-246. CHO-K1 рекомбинантные клетки для экспрессии энзима человека использовали в качестве исходного материала, а метиотепин - в качестве сравнительного соединения. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Связывание с серотониновым 5-НТ 6 рецептором выполняли, используя стандартную методику,описанную в Monsma F.J.Jr, Shen Y., Ward R.P., Hamblin M.W. and Sibley D.R. (1993) Cloning and expression of a novel serotonin receptor with high affinity fortricyclic psychotropic drugs. Mol. Pharmacol. 43: 320-327. HeLa рекомбинантные клетки для экспрессии энзима человека использовали в качестве исходного материала, а метиотепин - в качестве сравнительного соединения. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Связывание с серотониновым 5-НТ 7 рецептором выполняли, используя стандартную методику,описанную в Roth В. L, Craigo S. С, Choudhary M.S., Uluer S., Monsma F.J. Jr, Shen Y., Meltzer H.Y. andM.A., Jose P.A., Hamblin M.W. and Sibley D.R. (1993) "Molecular cloning and expression of a 5hydroxytryptamine 7 serotonin receptor subtype." J. Biol. Chem. 268: 18200-18204. CHO рекомбинантные клетки для экспрессии энзима человека использовали в качестве исходного материала, а метиотепин - в качестве сравнительного соединения. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Связывание с транспортером серотонина выполняли, используя стандартную методику, описанную в Shearman L.P., McReynolds A.M., Zhou F.C., Meyer J. S. (1998) "Relationship between [125I]RTI-55labeled cocaine binding sites and the serotonin transporter in rat placenta." Am. J. Physiol. 275 (6 Pt 1): C16211629 и в Wolf W.A. and Kuhn D.M. (1992) "Role of essential sulfhydryl groups in drug interactions at the neuronal 5-HT transporter. Differences between amphetamines and 5-HT uptake inhibitors." J. Biol. Chem. 267(29): 20820-20825. HEK293 рекомбинантные клетки для экспрессии энзима человека использовали в качестве исходного материала, а флуоксетин - в качестве сравнительного соединения. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Клеточный анализ введения серотонина выполняли, используя стандартную методику, описанную вinhibitor sensitivity, kinetics, and ion dependence" J. Biol. Chem. 269(10): 7124-7130. HEK293 клетки использовали в качестве исходного материала, а флуоксетин - в качестве сравнительного соединения. Тестируемые соединения анализировали дважды при единственной концентрации 1 мкМ. Тест С. Самцов CD-1 мышей, весящих 25-30 г, использовали для проведения теста судорожных движений головой. Животных обрабатывали интраперитонеально тестируемыми соединениями (5 мг/кг), суспендированными в метилцеллюлозе (МТС). Контрольных животных обрабатывали только наполнителем(МТС) тем же путем. Через полчаса после обработки тестируемыми соединениями животным вводили интраперитонеально инъекцию 5-гидрокситриптофана (5-НТР; 300 мг/кг), серотониновое исходное соединение, с целью индуцирования "судорожных движений головой", характерное потряхивание головой, вызванное у животных увеличением в центральных уровнях серотонина. Число судорожных движений головой, которое формирует параметр для оценки серотонинергиче- 24019217 ского отклика, измеряли в интервале от 24 до 26 мин после введения 5-НТР. Как показано на фиг. 1, соединения настоящего изобретения вызывают уменьшение числа судорожных движений головой по сравнению с животными, обработанными просто МТС, демонстрируя способность к антагонизму серотонинергическим эффектам, вызванным введением 5-НТР. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение 2-алкилиндазола общей формулы (I)Y обозначает СН или N;W обозначает СН или N; при условии, что по меньшей мере один из Y и W является атомом азота;R5 обозначает Н, метил, метоксигруппу или вместе с R6 образует 6-членное насыщенное кольцо,выбранное из группы, включающей пиперидин и пиперазин;R7 обозначает Н, этил или вместе с R6 образует 5- или 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолин, пиперидин и пиперазин. 2. Соединение по п.1, отличающееся тем, чтоR2 обозначает метил или метоксиэтил;R7 обозначает Н, этил или вместе с R6 образует 5- или 6-членное насыщенное кольцо, выбранное из группы, включающей пирролидин, имидазолин, пиразолин, пиперидин и пиперазин. 3. Способ получения (i) соединения 2-алкилиндазола общей формулы (I), в котором R1, R2, R3, R4,R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1 или (ii) его соли с фармацевтически приемлемой органической или неорганической кислотой, отличающийся тем, что включает в себя: 1 а) конденсацию амина формулы (II)Y, W, R3, R4, R5, R6, R7, n, m и р имеют значения, установленные ранее в п.1, с производным индазолкарбоновой кислоты формулы (III)Z выбирается из группы, включающей атом галогена, OR и OC(O)R группу, где R обозначает неразветвленную или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода,для получения соединения 2-алкилиндазола общей формулы (I), в которой R1, R2, R3, R4, R6, R7,X, Y, W, n, p и m имеют значения, установленные ранее в п.1; причем указанная конденсация выполняется в присутствии подходящего апротонного, полярного или неполярного растворителя при температуре от 0 до 140 С, в течение от 0,5 до 24 ч. 4. Способ получения (i) соединения 2-алкилиндазола общей формулы (I) по п.3, отличающийся тем,что Z выбирается из группы, включающей Cl или Br. 5. Способ получения (i) соединения 2-алкилиндазола общей формулы (I), в котором Rl, R2, R3, R4,R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1 или (ii) его соли с фармацевтически приемлемой органической или неорганической кислотой, отличающийся тем, что включает в себя: 1b) взаимодействие амина общей формулы (IV)Rl, R2, R5, R6, R7, Y, n и р имеют значения, установленные ранее в п.1,с соединением общей формулы (V)Q обозначает уходящую группу, выбранную из группы, включающей атом галогена, мезилатную группу (MeSO3-) и тозилатную группу (p-MePhSO3-),для получения соединения 2-алкилиндазола общей формулы (I), в которой R1, R2, R3, R4, R6, R7,X, Y, W, n, p и m имеют значения, установленные ранее в п.1; причем указанная реакция выполняется в присутствии подходящего апротонного, полярного или неполярного растворителя при температуре, находящейся в пределах от комнатной до 100 С, при этом время реакции составляет от 6 до 24 ч. 6. Способ получения (i) соединения 2-алкилиндазола общей формулы (I) по п.5, отличающийся тем,что Q обозначает уходящую группу, выбранную из группы, включающей Cl или Br. 7. Способ получения (i) соединения 2-алкилиндазола общей формулы (I), в котором R1, R2, R3, R4,R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1 или (ii) его соли с фармацевтически приемлемой органической или неорганической кислотой, отличающийся тем, что включает в себя: 1 с) конденсацию амина общей формулы (VI)R1, R2 и R5 имеют значения, установленные ранее в п.1,с производным карбоновой кислоты общей формулы (VII)Z имеет значения, установленные ранее в отношении соединения формулы (III),для получения соединения 2-алкилиндазола общей формулы (I), в которой R1, R2, R3, R4, R6, R7,- 26019217X, Y, W, n, p и m имеют значения, установленные ранее в п.1; и 2) возможное образование соли кислотного присоединения соединения 2-алкилиндазола общей формулы (I), полученного таким образом, с фармацевтически приемлемой органической или неорганической кислотой, причем указанная конденсация выполняется в присутствии подходящего апротонного,полярного или неполярного растворителя при температуре от 0 до 140 С, при этом время реакции составляет от 0,5 до 24 ч. 8. Промежуточное соединение формулы (II)R3, R4 и n имеют значения, установленные ранее в п.1,R5 обозначает C1-3 алкил,R6 и R7 вместе образуют 5- или 6-членное насыщенное кольцо, выбранное из группы, состоящей из пирролидина, имидазолина, пиразолидина, пиперидина и пиперазина,Y обозначает СН,W обозначает N,р является целым числом, выбранным из 0 и 1,m является 2. 9. Промежуточное соединение формулы (IV)R1, R2, R5, R6, R7, Y, n, p имеют значения, установленные ранее в п.1. 10. Промежуточное соединение формулы (VI)R5 обозначает Н, C1-3 алкил или C1-3 алкоксигруппу. 11. Промежуточное соединение общей формулы (XX)m является 2. 12. Промежуточное соединение общей формулы (XXX)Y обозначает СН. 13. Фармацевтическая композиция, содержащая эффективную дозировку по меньшей мере одного соединения 2-алкилиндазола формулы (I), в которой R1, R2, R3, R4, R6, R7, X, Y, W, n, p и m имеют значения, установленные ранее в п.1, или его соль с фармацевтически приемлемой органической или неорганической кислотой и по меньшей мере один фармацевтически приемлемый наполнитель. 14. Фармацевтическая композиция по п.13, отличающаяся тем, что содержит такое количество соединения 2-алкилиндазола формулы (I), или его фармацевтически приемлемую соль присоединения кислоты, чтобы обеспечить уровень введения от 0,0001 до 100 мг/кг/день. 15. Фармацевтическая композиция по п.13, отличающаяся тем, что содержит такое количество соединения 2-алкилиндазола формулы (I), или его фармацевтически приемлемую соль присоединения кислоты, чтобы обеспечить уровень введения от 0,001 до 50 мг/кг/день. 16. Фармацевтическая композиция по п.14, отличающаяся тем, что содержит такое количество соединения 2-алкилиндазола формулы (I), или его фармацевтически приемлемую соль присоединения кислоты, чтобы обеспечить уровень введения от 0,01 до 10 мг/кг/день. 17. Применение фармацевтической композиции по любому из пп.13-16 для лечения патологических состояний, выбранных из группы, включающей расстройства сна, шизофрению, беспокойство, нарушения в гладких мышцах как желудочно-кишечного тракта, так и сердечно-сосудистой системы.
МПК / Метки
МПК: A61P 25/00, C07D 401/06, C07D 401/12, A61K 31/435
Метки: лечения, расстройств, 2-алкилиндазола, некоторых, цнс, соединения
Код ссылки
<a href="https://eas.patents.su/29-19217-soedineniya-2-alkilindazola-dlya-lecheniya-nekotoryh-rasstrojjstv-cns.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения 2-алкилиндазола для лечения некоторых расстройств цнс</a>
Имидазольные соединения для лечения нейродегенеративных расстройств

Номер патента: 11689
Опубликовано: 28.04.2009
Авторы: Бродней Майкл Аарон, Коффман Карен Джин
МПК: A61P 25/28, A61K 31/4164, C07D 233/88...
Метки: соединения, лечения, расстройств, нейродегенеративных, имидазольные
Формула / Реферат:
1. Соединение формулы I где А отсутствует или выбран из и SO2; Z выбран из -СН2, -СН(ОН), групп -СН(С1-С6алкил), -СН(С1-С6алкокси), -CH(NR9R10), -CH(CH2(OH)), -СН(СН(С1-С4алкил)(ОН)) и -СН(С(С1-С4алкил)(С1-С4алкил)(ОН)); R1 выбран из С1-С20алкила, С2-С20алкенила, С2-С20алкинила, C1-С20алкокси, С2-С20алкенокси, C1-С20 гидроксиалкила, С3-С8циклоалкила, групп бензо(С3-С8циклоалкил), бензо(С3-С8гетероциклоалкил), С4-С8циклоалкенила, (С5-С11)би-...
Производные индол-2-она для лечения расстройств центральной нервной системы, желудочно-кишечных расстройств и сердечно-сосудистых расстройств

Номер патента: 10865
Опубликовано: 30.12.2008
Авторы: Капиллерне Дежёфи Рита, Эдьед Андраш, Сираи Нора, Сенаши Габор, Левелеки Чилла, Харсинг Ласло Габор, Мезеи Тибор, Гачальи Иштван, Мориц Кристина, Гиглер Габор, Леваи Дьёрдь, Баркоци Йожеф, Вольк Балаж, Паллаги Каталин, Шимиг Дьюла
МПК: A61K 31/404, A61P 25/00, C07D 401/06...
Метки: индол-2-она, производные, желудочно-кишечных, системы, сердечно-сосудистых, расстройств, центральной, лечения, нервной
Формула / Реферат:
1. 3,3-Дизамещенные производные индол-2-она общей формулы (I) где R1 и R2 независимо представляют собой водород, галоген, алкил, имеющий в своем составе от 1 до 7 атомов углерода, или сульфамоил; R3 представляет собой водород или алкил с прямой или разветвленной цепью, имеющий в своем составе от 1 до 7 атомов углерода; R4 означает алкил, имеющий в своем составе от 1 до 7 атомов углерода; R5 является водородом и R6 означает фенил, необязательно...
Соединения для лечения расстройств, опосредованных рецепторами адгезии cd11/cd18

Номер патента: 7852
Опубликовано: 27.02.2007
Авторы: Оаре Девид, Гадек Томас Р., Рейнолдс Марк, Марстерс Джеймс Си., Бурдик Даниел Дж., Стенли Марк Си., Макдауел Роберт С., Уиз Кеннет Дж.
МПК: A61K 31/17, A61K 31/166, A61K 31/4035...
Метки: лечения, адгезии, соединения, рецепторами, опосредованных, расстройств
Формула / Реферат:
1. Соединение общей формулы где D выбран из группы где Y1 выбран из группы, включающей СН и С-ОН; Y2, Y3, Y4 и Y5 выбраны из группы, включающей СН; Z1 выбран из группы, включающей NRn, О и S; n=0-3; LX выбран из группы, включающей замещенные или незамещенные С2-С5-алкилен, С3-С6-циклоалкилен, С0-С3-алкилен-NRn-(С=O)-С0-С3-алкилен, С0-С3-алкилен-(С=O)-NRn-С0-С3-алкилен, С0-С3-алкилен-О-С0-С3-алкилен, С0-С3-алкилен-NRn-С0-С3-алкилен,...
Аза-гетероциклические соединения, применяемые для лечения неврологических расстройств и выпадения волос

Номер патента: 8723
Опубликовано: 29.06.2007
Авторы: Норман Марк Х., Гамильтон Грегори С., Ву Йонг-Кайан
МПК: C07D 207/12, A61K 31/41, A61K 31/40...
Метки: неврологических, лечения, соединения, аза-гетероциклические, расстройств, применяемые, выпадения, волос
Формула / Реферат:
1. Фармацевтическая композиция, содержащая: 1) соединение формулы (I) или его фармацевтически приемлемые соли, сложный эфир или сольват, где n представляет собой 1 или 2, X представляет собой либо О, либо S, R1 представляет собой С1-С9алкил с прямой или разветвленной цепью, D представляет собой связь или С1-С10алкилен с прямой или разветвленной цепью, R2 представляет собой тетразол, -СООН, -CONH2, -CN, -SO3H или -ОН; и 2) фармацевтически...
Конденсированные гетероциклические соединения, применимые для лечения ожирения и расстройств центральной нервной системы

Номер патента: 11581
Опубликовано: 28.04.2009
Авторы: Йоханссон Гари, Йенмальм Йенсен Анника, Бейерлейн Катарина
МПК: A61P 3/10, A61K 31/496, A61P 3/04...
Метки: ожирения, системы, нервной, применимые, конденсированные, центральной, лечения, расстройств, гетероциклические, соединения
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль, где кольцо D представляет собой 5-членное гетероциклическое или гетероарильное кольцо, причем указанное кольцо включает один или два атома, выбранных из группы, включающей атомы азота, серы и кислорода, при условии, что, когда D содержит атом кислорода, D представляет собой гетероарил; каждый W независимо представляет собой -N-, -(СН)- или -С-, при условии, что по меньшей мере...
Предыдущий патент: Антикоррозионная упаковка
Следующий патент: Органический порошковый наполнитель, используемый для замены минерального наполнителя в композиционных материалах
Случайный патент: Восстановление оксидов металлов в электролизере