Бензоилсульфонамиды и сульфонилбензамидины для применения в качестве противоопухолевых агентов
Номер патента: 5810
Опубликовано: 30.06.2005
Авторы: Лин Хо-Шен, Хипскинд Филип Артур, Корбетт Томас Хьюз, Лобб Карен Линн, Гроссмэн Кора Сью, Ших Чуан


















Формула / Реферат
1. Соединение формулы I

где X представляет собой O или NH;
R1 представляет собой водород, галоген, C1-C6алкил, C1-C4алкокси, C1-C4алкилтио, CF3, OCF3, SCF3, (C1-C4алкокси)карбонил, нитро, азидо, O(SO2)CH3, N(CH3)2, гидрокси, фенил, замещенный фенил, пиридинил, тиенил, фурил, хинолинил или триазолил;
R2 представляет собой водород, галоген, циано, CF3, C1-C6алкил, (C1-C4алкокси)карбонил, C1-C4алкокси, фенил или хинолинил;
R2a представляет собой водород или C1-C4алкокси;
R2b представляет собой водород или C1-C6алкил при условии, что по крайней мере один из заместителей R2a и R2b является водородом;
R3 представляет собой водород, галоген, C1-C6алкил, CF3 или нитро;
R3a представляет собой водород, галоген или C1-C6алкил при условии, что если R3a является C1-C6алкилом, то R3 является водородом и R4 является галогеном; и
R4 представляет собой галоген, C1-C6алкил или CF3 при условии, что только один из заместителей R3 и R4 может являться C1-C6алкилом, и при условии, что если R4 является галогеном или C1-C6алкилом, то только один из заместителей R3 и R3a является водородом;
где вышеуказанный замещенный фенил обозначает монозамещенный фенил, где заместитель выбирают из группы, состоящей из C1-C4алкокси, C1-C4алкилтио, C1-C4ацила, трифторметила и галогена;
или его фармацевтически приемлемая соль присоединения основания, при условии, что
a) если оба заместителя R3 и R4 являются хлором и R2 является водородом, R1 является бромом, йодом, C1-C4алкокси, C1-C4алкилтио, CF3, OCF3, нитро, азидо, O(SO2)CH3, N(CH3)2, гидрокси, фенилом, замещенным фенилом, пиридинилом, тиенилом, фурилом или триазолилом;
b) если оба заместителя R3 и R4 являются хлором и R1 является водородом, R2 является бромом, фтором, CF3, C1-C6алкилом, C1-C4алкокси, фенилом или хинолинилом.
2. Соединение по п.1, где R2, R2a и R2b представляет собой водород и R1 выбирают из группы, состоящей из водорода, галогена, C1-C6алкила, C1-C4алкокси, C1-C4алкилтио, CF3, OCF3, SCF3, (C1-C4алкокси)карбонила, нитро, азидо, O(SO2)CH3, N(CH3)2, гидрокси, фенила, замещенного фенила, пиридинила, тиенила, фурила, хинолинила или триазолила, где вышеуказанный замещенный фенил обозначает монозамещенный фенил, где заместитель выбирают из группы, состоящей из C1-C4алкокси, C1-C4алкилтио, C1-C4ацила, трифторметила и галогена.
3. Соединение по п.1 или 2, представляющее собой фармацевтически приемлемую соль присоединения основания.
4. Соединение по п.3, где фармацевтически приемлемая соль присоединения основания является солью натрия.
5. Соединение по п.1, которое является N-[2-хлор-4-бромбензоил] -4-хлорфенилсульфонамидом или его солью присоединения основания.
6. Соединение по п.1, которое является N-[2-метил-4-хлорбензоил]-4-хлорфенилсульфонамидом или его солью присоединения основания.
7. Соединение по п.5 или 6, где соль присоединения основания является солью натрия.
8. Способ лечения восприимчивых новообразований у млекопитающих, включающий введение нуждающемуся в подобном лечении млекопитающему онколитически эффективного количества соединения формулы II

где X является O или NH;
R1 представляет собой водород, галоген, C1-C6алкил, C1-C4алкокси, C1-C4алкилтио, CF3, OCF3, SCF3, (C1-C4алкокси)карбонил, нитро, азидо, O(SO2)CH3, N(CH3)2, гидрокси, фенил, замещенный фенил, пиридинил, тиенил, фурил, хинолинил или триазолил;
R2 представляет собой водород, галоген, циано, CF3, C1-C6алкил, (C1-C4алкокси)карбонил, C1-C4алкокси, фенил или хинолинил;
R2a представляет собой водород или C1-C4алкокси;
R2b представляет собой водород или C1-C6алкил при условии, что по крайней мере один из заместителей R2a и R2b является водородом;
R3 представляет собой водород, галоген, C1-C6алкил, CF3 или нитро;
R3a представляет собой водород, галоген или C1-C6алкил при условии, что если R3a является C1-C6алкилом, то R3 является водородом и R4 является галогеном; и
R4 представляет собой галоген, C1-C6алкил или CF3 при условии, что только один из заместителей R3 и R4 может являться C1-C6алкилом, и при условии, что если R4 является галогеном или C1-C6алкилом, то только один из заместителей R3 и R3a является водородом;
где вышеуказанный замещенный фенил обозначает монозамещенный фенил, где заместитель выбирают из группы, состоящей из C1-C4алкокси, C1-C4алкилтио, C1-C4ацила, трифторметила и галогена;
или его фармацевтически приемлемой соли присоединения основания.
9. Способ по п.8, где восприимчивое новообразование является опухолью толстой или прямой кишки.
10. Фармацевтическая композиция, включающая соединение формулы II

где X представляет собой O или NH;
R1 представляет собой водород, галоген, C1-C6алкил, C1-C4алкокси, C1-C4алкилтио, CF3, OCF3, SCF3, (C1-C4алкокси)карбонил, нитро, азидо, O(SO2)CH3, N(CH3)2, гидрокси, фенил, замещенный фенил, пиридинил, тиенил, фурил, хинолинил или триазолил;
R2 представляет собой водород, галоген, циано, CF3, C1-C6алкил, (C1-C4алкокси)карбонил, C1-C4алкокси, фенил или хинолинил;
R2a представляет собой водород или C1-C4алкокси;
R2b представляет собой водород или C1-C6алкил при условии, что по крайней мере один из заместителей R2a и R2b является водородом;
R3 представляет собой водород, галоген, C1-C6алкил, CF3 или нитро;
R3a представляет собой водород, галоген или C1-C6алкил при условии, что если R3a является C1-C6алкилом, то R3 является водородом и R4 является галогенюь; и
R4 представляет собой галоген, C1-C6алкил или CF3 при условии, что только один из заместителей R3 и R4 может являться C1-C6алкилом, и при условии, что если R4 является галогеном или C1-C6алкилом, то только один из заместителей R3 и R3a является водородом;
где вышеуказанный замещенный фенил обозначает монозамещенный фенил, где заместитель выбирают из группы, состоящей из C1-C4алкокси, C1-C4алкилтио, C1-C4ацила, трифторметила и галогена;
или его фармацевтически приемлемую соль присоединения основания;
и фармацевтически приемлемый носитель, разбавитель или наполнитель.
Текст
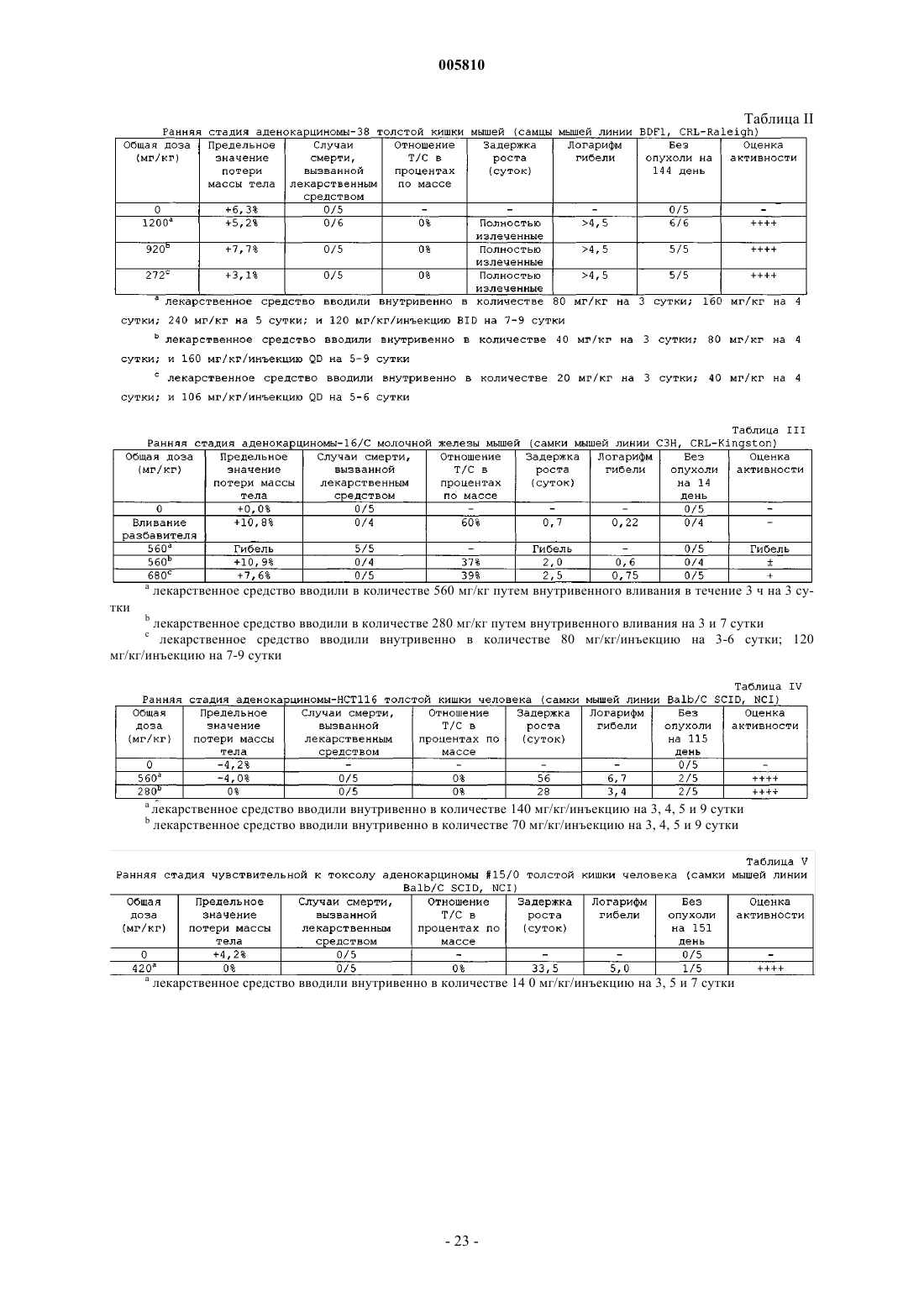
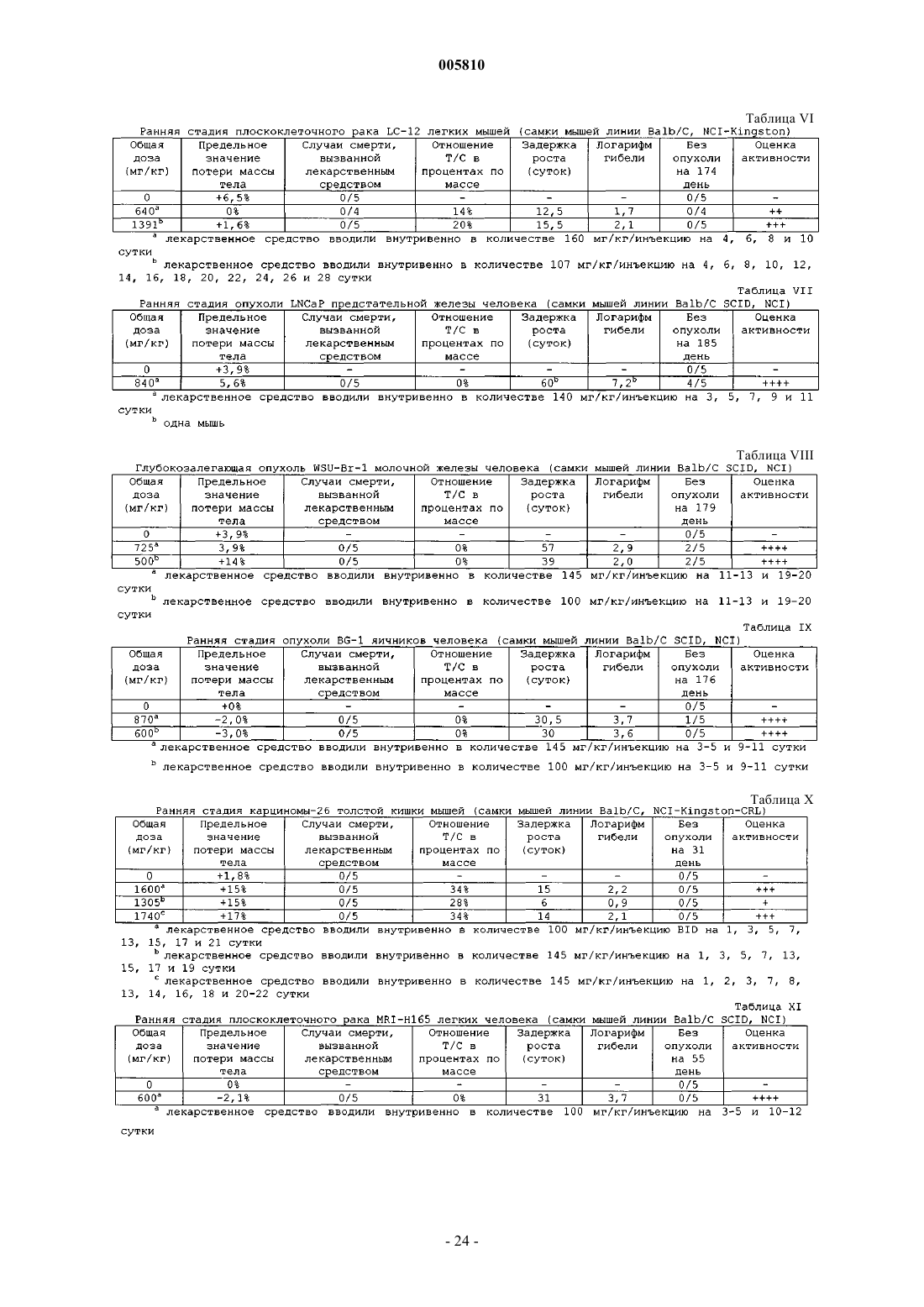
005810 В последние годы были достигнуты существенные успехи в разработке химических средств и схем лечения для борьбы с опухолевыми заболеваниями. Несмотря на успешное развитие в этой области, опухоли продолжают доставлять людям непереносимые боли и страдания. Потребность в новых и лучших способах лечения злокачественных новообразований и лейкозов остается двигателем усилий по созданию новых классов соединений, особенно в области неоперабельных или метастазирующих солидных опухолей. Новая лавина информации, относящейся к основным биологическим процессам, вовлеченным в развитие новообразований, привела к более глубокому пониманию гетерогенности опухолей. Именно вследствие данной чрезвычайной гетерогенности среди популяций опухолевых клеток новые химиотерапевтические средства должны обладать широким спектром активности и высоким терапевтическим индексом. Кроме того, такие средства должны быть химически устойчивы и совместимы с другими средствами. Также важным является то, чтобы любая схема химиотерапии была настолько удобна и безболезненна для пациента, насколько это возможно. Часто для лечения злокачественных опухолей применяют химиотерапию и облучение и, хотя они всегда оказывают определенный эффект на злокачественное заболевание, они редко бывают целительными. Большинство из солидных опухолей увеличивают свою массу за счет пролиферации злокачественных клеток и стромальных клеток, включая эпителиальные клетки. Для того чтобы увеличиться в размерах более чем на 2-3 мм в диаметре, опухоль должна образовывать сосудистую сеть в результате процесса, известного как ангиогенез. Сообщалось, что угнетение индуцированного опухолью ангиогенеза ангиостатином и эндостатином приводит к противоопухолевой активности (O'Reilly et al., Cell, 88,277-285 (1997. В связи с тем, что ангиогенез является крайне необходимым компонентом для увеличения массы большинством солидных опухолей, разработка новых средств для угнетения этого процесса представляет собой многообещающий подход к противоопухолевой терапии. Этот подход к противоопухолевой терапии может снизить токсические побочные эффекты или свойства, связанные с возникновением устойчивости к лекарствам, традиционной химиотерапии (Judah Folkman, Endogenous Inhibitors ofN-[Бензоил]фенилсульфонамиды известны в области сельскохозяйственной химии как инсектициды и гербициды (патент Германии 2744137). Применение N-[бензоил]фенилсульфонамидов в качестве противоопухолевых средств в целом или в качестве ингибиторов ангиогенеза в частности до сих пор не принимались во внимание. Краткое изложение сущности изобретения Настоящее изобретение относится к соединению формулы IR2a представляет собой водород или С 1-С 4 алкокси;R2b представляет собой водород или С 1-С 6 алкил при условии, что по крайней мере один из заместителей R2a и R2b является водородом;R4 представляет собой галоген, C1-С 6 алкил или CF3 при условии, что только один из заместителейR3 и R4 может являться C1-С 6 алкилом, и при условии, что если R4 является галогеном или C1-С 6 алкилом,то только один из заместителей R3 и R3a является водородом; или его фармацевтически приемлемой соли присоединения основания при условии, чтоa) если оба заместителя R3 и R4 являются хлором и R2 является водородом, то R1 является бромом,йодом, C1-С 4 алкокси, C1-С 4 алкилтио, CF3, OCF3, нитро, азидо, O(SO2)CH3, N(CH3)2, гидрокси, фенилом,замещенным фенилом, пиридинилом, тиенилом, фурилом или триазолилом;b) если оба заместителя R3 и R4 являются хлором и R1 является водородом, то R2 является бромом,фтором, CF3, C1-С 6 алкилом, C1-С 4 алкокси, фенилом или хинолинилом. Настоящее изобретение также относится к способу лечения восприимчивых новообразований у млекопитающих, включающему введение нуждающемуся в подобном лечении млекопитающему онко-1 005810 литически эффективного количества соединения формулы IIR2a представляет собой водород или С 1-С 4 алкокси;R2b представляет собой водород или C1-С 6 алкил при условии, что по крайней мере один из заместителей R2a и R2b является водородом;R4 представляет собой галоген, C1-С 6 алкил или CF3 при условии, что только один из заместителей 3R и R4 может являться C1-С 6 алкилом, и при условии, что если R4 является галогеном или C1-С 6 алкилом,то только один из заместителей R3 и R3a является водородом; или его фармацевтически приемлемой соли присоединения основания. Настоящее изобретение также относится к способу угнетения опухолевого ангиогенеза у млекопитающих, включающему введение нуждающемуся в подобном лечении млекопитающему соединения формулы II или его фармацевтически приемлемой соли присоединения основания в количестве, угнетающем опухолевый ангиогенез. Настоящее изобретение также относится к фармацевтической композиции, включающей соединение формулы II или его фармацевтически приемлемую соль присоединения основания, в комбинации с фармацевтически приемлемым носителем, разбавителем или наполнителем. Настоящее изобретение также относится к применению соединения формулы II для получения лекарственного средства для лечения восприимчивых новообразований. Дополнительно настоящее изобретение относится к содержащей соединение формулы II фармацевтической композиции для лечения восприимчивых новообразований. Кроме того, настоящее изобретение относится к способу лечения восприимчивых новообразований, который включает введение эффективного количества соединения формулыII. Подробное описание изобретения Применяемые в описанных выше формулах общие химические термины имеют следующие обычные значения. Например, термин C1-С 6 алкил включает метильный, этильный, пропильный, изопропильный, бутильный, изобутильный, втор-бутильный, трет-бутильный, пентильный и гексильный радикалы. Значение термина C1-С 4 алкил заключено внутри значения термина C1-С 6 алкил и применяется для обозначения метила, этила, пропила, изопропила, бутила, изобутила, втор-бутила и трет-бутила. Термин C1 С 4 алкокси применяется для обозначения С 1-С 4 алкильной группы, соединенной с основной частью молекулы через атом кислорода, и включает метоксильную, этоксильную и изопропоксильную группу. Подобно, термин C1-С 4 алкилтио применяется для обозначения C1-С 4 алкильной группы, соединенной с основной частью молекулы через атом серы, и включает метилтио, этилтио и изобутилтио. Термин галоген применяется для обозначения хлора, фтора, брома и йода. Термин замещенный фенил обозначает монозамещенный фенил, где заместитель выбирают из группы, состоящей из C1-С 4 алкокси, C1 С 4 алкилтио, C1-С 4 ацила, трифторметила и галогена. Термин ацил обозначает группу органической кислоты, в которой гидроксил карбоксигруппы замещен на какой-либо другой заместитель (RCO-). Если X=NH, то молекула может существовать в двух таутомерных формах Настоящее изобретение рассматривает обе эти формы. Хотя все соединения формулы II являются полезными противоопухолевыми средствами, предпочтительными являются конкретные классы соединений. Такие предпочтительные классы описаны далее:w) соединение формулы II, где соединение является фармацевтически приемлемой солью присоединения основания; х) соединение формулы II, где соединение является солью натрия; у) R1, R2a и R2b являются водородом и R2 выбирают из групп, состоящей из галогена, C1-С 4 алкила,C1-С 4 алкокси, циано, трифторметила и хинолинила;z) R2 и R2b являются водородом, R1 является галогеном или C1-С 4 алкилом, и R2a является C1 С 4 алкилом или С 1-С 4 алкокси; или аа) R2a является водородом, R1 является C1-С 4 алкокси, и R2 и R2b являются C1-С 4 алкилом. Кроме того, особенно предпочтительными являются следующие классы:b) R2a и R2b являются водородом и R1 выбирают из группы, состоящей из галогена и C1-С 4 алкила, и 2R выбирают из группы, состоящей из галогена, C1-С 4 алкила и C1-С 4 алкоксикарбонила. Следует понимать, что описанные выше предпочтительные и особенно предпочтительные классы могут быть объединены с образованием дополнительных предпочтительных и особенно предпочтительных классов. Соединения формулы II являются противоопухолевыми средствами. Поэтому настоящее изобретение также относится к способу лечения восприимчивого новообразования у млекопитающего, который включает введение нуждающемуся в подобном лечении млекопитающему онколитически эффективного количества соединения формулы II. Считается, что соединения по настоящему изобретению применимы для лечения злокачественных новообразований, таких как новообразования центральной нервной системы (полиморфная глиобластома, астроцитома, олигодендроглиальные опухоли, опухоли эпендимы и опухоли сосудистого сплетения глаза, опухоли шишковидного тела, нейрональные опухоли, медуллобластома, шваннома, менингиома, менингеальная саркома); новообразования глаза (базально-клеточный рак, плоскоклеточный рак, меланома, рабдомиосаркома, ретинобластома); новообразования эндокринных желез (новообразования гипофиза, новообразования щитовидной железы, новообразования коркового вещества надпочечников, новообразования нейроэндокринной системы, новообразования гастроэнтеропанкреатической эндокринной системы, новообразования половых желез); новообразований головы и шеи (рак головы и шеи, опухоли ротовой полости, глотки, гортани, одонтогенные опухоли); новообразования грудной клетки (крупноклеточный рак легких, мелкоклеточный рак легких, немелкоклеточный рак легких, злокачественная мезотелиома, тимомы, опухоли из первичных зародышевых клеток из грудного сегмента); новообразования пищеварительного тракта (новообразования пищевода, новообразования желудка, новообразования печени, новообразования желчного пузыря, новообразования экзокринной части поджелудочной железы, новообразования тонкого кишечника, червеобразного отростка и брюшины, аденокарцинома толстой и прямой кишки, новообразования заднего прохода); новообразования мочеполового тракта (рак из клеток почечного эпителия, новообразования почечной лоханки и мочеточников, новообразования мочевого пузыря, новообразования мочеиспускательного канала, новообразования предстательной железы, новообразования полового члена, новообразования яичек); новообразования-3 005810 женских репродуктивных органов (новообразования наружных женских половых органов и влагалища,новообразования шейки матки, аденокарцинома тела матки, рак яичников, гинекологические саркомы); новообразования молочной железы; новообразования кожи (базально-клеточный рак, плоскоклеточный рак, дерматофибросаркома, опухоль клеток Меркеля; злокачественная меланома); новообразования костей и мягких тканей (остеогенная саркома, злокачественная фиброзная гистиоцитома, хондросаркома,саркома Юинга, недифференцированная нейроэктодермальная опухоль, ангиосаркома); новообразования системы кроветворения (миелодиспластические синдромы, острый миелолейкоз, хронический миелолейкоз, острый лимфолейкоз, HTLV-1 и Т-клеточный лейкоз/лимфома, хронический лимфолейкоз, лейкозный ретикулоэндотелиоз, болезнь Ходжкина, неходжкинские лимфомы, тучноклеточный лейкоз); и новообразования у детей (острый лимфобластный лейкоз, острый миелолейкозы, нейробластома, опухоли костей, рабдомиосаркома, лимфомы, опухоли почек). В частности считается, что соединения по настоящему изобретению применимы для лечения солидных опухолей, особенно опухолей толстой и прямой кишки. Предпочтительно млекопитающим, подвергаемым лечению путем введения соединений формулы II, являлся человек. Соединения по настоящему изобретению являются кислотными по своей природе и, соответственно, могут взаимодействовать с любым из неорганических и органических оснований, включая амины и четвертичные соли аммония, с образованием фармацевтически приемлемых солей присоединения основания. Если для простоты введения необходимы водные растворы соединений по настоящему изобретению, предпочтительно преобразование соединений формулы II в их фармацевтически приемлемые соли присоединения основания. Соединения формулы II могут взаимодействовать с основными веществами,такими как гидроксиды щелочных и щелочно-земельных металлов, карбонаты и бикарбонаты, включая без ограничения гидроксид натрия, карбонат натрия, гидроксид калия, гидроксид кальция, гидроксид лития и так далее, с образованием фармацевтически приемлемых солей, таких как соответственная соль натрия, калия, лития или кальция. Особенно предпочтительными являются соли натрия и калия. Примерами аминов, пригодных для образования солей, являются первичные, вторичные и третичные алифатические и ароматические амины, такие как метиламин, этиламин, пропиламин, изопропиламин, четыре изомерных бутиламина, диметиламин, диэтиламин, диэтаноламин, дипропиламин, диизопропиламин, ди-н-бутиламин, пирролидин, пиперидин, морфолин, триметиламин, триэтиламин, трипропиламин, хинуклидин, пиридин, хинолин и изохинолин, особенно этил-, пропил-, диэтил- или триэтиламин, но особенно изопропиламин и диэтаноламин. Примерами четвертичных аммониевых оснований являются, в основном, катионы галогенаммониевых солей, например катион тетраметиламмония, катион триметилбензиламмония, катион триэтилбензиламмония, катион тетраэтиламмония или катион триметиэтиламмония, а также катион аммония. Соединения по настоящему изобретению могут быть получены при помощи способов, хорошо известных каждому специалисту в данной области техники. Обычно, N-[бензоил]фенилсульфонамиды формулы II получают путем сочетания замещенного подходящим образом фенилсульфонамида с замещенной подходящим образом бензойной кислотой или производным бензойной кислоты, как проиллюстрировано на следующей схеме. Значения переменных заместителей R1, R2, R2a, R2b, R3, R3a и R4 определены выше, a Z является ОН, Сl, Вr, метансульфонилокси или трифторметансульфонилокси. Если Z является гидроксилом, соответственную бензойную кислоту сочетают с фенилсульфонамидом в стандартных условиях пептидного сочетания, хорошо известных специалистам. Конкретно, фенилсульфонамид сочетают с бензойной кислотой в присутствие реагента пептидного сочетания, необязательно в присутствии катализатора. Подходящие пептидные сочетающие реагенты включают N,N'карбонилдиимидазол (CDI), N,N'-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (EDC) и 1-(3-(1-пирролидинил)пропил)-3-этилкарбодиимид (РЕРС). ФормыEDC (Tetrahedron Letters, 34(48), 7685 (1993 и РЕРС (патент США 5792763) на полимерных носителях были описаны и очень хорошо применимы для получения соединений по настоящему изобретению. Пригодные катализаторы для реакции сочетания включают N,N-диметил-4-аминопиридин (DMAP). Все реагенты объединяют в приемлемом растворителе, обычно в дихлорметане, хлороформе, тетрагидрофуране, диоксане или диэтиловом эфире, и перемешивают в течение 1-72 ч при температуре от температуры окружающей среды до температуры кипения растворителя с обратным холодильником. Желаемый продукт может быть выделен при помощи стандартных методик экстракции и кристаллизации и очищен методом хроматографии или кристаллизации, если это необходимо или желательно. В том случае, если применяются реагенты на полимерной основе, они могут быть удалены из реакционной смеси при помощи фильтрации. Альтернативно, может быть проведено взаимодействие сульфонамида с производным бензойной-4 005810 кислоты, такими как соединения, где Z является хлором, бромом, метансульфонилокси или трифторметансульфонилокси, в присутствие акцептора кислоты, такого как пиридин, триэтиламин или основная смола, необязательно в присутствии катализатора. Реагенты объединяют, а продукты выделяют, по существу, как описано выше. Специалист в данной области техники должен принять во внимание, что соединения формулы II,где X является NH, могут быть получены, как представлено на схеме II, где значения R1, R2, R2a, R2b, R3,R3a и R4 определены выше. Проводят взаимодействие замещенного подходящим образом бензамидина с производными сульфонила, такими как соединения, где Z' является хлором, бромом, метансульфонилокси или трифторметансульфонилокси, в присутствие акцептора кислоты, такого как пиридин, триэтиламин или основная смола, необязательно в присутствие катализатора. Реагенты объединяют, а продукты выделяют, по существу, как описано выше. Необходимые бензойные кислоты, производные бензойной кислоты, бензамидины, производные сульфонила и сульфонамиды являются или коммерчески доступными, или могут быть получены при помощи способов, известных специалистам. Получение 1. 2,4-Дибромбензонитрил. Раствор цианида меди(I) (2,32 г, 25,9 ммоль) в безводном диметилсульфоксиде (50 мл) перемешивали при 60 С и добавляли к этому раствору трет-бутилнитрит (7,1 мл, 59,7 ммоль) одной порцией. К смеси по каплям, через канюлю, добавляли раствор 2,4-диброманилина (5,0 г, 19,9 ммоль) в безводном диметилсульфоксиде (30 мл). После завершения добавления реакционную смесь перемешивали в течение 1 ч, охлаждали до 45 С и затем медленно обрабатывали 5 н. НСl (50 мл). Через 5 мин реакционную смесь охлаждали до температуры окружающей среды и экстрагировали этилацетатом/гексаном 1/1(2300 мл). Объединенные органические слои промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), сушили, концентрировали при пониженном давлении и очищали остаток хроматографией на силикагеле, элюируя гексаном, содержащим 0-5% этилацетата. Содержащие продукт фракции объединяли и концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,61 г, выход 31%). Т.пл.=76-78 С. FD МС: m/е=261 (М+). Получение 2. 2,4-Дибромбензойная кислота. Перемешанную суспензию 2,4-дибромбензонитрила (1,57 г, 6,0 ммоль) в серной кислоте (6 М, 150 мл) нагревали при кипении с обратным холодильником в течение 3 суток. Реакционную смесь охлаждали до температуры окружающей среды и затем экстрагировали этилацетатом (275 мл). Объединенные органические слои промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (50 мл),сушили, концентрировали, а затем очищали остаток хроматографией на силикагеле, элюируя хлороформом, содержащим 0,5% метанола и 0,1% уксусной кислоты. Содержащие продукт фракции объединяли и концентрировали при пониженном давлении с получением указанного в заголовке соединения (0,81 г,выход 48%). Т.пл.=171-172 С. ESI MC: m/е=279 (М+-1). Получение 3. 2-Бром-4-хлорбензойная кислота. Раствор нитрита натрия (2,21 г) в воде (15 мл) по каплям добавляли к охлажденной на льду перемешанной смеси 2-амино-4-хлорбензойной кислоты (5,00 г, 29,1 ммоль) и 48% бромисто-водородной кислоты (150 мл) в воде (150 мл). Реакционную смесь перемешивали в течение 2 ч при 0 С и затем по каплям обрабатывали раствором бромида меди(II) (7,81 г) в воде (20 мл). После завершения добавления реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешиваться в течение ночи. Затем реакционную смесь экстрагировали этилацетатом/гексаном 3/1 (2400 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (200 мл), сушили, концентрировали при пониженном давлении и очищали остаток хроматографией на силикагеле, элюируя хлороформом, содержащим 1% метанола и 0,5% уксусной кислоты. Содержащие продукт фракции объединяли и концентрировали при пониженном давлении с получением указанного в заголовке соединения (4,04 г, выход 59%). Т.пл.=154-155 С. ESI MC: m/e=233, 235 (М+-1). Получение 4. Метиловый эфир 4-сульфамоилбензойной кислоты. 4-Карбоксифенилсульфонамид (2,00 г, 9,9 ммоль) суспендировали в хлороформе/метаноле 3/1 (200 мл). При температуре окружающей среды добавляли (триметилсилил)диазометан в виде 2 М раствора в гексане (7,4 мл, 14,8 ммоль) и перемешивали в течение 5 мин. Раствор концентрировали в вакууме и очищали неочищенный продукт хроматографией на силикагеле, элюируя 0,5% МеОН/0,1% АсОН вCH2Cl2, с получением продукта в виде белого твердого вещества (2,11 г, выход 98%). Т.пл.=180 С. ESI-5 005810 Получение 5. 3,4-Дибромфенилсульфонамид. 3,4-Дибромфенилсульфонилхлорид (20 ммоль; Aldrich) суспендировали в 40 мл 30% водного раствора NH4OH и перемешивали смесь. Медленно, порциями, добавляли ацетон с получением гомогенной реакционной смеси (5-10 мл). Это добавление является экзотермическим и сопровождается интенсивным образованием пузырьков. Реакционную смесь перемешивали при комнатной температуре и анализировали при помощи ES IMS. Для удаления ацетона смесь концентрировали на роторном испарителе с получением твердого вещества. Твердое вещество собирали фильтрацией с отсасыванием, промывали водой и оставляли сушиться на воздухе. Полученное вещество использовали без дальнейшей очистки. ESI MC: 312, 314, 316 (М+-1). Т.пл.=169-171 С; Т.пл.=175-176 С (по литературным данным, Huntress, E.H., Carten,F.H., J. Am. Chem. Soc. 1940, 62, 511-514). Соединения получений 6-15 получали, по существу, как описано в процедуре получения 5. Получение 16. 2-Хлор-4-метилбензойная кислота. К 4-бром-3-хлортолуолу (4,97 г, 24,2 ммоль) в DMF (25 мл) добавляли Pd(OAc)2 (0,54 г, 2,42 ммоль), 1,3-бис(дифенилфосфино)пропан (0,998 г, 2,42 ммоль), триэтиламин (12,5 мл) и метанол (12,5 мл). Из реакционного сосуда откачивали воздух и трижды продували газообразным моноксидом углерода. Для поддержания атмосферы моноксида углерода применяли воздушный шар, наполненный газообразным моноксидом углерода. Реакционную смесь нагревали при 80 С в течение 8 ч. После охлаждения добавляли воду (50 мл). Смесь экстрагировали гексанами (250 мл). Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали и очищали хроматографией, элюируя 0-3% EtOAc в гексанах. Получали 1,24 г (28%) метил-2-хлор-4-метилбензоата в виде бесцветного масла. ESI MC:m/e=184 (М+; 35 Сl) и 186 (М+; 37 Сl). К метил-2-хлор-4-метилбензоату (1,00 г, 5,42 ммоль) в THF (10 мл), МеОН (5 мл) и Н 2 О (2,5 мл) добавляли 2 н. LiOH (8,12 мл, 16,2 ммоль). Реакционную смесь нагревали при 50 С в течение 2,5 ч, охлаждали до комнатной температуры, а затем тушили добавлением 5 н. НСl (3,24 мл). Для удаления THF и МеОН смесь концентрировали. Получали белый осадок и фильтровали его. После упаривания получали 0,922 г (100%) 2-хлор-4-метилбензойной кислоты. ESI MC: m/e=169 (М 1; 35 Сl) и 171 (М 1; 37 Сl). Получение 17. 4-(Трет-бутилдиметилсилилокси)фенилсульфонамид. 4-Гидроксифенилсульфонамид (3,46 г, 20 ммоль) растворяли в DMF (40 мл) и обрабатывали при комнатной температуре трет-бутилдиметилсилилхлоридом (3,31 г, 22,0 ммоль) и имидазолом (1,50 г, 22,0 ммоль). Через 20 ч реакционную смесь разбавляли EtOAc (100 мл) и промывали 1,0 н. НСl (250 мл). Органическую фазу сушили (MgSO4), фильтровали и концентрировали с получением масла. Неочищенное масло очищали колоночной (Biotage) хроматографией (колонка 40 М SiCO2, элюируя гексанами/EtOAc 1/1, 75 мл/мин). Получали белое твердое вещество (4,24 г, 15,4 ммоль, 77%). Т.пл.=117-118 С. ESI МС:EDC (1,25-1,5 экв.), а затем N,N-диметил-4-аминопиридин (1,2 экв.). Смесь интенсивно перемешивали в атмосфере азота в течение 16 ч, концентрировали при пониженном давлении и распределяли остаток между этилацетатом и водой. Органический слой промывали 1 н. соляной кислотой (4 раза, 20 мл/ммоль),а затем экстрагировали объединенные водные фазы этилацетатом (дважды, 20 мл/ммоль). В заключение,-6 005810 объединенные органические слои промывали водой и насыщенным водным раствором хлорида натрия,сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле или путем кристаллизации, если это необходимо или желательно. ESI МС: Общая процедура сочетания К перемешанному раствору бензойной кислоты (1,25 экв.) в безводном дихлорметане (10 мл/ммоль) добавляли фенилсульфонамид (1,0 экв.) одной порцией, затем EDC (1,25-1,5 экв.), а затем N,N[диметил]-4-аминопиридин (1,2 экв.). Смесь интенсивно перемешивали в атмосфере азота в течение 16 ч,концентрировали при пониженном давлении и распределяли остаток между этилацетатом и водой. Органический слой промывали 1 н. соляной кислотой (4 раза, 20 мл/ммоль), а затем экстрагировали объединенные водные фазы этилацетатом (дважды, 20 мл/ммоль). В заключение, объединенные органические слои промывали водой и насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали по методу хроматографии на силикагеле или путем кристаллизации, если это необходимо или желательно. Соединения примеров 1-84 получали, по существу, как описано в данной общей процедуре. Пример 85. N-[2-Хлор-4-бромбензоил]-4-хлорфенилсульфонамид. Восьмимиллилитровую реакционную пробирку загружали 2-хлор-4-бромбензойной кислотой (0,39 ммоль, 1,5 экв.) и 2,0 мл дихлорметана. Добавляли маточный раствор (4,0 мл), содержащий 4 хлорфенилсульфонамид (0,26 ммоль, 1 экв.) и N,N-[диметил]-4-аминопиридин (48 мг, 0,39 ммоль, 1,5 экв.) в дихлорметане, затем 0,261 г карбодиимидной полистироловой смолы (2,0 ммоль/г, 0,52 ммоль, 2,0 экв., Novabiochem), сосуд закупоривали и вращали. Через 72 ч добавляли 0,77 г сульфонированной пролистироловой смолы (MP-TsOH) (1,53 ммоль/г, 1,17 ммоль, Argonaut). Приблизительно через 18 ч реакционную смесь фильтровали и концентрировали под током азота. Остаток очищали ВЭЖХ с обращенной фазой (колонка CombiPrep, колонка YMC ODS-A 2050 мм с размером пор 5 мкм, С 18, размер пор 120), элюируя градиентом 5-95% CH3CN/0,01 водным раствором НСl. Содержащие продукт фракции объединяли и концентрировали при пониженном давлении с получением указанного в заголовке соединения.ESI MC: m/e=408 (М 1), 406 (М+-1), 410 (М 3). Соединения примеров 86-107 получали, по существу, как описано для примера 85. Пример 108. N-[2,4-Дихлорбензоил]-4-(1-метилсульфанилофен-4-ил)фенилсульфонамид. Этап А. Процедура активации смолы. Смолу на основе амида Ринка (СА Novabiochem, 0,53 ммоль/г) суспендировали в 30% растворе пиридина в DMF и перемешивали при комнатной температуре в течение 3 ч. Смесь фильтровали и дважды промывали смолу DMF, а затем, по выбору, СН 2 Сl2 или МеОН. Активированную смолу, содержащую активную аминогруппу, сушили и использовали без дополнительной очистки. Смолу на основе амида Ринка (0,53 ммоль/г) суспендировали в смеси (1:1) CH2Cl2/THF и Et3N (4 экв.), 4-йодфенилсульфонамиде (3 экв.) и DMAP (каталитическое количество). Раствор перемешивали в течение ночи при комнатной температуре. Смесь фильтровали и промывали смолу, по выбору, CH2Cl2 или МеОН. Смолу на основе 4-йодфенилсульфонамида Ринка сушили в вакууме. Соответствующую смолу на основе 4-йодфенилсульфонамида Ринка (0,26 ммоль, 0,53 ммоль/г), метилсульфанилбороновую кислоту (2 экв.), карбонат калия (6 экв.) и ацетат палладия (0,5 экв.) смешивали вместе и суспендировали в 7 мл смеси диоксана/воды 6:1. Смесь нагревали в Argovant QUEST 210 при 100 С в течение 24 ч. Затем смолу дважды промывали 5 мл смеси диоксан/вода 6:1 и затем шесть разCH2Cl2 (7 мл), промывая затем каждый раз МеОН (7 мл). К смоле, предварительно растворенной в 3 мл СН 2 Сl2, добавляли 3 мл 95% водного раствора трихлоруксусной кислоты. Смесь перемешивали в течение 30 мин при комнатной температуре и фильтровали, как описано выше. 4'-Метилсульфанилбифенил-4-сульфонамид использовали без дополнительной очистки. К перемешанному раствору 2,4-дихлорбензойной кислоты (1,25 экв.) в безводном СН 2 Сl2 (10 мл/ммоль) добавляли 4'-метилсульфанилбифенил-4-сульфонамид (1,0 экв.) одной порцией, затем EDC(1,25 или 1,5 экв.) и, наконец, DMAP (1,2 экв.). Смесь интенсивно перемешивали в атмосфере азота в течение 16 ч, затем упаривали в вакууме и распределяли остаток между EtOAc и водой. Органический слой промывали 1 н. НСl (4 раза, 20 мл/ммоль), а затем экстрагировали водную фазу EtOAc (дважды, 20 мл/ммоль). В завершение, объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный продукт очищали хроматографией на силикагеле, применяя подходящий элюент, с получением указанного в заголовке соединения.ESI MC: (М+-Н) 450,9870/450,0. Пример 109. N-[2,4-Дихлорбензоил]-4-3'-ацетилбифенилсульфонамид. Для получения указанного в заголовке соединения использовали суспензию смолы на основе 4 йодфенилсульфонамида Ринка (0,26 ммоль, 0,53 ммоль/г), 3-ацетилфенилбороновую кислоту (2 экв.) и 2,4-дихлорбензойную кислоту (1,25 экв.), по существу, как описано в примере 108. ESI МС: (М+-Н) 447,0099/446,0. Пример 110. N-[2,4-Дихлорбензоил]фенилсульфонамид. К смеси фенилсульфонамида (0,16 моль; 25,12 г) и карбоната калия (0,2 моль; 27,6 г) в 500 мл диоксана по каплям добавляли 2,4-дихлорбензоилхлорид (0,13 моль; 18,0 мл). Смесь нагревали в атмосфере азота при температуре возгонки в течение 16 ч. Затем реакционную смесь разбавляли водой (500 мл),нейтрализовывали до значения рН=5 добавлением концентрированной соляной кислоты и трижды экстрагировали этилацетатом. Объединенные этилацетатные слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением белого твердого вещества. Твердый остаток очищали хроматографией на силикагеле, элюируя дихлорметаном, содержащим 0-5% метанола. Содержащие продукт фракции объединяли и концентрировали при пониженном давлении с получением указанного в заголовке соединения. МС (ES): m/е=329,9m/e=363,9 (M+). Рассчитано для C13H8Cl3NO3S: теоретически: С, 42,82; Н, 21; N, 84. Обнаружено: С, 42,56; Н, 2,14,N, 3,76. Пример 112. N-[2-Хлор-4-бромбензоил]-4-хлорфенилсульфонамид. К реакционной смеси 4-хлорфенилсульфонамида (15,6 г, 81,4 ммоль), CDI (15,82 г, 97,7 ммоль) и этилацетата (300 мл) при комнатной температуре в течение 15 мин добавляли взвесь 2-хлор-4 бромбензойной кислоты (23,0 г, 97,7 ммоль) в этилацетате (100,0 мл) (примечание: наблюдается выделение газа, которое можно контролировать скоростью добавления взвеси; после окончания добавления взвеси реакционная смесь превращается в раствор; реакцию контролировали по методу ВЭЖХ или ТСХ,элюируя этилацетатом/гептаном 1:1). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем нагревали при 60 С в течение 90 мин или до тех пор, пока наблюдается выделение газа. Реакционную смесь охлаждали до 40 С и добавляли 1,8-диазабицикло[5,4,0]ундец-7-ен (14,63 мл) одной порцией. Температура реакционной смеси возрастала с 40 до 45 С. Перед тушением деионизованной водой (400 мл) реакционную смесь перемешивали до тех пор, пока она не достигала комнатной температуры. Верхний органический слой отделяли, промывали 1 н. НСl (300,0 мл), сушили над безводным MgSO4, фильтровали и промывали осадок этилацетатом (20,0 мл). Фильтрат концентрировали до состояния сиропа (50,0 г), а затем при интенсивном перемешивании добавляли гептан (250,0 мл). При нагревании образовывалась белая взвесь, которую нагревали с обратным холодильником и оставляли охлаждаться до комнатной температуры. Белый остаток фильтровали и промывали гептаном (20,0 мл). Осадок сушили в вакууме при 55 С в течение 18 ч (масса=29,12 г, выход 87,4 мас.%). Смесь 19,17 г продукта и этилацетата/гептана 1/2 (150,0 мл) нагревали с обратным холодильником в течение 30 мин и охлаждали до комнатной температуры. Не совсем белый осадок фильтровали и промывали гептаном (50,0 мл). Осадок сушили в вакууме при 50 С в течение 18 ч (масса=14,93 г; извлечение 78%). ESI МС: m/е=408 (М 1), 406 (М+-1), 410 (М 3). Пример 113. Натриевая соль N-[2-хлор-4-бромбензоил]-4-хлорфенилсульфонамида. К раствору N-[2-хлор-4-бромбензоил]-4-хлорфенилсульфонамида (5,2 г, 12,72 ммоль) и третбутилметилового эфира (88,0 мл) при комнатной температуре добавляли метоксид натрия (0,69 г, 12,72 ммоль) одной порцией. Реакционную смесь перемешивали в течение 5 ч, после чего добавляли гептан(88,0 мл) и интенсивно перемешивали в течение 60 мин. Полученный белый осадок отфильтровывали- 16005810 при положительном давлении азота и последовательно промывали осадок гептаном (244,0 мл). Осадок сушили до средней степени высушенности, а затем сушили в вакуумной печи при 130 С в течение 18 чH ЯМР (ДМСО d6) 7,8-7,85 (м, 1 Н), 7,81-7,82 (м, 1 Н), 7,58-7,59 (д, 1 Н, J=1,76 Гц), 7,51-7,52 (м, 1 Н),7,48-7,49 (м, 1 Н), 7,44-7,45 (д, 1 Н, J=1,76 Гц), 7,37-7,4 (д, 1 Н). Пример 114. N-[3-Хлор-4-фторфенилсульфонил]-3-фтор-4-метилбензамидин. К 3-хлор-4-фторфенилсульфонилхлориду (0,0304 г, 0,133 ммоль) добавляли 3-фтор-4-метилбензамидингидрохлорид (0,025 г, 0,133 ммоль) в THF (0,5 мл), а затем N-метилморфолин (0,2 мл). Реакционную смесь концентрировали и очищали по методу хроматографии с обращенной фазой, элюируя градиентом 5-95% (0,1% TFA в CH3CN) в (0,1% TFA в Н 2 О). Выделяли белое твердое вещество (16,4 мг,36%). ES масс-спектроскопия положительных ионов [М+Н]+ наблюдали ионы: m/z 345 (35 Сl) и m/z 347(0,0304 г, 0,133 ммоль) использовали, по существу, как описано в примере 114 для получения указанного в заголовке соединения. ES масс-спектроскопия положительных ионов [М+Н]+ наблюдали ионы: m/z 347(35 Сl, 35 Сl), m/z 349 (35 Сl, 37 Сl) и m/z 351 (37 Сl, 37 Сl). Пример 116. N-[3-Хлор-4-фторфенилсульфонил]-3-хлор-4-фторбензамидин. Смесь 3-хлор-4-фторбензамидингидрохлорида (0,025 г, 0,133 ммоль) и 3-хлор-4-фторфенилсульфонилхлорида (0,0304 г, 0,133 ммоль) использовали, по существу, как описано в примере 115 для получения указанного в заголовке соединения. ES масс-спектроскопия положительных ионов [М+Н]+ наблюдали ионы: m/z 365 (35 Сl, 35 Сl), m/z 367 (35 Сl, 37 Сl) и m/z 369 (37 Сl, 37 Сl). Пример 117. N-[2,4-Дихлорбензоил]-4-гидроксифенилсульфонамид. 4-Метоксифенил-4-сульфонамид (0,0608 г, 0,132 ммоль) растворяли в THF (1,25 мл) и при перемешивании обрабатывали фторидом тетрабутиламмония (1,0 н./THF; 200 мкл, 2,0 ммоль) при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли EtOAc (10 мл) и промывали насыщенным водным раствором NH4Cl (1 мл), водой (21 мл) и насыщенным солевым раствором (1 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали на роторном испарителе.(Лиофилизировали из Н 2 О/МеОН с получением прозрачного твердого вещества, 20 мг (0,058 ммоль, 58%. Очищали по методу препаративной ВЭЖХ. Т.пл.=155-157 С. ESI MC: m/e=344,0 (М+-Н); 1 Н ЯМР (ДМСО d6) 7,90 (д, 2 Н), 7,68 (с, 1 Н), 7,44 (с, 2 Н), 6,90 (д, 2 Н), 3,43 (шс, 3 Н). Пример 118. N-[2,4-Дихлорбензоил]-4-(тиен-3-ил)фенилсульфонамид. К раствору N-(2,4-дихлорбензоил)-4-йодфенилсульфонамида (0,10 ммоль) в толуоле/этаноле 20/1 (3 мл) добавляли 3-тиофенбороновую кислоту (0,18 ммоль, 0,18 мл, 1 М раствор в DMF) и тетракис(трифенилфосфин)палладия(0) (10 мол.%). Затем добавляли 2 М водный раствор Na2 СО 3 (0,3 мл) и нагревали перемешанную смесь до 100 С в течение ночи (17 ч) (Buchi Syncore system). Реакционную смесь концентрировали (на аппарате Genevac), затем добавляли воду (2,5 мл) и этилацетат (5 мл). Фазы разделяли и экстрагировали водный слой этилацетатом (35 мл). Этот процесс выполняли автоматически с использованием Tecan system. Растворители выпаривали и очищали соответственный неочищенный продукт по методу ВЭЖХ с получением указанного в заголовке соединения. ES MC m/e 410,96/410,0 Все указанные соединения являются пригодными для перорального введения и обычно вводятся перорально, и поэтому пероральное введение является предпочтительным. Однако пероральное введение не является единственным путем введения или даже единственным предпочтительным путем введения. Например, трансдермальный способ введения может быть очень желательным для пациентов, которые при пероральном приеме лекарственного средства являются забывчивыми или раздражительными, а внутривенный способ введения может быть предпочтительным с целью удобства введения или с целью избежания возникновения возможных осложнений, связанных с пероральным введением. В конкретных обстоятельствах соединения формулы II могут также вводиться чрескожным, внутримышечным, интраназальным или интраректальным путем. Способ введения может варьировать в любом направлении, ограничиваясь физическими свойствами лекарств, удобством для пациента и ухаживающего персонала и другими относящимися к делу обстоятельствами (Remington's Pharmaceutical Sciences, 18th Edition, MackPublishing Co. (1990. Фармацевтические композиции готовят способами, хорошо известными в фармации. Носитель или наполнитель может быть твердым, полутвердым или жидким материалом, который может служить растворителем или средой для активного ингредиента. Пригодные носители или наполнители хорошо известны в данной области техники. Фармацевтическая композиция может быть приспособлена для перорального, ингаляционного, парентерального или местного применения и может вводиться пациенту в виде таблеток, капсул, аэрозолей, лекарственных форм для ингаляций, свечей, растворов, суспензий и тому подобное. Соединения по настоящему изобретению могут вводиться перорально, например, с инертным разбавителем для капсул или спрессованы в таблетки. Для целей перорального терапевтического введения соединения могут быть смешаны с наполнителями и применяться в форме таблеток, пастилок, капсул,эликсиров, суспензий, сиропов, облаток, жевательных резинок, и тому подобное. Данные препараты должны содержать в качестве активного ингредиента по крайней мере 4% соединения по настоящему изобретению, но его содержание может варьировать в зависимости от конкретной формы, и может быть подходящим образом от 4% приблизительно до 70% от массы единицы. Количество содержащегося в препаратах соединения является таким, чтобы достигалась приемлемая дозировка. Предпочтительные композиции и препараты по настоящему изобретению могут быть определены при помощи способов,хорошо известных специалистам в данной области техники. Таблетки, пилюли, капсулы, пастилки и тому подобное, могут также содержать одну или несколько из следующих добавок: связующие вещества, такие как повидон, гидроксипропиловую целлюлозу, микрокристаллическую целлюлозу или желатин; наполнители или разбавители, такие как крахмал, лактозу,микрокристаллическую целлюлозу или двухкальциевый фосфат; разрыхляющие агенты, такие как поперечно-сшитая кармеллоза, поперечно-сшитый повидон, крахмалгликолят натрия, кукурузный крахмал и тому подобное; смазки, такие как стеарат мания, стеариновая кислота, тальк или гидрированное растительное масло; скользящие средства, такие как коллоидный диоксид кремния; увлажняющие средства,такие как лаурилсульфат натрия и полисорбат 80 (CAS No,9005-65-6); и подсластители, такие как сахароза, аспартам или сахарин, или ароматизирующие средства, такие как перечная мята, метилсалицилат или апельсиновый ароматизатор. Если дозированной формой является капсула, в дополнение к описанным выше типам материалов она может содержать жидкий носитель, такой как полиэтиленгликоль или нелетучее масло. Другие дозированные формы могут содержать другие различные материалы, которые изменяют физическую форму дозированных форм, например, таких как оболочки. Так, таблетки или пилюли- 18005810 могут быть покрыты сахаром, гидроксипропилметилцеллюлозой, полиметакрилатами или другими покрывающими средствами. В дополнение к соединениям по настоящему изобретению сиропы могут содержать в качестве подсластителя сахарозу и некоторые консерванты, красители и красящие вещества, и ароматизаторы. Материалы, применяемые для приготовления данных различных композиций, должны быть фармацевтически чистыми и нетоксичными в применяемых количествах. Инъекционные растворы для парентерального введения включают стерильные водные или неводные растворы, суспензии или эмульсии. Водные растворы или эмульсии могут включать дистиллированную воду для инъекций или физиологический солевой раствор. Неводные растворы или эмульсии могут включать пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, спирт,такой как этанол, или полисорбат 80. Инъекционные растворы могут включать дополнительные ингредиенты, отличные от инертных разбавителей, например консерванты, увлажняющие средства, эмульгирующие средства, диспергирующие средства, стабилизирующие средства (такие как лактоза), вспомогательные средства, такие как средства, способствующие растворению (например, глутаминовая кислота или аспарагиновая кислота). Они могут быть стерилизованы, например, путем фильтрации через задерживающий бактерии фильтр, путем введения в состав композиций стерилизующих средств или путем облучения. Они также могут производиться в форме стерильных твердых композиций, которые могут быть растворены в стерильной воде или некотором другом растворителе (растворителях) непосредственно перед инъекцией. Соединения формулы II обычно эффективны в широком диапазоне доз. Например, в норме ежедневная дозировка составляет приблизительно от 10 приблизительно до 300 мг/кг массы тела. В некоторых случаях дозировка, меньшая по сравнению с нижним пределом упомянутого выше диапазона, может быть более чем достаточной, в то время как в других случаях могут применяться еще большие дозы без возникновения каких-либо вредных побочных эффектов, и поэтому указанный выше диапазон доз никоим образом не призван ограничивать объем настоящего изобретения. Следует понимать, что фактически вводимое количество соединения определяется врачом в свете относящихся к делу обстоятельств, включая нуждающееся в лечении состояние, выбранный путь введения, фактическое вводимое соединение или соединения, возраст, вес и чувствительность конкретного пациента, и тяжесть симптомов заболевания у пациента. Ингибирование размножения HUVEC Эндотелиальные клетки из пупочной вены человека (HUVEC; BioWhittaker/Clonetics, Walkersville,MD) культивировали в среде для выращивания эндотелиальных клеток (EGM), содержащей минимальную среду (ЕВМ) с экстрактом бычьего мозга, эпидермальный фактор роста человека, гидрокортизон,гентамицин, амфотерицин В и 2% фетальную бычью сыворотку. Для анализа в лунки 96-луночного планшета для культивирования клеток помещали HUVEC (5103) в ЕВМ (200 мкл) с 0,5% фетальной бычьей сыворотки и инкубировали при 37 С в течение 24 ч в увлажненном воздухе, содержащем 5% углекислого газа. Тестируемые соединения последовательно разбавляли диметилсульфоксидом (DMSO) в концентрациях от 0,0013 до 40 мкМ и добавляли в лунки в количестве 20 мкл. Затем в лунки добавляли фактор роста эндотелия сосудов человека (VEGF) (20 нг/мл в лунках; RD Systems, Minneapolis, MN),приготовленный из маточного раствора (100 мкг/мл в фосфатном буферном солевом растворе, содержащий 0,1% бычьего сывороточного альбумина). HUVEC инкубировали при 37 С в течение 72 ч в увлажненном воздухе, содержащем 5% углекислого газа. В лунки добавляли реагент пролиферации клетокWST-1 (20 мкл, Boehringer Mannheim, Indianapolis, IN) и возвращали планшеты в инкубатор на 1 ч. В каждой лунке измеряли поглощение при 440 нм. Растущую фракцию определяли из значений поглощения в содержащих или не содержащих VEGF обработанных лунках, разделенных на значение поглощения в контрольных образцах, приятых за ноль и 1,0. Все приведенные в примерах соединения были протестированы по этой методике и для всех них получены значения IС 501,0 мкМ. Метод микрокармана роговицы крысы За 20 мин до начала ингаляции 2-3% изофлурана/кислорода самок крыс линии Fisher 344 (145-155 грамм; Taconic, Inc., Germantown, NY) анестезировали ацепромазином (2,5 мг/кг, внутрибрюшинно). Температуру тела поддерживали при помощи подушечки с циркулирующей горячей водой. Операцию проводили при помощи операционного микроскопа (OMS75 Operating Microscope, TopCon Corporation,Japan). Скальпелем (15) делали вертикальный линейный разрез роговицы на половину глубины чуть латеральнее центра глаза. При помощи кончика лезвия скальпеля осторожно делали надрыв верхнего слоя роговицы, ближайшего к краю. В роговице делали карман путем отслаивания при помощи ножниц для роговицы (Roboz, Rockville, MD). Фильтры из нитроцеллюлозы (0,45 мкм, Millipore, Bedford, MA) нарезали на маленькие диски при помощи игольчатого перфоратора 20 калибра. Диски вымачивали в 2 мкл раствора VEGF (0,82 мкг/мкл; RD Systems) или основного фактора роста фибробластов человека(0,20 мкг/мкл; RD Systems) в течение 10 мин на льду. При помощи пинцета диски с нанесенным на них ангиогенным фактором (VEGF или bFGF) помещали в карман на роговице таким образом, чтобы диск был плотно покрыт эпителием роговицы. Животных обрабатывали соединением примера 110 (160 мг/кг),вводимого перорально вместе с фосфатным буферным солевым раствором один раз в сутки с 1 по 10 сутки включительно после имплантации дисков. Глаза фотографировали на 7 и 14 сутки после импланта- 19005810 ции дисков. Для фотографирования животных обрабатывали местно сульфатом атропина (AmTechGroup, Inc., Phoenix Scientific, Inc., St. Joseph, МО) для расширения зрачка и анестезировали 2-3% изофлураном/кислородом. Глаза фотографировали при помощи офтальмологического микроскопа и сохраняли снимки при помощи программного обеспечения Image Pro-Plus. Снимки анализировали путем преобразования интересующей области снимка в высококонтрастный негативный черно-белый снимок, подсчитывая количество светлых пикселов как меру определения сосудистой зоны. Результатом являлись данные снимков по крайней мере 6 глаз. Соединение примера 110 являлось очень эффективным ингибитором VEGF-индуцированного неоангиогенеза, но не являлось эффективным ингибитором bFGFиндуцированного неоангиогенеза. Ингибирование роста клеток рака толстой кишки НСТ 116 Клетки рака толстой кишки НСТ 116 человека выращивали в монослойной культуре в среде RPMI 1640 с добавлением 10% фетальной бычьей сыворотки и 1% пенициллин-стрептомицина (GibcoBRL,Grand Island, NY). Клетки НСТ 116 в экспоненциальной фазе роста обрабатывали различными концентрациями тестируемых соединений при 37 С в течение 72 ч в воздухе, содержащем 5% углекислого газа. После обработки агентом клетки промывали 0,9% фосфатным буферным солевым раствором. Ингибирование роста определяли при помощи реагента пролиферации клеток WST-1 как описано выше. Результаты представляли в виде сравнения растущей фракции в обработанных клетках по отношению к контрольным культурам. Типичные представители соединений по настоящему изобретению были протестированы на их эффективность в отношении клеток рака толстой кишки НСТ 116 человека. Данные этих экспериментов просуммированы в табл. I. Традиционные методы анализа опухолей мышей и ксенотрансплантатов опухолей человека Ингибирование роста опухолей, пересаженных мышам, является распространенной методикой оценки эффективности противоопухолевых средств (Corbett et al., In vivo Methods for Screening и Preclinical Testing: Use of rodent solid tumors for drug discovery., In: Anticancer Drug Development Guide: PreclinicalScreening, Clinical Trials, and Approval, B. Teicher (ed), Humana Press Inc., Totowa, NJ, Chapter 5, pages 7599 (1997); (Corbett et al., Int. J. Pharmacol., 33, Supplement, 102-122 (1995. Опухоли мышей или ксенотрансплантаты опухолей человека имплантировали, по существу, как описано Corbett в In vivo Methods- 21005810 или ксенотрансплантат опухоли человека имплантировали подкожно при помощи троакар-имплантантов 12 калибра или подсчитанного количества клеток. Место введения троакара находится посередине между подмышечным и паховым участком вдоль бока мыши. Перед введением фрагмента опухоли троакар вводили подкожно приблизительно на 3/4 дюйма вверх по направлению к подмышечной ямке, защипывая кожу после удаления троакара. Альтернативно, клетки из опухоли человека получали из brie донорских опухолей (5106 клеток) имплантировали подкожно в заднюю лапу самца или самки голой мыши(Charles River). Или тестируемое соединение в носителе или только носитель вводили путем внутривенной (в/в) инъекции ударной дозы вещества, внутрибрюшинной инъекции (в/б) или перорального кормления (п/о). В каждом эксперименте как каждая получающая лечение группа животных, так и группа не получающих лечение животных, состояла из пяти животных. В течение хода эксперимента (60-120 суток) подкожное развитие опухоли отслеживали путем измерения опухоли дважды в неделю. В качестве показателя меры токсичности исследовали массу тела. Данные подкожного развития опухоли анализировали путем определения средней массы опухоли для каждой группы, получающей лечение, в течение хода эксперимента, и рассчитывая величину задержки роста опухоли как разность (в сутках) достижения объема 500 или 1000 мм 3 получающими лечение опухолями и контрольными опухолями. Соединение примера 110 было протестировано на различных опухолях мышей и человека, по существу, как описано выше. Полученные в этих тестах данные просуммированы в табл. II-XIII. Измеряемые в каждом эксперименте параметры просуммированы в следующих параграфах. Масса опухоли (мг) = (аb2)/2, где а=длина опухоли (мм); b=ширина опухоли (мм). Задержка роста опухоли = Т-С, где Т является средним временем (в сутках), которое требуется для достижения опухолями из получающей лечение группы определенного заранее размера, а С является средним временем (в сутках), которое требуется для достижения опухолями из контрольной группы того же самого размера. Выживших мышей без опухоли исключали из этого расчета и вносили в таблицу отдельно (без опухоли). Логарифм гибели = (задержка роста опухоли)/(3,32Td), где значение задержки роста опухоли определено выше, и Td является временем увеличения объема опухоли вдвое (в сутках), рассчитанным из линии полулогарифмического графика роста контрольной группы в экспоненциальной фазе роста (в диапазоне 100-800 мг), сглаженной наилучшим образом. Отношение Т/С в процентах по массе - получающую лечение и контрольную группы оценивали,когда опухоли из контрольной группы достигали в размере приблизительно 700-1200 мг (медианная группа). В каждой группе определяли среднюю массу 25 опухолей (включая нулевые массы). Значение отношения Т/С в процентах служило мерой противоопухолевой эффективности. Считали, что значение Т/С 42% отражает значительную противоопухолевую активность. Считали, что значение Т/С 10% отражает чрезвычайную противоопухолевую активность. Предельное значение потери массы тела - считали, что предельное значение потери массы тела(среднее группы), превышающее 20%, или случаи смерти, вызванные лекарственным средством, превышающие 20%, указывают на избыточную токсичность дозировки в испытаниях при однократной дозировке. Оценка активности - значение рейтинга активности получали в соответствие со значением логарифма гибели по следующей активности: лекарственное средство вводили в количестве 560 мг/кг путем внутривенного вливания в течение 3 ч на 3 су лекарственное средство вводили в количестве 280 мг/кг путем внутривенного вливания на 3 и 7 сутки лекарственное средство вводили внутривенно в количестве 80 мг/кг/инъекцию на 3-6 сутки; 120 мг/кг/инъекцию на 7-9 сутки с лекарственное средство вводили внутривенно в количестве 140 мг/кг/инъекцию на 3, 4, 5 и 9 сутки лекарственное средство вводили внутривенно в количестве 70 мг/кг/инъекцию на 3, 4, 5 и 9 сутки лекарственное средство вводили внутривенно в количестве 14 0 мг/кг/инъекцию на 3, 5 и 7 суткиR2a представляет собой водород или С 1-С 4 алкокси;R2b представляет собой водород или С 1-С 6 алкил при условии, что по крайней мере один из заместителей R2a и R2b является водородом;R4 представляет собой галоген, С 1-С 6 алкил или CF3 при условии, что только один из заместителей 3R и R4 может являться С 1-С 6 алкилом, и при условии, что если R4 является галогеном или С 1-С 6 алкилом,то только один из заместителей R3 и R3a является водородом; где вышеуказанный замещенный фенил обозначает монозамещенный фенил, где заместитель выбирают из группы, состоящей из С 1-С 4 алкокси, С 1-С 4 алкилтио, С 1-С 4 ацила, трифторметила и галогена; или его фармацевтически приемлемая соль присоединения основания, при условии, что а) если оба заместителя R3 и R4 являются хлором и R2 является водородом, R1 является бромом, йодом, С 1-С 4 алкокси, С 1-С 4 алкилтио, CF3, OCF3, нитро, азидо, О(SО 2)СН 3, N(СН 3)2, гидрокси, фенилом,замещенным фенилом, пиридинилом, тиенилом, фурилом или триазолилом;b) если оба заместителя R3 и R4 являются хлором и R1 является водородом, R2 является бромом,фтором, CF3, C1-С 6 алкилом, C1-С 4 алкокси, фенилом или хинолинилом.- 25005810 2. Соединение по п.1, где R2, R2a и R2b представляет собой водород и R1 выбирают из группы, состоящей из водорода, галогена, C1-С 6 алкила, C1-С 4 алкокси, C1-С 4 алкилтио, CF3, OCF3, SCF3, (C1 С 4 алкокси)карбонила, нитро, азидо, О(SО 2)СН 3, N(СН 3)2, гидрокси, фенила, замещенного фенила, пиридинила, тиенила, фурила, хинолинила или триазолила, где вышеуказанный замещенный фенил обозначает монозамещенный фенил, где заместитель выбирают из группы, состоящей из С 1-С 4 алкокси, С 1 С 4 алкилтио, С 1-С 4 ацила, трифторметила и галогена. 3. Соединение по п.1 или 2, представляющее собой фармацевтически приемлемую соль присоединения основания. 4. Соединение по п.3, где фармацевтически приемлемая соль присоединения основания является солью натрия. 5. Соединение по п.1, которое является N-[2-хлор-4-бромбензоил] -4-хлорфенилсульфонамидом или его солью присоединения основания. 6. Соединение по п.1, которое является N-[2-метил-4-хлорбензоил]-4-хлорфенилсульфонамидом или его солью присоединения основания. 7. Соединение по п.5 или 6, где соль присоединения основания является солью натрия. 8. Способ лечения восприимчивых новообразований у млекопитающих, включающий введение нуждающемуся в подобном лечении млекопитающему онколитически эффективного количества соединения формулы IIR2a представляет собой водород или C1-С 4 алкокси;R2b представляет собой водород или C1-С 6 алкил при условии, что по крайней мере один из заместителей R2a и R2b является водородом;R4 представляет собой галоген, C1-С 6 алкил или CF3 при условии, что только один из заместителей 3R и R4 может являться C1-С 6 алкилом, и при условии, что если R4 является галогеном или C1-С 6 алкилом,то только один из заместителей R3 и R3a является водородом; где вышеуказанный замещенный фенил обозначает монозамещенный фенил, где заместитель выбирают из группы, состоящей из C1-С 4 алкокси, С 1-С 4 алкилтио, С 1-С 4 ацила, трифторметила и галогена; или его фармацевтически приемлемой соли присоединения основания. 9. Способ по п.8, где восприимчивое новообразование является опухолью толстой или прямой кишки. 10. Фармацевтическая композиция, включающая соединение формулы IIR2a представляет собой водород или C1-С 4 алкокси;R2b представляет собой водород или C1-С 6 алкил при условии, что по крайней мере один из заместителей R2a и R2b является водородом;R4 представляет собой галоген, C1-С 6 алкил или CF3 при условии, что только один из заместителей 3R и R4 может являться C1-С 6 алкилом, и при условии, что если R4 является галогеном или C1-С 6 алкилом,то только один из заместителей R3 и R3a является водородом; где вышеуказанный замещенный фенил обозначает монозамещенный фенил, где заместитель выбирают из группы, состоящей из C1-С 4 алкокси, С 1-С 4 алкилтио, С 1-С 4 ацила, трифторметила и галогена; или его фармацевтически приемлемую соль присоединения основания; и фармацевтически приемлемый носитель, разбавитель или наполнитель.
МПК / Метки
МПК: A61K 31/18, C07D 213/34, C07C 311/51, A61P 35/00
Метки: качестве, сульфонилбензамидины, применения, противоопухолевых, бензоилсульфонамиды, агентов
Код ссылки
<a href="https://eas.patents.su/28-5810-benzoilsulfonamidy-i-sulfonilbenzamidiny-dlya-primeneniya-v-kachestve-protivoopuholevyh-agentov.html" rel="bookmark" title="База патентов Евразийского Союза">Бензоилсульфонамиды и сульфонилбензамидины для применения в качестве противоопухолевых агентов</a>
Производные дистамицина, способ их получения и применение в качестве противоопухолевых агентов

Номер патента: 2273
Опубликовано: 28.02.2002
Авторы: Кальдарелли Марина, Коцци Паоло, Берия Итало, Джерони Мария Кристина, Песенти Энрико
МПК: A61K 31/40, C07D 207/34, A61P 35/00...
Метки: способ, получения, производные, качестве, противоопухолевых, применение, агентов, дистамицина
Формула / Реферат:
1. Соединение, которое представляет собой производное дистамицина формулы (I) где n равен 2, 3 или 4; R0 представляет С1-С4-алкил или C1-С3-галогеналкил; R1 и R2, которые могут быть одинаковыми или различными, выбирают, каждый, из водорода, C1-C4-алкила, необязательно замещенного одним или несколькими атомами фтора, и C1-C4-алкокси; Х представляет атом галогена; В выбирают из групп следующих формул: где R3, R4, R5, R6, R7, R8 и R9, которые...
Комплексы лимфотоксина альфа/бета и антител против рецептора лимфотоксина-бета в качестве противоопухолевых агентов

Номер патента: 96
Опубликовано: 27.08.1998
Авторы: Бенджамин Кристофер, Броунинг Джеффри Л., Мейер Вернер
МПК: G01N 33/53, A61K 38/19
Метки: агентов, противоопухолевых, качестве, лимфотоксина-бета, рецептора, лимфотоксина, комплексы, против, антител
Формула / Реферат:
1. Способ лечения или уменьшения прогрессирования, тяжести или эффектов неоплазии, включающий введение терапевтически эффективного количества гетеромерного комплекса лимфотоксина a/b в присутствии терапевтически эффективного количества антитела к рецептору лимфотоксина b и/или g-интерферона. 2. Способ лечения или уменьшения прогрессирования, тяжести или эффектов неоплазии, включающий введение терапевтически эффективного количества антитела к...
Производные арилметилкарбониламинотиазола и их применение в качестве противоопухолевых средств

Номер патента: 5375
Опубликовано: 24.02.2005
Авторы: Салом Барбара, Варази Марио, Вульпетти Анна, Певарелло Паоло, Вилла Мануэла, Амичи Раффаэлла
МПК: A61P 35/00, C07D 277/46, A61K 31/426...
Метки: противоопухолевых, арилметилкарбониламинотиазола, применение, качестве, производные, средств
Формула / Реферат:
1. Производное 2-амино-1,3-тиазола, представленное формулой (I) где L представляет собой фенил или 1,3-тиазолильную группу; R представляет собой C1-C6алкил или моно- или дизамещенную аминогруппу, где заместитель выбран из C1-C6алкила, необязательно замещенного гидрокси или C1-C6алканоилокси; R2 и R3 независимо выбраны из H; гидрокси; C1-C6алкила, необязательно замещенного ди(C1-C6алкил)аминокарбонилом или C1-C6алкилпиперазинилом;...
Производные 3 (5)-аминопиразола, способ их получения и их применение в качестве противоопухолевых средств

Номер патента: 5373
Опубликовано: 24.02.2005
Авторы: Фритцен Эдвард Л., Браска Мария Габриелла, Певарелло Паоло, Орсини Паоло, Варази Марио, Тракуанди Габриэлла, Пирс Бетси С., Варпехоски Марта А.
МПК: C07D 231/40, A61K 31/415
Метки: производные, 5)-аминопиразола, противоопухолевых, способ, средств, применение, качестве, получения
Формула / Реферат:
1. Производное 3-аминопиразола, представленное формулой (I) где R представляет собой циклоалкильную группу, необязательно замещенную линейной или разветвленной алкильной или арилалкильной группой; R1 представляет собой линейную или разветвленную алкильную, алкенильную, циклоалкильную, циклоалкенильную, гетероциклическую, арильную, арилалкильную, арилкарбонильную, арилоксиалкильную или арилалкенильную группу, каждая из которых необязательно...
Производные 2-аминотиазола, способ их получения, фармкомпозиция и применение указанных веществ в качестве противоопухолевых средств

Номер патента: 5575
Опубликовано: 28.04.2005
Авторы: Тракуанди Габриэлла, Певарелло Паоло, Вульпетти Анна, Изакки Антонелла, Вилла Мануэла, Амичи Рафаэлла
МПК: A61K 31/426, C07D 277/46, A61P 35/00...
Метки: применение, средств, производные, указанных, получения, способ, качестве, 2-аминотиазола, противоопухолевых, фармкомпозиция, веществ
Формула / Реферат:
1. Применение производного 2-амино-1,3-тиазола формулы (I) где R представляет собой изопропил; R1 представляет собой необязательно дополнительно замещенную группу, выбранную из следующих групп: i) C1-C8 алкил или C2-C6 алкенил с прямой или разветвленной цепью; ii) 3-6-членное карбоциклическое или 5-7-членное гетероциклическое кольцо; iii) арил или арилкарбонил; iv) арилалкил, имеющий 1-8 атомов углерода в прямой или разветвленной алкильной...
Предыдущий патент: Производные хиназолина в качестве ингибиторов киназы
Следующий патент: Способ получения циталопрама