Дейтерированные производные изоиндолин-1,3-диона






Формула / Реферат
1. Соединение формулы I

в которой R1 выбран из СН3, CH2D, CHD2 и CD3;
R2 выбран из группы, состоящей из метила, изопропила, циклопентила, циклопропила, 2-фуранила, трифторметила, метоксиметила, аминометила, диметиламинометила, диметиламино-1-этила, 1-диметиламиноэтила и 2-диметиламиноэтила, где R2 необязательно замещен дейтерием;
R3 представляет собой CD3;
R4 представляет собой этильную группу, замещенную 0-5 атомами дейтерия, или представляет собой циклопентильную группу, замещенную 5-9 атомами дейтерия;
X выбран из С=О;
каждый из Y1a, Y1b, Y2, Y3, Y4, Y5, Y7 и Y8 независимо выбран из Н и D;
Y6 выбран из Cl, H и D;
где любой атом, не обозначенный как дейтерий, присутствует в его природном изотопном составе,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором
R2 представляет собой СН3 или CD3;
Y6, Y7 и Y8 являются одинаковыми и каждый представляет собой либо Н, либо D;
Y1a и Y1b являются одинаковыми и каждый представляет собой либо Н, либо D;
Y3, Y4 и Y5 являются одинаковыми и каждый представляет собой либо Н, либо D.
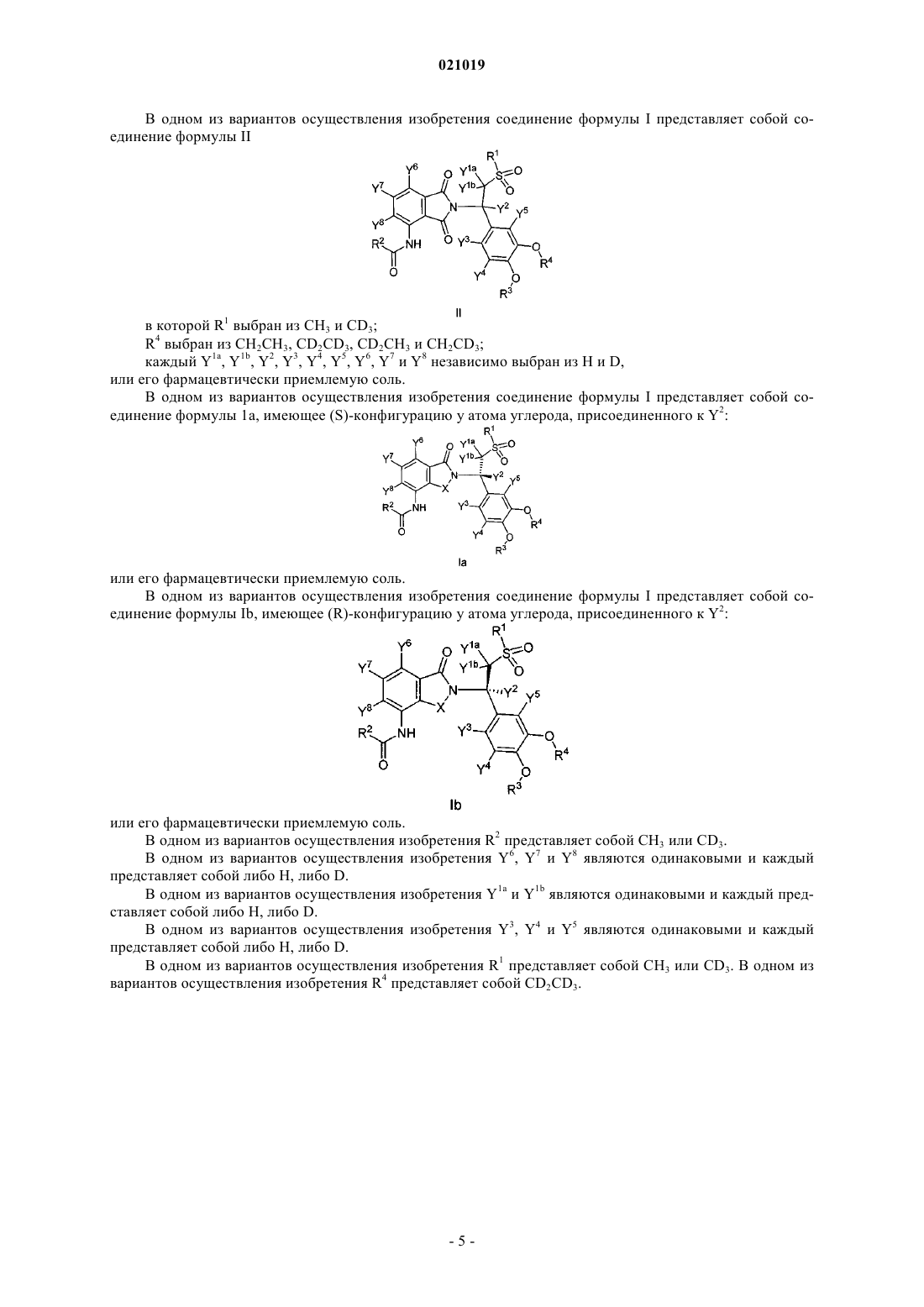
3. Соединение по п.1, в котором соединение формулы I представляет собой соединение формулы II

в которой R1 выбран из СН3 и CD3;
R4 выбран из СН2СН3, CD2CD3, CD2CH3 и CH2CD3;
каждый Y1a, Y1b, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 независимо выбран из Н и D,
или его фармацевтически приемлемую соль.
4. Соединение по п.2, в котором соединение формулы I представляет собой соединение формулы Ia, имеющее (S)-конфигурацию у атома углерода, присоединенного к Y2:

или его фармацевтически приемлемую соль.
5. Соединение по п.2, в котором соединение формулы I представляет собой соединение формулы Ib, имеющее (R)-конфигурацию у атома углерода, присоединенного к Y2:

или его фармацевтически приемлемую соль.
6. Соединение по п.1, в котором R2 представляет собой СН3 или CD3.
7. Соединение по п.1, в котором Y6, Y7 и Y8 являются одинаковыми и каждый представляет собой либо Н, либо D.
8. Соединение по п.1, в котором Y1a и Y1b являются одинаковыми и каждый представляет собой либо Н, либо D.
9. Соединение п.1, в котором Y3, Y4 и Y5 являются одинаковыми и каждый представляет собой либо Н, либо D.
10. Соединение по любому из пп.1, 2 или 4-9, в котором R1 представляет собой СН3 или CD3.
11. Соединение по любому из пп.1-10, в котором R4 представляет собой CD2CD3.
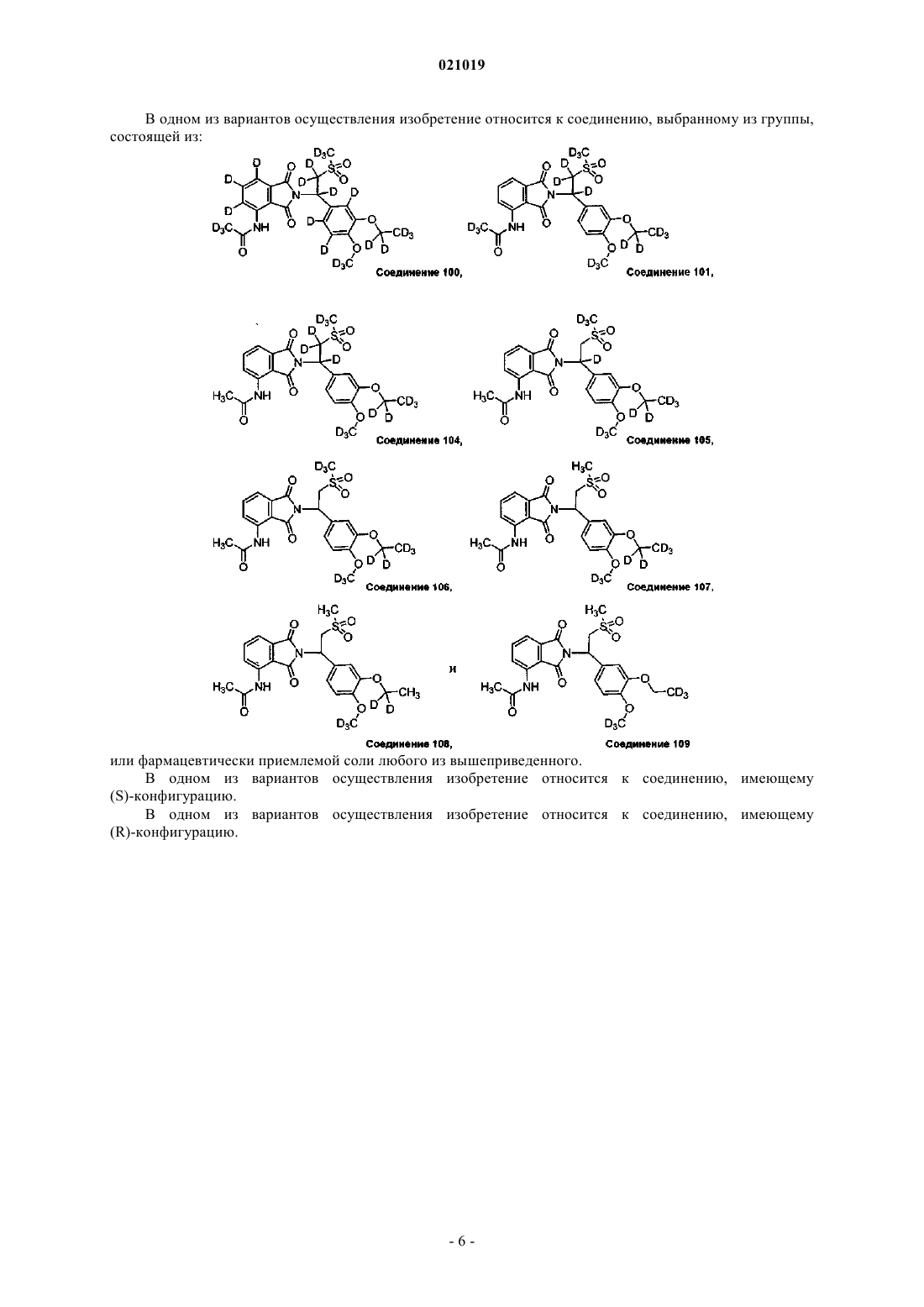
12. Соединение по п.1, выбранное из группы, состоящей из

или фармацевтически приемлемая соль любого из вышеприведенного.
13. Соединение по п.12, имеющее (S)-конфигурацию.
14. Соединение по п.12, имеющее (R)-конфигурацию.
15. Соединение по п.1, выбранное из группы, состоящей из

или фармацевтически приемлемая соль любого из вышеприведенного.
16. Фармацевтическая композиция для ингибирования PDE4 (фосфодиэстеразы 4 типа) или снижения уровней TNF-α у субъекта, нуждающегося в этом, содержащая эффективное количество соединения по п.1 или его фармацевтически приемлемую соль и приемлемый носитель.
17. Фармацевтическая композиция, содержащая эффективное количество соединения по п.1 или его фармацевтически приемлемую соль и приемлемый носитель, отличающаяся тем, что композиция подходит для лечения заболевания, выбранного из группы, состоящей из септического шока, сепсиса, эндотоксического шока, гемодинамического шока и сепсисоподобного синдрома, постишемического реперфузионного повреждения, малярии, микобактериальной инфекции, менингита, псориаза, саркоидоза, псориатического артрита, болезни Бехчета, узелковой почесухи, волчанки, увеита, застойной сердечной недостаточности, фиброза, кахексии, отторжения трансплантата, рака, аутоиммунного заболевания, оппортунистических инфекций при СПИДе, ревматоидного артрита, ревматоидного спондилита, остеоартроза, других артрогенных заболеваний, болезни Крона, неспецифического язвенного колита, рассеянного склероза, системного волчаночного эритрематоза, эритемы лепрозной узловатой при проказе, радиационного повреждения, альвеолярного повреждения, вызванного гипероксией, нежелательного ангиогенеза, воспалительного заболевания, артрита, воспалительного заболевания кишечника, афтозных язв, астмы, дистресс-синдрома дыхательных путей у взрослых и СПИДа.
18. Способ ингибирования PDE4 (фосфодиэстеразы 4 типа) у пациента, нуждающегося в этом, содержащий введение пациенту эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
19. Способ снижения уровней TNF-α (фактора некроза опухолей альфа), содержащий введение пациенту эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
20. Способ лечения заболевания, выбранного из группы, состоящей из септического шока, сепсиса, эндотоксического шока, гемодинамического шока и сепсисоподобного синдрома, постишемического реперфузионного повреждения, малярии, микобактериальной инфекции, менингита, псориаза, саркоидоза, псориатического артрита, болезни Бехчета, узелковой почесухи, волчанки, увеита, застойной сердечной недостаточности, фиброза, кахексии, отторжения трансплантата, рака, аутоиммунного заболевания, оппортунистических инфекций при СПИДе, ревматоидного артрита, ревматоидного спондилита, остеоартроза, других артрогенных заболеваний, болезни Крона, неспецифического язвенного колита, рассеянного склероза, системного волчаночного эритрематоза, эритемы лепрозной узловатой при проказе, радиационного повреждения, альвеолярного повреждения, вызванного гипероксией, нежелательного ангиогенеза, воспалительного заболевания, артрита, воспалительного заболевания кишечника, афтозных язв, астмы, дистресс-синдрома дыхательных путей у взрослых и СПИДа, у пациента, нуждающегося в этом, содержащий введение пациенту эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
21. Способ по п.20, отличающийся тем, что заболевание представляет собой псориаз или саркоидоз.
22. Способ по п.21, отличающийся тем, что псориаз представляет собой псориаз бляшечного типа или рефракторный псориаз.
23. Способ по п.21, отличающийся тем, что саркоидоз представляет собой кожный саркоидоз.
24. Способ по п.20, отличающийся тем, что волчанка представляет собой кожную волчанку.
25. Способ по п.20, отличающийся тем, что заболевание представляет собой псориатический артрит.
26. Способ по п.20, отличающийся тем, что заболевание представляет собой ревматоидный артрит.
27. Способ по п.20, отличающийся тем, что заболевание представляет собой болезнь Бехчета.
28. Способ по п.20, отличающийся тем, что заболевание представляет собой болезнь Крона.
29. Способ по п.20, отличающийся тем, что заболевание представляет собой системный волчаночный эритрематоз.
30. Способ по п.20, отличающийся тем, что заболевание представляет собой воспалительное заболевание кишечника.
Текст
и к его фармацевтически приемлемым солям. Более конкретно, изобретение относится к дейтерированным производным изоиндолин-1,3-диона, которые являются аналогами апремиласта. Это изобретение также относится к композициям, содержащим соединение по данному изобретению и носитель, и к использованию соединений и композиций в способах лечения заболеваний, расстройств и состояний, которые успешно лечатся посредством введения апремиласта. Уровень техники Многие современные лекарственные средства характеризуются неудовлетворительными свойствами в отношении всасывания, распределения, метаболизма и/или выведения (ADME), которые препятствуют их более широкому применению или ограничивают их использование при некоторых показаниях. Неудовлетворительные свойства ADME являются также главной причиной неудачи кандидатов лекарственных средств при клинических испытаниях. Хотя в некоторых случаях для улучшения свойствADME могут использоваться определенные технологии составления рецептур и пролекарственные стратегии, эти подходы часто не решают основных проблем в отношении ADME, которые существуют для многих лекарственных средств и их кандидатов. Одной из таких проблем является быстрый обмен веществ, который способствует слишком быстрому выведению из организма некоторых препаратов, которые в противном случае были бы весьма эффективны при лечении заболевания. Возможным решением проблемы быстрого выведения лекарственного средства являются частые или высокие дозировки для достижения достаточно высокого уровня лекарственного средства в плазме. Однако это вводит ряд потенциальных проблем при лечении, таких как плохое соблюдение пациентом режима дозирования, побочные эффекты, которые обостряются при более высоких дозах, и увеличение стоимости лечения. Быстро метаболизируемое в организме лекарственное средство может также подвергнуть пациентов действию нежелательных токсичных или активных метаболитов. Другое ограничение в отношении ADME, которому подвергаются многие лекарственные средства,состоит в образовании токсических или биологически активных метаболитов. В результате, некоторые пациенты, принимающие препарат, могут испытывать интоксикацию или безопасная дозировка такого лекарственного средства может оказаться недостаточной, так что пациенты получат недостаточное количество активного вещества. В некоторых случаях изменение интервалов дозирования или рецептурные подходы могут помочь уменьшить клинические побочные эффекты, однако часто образование таких нежелательных метаболитов свойственно метаболизму этого соединения. В некоторых отдельных случаях одновременно с лекарственным средством, которое выводится слишком быстро, вводится ингибитор метаболизма. Так обстоит дело в случае с классом препаратов,ингибирующих протеазу, которые используются для лечения ВИЧ-инфекции. FDA (Управление по контролю за лекарственными средствами и пищевыми продуктами) рекомендует, чтобы эти препараты дозировали совместно с ритонавиром, ингибитором фермента 3 А 4 (CYP3A4) семейства цитохрома Р 450,который обычно отвечает за их метаболизм (см. Kempf, D.J. et al., Antimicrobial agents and chemotherapy,1997, 41(3):654-60). Ритонавир, однако, вызывает побочные эффекты и к этой таблетке добавляет вторичную нагрузку для ВИЧ-инфицированных пациентов, которые должны принимать уже комбинацию различных лекарственных средств. Подобным образом, ингибитор CYP2D6 хинидин был добавлен к декстрометорфану с целью снижения опосредованного CYP2D6 быстрого метаболизма декстрометорфана при лечении аффективной лабильности. Хинидин, однако, имеет нежелательные побочные эффекты, что в значительной степени ограничивает его использование в потенциальной комбинированной терапии (см. Wang, L. et al.,Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt. 1):659-67 и FDA этикетку для хинидина наwww.accessdata.fda.gov). В общем, сочетание препаратов с ингибиторами цитохрома Р 450 не является удовлетворительной стратегией для снижения выведения лекарственного средства из организма. Ингибирование активности фермента CYP может влиять на метаболизм и выведение других лекарственных веществ, которые метаболизируются тем же самым ферментом. Ингибирование CYP может привести к тому, что в организме будут накапливаться до токсичного уровня другие лекарственные вещества. Потенциально привлекательной стратегией улучшения метаболических свойств лекарственного вещества является его модификация дейтерием. При таком подходе пытаются замедлитьCYP-опосредованный метаболизм лекарственного средства или снизить образование нежелательных метаболитов путем замены одного или нескольких атомов водорода на атомы дейтерия. Дейтерий является безопасным, стабильным, нерадиоактивным изотопом водорода. По сравнению с водородом, дейтерий образует более прочные связи с углеродом. В отдельных случаях увеличенная прочность связи, обеспеченная дейтерием, может положительно отразиться на ADME свойствах лекарственного средства, создавая потенциал для повышения эффективности препарата, безопасности и/или переносимости. В то же время, поскольку размер и форма дейтерия в основном идентичны размеру и форме атомов водорода,замена водорода на дейтерий, как ожидается, не будет влиять на биохимическую активность и избирательность лекарственного средства по сравнению с оригинальной химической формой, которая содержит только водород. За последние 35 лет влияние замещения водорода дейтерием на скорость метаболизма отмечалось для очень небольшого процента утвержденных лекарственных средств (см., например, Blake, M.I. et al., J.("Fisher". Результаты были непостоянны и не предсказуемы. Дейтерирование некоторых соединений приводило к уменьшенному метаболическому выведению in vivo. В других случаях не было никаких изменений метаболизма. Прочие демонстрировали увеличение метаболического выведения. Изменчивость воздействий дейтерия привела также экспертов к вопросу, следует ли отвергать модификацию посредством дейтерия как жизнеспособную стратегию при разработке лекарственных препаратов для ингибирования побочных эффектов метаболизма (см. Foster at p. 35 and Fisher at p. 101). Влияние дейтериевой модификации на метаболические свойства лекарственного вещества не предсказуемы даже тогда, когда атомы дейтерия включаются в известные места метаболизма. Только путем фактического получения и исследования дейтерированного лекарственного средства можно определить,изменится ли скорость метаболизма и как она будет отличаться от своего недейтерированного аналога. См., например, Fukuto et al. (J. Med. Chem. 1991, 34, 2871-76). Многие лекарственные вещества имеют многочисленные места, где возможен метаболизм. Место(а), где требуется замещение водорода дейтерием, и степень необходимого дейтерирования, чтобы увидеть его влияние на метаболизм, если таковое существует, будут различными для каждого лекарственного средства. Апремиласт,известный также какPDE4 (фосфодиэстеразы), а также способствует снижению уровней TNF- (фактора некроза опухолей альфа). Апремиласт проходит клинические испытания для лечения псориаза, бляшечного псориаза, рефракторного псориаза, кожного саркоидоза, псориатического артрита, болезни Бехчета, узелковой почесухи, кожной волчанки, увеита и др. Общие побочные явления, связанные с ингибиторами PDE4, обычно включают в себя головную боль, тошноту, рвоту и желудочно-кишечные расстройства. Было бы желательным обеспечить соединение, которое имеет полезные функции апремиласта и другие преимущества, например снижение негативных побочных эффектов с уменьшением подверженности метаболизму, с целью расширения его фармакологической эффективной жизни, повышения подверженности пациента к лечению и, потенциально, уменьшения совокупной фармакокинетической изменчивости и/или уменьшения его возможности опасных лекарственных взаимодействий. Сущность изобретения Настоящее изобретение относится к новым замещенным производным изоиндолин-1,3-диона и к его фармацевтически приемлемым солям. Более конкретно, изобретение относится к новым замещенным производным изоиндолин-1,3-диона,которые являются аналогами апремиласта. Это изобретение также относится к композициям, содержащим соединение по данному изобретению, и носителю, а также к использованию раскрытых соединений и композиций в способах лечения заболеваний, расстройств и состояний, которые успешно лечатся посредством введения апремиласта. Подробное описание изобретения Термин "лечение" означает снижение, подавление, ослабление, уменьшение, остановку или стабилизацию развития или прогрессирования заболевания (например, заболевания или расстройства описанных здесь), уменьшение тяжести заболевания или улучшение симптомов, связанных с болезнью. Термин "заболевание" означает любое состояние или расстройство, которое повреждает или препятствует нормальному функционированию клетки, ткани или органу. Следует отметить, что в синтезированном соединении наблюдается некоторое варьирование содержания природных изотопов в зависимости от происхождения химических материалов, используемых при синтезе. Так, препарат апремиласт будет изначально содержать небольшое количество дейтерированных изотопологов. Концентрация распространенных в природе стабильных изотопов водорода и углерода,невзирая на это варьирование, невелика и не существенна, по сравнению со степенью изотопного замещения соединений по настоящему изобретению. См., например, Wada E. et al., Seikagaku 1994, 66:15;Gannes L.Z. et al., Comp. Biochem. Physiol. Mol. Integr. Physiol. 1998, 119:725. В соединениях по данному изобретению в случае если какой-либо атом не обозначен специально как определенный изотоп, то это означает, что он представляет собой любой стабильный изотоп этого атома. Если не указано иное, то, когда позиция обозначена конкретно как "Н" или "водород", понимается, что на этой позиции находится водород, в его природном изотопном составе. А также, если не указано иное и если позиция обозначена конкретно как "D" или "дейтерий", то понимается, что на этой позиции находится дейтерий, состав которого по меньшей мере в 3340 раз больше, чем природный изотопный состав дейтерия, который составляет 0,015% (т.е. включение дейтерия составляет по меньшей мере 50,1%). Термин "фактор изотопного обогащения", который используется здесь, означает соотношение между изотопным составом и природным изотопным составом определенного изотопа. В других вариантах осуществления соединение по настоящему изобретению имеет фактор изотопного обогащения для каждого указанного атома дейтерия по меньшей мере 3500 (включение дейтерия на каждый указанный атом дейтерия составляет 52,5%), по меньшей мере 4000 (60% включение дейтерия),по меньшей мере 4500 (67,5% включение дейтерия), по меньшей мере 5000 (75% дейтерия), по меньшей мере 5500 (82,5% включение дейтерия), по меньшей мере 6000 (90% включение дейтерия), по меньшей мере 6333,3 (95% включение дейтерия), по меньшей мере 6466,7 (97% включение дейтерия), по меньшей мере 6600 (99% включение дейтерия) или по меньшей мере 6633,3 (99,5% включение дейтерия). Термин "изотополог" относится к видам, химическая структура которых отличается от конкретного соединения по настоящему изобретению только их изотопным составом. Термин "соединение", относящийся к соединению по настоящему изобретению, относится к совокупности молекул, имеющих идентичную химическую структуру, за исключением того, что могут быть изотопные изменения среди атомов, составляющих молекулу. Таким образом, специалисту в данной области будет ясно, что соединение, представленное конкретной химической структурой, содержащей указанные атомы дейтерия, будет также содержать меньшее количество изотопологов, имеющих водородные атомы на одной или более позициях в этой структуре, обозначенных как дейтерий. Относительное количество таких изотопологов в соединении по данному изобретению будет зависеть от ряда факторов,включая изотопную чистоту дейтерированных реагентов, используемых для получения соединения, и эффективность включения дейтерия на различных этапах синтеза, используемого для получения соединения. Однако, как указано выше, относительное количество таких изотопопогов in toto будет менее чем 49,9% от соединения. В других вариантах осуществления относительное количество таких изотопологовin toto будет менее чем 47,5%, менее чем 40%, менее чем 32,5%, менее чем 25%, менее чем 17,5%, менее чем 10%, менее чем 5%, менее чем 3%, менее чем 1% или менее чем 0,5% от соединения. Изобретение относится также к солям соединений по изобретению. Соль соединения по настоящему изобретению образуется путем взаимодействия кислоты с основной группой соединения, такой как функциональная аминогруппа, или путем взаимодействия основания с кислотной группой соединения, такой как функциональная карбоксильная группа. Согласно другому варианту осуществления изобретения соединение представляет собой фармацевтически приемлемую соль, полученную путем взаимодействия соединения с кислотой. Термин "фармацевтически приемлемый", используемый здесь, относится к компоненту, который по результатам тщательной медицинской оценки пригоден для использования в контакте с тканями человека и других млекопитающих без неоправданных токсических явлений, раздражения, аллергических реакций и т.п. и применение которого соизмеримо с разумным соотношением польза/риск. "Фармацевтически приемлемая соль" означает любую нетоксичную соль, которая при введении пациенту способна обеспечить либо прямо, либо косвенно соединение по данному изобретению. "Фармацевтически приемлемый противоион" представляет собой ионную часть соли, которая не является токсичной, когда освобождается от соли после введения пациенту. Кислоты, обычно используемые для образования фармацевтически приемлемых солей, включают неорганические кислоты, такие как сероводородная, соляная, бромисто-водородная, йодисто-водородная,серная и фосфорная кислоты; органические кислоты, такие как паратолуолсульфоновая, салициловая,винная, дивинная, аскорбиновая, малеиновая, безиловая, фумаровая, глюконовая, глюкуроновая, муравьиная, глутаминовая, метансульфоновая, этансульфоновая, бензолсульфоновая, молочная, щавелевая, парабромфенилсульфоновая, угольная, янтарная, лимонная, бензойная и уксусная кислоты; а также соли неорганических и органических кислот. Такие фармацевтически приемлемые соли, таким образом,включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себацат,фумарат, малеат, бутин-1,4-диоат, гексин-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, терефталат, сульфонат, ксилолсульфонат, фенилацетат,фенилпропионат, фенилбутират, цитрат, лактат, -гидроксибутират, гликолят, малеат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и другие соли. В одном варианте осуществления изобретения фармацевтически приемлемые соли, полученные взаимодействием с кислотой, включают соли минеральных кислот, таких как соляная и бромисто-водородная кислоты, и особенно соли органических кислот, таких как малеиновая кислота. Фармацевтически приемлемой солью также может быть соль соединения по настоящему изобретению, имеющая кислотную функциональную группу, такую как функциональная группа, карбоновой кислоты, а также основания. Примеры оснований включают, не ограничиваясь таковыми, гидроксид щелочных металлов, включая натрий, калий и литий; гидроксиды щелочно-земельных металлов, таких как кальций и магний; гидроксиды других металлов, таких как алюминий и цинк; аммиак, органические амины, такие как незамещенные или гидроксилзамещенные моно-, ди- или триалкиламины, дициклогексиламин; трибутиламин; пиридин; N-метил, N-этиламин; диэтиламин; триэтиламин; моно-,бис- или трис-(2-ОН-(С 1-С 6)алкиламин), такие как N,N-диметил-N-(2-гидроксиэтил)амин или три(2-гидроксиэтил)амин, N-метил-D-глюкамин, морфолин; тиоморфолин; пиперидин, пирролидин и аминокислоты, такие как аргинин, лизин, и т.п. Соединения по настоящему изобретению (например, соединения формулы I) могут содержать асимметричный атом углерода, например, в результате замещения дейтерия или иным путем. Как таковые, соединения по настоящему изобретению могут существовать либо в виде отдельных энантиомеров,либо в виде смеси двух энантиомеров. Соответственно, соединение по настоящему изобретению может существовать либо как рацемическая смесь, либо как скалемическая смесь, такая как смесь, содержащая преимущественно один стереоизомер, или в виде отдельных соответствующих стереоизомеров, которые,по существу, являются свободными от другого возможного стереоизомера. Термин "по существу, свободны от других стереоизомеров", используемый здесь, означает менее чем от 25% других стереоизомеров, предпочтительно менее чем от 10% других стереоизомеров, более предпочтительно менее чем от 5% других стереоизомеров и наиболее предпочтительно менее чем от 2% других стереоизомеров, о которых идет речь. Способы получения или синтеза отдельных энантиомеров для данного соединения являются известными в данной области и могут применяться на практике для конечных соединений, или исходного материала, или промежуточных продуктов. Если не указано иное, то, когда раскрытое соединение называется или изображается в виде структуры, без указания стереохимии, и имеет один или несколько хиральных центров, следует понимать, что оно представляет все возможные стереоизомеры этого соединения. Термин "устойчивые соединения", используемый здесь, относится к соединениям, которые обладают устойчивостью, достаточной для того, чтобы позволить их изготовление, и которые сохраняют целостность в течение достаточного периода времени, чтобы быть полезными для целей, изложенных в настоящем документе (например, для составления рецептуры терапевтических продуктов, промежуточных продуктов для использования в производстве терапевтических соединений, возможности выделения или длительного хранения промежуточных соединений, лечения заболеваний или расстройств, реагирующих на терапевтические агенты). Символы "D" и "d", оба, относятся к дейтерию; "стереоизомер" относится как к энантиомерам, так и диастереомерам; "трет" и "т-", каждый, относятся к третичному; "США" относится к Соединенным Штатам Америки."Замещенный дейтерием" относится к замене одного или нескольких атомов водорода соответствующим числом атомов дейтерия. По всему описанию переменная может быть отнесена к "обычно" (например, "каждый R") или может быть отнесена к "конкретно" (например, R1, R2, R3 и др.). Если не указано иное, то, когда переменная отнесена к "обычно", это означает, что включены все конкретные варианты осуществления этой конкретной переменной. Терапевтические соединения Настоящее изобретение относится к соединению формулы I в которой R1 выбран из СН 3, CH2D, CHD2 и CD3;R4 представляет собой этильную группу, замещенную 0-5 атомами дейтерия, или представляет собой циклопентильную группу, замещенную 0-9 атомами дейтерия;X выбран из С=О; каждый из Y1a, Y1b, Y2, Y3, Y4, Y5, Y7 и Y8 независимо выбран из Н и D;Y6 выбран из Cl, H и D,или его фармацевтически приемлемой соли. В одном из вариантов осуществления изобретенияR2 представляет собой СН 3 или CD3;Y6, Y7 и Y8 являются одинаковыми и каждый представляет собой либо Н, либо D;Y1a и Y1b являются одинаковыми и каждый представляет собой либо Н, либо D;Y3, Y4 и Y5 являются одинаковыми и каждый представляет собой либо Н, либо D. В одном из вариантов осуществления изобретения соединение формулы I представляет собой соединение формулы II в которой R1 выбран из СН 3 и CD3;R4 выбран из СН 2 СН 3, CD2CD3, CD2CH3 и CH2CD3; каждый Y1a, Y1b, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 независимо выбран из Н и D,или его фармацевтически приемлемую соль. В одном из вариантов осуществления изобретения соединение формулы I представляет собой соединение формулы 1 а, имеющее (S)-конфигурацию у атома углерода, присоединенного к Y2: или его фармацевтически приемлемую соль. В одном из вариантов осуществления изобретения соединение формулы I представляет собой соединение формулы Ib, имеющее (R)-конфигурацию у атома углерода, присоединенного к Y2: или его фармацевтически приемлемую соль. В одном из вариантов осуществления изобретения R2 представляет собой СН 3 или CD3. В одном из вариантов осуществления изобретения Y6, Y7 и Y8 являются одинаковыми и каждый представляет собой либо Н, либо D. В одном из вариантов осуществления изобретения Y1a и Y1b являются одинаковыми и каждый представляет собой либо Н, либо D. В одном из вариантов осуществления изобретения Y3, Y4 и Y5 являются одинаковыми и каждый представляет собой либо Н, либо D. В одном из вариантов осуществления изобретения R1 представляет собой СН 3 или CD3. В одном из вариантов осуществления изобретения R4 представляет собой CD2CD3. В одном из вариантов осуществления изобретение относится к соединению, выбранному из группы,состоящей из: или фармацевтически приемлемой соли любого из вышеприведенного. В одном из вариантов осуществления изобретение относится к соединению, имеющему(S)-конфигурацию. В одном из вариантов осуществления изобретение относится к соединению, имеющему В одном из вариантов осуществления изобретение относится к соединению, выбранному из группы,состоящей из: или фармацевтически приемлемой соли любого из вышеприведенного. В одном из вариантов осуществления изобретение относится к соединению, в котором любой атом,не обозначенный как дейтерий, присутствует в его природном изотопном составе. Синтез соединений формулы I может быть легко осуществлен специалистом в данной области. Соответствующие методики и промежуточные продукты раскрыты, например, в Man, H.W. et al., Journal ofMedicinal Chemistry (2009), 52(6), 1522-1524; Muller, G.W. et al. Journal of Medicinal Chemistry (1996),39(17), 3238-3240; WO 2006/025991; AU 2006/200033; WO 2001/034606; патентах США 6020358 и U.S. 6667316. Такие способы могут выполняться с использованием соответствующих дейтерированных и, необязательно, других, содержащих изотопы реагентов и/или промежуточных химических соединений, чтобы получить описанные здесь соединения, или с использованием стандартных протоколов синтеза, известных в данной области техники, для введения изотопных атомов в химическую структуру. Некоторые промежуточные соединения могут быть использованы с очисткой или без очистки (например, фильтрации, дистилляции, сублимации, кристаллизации, тритурации, твердофазной экстракции и хроматографии). Примерный синтез Схема 1 Общий способ получения соединений формулы I На схеме 1 показан общий способ получения соединений формулы I в соответствии с общими способами, Man H.W., et al. Journal of Medicinal Chemistry (2009), 52(6), 1522-1524. Так, соответственно замещенный альдегид 10 обрабатывали гексаметилдисилазидом лития, затем диметилсульфоном лития и бортрифторидэфиратом, чтобы обеспечить рацемический амин 11, который имеет стереоцентр на атоме углерода, связанном с Y2. При желании, рацемический амин 11 может быть разделен путем обработки энантиочистой кислотой в метаноле. Например, обработка рацемического амина 11 N-ацетил-Lлейцином обеспечивает амин 11, как S-энантиомер, в то время как обработка N-ацетил-D-лейцином обеспечивает амин 11, как R-энантиомер. Амин 11 может быть использован как рацемическая смесь, какS-энантиомер или как R-энантиомер, для получения соединений формулы I после обработки ангидридом 12 либо в чистом виде, либо в растворителе, таком как уксусная кислота. Специалист в данной области оценит, какое использование соответствующим образом дейтерированных промежуточных соединений и реагентов по схеме 1 приведет в результате к получению соединений формулы I, имеющих различные схемы замещения дейтерием. Схема 2 Получение альдегида 10 На схеме 2 показано получение альдегида 10, который является подходящим исходным материалом для схемы 1. Как в общем описано Li, Juren, et al. Hecheng Huaxue (1993), 1(4), 333-40, дейтерированный диол 13 обрабатывали соответственно дейтерированным этилбромидом 14 в условиях фазового перехода, чтобы обеспечить фенол 15. Реакция Раймера-Тимана фенола 15 с хлороформом обеспечивает альдегид 16. На этом этапе могут быть использованы дейтерированные реагенты и растворители, чтобы максимально повысить уровни изотопного включения. В альтернативном варианте, могут быть использованы свойства тетрабутиламмония бромида, в общем описанные Li, Ying-chun, et al. Yingyong Huagong(2004), 33(1), 26-27, чтобы превратить 15 в 16. Согласно общим способам Kiehlmann, E., et al., OrganicPreparations and Procedures International (1982), 14(5), 337-42 обработка 16 соответственно дейтерированным диметилсульфатом 17 обеспечивает желаемое промежуточное соединение 10. Например, коммерчески доступный диметил-d6 сульфат может быть использован в качестве реагента 17 по схеме 2, чтобы в конечном счете получить соединения формулы I, в которых R3 представляет собой CD3. В другом примере коммерчески доступный бромэтан-d5 может быть использован в качестве реагента 14 по схеме 2, чтобы в конечном счете получить соединения формулы I, в которых R4 представляет собой CD2CD3. Подобным образом, коммерчески доступный бромэтан-2,2,2-d3 и бромэтан-1,1-d2 также могут быть использованы по схеме 2, чтобы в конечном счете получить соединения формулы I,имеющие другие различные схемы замещения дейтерием на R4. Схема 3 Получение промежуточных соединений 12 а и 12b На схеме 3 показано получение промежуточного соединения 12 а, пример промежуточного соединения 12, в котором X представляет собой C=O, и промежуточного соединения 12b, пример промежуточного соединения 12, в котором X представляет собой CH2, CHD или CD2. Нитрование ангидридного каркаса 18 хорошо известно в литературе, например в патентных заявках WO 2005/051870, CN 1740138 и(2002), 76(4), 511-517 и Culhane, P.I., et al. Organic Syntheses (1927), 7; номера страниц нигде не указаны. Использование соответственно дейтерированных исходных материалов и реагентов приводит к получению дейтерированных версий 19. Согласно общим способам, описанным в заявке к патенту СШАUS 2008/234359, гидрирование 19 в присутствии палладия на угле обеспечивает 20, который затем обрабатывается соответственно дейтерированным уксусным ангидридом 21, чтобы обеспечить промежуточное соединение 12 а. Согласно общим способам Wamser, С.С., et al. J. Org. Chem. (1976), 41(17), 2929-31 промежуточное соединение 12 а может быть восстановлено цинком и кислотой, чтобы обеспечить промежуточное соединение 12b. Коммерчески доступные DCl, уксусная кислота-d4 и уксусный ангидрид-d6 могут быть использованы на заключительном этапе для обеспечения альтернативных схем введения дейтерия. Например, коммерчески доступная 3-аминофталевая кислота может быть использована по схеме 3 в качестве промежуточного соединения 20, чтобы в конечном счете получить соединения формулы I, в которых Y6, Y7 и Y8, все, представляют собой водород. В другом примере коммерчески доступный уксусный ангидрид-d6 может быть использован по схеме 3 в качестве реагента 21, чтобы в конечном счете получить соединения формулы I, в которых R2 представляет собой CD3. Еще в другом примере коммерчески доступный фталевый ангидрид-d4 может быть использован по схеме 3, как ангидрид 18, чтобы в конечном счете обеспечить соединения формулы I, в которых Y6, Y7 и Y8, все, представляют собой дейтерий. В настоящем изобретении предусмотрены только те комбинации заместителей и переменные факторы, которые приводят к образованию устойчивых соединений. Композиции Настоящее изобретение относится также к композициям, содержащим эффективное количество соединения формулы I (например, включая любую из формул в этом документе) или его фармацевтически приемлемой соли, а также приемлемый носитель. Носитель(и) является "приемлемым" в смысле совместимости с другими ингредиентами лекарственного средства, и в случае фармацевтически приемлемого носителя он не наносит вред пациенту, получающему его вместе с используемым лекарственным средством. Фармацевтически приемлемые носители, вспомогательные вещества и наполнители, которые могут быть использованы в фармацевтических композициях по этому изобретению, включают, не ограничиваясь таковыми, иониты, алюминия оксид, алюминия стеарат, лецитин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота, сорбат калия, смеси частичных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протамина сульфат, динатрия гидрофосфат, калия гидрофосфат, хлорид натрия, соли цинка, коллоидный диоксид кремния, магния трисиликат, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрия карбоксиметилцеллюлоза, полиакрилаты, воски, полиэтилен-полиоксипропилен-блок-сополимеры, полиэтиленгликоль и ланолин. Фармацевтические композиции по изобретению включают композиции, пригодные для перорального, ректального, назального, местного (в том числе внутриротового и под язык), вагинального или парентерального (включая подкожные, внутримышечные, внутривенные и внутрикожные) введения. В некоторых вариантах осуществления соединения формулы по этому изобретению вводятся через кожу (например, с помощью трансдермальных пластырей или с использованием ионтофоретического метода). Другие лекарственные средства могут быть для удобства представлены в виде стандартной лекарственной формы, например таблеток, капсул с замедленным высвобождением и в форме липосом, и могут быть изготовлены любыми способами, хорошо известными в области фармацевтики. См., например,Remington: The Science and Practice of Pharmacy, Lippincott WilliamsWilkins, Baltimore, MD (20th ed. 2000). Такие способы изготовления включают этап ассоциации с молекулой для введения ингредиентов,таких как носитель, который состоит из одного или более вспомогательных ингредиентов. В общем,композиции готовят путем однородного и тщательного соединения активных ингредиентов с жидкими носителями, липосомами или тонкоизмельченными твердыми носителями или обоими и затем, при необходимости, формирования продукта. В некоторых вариантах осуществления соединение вводится перорально. Композиции по настоящему изобретению, пригодные для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, саше или таблетки, каждая из которых содержит предопределенное количество активного ингредиента; порошка или гранул; раствора или суспензии в водной или неводной жидкости; жидкой эмульсии масло в воде; жидкой эмульсии вода в масле; упакованными в липосомы либо в виде болюса и т.д. Мягкие желатиновые капсулы могут использоваться для включения таких суспензий,которые могут существенно увеличить скорость абсорбции соединения. В случае таблеток для перорального введения носители, которые обычно используются, включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в форме капсул пригодные разбавители включают лактозу и сухой кукурузный крахмал. Когда вводят перорально водные суспензии, активный ингредиент объединяется с эмульгирующими и суспендирующими веществами. При желании, могут быть добавлены некоторые подслащивающие и/или ароматизирующие вещества и/или красители. Композиции, пригодные для перорального введения, включают таблетки для рассасывания, содержащие ингредиенты в ароматизированной основе, обычно сахарозе и камеди или трагаканта; и пастилки,содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и камедь. Композиции, подходящие для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические средства и растворенные вещества, которые делают лекарственное средство изотоническим с кровью предполагаемого пациента, а также водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут быть представлены в форме однодозовых или многодозовых контейнеров, например запаянных ампул и флаконов, и могут храниться в сублимированном(лиофилизированном) состоянии, требуя лишь добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием. Приготовленные для немедленного введения растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток. Такие растворы для инъекций могут быть, например, в форме стерильной инъекционной водной или масляной суспензии. Эта суспензия может быть получена в соответствии с методами, известными в данной области, с использованием подходящих диспергирующих или смачивающих агентов (таких как,например, Твин 80) и суспендирующих агентов. Стерильное инъекционное лекарственное средство может также представлять стерильный инъекционный раствор или суспензию в нетоксичном парентерально-приемлемом разбавителе или растворителе, например в виде раствора в 1,3-бутандиоле. Среди приемлемых разбавителей и растворителей, которые могут быть использованы, находятся маннитол, вода,раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. Для этой цели может быть использовано любое легкое жирное масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, пригодны при подготовке инъекционных лекарственных средств, как и природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Эти масляные растворы или суспензии могут также содержать длинноцепочечный спирт в качестве разбавителя или диспергирующего вещества. Фармацевтические композиции по настоящему изобретению могут вводиться в форме суппозиториев для ректального введения. Эти композиции могут быть получены путем смешивания соединения по данному изобретению с подходящим не раздражающим формообразующим наполнителем, который является твердым при комнатной температуре, но становится жидким при ректальной температуре и, следовательно, будет таять в прямой кишке, чтобы освободить активные компоненты. Такие материалы включают, не ограничиваясь таковыми, какао-масло, пчелиный воск и полиэтиленгликоли. Фармацевтические композиции по настоящему изобретению можно вводить в виде назального аэрозоля или ингаляцией. Такие композиции получают в соответствии с методами, хорошо известными в области фармацевтических препаратов. Они могут быть приготовлены в виде растворов в физиологическом растворе с использованием бензилового спирта или других подходящих консервантов, абсорбционных стимуляторов для повышения биодоступности, фторуглеродов и/или других солюбилизирующих или диспергирующих добавок, известных в данной области. См., например: Rabinowitz J.D. andZaffaroni A.C., US Patent 6803031, assigned to Alexza Molecular Delivery Corporation. Местное применение фармацевтических композиций по настоящему изобретению является особенно полезным, когда желаемое лечение включает области или органы, которые легкодоступны для местного применения. Для применения местно на коже фармацевтическая композиция должна быть приготовлена с использованием подходящей мази, содержащей активные компоненты, суспендированные или растворенные в носителе. Носители для местного применения соединений по изобретению включают, не ограничиваясь таковыми, минеральные масла, жидкие нефтепродукты, белую нефть, пропиленгликоль,полиоксиэтилен полиоксипропилена, эмульсионный воск и воду. Кроме того, фармацевтическая композиция может быть приготовлена с подходящим лосьоном или кремом, содержащим активное соединение,суспендированное или растворенное в носителе. Подходящие носители включают, не ограничиваясь таковыми, минеральное масло, моностеарат сорбита, полисорбат 60, цетиловый сложный эфир воска, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду. Фармацевтические композиции по изобретению могут также применяться местно к нижней части кишечника путем лекарственных ректальных свечей или соответствующей лекарственной клизмы. Поверхностные трансдермальные пластыри и ионтофоретический способ введения также включены в настоящее изобретение. Применение лекарственных средств может быть локальным, так чтобы вводить препарат в нужном месте. Чтобы обеспечить введение композиции в нужном месте, могут быть использованы различные способы, такие как инъекции, использование катетеров, трокаров, направленных частиц, геля плуроника,стентов, полимеров с замедленным высвобождением лекарственного средства или других устройств,которые обеспечивают внутренний доступ. Соединения по настоящему изобретению могут быть включены в композиции для покрытия имплантируемых медицинских устройств, таких как протезы, искусственные клапаны, сосудистые трансплантаты, стенты или катетеры. Подходящие покрытия и общая подготовка покрытых имплантируемых устройств известны в данной области, и их примеры имеются в патентах США 6099562, 5886026 и 5304121. Для покрытия, как правило, применяются биосовместимые полимерные материалы, такие как гидрогельный полимер, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимер молочной кислоты, этиленвинилацетат и их смеси. Покрытия могут быть, необязательно, дополнительно покрыты подходящим верхним слоем фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидами или их комбинациями, чтобы придать способность контролированного высвобождения композиции. Покрытия для инвазивных устройств должны быть включены в определение фармацевтически приемлемый носитель, вспомогательное вещество или разбавитель, поскольку эти термины используются в настоящем документе. Может быть осуществлен способ покрытия имплантируемого медицинского устройства, включающий этап контактирования указанного устройства с композицией покрытия, описанной выше. Для специалиста в данной области будет очевидно, что покрытие устройства будет осуществляться до его имплантации в тело млекопитающего. Может быть осуществлен способ пропитки имплантируемого устройства, способного высвобождать лекарственное средство, содержащий этап контактирования названного устройства для высвобождения лекарственного средства с соединением или композицией данного изобретения. Имплантируемые устройства для высвобождения лекарственного средства включают, не ограничиваясь таковыми, биоразлагаемые полимерные капсулы или пули, неразлагаемые, способные диффундировать полимерные капсулы и биоразлагаемые полимерные облатки (капсулы). Может быть обеспечено имплантируемое медицинское устройство, покрытое соединением или композицией, содержащей соединение по настоящему изобретению, так, что указанное соединение является терапевтически активным. Могут быть обеспечены имплантируемые устройства для высвобождения лекарственного средства,пропитанные или содержащие соединение или композицию, содержащую соединение по настоящему изобретению, так, что указанное соединение высвобождается из указанного устройства и является терапевтически активным. Если орган или ткань, являются доступными из-за того, что удалены из тела пациента, то такой орган или ткань могут быть обмыты в среде, содержащей композицию по настоящему изобретению. Композиция по изобретению может быть нанесена на орган или композиция по изобретению может быть использована любым другим удобным способом. Композиция по настоящему изобретению может дополнительно содержать второе терапевтическое средство. Второе терапевтическое средство может быть выбрано из любого соединения или терапевтического средства, о котором известно, что оно проявляет полезные свойства, когда вводится с соединением,имеющим тот же самый механизм действия, как и апремиласт. Такие средства включают препараты, на которые указывается как на полезные в сочетании с апремиластом, включая, не ограничиваясь, те препараты, которые используются для лечения псориаза, в том числе псориаза бляшечного типа и рефракторного псориаза; саркоидоза, в том числе кожного саркоидоза; псориатического артрита; болезни Бехчета; узелковой почесухи; волчанки, в том числе кожной волчанки; и увеита, среди других. Второе терапевтическое средство может представлять собой препарат, используемый для лечения псориаза или саркоидоза. Могут быть получены отдельные лекарственные формы соединения по настоящему изобретению и одно или более из любых описанных выше дополнительных терапевтических средств, когда соединение и второе терапевтическое средство объединены друг с другом. Термин "объединены друг с другом", используемый здесь, означает, что отдельные лекарственные формы упакованы совместно или иным образом объединены друг с другом. При этом можно легко увидеть, что отдельные лекарственные формы предназначены для реализации и введения совместно (в течение менее чем через 24 ч друг от друга, последовательно или одновременно). В фармацевтической композиции по изобретению соединения настоящего изобретения присутствуют в эффективном количестве. Используемый здесь термин "эффективное количество" относится к количеству, которое при введении в надлежащем режиме дозирования достаточно для лечения (терапевтического или профилактического) определенного расстройства, например, чтобы уменьшить тяжесть,продолжительность или прогрессирование расстройства, подлежащего лечению, предотвратить развитие расстройства, подлежащего лечению, вызвать регрессию расстройства, подлежащего лечению, или расширить или улучшить профилактический или лечебный эффект(ы) другой терапии. Взаимосвязь рекомендованных дозировок для животных и людей (основанных на количестве миллиграммов на квадратный метр поверхности тела) описывается в Freireich et al., Cancer Chemother. Rep.,1966, 50:219. Площадь поверхности тела может быть приблизительно определена в зависимости от высоты и веса пациента. См., например, Scientific Tables, Geigy Pharmaceuticals, Ardsley, N.Y., 1970, 537. В одном из вариантов осуществления эффективное количество соединения по настоящему изобретению может составлять от 0,2 до 2000 мг на курс лечения, в более конкретных вариантах осуществления от 2 до 1000 мг и от 4 до 400 мг или более конкретно от 20 до 200 мг на курс лечения. Лечение обычно представляет собой введение со скоростью от 0,625 до 1,25 нг/кг/мин. Скорость инфузии может быть увеличена с шагом не более 1,25 нг/кг/мин в неделю в течение первых четырех недель, а затем не более 2,5 нг/кг/мин в неделю в течение всего оставшегося периода инфузии. Как это признано специалистами в данной области, эффективные дозы будут также варьироваться в зависимости от заболевания, тяжести заболевания, способа введения препарата, пола, возраста и общего состояния здоровья пациента, использующего лекарственное средство, возможности его совместного использования с другими терапевтическими приемами, такими как использование других препаратов, и решений лечащего врача. Например, рекомендации для выбора эффективной дозы могут быть найдены при обращении к инструкции по медицинскому применению препарата апремиласта. В случае фармацевтических композиций, которые содержат второе терапевтическое средство, эффективное количество этого второго терапевтического средства составляет от около 20 до 100% от дозировки, обычно применяемой в режиме монотерапии, использующей только это средство. Предпочтительно эффективное количество второго терапевтического средства составляет от около 70 до 100% от обычной дозы монотерапии. Обычные монотерапевтические дозы этих вторых терапевтических средств хорошо известны в данной области. См., например, работы Wells et al., eds., Pharmacotherapy Handbook,2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), которые вводятся здесь ссылкой во всей полноте. Ожидается, что некоторые из вторых терапевтических средств, указанных выше, будут действовать синергически с соединениями по данному изобретению. Когда это происходит, то позволяет снизить эффективную дозу второго терапевтического средства и/или соединения по данному изобретению, по сравнению с дозой, которая требуется при монотерапии. Это способствует минимизации токсических побочных эффектов либо второго терапевтического средства, либо соединения по настоящему изобретению,синергическим улучшениям в эффективности, повышению легкости введения или использования и/или снижению общих расходов на изготовление соединения или композиции. Способы лечения Еще в одном варианте осуществления изобретение относится к способу ингибирования PDE4 (фосфодиэстеразы) у пациента, включающему введение пациенту соединения формулы I по изобретению или его фармацевтически приемлемой соли. В другом варианте осуществления изобретение относится к способу снижения уровней TNF- у пациента, содержащему введение пациенту соединения формулы I по изобретению или его фармацевтически приемлемой соли. Согласно следующему варианту осуществления изобретение относится к способу лечения заболевания, которое успешно лечится апремиластом, содержащему этап введения пациенту, нуждающемуся в этом, эффективного количества соединения формулы I или его фармацевтически приемлемой соли или композиции по настоящему изобретению. Такие заболевания хорошо известны в данной области и раскрыты, не ограничиваясь таковыми, в следующих патентах и опубликованных заявках: WO 2006/025991;AU 2006/200033; WO 2001/034606; патенты США 6020358 и 6667316. К таким заболеваниям, не ограничиваясь таковыми, относятся септический шок, сепсис, эндотоксический шок, гемодинамический шок и сепсисоподобный синдром, постишемическое реперфузионное повреждение, малярия, микобактериальная инфекция, менингит, псориаз, в том числе бляшечный псориаз и рефракторный псориаз; саркоидоз, в том числе кожный саркоидоз; псориатический артрит; болезнь Бехчета; узелковая почесуха; волчанка, в том числе кожная волчанка; увеит; застойная сердечная недостаточность, фиброз, кахексия, отторжение трансплантата, рак, аутоиммунное заболевание, оппортунистические инфекции при СПИДе, ревматоидный артрит, ревматоидный спондилит, остеоартроз, другие артрогенные заболевания, болезнь Крона, неспецифический язвенный колит, рассеянный склероз, системный волчаночный эритрематоз, ENL при проказе, радиационное повреждение, альвеолярное повреждение, вызванное гипероксией, нежелательный ангиогенез, воспалительное заболевание, артрит, воспалительное заболевание кишечника, афтозные язвы, астма, дистресс-синдром дыхательных путей у взрослых и СПИД. В одном конкретном варианте осуществления способ по настоящему изобретению используется для лечения псориаза и саркоидоза. Способы, описанные здесь, включают также те, в которых пациент идентифицируется как нуждающийся в конкретно установленном лечении. Выявление пациентов, нуждающихся в таком лечении,может происходить по решению пациента или медицинского работника и может быть субъективным(например, мнение) или объективным (например, по данным тестирования или диагностического исследования). Любой из перечисленных выше способов лечения может содержать дополнительный этап совместного введения пациенту одного или более вторых лекарственных средств. Выбор второго лекарственного средства может быть сделан из любого второго лекарственного средства, про которое известно, что оно пригодно для одновременного применения с апремиластом. Выбор второго лекарственного средства также зависит от конкретного заболевания или расстройства, подлежащего лечению. Примерами вторых лекарственных средств, которые могут быть использованы в способах по этому изобретению, являются лекарственные средства, описанные выше, для использования в сочетании с композициями, содержащими соединения по настоящему изобретению и второе лекарственное средство. В частности, комбинированное лечение включает совместное введение соединения формулы I или его фармацевтически приемлемой соли и второго лекарственного средства для лечения следующих заболеваний: псориаза, в том числе бляшечного псориаза и рефракторного псориаза; саркоидоза, в том числе кожного саркоидоза; псориатического артрита; болезни Бехчета; узелковой почесухи; волчанки, в том числе кожной волчанки; и увеита. Термин "совместное введение", используемый здесь, означает, что второе терапевтическое средство может вводиться вместе с соединением по настоящему изобретению как часть единой лекарственной формы (например, композиции по настоящему изобретению, содержащей соединение по изобретению и второе терапевтическое средство, как описано выше) либо отдельно в виде нескольких лекарственных форм. Дополнительное средство может быть введено до, подряд или после введения соединения по настоящему изобретению. При такой комбинированной терапии оба соединения по настоящему изобретению и второе терапевтическое средство(а) вводятся обычными способами. Введение пациенту композиции по настоящему изобретению, содержащей как соединение по изобретению, так и второе терапевтическое средство, не исключает отдельного введения того же терапевтического средства, любого другого второго терапевтического средства или любого соединения по настоящему изобретению указанному пациенту в другое время в течение курса лечения. Эффективные количества этих вторых терапевтических средств хорошо известны специалистам в данной области, и рекомендации по дозированию могут быть найдены в патентах и опубликованных заявках на патент, упоминаемых в настоящем документе, а также в Wells et al., eds., PharmacotherapyPharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), и других медицинских текстах. Однако любой специалист в данной области способен определить диапазон оптимального эф- 13021019 фективного количества второго терапевтического средства. Когда пациенту вводится второе терапевтическое средство, эффективное количество соединения по настоящему изобретению будет меньше, чем было бы его эффективное количество в случае, когда не вводится второе терапевтическое средство. Эффективное количество второго терапевтического средства будет меньше его эффективного количества, когда не вводится соединение по данному изобретению. В этом случае нежелательные побочные эффекты, связанные с высокими дозами любого средства, могут быть сведены до минимума. Другие потенциальные преимущества (включая, без ограничения, улучшенные режимы дозирования и/или снижение стоимости лекарственных средств) будут очевидны для специалиста в данной области. Возможно применение соединения формулы I или его фармацевтической соли самостоятельно или совместно с одним или несколькими из описанных выше вторых терапевтических средств при изготовлении лекарственного средства либо в виде единой композиции, либо в виде отдельных лекарственных форм для лечения у больного заболевания, расстройства или симптома, изложенных выше. Другой аспект изобретения представляет соединение формулы I или его фармацевтическую соль для использования в лечении у больного заболевания, расстройства или их симптома, описанных в настоящем документе. Примеры Пример 1. Синтез (S)-N-(2-(1-d-2-(метилсульфонил)-1-(3-(этокси-d5)-4-(метокси-d3)фенил)этил)-1,3 диоксоизоиндолин-4-ил)ацетамид (соединение 113 а). Схема 4 Получение соединения 113 а Этап 1. Этил 3-гидрокси-4-(метокси-d3)бензоат (23). Коммерчески доступный сложный эфир 22 (10 г, 55 ммоль) смешивали с CD3I (99 атом.% D,Cambridge Isotopes; 8,1 г, 55 моль) и K2CO3 (7,59 г) в DMF (диметилформамид) и перемешивали при комнатной температуре в течение выходных дней (weekend). LCMS (жидкостная хромато-массспектрометрия) показала три пика с массами, соответствующими исходному материалу (20%), целевому моноалкилированному 23 (55%) продукту реакции и бисалкилированному (23%) побочному продукту. Реакционную смесь фильтровали через целит, промывая EtOAc (этилацетат), и фильтрат концентрировали почти досуха. Остаток растворяли в CH2Cl2 (300 мл) и раствор промывали водой (550 мл), рассолом,сушили (Na2SO4) и концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле, элюируя смесью EtOAc/гептан (1:9 до 1:6), затем дополнительно растирали с гептаном, чтобы получить 4,1 г (36%) целевого 23. Этап 2. Этил 3-(этокси-d5)-4-(метокси-d3)бензоат (24). 23 (4,1 г, 20 ммоль) растворяли в DMF (10 мл) и K2CO3 (2,5 г) и добавляли CD3CD2Br (99 атом.% D,Cambridge Isotopes; 4,7 г, 41 ммоль). Реакционную колбу запечатывали и смесь перемешивали при комнатной температуре в течение 24 ч. LCMS показала, что реакция была доведена до конца. Смесь фильтровали через целит, промывая МТВЕ (метил-трет-бутиловый эфир). Фильтрат концентритровали для удаления летучих компонентов и добавляли (100 мл) воды. Твердые вещества отбирали под вакуумом и промывали водой (50 мл). Собранное твердое вещество снова растворяли в МТВЕ (200 мл) и раствор промывали рассолом, сушили (Na2SO4) и концентрировали, чтобы получить примерно 3,8 г (81%) 24 (чистота приблизительно 90% согласно LCMS). Этап 3. (3-(Этокси-d5)-4-(метокси-d3)фенил)-1,1-d2-метанол (25). 24 (3,8 г, 16,3 ммоль) растворяли в МТВЕ (50 мл) и добавляли LiAlD4 (98 атом.% D, CambridgeIsotopes; 0,7 г, 17 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи.LCMS показала, что реакция была доведена до конца. Водный NH4Cl (20 мл) добавляли осторожно, чтобы остановить реакцию, и смесь фильтровали через целлит. Фазы разделяли и водную фазу экстрагировали EtOAc (220 мл). Объединенные органические фазы сушили (Na2SO4) и концентрировали, чтобы получить 2,8 г 25 в виде светло желтого масла. Этот материал использовали непосредственно в следующем этапе. Этап 4. 3-(Этокси-d5)-4-(метокси-d3)бензальдегид-d (10 а). 25 (2,8 г, 16 ммоль) растворяли в EtOAc (30 мл). Добавляли MnO2 (14 г, 160 ммоль) и темную смесь перемешивали при комнатной температуре в течение ночи. LCMS показала полный расход исходного материала. Смесь пропускали через целит, промывая EtOAc, и фильтрат концентрировали, чтобы получить желтое масло. Масло очищали посредством хроматографии на силикагеле, элюируя 20% смесьюEtOAc/гептан, чтобы получить 2,05 г (68% для 2 этапов) 10 а в виде белого твердого вещества. Этап 5. 1-(3-(Этокси-d5)-4-(метокси-d3)фенил)-1-d-2-(метилсульфонил)этанамин (11 а). Метилсульфон (1 г, 10,7 ммоль) суспендировали в THF (тетрагидрофуран) (70 мл) и охлаждали на бане, заполненной смесью ацетон/сухой лед, до температуры ниже -70C. Добавляли n-BuLi (2,5 М в гексане, 4,6 мл, 11,5 ммоль) и смесь перемешивали в течение 30 мин. В отдельной колбе раствор 10 а (1,9 г,10,0 ммоль) в THF (20 мл) охлаждали до 0C. Добавляли раствор гексаметилдисилазида лития (LHMDS)(1M в THF, 12 мл). Спустя 15 мин добавляли эфират трифторида бора (2,8 мл, 22 ммоль) и перемешивание продолжали еще в течение 5 мин. Этот раствор затем посредством шприца добавляли к раствору метил сульфон/n-BuLi с охлаждением на бане со смесью ацетон/сухой лед до температуры ниже -70C. Наблюдали экзотермический эффект. Этой смеси позволяли нагреваться до комнатной температуры и перемешивали в течение ночи. После охлаждения в водно-ледяной бане добавляли K2CO3 (8 г) и затем воду(50 мл). Слои разделяли и водную фазу экстрагировали EtOAc (220 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали, чтобы получить вязкое масло. Добавляли МТВЕ(30 мл) и водный HCl (4 н., 30 мл) и смесь перемешивали при комнатной температуре в течение 2 ч, чтобы получить прозрачный двухфазный раствор. Фазы разделяли и органический раствор экстрагировали водным HCl (4 н., 25 мл). К объединенным водным фазам добавляли водный NaOH (24%), доводя доpH12. Водную фазу экстрагировали EtOAc (350 мл), органическую фазу сушили (Na2SO4) и концентрировали, чтобы получить желтое твердое вещество. Это твердое вещество суспендировали в МТВЕ(20 мл) и перемешивали в течение 1 ч. После фильтрации под вакуумом получали 1,2 г (36%) 11 а. Этап 6. Соль (S)-1-(3-(этокси-d5)-4-(метокси-d3)фенил)-1-d-2-(метилсульфонил)этанамин N-ацетил лейцина S)-11 а). 11 а (1,2 г, 4,25 ммоль) смешивали с N-ацетил-L-лейцином (0,44 г, 2,55 ммоль) в МеОН (метанол)(10 мл). Эту смесь нагревали при 70C в течение 3 ч, затем перемешивали при комнатной температуре в течение ночи. Твердое вещество отбирали посредством вакуумной фильтрации и суспендировали в метаноле (15 мл). Смесь перемешивали при 70C в течение 2 ч, затем при комнатной температуре в течение ночи. Твердое вещество отбирали и растирание с МеОН повторяли. Долю 600 мг (31%) от (S)-11a выделяли с 99% ее (энантиомерный избыток). Этап 7.(S)-11a (380 мг, 0,88 моль) перемешивали с известным 12 а (200 мг, 1 ммоль; см. US 20080234359) в уксусной кислоте (6 мл) и нагревали с обратным холодильником в течение 24 ч, чтобы привести реакцию к завершению. Смесь концентрировали, бесцветное масло снова растворяли в EtOAc (100 мл). Раствор промывали насыщенным водным раствором NaHCO3 (20 мл), сушили (Na2SO4) и концентрировали. Неочищенный продукт очищали колоночной хроматографией на Analogix, элюируя 0-3% MeOH/CH2Cl2,чтобы обеспечить 360 мг (87%) 113 а. 1 129,18, 131,07, 136,14, 137,66, 148,70, 167,51, 169,16. ВЭЖХ (высокоэффективная жидкостная хроматография) (метод: 50 мм 3 мкм Atlantis (Waters) Т 3 2.1 колонка - градиентный метод 5-95% ACN (ацетонитрил) + 0,1% муравьиная кислота 14 мин с 4 мин удерживания при 95% ACN + 0,1% муравьиная кислота; длина волны: 305 нм): время удерживания: 5,96 мин; 99,5% чистота.(99 атом.% D, Cambridge Isotopes; 12,4 г, 84 ммоль), затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли EtOAc (200 мл) и фильтровали через целит. Фильтрат концентрировали до получения темного масла. Добавляли EtOAc (150 мл) и воду(50 мл) и слои разделяли. Водную фазу доводили до pH 6 путем медленного добавления 1 н. HCl и смесь экстрагировали EtOAc (2100 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали. Неочищенный материал очищали с помощью колоночной хроматографии на силикагеле с элюированием смесью EtOAc/гептан (1:6 до 1:2), чтобы получить больше 5 г 27 около 90% чистоты. Этот материал затем очищали хроматографией на Analogix, элюируя 0-30% EtOAc/гептан, чтобы получить 4,3 г(35%) 27. Этап 2. 3-(Этокси-d5)-4-(метокси-d3)бензальдегид (10b). 27 (4,3 г, 27,7 ммоль) смешивали с Cs2CO3 (15 г, 46 ммоль) в ацетоне и охлаждали в ледяной бане. Добавляли бромэтан-d5 (99 атом.% D, Cambridge Isotopes; 3,8 г, 33,6 ммоль) и реакционную смесь перемешивали в течение ночи. Добавляли МТБЭ и смесь фильтровали через целит. После концентрирования неочищенный продукт очищали хроматографией на силикагеле, элюируя смесью EtOAc/гептан 1:4, чтобы получить 2 г (38%) желаемого 10b. Этап 3. 1-(3-(Этокси-d5)-4-(метокси-d3)фенил)-2-(метилсульфонил)этанамин (11b). Метилсульфон (1 г, 10,7 ммоль) суспендировали в THF (70 мл) и охлаждали на бане, заполненной смесью ацетон/сухой лед, до температуры ниже -70C. Добавляли n-BuLi (2,5 М в гексане, 4,8 мл,11,9 ммоль) и смесь перемешивали около 30 мин. В отдельной колбе раствор альдегида 10b (2 г,10,6 ммоль) в ТГФ (20 мл) охлаждали до 0C. Добавляли LHMDS (1 M в THF, 12 мл). Через 15 мин до- 16021019 бавляли эфират трифторида бора (2,8 мл, 22 ммоль) и перемешивание продолжали в течение еще 5 мин. Затем этот раствор с помощью шприца добавляли к раствору метилсульфон/n-BuLi с охлаждением на бане, содержащей ацетон/сухой лед при температуре ниже -70C. Наблюдали экзотермический эффект. Этой смеси позволяли нагреваться до комнатной температуры и перемешивали в течение ночи. После охлаждения на водно-ледяной бане добавляли K2CO3 (8 г) и затем воду (50 мл). Слои разделяли и водную фазу экстрагировали EtOAc (220 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали, получая вязкое масло. Добавляли МТВЕ (30 мл) и водный HCl (4 н., 30 мл) и смесь перемешивали при комнатной температуре в течение 2 ч, чтобы дать прозрачный двухфазный раствор. Фазы разделяли и органическую фазу экстрагировали водным HCl (4 н., 25 мл). К объединенным водным фазам добавляли водный NaOH (24%) для увеличения pH выше 12. Раствор экстрагировали EtOA (350 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали для получения желтого твердого вещества. Твердое вещество суспендировали в МТВЕ (20 мл) и перемешивали в течение 1 ч. Фильтрация под вакуумом обеспечила 1,2 г (38%) 11b в виде светло-желтого твердого вещества. Этап 4.N-ацетил-L-лейцина соль S)-11b). 11b (1,05 г, 3,73 ммоль) смешивали с N-ацетил-L-лейцином (0,39 г, 2,24 ммоль) в МеОН (6 мл). Эту смесь нагревали при 70C в течение 3 ч, затем перемешивали при комнатной температуре в течение ночи. Твердое вещество отбирали путем вакуумной фильтрации и суспендировали в МеОН (15 мл) Суспензию перемешивали при 70C в течение 2 ч, затем при комнатной температуре в течение ночи. Твердое вещество собирали и повторяли растирание с МеОН. Доля 400 мг (23%) (S)-11b соли(S)-11b N-Ацетил-L-лейцина соль (220 мг, 0,5 ммоль) смешивали с известным 12 а (123 мг,0,6 ммоль) в уксусной кислоте (5 мл) и нагревали с обратным холодильником в течение 24 ч, доводя реакцию почти до завершения. Смесь концентрировали, бесцветное масло растворяли в EtOAc (100 мл) и раствор промывали насыщенным водным раствором NaHCO3 (20 мл). Органическую фазу сушили(Na2SO4) и концентрировали. Неочищенный продукт очищали колоночной хроматографией на Analogix,элюируя 0-70% смесью EtOAc/гептан, чтобы получить 210 мг (89%) 107 а. 1 Н-ЯМР (300 МГц, CDCl3):1,59 (s, 1H), 2,27 (s, 3H), 2,87 (s, 3H), 3,72 (dd, J=4,6, 14,4 Гц, 1H), 3,85(s, 3H), 4,56 (dd, J=10,8, 14,4 Гц, 1H), 5,87 (dd, J=4,4, 10,6 Гц), 6,84 (d, J=8,8 Гц, 1H), 7,10 (d, J=7,0 Гц, 2H),7,49 (d, J=6,6 Гц, 1H), 7,65 (t, J=7,3 Гц, 1H), 8,76 (d, J=8,0 Гц, 1H), 9,46 (s, 1H). 13 С-ЯМР (75 МГц, CDCl3):24,97, 41,66, 48,60, 54,55, 55,96, 76,58, 77,01, 77,43, 111,48, 112,40,115,14, 118,25, 120,29, 125,00, 129,26, 131,07, 136,14, 137,66, 148,70, 149,79, 167,51, 169,17, 169,53. ВЭЖХ (метод: 50 мм 3 мкм Atlantis (Waters) T3 2.1 колонка-градиентный метод 5-95% ACN + 0,1% муравьиная кислота 14 мин с 4 мин удерживания при 95% ACN + 0,1% муравьиная кислота; длина волны: 305 нм): время удерживания: 6,02 мин; 98,0% чистоты. Хиральная ВЭЖХ (метод: Chiralpak AD 25 см колонка - изократический метод 78% гексан/22% изопропанол/0,01% диэтиламин в течение 40 мин при 1,00 мл/мин; длина волны: 254 нм): время удерживания: 12,73 мин (основной энантиомер); 99% ее чистоты. Этап 1. 3-Этокси-4-(метокси-d3)бензальдегид (10 с). Смесь коммерчески доступного 16 а (5 г, 30 ммоль) и Cs2CO3 (15 г, 46 ммоль) в ацетоне охлаждали на водяно-ледяной бане. Добавляли (CD3)2SO4 (99 атом.% D, Cambridge Isotopes; 2,7 мл, 30 ммоль), реакционной смеси позволяли медленно нагреваться до комнатной температуры и перемешивали в течение ночи. Смесь фильтровали через целит и концентрировали, получая 5,7 г (около 100%) 10 с. Этап 2. 1-(3-Этокси-4-(метокси-d3)фенил)-2-(метилсульфонил)этанамин (11 с). Метилсульфон (3 г, 32,1 ммоль) суспендировали в THF (280 мл) и охлаждали на бане со смесью ацетон/сухой лед до температуры ниже -70C. Добавляли n-BuLi (2,5 М в гексане, 13,6 мл, 35,7 ммоль) и смесь перемешивали около 30 мин. В отдельной колбе раствор 10 с (5,7 г, 30,2 ммоль) в THF (60 мл) охлаждали до 0C. Добавляли LHMDS (1 M в THF, 34,4 мл). Через 15 мин добавляли эфират трифторида бора (8 мл, 62,9 ммоль) и смесь перемешивали в течение 5 мин. Этот раствор с помощью шприца добавляли к раствору метилсульфон/n-BuLi с охлаждением на бане ацетон/сухой лед до температуры ниже-70C. Наблюдался экзотермический эффект. Этой смеси позволяли нагреваться до комнатной температуры и перемешивали в течение ночи. После охлаждения на водно-ледяной бане, добавляли K2CO3 (24 г) с последующим добавлением воды (150 мл). Слои разделяли и водную фазу экстрагировали EtOAc(360 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали, получая вязкое масло. К остатку добавляли МТВЕ (90 мл) и водный HCl (4 н., 90 мл) и смесь перемешивали при комнатной температуре в течение 2 ч, чтобы получить прозрачный двухфазный раствор. Фазы разделяли и органическую фазу экстрагировали водным HCl (4 н., 75 мл). К объединенным водным фазам добавляли водный NaOH (24%) для увеличения pH выше 12. Водный слой экстрагировали EtOAc (3150 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали, чтобы получить желтое твердое вещество. Твердое вещество суспендировали в МТВЕ (60 мл) и перемешивали в течение 1 ч. Фильтрация под вакуумом дала 2,7 г (31,4%) 11 с в виде светло-желтого твердого вещества. Этап 3. (S)-1-(3-Этокси-4-(метокси-d3)фенил)-2-(метилсульфонил)этанамин N-ацетил-L-лейциновая соль S)-11c). 11 с (2,3 г, 8,17 ммоль) смешивали с N-ацетил-L-лейцином (0,78 г, 4,48 ммоль) в метаноле (12 мл). Смесь нагревали при 70C в течение 3 ч, затем перемешивали при комнатной температуре в течение ночи. Твердое вещество собирали посредством вакуумной фильтрации, суспендировали в МеОН (12 мл) и перемешивали при 70C в течение 2 ч, затем при комнатной температуре в течение ночи. Твердое вещество собирали и растирание с МеОН повторяли. Часть 1 г (28,8%) из (S)-11c соли N-ацетил-L-лейцина получали с 98% ее. Этап 4.(20 мл) и нагревали с обратным холодильником в течение 24 ч, почти доводя реакцию до завершения. Смесь концентрировали, бесцветное масло растворяли в EtOAc (200 мл) и раствор промывали насыщенным NaHCO3 (40 мл). Органическую фазу сушили (Na2SO4) и концентрировали, неочищенный продукт очищали с помощью колоночной хроматографии на Analogix, элюируя 0-70% смесью EtOAc/гептан, чтобы получить 0,7 г (68%) 114 а. 1J=4,6, 14,4 Гц, 1H), 4,11 (q, J=6,9, 14,0 Гц, 2 Н), 4,55 (dd, J=10,5, 14,4 Гц, 1 Н), 5,87 (dd, J=4,4, 10,6 Гц, 1H),6,84 (d, J=8,7 Гц, 1H), 7,10 (d, J=6,5 Гц, 2H), 7,49 (d, J=7,3 Гц, 1H), 7,65 (t, J=7,7 Гц, 1H), 8,76 (d, J=8,5 Гц,1H), 9,46 (s, 1H). 13 С-ЯМР (75 МГц, CDCl3):14,70, 24,96, 41,65, 48,59, 54,54, 64,55, 111,46, 112,44, 115,14, 118,25,120,32, 125,00, 129,24, 131,07, 136,14, 137,66, 148,67, 149,79, 167,51, 169,17, 169,53. ВЭЖХ (метод: 50 мм 3 мкм Atlantis (Waters) T3 2.1 колонка - градиентный метод 5-95% ACN + 0,1% муравьиная кислота 14 мин с 4 мин удерживания при 95% ACN + 0,1% муравьиная кислота; длина волны: 305 нм): время удерживания: 6,03 мин; 97,4% чистоты. Хиральная ВЭЖХ (метод: Chiralpak AD 25 см колонка - изократический метод 78% гексан/22% изопропанол/0,01% диэтиламин в течение 40 мин при 1,00 мл/мин, длина волны: 254 нм): время удерживания: 12,69 мин (основной энантиомер); 39,03 мин (второстепенный энантиомер); 99% ее чистота. Этап 1. 3-(Этокси-d5)-4-метоксибензальдегид (10d). Коммерчески доступный 29 (5 г, 30 ммоль) смешивали с Cs2CO3 (15 г, 46 ммоль) в ацетоне и охлаждали на водно-ледяной бане. Добавляли бромэтан-d5 (99 атом.% D, Cambridge Isotopes; 3,8 г,33,6 ммоль) и реакционной смеси позволяли медленно нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли МТВЕ, фильтровали через целит и концентрировали, получая 5,5 г (около 100%) 10d. Этап 2. 1-(3-(Этокси-d5)-4-метоксифенил)-2-(метилсульфонил)этанамин (11d). Метилсульфон (2,76 г, 29,5 ммоль) суспендировали в THF (250 мл) и охлаждали на бане ацетон/сухой лед до температуры ниже -70C. Добавляли n-BuLi (2,5 М в гексане, 12,5 мл, 31 ммоль) и смесь перемешивали в течение примерно 30 мин. В отдельной колбе раствор альдегида 10d (5,25 г,27,6 ммоль) в THF (50 мл) охлаждали до 0C. Добавляли LHMDS (1 M в THF, 31,7 мл). Через 15 мин добавляли эфират трифторида бора (7,36 мл, 57,8 ммоль) и смесь перемешивали еще 5 мин. Затем этот раствор с помощью шприца добавляли к раствору метилсульфон/n-BuLi с охлаждением на бане ацетон/сухой лед при температуре ниже -70C. Наблюдали выделение тепла. Этой смеси позволяли нагреваться до комнатной температуры и перемешивали в течение ночи. После охлаждения на водно-ледяной бане добавляли K2CO3 (24 г) с последующим добавлением воды (150 мл). Слои разделяли и водную фазу экстрагировали EtOAc (3 60 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали, получая вязкое масло. Добавляли МТВЕ (90 мл) и водный HCl (4 н., 90 мл) и смесь перемешивали при комнатной температуре в течение 2 ч, чтобы получить прозрачный двухфазный раствор. Фазы разделяли и органическую фазу экстрагировали водным HCl (4 н., 75 мл). К объединенным водным фазам добавляли водный NaOH (24%), чтобы увеличить pH выше 12. Смесь экстрагировали EtOAc (3150 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали, чтобы получить желтое твердое вещество. Твердое вещество суспендировали в МТВЕ (60 мл) и перемешивали в течение 1 ч. Фильтрация под вакуумом обеспечивала 2,7 г (34,2%) 11d в виде светло-желтого твердого вещества. Этап 3. S)-1-(3-(Этокси-d5)-4-метоксифенил)-2-(метилсульфонил)этанамин N-ацетил L-лейцина соль S)-11d). 11d (2,6 г, 9,33 ммоль) смешивали с N-ацетил-L-лейцином (0,98 г, 5,6 ммоль) в МеОН (15 мл). Эту смесь нагревали при 70C в течение 3 ч, затем перемешивали при комнатной температуре в течение ночи. Твердое вещество собирали посредством вакуумной фильтрации и суспендировали в МеОН (15 мл). Суспензию перемешивали при 70C в течение 2 ч, затем при комнатной температуре в течение ночи. Твердое вещество собирали и растирание с МеОН повторяли. Часть в 1 г (23%) (S)-11d соли N-ацетил-Lлейцина была получена с 98% ее. Этап 4.(20 мл) и нагревали с обратным холодильником в течение 24 ч, чтобы довести реакцию почти до завершения. Смесь концентрировали, бесцветное масло растворяли в EtOAc (200 мл) и раствор промывали насыщенным NaHCO3 (40 мл). Органический слой сушили (Na2SO4) и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии на Analogix, элюируя 0-70% EtOAc/гептанMS (M+Na): 488,1. Элементный анализ (C22H21D3 N2O7S): Рассчитано: С=56,76, Н=5,20, N=6,02, S=6,89. Найдено: С=56,74, Н=5,43, N=5,70, S=6,51. Пример 5. Синтез промежуточного соединения 12b. Схема 8 Получение 12bN-(1,3-Диоксо-1,3-дигидроизобензофуран-4-ил)ацетамид-d3 (12b). Коммерчески доступный 4-аминоизобензофуран-1,3-дион 30 (5 г, 30,6 ммоль) суспендировали в уксусном ангидриде-d6 (98 атом.% D, Cambridge Isotopes; 10 г) и нагревали с обратным холодильником в течение 3 ч, затем перемешивали при комнатной температуре в течение ночи. Раствор охлаждали до 0C и фильтровали, затем твердое вещество промывали МТВЕ и сушили, чтобы обеспечить 2,5 г 12b. Пример 6. Синтез (S)-N-(2-(1-(3-(этокси-d5)-4-(метокси-d3)фенил)-2-(метилсульфонил)этил)-1,3 диоксоизоиндолин-4-ил)ацет-d3-амида (соединение 115 а).(S)-11b N-Ацетил-L-лейциновая соль (200 мг, 0,44 ммоль, см. схему 5) смешивали с 12b (130 мг, см. схему 8) в уксусной кислоте (5 мл) и раствор нагревали при 80C в течение 20 ч. Смесь концентрировали и бесцветное масло снова растворяли в EtOAc (100 мл). Раствор промывали насыщенным водным раствором NaHCO3 (20 мл), сушили (Na2SO4) и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии на Analogix, элюируя 0-70% EtOAc/гептан, чтобы обеспечить 174 мг(73%) 115 а. 1 Н-ЯМР (300 МГц, CDCl3):1,55 (s, 1H), 2,87 (s, 3H), 3,72 (dd, J=4,4, 14,3 Гц, 1H), 4,56 (dd, J=10,5,14,4 Гц, 1H), 5,87 (dd, J=4,4, 10,5 Гц, 1H), 6,84 (d, J=8,5 Гц, 1H), 7,10 (d, J=7,0 Гц, 2H), 7,49 (d, J=7,3 Гц,1H), 7,66 (t, J=7,5 Гц, 1 Н), 8,76 (d, J=8,3 Гц, 1H), 9,46 (s, 1H). 13 С-ЯМР (75 МГц, CDCl3):41,66, 48,61, 54,56, 76,58, 77,00, 77,21, 77,43, 111,45, 112,40, 115,14,118,26, 120,29, 125,01, 129,24, 131,07, 136,15, 137,66, 148,70, 167,52, 169,54. ВЭЖХ (метод: 50 мм 3 мкм Atlantis (Waters) T3 2.1 колонка - градиентный метод 5-95% ACN + 0,1% муравьиная кислота 14 мин с 4 мин удерживания при 95% ACN + 0,1% муравьиная кислота; длина волны: 305 нм): время удерживания: 5,96 мин; 99,1% чистота. MS (М+Н): 472,0. Элементный анализ (C22H16D8N2O7S): Рассчитано: С=56,04, Н=5,13, N=5,94. Найдено: С=55,90,Н=5,23, N=5,85. Пример 7. (S)-N-(2-(1-(3-(Этокси-d5)-4-(метокси-d3)фенил)-2-метил-d3)сульфонил)-2,2-d2-этил)1,3-диоксоизоиндолин-4-ил)ацетамид (соединение 116 а). Схема 9 Получение соединения 116 а Этап 1. 1-(3-(Этокси-d5)-4-(метокси-d3)фенил)-2-метил-d3)сульфонил)-2,2-d2-этанамин (11e). Метилсульфон-d6 (99 атом.% D, Isotec; 1 г, 10,0 ммоль) суспендировали в THF (70 мл) и охлаждали на бане ацетон/сухой лед до температуры ниже -70C. Добавляли n-BuLi (2,5 М в гексане, 4,4 мл,11 ммоль) и смесь перемешивали около 30 мин. В отдельной колбе, раствор альдегида 10b (1,91 г,10,0 ммоль; см. схему 5) в THF (20 мл) охлаждали до 0C. Добавляли LHMDS (1 M в THF, 11 мл). Через 15 мин добавляли эфират трифторида бора (2,8 мл, 22 ммоль) и перемешивание продолжали в течение еще 5 мин. Этот раствор с помощью шприца добавляли к раствору метилсульфон-d6/n-BuLi, с охлаждением на бане ацетон/сухой лед до температуры ниже -70C. Наблюдался экзотермический эффект. Смеси позволяли нагреваться до комнатной температуры и перемешивали в течение ночи. После охлаждения на водно-ледяной бане, добавляли K2CO3 (8 г) с последующим добавлением воды (50 мл). Слои разделяли, и водную фазу экстрагировали EtOAc (220 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали, чтобы получить вязкое масло. Добавляли МТВЕ (30 мл) и водный HCl (4 н., 30 мл) и смесь перемешивали при комнатной температуре в течение 2 ч, получая прозрачный двухфазный раствор. Фазы разделяли и органическую фазу экстрагировали водным HCl (4 н., 25 мл). К объединенным водным фазам добавляли водный NaOH (24%) для увеличения pH выше 12. Раствор экстрагировалиEtOAc (350 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали, чтобы получить желтое твердое вещество. Твердое вещество суспендировали в МТВЕ (20 мл) и перемешивали в течение 1 ч. Фильтрация под вакуумом обеспечила 1,2 г (37%) 11e в виде светло-желтого твердого вещества. 1 Н ЯМР и LCMS показали некоторую потерю изотопной чистоты alpha на сульфоне. Этот D-c-H обмен, вероятно, происходит во время экстракции кислоты/основания. Использование дейтерированных растворителей является предпочтительным во время всего процесса получения продукта. Менее изотонически чистый материал растворяли в MeOD (99 атом.% D, Cambridge Isotopes; 30 мл) и добавляли K2CO3 (0,5 г). Эту смесь нагревали при 70C в течение 6 ч и затем концентрировали досуха. Добавляли свежий MeOD (30 мл) и смесь нагревали до 70C в течение ночи. Охлажденный раствор разбавляли EtOAc (100 мл) и смесь фильтровали. Фильтрат концентрировали и повторно растворяли вEtOAc (100 мл). Раствор промывали D2O (99,9 атом.% D, Cambridge Isotopes; 20 мл). Органическую фазу сушили (Na2SO4) и концентрировали, чтобы получить примерно 1 г 11e с восстановленной высокой изотонической чистотой. Этап 2.D, Cambridge Isotopes; 6 мл). Эту смесь нагревали при 70C в течение 3 ч, затем перемешивали при комнатной температуре в течение ночи. Твердое вещество собирали посредством вакуумной фильтрации и суспендировали в МеОН (6 мл). Смесь перемешивали при температуре 70C в течение 2 ч, затем при комнатной температуре в течение ночи. Твердое вещество собирали и растирание с МеОН повторяли. Порция 300 мг (29%) (S)-11 е соли N-ацетил-L-лейцина получали с 99% ее. Этап 3. (S)-N-(2-(1-(3-(Этокси-d5)-4-(метокси-d3)фенил)-2-метил-d3)сульфонил)-2,2-d2-этил)-1,3 диоксоизоиндолин-4-ил)ацетамид (116 а).(S)-11e N-Ацетил-L-лейцина соль (280 мг, 0,62 ммоль) смешивали с известным 12 а (145 мг,0,7 ммоль) в уксусной кислоте-d (99 атом.% D, Aldrich; 5 мл) и нагревали с обратным холодильником в течение 24 ч, чтобы привести реакцию почти к завершению. Смесь концентрировали и бесцветное масло растворяли в EtOAc (100 мл). Раствор промывали NaHCO3 (20 мл), сушили (Na2SO4) и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии на Analogix, элюируя 0-3%MeOH/CH2Cl2, чтобы обеспечить 245 мг (84%) 116 а. 1 Н-ЯМР (300 МГц, CDCl3):1,57 (s, 1 Н), 2,26 (s, 3H), 5,86 (s, 1H), 6,84 (d, J=6,8 Гц, 1H), 7,10 (d,J=6,8 Гц, 2H), 7,49 (d, J=6,4 Гц, 1H), 7,65 (t, J=7,9 Гц, 1H), 8,76 (d, J=8,5 Гц, 1H), 9,46 (s, 1H). 13 С-ЯМР (75 МГц, CDCl3):24,97, 48,43, 111,45, 112,40, 115,14, 118,25, 120,28, 125,00, 129,22,131,07, 136,14, 137,66, 148,70, 149,79, 167,52, 169,17, 169,54. ВЭЖХ (метод: 50 мм 3 мкм Atlantis (Waters) T3 2.1 колонка - градиентный метод 5-95% ACN + 0,1% муравьиная кислота 14 мин, 4 мин удерживания при 95% ACN + 0,1% муравьиная кислота; длина волны: 305 нм): время удерживания: 5,97 мин; 99,7% чистоты.MS (М+Н): 474,3. Элементный анализ (C22H11D13 N2O7S): Рассчитано: С=55,80, Н=5,11, N=5,92. Найдено: С=52,73,Н=4,73, N=5,43. Пример. Оценка метаболической устойчивости. Микросомный анализ. Микросомы печени человека (20 мг/мл) получали из Xenotech, LLC (Lenexa, KS).-никотинамидадениндинуклеотидфосфат, восстановленная форма (NADPH), хлорид магния (MgCl2) и диметилсульфоксид (DMSO) закупали у Sigma-Aldrich. Определение метаболической устойчивости. 7,5 мМ исходные растворы испытуемых соединений готовили в ДМСО. 7,5 мМ исходные растворы разбавляли до 12,5-50 мкМ в ацетонитриле (ACN). 20 мг/мл микросомов печени человека разбавляли до 0,625 мг/мл в 0,1 М буферном растворе фосфата калия, pH 7,4, содержащем 3 мМ MgCl2. Разбавленные микросомы добавляли в глубокие лунки 96-луночного полипропиленового планшета в трех экземплярах. 10 мкл аликвоту 12,5-50 мкМ тестируемого соединения добавляли к микросомам и смесь предварительно нагревали в течение 10 мин. Реакции инициировали путем добавления предварительно нагретого раствора NADPH. Окончательный объем реакционной смеси составлял 0,5 мл и содержал 0,5 мг/мл микросомов печени человека, 0,25-1,0 мкМ тестируемого соединения и 2 мМ NADPH в 0,1 М калий фосфатном буфере, pH 7,4 и 3 мМ MgCl2. Реакционные смеси инкубировали при 37C и 50 мкл аликвот удаляли на 0,- 22021019 5, 10, 20 и 30 мин и добавляли в мелкие лунки 96-луночных планшетов, которые содержали 50 мкл охлажденного до 0C ACN, с внутренним стандартом, чтобы остановить реакцию. Планшеты сохраняли при температуре 4C в течение 20 мин, после чего добавляли 100 мкл воды в лунки планшета перед центрифугированием для получения осажденных белков. Супернатанты переводили в другой 96-луночный планшет и проводили анализ количества оставшихся исходных материалов посредством LC-MS/MS (метод жидкостной хроматографии/тандемной масс-спектрометрии), используя Applied Bio-systemsAPI 4000 масс-спектрометр. Такую же процедуру выполняли для апремиласта и положительного контроля, 7-этоксикумарина (1 мкМ). Тестирование проводили в трех экземплярах. Анализ полученных данных. Периоды полураспада t1/2 in vitro для тестируемых соединений рассчитывали из наклонов линейной регрессии % оставшихся исходных материалов (ln) в зависимости от времени инкубацииin vitro t1/2 = 0,693/k,где k = -[наклон линейной регрессии % оставшихся исходных материалов (ln) в зависимости от времени инкубации]. Анализ данных выполняли с помощью программного обеспечения Microsoft Exel. Без дальнейшего описания понятно, что любой специалист в данной области может, используя предыдущее описание и иллюстративные примеры, изготавливать и использовать соединения по настоящему изобретению и осуществлять на практике заявленные способы. Следует понимать, что приведенные выше обсуждение и примеры представляют подробное описание только некоторых предпочтительных вариантов осуществления. Специалисту в данной области будет очевидно, что различные модификации и эквивалентные варианты могут быть выполнены без отступления от объема и сущности изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I в которой R1 выбран из СН 3, CH2D, CHD2 и CD3;R4 представляет собой этильную группу, замещенную 0-5 атомами дейтерия, или представляет собой циклопентильную группу, замещенную 5-9 атомами дейтерия;X выбран из С=О; каждый из Y1a, Y1b, Y2, Y3, Y4, Y5, Y7 и Y8 независимо выбран из Н и D;Y6 выбран из Cl, H и D; где любой атом, не обозначенный как дейтерий, присутствует в его природном изотопном составе,или его фармацевтически приемлемая соль. 2. Соединение по п.1, в которомR2 представляет собой СН 3 или CD3;Y6, Y7 и Y8 являются одинаковыми и каждый представляет собой либо Н, либо D;Y1a и Y1b являются одинаковыми и каждый представляет собой либо Н, либо D;Y3, Y4 и Y5 являются одинаковыми и каждый представляет собой либо Н, либо D. 3. Соединение по п.1, в котором соединение формулы I представляет собой соединение формулы II в которой R1 выбран из СН 3 и CD3;R4 выбран из СН 2 СН 3, CD2CD3, CD2CH3 и CH2CD3;- 23021019 каждый Y1a, Y1b, Y2, Y3, Y4, Y5, Y6, Y7 и Y8 независимо выбран из Н и D,или его фармацевтически приемлемую соль. 4. Соединение по п.2, в котором соединение формулы I представляет собой соединение формулы Ia,имеющее (S)-конфигурацию у атома углерода, присоединенного к Y2: или его фармацевтически приемлемую соль. 5. Соединение по п.2, в котором соединение формулы I представляет собой соединение формулы Ib,имеющее (R)-конфигурацию у атома углерода, присоединенного к Y2: или его фармацевтически приемлемую соль. 6. Соединение по п.1, в котором R2 представляет собой СН 3 или CD3. 7. Соединение по п.1, в котором Y6, Y7 и Y8 являются одинаковыми и каждый представляет собой либо Н, либо D. 8. Соединение по п.1, в котором Y1a и Y1b являются одинаковыми и каждый представляет собой либо Н, либо D. 9. Соединение п.1, в котором Y3, Y4 и Y5 являются одинаковыми и каждый представляет собой либо Н, либо D. 10. Соединение по любому из пп.1, 2 или 4-9, в котором R1 представляет собой СН 3 или CD3. 11. Соединение по любому из пп.1-10, в котором R4 представляет собой CD2CD3. 12. Соединение по п.1, выбранное из группы, состоящей из или фармацевтически приемлемая соль любого из вышеприведенного. 13. Соединение по п.12, имеющее (S)-конфигурацию. 14. Соединение по п.12, имеющее (R)-конфигурацию. 15. Соединение по п.1, выбранное из группы, состоящей из или фармацевтически приемлемая соль любого из вышеприведенного. 16. Фармацевтическая композиция для ингибирования PDE4 (фосфодиэстеразы 4 типа) или снижения уровней TNF- у субъекта, нуждающегося в этом, содержащая эффективное количество соединения по п.1 или его фармацевтически приемлемую соль и приемлемый носитель. 17. Фармацевтическая композиция, содержащая эффективное количество соединения по п.1 или его фармацевтически приемлемую соль и приемлемый носитель, отличающаяся тем, что композиция подходит для лечения заболевания, выбранного из группы, состоящей из септического шока, сепсиса, эндотоксического шока, гемодинамического шока и сепсисоподобного синдрома, постишемического реперфузионного повреждения, малярии, микобактериальной инфекции, менингита, псориаза, саркоидоза, псориатического артрита, болезни Бехчета, узелковой почесухи, волчанки, увеита, застойной сердечной недостаточности, фиброза, кахексии, отторжения трансплантата, рака, аутоиммунного заболевания, оппортунистических инфекций при СПИДе, ревматоидного артрита, ревматоидного спондилита, остеоартроза,других артрогенных заболеваний, болезни Крона, неспецифического язвенного колита, рассеянного склероза, системного волчаночного эритрематоза, эритемы лепрозной узловатой при проказе, радиационного повреждения, альвеолярного повреждения, вызванного гипероксией, нежелательного ангиогенеза, воспалительного заболевания, артрита, воспалительного заболевания кишечника, афтозных язв, астмы, дистресс-синдрома дыхательных путей у взрослых и СПИДа. 18. Способ ингибирования PDE4 (фосфодиэстеразы 4 типа) у пациента, нуждающегося в этом, содержащий введение пациенту эффективного количества соединения по п.1 или его фармацевтически приемлемой соли. 19. Способ снижения уровней TNF- (фактора некроза опухолей альфа), содержащий введение пациенту эффективного количества соединения по п.1 или его фармацевтически приемлемой соли. 20. Способ лечения заболевания, выбранного из группы, состоящей из септического шока, сепсиса,эндотоксического шока, гемодинамического шока и сепсисоподобного синдрома, постишемического реперфузионного повреждения, малярии, микобактериальной инфекции, менингита, псориаза, саркоидоза, псориатического артрита, болезни Бехчета, узелковой почесухи, волчанки, увеита, застойной сердечной недостаточности, фиброза, кахексии, отторжения трансплантата, рака, аутоиммунного заболевания,оппортунистических инфекций при СПИДе, ревматоидного артрита, ревматоидного спондилита, остеоартроза, других артрогенных заболеваний, болезни Крона, неспецифического язвенного колита, рассеянного склероза, системного волчаночного эритрематоза, эритемы лепрозной узловатой при проказе, радиационного повреждения, альвеолярного повреждения, вызванного гипероксией, нежелательного ангиогенеза, воспалительного заболевания, артрита, воспалительного заболевания кишечника, афтозных язв, астмы, дистресс-синдрома дыхательных путей у взрослых и СПИДа, у пациента, нуждающегося в этом, содержащий введение пациенту эффективного количества соединения по п.1 или его фармацевти- 26021019 чески приемлемой соли. 21. Способ по п.20, отличающийся тем, что заболевание представляет собой псориаз или саркоидоз. 22. Способ по п.21,отличающийся тем, что псориаз представляет собой псориаз бляшечного типа или рефракторный псориаз. 23. Способ по п.21, отличающийся тем, что саркоидоз представляет собой кожный саркоидоз. 24. Способ по п.20, отличающийся тем, что волчанка представляет собой кожную волчанку. 25. Способ по п.20, отличающийся тем, что заболевание представляет собой псориатический артрит. 26. Способ по п.20, отличающийся тем, что заболевание представляет собой ревматоидный артрит. 27. Способ по п.20, отличающийся тем, что заболевание представляет собой болезнь Бехчета. 28. Способ по п.20, отличающийся тем, что заболевание представляет собой болезнь Крона. 29. Способ по п.20, отличающийся тем, что заболевание представляет собой системный волчаночный эритрематоз. 30. Способ по п.20, отличающийся тем, что заболевание представляет собой воспалительное заболевание кишечника.
МПК / Метки
МПК: C07D 405/12, C07D 209/48, A61K 31/4035, C07D 209/46, A61P 17/06
Метки: дейтерированные, производные, изоиндолин-1,3-диона
Код ссылки
<a href="https://eas.patents.su/28-21019-dejjterirovannye-proizvodnye-izoindolin-13-diona.html" rel="bookmark" title="База патентов Евразийского Союза">Дейтерированные производные изоиндолин-1,3-диона</a>
Дейтерированные производные 1,3-бензодиоксола

Номер патента: 14432
Опубликовано: 30.12.2010
Автор: Танг Роджер
МПК: C07D 295/00
Метки: 1,3-бензодиоксола, дейтерированные, производные
Формула / Реферат:
1. Выделенное соединение формулы (I)или его соль; или пролекарство или соль его пролекарства; или его гидрат, сольват или полиморф; в которомD-является дейтерием;каждый Y независимо выбран из дейтерия или водорода;каждый водород независимо необязательно замещен дейтерием икаждый углерод независимо необязательно замещен 13С.2. Соединение или его пролекарство по п.1, в котором Y1является дейтерием.3. Соединение по п.2, в котором до 4 атомов...
Дейтерированные производные катехоламина, а также лекарственные средства, содержащие эти соединения

Номер патента: 9258
Опубликовано: 28.12.2007
Автор: Алкен Рудольф-Гизберт
МПК: C07B 59/00, A61P 25/00, A61K 31/198...
Метки: соединения, содержащие, эти, катехоламина, лекарственные, также, производные, средства, дейтерированные
Формула / Реферат:
1. Дейтерированные производные катехоламина общей формулы I причем R1 обозначает H или D, R2 обозначает H или D, R3 обозначает H, D, C1-С6алкил или С5-С6циклоалкил, дейтерированный C1-С6алкил или C5-C6циклоалкил, R4 обозначает H или D, R5 обозначает H или D. 2. Дейтерированные производные катехоламина по п.1, причем R1 обозначает H или D, R2 обозначает H или D, R3 обозначает H, D, C1-С6алкил или С5-С6циклоалкил, дейтерированный C1-С6алкил или...
Дейтерированные производные катехоламина и лекарственные средства, содержащие указанные соединения

Номер патента: 17983
Опубликовано: 30.04.2013
Авторы: Шнайдер Франк, Алкен Рудольф-Гизберт
МПК: C07C 229/26, C07B 59/00
Метки: соединения, средства, дейтерированные, лекарственные, производные, содержащие, указанные, катехоламина
Формула / Реферат:
1. Дейтерированные производные катехоламина общей формулы Iгде R1 представляет собой H;R2 представляет собой D;R3 представляет собой H, метил или этил;R4 представляет собой H;R5 представляет собой Н или D;R6 представляет собой Н или D, где оба остатка R6 не являются одновременно D и где оба остатка R6 не являются одновременно H,их стереоизомеры, энантиомеры или диастереомеры в оптически чистой форме, а также их физиологически приемлемые соли.2....
Производные пиримидин-2,4-диона в качестве антагонистов рецептора гонадотропин-высвобождающего гормона

Номер патента: 10370
Опубликовано: 29.08.2008
Авторы: Дуайт Уэсли Дж., У Дунпэй, Го Чжицян, Хуанг Чарльз К., Чень Юншэн, Уэйд Уоррен, Туччи Фабио К., Чен Чен
МПК: C07D 239/54, A61P 5/02, A61K 31/513...
Метки: пиримидин-2,4-диона, антагонистов, рецептора, гонадотропин-высвобождающего, качестве, гормона, производные
Формула / Реферат:
Соединение, имеющее следующую структуру: или его стереоизомер или фармацевтически приемлемая соль, где R1a, R1b, R1c являются одинаковыми или разными и независимо представляют собой водород, галоген, С1-4алкил, гидрокси или алкокси, или R1a и R1b, взятые вместе, образуют -OCH2O- или -ОСН2СН2-; R2a и R2b являются одинаковыми или разными и независимо представляют собой водород, галоген, трифторметил, циано или -SO2CH3; R3 представляет собой...
Аминокислотные производные хиноксалин-2,3-диона, фармацевстическая композиция, обладающая антагонистической в отношении рецептора глутамата и противосудорожной активностью, способ лечения заболеванийсосудов мозга или судорог и промежуточный продукт

Номер патента: 605
Опубликовано: 29.12.1999
Автор: Никам Шам
МПК: C07F 9/6509, C07C 229/18, C07D 241/44...
Метки: глутамата, способ, продукт, активностью, мозга, рецептора, судорог, антагонистической, производные, хиноксалин-2,3-диона, аминокислотные, отношении, лечения, противосудорожной, композиция, фармацевстическая, промежуточный, обладающая, заболеванийсосудов
Формула / Реферат:
1. Аминокислотные производные хиноксалин-2,3-диона общей формулы (I) где R означает где R3 - водород или метил, R4 - группа формулы где R5 -гидроксил, низший, алкоксил, амид, R6 -низший, алкил или аралкил, R1 - водород, бром, нитро, R2 - низший алкил, винил, при этом R2 может находиться в b-положении, а группа R-CH2 - в а-положении кольца, или их фармацевтически приемлемые соли. 2. Аминокислотные производные...
Предыдущий патент: Многоканальная система радиосвязи
Следующий патент: Газоочистное устройство и способ очистки газа
Случайный патент: Устройство экспонирования, способ экспонирования и печатающее устройство