Трипептиды, несущие простой гидроксипролиновый эфир замещённого хинолина, предназначенные для ингибирования протеазы ns3 (гепатит с)









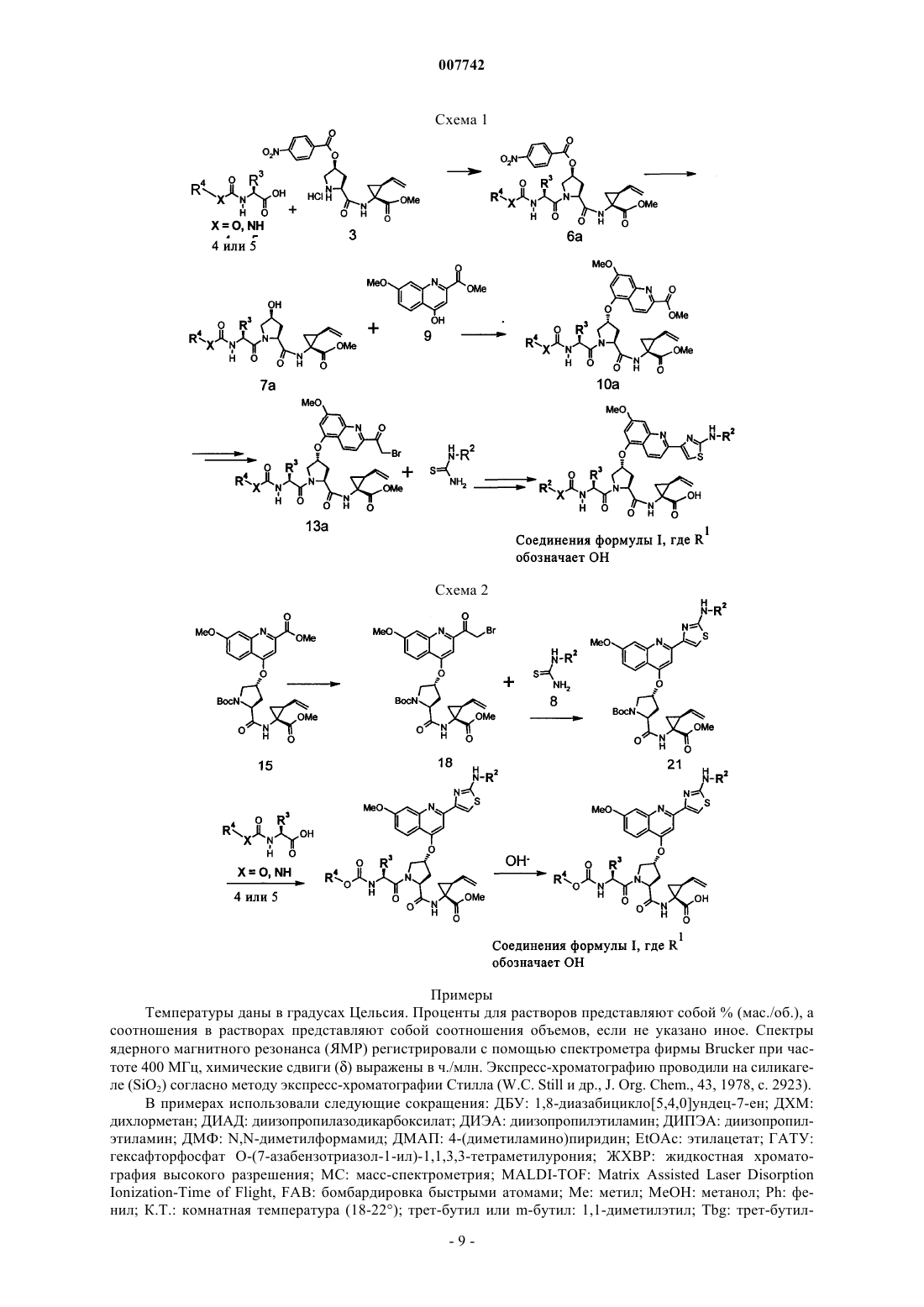




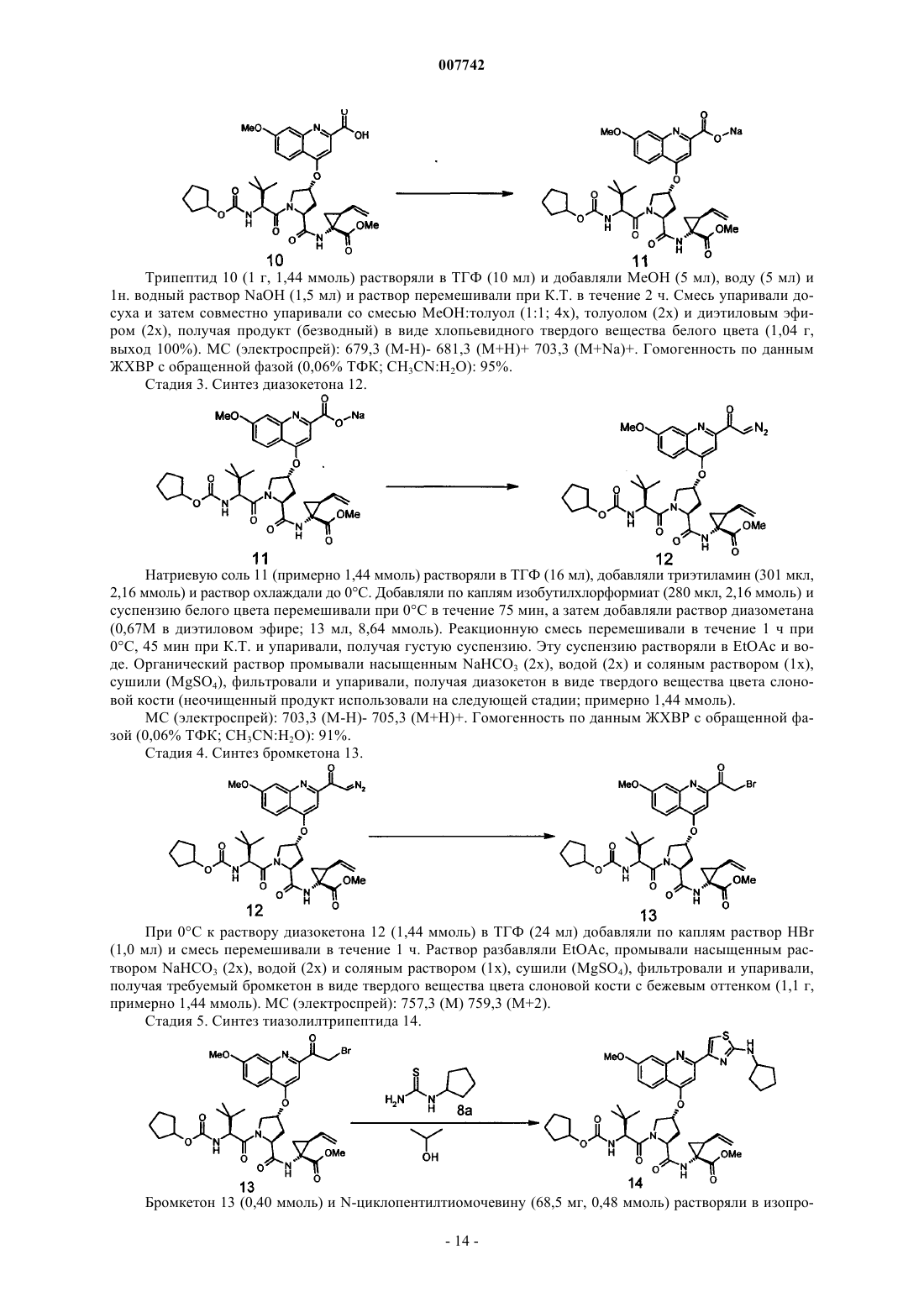

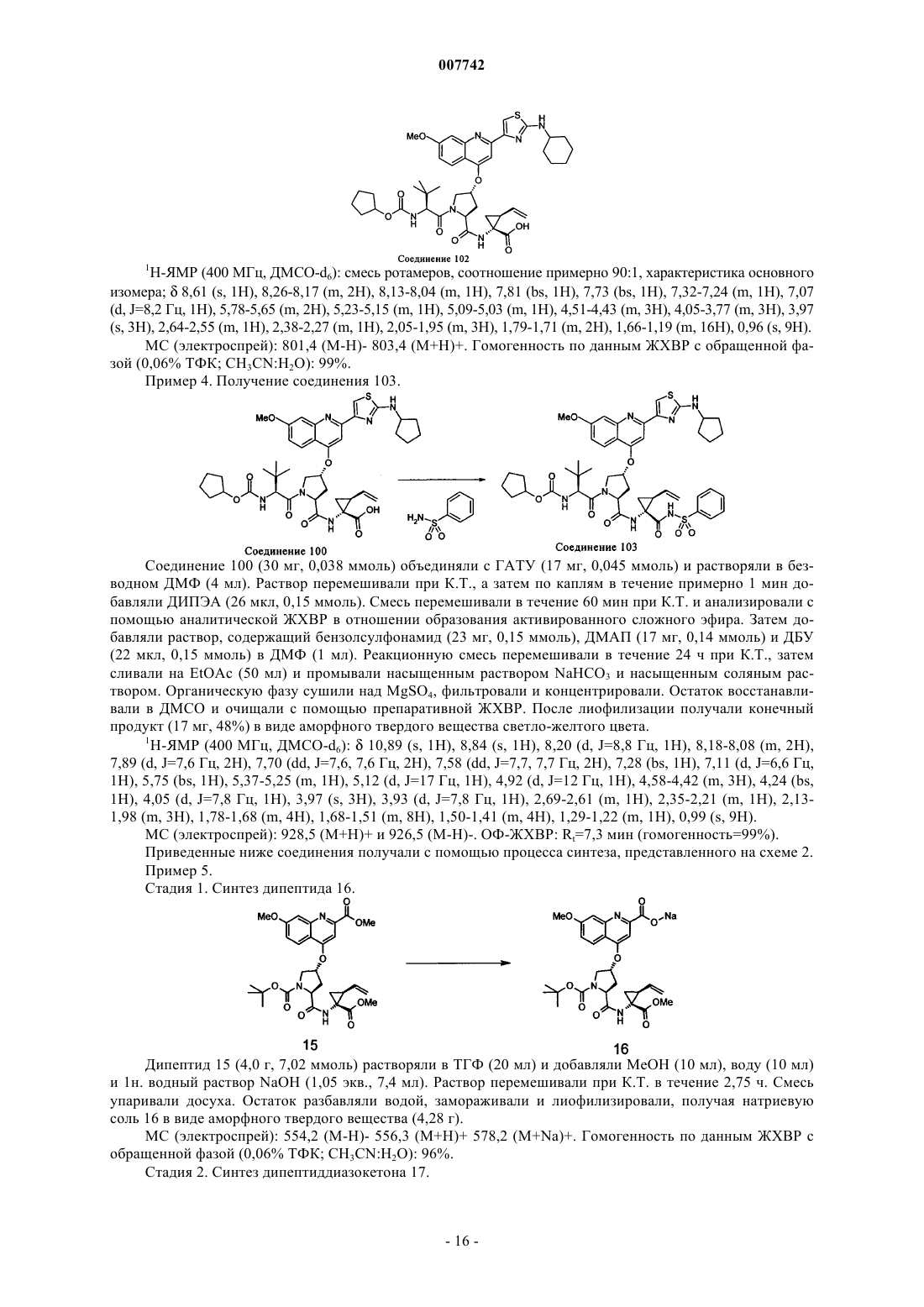
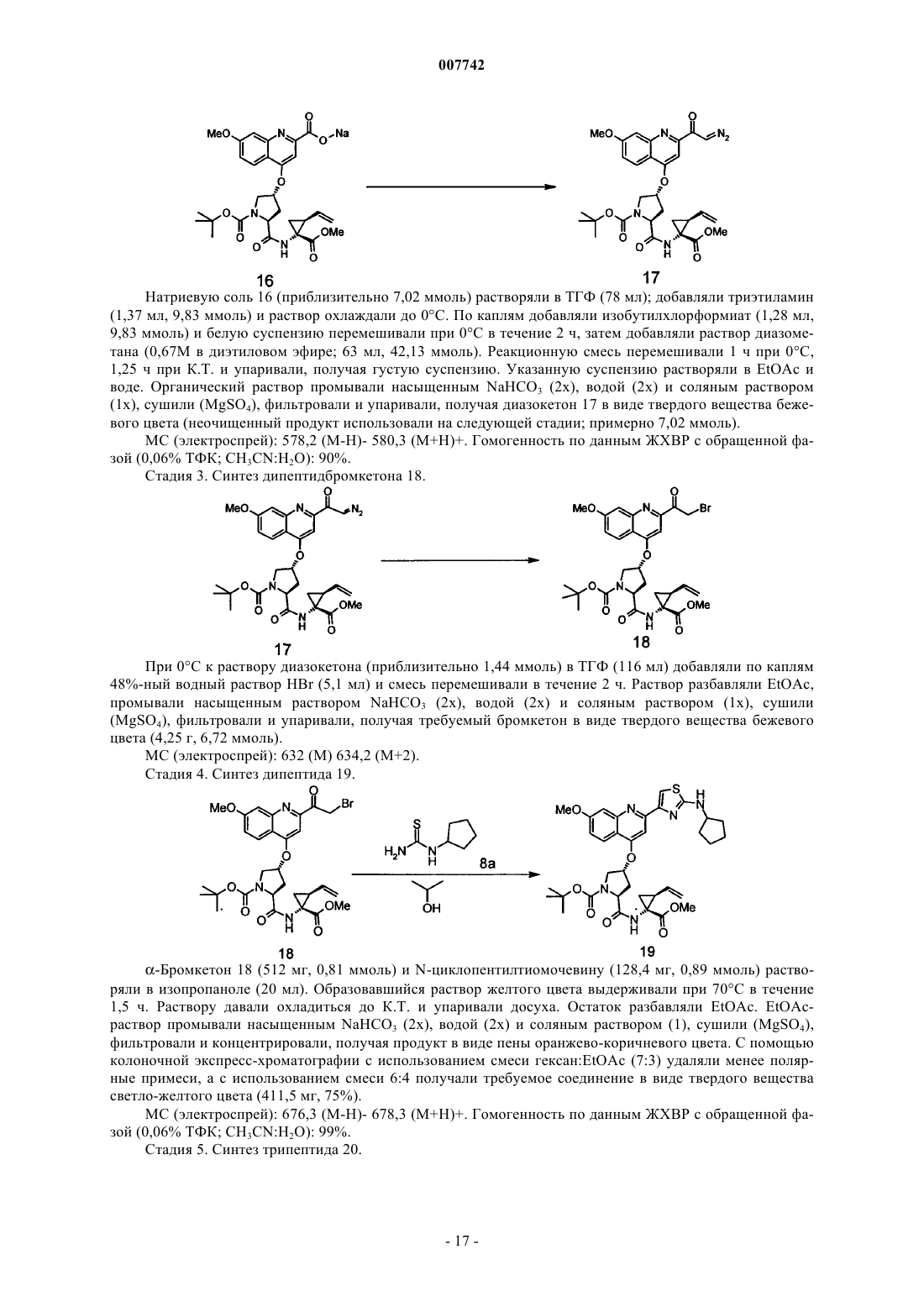


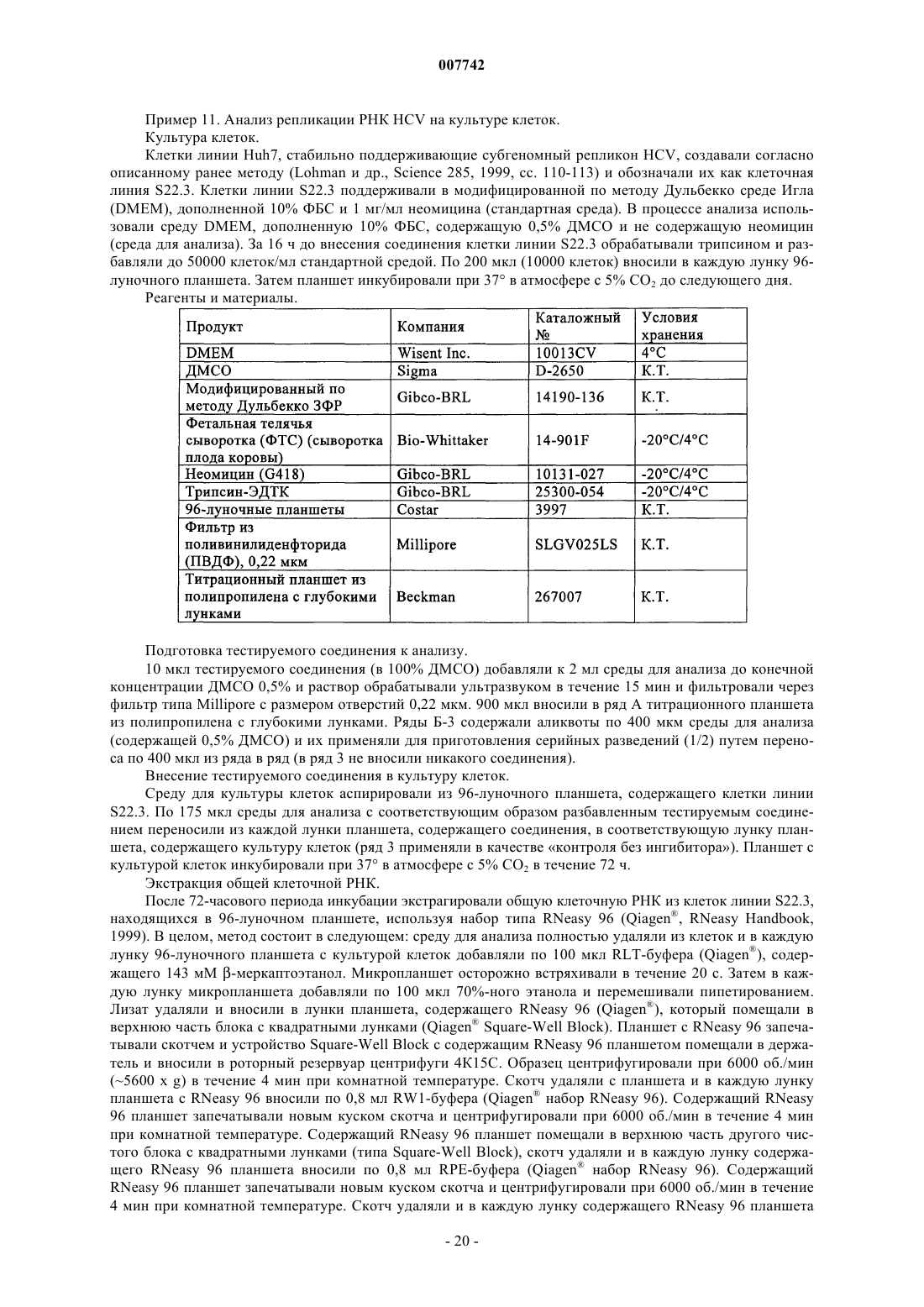
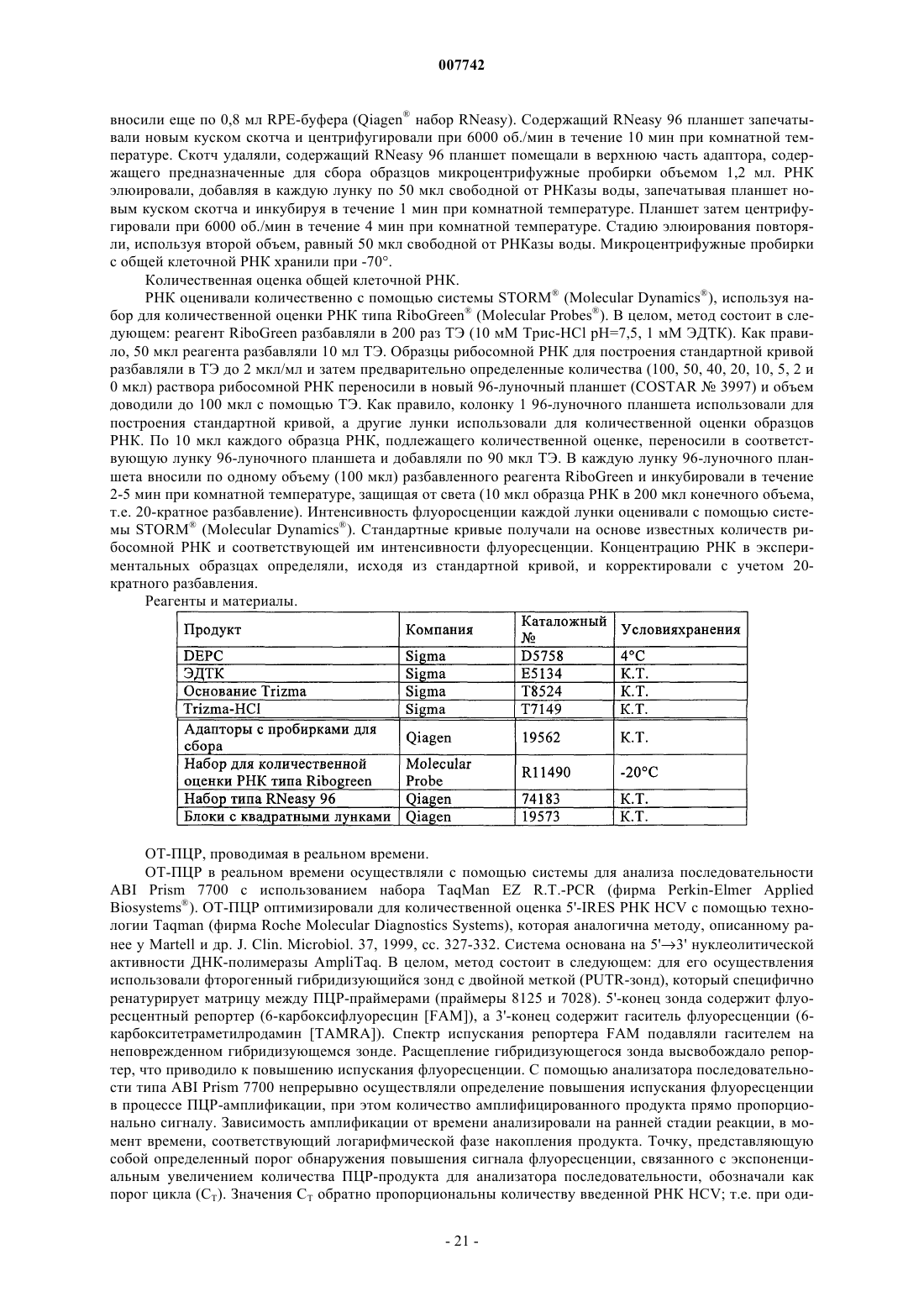
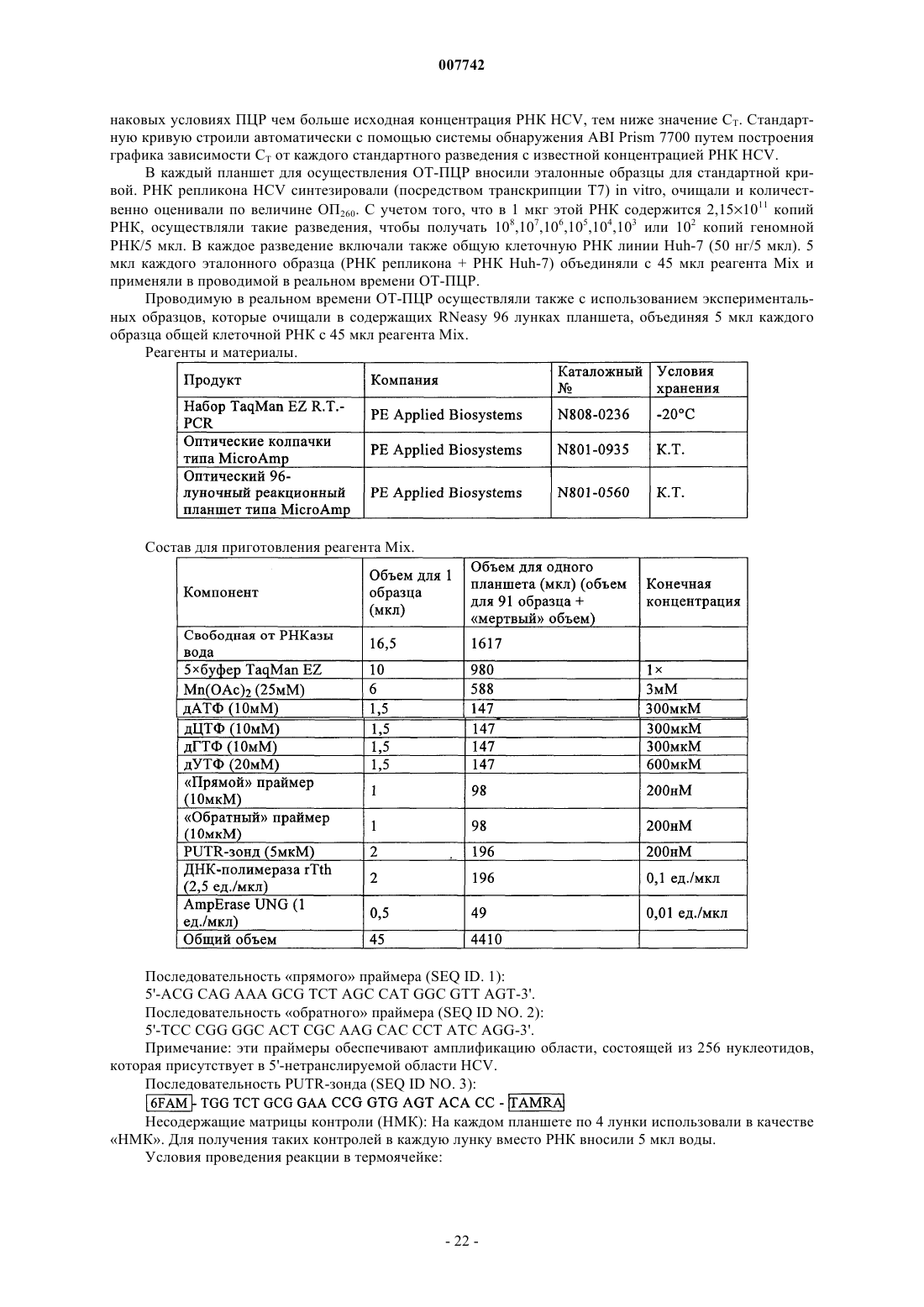





Формула / Реферат
1. Соединение формулы (I)

в котором R1 обозначает гидрокси или NHSO2Ph; R2 обозначает С4-С6циклоалкил; R3 обозначает трет-бутил или С5-С6циклоалкил и R4 обозначает С4-С6циклоалкил; или его фармацевтически приемлемая соль.
2. Соединение формулы (I) по п.1, в котором R1 обозначает NHSO2Ph.
3. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси.
4. Соединение формулы (I) по одному из пп.1-3, в котором R2 обозначает циклопентил или циклогексил.
5. Соединение формулы (I) по п.4, в котором R2 обозначает циклопентил.
6. Соединение формулы (I) по одному из пп.1-5, в котором R3 обозначает трет-бутил или циклогексил.
7. Соединение формулы (I) по п.6, в котором R3 обозначает трет-бутил.
8. Соединение формулы (I) по одному из пп.1-7, в котором R4 обозначает циклобутил или циклопентил.
9. Соединение формулы (I) по п.8, в котором R4 обозначает циклопентил.
10. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 и R4 каждый обозначает циклопентил и R3 обозначает трет-бутил.
11. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 обозначает циклобутил, R3 обозначает трет-бутил и R4 обозначает циклопентил.
12. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 обозначает циклогексил, R3 обозначает трет-бутил и R4 обозначает циклопентил.
13. Соединение формулы (I) по п.1, в котором R1 обозначает NHSO2Ph, R2 и R4 каждый обозначает циклопентил и R3 обозначает трет-бутил.
14. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 обозначает циклопентил, R3 обозначает трет-бутил и R4 обозначает циклобутил.
15. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 обозначает циклопентил, R3 обозначает тpeт-бутил и R4 обозначает циклогексил.
16. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 и R4 каждый обозначает циклопентил и R3 обозначает циклогексил.
17. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2, R3 и R4 каждый обозначает циклопентил.
18. Способ лечения или предупреждения инфекции, вызываемой вирусом гепатита С у млекопитающего, заключающийся в том, что млекопитающему вводят эффективное в отношении вируса гепатита С количество соединения формулы (I) по одному из пп.1-17 или его фармацевтически приемлемой соли.
19. Способ лечения или предупреждения инфекции, вызываемой вирусом гепатита С у млекопитающего, заключающийся в том, что млекопитающему вводят эффективное в отношении вируса гепатита С количество соединения формулы (I) по одному из пп.1-17 или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемой средой носителя или вспомогательным веществом.
20. Способ лечения или предупреждения инфекции, вызываемой вирусом гепатита С у млекопитающего, заключающийся в том, что млекопитающему вводят эффективное в отношении вируса гепатита С количество соединения формулы (I) по одному из пп.1-17 или его терапевтически приемлемой соли в сочетании с одним или несколькими другими агентами, обладающими активностью в отношении HCV.
21. Способ по п.20, заключающийся в том, что другой агент, обладающий активностью в отношении HCV, выбирают из a -интерферона или пэгилированного a -интерферона.
22. Способ по п.20 или п.21, заключающийся в том, что другой агент, обладающий активностью в отношении HCV, представляет собой рибавирин.
23. Способ по одному из пп.20-22, заключающийся в том, что другой агент, обладающий активностью в отношении HCV, выбирают из ингибиторов геликазы, NS2/3-протеазы и внутреннего сайта входа в рибосому (IRES).
24. Способ по одному из пп.20-22, заключающийся в том, что другой агент, обладающий активностью в отношении HCV, представляет собой ингибитор HCV-полимеразы.
25. Применение соединения формулы I по одному из пп.1-17 для приготовления лекарственного средства, предназначенного для лечения или предупреждения инфекции, вызываемой вирусом гепатита С.
Текст
007742 Область техники, к которой относится изобретение Настоящее изобретение относится к соединениям, способам их синтеза, композициям и способам лечения инфекции, вызываемой вирусом гепатита С (HCV). В частности, настоящее изобретение относится к новым пептидным аналогам, фармацевтическим композициям, содержащим такие аналоги, и к способам применения этих аналогов для лечения инфекции, вызываемой HCV. Предпосылки создания изобретения Вирус гепатита С (HCV) является основным возбудителем гепатита, относящегося к не-А, не-B гепатиту, который передается при переливании крови и который поражает людей во всем мире. Установлено, что свыше 200 миллионов людей в мире заражено вирусом. Большой процент носителей становится хронически инфицированным и у многих пациентов это приводит к хроническому заболеванию печени, так называемому хроническому гепатиту С. В свою очередь, эта группа больных имеет высокий риск заболевания такой серьезной болезнью печени, как цирроз печени, печеночно-клеточный рак и последняя стадия болезни печени, приводящая к смерти. Механизм, с помощью которого происходит сохранение вируса HCV в организме и обеспечивается высокий коэффициент заболеваемости хронической болезнью печени, пока недостаточно изучен. Неизвестно, как HCV взаимодействует с иммунной системой хозяина и преодолевает ее действие. Кроме того,также еще не выявлены роли клеточных и гуморальных иммунных ответов в защите от HCV-инфекции и при заболевании гепатитом. Имеются данные о том, что для профилактики связанного с переливанием крови вирусного гепатита можно применять иммуноглобулины, однако, Центр по контролю заболеваемости в настоящее время не рекомендует для этой цели применять лечение с использованием иммуноглобулинов. Отсутствие эффективного защитного иммунного ответа затрудняет разработку вакцины или адекватных профилактических мер после контакта, поэтому в ближайшее время надежды в основном возлагаются на антивирусные средства. С целью выявления фармацевтических агентов, обладающих эффективностью в отношении леченияHCV-инфекции, были проведены различные клинические исследования с участием пациентов, страдающих хроническим гепатитом С. В этих исследованиях применяли интерферон-альфа индивидуально или в сочетании с другими антивирусными агентами. Эти исследования позволили установить, что у основного большинства участников эксперимента не было обнаружено реакции на такие схемы лечения, а из тех участников, которые оказались чувствительными к лечению, у большей части после окончания лечения происходил рецидив. Таким образом, до последнего времени терапия с использованием интерферона (IFN) оставалась единственным доступным методом лечения пациентов, страдающих хроническим гепатитом С, которая обладала доказанной в клинических условиях эффективностью. Однако эффективность такого лечения сохраняется в течение непродолжительного периода времени, и, кроме того, лечение интерфероном вызывает серьезные побочные действия (т.е. ретинопатию, тиреоидит, острый панкреатит, депрессию), что снижает качество жизни пациентов, подвергающихся лечению. В настоящее время интерферон в сочетании с рибавирином предложен для лечения пациентов, нечувствительных к IFN при его индивидуальном применении. Однако побочные действия, вызываемые IFN, не уменьшаются при такой совместной терапии. Установлено, что пэгилированные формы интерферонов, такие как PEG-Intron и Pegasys, могут частично снижать указанные вредные побочные действия, однако антивирусные лекарственные препараты все еще остаются основным средством для орального лечения HCV. Таким образом, существует необходимость в разработке эффективных антивирусных агентов для лечения HCV-инфекции, которые были бы лишены недостатков существующих методов лечения, основанных на применении фармацевтических средств.HCV представляет собой заключенную в оболочку положительную цепь РНК-ового вируса семейства Flaviviridae. Геном HCV, представленный одноцепочечной РНК, состоит приблизительно из 9500 нуклеотидов и имеет одну открытую рамку считывания (ОРС), которая кодирует один большой полипротеин, состоящий примерно из 3000 аминокислот. В зараженных клетках этот полипротеин расщепляется во многих сайтах клеточными и вирусными протеазами с образованием структурных и неструктурных (NS) протеинов. В случае HCV под действием двух вирусных протеаз образуются зрелые неструктурные протеины (NS2, NS3, NS4A, NS4B, NS5A и NS5B). Первая из указанных протеаз, которая пока еще недостаточно охарактеризована, расщепляет связь NS2-NS3 (далее в контексте настоящего описания обозначена как NS2/3-пpoтeaзa); вторая представляет собой сериновую протеазу, содержащуюся в Nконцевой области NS3 (далее обозначена как NS3-пpoтeaзa), и она опосредует все последующие расщепления, происходящие по ходу транскрипции NS3, как в цис-ориентации в сайте расщепления NS3-NS4A,так и в транс-ориентации в остальных сайтах NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. Протеин NS4A,вероятно, обладает множественными функциями, действуя в качестве кофактора для NS3-протеазы и возможно способствуя мембранной локализации NS3 и других компонентов вирусных репликаз. Образование комплекса протеазы NS3 с NS4A, вероятно, необходимо для процессирования, усиления протеолитической эффективности во всех сайтах. Протеин NS3 обладает также нуклеозидтрифосфатазной активностью и РНК-геликазной активностью. NS5B представляет собой РНК-зависимую РНК-полимеразу,принимающую участие в репликации HCV.-1 007742 Общая стратегия разработки антивирусных агентов предусматривает инактивацию кодируемых вирусом ферментов, которые играют основную роль в репликации вируса. Ниже приведен перечень заявок на патенты, опубликованных в течение последних нескольких лет,в которых описаны пептидные аналоги, являющиеся ингибиторами NS3-пpoтeaзы HCV, которые по строению отличаются от соединений, предлагаемых в настоящем изобретении: GB 2337262; JP 10298151;WO 02/18369; WO 02/60926 и WO 02/79234. Одним из преимуществ настоящего изобретения является то, что в нем заявлены трипептидные производные, которые обладают ингибирующей активностью в отношении NS3-пpoтeaзы, фермента,который играет важную роль в репликации вируса гепатита С. Кроме того, соединения обладают способностью ингибировать репликацию РНК HCV, что установлено с использованием клеточной модели репликона. Еще одним преимуществом настоящего изобретения является тот факт, что эти соединения специфически ингибируют NS3-пpoтeaзy и не проявляют заметной ингибирующей активности в отношении других сериновых протеаз, таких как эластаза лейкоцитов человека (HLE), эластаза панкреатической железы свиньи (РРЕ) или бычий химотрипсин поджелудочной железы, или в отношении цистеиновых протеаз, таких как катепсин В печени человека (Cat В). Краткое изложение сущности изобретения Изобретение относится к соединению формулы (I) в котором R1 обозначает гидроксй или NHSO2R1A, где R1A обозначает C1-С 8 алкил, С 3-С 7 циклоалкил или С 1-С 6 алкил-С 3-С 7 циклоалкил, которые все необязательно замещены 1-3 заместителями, выбранными из ряда, включающего галоген, циано, нитро, ОС 1-С 6 алкил, амидо, амино или фенил, или R1A обозначает С 6 или С 10 арил, необязательно замещенный 1-3 заместителями, выбранными из ряда, включающего галоген,циано, нитро, С 1-С 6 алкил, OC1-С 6 алкил, амидо, амино или фенил;R4 обозначает С 4-С 6 циклоалкил; или его фармацевтически приемлемой соли. Изобретение относится также к фармацевтической композиции, содержащей эффективное в отношении вируса гепатита С количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемой средой носителя или вспомогательным веществом. Согласно одному из вариантов осуществления изобретения фармацевтическая композиция, предлагаемая в настоящем изобретении, дополнительно содержит интерферон (пэгилированный или непэгилированный) или рибавирин, или один или несколько других агентов, обладающих активностью в отношении HCV, или их любую комбинацию. Еще одним важным объектом изобретения является способ лечения инфекции, вызываемой вирусом гепатита С у млекопитающего, заключающийся в том, что млекопитающему вводят эффективное в отношении вируса гепатита С количество соединения формулы (I) или его фармацевтически приемлемой соли, или описанной выше композиции, индивидуально или в сочетании с одним или несколькими следующими компонентами: интерферон (пэгилированный или непэгилированный) или рибавирин, или один или несколько других агентов, обладающих активностью в отношении HCV, которые в каждом случае вводят совместно или раздельно. Следующим важным объектом изобретения является способ предупреждения инфекции, вызываемой вирусом гепатита С у млекопитающего, заключающийся в том, что млекопитающему вводят эффективное в отношении вируса гепатита С количество соединения формулы (I) или его терапевтически приемлемой соли, или описанной выше композиции, индивидуально или в сочетании с одним или несколькими следующими компонентами: интерферон (пэгилированный или непэгилированный) или рибавирин,-2 007742 или один или несколько других агентов, обладающих активностью в отношении HCV, которые вводят совместно или раздельно. Настоящее изобретение относится также к применению описанного выше соединения формулы (I) для приготовления лекарственного средства, предназначенного для лечения или предупреждения инфекции, вызываемой вирусом гепатита С. В настоящем описании сделаны ссылки на многочисленные документы, содержание которых включено в качестве ссылки. Подробное описание предпочтительных вариантов осуществления изобретения Определения Если не указано иное, то в контексте настоящего описания используемые понятия имеют следующие значения. В тех случаях, когда для обозначения абсолютной конфигурации заместителя или асимметричного центра соединения формулы (I) используют дескрипторы (R) или (S), то обозначение относится ко всему соединению, а не только к заместителю или асимметричному центру. Обозначение P1, P2 и Р 3 в контексте настоящего описания относится к положению аминокислотных остатков, начиная с С-конца пептидных аналогов и простираясь к N-концу [т.е. Р 1 обозначает положение 1 относительно С-конца, Р 2 обозначает положение 2 относительно С-конца и т.д. (см. Berger А. иSchechter I., Transactions of the Royal Society London series, B257, 1970, cc. 249-264). В контексте настоящего описания понятие винил-АЦКК относится к соединению формулы т.е. (1R, 2S)-1-амино-2-этенилциклопропилкарбоновой кислоте. Понятие С 1-С 8 алкил в контексте настоящего описания, индивидуально или в сочетании с другим заместителем, обозначает ациклические алкильные заместители с прямой или разветвленной цепью, содержащие от 1 до 8 атомов углерода, и включает, например, метил, этил, 2-метилгексил, 1,1-диметилгексил (или трет-бутил) и октил. Понятие С 3-С 7 циклоалкил в контексте настоящего описания, индивидуально или в сочетании с другим заместителем, обозначает циклоалкильный заместитель, содержащий от 3 до 7 атомов углерода,и включает циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Понятие C1-С 6 алкил-С 3-С 6 циклоалкил в контексте настоящего описания обозначает циклоалкильный радикал, содержащий от 3 до 6 атомов углерода, который непосредственно связан с алкиленовым радикалом, содержащим от 1 до 6 атомов углерода; например, циклопропилметил, циклопентилэтил,циклогексилметил и циклогексилэтил. Например, когда R1A обозначает С 1-С 6 алкил-С 3-С 6 циклоалкил, то эта группа связана с SО 2-группой через С 1-С 6 алкил (т.е. алкиленовый фрагмент). Понятие С 6- или С 10 арил в контексте настоящего описания, индивидуально или в сочетании с другим радикалом, обозначает либо ароматическую моноциклическую группу, содержащую 6 атомов углерода, либо ароматическую бициклическую группу, содержащую 10 атомов углерода. Например, арил представляет собой фенил, 1-нафтил или 2-нафтил. Понятие ОС 1-С 6 алкил в контексте настоящего описания, индивидуально или в сочетании с другим радикалом, обозначает радикал -ОС 1-С 6 алкил, в котором алкил, как указано выше, содержит вплоть до 6 атомов углерода. ОС 1-С 6 алкил обозначает метокси, этокси, пропокси, 1-метилэтокси, бутокси и 1,1 диметилэтокси. Последний радикал обычно обозначают как трет-бутокси. Понятие гало в контексте настоящего описания обозначает галогеновый заместитель, выбранный из брома, хлора, фтора или йода. Понятие фармацевтически приемлемая соль обозначает соль соединения формулы (I), которую с медицинской точки зрения можно использовать в контакте с тканями человека и низших животных без признаков повышенной токсичности, раздражения, аллергической реакции и т.п., которая соответствует приемлемому соотношению польза/риск и, как правило, может растворяться или диспергироваться в воде или масле и обладает эффективностью при целевом использовании. Понятие включает фармацевтически приемлемые кислотно-аддитивные соли и фармацевтически приемлемые соли присоединения оснований. Перечень приемлемых солей приведен, например, у S.M. Birge и др., J. Pharm. Sci., 66, 1977, сс. 119, публикация полностью включена в настоящее описание в качестве ссылки. Понятие фармацевтически приемлемая кислотно-аддитивная соль обозначает соли, которые сохраняют свою биологическую эффективность и свойства свободных оснований и которые не являются нежелательными с позиций биологии или по другим причинам, образованные с неорганическими кислотами, такими как соляная кислота, бромисто-водородная кислота, йодисто-водородная кислота, серная кислота, сульфаминовая кислота, азотная кислота, фосфорная кислота и т.п., и с органическими кислотами, такими как уксусная кислота, трифторуксусная кислота, адипиновая кислота, альгиновая кислота,аскорбиновая кислота, аспарагиновая кислота, бензолсульфоновая кислота, бензойная кислота, 2-3 007742 ацетоксибензойная кислота, масляная кислота, камфорная кислота, камфорсульфоновая кислота, коричная кислота, лимонная кислота, диглюконовая кислота, этансульфоновая кислота, глутаминовая кислота,гликолевая кислота, глицерофосфорная кислота, полусерная кислота, энантовая кислота, капроновая кислота, муравьиная кислота, фумаровая кислота, 2-гидроксиэтансульфоновая кислота (изэтионовая кислота), молочная кислота, малеиновая кислота, гидроксималеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, мезитиленсульфоновая кислота, метансульфоновая кислота, нафталинсульфоновая кислота, никотиновая кислота, 2-нафталинсульфоновая кислота, щавелевая кислота, памовая кислота, пектиновая кислота, фенилуксусная кислота, 3-фенилпропионовая кислота, пикриновая кислота, пивалиновая кислота, пропионовая кислота, пировиноградная кислота, салициловая кислота, стеариновая кислота, янтарная кислота, сульфаниловая кислота, винная кислота, паратолуолсульфоновая кислота, ундекановая кислота и т.п. Понятие фармацевтически приемлемая соль присоединения основания обозначает соли, которые сохраняют свою биологическую эффективность и свойства свободных кислот и которые не являются нежелательными с позиций биологии или по другим причинам, образованные с неорганическими основаниями, такими как аммиак или гидроксид, карбонат или бикарбонат аммония, или катионами металла,такого как натрий, калий, литий, кальций, магний, железо, цинк, медь, марганец, алюминий и т.п. Наиболее предпочтительными являются соли аммония, калия, натрия, кальция и магния. Соли, полученные из фармацевтически приемлемых органических нетоксических оснований, включают соли первичных, вторичных и третичных аминов, четвертичных аминовых соединений, замещенных аминов, включая встречающиеся в естественных условиях замещенные амины, циклические амины и основные ионообменные смолы, такие как метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, изопропиламин, трипропиламин, трибутиламин, этаноламин, диэтаноламин, 2-диметиламиноэтанол, 2 диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, гидрабамин, холин, бетаин,этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, Nэтилпиперидин, производные тетраметиламмония, производные тетраэтиламмония, пиридин, N,Nдиметиланилин, N-метилпиперидин, N-метилморфолин, дициклогексиламин, дибензиламин, N,Nдибензилфенэтиламин, 1-эфенамин, N,N'-дибензилэтилендиамин, полиаминовые смолы и т.п. Наиболее предпочтительными органическими нетоксическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин. Понятие антивирусный агент в контексте настоящего описания обозначает агент (химическое соединение или биологический агент), который обладает эффективностью в отношении ингибирования образования и/или репликации вируса в организме млекопитающего. Оно включает агенты, которые оказывают воздействие на связанные либо с хозяином, либо с вирусом механизмы, необходимые для образования и/или репликации вируса в организме млекопитающего. Антивирусные агенты включают, например, рибавирин, амантадин, VX-497 (меримеподиб, фирма Vertex Pharmaceuticals), VX-498 (фирмаBiopharmaceuticals). Понятие другой агент, обладающий активностью в отношении HCV в контексте настоящего описания обозначает агенты, эффективные в отношении снижения или предупреждения развития связанных с гепатитом С симптомов болезни. Такие агенты выбирают из: антивирусных агентов, иммуномодуляторов, ингибиторов NS3-протеазы HCV, ингибиторов полимеразы HCV или ингибиторов другой мишени в жизненном цикле HCV. Понятие иммуномодулятор в контексте настоящего описания обозначает агенты (химические соединения или биологические агенты), которые обладают эффективностью в отношении повышения или потенциирования реакции иммунной системы у млекопитающего. Иммуномодуляторы включают, например, интерфероны класса I (такие как -, - и -интерфероны, -интерфероны, консенсусные интерфероны и азиало-интерфероны), интерфероны класса II (такие как -интерфероны) и пэгилированные интерфероны. Понятие ингибитор NS3-протеазы HCV в контексте настоящего описания обозначает агент (химическое соединение или биологический агент), который обладает эффективностью в отношении ингибирования функции NS3-пpoтeaзы HCV у млекопитающего. Ингибиторы NS3-протеазы HCV включают,например, соединения, описанные в WO 99/07733, WO 99/07734, WO 00/09558, WO 00/09543, WO 00/59929 или WO 02/060926, перспективное соединение, разработанное фирмой Boehringer Ingelheim,обозначенное как BILN 2061, которое изучено в клиническом исследовании, и перспективные соединения, разработанные фирмой Vertex/Eli Lilly, обозначенные как VX-950 или LY-570310, которые еще не окончательно исследованы. В частности, соединения 2, 3, 5, 6, 8, 10, 11, 18, 19, 29, 30, 31, 32, 33, 37,38, 55, 59, 71, 91, 103, 104, 105, 112, 113, 114, 115, 116, 120, 122, 123, 124, 125, 126 и 127, представленные в таблице на сс. 224-226 WO 02/060926, можно применять в сочетании с соединениями, предлагаемыми в настоящем изобретении. Понятие ингибитор полимеразы HCV в контексте настоящего описания обозначает агент (химическое соединение или биологический агент), который обладает эффективностью в отношении ингиби-4 007742 рования функции полимеразы HCV у млекопитающего. Оно относится, например, к ингибиторам NS5Bполимеразы HCV. Ингибиторы полимеразы HCV включают не относящиеся к нуклеозидам соединения,например, описанные в заявке на патент США 10/198680, которая соответствует РСТ/СА 02/01127,оба документа поданы 18 июля 2002 г. (фирма Boehringer Ingelheim), заявке на патент США 10/198384, которая соответствует РСТ/СА 02/01128, оба документа поданы 18 июля 2002 г. (фирма Boehringer Ingelheim), заявке на патент США 10/198259, которая соответствует РСТ/СА 02/01129, оба документа поданы 18 июля 2002 г. (фирма Boehringer Ingelheim), WO 02/100846 А 1 и WO 02/100851 A2(обе на имя фирмы Shire), WO 01/85172 А 1 и WO 02/098424 А 1 (обе на имя фирмы GSK), WO 00/06529 иWO 02/06246 А 1 (обе на имя фирмы Merck), WO 01/47883 и WO 03/000254 (обе на имя фирмы Japan Tobacco) и ЕР 1256628 А 2 (на имя фирмы Agouron). Кроме того, другие ингибиторы полимеразы HCV включают также нуклеозидные аналоги, например, соединения, описанные в WO 01/90121 А 2 (фирма Idenix), WO 02/069903 А 2 (Biocryst Pharmaceuticals Inc.) и WO 02/057287 A2 и WO 02/057425 A2 (обе на имя фирмы Merck/Isis). Конкретными примерами ингибиторов полимеразы HCV являются JTK-002, JTK-003 и JTK-109(фирма Japan Tobacco). Понятие ингибитор другой мишени в жизненном цикле HCV в контексте настоящего описания обозначает агент (химическое соединение или биологический агент), который обладает эффективностью в отношении ингибирования образования и/или репликации HCV у млекопитающего, отличной от ингибирования функции NS3-пpoтeaзы HCV. Они включают агенты, которые оказывают воздействие на связанные либо с хозяином, либо с вирусом HCV механизмы, необходимые для образования и/или репликации HCV в организме млекопитающего. Ингибиторы другой мишени в жизненном цикле HCV включают, например, агенты, которые ингибируют мишень, выбранную из ряда, включающего геликазу, NS2/3 пpoтeaзy HCV и внутренний сайт входа в рибосому (IRES). Конкретным примером ингибиторов другой мишени в жизненном цикле HCV является ISIS-14803 (ISIS Pharmaceuticals). Понятие ингибитор ВИЧ в контексте настоящего описания обозначает агент (химическое соединение или биологический агент), который обладает эффективностью в отношении ингибирования образования и/или репликации ВИЧ в организме млекопитающего. Оно включает агенты, которые оказывают воздействие на связанные либо с хозяином, либо с вирусом механизмы, необходимые для образования и/или репликации ВИЧ в организме млекопитающего. Ингибиторы ВИЧ включают, например, нуклеозидные ингибиторы, ненуклеозидные ингибиторы, ингибиторы протеаз, ингибиторы слияния и ингибиторы интегразы. Понятие ингибитор HAV (вирус гепатита А) в контексте настоящего описания обозначает агент(химическое соединение или биологический агент), который обладает эффективностью в отношении ингибирования образования и/или репликации HAV в организме млекопитающего. Оно включает агенты,которые оказывают воздействие на связанные либо с хозяином, либо с вирусом механизмы, необходимые для образования и/или репликации HAV в организме млекопитающего. Ингибиторы HAV включают вакцины, содержащие вирус гепатита А, например, Havrix (фирма GlaxoSmithKline), VAQTA (фирмаMerck) и Avaxim (фирма Aventis Pasteur). Понятие ингибитор HBV (вирус гепатита В) в контексте настоящего описания обозначает агент(химическое соединение или биологический агент), который обладает эффективностью в отношении ингибирования образования и/или репликации HBV в организме млекопитающего. Оно включает агенты,которые оказывают воздействие на связанные либо с хозяином, либо с вирусом механизмы, необходимые для образования и/или репликации HBV в организме млекопитающего. Ингибиторы HBV включают, например, агенты, которые ингибируют ДНК-полимеразу вируса HBV или вакцины на основе HBV. Конкретными примерами ингибиторов HBV являются ламивудин (Epivir-HBV), адефовирдипивоксил,энтекавир, FTC (Coviracil), DAPD (DXG), L-FMAU (Clevudine), АМ 365 (амрад), Ldt (телбивудин), моновалентный-LdC (валторцитабин), АСН-126,443 (L-Fd4C) (ахиллион), МСС 478 (фирма Eli Lilly), рацивир (RCV), фтор-L- и -D-нуклеозиды, робустафлавон, ICN 2001-3 (ICN), Ваm 205 (Novelos), XTL-001(XTL), иминосахара (Nonyl-DNJ) (фирма Synergy), HepBzyme; иммуномодуляторы, такие как: интерферон альфа 2b, HE2000 (фирма Hollis-Eden), Theradigm (эпиммун), ЕНТ 899 (фирма Enzo Biochem), тимозин альфа-1 (Zadaxin), вакцину на основе ДНК HBV (фирма PowderJect), вакцину на основе ДНК HBV(фирма Jefferon Center), антиген HBV (фирма OraGen), BayHep В (фирма Bayer), Nabi-HB (фирма Nabi) и антитела к вирусу гепатита В (фирма Cangene); и вакцины на основе HBV, такие, например, как Engerix В, Recombivax HB, GenHevac В, Нерасаrе, Bio-Hep В, TwinRix, Comvax, Hexavac. Понятие интерферон класса I в контексте настоящего описания обозначает интерферон, выбранный из группы интерферонов, которые все связываются с рецептором типа I. Оно включает как встречающиеся в естественных условиях, таких и полученные синтетическим путем интерфероны класса I. Примеры интерферонов класса I включают -, -, -интерфероны, -интерфероны, консенсусные интерфероны, азиало-интерфероны. Понятие интерферон класса II в контексте настоящего описания обозначает интерферон, выбранный из группы интерферонов, которые все связываются с рецептором типа II. Примеры интерферонов-5 007742 класса II включают -интерфероны. Фармацевтические композиции, предлагаемые в изобретении, могут содержать одно или несколько дополнительных действующих веществ, например, выбранных из антивирусных агентов, иммуномодуляторов, других ингибиторов NS3-пpoтeaзы HCV, ингибиторов полимеразы HCV, ингибиторов другой мишени в жизненном цикле HCV, ингибиторов ВИЧ, ингибиторов HAV и ингибиторов HBV. Примеры таких агентов приведены выше в разделе Определения. Ниже представлены конкретные предпочтительные примеры некоторых таких агентов антивирусные агенты: рибавирин и амантадин; иммуномодуляторы: интерфероны класса I, интерфероны класса II и пэгилированные интерфероны; ингибитор другой мишени в жизненном цикле HCV, мишенью такого ингибитора являются NS3 геликаза, NS2/3-пpoтeaзa HCV или внутренний сайт входа в рибосому (IRES); ингибиторы ВИЧ: нуклеозидные ингибиторы, ненуклеозидные ингибиторы, ингибиторы протеаз,ингибиторы слияния и ингибиторы интеграз; или ингибиторы HBV: агенты, которые ингибируют вирусную ДНК-полимеразу HBV, или вакцина на основе HBV. Как указано выше, совместная терапия предусматривает совместное введение соединения формулы(I) или его фармацевтически приемлемой соли по меньшей мере с одним дополнительным агентом, выбранным из следующего ряда: антивирусный агент, иммуномодулятор, другой ингибитор NS3-протеазыHCV, ингибитор полимеразы HCV, ингибитор другой мишени в жизненном цикле HCV, ингибитор ВИЧ,ингибитор HAV и ингибитор HBV. Примеры таких агентов приведены выше в разделе Определения. Указанные дополнительные агенты можно объединять с соединениями, предлагаемыми в изобретении,получая лекарственное средство, которое содержит однократную фармацевтическую дозу. В другом варианте указанные дополнительные агенты можно вводить пациенту индивидуально в виде составной части многокомпонентного лекарственного средства, например, используя набор. Указанные дополнительные агенты можно вводить пациенту до, одновременно или после введения соединения формулы (I) или его фармацевтически приемлемой соли. В контексте настоящего описания понятие лечение обозначает введение соединения или композиции, предлагаемых в настоящем изобретении, для облегчения или устранения симптомов гепатита С и/или снижения вирусного титра у пациента. В контексте настоящего описания понятие предупреждение обозначает введение соединения или композиции, предлагаемых в настоящем изобретении, после контакта индивидуума с вирусом, но до проявления симптомов болезни и/или до обнаружения вируса в крови. Предпочтительные варианты осуществления изобретения Предпочтительными являются соединения формулы (I), как они определены выше, в которых R1 обозначает гидрокси, NHSO2Me, NHSO2-циклопропил или NHSO2Ph. Более предпочтительно R1 обозначает NHSO2-циклопропил или NHSO2Ph. В другом варианте наиболее предпочтительно R1 обозначает гидрокси. Предпочтительными являются соединения формулы (I), как они определены выше, в которых R2 обозначает циклопентил или циклогексил. Наиболее предпочтительно R2 обозначает циклопентил. Предпочтительно R3 обозначает тpeт-бутил или циклогексил. Наиболее предпочтительно R3 обозначает тpeт-бутил. Предпочтительными являются соединения формулы (I), как они определены выше, в которых R4 обозначает циклобутил или циклопентил. Наиболее предпочтительно R4 обозначает циклопентил. Более предпочтительным является соединение формулы (I), как оно определено выше, в котором R1 обозначает гидрокси, R2 и R4 каждый обозначает циклопентил и R3 обозначает тpeт-бутил. Более предпочтительным является соединение формулы (I), в котором R1 обозначает гидрокси, R2 обозначает циклобутил, R3 обозначает тpeт-бутил и R4 обозначает циклопентил. Более предпочтительным является соединение формулы (I), в котором R1 обозначает гидрокси, R2 обозначает циклогексил, R3 обозначает тpeт-бутил и R4 обозначает циклопентил. Более предпочтительно R1 обозначает NHSO2Ph, R2 и R4 каждый обозначает циклопентил и R3 обозначает тpeт-бутил. Более предпочтительным является соединение формулы (I), в котором R1 обозначает гидрокси, R2 обозначает циклопентил, R3 обозначает тpeт-бутил и R4 обозначает циклобутил. Более предпочтительным является соединение формулы (I), в котором R1 обозначает гидрокси, R2 обозначает циклопентил, R3 обозначает тpeт-бутил и R4 обозначает циклогексил. Более предпочтительным является соединение формулы (I), в котором R1 обозначает гидрокси, R2 и 4R каждый обозначает циклопентил и R3 обозначает циклогексил. Более предпочтительным является соединение формулы (I), в котором R1 обозначает гидрокси, R2,3R и R4 каждый обозначает циклопентил. Согласно другому варианту осуществления изобретения фармацевтическая композиция, предлагаемая в настоящем изобретении, может дополнительно содержать другой агент, обладающий активностью-6 007742 в отношении HCV. Примеры других агентов, обладающих активностью в отношении HCV, включают (альфа), - (бета), - (дельта), - (гамма) или - (омега) интерферон, рибавирин и амантадин. Согласно другому варианту осуществления изобретения фармацевтическая композиция, предлагаемая в настоящем изобретении, может дополнительно содержать другой ингибитор NS3-пpoтeaзы HCV. Согласно еще одному варианту осуществления изобретения фармацевтическая композиция, предлагаемая в настоящем изобретении, может дополнительно содержать ингибитор полимеразы HCV. Согласно следующему варианту осуществления изобретения фармацевтическая композиция, предлагаемая в настоящем изобретении, может дополнительно содержать ингибитор других мишеней жизненного цикла HCV, включая (но не ограничиваясь ими) геликазу, NS2/3-пpoтeaзy или внутренний сайт входа в рибосому (IRES). Фармацевтическую композицию, предлагаемую в настоящем изобретении, можно вводить орально,парентерально или с помощью устройства для имплантации. Предпочтительным является оральное введение или введение с помощью инъекции. Фармацевтическая композиция, предлагаемая в настоящем изобретении, может содержать любые общепринятые нетоксические фармацевтически приемлемые носители, вспомогательные вещества или наполнители. В некоторых случаях значение рН композиции можно регулировать с помощью фармацевтически приемлемых кислот, оснований или буферов с целью повышения стабильности соединения, включенного в композицию или в форму для его введения. В контексте настоящего описания понятие парентеральный включает подкожный, внутрикожный, внутривенный, внутримышечный, внутрисуставной, интрасиновиальный, интрастернальный, интратекальный путь введения, введение с помощью инъекции или инфузии, а также введение в область повреждения. Фармацевтическая композиция может иметь форму стерильного препарата для инъекции, например стерильной инъецируемой водной или жирорастворимой суспензии. Эту суспензию можно получать с помощью хорошо известных методов с использованием диспергирующих или смачивающих агентов (таких, например, как Твин 80) и суспендирующих агентов. Фармацевтическую композицию, предлагаемую в настоящем изобретении, можно вводить орально в виде любой приемлемой формы лекарственного средства, предназначенной для орального введения, включая (но не ограничиваясь ими) капсулы, таблетки и водные суспензии и растворы. В случае таблеток для орального введения обычно применяемые носители представляют собой лактозу и кукурузный крахмал. Как правило, добавляют также замасливатели, такие как стеарат магния. Для орального введения в форме капсул приемлемые разбавители представляют собой лактозу и безводный кукурузный крахмал. Когда оральным путем вводят водные суспензии, действующее вещество объединяют с эмульгирующими и суспендирующими агентами. При необходимости можно добавлять определенные подслащивающие вещества, и/или корригенты, и/или красители. Другие пригодные наполнители или носители для указанных выше препаративных форм и композиций приведены в обычных фармацевтических справочниках, например, в Remington's PharmaceuticalSciences, The Science и Practice of Pharmacy, 19-е изд. Mack Publishing Company, Easton, Penn., (1995). Для монотерапии с целью предупреждения и лечения опосредованной HCV болезни можно применять дозы в диапазоне от примерно 0,01 до примерно 100 мг/кг веса тела в день, предпочтительно от примерно 0,1 до примерно 50 мг/кг веса тела в день предлагаемого соединения, которое является ингибитором протеазы. Как правило, фармацевтическую композицию, предлагаемую в изобретении, можно вводить от примерно 1 до примерно 5 раз в день или в другом варианте путем непрерывной инфузии. Такое введение можно применять как для длительного, так и для экстренного лечения. Количество действующего вещества, которое можно объединять с носителями для получения однократной дозы лекарственного средства, должно варьироваться в зависимости от хозяина, подлежащего лечению, и конкретного пути введения. Обычная композиция может содержать от примерно 5 до примерно 95% действующего вещества (мас.%). Предпочтительно такие композиции содержат от примерно 20 до примерно 80% действующего вещества. Как должно быть очевидно специалисту в данной области, можно применять и более низкие или более высокие дозы по сравнению с указанными выше. Конкретные дозы и схемы лечения любого конкретного пациента могут зависеть от различных факторов, включая активность конкретного применяемого соединения, возраст, вес тела, общее состояние здоровья, пол, рацион, время введения, скорость выведения, сочетание лекарственных средств, серьезность и особенности развития болезни, предрасположенность пациента к инфекции, и их определяет лечащий врач. Как правило, лечение начинают с небольших доз, существенно более низких, чем оптимальная доза пептида. Затем дозу повышают небольшими порциями до достижения оптимального действия в конкретных условиях. Как правило, наиболее предпочтительно вводить соединение в таком диапазоне концентраций, которые, в целом, обеспечивают антивирусную активность, но не обладают какими-либо вредными или опасными побочными действиями. Когда композицию, предлагаемую в настоящем изобретении, включают в комбинацию, содержащую соединение формулы (I) и один или несколько дополнительных терапевтических или профилактических агентов, то соединение и дополнительный агент должны присутствовать в дозе, составляющей примерно от 10 до 100% и более предпочтительно примерно от 10 до 80% от дозы, обычно применяемой-7 007742 в режиме монотерапии. Когда эти соединения или их фармацевтически приемлемые соли объединяют в препаративной форме с фармацевтически приемлемым носителем, то полученную композицию можно вводить in vivo млекопитающему, такому как человек, для ингибирования NS3-протеазы HCV или для лечения или предупреждения инфекции, вызываемой вирусом HCV. Такое лечение можно осуществлять также при использовании соединения, предлагаемого в изобретении, в сочетании с агентами, включающими (но не ограничиваясь ими): -, -, -, -, - или -интерферон, рибавирин, амантадин; другие ингибиторы NS3 пpoтeaзы HCV; ингибиторы полимеразы HCV; ингибиторы других мишеней жизненного цикла HCV,которые включают (но не ограничиваясь ими) геликазу, NS2/3-пpoтeaзy или внутренний сайт входа в рибосому (IRES); или их комбинации. С соединениями, предлагаемыми в настоящем изобретении, можно объединять другие агенты, получая однократную дозу лекарственного средства. В другом варианте эти дополнительные агенты можно вводить млекопитающему по отдельности в виде составной части многокомпонентного лекарственного средства. Еще одним вариантом осуществления настоящего изобретения является способ ингибирования NS3 протеазной активности HCV у млекопитающего, заключающийся во введении соединения формулы (I). Согласно предпочтительному варианту осуществления изобретения этот способ предназначен для снижения NS3-протеазной активности у зараженного вирусом гепатита С млекопитающего. Если фармацевтическая композиция содержит в качестве действующего вещества только соединение, предлагаемое в настоящем изобретении, то такой способ может дополнительно предусматривать введение указанному млекопитающему агента, выбранного из ряда, включающего иммуномодулятор,антивирусный агент, ингибитор NS3-пpoтeaзы HCV, ингибитор полимеразы HCV или ингибитор других мишеней жизненного цикла HCV, таких как геликаза, NS2/3-пpoтeaзa или IRES. Такой дополнительный агент можно вводить млекопитающему до, одновременно или после введения композиции, предлагаемой в настоящем изобретении. Соединение формулы (I), предлагаемое в настоящем изобретении, можно применять также в качестве лабораторного реагента. Соединение, предлагаемое в настоящем изобретении, можно применять также для устранения или предупреждения вирусного загрязнения материалов, снижая тем самым риск заражения вирусом персонала лабораторий или медицинских учреждений или пациентов, которые имеют контакт с такими материалами (например, кровью, тканями, хирургическими инструментами и предметами одежды, лабораторными инструментами и предметами одежды, приборами и материалами для сбора крови). Соединение формулы (I), предлагаемое в настоящем изобретении, можно применять также в качестве реагента для исследований. Соединение формулы (I) можно использовать в качестве позитивного контроля для обоснования модельных анализов на клетках или анализов in vitro или in vivo репликации вирусов. Ниже изобретение проиллюстрировано на примерах, не ограничивающих его объем, заявляемый в приведенной ниже формуле изобретения. Методология В целом, соединения формулы (I) и их промежуточные продукты получают известными методами,используя реакционные условия, которые, как известно, являются приемлемыми для реактантов. Некоторые такие методы описаны в WO 00/09543, WO 00/09558 и US 6323180, которые включены в настоящее описание в качестве ссылки. Соединения формулы (I), в которых R1 обозначает NHSO2R1A, как они определены выше, получают путем сочетания соответствующей кислоты формулы (I) (т.е. R1 обозначает гидрокси) с соответствующим сульфонамидом формулы R1A SO2NH2 в присутствии связывающего агента в стандартных условиях. Хотя можно применять несколько общепринятых связующих агентов, предпочтительными для практики являются ТБТУ и ГАТУ. Сульфонамиды поступают в продажу или их можно получать известными методами. На приведенных ниже схемах проиллюстрировано два основанных на известных методах приемлемых процесса получения соединений формулы (I), в которых R1 обозначает ОН. Примеры Температуры даны в градусах Цельсия. Проценты для растворов представляют собой % (маc./об.), а соотношения в растворах представляют собой соотношения объемов, если не указано иное. Спектры ядерного магнитного резонанса (ЯМР) регистрировали с помощью спектрометра фирмы Brucker при частоте 400 МГц, химические сдвигивыражены в ч./млн. Экспресс-хроматографию проводили на силикагеле (SiO2) согласно методу экспресс-хроматографии Стилла (W.C. Still и др., J. Org. Chem., 43, 1978, с. 2923). В примерах использовали следующие сокращения: ДБУ: 1,8-диазабицикло[5,4,0]ундец-7-ен; ДХМ: дихлорметан; ДИАД: диизопропилазодикарбоксилат; ДИЭА: диизопропилэтиламин; ДИПЭА: диизопропилэтиламин; ДМФ: N,N-диметилформамид; ДМАП: 4-(диметиламино)пиридин; EtOAc: этилацетат; ГАТУ: гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония; ЖХВР: жидкостная хроматография высокого разрешения; МС: масс-спектрометрия; MALDI-TOF: Matrix Assisted Laser DisorptionIonization-Time of Flight, FAB: бомбардировка быстрыми атомами; Me: метил; МеОН: метанол; Ph: фенил; К.Т.: комнатная температура (18-22); трет-бутил или m-бутил: 1,1-диметилэтил; Tbg: тpeт-бутил-9 007742 глицин: трет-лейцин; ТБТУ: тетрафторборат 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония; ТФК: трифторуксуная кислота; и ТГФ: тетрагидрофуран. Синтез соединений формулы (I). Представляющий собой дипептид промежуточный продукт 15 (схема 2) и 2-карбометокси-4 гидрокси-7-метоксихинолин 9 (схема 1) синтезировали с помощью методов, описанных в WO 00/09543. Синтез дипептида 1.(800 мл), перемешивали при К.Т. в атмосфере азота. Через 3,5 ч растворитель выпаривали и остаток экстрагировали EtOAc. Экстракт промывали соляной кислотой (10%), насыщенным раствором бикарбоната натрия и соляным раствором. Затем органическую фазу сушили над сульфатом магния, фильтровали и упаривали, получая масло. После сушки масла в течение ночи в глубоком вакууме получали дипептид 1 в виде пены желтого цвета (72,0 г, 94%, чистота 95% по данным ЖХВР). Получение дипептида 3. Дипептид 1 (72,0 г, 203 ммоль), трифенилфосфин (63,94 г, 243,8 ммоль, 1,2 экв.) и 4 нитробензойную кислоту (41,08 г, 245,8 ммоль, 1,2 экв.) растворяли в безводном ТГФ (1,4 л). Раствор при перемешивании охлаждали до 0 в атмосфере азота. Затем по каплям в течение 45 мин добавляли диэтилазодикарбоксилат (38,4 мл, 244 ммоль, 1,2 экв.) и реакционной смеси давали нагреться до К.Т. Через 4 ч растворитель выпаривали. Остаток разделяли на 4 порции. Каждую из них хроматографировали на тонкоизмельченном силикагеле (зернистость 10-40 мкм, диаметр колонки 12 см, длина колонки 16 см), используя градиент от 2:1 гексан/EtOAc до 1:1 гексан/ЕtOАс и до чистого (100%) ЕtOАс. После выпаривания растворителей и сушки в глубоком вакууме при 70 в течение 1 ч получали сложный эфир защищенного с помощью Воc дипептида (Вос-дипептид) 2 в виде аморфного твердого вещества белого цвета (108,1 г, количественный выход-105%). К сложному эфиру Вос-дипептида 2 (108 г, 243 ммоль) добавляли раствор 4 н. хлористого водорода в диоксане, получая бесцветный раствор. Раствор перемешивали при К.Т. в течение 1 ч. Растворитель выпаривали и остаток помещали в глубокий вакуум на 3 ч, получая гидрохлорид соединения 3 в виде аморфного твердого вещества. Твердое вещество применяли без дополнительной очистки. Синтез карбаматов 4. Получение карбамата 4 а. При 0 С к раствору метилового эфира тpeт-бутилглицина (590 мг, 3,25 ммоль) в ТГФ (8 мл) добавляли винилхлорформиат (0,55 мл, 6,47 ммоль) и триэтиламин (1,15 мл, 8,25 ммоль). Температуре давали подняться до К.Т. Раствор перемешивали в течение 16 ч. Раствор концентрировали и остаток растворяли в ЕtOAc. EtOАс-раствор промывали 10%-ным водным раствором лимонной кислоты (2 х), насыщенным водным раствором NаНСО 3 (2 х) и соляным раствором, сушили и концентрировали, получая соответствующий винилкарбамат (608 мг) в виде бесцветного масла. Масло растворяли в ДХМ (4 мл), охлаждали до 0 С и добавляли дийодметан (0,15 мл, 1,86 ммоль) и диэтилцинк (95 мкл, 0,93 ммоль). Сначала появлялось твердое вещество белого цвета, но оно со временем (1 ч) растворялось. Суспензию/раствор перемешивали при К.Т. в течение 5 ч. Добавляли насыщенный раствор хлорида аммония и раствор экстрагировали EtOАс (2 х). Органический экстракт сушили (MgSO4) и концентрировали. Остаток очищали колоночной экспресс-хроматографией. Поле элюирования гексаном:ЕtOАс (95:5) получали соответствующий сложный метиловый эфир в виде бесцветного масла (96 мг, выход 90%). Раствор сложного метилового эфира (93 мг, 0,41 ммоль) в ТГФ (5 мл), МеОН (1 мл) и водный раствор LiOH (45 мг, 1,81 ммоль) в воде (2 мл) перемешивали в течение 4 ч. Раствор разбавляли водой и- 10007742 экстрагировали EtOАс (2 х). Водный раствор подкисляли, добавляя 1 н. НСl, и кислый раствор экстрагировали EtOAc (2x). Объединенные органические экстракты сушили (MgSO4), фильтровали и упаривали,получая требуемый карбамат 4 а в виде твердого вещества белого цвета (53 мг; выход 60%). Получение карбамата 4b. ТГФ (350 мл) добавляли в колбу, содержащую 2,5-диоксопирролидин-1-иловый эфир циклопентилового эфира угольной кислоты (9,00 г, 39,6 ммоль) и трет-лейцин (6,24 г, 47,5 ммоль), получая суспензию. При интенсивном перемешивании добавляли дистиллированную воду (100 мл). Небольшое количество твердого вещества оставалось нерастворенным. Затем добавляли триэтиламин (16,6 мл, 119 ммоль),получая гомогенный раствор, который перемешивали при К.Т. Через 2,5 ч ТГФ выпаривали и водный остаток разбавляли водой (100 мл). Реакционную смесь подщелачивали, добавляя 1 н. NaOH (25 мл - конечное значение рН 10). Раствор промывали ЕtOАс (2200 мл) и затем водную фазу подкисляли с помощью 1 н. НСl (примерно 70 мл - конечное значение рН 2). Мутный раствор экстрагировали ЕtOАс(200+150 мл). Экстракт сушили (MgSO4) и упаривали, получая карбамат 4b в виде твердого вещества белого цвета (8,68 г). Получение других карбаматов. С помощью описанного выше процесса и используя соответствующие комбинации третбутилглицина, циклопентилглицина или циклогексилглицина и 2,5-диоксопирролидинового эфира циклобутилового, циклопентилового или циклогексилового эфира угольной кислоты, получали карбаматы,представленные следующими формулами: Раствор, содержащий гидрохлорид бензилового эфира трет-бутилглицина (2,55 г, 9,89 ммоль) в ТГФ (20 мл) и пиридин (2,0 мл, 24,73 ммоль), охлаждали до 0 С. К охлажденному раствору по каплям добавляли хлорформиат (1,30 мл, 10,19 ммоль). Образовавшуюся суспензию перемешивали в течение 5 мин при 0 С, затем в течение 1,5 ч при К.Т. Реакционную смесь разбавляли ЕtOАс, промывали 10%-ной лимонной кислотой (2 х), водой (2 х), насыщенным раствором NaHCO3 (2x), водой (2x) и соляным раствором (1x), сушили (MgSO4), фильтровали и упаривали, получая неочищенное соединение в виде практически бесцветного масла (3,73 г; 100%; примерно 9,89 ммоль). Неочищенный продукт (1,01 г, 2,97 ммоль) растворяли в ДМСО (6,5 мл) и по каплям добавляли циклопентиламин. Реакционную смесь перемешивали при К.Т. в течение 45 мин. Реакционную смесь разбавляли ЕtOАс. Органическую фазу промывали 10%-ной лимонной кислотой (2x), водой (2 х), насыщенным раствором NаНСО 3 (2 х), водой (2 х) и соляным раствором (1x), сушили (MgSO4), фильтровали и упаривали, получая неочищенный продукт, представляющий собой циклопентилмочевину-Tbg-Obn, в виде практически бесцветного масла. Неочищенный продукт очищали колоночной экспресс-хроматографией на силикагеле, используя в качестве элюента смесь гексан:ЕtOАс в соотношении 9:1 для удаления менее полярных примесей и в соотношении 7:3 для элюирования очищенного продукта в виде густого бесцветного масла (936 мг, 95%). У продукта,представляющего собой сложный эфир бензилового эфира (936 мг, 2,82 ммоль), удаляли защитную группу в атмосфере поступающего из баллона водорода, при К.Т., в абсолютном этаноле (15 мл) путем перемешивания раствора с 10% Pd/C (93,6 мг) в течение 5,5 ч. Реакционную смесь фильтровали через фильтр с размером отверстий 0,45 мкм и упаривали досуха, получая мочевину 5 в виде твердого вещества белого цвета (668,8 мг, 98%). Н-ЯМР (400 МГц, ДМСО-d6):12,39 (s, 1 Н), 6,09 (d, J=7,4 Гц, 1 Н), 5,93 (d, J=9,4 Гц, 1 Н), 3,90 (d,J=9,4 Гц, 1 Н), 3,87-3,77 (m, 1H), 1,84-1,72 (m, 2 Н), 1,63-1,42 (m, 4 Н), 1,30-1,19 (m, 2 Н), 0,89 (s, 9H). МС (электроспрей): 241,0 (М-Н)- 243,0 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2O): 99%. Синтез трипептида 6. Карбамат 4b (6,15 г, 22,5 ммоль) и ТБТУ (7,72 г, 24,7 ммоль) суспендировали в ДХМ и суспензию быстро перемешивали. Добавляли ДИПЭА (3,92 мл, 22,5 ммоль) при К.Т. и через 10 мин реакционная смесь становилась практически гомогенной. Затем в реакционную смесь сливали раствор дипептида 3(10,39 г, 23,6 ммоль) в безводном ДХМ (100 мл), содержащем ДИПЭА (4,11 мл, 23,62 ммоль). Образовавшемуся раствору желтого цвета давали перемешиваться в течение 14 ч. Затем растворитель выпаривали, получая сироп желтого цвета, который экстрагировали ЕtOАс (300+150 мл) и промывали 0,05 н. НСl (2200 мл), насыщенным раствором Na2 СО 3 (300 мл) и соляным раствором (150 мл). Объединенные экстракты сушили над MgSO4 и упаривали, получая трипептид 6 в виде пены светло-желтого цвета Трипептид 6 (15,68 г) растворяли в ТГФ (200 мл) и добавляли воду (30 мл). Образовавшийся раствор охлаждали до 0 С и в течение 3 мин при интенсивном перемешивании добавляли раствор моногидрата гидроксида лития (1,18 г, 28,12 ммоль). После выдерживания в течение 3 ч при 0 С избыток основания нейтрализовали с помощью 1 н. НСl (конечное значение рН примерно 6) и ТГФ выпаривали, получая водную суспензию (смола желтого цвета). Смесь экстрагировали EtOAc (2200 мл) и промывали насыщенным раствором NaHCO3 (2300 мл). Объединенные экстракты сушили над MgSO4 и упаривали, получая пену светло-желтого цвета. С помощью экспресс-хроматографии пены на силикагеле с использованием ЕtOАс в качестве элюента получали соединение 7 в виде аморфного твердого вещества белого цвета (9,77 г, 91%). Получение тиомочевин 8. Синтез тиомочевины 8 а. К раствору трет-бутиизотиоцианата (5,0 мл, 39,4 ммоль) в ДХМ (200 мл) добавляли циклопентиламин (4,67 мл, 47,3 ммоль), а затем ДИЭА и реакционную смесь перемешивали при К.Т. в течение 2 ч. Смесь разбавляли ЕtOАс, промывали 10%-ным водным раствором лимонной кислоты (2 х), насыщенным раствором NaHCO3 (2x), Н 2 О (2x) и соляным раствором (1x). Органический слой сушили над безводнымMgSO4, фильтровали и упаривали, получая N-тpeт-бутил-N'-циклопентилтиомочевину в виде твердого вещества белого цвета (3,70 г, выход 47%). N-тpeт-Бутил-N'-циклопентилтиомочевину (3,70 г) растворяли в концентрированной НСl (46 мл). Раствор темно-желтого цвета осторожно кипятили с обратным холодильником. Через 40 мин реакционной смеси давали охладиться до К.Т. и затем охлаждали на льду и подщелачивали до рН 9,5 с помощью твердого и насыщенного водного раствора NаНСО 3. Продукт экстрагировали ЕtOАс (3x). Объединенные ЕtOАс-экстракты промывали Н 2 О (2x) и соляным раствором (1x). Органический слой сушили (MgSO4), фильтровали и концентрировали, получая твердое вещество бежевого цвета (2,46 г неочищенного продукта). Растирали неочищенный продукт в смеси гексан/ЕtOАс(95/5) и после фильтрации получали N-циклопентилтиомочевину 8 а в виде твердого вещества белого цвета (2,38 г; выход 90%). 1 Н -ЯМР (400 МГц, ДМСО-d6):7,58 (bs, 1H), 7,19 (bs, 1H), 6,76 (bs, 1H), 4,34 (bs, 1 Н), 1,92-1,79 (m,2 Н), 1,66-1,55 (m, 2 Н), 1,55-1,30 (m, 4 Н). МС; es+ 144,9 (М+Н)+; es-: 142,8 (М-Н)-. Получение тиомочевины 8b.- 12007742 С помощью описанного выше процесса и используя поступающий в продажу циклобутиламин вместо циклопентиламина, получали тиомочевину 8b. Получение тиомочевины 8 с. С помощью описанного выше процесса и используя поступающий в продажу циклогексиламин вместо циклопентиламина, получали тиомочевину 8 с. К раствору KSCN (4,60 г, 47,33 ммоль) в ацетоне (35 мл) при 0 С добавляли по каплям бензоилхлорид (5,0 мл, 43,03 ммоль). Раствор молочного цвета перемешивали в ледяной бане в течение 1,5 ч, затем по каплям добавляли циклопропиламин (3,2 мл, 46,0 ммоль). Реакционную смесь перемешивали в течение 1,5 ч при 0 С, затем добавляли новую порцию циклопропиламина (0,50 мл, 7,22 ммоль) и реакционную смесь перемешивали при К.Т. в течение еще 30 мин. Реакционную смесь сливали на смесь лед/Н 2 О(300 мл), перемешивали в течение 5 мин и твердое вещество светло-желтого цвета фильтровали, промывали несколько раз Н 2 О и сушили, получая N-бензилокси-N'-циклопропилтиомочевину (6,62 г). Указанную тиомочевину суспендировали в растворе 2 н. NaOH (50 мл) и выдерживали при температуре дефлегмации в течение 15 мин. Раствор охлаждали до К.Т., насыщали твердым NaCl и экстрагировали ЕtOАс(3 х). Объединенные EtOАс-экстракты промывали Н 2 О (2 х) и соляным раствором (1x), сушили (MgSO4),фильтровали и упаривали, получая неочищенный продукт в виде беловатого твердого вещества. Твердый продукт растирали в смеси гексан/EtOAc (5/5), получая N-циклопропилтиомочевину 8d в виде кристаллического твердого вещества белого цвета (2,5 г, выход 50%). 1 Н-ЯМP (400 МГц, ДМСО-d6):7,92 (bs, 1H), 7,61 (bs, 1H), 7,13 (bs, 1H), 2,39 (bs, 1 Н), 0,67-0,63 (m,2 Н), 0,51-0,44 (m, 2 Н). МС es+ 116,9 (М+Н)+; es-: 114,8 (М-Н)-. Пример 1. Получение соединения 100. Стадия 1. Синтез трипептида 10.(729 мг, 3,13 ммоль) и трифенилфосфин (1,1 г, 4,2 ммоль). Суспензию желтого цвета охлаждали в ледяной бане и по каплям добавляли ДИАД (821 мкл, 4,2 ммоль). Раствор перемешивали при температуре ледяной бани в течение 30 мин и при К.Т. в течение 16 ч. Раствор упаривали досуха и остаток растворяли в EtOАс, промывали насыщенным раствором бикарбоната натрия (2 х), водой (2 х) и соляным раствором(1x), сушили (MgSO4), фильтровали и упаривали, получая масло желтого цвета, которое осаждалось при выстаивании. Неочищенное твердое вещество суспендировали в ДХМ и нерастворившийся продукт отфильтровывали. Раствор концентрировали и остаток очищали экспресс-хроматографией с использованием смеси гексан:ЕtOАс (5:5) для удаления всех менее полярных примесей и смеси СНСl3:ЕtOАс (80:20) для элюирования всего Рh3 Р=O. Требуемое соединение элюировали с помощью смеси СНСl3:ЕtOАс(65:35), получая твердое вещество белого цвета (1 г, выход 70%). МС (электроспрей): 693,3 (М-Н)- 695,4 (М+Н)+ 717,4 (M+Na)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2 О): 99%. Стадия 2. Избирательный моногидролиз сложного эфира 10. Трипептид 10 (1 г, 1,44 ммоль) растворяли в ТГФ (10 мл) и добавляли МеОН (5 мл), воду (5 мл) и 1 н. водный раствор NaOH (1,5 мл) и раствор перемешивали при К.Т. в течение 2 ч. Смесь упаривали досуха и затем совместно упаривали со смесью МеОН:толуол (1:1; 4 х), толуолом (2 х) и диэтиловым эфиром (2x), получая продукт (безводный) в виде хлопьевидного твердого вещества белого цвета (1,04 г,выход 100%). МС (электроспрей): 679,3 (М-Н)- 681,3 (М+Н)+ 703,3 (M+Na)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2 О): 95%. Стадия 3. Синтез диазокетона 12. Натриевую соль 11 (примерно 1,44 ммоль) растворяли в ТГФ (16 мл), добавляли триэтиламин (301 мкл,2,16 ммоль) и раствор охлаждали до 0 С. Добавляли по каплям изобутилхлорформиат (280 мкл, 2,16 ммоль) и суспензию белого цвета перемешивали при 0 С в течение 75 мин, а затем добавляли раствор диазометана(0,67 М в диэтиловом эфире; 13 мл, 8,64 ммоль). Реакционную смесь перемешивали в течение 1 ч при 0 С, 45 мин при К.Т. и упаривали, получая густую суспензию. Эту суспензию растворяли в EtOAc и воде. Органический раствор промывали насыщенным NаНСО 3 (2 х), водой (2x) и соляным раствором (1 х),сушили (MgSO4), фильтровали и упаривали, получая диазокетон в виде твердого вещества цвета слоновой кости (неочищенный продукт использовали на следующей стадии; примерно 1,44 ммоль). МС (электроспрей): 703,3 (М-Н)- 705,3 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:H2O): 91%. Стадия 4. Синтез бромкетона 13. При 0 С к раствору диазокетона 12 (1,44 ммоль) в ТГФ (24 мл) добавляли по каплям раствор НВr(1,0 мл) и смесь перемешивали в течение 1 ч. Раствор разбавляли EtOAc, промывали насыщенным раствором NаНСО 3 (2x), водой (2 х) и соляным раствором (1x), сушили (MgSO4), фильтровали и упаривали,получая требуемый бромкетон в виде твердого вещества цвета слоновой кости с бежевым оттенком (1,1 г,примерно 1,44 ммоль). МС (электроспрей): 757,3 (М) 759,3 (М+2). Стадия 5. Синтез тиазолилтрипептида 14. Бромкетон 13 (0,40 ммоль) и N-циклопентилтиомочевину (68,5 мг, 0,48 ммоль) растворяли в изопро- 14007742 паноле (15 мл) и раствор желтого цвета выдерживали при 70 С в течение 75 мин. Раствору давали охладиться до К.Т. и упаривали досуха. Остаток растворяли в EtOAc. Раствор промывали насыщенным NаНСО 3(2x), водой (2x) и соляным раствором (1x), сушили (MgSO4), фильтровали и концентрировали, получая продукт в виде пены оранжево-коричневого цвета. С помощью колоночной экспресс-хроматографии с использованием указанной смеси гексан:ЕtOАс (7:3) удаляли менее полярные примеси, а с использованием смеси в соотношении 6:4 получали требуемое соединение в виде пены светло-желтого цвета (218 мг, 69%). МС (электроспрей): 801,4 (М-Н)- 803,4 (М+Н)+ 825 (M+Na)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2O): 99%. Стадия 6. Гидролиз сложного эфира 14. Раствор сложного метилового эфира 14 (145 мг, 0,181 ммоль) в ТГФ (3 мл), МеОН (1,5 мл) и водный раствор LiOH (75,8 мг, 1,81 ммоль) в воде (1,5 мл) перемешивали в течение 18 ч. Органический раствор концентрировали, получая беловатую суспензию, которую разбавляли EtOAc и соляным раствором до полного растворения. Значение рН доводили до 6, добавляя 1 н. НСl и органический слой экстрагировали дополнительно ЕtOАс (2 х). Объединенные органические экстракты промывали водой (2x), соляным раствором (1x), сушили (MgSO4), фильтровали и упаривали, получая требуемое соединение в виде твердого вещества желтого цвета (138,2 мг, выход 97%). Превращение в Na-соль. Соединение 100 (138,2 мг, 0,175 ммоля) растворяли в МеОН (30 мл) и добавляли 1 экв. 0,01 н. NaOH(17,5 мл). Прозрачный раствор желтого цвета концентрировали, удаляя МеОН, и разбавляли водой, замораживали и лиофилизировали, получая продукт (Na-соль) в виде аморфного твердого вещества желтого цвета (139 мг; теоретический выход: 142 мг; ММ Na-соли: 810,95). МС (электроспрей): 787,2 (М-Н)- 789,3 (М+Н)+ 811,3 (M+Na)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2O): 98%. 1 Н-ЯМР (400 МГц, ДМСО-d6): смесь ротамеров, соотношение примерно 5:1;8,14 (bs, 1H), 8,02 (d,J=9,2 Гц, 1H), 7,89 (d, J=6,7 Гц, 1H), 7,49-7,36 (m, 2H), 7,27 (bs, 1 Н), 7,06-6,96 (m, 2 Н), 6,10-5,90 (m, 1 Н),5,33 (s, 1H), 5,01 (d, J=16,8 Гц, 1H), 4,84 (d, J=10,6 Гц, 1H), 4,79-4,65 (m, 1 Н), 4,47-4,40 (m, 1H), 4,30 (d,J=11,5 Гц, 1 Н), 4,15 (d, J=8,8 Гц, 1H), 4,00-3,85 (m, 2 Н), 3,90 (s, 3H), 2,37-2,26 (m, 1 Н), 2,15-1,91 (m, 2 Н),1,80-1,23 (m, 18 Н), 0,96 и 0,86 (2 x s, 9H). Пример 2. Получение соединения 101. С помощью процесса, описанного в примере 1, используя на стадии 5 N-циклобутилтиомочевину 8b вместо N-циклопентилтиомочевины 8 а, получали соединение 101 в виде соли ТФК.(m, 1H), 7,01 (d, J=8,3 Гц, 1 Н), 5,78-5,66 (m, 2 Н), 5,20 (dd, J=17,0, 1,6 Гц, 1 Н), 5,09-5,04 (m, 1 Н), 4,53-4,36 (m, 4 Н),4,05-3,92 (m, 2 Н), 3,97 (s, 3 Н), 2,63-2,55 (m, 1 Н), 2,44-2,29 (m, 3 Н), 2,07-1,95 (m, 3 Н), 1,79-1,23 (m, 12 Н), 0,96 (s, 9H). МС (электроспрей): 773,4 (М-Н)- 775,4 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2O): 98%. Пример 3. Получение соединения 102. С помощью процесса, описанного в примере 1, и используя на стадии 5 N-циклогексилтиомочевину 8 с вместо N-циклопентилтиомочевины 8 а, получали соединение 102 в виде соли ТФК.(s, 3 Н), 2,64-2,55 (m, 1 Н), 2,38-2,27 (m, 1 Н), 2,05-1,95 (m, 3 Н), 1,79-1,71 (m, 2 Н), 1,66-1,19 (m, 16 Н), 0,96 (s, 9H). МС (электроспрей): 801,4 (М-Н)- 803,4 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2O): 99%. Пример 4. Получение соединения 103. Соединение 100 (30 мг, 0,038 ммоль) объединяли с ГАТУ (17 мг, 0,045 ммоль) и растворяли в безводном ДМФ (4 мл). Раствор перемешивали при К.Т., а затем по каплям в течение примерно 1 мин добавляли ДИПЭА (26 мкл, 0,15 ммоль). Смесь перемешивали в течение 60 мин при К.Т. и анализировали с помощью аналитической ЖХВР в отношении образования активированного сложного эфира. Затем добавляли раствор, содержащий бензолсулфонамид (23 мг, 0,15 ммоль), ДМАП (17 мг, 0,14 ммоль) и ДБУ(22 мкл, 0,15 ммоль) в ДМФ (1 мл). Реакционную смесь перемешивали в течение 24 ч при К.Т., затем сливали на EtOAc (50 мл) и промывали насыщенным раствором NаНСО 3 и насыщенным соляным раствором. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Остаток восстанавливали в ДМСО и очищали с помощью препаративной ЖХВР. После лиофилизации получали конечный продукт (17 мг, 48%) в виде аморфного твердого вещества светло-желтого цвета. 1 Н-ЯМР (400 МГц, ДМСО-d6):10,89 (s, 1H), 8,84 (s, 1H), 8,20 (d, J=8,8 Гц, 1 Н), 8,18-8,08 (m, 2 Н),7,89 (d, J=7,6 Гц, 2 Н), 7,70 (dd, J=7,6, 7,6 Гц, 2 Н), 7,58 (dd, J=7,7, 7,7 Гц, 2 Н), 7,28 (bs, 1 Н), 7,11 (d, J=6,6 Гц,1 Н), 5,75 (bs, 1H), 5,37-5,25 (m, 1 Н), 5,12 (d, J=17 Гц, 1 Н), 4,92 (d, J=12 Гц, 1 Н), 4,58-4,42 (m, 3 Н), 4,24 (bs,1 Н), 4,05 (d, J=7,8 Гц, 1 Н), 3,97 (s, 3 Н), 3,93 (d, J=7,8 Гц, 1 Н), 2,69-2,61 (m, 1H), 2,35-2,21 (m, 1H), 2,131,98 (m, 3 Н), 1,78-1,68 (m, 4 Н), 1,68-1,51 (m, 8 Н), 1,50-1,41 (m, 4 Н), 1,29-1,22 (m, 1 Н), 0,99 (s, 9H). МС (электроспрей): 928,5 (М+Н)+ и 926,5 (М-Н)-. ОФ-ЖХВР: Rt=7,3 мин (гомогенность=99%). Приведенные ниже соединения получали с помощью процесса синтеза, представленного на схеме 2. Пример 5. Стадия 1. Синтез дипептида 16. Дипептид 15 (4,0 г, 7,02 ммоль) растворяли в ТГФ (20 мл) и добавляли МеОН (10 мл), воду (10 мл) и 1 н. водный раствор NaOH (1,05 экв., 7,4 мл). Раствор перемешивали при К.Т. в течение 2,75 ч. Смесь упаривали досуха. Остаток разбавляли водой, замораживали и лиофилизировали, получая натриевую соль 16 в виде аморфного твердого вещества (4,28 г). МС (электроспрей): 554,2 (М-Н)- 556,3 (М+Н)+ 578,2 (M+Na)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2 О): 96%. Стадия 2. Синтез дипептиддиазокетона 17.(1,37 мл, 9,83 ммоль) и раствор охлаждали до 0 С. По каплям добавляли изобутилхлорформиат (1,28 мл,9,83 ммоль) и белую суспензию перемешивали при 0 С в течение 2 ч, затем добавляли раствор диазометана (0,67 М в диэтиловом эфире; 63 мл, 42,13 ммоль). Реакционную смесь перемешивали 1 ч при 0 С,1,25 ч при К.Т. и упаривали, получая густую суспензию. Указанную суспензию растворяли в EtOAc и воде. Органический раствор промывали насыщенным NаНСО 3 (2 х), водой (2 х) и соляным раствором(1x), сушили (MgSO4), фильтровали и упаривали, получая диазокетон 17 в виде твердого вещества бежевого цвета (неочищенный продукт использовали на следующей стадии; примерно 7,02 ммоль). МС (электроспрей): 578,2 (М-Н)- 580,3 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2O): 90%. Стадия 3. Синтез дипептидбромкетона 18. При 0 С к раствору диазокетона (приблизительно 1,44 ммоль) в ТГФ (116 мл) добавляли по каплям 48%-ный водный раствор НВr (5,1 мл) и смесь перемешивали в течение 2 ч. Раствор разбавляли EtOAc,промывали насыщенным раствором NaHCO3 (2x), водой (2 х) и соляным раствором (1x), сушили(MgSO4), фильтровали и упаривали, получая требуемый бромкетон в виде твердого вещества бежевого цвета (4,25 г, 6,72 ммоль). МС (электроспрей): 632 (М) 634,2 (М+2). Стадия 4. Синтез дипептида 19.-Бромкетон 18 (512 мг, 0,81 ммоль) и N-циклопентилтиомочевину (128,4 мг, 0,89 ммоль) растворяли в изопропаноле (20 мл). Образовавшийся раствор желтого цвета выдерживали при 70 С в течение 1,5 ч. Раствору давали охладиться до К.Т. и упаривали досуха. Остаток разбавляли ЕtOAc. EtOАсраствор промывали насыщенным NaHCO3 (2x), водой (2x) и соляным раствором (1), сушили (MgSO4),фильтровали и концентрировали, получая продукт в виде пены оранжево-коричневого цвета. С помощью колоночной экспресс-хроматографии с использованием смеси гексан:ЕtOАс (7:3) удаляли менее полярные примеси, а с использованием смеси 6:4 получали требуемое соединение в виде твердого вещества светло-желтого цвета (411,5 мг, 75%). МС (электроспрей): 676,3 (М-Н)- 678,3 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2O): 99%. Стадия 5. Синтез трипептида 20. Защищенный с помощью Воc дипептид (32,6 г, 0,048 ммоль) растворяли в смеси 4 н. НСl/диоксан (3 мл) и перемешивали при К.Т. Через 2 ч реакционную смесь обрабатывали, упаривая досуха. Получали гидрохлорид в виде беловатого твердого вещества. Гидрохлорид помещали в глубокий вакуум на 30 мин. К раствору гидрохлорида в ДХМ (2 мл) и ДИЭА (33 мкл, 0,192 ммоль) добавляли мочевину 5 (13,96 мг,0,058 ммоль), а затем связывающий агент ГАТУ (21,90 мг, 0,058 ммоль). Реакционную смесь перемешивали при К.Т. в течение 3 ч. Реакционную смесь разбавляли ЕtOАс, промывали насыщенным NаНСО 3(2x), водой (2 х) и соляным раствором (1x), сушили (MgSO4), фильтровали и упаривали, получая неочищенный продукт в виде густого масла желтого цвета (приблизительно 0,048 ммоль). МС (электроспрей): 800,4 (М-Н)- 802,4 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2O): 95%. Стадия 6. Гидролиз сложного метилового эфира трипептида 20. Раствор сложного метилового эфира 20 (77,80 мг, 0,097 ммоль) в ТГФ (2 мл) и МеОН (1 мл) и водный раствор LiOH (40,7 мг, 0,97 ммоль) в воде (1 мл) перемешивали в течение 16 ч. Органический раствор концентрировали, получая беловатую суспензию. Неочищенный продукт очищали с помощью препаративной ЖХВР (YMC Combiscreen ODS-AQ, 5020 мм, ID S-5 мкм,120 ; =220 нм), используя линейный градиент и 0,06% ТФК CH3CN/H2O. Чистые фракции объединяли, концентрировали и превращали в натриевую соль. Превращение в Na-соль. Концентрированные фракции разбавляли EtOAc и несколько мл соляного раствора (который подщелачивали до рН 13 с помощью 5 н. NaOH, затем нейтрализовали до рН 5,5-6,0 с помощью 1 н. НСl). Продукт экстрагировали EtOAc (3 х), промывали водой (2 х) и соляным раствором (1 х), сушили (MgSO4),фильтровали и упаривали досуха, получая нейтральный продукт в виде твердого вещества желтого цвета(52,7 мг, 70%). Нейтральный продукт (49,4 мг, 0,0627 ммоль) растворяли в МеОН (10 мл) и добавляли 1 экв. 0,01 н. NaOH (6,27 мл). Прозрачный раствор желтого цвета концентрировали для удаления МеОН и разбавляли водой, замораживали и лиофилизировали, получая продукт (Na-соль) в виде аморфного твердого вещества желтого цвета (50,8 мг; теоретический выход: 50,8 мг; ММ Na-соли: 809,75). 1 Н-ЯМР (400 МГц, ДМСО-d6): смесь ротамеров, соотношение примерно 8:1;8,20 (bs, 1H), 7,96 (d,J=8,8 Гц, 1 Н), 7,86 (d, J=5,7 Гц, 1 Н), 7,51-7,47 (m, 2H), 7,26 (s, 1 Н), 6,97 (d, J=8,6 Гц, 1 Н), 6,19-6,02 (m,1H), 5,33 (bs, 1H), 4,99 (d, J=16,8 Гц, 1H), 4,77 (d, J=10,0 Гц, 1H), 4,52-4,41 (m, 2 Н), 4,34 (d, J=11,0 Гц, 1H),4,04-3,96 (m, 2 Н), 3,89 (s, 3 Н), 3,79-3,68 (m, 1 Н), 3,68-3,15 (под пиком, соответствующим воде, 2 Н), 2,452,36 (m, 1 Н), 2,05-1,92 (m, 2 Н), 1,82-1,35 (m, 16 Н), 1,35-1,12 (m, 2 Н), 0,91 и 0,84 (2 x s, 9H). МС (электроспрей): 786,4 (М-Н)- 788,3 (М+Н)+ 810 (M+Na)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:Н 2O): 99%. Пример 6. Соединение 104. С помощью процесса, описанного в примере 5, используя на стадии 5 циклобутилкарбамат третбутилглицина вместо мочевины 5 и осуществляя очистку неочищенной карбоновой кислоты после стадии 6 с помощью препаративной ЖХВР, получали указанное в заголовке соединение в виде соли ТФК.(m, 1 Н), 4,48-4,41 (m, 1 Н), 4,29 (d, J=11,7 Гц, 1 Н), 4,12 (d, J=8,6 Гц, 1H), 3,97-3,85 (m, 2 Н), 3,91 (s, 3 Н), 2,362,27 (m, 1H), 2,18-2,04 (m, 2 Н), 2,03-1,81 (m, 6 Н), 1,77-1,43 (m, 9 Н), 1,41-1,34 (m, 1H), 0,96 и 0,85 (2 x s, 9H). МС (электроспрей): 773,3 (М-Н)- 775.4 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:H2O): 99%. Пример 7. Соединение 105. С помощью процесса, описанного в примере 5, используя на стадии 5 циклогексилкарбамат третбутилглицина вместо мочевины 5, получали указанное в заголовке соединение в виде натриевой соли.(m, 1 Н), 2,04-1,91 (m, 5 Н), 1,79-1,41 (m, 10 Н), 1,39-1,08 (m, 7 Н), 0,97 и 0,86 (2 x s, 9H). МС (электроспрей): 801,4 (М-Н)- 803,4 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:H2O): 98%. Пример 8. Соединение 106. С помощью процесса, описанного в примере 5, и используя на стадии 5 циклопентилкарбамат циклогексилглицина вместо мочевины 5, получали указанное в заголовке соединение в виде натриевой соли. 1 Н-ЯМР (400 МГц, ДМСО-d6): смесь ротамеров, соотношение примерно 1:4;8,22 и 8,04 (2 x s,1H), 8,00 (d, J=9,2 Гц, 1H), 7,91 (d, J=6,5 Гц, 1 Н), 7,46 (s, 1H), 7,41 (s, 1H), 7,27 (d, J=2,4 Гц, 1H), 7,20 (d,J=8,4 Гц, 1H), 7,01 (dd, J=2,4, 9,0 Гц, 1H), 6,09-5,98 (m, 1H), 5,34 (s, 1H), 4,98 (dd, J=1,6, 17,4 Гц, 1 Н), 4,80(d, J=11,9 Гц, 1H), 4,73-4,67 (m, 1 Н), 4,43-4,31 (m, 2 Н), 4,05-3,95 (m, 2 Н), 3,95-3,84 (m, 1H), 3,90 (s, 3 Н),2,38-2,29 (m, 1H), 2,03-1,92 (m, 2 Н), 1,83-1,21 (m, 24 Н), 1,21-0,83 (m, 5 Н). МС (электроспрей): 813,4 (М-Н)- 815,4 (М+Н)+. Гомогенность по данным ЖХВР с обращенной фазой (0,06% ТФК; CH3CN:H2O): 99%. Пример 9. Соединение 107. С помощью процесса, описанного в примере 5, и используя на стадии 5 циклопентилкарбамат циклопентилглицина вместо мочевины 5, получали указанное в заголовке соединение, которое превращали в соответствующую Na-соль. Пример 10. Анализ NS3-NS4A-протеазы. Ферментативный анализ, который применяли для оценки соединений, предлагаемых в настоящем изобретении, описан в WO 00/09543 и WO 00/59929.- 19007742 Пример 11. Анализ репликации РНК HCV на культуре клеток. Культура клеток. Клетки линии Huh7, стабильно поддерживающие субгеномный репликон HCV, создавали согласно описанному ранее методу (Lohman и др., Science 285, 1999, сс. 110-113) и обозначали их как клеточная линия S22.3. Клетки линии S22.3 поддерживали в модифицированной по методу Дульбекко среде Игла(DMEM), дополненной 10% ФБС и 1 мг/мл неомицина (стандартная среда). В процессе анализа использовали среду DMEM, дополненную 10% ФБС, содержащую 0,5% ДМСО и не содержащую неомицин(среда для анализа). За 16 ч до внесения соединения клетки линии S22.3 обрабатывали трипсином и разбавляли до 50000 клеток/мл стандартной средой. По 200 мкл (10000 клеток) вносили в каждую лунку 96 луночного планшета. Затем планшет инкубировали при 37 в атмосфере с 5% СО 2 до следующего дня. Реагенты и материалы. Подготовка тестируемого соединения к анализу. 10 мкл тестируемого соединения (в 100% ДМСО) добавляли к 2 мл среды для анализа до конечной концентрации ДМСО 0,5% и раствор обрабатывали ультразвуком в течение 15 мин и фильтровали через фильтр типа Millipore с размером отверстий 0,22 мкм. 900 мкл вносили в ряд А титрационного планшета из полипропилена с глубокими лунками. Ряды Б-3 содержали аликвоты по 400 мкм среды для анализа(содержащей 0,5% ДМСО) и их применяли для приготовления серийных разведений (1/2) путем переноса по 400 мкл из ряда в ряд (в ряд 3 не вносили никакого соединения). Внесение тестируемого соединения в культуру клеток. Среду для культуры клеток аспирировали из 96-луночного планшета, содержащего клетки линииS22.3. По 175 мкл среды для анализа с соответствующим образом разбавленным тестируемым соединением переносили из каждой лунки планшета, содержащего соединения, в соответствующую лунку планшета, содержащего культуру клеток (ряд 3 применяли в качестве контроля без ингибитора). Планшет с культурой клеток инкубировали при 37 в атмосфере с 5% СО 2 в течение 72 ч. Экстракция общей клеточной РНК. После 72-часового периода инкубации экстрагировали общую клеточную РНК из клеток линии S22.3,находящихся в 96-луночном планшете, используя набор типа RNeasy 96 (Qiagen, RNeasy Handbook,1999). В целом, метод состоит в следующем: среду для анализа полностью удаляли из клеток и в каждую лунку 96-луночного планшета с культурой клеток добавляли по 100 мкл RLT-буфера (Qiagen), содержащего 143 мМ -меркаптоэтанол. Микропланшет осторожно встряхивали в течение 20 с. Затем в каждую лунку микропланшета добавляли по 100 мкл 70%-ного этанола и перемешивали пипетированием. Лизат удаляли и вносили в лунки планшета, содержащего RNeasy 96 (Qiagen), который помещали в верхнюю часть блока с квадратными лунками (Qiagen Square-Well Block). Планшет с RNeasy 96 запечатывали скотчем и устройство Square-Well Block с содержащим RNeasy 96 планшетом помещали в держатель и вносили в роторный резервуар центрифуги 4 К 15 С. Образец центрифугировали при 6000 об./мин(5600 х g) в течение 4 мин при комнатной температуре. Скотч удаляли с планшета и в каждую лунку планшета с RNeasy 96 вносили по 0,8 мл RW1-буфера (Qiagen набор RNeasy 96). Содержащий RNeasy 96 планшет запечатывали новым куском скотча и центрифугировали при 6000 об./мин в течение 4 мин при комнатной температуре. Содержащий RNeasy 96 планшет помещали в верхнюю часть другого чистого блока с квадратными лунками (типа Square-Well Block), скотч удаляли и в каждую лунку содержащего RNeasy 96 планшета вносили по 0,8 мл RPE-буфера (Qiagen набор RNeasy 96). СодержащийRNeasy 96 планшет запечатывали новым куском скотча и центрифугировали при 6000 об./мин в течение 4 мин при комнатной температуре. Скотч удаляли и в каждую лунку содержащего RNeasy 96 планшета- 20007742 вносили еще по 0,8 мл RPE-буфера (Qiagen набор RNeasy). Содержащий RNeasy 96 планшет запечатывали новым куском скотча и центрифугировали при 6000 об./мин в течение 10 мин при комнатной температуре. Скотч удаляли, содержащий RNeasy 96 планшет помещали в верхнюю часть адаптора, содержащего предназначенные для сбора образцов микроцентрифужные пробирки объемом 1,2 мл. РНК элюировали, добавляя в каждую лунку по 50 мкл свободной от РНКазы воды, запечатывая планшет новым куском скотча и инкубируя в течение 1 мин при комнатной температуре. Планшет затем центрифугировали при 6000 об./мин в течение 4 мин при комнатной температуре. Стадию элюирования повторяли, используя второй объем, равный 50 мкл свободной от РНКазы воды. Микроцентрифужные пробирки с общей клеточной РНК хранили при -70. Количественная оценка общей клеточной РНК. РНК оценивали количественно с помощью системы STORM (Molecular Dynamics), используя набор для количественной оценки РНК типа RiboGreen (Molecular Probes). В целом, метод состоит в следующем: реагент RiboGreen разбавляли в 200 раз ТЭ (10 мМ Трис-HCl рН=7,5, 1 мМ ЭДТК). Как правило, 50 мкл реагента разбавляли 10 мл ТЭ. Образцы рибосомной РНК для построения стандартной кривой разбавляли в ТЭ до 2 мкл/мл и затем предварительно определенные количества (100, 50, 40, 20, 10, 5, 2 и 0 мкл) раствора рибосомной РНК переносили в новый 96-луночный планшет (COSTAR3997) и объем доводили до 100 мкл с помощью ТЭ. Как правило, колонку 1 96-луночного планшета использовали для построения стандартной кривой, а другие лунки использовали для количественной оценки образцов РНК. По 10 мкл каждого образца РНК, подлежащего количественной оценке, переносили в соответствующую лунку 96-луночного планшета и добавляли по 90 мкл ТЭ. В каждую лунку 96-луночного планшета вносили по одному объему (100 мкл) разбавленного реагента RiboGreen и инкубировали в течение 2-5 мин при комнатной температуре, защищая от света (10 мкл образца РНК в 200 мкл конечного объема,т.е. 20-кратное разбавление). Интенсивность флуоросценции каждой лунки оценивали с помощью системы STORM (Molecular Dynamics). Стандартные кривые получали на основе известных количеств рибосомной РНК и соответствующей им интенсивности флуоресценции. Концентрацию РНК в экспериментальных образцах определяли, исходя из стандартной кривой, и корректировали с учетом 20 кратного разбавления. Реагенты и материалы. ОТ-ПЦР, проводимая в реальном времени. ОТ-ПЦР в реальном времени осуществляли с помощью системы для анализа последовательностиBiosystems). ОТ-ПЦР оптимизировали для количественной оценка 5'-IRES РНК HCV с помощью технологии Taqman (фирма Roche Molecular Diagnostics Systems), которая аналогична методу, описанному ранее у Martell и др. J. Clin. Microbiol. 37, 1999, сс. 327-332. Система основана на 5'3' нуклеолитической активности ДНК-полимеразы AmpliTaq. В целом, метод состоит в следующем: для его осуществления использовали фторогенный гибридизующийся зонд с двойной меткой (PUTR-зонд), который специфично ренатурирует матрицу между ПЦР-праймерами (праймеры 8125 и 7028). 5'-конец зонда содержит флуоресцентный репортер (6-карбоксифлуоресцин [FAM]), а 3'-конец содержит гаситель флуоресценции (6 карбокситетраметилродамин [TAMRA]). Спектр испускания репортера FAM подавляли гасителем на неповрежденном гибридизующемся зонде. Расщепление гибридизующегося зонда высвобождало репортер, что приводило к повышению испускания флуоресценции. С помощью анализатора последовательности типа ABI Prism 7700 непрерывно осуществляли определение повышения испускания флуоресценции в процессе ПЦР-амплификации, при этом количество амплифицированного продукта прямо пропорционально сигналу. Зависимость амплификации от времени анализировали на ранней стадии реакции, в момент времени, соответствующий логарифмической фазе накопления продукта. Точку, представляющую собой определенный порог обнаружения повышения сигнала флуоресценции, связанного с экспоненциальным увеличением количества ПЦР-продукта для анализатора последовательности, обозначали как порог цикла (СT). Значения CT обратно пропорциональны количеству введенной РНК HCV; т.е. при оди- 21007742 наковых условиях ПЦР чем больше исходная концентрация РНК HCV, тем ниже значение СT. Стандартную кривую строили автоматически с помощью системы обнаружения ABI Prism 7700 путем построения графика зависимости СT от каждого стандартного разведения с известной концентрацией РНК HCV. В каждый планшет для осуществления ОТ-ПЦР вносили эталонные образцы для стандартной кривой. РНК репликона HCV синтезировали (посредством транскрипции Т 7) in vitro, очищали и количественно оценивали по величине ОП 260. С учетом того, что в 1 мкг этой РНК содержится 2,151011 копий РНК, осуществляли такие разведения, чтобы получать 108,107,106,105,104,103 или 102 копий геномной РНК/5 мкл. В каждое разведение включали также общую клеточную РНК линии Huh-7 (50 нг/5 мкл). 5 мкл каждого эталонного образца (РНК репликона + РНК Huh-7) объединяли с 45 мкл реагента Mix и применяли в проводимой в реальном времени ОТ-ПЦР. Проводимую в реальном времени ОТ-ПЦР осуществляли также с использованием экспериментальных образцов, которые очищали в содержащих RNeasy 96 лунках планшета, объединяя 5 мкл каждого образца общей клеточной РНК с 45 мкл реагента Mix. Реагенты и материалы. Состав для приготовления реагента Mix. Последовательность прямого праймера (SEQ ID. 1): 5'-ACG CAG AAA GCG ТСТ AGC CAT GGC GTT AGT-3'. Последовательность обратного праймера (SEQ ID NO. 2): 5'-TCC CGG GGC ACT CGC AAG САС ССТ АТС AGG-3'. Примечание: эти праймеры обеспечивают амплификацию области, состоящей из 256 нуклеотидов,которая присутствует в 5'-нетранслируемой области HCV. Последовательность PUTR-зонда (SEQ ID NO. 3): Несодержащие матрицы контроли (НМК): На каждом планшете по 4 лунки использовали в качестве НМК. Для получения таких контролей в каждую лунку вместо РНК вносили 5 мкл воды. Условия проведения реакции в термоячейке: После завершения ОТ-ПЦР для анализа данных необходимо определять порог обнаружения сигнала флуоресценции для ПЦР-планшета, и для этого получали стандартную кривую путем построения графика зависимости значения Ct от количества копий РНК, применяемых в каждой эталонной реакции. Значения Ct, полученные для анализируемых образцов, использовали для определения путем интерполяции количества копий РНК на основе стандартной кривой. И, наконец, количество копий РНК стандартизовали (на основе количественной оценки РНК, экстрагируемой из лунки с клеточной культурой, с помощью набора RiboGreen RNA) и выражали в виде геномных эквивалентов/мкг общей РНК [г.э./мкг]. Количество копий РНК [г.э./мкг] для каждой лунки планшета с культурой клеток принимали за меру количества реплицирующейся РНК HCV в присутствии различных концентраций ингибитора. Ингибирование в % рассчитывали с помощью следующего уравнения: 100-[(г.э./мкг инг.)/(г.э./мкг контр.)100] Для обработки данных зависимости ингибирования от концентрации применяли основанную на модели Хилла аппроксимацию нелинейной кривой, и значения концентрации, обеспечивающей 50%-ную эффективность (50%-ное ингибирование) (ЕС 50) рассчитывали с помощью программного обеспеченияSAS (Statistical Software System; SAS Institute, Inc. Кэри, шт.Северная Каролина). При оценке соединений,предлагаемых в настоящем изобретении, с помощью описанных выше ферментативных анализов и анализов клеточных культур, выявлена их высокая активность. Более конкретно, установлено, что значенияIC50 соединений составляли менее 0,1 мкМ при анализе NS3-NS4A-протеазы, и значения ЕС 50 составляли менее 0,5 мкМ при анализе репликации РНК HCV на культуре клеток. Пример 12. Анализы специфичности. Анализы специфичности, применяемые для оценки избирательности действия указанного соединения, соответствуют описанным в WO 00/09543. Когда соединения оценивали в опытах по определению специфичности, то было установлено, что соединения формулы (I) обладали селективностью, т.е. они не вызывали существенного ингибирования эластазы лейкоцитов человека и катепсина В. Пример 13. Фармакокинетические свойства. Соединения, предлагаемые в настоящем изобретении, обладают также удовлетворительными фармакокинетическими свойствами, такими как значительные уровни в плазме крыс через 1 и 2 ч после орального введения дозы 5 мг/кг. Более конкретно, для оценки уровней в плазме крыс тестируемых соединений после орального введения применяли следующий анализ, основанный на оценке in vivo абсорбции после орального введения: Материалы и методы. 1. Метод, применяемый для группировки соединений (отбор с помощью кассет). Отбор соединений, предназначенных для объединения в кассету, был основан на их структурном сходстве и физикохимических свойствах. Был разработан метод твердофазной экстракции, применимый для всех отобранных соединений. Данные начального тестирования, при котором каждое соединение вводили в плазму крыс и с помощью ЖХВР или ЖХВР-МС анализировали каждое соединение, взятое в концентрации 0,5 мкМ, с определением времени удерживания, ионной массы и возможности разделения соединений с помощью ЖХВР и/или ЖХВР/МС, использовали для объединения 3-4 соединений в одну кассету. 2. Подготовка носителя и соединения для орального введения. Каждая кассета содержала 3-4 соединения, каждое в концентрации 5 или 4 мг/кг. Кассету приготавливали в виде суспензии для орального введения, содержащей 0,5% водной метилцеллюлозы и 0,3% полиоксиэтилен(20)сорбитанмоноолеата (Твин-80). Объем вводимой дозы составлял 10 мл/кг и ее вводили через оральный желудочный зонд. 3. Применяемые дозы и отбор образцов плазмы. Самцов крыс Sprague Dawley содержали в индивидуальных клетках в течение ночи, и животные имели доступ к водной 10%-ной декстрозе.Каждую кассету вводили 2 крысам. Образцы плазмы (1 мл) получали через 1 и 2 ч после обработки у каждой из двух крыс и объединяли для экстракции и анализа. 4. Экстракция и анализ соединений. Для каждой кассеты образцы плазмы, полученные через 1 и 2 ч, образцы контрольной плазмы, образцы контрольной плазмы, в которую вводили все соединения, каждое в концентрации 0,5 мкМ, экстрагировали с помощью метода твердофазной экстракции. Для сравнения образцы анализировали с помощью ЖХВР и ЖХВР/- 23007742 МС. Концентрации в плазме определяли на основе одной концентрации стандарта, составляющей 0,5 мкМ. При оценке предлагаемых в настоящем изобретении соединений, описанных в примерах 1-9, с помощью указанного выше метода отбора выявлены их высокие уровни в плазме через 1 и 2 ч после орального введения, при этом усредненные уровни в плазме крови составляли 1,23 и 1,16 мкМ соответственно. Эти результаты демонстрируют, что предлагаемые в настоящем изобретении соединения обладали выраженным уровнем абсорбции in vivo после орального введения в сравнении с низким уровнем абсорбции после орального введения, характерным в целом для этого класса пептидов. Способность к абсорбции после орального введения делает указанные соединения перспективными для лечения инфекции, вызываемой HCV. Ниже в таблице представлены репрезентативные предлагаемые в изобретении соединения. В дополнении к указанному ранее в описании следует отметить, что для всех соединений значения IC50 составляли менее 0,1 мкМ при анализе NS3-NS4A-протеазы, и значения ЕС 50 составляли менее 0,5 мкМ при анализе репликации РНК HCV на культуре клеток. Таблица 1 в котором R1 обозначает гидрокси или NHSO2Ph; R2 обозначает С 4-С 6 циклоалкил; R3 обозначает третбутил или С 5-С 6 циклоалкил и R4 обозначает С 4-С 6 циклоалкил; или его фармацевтически приемлемая соль. 2. Соединение формулы (I) по п.1, в котором R1 обозначает NHSO2Ph. 3. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси. 4. Соединение формулы (I) по одному из пп.1-3, в котором R2 обозначает циклопентил или циклогексил. 5. Соединение формулы (I) по п.4, в котором R2 обозначает циклопентил. 6. Соединение формулы (I) по одному из пп.1-5, в котором R3 обозначает трет-бутил или циклогексил. 7. Соединение формулы (I) по п.6, в котором R3 обозначает трет-бутил. 8. Соединение формулы (I) по одному из пп.1-7, в котором R4 обозначает циклобутил или циклопентил. 9. Соединение формулы (I) по п.8, в котором R4 обозначает циклопентил. 10. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 и R4 каждый обозначает циклопентил и R3 обозначает трет-бутил. 11. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 обозначает циклобутил,3R обозначает трет-бутил и R4 обозначает циклопентил. 13. Соединение формулы (I) по п.1, в котором R1 обозначает NHSO2Ph, R2 и R4 каждый обозначает циклопентил и R3 обозначает трет-бутил. 14. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 обозначает циклопентил,3R обозначает тpeт-бутил и R4 обозначает циклогексил. 16. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2 и R4 каждый обозначает циклопентил и R3 обозначает циклогексил. 17. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси, R2, R3 и R4 каждый обозначает циклопентил. 18. Способ лечения или предупреждения инфекции, вызываемой вирусом гепатита С у млекопитающего, заключающийся в том, что млекопитающему вводят эффективное в отношении вируса гепатита С количество соединения формулы (I) по одному из пп.1-17 или его фармацевтически приемлемой соли. 19. Способ лечения или предупреждения инфекции, вызываемой вирусом гепатита С у млекопитающего, заключающийся в том, что млекопитающему вводят эффективное в отношении вируса гепатита С количество соединения формулы (I) по одному из пп.1-17 или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемой средой носителя или вспомогательным веществом. 20. Способ лечения или предупреждения инфекции, вызываемой вирусом гепатита С у млекопитающего, заключающийся в том, что млекопитающему вводят эффективное в отношении вируса гепатита С количество соединения формулы (I) по одному из пп.1-17 или его терапевтически приемлемой соли в сочетании с одним или несколькими другими агентами, обладающими активностью в отношении HCV. 21. Способ по п.20, заключающийся в том, что другой агент, обладающий активностью в отношении HCV, выбирают из -интерферона или пэгилированного -интерферона. 22. Способ по п.20 или п.21, заключающийся в том, что другой агент, обладающий активностью в отношении HCV, представляет собой рибавирин. 23. Способ по одному из пп.20-22, заключающийся в том, что другой агент, обладающий активностью в отношении HCV, выбирают из ингибиторов геликазы, NS2/3-протеазы и внутреннего сайта входа в рибосому (IRES).- 26007742 24. Способ по одному из пп.20-22, заключающийся в том, что другой агент, обладающий активностью в отношении HCV, представляет собой ингибитор HCV-полимеразы. 25. Применение соединения формулы I по одному из пп.1-17 для приготовления лекарственного средства, предназначенного для лечения или предупреждения инфекции, вызываемой вирусом гепатита С.
МПК / Метки
МПК: A61P 31/14, C07K 5/08
Метки: трипептиды, замещённого, гидроксипролиновый, простой, эфир, протеазы, несущие, гепатит, ингибирования, предназначенные, хинолина
Код ссылки
<a href="https://eas.patents.su/28-7742-tripeptidy-nesushhie-prostojj-gidroksiprolinovyjj-efir-zameshhyonnogo-hinolina-prednaznachennye-dlya-ingibirovaniya-proteazy-ns3-gepatit-s.html" rel="bookmark" title="База патентов Евразийского Союза">Трипептиды, несущие простой гидроксипролиновый эфир замещённого хинолина, предназначенные для ингибирования протеазы ns3 (гепатит с)</a>
Соединения пиразинона, фармацевтическая композиция (варианты) и лекарственная форма, способ ингибирования сериновой протеазы, способ снижения тромбообразующей способности поверхности, способ лечения нарушенного протеолиза и тромбоза, связанного с различными заболеваниями, способ снижения свертывания крови у млекопитающего

Номер патента: 4884
Опубликовано: 26.08.2004
Авторы: Лу Тианбао, Томкзук Брюс Э., Маркоутэн Томас П.
МПК: A61K 38/05, C07K 5/078, A61P 7/02...
Метки: протеазы, фармацевтическая, различными, связанного, нарушенного, пиразинона, сериновой, свертывания, способ, ингибирования, протеолиза, снижения, форма, способности, лечения, варианты, тромбообразующей, млекопитающего, крови, заболеваниями, композиция, соединения, поверхности, лекарственная, тромбоза
Формула / Реферат:
1. Соединение формулы I или его сольват, гидрат или фармацевтически приемлемая соль, где W означает R1; R1 означает R2, R2(CH2)tC(R12)2, где t равно 0-3 и оба R12 одинаковые или различные, (R2)(OR12)CH(CH2)p, где p равно 1-4, R2C(R12)2(CH2)t, где t равно 1-3 и оба R12 одинаковые или различные, причем (R12)2 может быть объединен с атомом углерода, к которому он присоединен, с образованием от 3- до 7-членного циклоалкильного кольца; ...
Гетероциклические трипептиды в качестве ингибиторов вируса гепатита с

Номер патента: 7739
Опубликовано: 29.12.2006
Авторы: Ллина-Брюне Монсе, Гиро Элиза, Бейли Марри Д.
МПК: A61P 31/00, A61K 31/47, C07D 417/14...
Метки: вируса, трипептиды, качестве, ингибиторов, гепатита, гетероциклические
Формула / Реферат:
1. Соединение формулы (I) в котором R1 обозначает гидрокси или NHSO2R1A, где R1A обозначает C1-С8алкил, С3-С7циклоалкил или С1-С6алкил-С3-С7циклоалкил, которые все необязательно замещены 1-3 заместителями, выбранными из ряда, включающего галоген, циано, нитро, OC1-С6алкил, амидо, амино или фенил, или R1A обозначает C6- или С10арил, необязательно замещенный 1-3 заместителями, выбранными из ряда, включающего галоген, циано, нитро, С1-С6алкил,...
Фармацевтически приемлемые тельца, несущие фосфат глицерина

Номер патента: 7426
Опубликовано: 27.10.2006
Авторы: Мэндел Аркадий, Болтон Энтони
МПК: A61K 31/683, A61K 9/127
Метки: тельца, фосфат, глицерина, несущие, фармацевтически, приемлемые
Формула / Реферат:
1. Фармацевтическая композиция в форме разовых дозировок для введения пациенту-млекопитающему, содержащая фармацевтически приемлемые тельца и фармацевтически приемлемый носитель, характеризующаяся тем, что по меньшей мере часть телец имеет размер в интервале приблизительно от 20 нм до 500 мкм, и при этом поверхности указанных телец содержат фосфоглицеридные группы или группы, способные к преобразованию в фосфоглицеридные группы, а указанная...
Композиция, включающая ингибитор il-1 и гидрогелевый эфир, и способы ее применения

Номер патента: 1792
Опубликовано: 27.08.2001
Авторы: Бевилаква Майкл П., Коллинс Дэвид С.
МПК: A61K 47/36, A61P 29/00
Метки: способы, эфир, композиция, применения, ингибитор, гидрогелевый, включающая
Формула / Реферат:
1. Фармацевтическая композиция, включающая гидрогелевый эфир, контролирующий высвобождение, и белковый ингибитор интерлейкина-1 (IL-1). 2. Композиция по п.1, отличающаяся тем, что гидрогелевый эфир является гиалуронаном или его солью. 3. Композиция по п.2, отличающаяся тем, что гиалуронан является гиалуроновой кислотой. 4. Композиция по п.2, отличающаяся тем, что гиалуронан является гиланом. 5. Композиция по п.2, отличающаяся тем, что гиалуронан...
Производные хинолина

Номер патента: 2124
Опубликовано: 24.12.2001
Авторы: Равелиа Лука Франческо, Гьярдина Джузеппе Арнальдо Мариа, Фарино Карло, Груни Марио
МПК: A61P 25/00, C07D 215/52, A61K 31/47...
Метки: производные, хинолина
Формула / Реферат:
1. Соединение формулы (I) или его соль, или его сольват, где Аr представляет собой фенил; R представляет собой C1-C6 алкил; R1 представляет собой водород; R2 представляет собой группу -O-(СН2)n-Х, где Х представляет собой C1-С6 алкил, необязательно замещенный одной или двумя группами, выбранными из гидрокси и амино; карбокси, циано, C1-6 алкоксикарбонила, аминокарбонила, моно- или ди-С1-6 алкиламинокарбонила, амино-С1-6 алкиламинокарбонила...
Предыдущий патент: Способ получения 2-(замещенный фенил)-2-гидроксиэтилкарбаматов
Следующий патент: Способ получения производных 1,3,4,5 – тетрагидро -2н-3- бензазепин -2- она
Случайный патент: Конъюгаты e-полилизина и их применение