Агонисты mglu2
Номер патента: 20229
Опубликовано: 30.09.2014
Авторы: Монтеро Сальгадо Карлос, Табоада Мартинес Лорена, Монн Джеймс Аллен, Пьето Лурдес, Шоу Брюс Вилльям

























Формула / Реферат
1. Соединение формулы

где R1 представляет собой водород, R2 представляет собой водород и R3 представляет собой водород;
R1 представляет собой водород, R2 представляет собой (2S)-2-аминопропаноил и R3 представляет собой водород;
R1 представляет собой водород, R2 представляет собой (2S)-2-амино-4-метилсульфанилбутаноил и R3 представляет собой водород;
R1 представляет собой водород, R2 представляет собой (2S)-2-амино-4-метилпентаноил и R3 представляет собой водород;
R1 представляет собой водород, R2 представляет собой 2-аминоацетил и R3 представляет собой водород;
R1 представляет собой бензил, R2 представляет собой водород и R3 представляет собой бензил; или
R1 представляет собой (2-фторфенил)метил, R2 представляет собой водород и R3 представляет собой (2-фторфенил)метил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, представляющее собой (1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль.
3. Соединение по п.2, представляющее собой (1R,2S,4R,5R,6R)-2-амино-4-(4Н-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту.
4. Соединение по п.1, представляющее собой (1R,2S,4R,5R,6R)-2-[[(2S)-2-аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль.
5. Соединение по п.4, представляющее собой гидрохлорид (1R,2S,4R,5R,6R)-2-[[(2S)-2-аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты.
6. Соединение по п.1, представляющее собой (1R,2S,4R,5R,6R)-2-[[(2S)-2-амино-4-метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль.
7. Соединение по п.6, представляющее собой гидрохлорид (1R,2S,4R,5R,6R)-2-[[(2S)-2-амино-4-метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты.
8. Соединение по п.1, представляющее собой (1R,2S,4R,5R,6R)-2-[[(2S)-2-амино-4-метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль.
9. Соединение по п.8, представляющее собой гидрохлорид (1R,2S,4R,5R,6R)-2-[[(2S)-2-амино-4-метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты.
10. Соединение по п.1, представляющее собой (1R,2S,4R,5R,6R)-2-[(2-аминоацетил)амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль.
11. Соединение по п.10, представляющее собой гидрохлорид (1R,2S,4R,5R,6R)-2-[(2-аминоацетил)амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты.
12. Соединение по п.1, представляющее собой дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0] гексан-2,6-дикарбоксилат или его фармацевтически приемлемую соль.
13. Соединение по п.12, представляющее собой дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат.
14. Соединение по п.1, представляющее собой бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемую соль.
15. Соединение по п.14, представляющее собой гидрохлорид бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата.
16. Соединение, представляющее собой моногидрат моноаммониевой соли (1R,2S,4R,5R,6R)-2-[[(2S)-2-амино-1-оксопропил]амино]-4-(4H-1,2,4-триазол-3-илтио)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты.
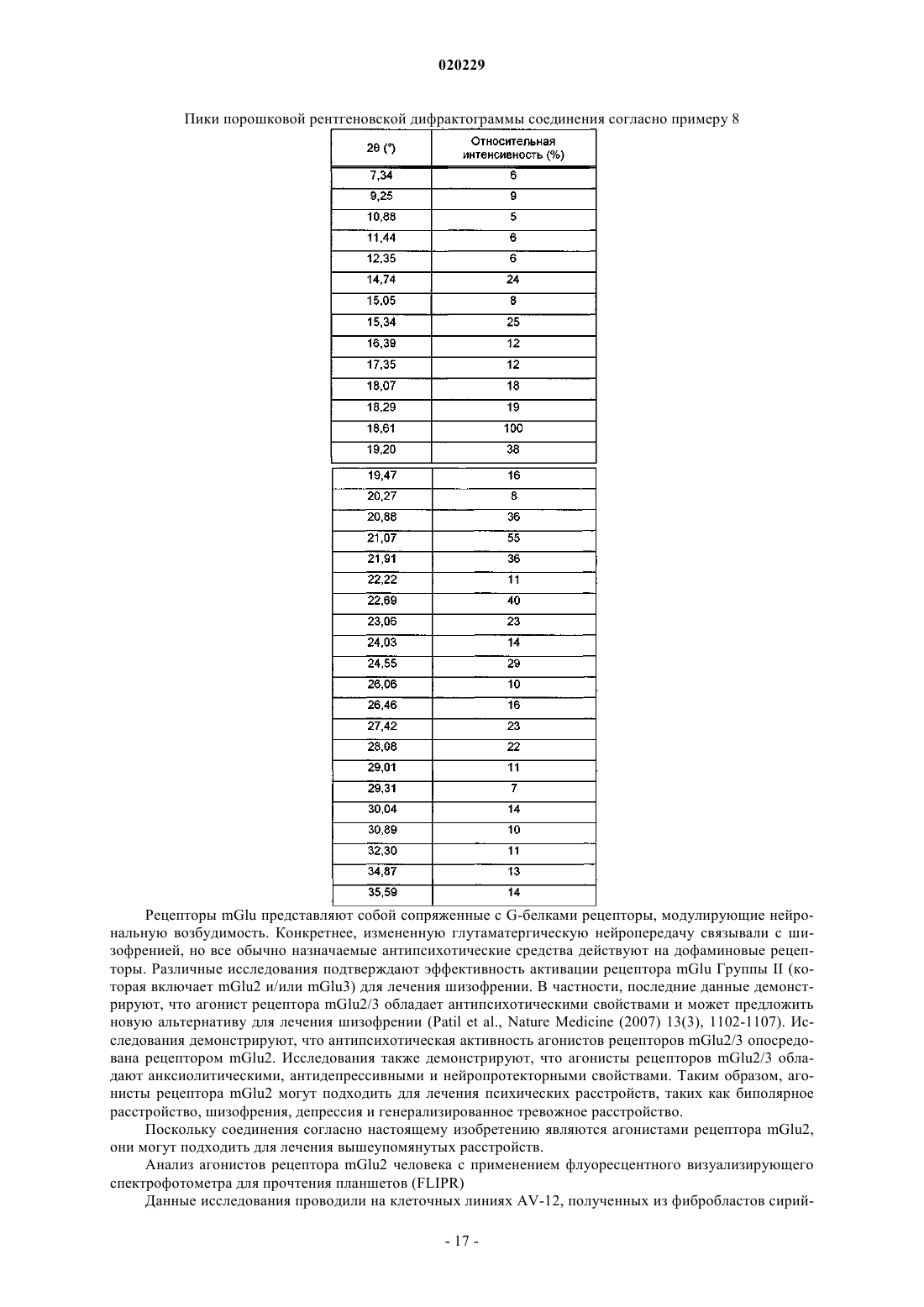
17. Соединение по п.16, представляющее собой моногидрат моноаммониевой соли (1R,2S,4R,5R,6R)-2-[[(2S)-2-амино-1-оксопропил]амино]-4-(4H-1,2,4-триазол-3-илтио)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты в кристаллической форме, характеризующейся порошковой рентгеновской дифрактограммой, имеющей дифракционные пики по шкале 2q±0,2, соответствующие 18,61 и 21,07.
18. Фармацевтическая композиция, содержащая соединение или соль по любому из пп.1-17 и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
19. Применение соединения или соли по любому из пп.1-17 в терапии.
20. Применение соединения или соли по любому из пп.1-17 для лечения психического расстройства, выбранного из группы, состоящей из биполярного расстройства, шизофрении, депрессии и генерализированного тревожного расстройства.
21. Применение соединения или соли по п.20, где психическое расстройство представляет собой биполярное расстройство.
22. Применение соединения или соли по п.20, где психическое расстройство представляет собой шизофрению.
23. Применение соединения или соли по п.20, где психическое расстройство представляет собой депрессию.
24. Применение соединения или соли по п.20, где психическое расстройство представляет собой генерализированное тревожное расстройство.
Текст
Согласно настоящему изобретению предложены новые агонисты mGlu2, подходящие для лечения биполярного расстройства, шизофрении, депрессии и генерализированного тревожного расстройства. Новые агонисты представлены формулой(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Настоящее изобретение относится к соединениям-агонистам mGlu2, их конкретным пролекарствам и их солям и сольватам, более конкретно к новым 4-замещенным бицикло[3.1.0]гексановым соединениям, их конкретным пролекарствам и их солям и сольватам, а также фармацевтическим композициям и терапевтическим применениям таких соединений, конкретных пролекарств и их солей и сольватов.L-глутамат является основным возбуждающим нейромедиатором в центральной нервной системе, и его называют возбуждающей аминокислотой. Метаботропные глутаматные (mGlu) рецепторы представляют собой сопряженные с G-белками рецепторы, модулирующие нейрональную возбудимость. Лечение неврологических или психических расстройств было связано с селективной активацией рецепторов mGlu возбуждающей аминокислоты. Различные исследования подтверждают эффективность активации рецептора mGlu Группы II (которая включает рецепторы mGlu2 и/или mGlu3) для лечения шизофрении. Конкретнее, последние данные демонстрируют, что агонист рецептора mGlu2/3 обладает антипсихотическими свойствами и может представлять собой новую альтернативу для лечения шизофрении. Исследования демонстрируют, что антипсихотическая активность агонистов mGlu2/3 опосредована mGlu2. Исследования также демонстрируют, что агонисты mGlu2/3 обладают анксиолитическими, антидепрессивными и нейропротекторными свойствами. Таким образом, агонисты mGlu2 могут подходить для лечения психических расстройств, таких как биполярное расстройство, также известное как маниакально-депрессивное расстройство, шизофрения, депрессия и генерализированное тревожное расстройство. В WO 9717952 предложены некоторые 4-замещенные бицикло[3.1.0]гексановые соединения, являющиеся, по утверждению авторов, антагонистами или агонистами метаботропных глутаматных рецепторов. В WO 03104217 предложены бицикло[3.1.0]гексановые и гетеробицикло[3.1.0]гексановые соединения, представляющие собой, по утверждению авторов, пролекарственные формы соединенийагонистов рецептора mGluR2. Повышенный глутаматергический тонус вовлечен во многие болезненные состояния центральной нервной системы, однако эффективные агенты для коррекции таких патофизиологических состояний отсутствуют в клинической практике. В частности, клиническое применение не было реализовано из-за отсутствия агонистов mGlu2 с подходящими лекарственно-подобными свойствами. Таким образом, попрежнему существует потребность в высокоактивных эффективных агонистах mGlu2. Согласно настоящему изобретению предложены новые 4-замещенные бицикло[3.1.0]гексаны, включая их конкретные пролекарства, обеспечивающие повышенную биодоступность, подходящую для клинической разработки,которые являются высокоактивными и эффективными агонистами рецептора mGlu2. Такие новые соединения согласно настоящему изобретению могли бы удовлетворить потребность в мощных эффективных способах лечения психических расстройств, таких как биполярное расстройство, шизофрения, депрессия и генерализированное тревожное расстройство. Согласно настоящему изобретению предложено соединение формулы где R1 представляет собой водород, R2 представляет собой водород и R3 представляет собой водород;R1 представляет собой бензил, R2 представляет собой водород и R3 представляет собой бензил; илиR1 представляет собой (2-фторфенил)метил, R2 представляет собой водород и R3 представляет собой (2-фторфенил)метил; или его фармацевтически приемлемая соль или сольват этой соли. Согласно настоящему изобретению предложены (1R,2S,4R,5R,6R)-2-амино-4-(4 Н-1,2,4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль. В качестве конкретного варианта реализации согласно настоящему изобретению предложена(1R,2S,4R,5R,6R)-2-амино-4-(4 Н-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота. Согласно настоящему изобретению предложены(1R,2S,4R,5R,6R)-2-(2S)-2-аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль. В качестве конкретного варианта реализации согласно настоящему изобретению предложен гидрохлорид (1R,2S,4R,5R,6R)-2-(2S)-2-аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты. Согласно настоящему изобретению предложены (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль. В качестве конкретного варианта реализации согласно настоящему изобретению предложен гидрохлорид (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты. Согласно настоящему изобретению предложены (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль. В качестве конкретного варианта реализации согласно настоящему изобретению предложен гидрохлорид(1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты. Согласно настоящему изобретению предложены (1R,2S,4R,5R,6R)-2-[(2-аминоацетил)амино]-4-(4H1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль. В качестве конкретного варианта реализации согласно настоящему изобретению предложен гидрохлорид (1R,2S,4R,5R,6R)-2-[(2-аминоацетил)амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0] гексан-2,6-дикарбоновой кислоты. Согласно настоящему изобретению предложены дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемая соль. В качестве конкретного варианта реализации согласно настоящему изобретению предложен дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат. Согласно настоящему изобретению предложены бис[(2-фторфенил)метил]-[1R,2S,4R,5R,6R)-2 амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемая соль. В качестве конкретного варианта реализации согласно настоящему изобретению предложен гидрохлорид бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата. Согласно настоящему изобретению предложена бицикло[3.1.0]гексан-2,6-дикарбоновая кислота, 2(2S)-2-амино-1-оксопропил]амино]-4-(4 Н-1,2,4-триазол-3-илтио)-, моноаммониевая соль, (1R,2S,4R,5R,6R)-,моногидрат. Согласно настоящему изобретению предложена бицикло[3.1.0]гексан-2,6-дикарбоновая кислота, 2(2S)-2-амино-1-оксопропил]амино]-4-(4 Н-1,2,4-триазол-3-илтио)-, моноаммониевая соль, (1R,2S,4R,5R,6R)-,моногидрат в кристаллической форме. Согласно настоящему изобретению предложена бицикло[3.1.0]гексан-2,6-дикарбоновая кислота, 2(2S)-2-амино-1-оксопропил]амино]-4-(4 Н-1,2,4-триазол-3-илтио)-, моноаммониевая соль, (1R,2S,4R,5R,6R)-,моногидрат в кристаллической форме, характеризующейся порошковой рентгеновской дифрактограммой,имеющей пики по шкале 2 0,2, соответствующие 18,61 и 21,07. Согласно настоящему изобретению также предложена бицикло[3.1.0]гексан-2,6-дикарбоновая кислота,2-(2S)-2-амино-1-оксопропил]амино]-4-(4 Н-1,2,4-триазол-3-илтио)-,моноаммониевая соль,(1R,2S,4R,5R,6R)-, моногидрат в кристаллической форме, характеризующейся порошковой рентгеновской дифрактограммой, имеющей пики по шкале 20,2, при 18,61 в комбинации с одним или более пиками, выбранными из группы, состоящей из 21,07, 15,34, 14,74 и 19,20. Соединения согласно настоящему изобретению представляют собой (1R,2S,4R,5R,6R)-2-амино-4-(4 Н 1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту, (1R,2S,4R,5R,6R)-2-(2S)-2 аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту, (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту, (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту,(1R,2S,4R,5R,6R)-2-[(2-аминоацетил)амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6 дикарбоновую кислоту, дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат и/или бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или фармацевтически приемлемую соль указанных соединений. Согласно настоящему изобретению предложена фармацевтическая композиция, содержащая кислоту или ее фармацевтически приемлемую соль,(1R,2S,4R,5R,6R)-2-(2S)-2 аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль, (2R,2S,4R,5R,6R)-2-(2S)-2-амино-4 метилсульфанилбутаноил]амино]-4-(4 Н-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль, (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4 метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль, [1R,2S,4R,5R,6R)-2-[(2-аминоацетил)амино]-4-(4H-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль, дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемую соль, бис[(2-фторфенил)метил](1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемую соль или бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту, 2(2S)-2-амино-1-оксопропил]амино]-4-(4 Н-1,2,4-триазол-3-илтио)-,моноаммониевую соль,(1R,2S,4R,5R,6R)-, моногидрат совместно с фармацевтически приемлемым носителем и возможно другими терапевтическими ингредиентами. Согласно настоящему изобретению предложена фармацевтическая композиция, содержащая(1R,2S,4R,5R,6R)-2-амино-4-(4 Н-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль, (1R,2S,4R,5R,6R)-2-(2S)-2-аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль, (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилсульфанилбутаноил]амино]4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль, (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилпентаноил]амино]-4-(4H-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль, (1R,2S,4R,5R,6R)-2-[(2-аминоацетил)амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль, дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемую соль, бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино-4(4H-1,2,4-триазол-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемую соль или бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту,2-(2S)-2-амино-1 оксопропил]амино]-4-(4 Н-1,2,4-триазол-3-илтио)-, моноаммониевую соль, (1R,2S,4R,5R,6R)-, моногидрат, и фармацевтически приемлемые носитель, разбавитель или вспомогательное вещество. Согласно настоящему изобретению предложен способ лечения психического расстройства, включающий введение пациенту, нуждающемуся в этом, эффективного количества (1R,2S,4R,5R,6R)-2-амино 4-(4 Н-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли, (1R,2S,4R,5R,6R)-2-(2S)-2-аминопропаноил]амино]-4-(1H-1,2,4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли, (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли,(1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли, (1R,2S,4R,5R,6R)2-[(2-аминоацетил)амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли, дибензил[1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата или его фармацевтически приемлемой соли,бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата или его фармацевтически приемлемой соли или бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты, 2-(2S)-2-амино-1-оксопропил]амино]-4-(4 Н-1,2,4-триазол-3 илтио)-, моноаммониевой соли, (1R,2S,4R,5R,6R)-, моногидрата. Согласно настоящему изобретению предложено применение (1R,2S,4R,5R,6R)-2-амино-4-(4 Н-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли, (1R,2S,4R,5R,6R)-2-(2S)-2-аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли,(1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли,(1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли, (1R,2S,4R,5R,6R)2-[(2-аминоацетил)амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты или ее фармацевтически приемлемой соли, дибензил(2R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата или его фармацевтически приемлемой соли, бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата или его фармацевтически приемлемой соли или бицикло[3.1.0]гексан 2,6-дикарбоновой кислоты, 2-(2S)-2-амино-1-оксопропил]амино]-4-(4 Н-1,2,4-триазол-3-илтио)-, моноаммониевой соли, (1R,2S,4R,5R,6R)-, моногидрата для получения лекарственного средства для лечения психического расстройства. Согласно настоящему изобретению предложены (1R,2S,4R,5R,6R)-2-амино-4-(4 Н-1,2, 4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль,(1R,2S,4R,5R,6R)-2-(2S)-2-аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль, (1R,2S,4R,5R,6R)-2-(2S)-2 амино-4-метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6 дикарбоновая кислота или ее фармацевтически приемлемая соль, (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4 метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль, (1R,2S,4R,5R,6R)-2-[(2-аминоацетил)амино]-4-(4H1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль, дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемая соль, бис[(2-фторфенил)метил](1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемая соль или бицикло[3.1.0]гексан-2,6-дикарбоновая кислота, 2-(2S)2-амино-1-оксопропил]амино]-4-(4 Н-1,2,4-триазол-3-илтио)-, моноаммониевая соль, (1R,2S,4R,5R,6R)-,моногидрат для применения в терапии. Согласно настоящему изобретению также предложены (1R,2S,4R,5R,6R)-2-амино-4-(4 Н-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль,(1R,2S,4R,5R,6R)-2-(2S)-2-аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль,(1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль,(1R,2S,4R,5R,6R)-2-(2S)-2-амино-4-метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль, (1R,2S,4R,5R,6R)-2[(2-аминоацетил)амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота или ее фармацевтически приемлемая соль, дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол 3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемая соль,бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемая соль или бицикло[3.1.0]гексан-2,6 дикарбоновая кислота, 2-(2S)-2-амино-1-оксопропил]амино]-4-(4 Н-1,2,4-триазол-3-илтио)-, моноаммониевая соль, (1R,2S,4R,5R,6R)-, моногидрат для применения для лечения психического расстройства. Кроме того, согласно настоящему изобретению предложены предпочтительные варианты реализации способов и применений, описанные в настоящей заявке, в которых психическое расстройство выбрано из группы, состоящей из биполярного расстройства, шизофрении, депрессии и генерализированного тревожного расстройства. В вышеприведенном тексте и по всему описанию изобретения следующие термины, если не указано иное, имеют следующие значения."Фармацевтически приемлемые соли" относятся к относительно нетоксичным неорганическим и органическим солям соединений согласно настоящему изобретению."Терапевтически эффективное количество" или "эффективное количество" означает количество соединения или его фармацевтически приемлемой соли или сольвата этой соли согласно настоящему изобретению или фармацевтической композиции, содержащей соединение или его фармацевтически приемлемую соль или сольват этой соли согласно настоящему изобретению, которое вызовет биологический или терапевтический ответ или желаемый терапевтический эффект на ткань, систему, животное, млекопитающее или человека, к которому стремится исследователь, ветеринар, врач или другой клиницист. Термины "лечение", "лечить", "лечащий" и т.п. включают замедление или обращение развития расстройства. Эти термины также включают облегчение, уменьшение интенсивности, ослабление, устранение или сокращение одного или более симптомов расстройства или состояния, даже если расстройство или состояние в действительности не устранено и даже если само развитие расстройства или состояния не замедлено или не обращено. Согласно стандартной номенклатуре, применяемой по всему настоящему описанию, концевую часть обозначаемой боковой цепи описывают первой, а вслед за ней - соседнюю функциональную группу по направлению к точке присоединения. Например, метилсульфонильный заместитель эквивалентенCH3-SO2-. Соединения согласно настоящему изобретению способны вступать в реакцию, например, с рядом неорганических и органических кислот с образованием фармацевтически приемлемых солей присоединения кислоты или солей присоединения основания. Такие фармацевтически приемлемые соли и общая методология для их получения хорошо известны в данной области техники (см., например, P. Stahl et al.,HANDBOOK OF PHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE (VCHA/WileyVCH, 2002); S.M. Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January 1977). Соединения согласно настоящему изобретению предпочтительно готовят в виде фармацевтических композиций, применяя один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ, и вводят с помощью множества способов. Предпочтительно такие композиции предназначены для перорального или внутривенного введения. Такие фармацевтические композиции и способы для их получения хорошо известны в данной области техники (см., например, Remington:The Science and Practice of Pharmacy (A. Gennaro et al., eds., 21st ed., Mack Publishing Co., 2005. Фактически вводимое соединение или соединения согласно настоящему изобретению будут определены врачом при соответствующих обстоятельствах, включающих состояние, подлежащее лечению,выбранный способ введения, конкретное вводимое соединение или соединения согласно настоящему изобретению, возраст, вес и реакцию отдельного пациента и тяжесть симптомов пациента. Суточные дозировки, как правило, попадают в диапазон от примерно 0,1 до примерно 300 мг. В некоторых случаях уровни дозировки меньше нижнего предела вышеуказанного диапазона могут быть более чем достаточными, в то время как в других случаях могут быть применены еще большие дозы. Соединения согласно настоящему изобретению могут быть получены с помощью множества способов, известных в данной области техники, а также способов, описанных в синтезах и примерах ниже. Конкретные стадии синтеза для каждого из описанных путей синтеза можно по-разному комбинировать для получения соединений согласно настоящему изобретению. Заместители, если не указано иное, имеют значения, определенные ранее. Реагенты и исходные вещества в общем случае легко доступны специалисту в данной области техники. Другие реагенты могут быть получены с помощью стандартных методик органической химии и химии гетероциклических соединений, методик, аналогичных синтезам известных структурно родственных соединений, и способов,описанных в последующих синтезах и примерах, включая любые новые способы. Соединения в последующих синтезах и примерах 1-7 названы с применением программы Symyx Draw 3.1. Соединение в примере 8 названо с применением названия по классификатору CAS из программы ACD Labs. В настоящей заявке следующие термины имеют указанные значения: "ВЭЖХ" относится к высокоэффективной жидкостной хроматографии; "ЖХ" относится к жидкостной хроматографии; "МС" относится к масс-спектрометрии; "ЯМР" относится к ядерному магнитному резонансу; "ТСХ" относится к тонкослойной хроматографии; "ЭДТУ" относится к этилендиаминтетрауксусной кислоте; "ФСБ" относится к фосфатно-солевому буферу; "ПЦР" относится к полимеразной цепной реакции; "SCX" относится к сильному катионообменному; "ГЛБ" относится к гидрофильно-липофильному балансу. Синтез 1. ди-трет-Бутил(1S,2S,4S,5R,6R)-2-(трет-бутоксикарбониламино)-4-(п-толилсульфонилокси)бицикло[3.1.0]гексан-2,6-дикарбоксилат В двугорлую круглодонную колбу помещали в атмосфере азота ди-трет-бутил(1S,2S,4S,5R,6R)-2(трет-бутоксикарбониламино)-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат (20,7 г, 0,5 моль, подробнее детали синтеза см. в WO 03/104217/A2), 4-диметиламинопиридин (10,4 г, 0,85 моль), триэтиламин(6,98 мл, 0,5 ммоль) и п-толуолсульфонилхлорид (10,6 г, 0,55 моль) в дихлорметане (200 мл) и перемешивали смесь при комнатной температуре в течение ночи. Добавляли 1 н. раствор гидросульфата калия(200 мл), воду (100 мл) и экстрагировали органический слой. Промывали водой (200 мл), солевым раствором (200 мл), высушивали над сульфатом магния, фильтровали и упаривали досуха. Добавляли тетрагидрофуран (30 мл), затем гептаны (90 мл). Смесь нагревали при 60 С и медленно добавляли дополнительное количество гептанов (200 мл). Охлаждали смесь до комнатной температуры. Твердое вещество отфильтровывали и сушили под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (24,6 г, 87%). МС (m/z): 590 (М+23). Способ 1. В круглодонную колбу помещали в атмосфере азота ди-трет-бутил(1S,2S,4S,5R,6R)-2-(третбутоксикарбониламино)-4-(п-толилсульфонилокси)бицикло[3.1.0]гексан-2,6-дикарбоксилат (462 г, 813,8 ммоль), 1H-1,2,4-триазол-3-тиол (88,2 г, 854,5 ммоль), карбонат калия (123,7 г, 895,2 ммоль) в N,Nдиметилформамиде (2,3 л) и перемешивали смесь при 80 С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, затем добавляли метил-трет-бутиловый эфир (2,3 л) и воду (4,6 л). Наблюдали выделение газа, происходящее после медленного добавления 1 М гидросульфата калия (1,85 л). Смесь экстрагировали метил-трет-бутиловым эфиром (2,3 л) и удаляли водную фазу. Промывали последовательно водой (2,5 л), солевым раствором (2 л) и удаляли водные фазы. Концентрировали досуха с получением твердого вещества (440 г). Очистку проводили посредством флэш-хроматографии, элюируя смесью этилацетат:гексан (от 20:80 до 70:30) с получением указанного в заголовке соединения в виде белого твердого вещества (305,6 г, 76%). МС (m/z): 497 (М+1). Способ 2. ди-трет-Бутил(1S,2S,4S,5R,6R)-2-(трет-бутоксикарбониламино)-4-(п-толилсульфонилокси)бицикло[3.1.0]гексан-2,6-дикарбоксилат (1,31 г, 2,31 ммоль) и 1H-1,2,4-триазол-3-тиол (0,31 г, 3 ммоль) растворяли в N,N-диметилформамиде (7 мл), затем добавляли карбонат калия (639 мг, 4,62 ммоль) и перемешивали смесь в течение ночи при 80 С. Концентрировали досуха, снова растворяли в этилацетате и промывали 10%-ным раствором лимонной кислоты и 10%-ным солевым раствором. Сушили над сульфатом натрия, отфильтровывали и концентрировали досуха. Очистку проводили посредством флэшхроматографии, элюируя смесью этилацетат:гексан (от 10:90 до 80:20) с получением указанного в заголовке соединения (830 мг, 72%). МС (m/z): 497 (М+1). Синтез 3. Гидрохлорид диэтил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата Способ 1. В круглодонную колбу помещали ди-трет-бутил(1R,2S,4R,5R,6R)-2-(трет-бутоксикарбониламино)4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат (305,6 г, 0,61 моль) и этанол(1,53 л). Медленно добавляли тионилхлорид (179,3 мл, 2,46 моль) (экзотермическая реакция до 45 С) и перемешивали смесь при 80 С в течение ночи. Растворитель удаляли под вакуумом с получением белой пены. Добавляли метил-трет-бутиловый эфир (2,5 л) и удаляли растворитель под вакуумом. Добавляли метил-трет-бутиловый эфир (2,5 л) и перемешивали в течение ночи. Твердое вещество отфильтровывали и промывали метил-трет-бутиловым эфиром. Сушили в атмосфере азота с получением указанного в заголовке соединения в виде белого твердого вещества (264,6 г, 0,7 моль). МС (m/z): 341 (М+1). Способ 2. Растворяли ди-трет-бутил(1R,2S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-4-(4H-1,2,4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат (830 мг, 1,67 ммоль) в этаноле (6,7 мл), смесь охлаждали до 5 С и добавляли тионилхлорид (487 мкл, 6,69 ммоль). Смесь нагревали при 80 С в течение ночи. Растворитель удаляли под вакуумом с получением белого твердого вещества, затем добавляли диэтиловый эфир и концентрировали досуха. Вещество сушили дополнительно в течение 48 ч с получением указанного в заголовке соединения (714 мг, 1,89 ммоль). МС (m/z): 341 (М+1). Синтез 4. Диэтил(1R,2S,4R,5R,6R)-2-(2S)-2-(трет-бутоксикарбониламино)пропаноил]амино]-4-(4H-1,2,4-6 020229 Способ 1. В 5-литровый реактор добавляли в атмосфере азота гидрохлорид диэтил(1R,2S,4R,5R,6R)-2-амино 4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата (264 г, 0,7 моль) и тетрагидрофуран (1,32 л) и охлаждали смесь до 0-5 С на водно-ледяной бане. Затем добавляли хлордиметокситриазин (125,5 г, 0,7 моль) и (2S)-2-(трет-бутоксикарбониламино)пропановую кислоту (141,3 г, 0,73 моль). Медленно добавляли N-метилморфолин (231,8 мл, 2,1 моль) и перемешивали в течение 3 ч. Смесь отфильтровывали и промывали белое твердое вещество тетрагидрофураном. Твердое вещество удаляли,а раствор концентровали досуха. Очистку проводили посредством флэш-хроматографии, элюируя смесью этилацетат:гексан (от 60:40 до 100:0) с получением указанного в заголовке соединения (195 г, 54%). МС (m/z): 512 (М+1), 534 (М+23). Способ 2. Объединяли гидрохлорид диэтил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата (354 мг, 0,94 ммоль), (2S)-2-(трет-бутоксикарбониламино)пропановую кислоту (271 мг, 1,41 ммоль), 4-диметиламинопиридин (11,5 мг, 94 мкмоль), 1 гидроксибензотриазолгидрат (219 мг, 1,41 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3 этилкарбодиимида (274 мг, 1,41 ммоль) в дихлорметане (9,4 мл), затем добавляли триэтиламин (393 мкл,2,82 ммоль) и перемешивали смесь при комнатной температуре в течение ночи в атмосфере азота. Промывали 10%-ным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и солевым раствором. Водные слои удаляли, отфильтровывали органический слой через кизельгуровый фильтр и удаляли растворитель под вакуумом. Очистку проводили посредством флэш-хроматографии, элюируя смесью этилацетат:гексан (от 20:80 до 100:0) с получением указанного в заголовке соединения (319 мг, 66%). MC Способ 1. В двугорлую круглодонную колбу помещали диэтил(1R,2S,4R,5R,6R)-2-[(2S)-2-(третбутоксикарбониламино)пропаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6 дикарбоксилат (195 г, 0,38 моль) и тетрагидрофуран (1,17 л) и охлаждали смесь до 4 С с помощью водно-ледяной бани. Медленно добавляли холодный (4 С) раствор водного 50%-ного раствора гидроксида натрия (81 мл) в воде (1,17 л) в течение 30 мин (небольшой экзотермический эффект до 8 С). Водноледяную баню удаляли и перемешивали смесь при 15 С. Через 3 ч смесь подкисляли до рН 3 раствором концентрированной соляной кислоты в воде (1:3, приблизительно 500 мл), поддерживая температуру ниже 20 С. Проводили экстракцию мутного раствора этилацетатом (600 мл, затем 2100 мл). Объединяли органические слои, промывали солевым раствором (100 мл) и удаляли водную фазу. Сушили над сульфатом магния, фильтровали и концентрировали досуха с получением указанного в заголовке соединения в виде белого твердого вещества (187 г, 0,41 моль). МС (m/z): 456 (М+1). Способ 2. Растворяли диэтил(1R,2S,4R,5R,6R)-2-(2S)-2-(трет-бутоксикарбониламино)пропаноил]амино]-4(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат (319 мг, 0,62 ммоль) в тетрагидрофуране (4,2 мл), затем добавляли 2 М гидроксид лития (2,5 мл, 5 ммоль). Перемешивали смесь при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой и промывали этилацетатом. Удаляли органический слой. Доводили рН водной фазы до 21 н. соляной кислотой и проводили экстракцию этилацетатом. Органическую фазу пропускали для сушки через диатомовую землю и концентрировали досуха с получением указанного в заголовке соединения (244 мг, 86%). МС (m/z): 456 (М+1). Следующие соединения получены, главным образом, согласно способу 2 синтеза 7.: В качестве основания применяли 2,5 М LiOH.(40 мл) при 0-5 С осторожно (экзотермическая реакция) добавляли по каплям в течение 5 мин тионилхлорид (2,5 мл, 34,32 ммоль). Охлаждающую баню удаляли и нагревали реакционную смесь при температуре кипения с обратным холодильником в течение 16 ч. Выпаривали летучие вещества, а остаток сушили под высоким вакуумом в течение 7 ч с получением бесцветной пены гидрохлорида диэтил(1R,2S,4R,5R,6R)-2-амино-4-(4 Н-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата. Затем добавляли 2-(трет-бутоксикарбониламино)уксусную кислоту (0,89 г, 5,07 ммоль) и гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (1,93 г, 5,07 ммоль) к раствору гидрохлорида диэтил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата в безводном N,N-диметилформамиде (20 мл) при комнатной температуре,затем добавляли диизопропилэтиламин (2 мл, 11,47 ммоль) и перемешивали полученный желтый раствор в атмосфере азота в течение 18 ч. Растворитель концентрировали и распределяли остаток между этилацетатом (40 мл) и насыщенным раствором гидрокарбоната натрия в воде (40 мл). Смесь перемешивали в течение 20 мин, разделяли слои и экстрагировали водный слой дополнительным объемом этилацетата(40 мл). Органические слои объединяли, высушивали над сульфатом натрия и концентрировали досуха. Остаток очищали посредством флэш-хроматографии, элюируя смесью этилацетат:изогексан (от 80:20 до 100:0) с получением указанного в заголовке соединения в виде белого твердого вещества (2,42 г, 4,86 ммоль). МС (m/z): 498 (М+1), 520 (М+23). Синтез 11. К перемешиваемому раствору диэтил(1R,2S,4R,5R,6R)-2-2-(трет-бутоксикарбониламино)ацетил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата (1,4 г, 2,81 ммоль) в тетрагидрофуране (8 мл) добавляли 2 М гидроксид натрия (4,5 мл, 9 ммоль) при комнатной температуре. Перемешивали двухфазную смесь в течение 2 ч, разбавляли реакционную смесь водой (50 мл) и промывали диэтиловым эфиром (50 мл). Водный слой охлаждали до 0-5 С, подкисляли до рН 2 2 М соляной кислотой и экстрагировали этилацетатом (260 мл). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения в виде белого твердого вещества (0,75 г, 1,69 ммоль). 1 Н ЯМР (D2O)1,18-1,27 (м, 1 Н), 1,32 (с, 9 Н), 1,44-1,49 (м, 1 Н), 2,0-2,6 (м, 1 Н), 2,12-2,19 (м, 1 Н),2,65-2,75 (м, 2 Н), 3,57-3,68 (м, 2 Н), 4,12-4,21 (м, 1 Н), 8,23 (с, 1 Н), а также некоторого количества указанного в заголовке соединения в оставшемся водном слое (приблизительно 1,12 ммоль). МС (m/z): 442 К раствору ди-трет-бутил(1R,2S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-4-(4H-1,2,4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата (1,97 г, 3,97 ммоль) в 1,4-диоксане (10 мл) добавляли 4 М раствор хлороводорода в 1,4-диоксане (10 мл, 40 ммоль). Смесь нагревали до 50 С с перемешиванием в течение ночи (белое твердое вещество начинает осаждаться из раствора вскоре после начала нагревания). Добавляли этилацетат (50 мл) к реакционной смеси (белый мутный раствор). Смесь оставляли охлаждаться до комнатной температуры, затем твердое вещество собирали фильтрованием и высушивали с получением белого твердого вещества (1,97 г). Твердое вещество высушивали в течение выходных при пониженном давлении при 40 С с получением указанного в заголовке соединения в виде белого твердого вещества, загрязненного некоторым количеством 1,4-диоксана и небольшим количеством моно-трет-бутилового сложного эфира (1,936 г, 65% чистоты % мас./мас., 3,95 ммоль). МС (m/z): 285(9,72 мл) добавляли 2,5 М гидроксид натрия (15,55 мл, 38,88 ммоль). Реакционную смесь нагревали до 60 С и поддерживали перемешивание в течение ночи. Нагревание продолжали в течение 4 ч, затем промывали этилацетатом. Водную фазу охлаждали на ледяной бане и подкисляли до рН 2-3 1 н. раствором соляной кислоты. Проводили экстракцию этилацетатом (3 раза), сушили органический слой над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения в виде оранжевого твердого вещества (1,4 г, 96%). МС (m/z): 322 (М+23). Синтез 14. бис[(2-Фторфенил)метил]-(1S,2S,5R,6R)-2-(трет-бутоксикарбониламино)-4-оксобицикло[3.1.0]гексан-2,6-дикарбоксилат[3.1.0]гексан-2,6-дикарбоновой кислоты (0,17 г, 0,57 ммоль) и карбоната цезия (0,37 г, 1,14 ммоль) в сухом N,N-диметилформамиде (1,42 мл) добавляли по каплям 2-фторбензилбромид (0,21 мл, 1,7 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи в атмосфере азота. Реакцию гасили водой и проводили разбавление этилацетатом. Водную фазу экстрагировали этилацетатом (3 раза) и промывали органические слои солевым раствором и водой. Сушили над сульфатом натрия, фильтровали и концентрировали с получением неочищенного вещества в виде бледно-коричневого масла. Очистку проводили посредством флэш-хроматографии, элюируя смесью этилацетат:гексан (от 20:80 до 30:70) с получением указанного в заголовке соединения в виде бледно-желтого масла (229 мг, 79%). МС(20,3 мл) добавляли по каплям 1 М раствор L-селектрида в ТГФ (6,78 мл, 6,78 ммоль) при -78 С. Перемешивали полученную оранжевую смесь в атмосфере азота в течение 1 ч 45 мин. Реакцию гасили насыщенным раствором гидрокарбоната натрия при -78 С. Разбавляли водой и этилацетатом. Разделяли слои и промывали органическую фазу солевым раствором и водой. Сушили над сульфатом натрия, фильтровали и концентрировали досуха с получением неочищенного вещества в виде бледножелтого масла, загрязненного приблизительно 6% минорного изомера бис[(2-фторфенил)метил](1S,2S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилата,обнаруживаемого методом МС. Данное неочищенное вещество объединяли со второй порцией, полученной аналогичным образом из бис[(2-фторфенил)метил]-(1S,2S,5R,6R)-2-(трет-бутоксикарбониламино)-4 оксобицикло[3.1.0]гексан-2,6-дикарбоксилата (0,23 г, 0,44 ммоль). Объединенные продукты очищали посредством флэш-хроматографии, элюируя смесью этилацетат:гексан (от 20:80 до 50:50) с получением указанного в заголовке продукта в виде единственного изомера (2,49 г, 4,81 ммоль). МС (m/z): 540 К раствору бис[(2-фторфенил)метил]-[1S,2S,4S,5R,6R)-2-(трет-бутоксикарбониламино)-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилата (2,5 г, 4,83 ммоль) в дихлорметане (19,32 мл) добавляли птолуолсульфонилхлорид (1,02 г, 5,31 ммоль) при комнатной температуре. Охлаждали с помощью водноледяной бани, затем добавляли триэтиламин (0,74 мл, 5,31 ммоль) и порциями - N,N-диметил-4 аминопиридин (1 г, 8,21 ммоль). Смесь оставляли нагреваться до комнатной температуры и поддерживали перемешивание в течение ночи в атмосфере азота. Реакцию гасили водой и разделяли слои. Органические слои промывали 1 М раствором гидросульфата калия, солевым раствором и водой. Органическую фазу сушили над сульфатом натрия, фильтровали, концентрировали досуха и сушили под вакуумом с получением бледно-желтой пены (2,72 г). Очистку проводили посредством флэш-хроматографии, элюируя смесью этилацетат:гексан (от 20:80 до 40:60) с получением указанного в заголовке продукта в виде бесцветного масла (2,7 г, 4,02 ммоль). МС (m/z): 672 (М+1).(0,81 г, 5,87 ммоль). Полученную смесь нагревали при 70 С в атмосфере азота в запаянной ампуле и поддерживали перемешивание в течение ночи. Реакцию гасили водой и разбавляли этилацетатом. Органический слой промывали раствором лимонной кислоты (водным 10%-ным), солевым раствором. Сушили над сульфатом натрия, фильтровали и концентрировали с получением неочищенного вещества в виде бледно-коричневого масла (2,3 г). Очистку проводили посредством флэш-хроматографии, элюируя смесью этилацетат:гексан (от 30:70 до 70:30) с получением указанного в заголовке продукта в виде бесцветного масла (1,84 г, 3,06 ммоль). МС (m/z): 601 (М+1), 623 (М+23). Пример 1. Способ 1. К раствору ди-трет-бутил(1R,2S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-4-(4H-1,2,4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата (3,66 г, 7,37 ммоль) в 1,4-диоксане (17,69 мл) добавляли 4 М раствор хлороводорода в 1,4-диоксане (18,42 мл, 7,37 ммоль) и перемешивали смесь в течение выходных при 50 С. Добавляли этилацетат и охлаждали смесь в ледяной бане. Белое твердое вещество собирали декантацией и промывали несколько раз этилацетатом. Твердый продукт высушивали под вакуумом и очищали посредством ионообменной хроматографии. Ионообменную колонку (колонкуSCX-2) предварительно подготавливали с помощью ацетонитрила. Растворяли вещество в минимальном количестве воды и наносили на колонку. Элюирование проводили ацетонитрилом (2 объема колонки),смесью 2 н. раствор аммиака в метаноле:ацетонитрил (1:1) (2 объема колонки), раствором аммиака в метаноле и 7 н. раствором аммиака в метаноле. Фракцию, полученную при элюировании 2 н. раствором аммиака в метаноле, концентрировали досуха с получением целевого продукта (1,39 г) в виде белого твердого вещества. Твердое вещество сушили в течение 48 ч при 40 С с получением указанного в заголовке соединения (1,24 г, 66%). 1H ЯМР (D2O)1,41-1,55 (м, 2 Н), 2,02 (дд, J=3,1 и 6,4 Гц, 1 Н), 2,15-2,20 (м, 1 Н), 2,40 (дд, J=8,1 и 14,1 Гц, 1 Н), 3,22 (с, 1 Н), 4,20-4,28 (м, 1 Н), 8,30 (с, 1 Н). Способ 2. Растворяли ди-трет-бутил(1R,2S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-4-(4H-1,2,4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат (433 мг, 0,87 ммоль) в 1,4-диоксане (7 мл) и добавляли 4 М раствор хлороводорода в 1,4-диоксане (7 мл, 2,8 ммоль). Смесь оставляли перемешиваться при 50 С в течение ночи. Концентрировали досуха. Очистку проводили катионным ионным обменом (DowexMarathon С, сильнокислотный катионит в Na+-форме). Растворяли остаток в минимальном количестве воды для солюбилизации вещества и нанесения его на смолу. Промывали смолу последовательно 2 объемами колонки воды, затем 2 объемами колонки смеси вода:тетрагидрофуран (1:1) и 2 объемами колонки воды. Целевой продукт элюировали 2 объемами колонки 10%-го раствора пиридина в воде. Концентрировали досуха с получением указанного в заголовке соединения (202 мг, 82%). МС (m/z): 285 (М+1). Способ 1. В трехгорлую круглодонную колбу вместимостью 5 л, снабженную механической мешалкой, помещали(1R,2S,4R,5R,6R)-2-(2S)-2-(трет-бутоксикарбониламино)пропаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту (186 г, 0,41 моль) в ацетоне (1,12 л). К этой суспензии добавляли по каплям в течение 15 мин 37%-ный водный раствор соляной кислоты(100 мл). Перемешивали смесь при 40 С в течение 1 ч. Смесь охлаждали до комнатной температуры и добавляли ацетон (3,72 л). Перемешивали смесь в течение 2 ч до завершения образования белой смолы. Повторно добавляли ацетон (3 л) до образования белой смолы. Ацетон удаляли, добавляли толуол и концентрировали с получением масла. Снова растворяли неочищенный продукт в воде (400 мл) и высушивали вещество сублимационной сушкой с получением указанного в заголовке соединения в виде белого твердого вещества (143 г, 89%). МС (m/z): 356 (М+1), 378 (М+23). 1(1R,2S,4R,5R,6R)-2-(2S)-2-(трет-бутоксикарбониламино)пропаноил]амино]-4-(4H1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту (244 мг, 0,54 ммоль) в этилацетате (24,5 мл) и пропускали газообразный хлороводород в течение 1-2 мин при 0 С. После 5 мин при 0 С перемешивали реакционную смесь при комнатной температуре в течение 90 мин. Растворитель удаляли под вакуумом и снова растворяли твердый продукт в воде. Раствор высушивали сублимационной сушкой с получением указанного в заголовке соединения (180 мг, 86%) в виде белого твердого вещества. МС (m/z): 356 (М+1), 378 (М+23). Пример 3. Гидрохлорид К раствору (1R,2S,4R,5R,6R)-2-(2S)-2-(трет-бутоксикарбониламино)-4-метилсульфанилбутаноил]амино]-4-(4 Н-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (2,66 г,5,16 ммоль) в 1,4-диоксане (26,6 мл) добавляли по каплям 4 М раствор хлороводорода в 1,4-диоксане(19,35 мл, 77,38 ммоль) при комнатной температуре на водяной бане и поддерживали перемешивание в течение ночи при комнатной температуре. Добавляли метил-трет-бутиловый эфир (200 мл) и перемешивали смесь в течение 2 ч. Твердое вещество отфильтровывали и высушивали под вакуумом в течение ночи. Снова растворяли вещество в воде и высушивали сублимационной сушкой в течение ночи. Твердое вещество измельчали в ацетоне (10 объемов) при температуре кипения с обратным холодильником, отфильтровывали и высушивали под вакуумом в атмосфере азота в течение 48 ч с получением указанного в заголовке соединения (2,33 г, 5,16 ммоль) в виде белого твердого вещества. МС (m/z): 416 (М+1). К раствору (1R,2S,4R,5R,6R)-2-(2S)-2-(трет-бутоксикарбониламино)-4-метилпентаноил]амино]-4(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (0,4 г, 0,8 ммоль) в 1,4-диоксане (4 мл) добавляли 4 М раствор хлороводорода в 1,4-диоксане (3,01 мл, 12,06 ммоль). Белый осадок, появляющийся сразу после добавления, перемешивали в течение ночи. Добавляли метил-третбутиловый эфир (16 мл) и собирали белый твердый продукт декантацией, промывая несколько раз метил-трет-бутиловым эфиром с получением 330 мг целевого вещества. Объединяли со второй партией вещества, полученной аналогичным образом из (1R,2R,4S,5R,6R)-4-(2S)-2-(трет-бутоксикарбониламино)-4-метилпентаноил]амино]-2-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-4,6-дикарбоновой кислоты (100 мг, 0,2 ммоль) и 4 М раствора хлороводорода в 1,4-диоксане (0,75 мл, 3,01 ммоль) с получением 374 мг вещества. Очищали половину вещества (190 мг), используя картридж с сорбентомOASIS HLB (1 г). Элюент, содержащий целевое вещество, подкисляли до рН 2-3 1 н. раствором соляной кислоты и проводили сушку под вакуумом в течение ночи при 45 С с получением 170 мг белого твердого вещества. Аналогичным образом очищали оставшееся вещество и объединяли продукты очистки с получением указанного в заголовке соединения (290 мг, 0,67 ммоль). МС (m/z): 398 (М+1), 795 (2 М+1). Пример 5. Гидрохлорид К водному раствору (1R,2S,4R,5R,6R)-2-2-(трет-бутоксикарбониламино)ацетил]амино]-4-(4H1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (приблизительно 1,12 ммоль) добавляли 5 М соляную кислоту (10 мл, 50 ммоль) и перемешивали при комнатной температуре в течение 5 ч. Концентрировали досуха и очищали остаток посредством катионообменной хроматографии(смола DOWEX 50WX8-100). Растворяли соединение в воде и доводили рН до 2. Обеспечивали прохождение соединения через колонку со скоростью падения капель, составляющей примерно 1 каплю в 1-2 с. После того как исходный загрузочный объем был нанесен на поверхность смолы, промывали водой (от 5 до 10 мл) и повторяли 3 раза. рН элюата контролировали и продолжали промывание водой до полного нанесения вещества (наблюдаемое циклическое изменение рН: величина рН элюата из колонки изначально равна 7, затем падает до 1 и возвращается до 7). Промывали колонку по меньшей мере одним объемом колонки воды, одним объемом смеси вода:тетрагидрофуран (1:1), затем одним объемом воды. Продукт удаляли со смолы 10%-ным водным раствором пиридина. Продолжали элюирование 10%-ным водным раствором пиридина до тех пор, пока отсутствие продукта не будет показано посредством ТСХ. Фракции, содержащие продукт, концентрировали с получением бесцветного твердого вещества (173 мг). Растворяли твердое вещество в воде (8 мл), добавляли 5 М соляную кислоту (0,11 мл, 0,55 ммоль) и высушивали раствор сублимационной сушкой в течение 48 ч с получением указанного в заголовке соединения в виде белого твердого вещества (195 мг, 0,52 ммоль). МС (m/z): 342 (М+1). К смеси бензилового спирта (18,18 мл, 175,68 ммоль) и гидрохлорида (1R,2S,4R,5R,6R)-2-амино-4(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (1,61 г, 3,51 ммоль) добавляли ацетилхлорид (2,5 мл, 35,14 ммоль) при комнатной температуре. Мутную реакционную смесь нагревали при 60 С в атмосфере азота. Через 3 дня реакционную смесь охлаждали до комнатной температуры, осторожно разбавляли ацетонитрилом (10 мл) и очищали на колонке SCX-2 (20 г). Реакционную смесь наносили на колонку, предварительно обработанную ацетонитрилом, промывали ацетонитрилом(2), затем элюировали смесью 2 н. раствор аммиака в метаноле:ацетонитрил (2 объема колонки), затем растворитель упаривали под вакуумом с получением белой смолы (576 мг), соответствующей целевому неочищенному продукту. Затем проводили элюирование 7 н. раствором аммиака в метаноле (1 объем колонки) и удаляли растворитель под вакуумом с получением белого твердого вещества, соответствующего(1R,2S,4R,5R,6R)-2-амино-6-бензилоксикарбонил-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2-карбоновой кислоте (750 мг). МС (m/z): 372 (М+1). Неочищенный продукт очищали посредством флэш-хроматографии, дважды элюируя смесью этилацетат:циклогексан (от 60:40 до 100:0) с получением указанного в заголовке соединения в виде белого твердого вещества (430 мг). Растворяли продукт в смеси дихлорметан:этилацетат:ацетонитрил, концентрировали досуха и высушивали в высоком вакууме при комнатной температуре в течение ночи с получением указанного в заголовке соединения в виде белого твердого вещества (402 мг, 25%). МС (m/z): 465 (М+1). Вторую порцию указанного в заголовке соединения получали аналогичным образом путем добавления при комнатной температуре ацетилхлорида (998 мкл, 14,02 ммоль) к смеси бензилового спирта(7,26 мл, 70,11 ммоль) и (1R,2S,4R,5R,6R)-2-амино-6-бензилоксикарбонил-4-(4H-1,2,4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2-карбоновой кислоты (0,75 г, 1,40 ммоль). Реакционную смесь нагревали при 60 С в атмосфере азота. Через 3,5 дня осторожно добавляли тионилхлорид (204,31 мкл, 2,8 ммоль) к реакционной смеси и поддерживали нагревание в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли ацетонитрилом (10 мл). Очистку проводили с помощью колонки SCX-2 (20 г) с получением белого твердого вещества (567 мг). Очистку неочищенного продукта проводили посредством флэш-хроматографии, элюируя смесью этилацетат:циклогексан (от 60:40 до 100:0). Дополнительную очистку посредством флэш-хроматографии проводили, элюируя смесью этилацетат:циклогексан (от 60:40 до 80:20) с получением после сушки дополнительной порции указанного в заголовке соединения (140 мг, 21%). МС (m/z): 465 (М+1). Пример 7. Гидрохлорид бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата К раствору бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-(трет-бутоксикарбониламино)-4-(4H-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата (0,48 г, 0,79 ммоль) в этилацетате (7,91 мл) добавляли раствор концентрированного хлороводорода в этилацетате (7,91 мл). Перемешивали полученную смесь при комнатной температуре в течение ночи. Хлороводород удаляли, пропуская азот через реакционную смесь. Концентрировали и высушивали полученное бесцветное масло в вакуумном сушильном шкафу при 40 С в течение ночи. Снова растворяли вещество в воде, оставляли в морозильной камере на 48 ч и высушивали сублимационной сушкой с получением указанного в заголовке соединения К очищенной воде (28,0 кг) добавляли при перемешивании (1R,2S,4R,5R,6R)-2-(2S)-2-(третбутоксикарбониламино)пропаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6 дикарбоновую кислоту (11,05 кг). Добавляли к смеси соляную кислоту (5 н., 8,5 кг) при температуре ниже 50 С. Смесь нагревали при 50-55 С в течение 1,5-2,0 ч. Смесь охлаждали до 15-25 С. Доводили рН до 9 путем добавления по каплям гидроксида аммония (25%, 4,8 кг), поддерживая температуру 45-50 С. Затем добавляли этанол (34 кг) и охлаждали до 20-25 С. В отдельную емкость помещали очищенную воду(6,5 кг), отфильтрованный на фильтре тонкой очистки (polish filtered) абсолютный этанол (45 кг) и вносили кристалл бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты, 2-(2S)-2-амино-1-оксопропил]амино]4-(4 Н-1,2,4-триазол-3-илтио)-, моноаммониевой соли, (1R,2S,4R,5R,6R)-, моногидрата, полученной отдельно (0,1 кг). Данную смесь нагревали до 45-50 С, затем добавляли по каплям отфильтрованный на фильтре тонкой очистки (polish filtered) реакционный раствор с гидроксидом аммония. Смесь нагревали при 50-55 С в течение 2-3 ч и добавляли по каплям абсолютный этанол (72 кг) и поддерживали температуру при 50-55 С. Температуру реакционной смеси доводили до 40-45 С в атмосфере азота в течение 11,5 ч. Смесь охлаждали до 30-35 С и поддерживали при этой температуре в течение 1-1,5 ч. Смесь охлаждали до 20-25 С и поддерживали при этой температуре в течение 3-4 ч все еще в атмосфере азота. Осадки отфильтровывали и промывали их смесью очищенная вода/этанол (1:10). Осадок высушивали под вакуумом при 50 С-55 С с получением указанного в заголовке соединения в виде белого твердого вещества (6,82 кг). МС (m/z): 391 (М+1). Порошковые рентгеновские дифрактограммы кристаллических твердых веществ получали с применением порошкового рентгеновского дифрактометра Bruker D4 Endeavor, снабженного источником излучения CuK (=1,54060 ) и детектором Vantec, работающим при 35 кВ и 50 мА. Образец сканировали между 4 и 40 по шкале 2 с размером шага, составляющим 0,009 по шкале 2. Сухой порошок укладывали на кварцевый держатель образца и получали гладкую поверхность, используя предметное стекло. Рентгеновские дифрактограммы кристаллических форм получали при комнатной температуре и относительной влажности. В данном случае вариабельность положения пика, составляющая 0,2 по шкале 2,учитывает возможные колебания, не препятствуя бесспорной идентификации указанной кристаллической формы. Подтверждение кристаллической формы может быть выполнено на основании любой уникальной комбинации характерных пиков (в единицах 2), как правило, более выраженных пиков. Рентгеновские дифрактограммы кристаллических форм, полученные при комнатной температуре и относительной влажности, корректировали на основании пиков стандарта NIST 675, при 8,85 и 26,77 по шкале 2. Таким образом, кристаллическую форму образца соединения характеризовали с помощью рентгеновской дифрактограммы, полученной с применением излучения CuK, имеющей дифракционные пики(значения по шкале 2), описанные в таблице ниже, и, в частности, имеющей пик при 18,61 в сочетании с одним или более пиками, выбранными из группы, состоящей из 21,07, 15,34, 14,74 и 19,20; с допустимым отклонением для дифракционных углов, составляющим 0,2. Пики порошковой рентгеновской дифрактограммы соединения согласно примеру 8 Рецепторы mGlu представляют собой сопряженные с G-белками рецепторы, модулирующие нейрональную возбудимость. Конкретнее, измененную глутаматергическую нейропередачу связывали с шизофренией, но все обычно назначаемые антипсихотические средства действуют на дофаминовые рецепторы. Различные исследования подтверждают эффективность активации рецептора mGlu Группы II (которая включает mGlu2 и/или mGlu3) для лечения шизофрении. В частности, последние данные демонстрируют, что агонист рецептора mGlu2/3 обладает антипсихотическими свойствами и может предложить новую альтернативу для лечения шизофрении (Patil et al., Nature Medicine (2007) 13(3), 1102-1107). Исследования демонстрируют, что антипсихотическая активность агонистов рецепторов mGlu2/3 опосредована рецептором mGlu2. Исследования также демонстрируют, что агонисты рецепторов mGlu2/3 обладают анксиолитическими, антидепрессивными и нейропротекторными свойствами. Таким образом, агонисты рецептора mGlu2 могут подходить для лечения психических расстройств, таких как биполярное расстройство, шизофрения, депрессия и генерализированное тревожное расстройство. Поскольку соединения согласно настоящему изобретению являются агонистами рецептора mGlu2,они могут подходить для лечения вышеупомянутых расстройств. Анализ агонистов рецептора mGlu2 человека с применением флуоресцентного визуализирующего спектрофотометра для прочтения планшетов (FLIPR) Данные исследования проводили на клеточных линиях AV-12, полученных из фибробластов сирий- 17020229 ского хомяка, устойчиво экспрессирующих рецептор mGlu2 человека и ко-трансфицированных глутаматным транспортером ЕААТ 1 (транспортер 1 возбуждающих аминокислот) крысы и G15 субъединицей. Экспрессия субъединицы G15 позволяет рецепторам, сопряженным с Gi-белками, передавать сигнал через путь, опосредованный фосфолипазой С, что позволяет измерить активацию рецептора с помощью флуориметрического анализа кальциевого ответа. Клеточные линии поддерживали культивированием в модифицированной по способу Дульбекко среде Игла (DMEM) с высоким содержанием глюкозы и гидрохлорида пиридоксина с добавлением 5% диализованной фетальной телячьей сыворотки,1 мМ пирувата натрия, 10 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоты), 1 мМL-глутамина и 5 мкг/мл бластицидина (все среды приобретены в Invitrogen). Образующие конфлюентный монослой культуры пересевали два раза в неделю с применением не содержащего ферменты диссоциирующего раствора (Chemicon S-004-B). Клетки собирали за 24 ч до анализа и распределяли с помощью устройства для посева клеток Matrix Well-Mate в концентрации 85000 (mGlu2) или 115000 (mGlu3) клеток/лунка в 96-луночные планшеты с черными стенками, покрытые поли-D-лизином (BD BioCoat 354640) в среде, содержащей только 250 (mGlu2) или 125 (mGlu3) мкМ L-глутамина (свежедобавленного). Внутриклеточные уровни кальция контролировали до и после добавления соединений, применяя флуоресцентный визуализирующий спектрофотометр для прочтения планшетов (FLIPR, Molecular Devices). Аналитический буфер состоял из сбалансированного солевого раствора Хенка (HBSS; Sigma) с добавлением 20 мМ HEPES. Среду удаляли и клетки инкубировали с 8 мкМ флуоресцентного зонда Fluo3 АМ (Molecular Probes, F-1241; 50 мкл в лунку) в аналитическом буфере в течение 90 мин при 25 С. Раствор красителя удаляли и замещали свежим аналитическим буфером (50 мкл в лунку). Одностадийный анализ FLIPR, дающий кривую концентрация-эффект из 11 точек (трехкратные разведения, начиная с 10 мкМ), для агониста глутамата (Fisher A125-100) проводили перед каждым экспериментом для подтверждения типичного эффекта EC50. Результаты анализировали, применяя программу Prism v4.03 (GraphPadSoftware). Соединения согласно настоящему изобретению исследовали путем одностадийного анализаFLIPR, с применением профиля концентрация-эффект из 10 точек, полученных трехкратными разведениями, начиная с последней концентрации 25 мкМ. Соединения согласно настоящему изобретению растворяли в виде 10 мМ стоков в 0,1 н. NaOH и хранили при -20 С. Их разбавляли посредством трехкратных серийных разведений в аналитическом буфере. После выполнения первоначального пятисекундного измерения флуоресценции на приборе FLIPR соединения согласно настоящему изобретению добавляли в клеточный планшет (50 мкл в лунку). Данные собирали каждую секунду в течение первых 30 с и затем каждые 3 с до общего времени 90 с для обнаружения активности агониста. Максимальный эффект определяли как эффект, вызываемый концентрацией ECmax (100 мкМ глутамата). Эффект соединения измеряли как разность максимальной и минимальной высот пика в относительных единицах флуоресценции(ОЕФ), скорректированную на фоновую флуоресценцию, измеряемую в отсутствие глутамата. Определения выполняли с применением отдельных планшетов. Эффекты агониста определяли количественно как процент стимуляции, вызванной только соединением, относительно максимального ответа на воздействие глутамата. Все данные рассчитывали как относительные значения ЕС 50 с применением программы, подбирающей четырехпараметрическую логистическую кривую (ActivityBase V5.3.1.22). Соединение согласно примеру 1, согласно измерениям посредством анализа FLIPR рецептораmGlu2 человека, проведенного, в основном, как описано выше, имело значение EC50, равное 23,0 нМ 3,9 (n=5, ошибку рассчитывали как среднеквадратичное отклонение). Этот результат демонстрирует, что соединение согласно примеру 1, активное соединение для примеров 2-8, является агонистом рецептораmGlu2. Соединение 1SR,2RS,4SR,5SR,6SR-2-амино-4-(фенилтио)бицикло[3.1.0]гексан-2,6-дикарбоновая кислота, описанное в публикации WO 9717952, активность которого измерили посредством анализаFLIPR рецептора mGlu2 человека, проведенного, в основном, как описано выше, имело значение EC50,превышающее 25000 нМ. Устранение индуцируемой фенциклидином (РСР) гиперлокомоторной активности у крыс Введение антагонистов рецептора N-метил-D-аспартата (NMDA), таких как кетамин или фенциклидин (РСР), вызывает психотомиметическиподобные эффекты у человека, похожие на симптомы, наблюдаемые у пациентов с шизофренией. Способность агентов устранять стимулирующие локомоторную активность эффекты антагонистов рецептора NMDA часто применяют в качестве животной модели психоза, демонстрирующей хорошую прогностическую ценность для обнаружения клинической эффективности лекарственных препаратов для лечения шизофрении и биполярного расстройства. Моторную активность контролировали, помещая отдельных самцов крыс линии Sprague-Dawley(Harlan, Indianapolis, IN) в прозрачные пластиковые клетушки размерами 452520 см со слоем древесных стружек высотой 1 см в качестве подстилки и металлической решеткой на верху клетки. Мониторы для отслеживания моторной активности (Kinder Scientific) состояли из прямоугольной рамки из 12 датчиков фотолучей, расположенных в порядке 84 (или группы высокой плотности из 22 датчиков фотолучей, расположенных по схеме 157) на высоте 5 см со второй рамкой (для измерения вертикальной активности крыс) на высоте 15 см. Клетку помещали внутрь этих рамок, при этом рамки находились на поверхности стола высотой три фута в изолированной комнате. Соединение согласно настоящему изо- 18020229 бретению вводили в дозе (интраперитонеальный путь введения (и.п.), не пролекарство) в диапазоне 0,310 мг/кг за 30 мин до введения провоцирующей дозы (5 мг/кг) фенциклидина (РСР). Соединение согласно настоящему изобретению вводили в дозе (пероральный путь введения, пролекарство) в диапазоне 0,330 мг/кг крысам, не получавшим пищу в течение ночи, за 4 ч до введения провоцирующей дозы (5 мг/кг) РСР. В день эксперимента крыс помещали в клетку для исследования и оставляли осваиваться в течение 30 мин до воздействия РСР; за крысами наблюдали в течение дополнительных 60 мин, следующих за введением РСР. Анализ данных и вычисления ED50 проводили с применением программы GraphPad Prism (San Diego, CA. USA). Анализы мощности показали, что наличие 8-10 крыс в каждой группе необходимо для обеспечения подходящей статистической мощности для обнаружения разностей, обусловленных обработкой препаратами (мощность = 0,8). Однофакторный дисперсионный анализ (ANOVA) с применением ретроспективного критерия множественного сравнения Даннетта проводили согласно итоговой 60 минутной локомоторной активности. Вычисления ED50 выполняли с применением нелинейной кривой регрессии, подобранной к проценту обратно преобразованных данных для каждой дозы. Соединение согласно примеру 1, как измерили посредством этого анализа, проведенного, в основном, как описано выше, имело значение ED50, равное 1,23 мг/кг (и.п.). Соединения согласно примерам 2 и 3, как измерили посредством этого анализа, проведенного, в основном, как описано выше, имели значения ED50, равные 2,94 мг/кг (п.о.) и 5,46 мг/кг (п.о.), соответственно. Эти результаты демонстрируют,что соединения согласно настоящему изобретению подходят в качестве лекарственных средств для лечения шизофрении и биполярного расстройства. Устранение индуцируемой фенциклидином (РСР) гиперлокомоторной активности у мышей Этот анализ на устранение вызываемой фенциклидином (РСР) гиперлокомоторной активности у мышей проводили, в основном, так же, как анализ устранения вызываемой фенциклидином (РСР) гиперлокомоторной активности у крыс, представленный выше, с применением мышей вместо крыс и с изменениями, отмеченными ниже. Моторную активность контролировали, помещая отдельных самцов мышей линии ICR (CD-1) (Harlan, Indianapolis, IN) в прозрачные пластиковые клетушки размерами 452520 см со слоем древесных стружек высотой 0,5 см в качестве подстилки и пластиковой крышкой на верху клетки. Мониторы для отслеживания моторной активности (Kinder Scientific) состоли из прямоугольной рамки из 12 датчиков фотолучей, расположенных в порядке 84 (или группы высокой плотности из 22 датчиков фотолучей,расположенных по схеме 157) на высоте 2,5 см. Клетку помещали внутрь этих рамок, при этом рамки находятся на поверхности стола высотой три фута в изолированной комнате. Соединение согласно настоящему изобретению дозировали (интраперитонеальный путь введения, не пролекарство), как правило,в диапазоне 0,3-30 мг/кг, хотя могут быть применены более высокие дозы, за 30 мин до введения провоцирующей дозы (7,5 мг/кг) фенциклидина (РСР). В день эксперимента мышей помещали в клетку для исследования и оставляли осваиваться в течение 45 мин до воздействия РСР; за мышами наблюдали в течение дополнительных 60 мин, следующих за введением РСР. Анализы мощности показали, что наличие 7-8 мышей в каждой группе необходимо для обеспечения подходящей статистической мощности для обнаружения разностей, обусловленных обработкой препаратами (мощность = 0,8). В эксперименте с применением единичной дозы, составляющей 10 мг/кг, соединение согласно примеру 1, как измерили посредством этого анализа, проведенного, в основном, как описано выше, вызывало 789%-ное ингибирование индуцированных РСР передвижений (и.п.). В эксперименте с применением многократной дозы соединение согласно примеру 1, как измерили посредством этого анализа, проведенного, в основном, как описано выше, вызывало 815%-ное ингибирование индуцированных РСР передвижений (и.п.) в дозе 10 мг/кг. Эти результаты демонстрируют, что соединения согласно настоящему изобретению подходят в качестве лекарственных средств для лечения шизофрении и биполярного расстройства. Соединение 1SR,2RS,4SR,5SR,6SR-2-амино-4-(фенилтио)бицикло[3.1.0]гексан-2,6 дикарбоновая кислота, активность которого измерили посредством этого анализа, проведенного, в основном, как описано выше, вызывало 1811%-ное ингибирование индуцированных РСР передвижений(и.п.) в эксперименте с применением единичной дозы, составляющей 10 мг/кг, 510%-ное ингибирование индуцированных РСР передвижений (и.п.) в эксперименте с применением многократной дозы для дозы 10 мг/кг и 198%-ное ингибирование индуцированных РСР передвижений (и.п.) в эксперименте с применением многократной дозы для дозы 100 мг/кг. Ослабление индуцируемой стрессом гипертермии у крыс Гипертермия, повышение центральной температуры тела, представляет собой общий синдром, достоверно продемонстрированный у многих млекопитающих, включая людей, который возникает в ответ на стресс. При многих тревожных расстройствах гипертермия возникает как часть патологии, и ее считают симптомом болезни. Соединения, ослабляющие индуцируемую стрессом гипертермию у животных,как полагают, подходят для лечения тревожных расстройств у людей. Генерализованное тревожное расстройство является примером таких расстройств, которые можно лечить с помощью таких соединений. Традиционный и минимально инвазивный способ для анализа индуцируемой стрессом гипертермии заключается в измерении температуры тела и вызываемых стрессом повышений температуры тела с помощью ректального термометра. Исследовали самцов крыс линии Fischer F-344 (Harlan, Indianapolis, IN,USA), имеющих вес в пределах 275-350 г. Всех животных размещали отдельно с неограниченным доступом к пище и воде из автоматической поилки и поддерживали в условиях 12-часового цикла свет/темнота (начало световой фазы в 06:00). Животные голодали в течение приблизительно 12-18 ч перед экспериментом, который проводили во время световой фазы. Препараты вводили крысам за 2 ч до эксперимента либо интраперитонеальным (и.п., не пролекарство), либо пероральным (п.о., пролекарство) путем в дозированном объеме, составляющем 1 мл/кг. Соединения согласно настоящему изобретению вводили в дозах 0,3, 1, 3 и 10 мг/кг (и.п.) и 4,13, 13,78 и 41,35 мг/кг (п.о.). Эти пероральные дозы соответствуют дозам активного лекарственного вещества, равным 3, 10 и 30 мг/кг, соответственно. В качестве носителя в этих исследованиях применяли раствор хлорида натрия с добавлением NaOH в количестве,достаточном для обеспечения рН в пределах 5-7. Антагонист рецептора mGluR5 MTEP (3-[(2-метил-1,3 тиазол-4-ил)этинил]пиридин), который продемонстрировал сильную подобную анксиолитической активность в доклинических моделях, применяли в качестве препарата сравнения (5 мг/кг, и.п. путь введения, раствор в воде). Вслед за введением препаратов крыс немедленно возвращали в их "домашние" клетки, и экспериментатор выключал свет и покидал комнату. Комната, в которой выполняли введение препаратов, оставалась затемненной в течение остатка 1-часового подготовительного периода. После подготовительного периода крыс индивидуально помещали в ярко освещенную соседнюю комнату, где определяли начальную температуру тела путем введения ректального зонда, смазанного минеральным маслом. Температуру тела оценивали с помощью термометра PHYSITEMP BAT-12 Microprobe Thermometer с ректальным зондом для крыс PHYSITEMP RET-2 (Physitemp Instruments Inc.,Clifton, NJ, USA). Зонд вводили приблизительно на 2 см в прямую кишку для измерения центральной температуры тела (это начальная температура тела, Т 1, в градусах Цельсия). Через 10 мин регистрировали второе измерение температуры тела (Т 2). Разницу температур тела (Т 2-Т 1) обозначали как индуцируемый стрессом гипертермический ответ. Дозу, при которой соединение согласно настоящему изобретению вызывает 35%-ное уменьшение индуцируемого стрессом гипертермического ответа относительно ответа, возникающего при введении носителя, обозначали как доза Т 35. Соединение согласно примеру 1, как измерили посредством этого анализа, проведенного, в основном, как описано выше, имело значение Т 35, равное 1,27 мг/кг (и.п.). Соединение согласно примеру 2, как измерили посредством этого анализа, проведенного, в основном, как описано выше, имело значение T35,равное 16,2 мг/кг (п.о.). Эти результаты демонстрируют, что соединения согласно настоящему изобретению подходят в качестве лекарственных средств для лечения тревожных расстройств. Конкретнее, соединения согласно настоящему изобретению могут подходить в качестве лекарственных средств для лечения генерализованного тревожного расстройства. Тест вынужденного плавания на грызунах Тест вынужденного плавания у грызунов хорошо изучен и показывает хорошую прогностическую ценность для обнаружения активности, подобной антидепрессивной, современных лекарственных препаратов для лечения большого депрессивного расстройства. В этом анализе воздействия с предполагаемой активностью, подобной антидепрессивной, снижают неподвижность в коротком неизбежном вынужденном эпизоде плавания. Тест вынужденного плавания проводили как на мышах (самцы, мыши линии NIH-Swiss, 20-25 г,Harlan Sprague-Dawley, Indianapolis, IN), так и на крысах (самцы, крысы линии Sprague-Dawley, 250-350 г, Harlan Sprague-Dawley, Indianapolis, IN). Мышей помещали на 6 мин в прозрачные пластиковые цилиндры (диаметр: 10 см; высота: 25 см), наполненные на 6 см водой с температурой 22-25 С. Длительность неподвижного поведения регистрировали в течение последних 4 мин 6-минутного исследования. Аналогично проводили опыт на крысах, но применяли прозрачные пластиковые цилиндры с большими размерами (диаметр: 18 см; высота: 40 см), наполненные водой (22-25 С) до высоты 16 см, на 15 мин. Кроме того, мышей подвергали единичному эпизоду вынужденного плавания; крыс подвергали двум 5 минутным сеансам, разделенным промежутком времени 24 ч (данные регистрировали только на второй день). Соединение согласно примеру 1 исследовали на мышах с применением интраперитонеального дозирования (1, 3 или 10 мг/кг) за 60 мин до опыта. Соединение согласно примеру 2 исследовали на крысах, придерживаясь перорального дозирования (1,4, 4,1 или 13,8 мг/кг; что соответствует 1, 3 или 10 мг/кг активного эквивалента). Соединение согласно примеру 2 вводили через 5 мин вслед за первым эпизодом вынужденного плавания и за 120 мин перед экспериментальным сеансом на второй день. В качестве положительного контроля для этих исследований применяли имипрамин. Соединения получали в водной среде-носителе с добавлением минимального количества NaOH к соединениям согласно примерам 1 и 2. Соединения находились в прозрачном растворе. Количество времени, проведенное в неподвижном состоянии (которое определяли как движения, необходимые только для поддержания головы субъекта над водой), является зависимым показателем, регистрируемым наблюдателем, не осведомленным об обработке субъектов лекарственными препаратами. Данные анализировали с применением рет- 20020229 роспективного критерия Даннетта с уровнем , принятым 0,05. Наиболее часто применяли вариант теста вынужденного плавания на мышах. Для оценки мощности исследуемых соединений вычисляли значениеED60 (60% времени в неподвижном состоянии относительно носителя). При этих условиях исследования крысы не отвечали на воздействие препаратов так сильно, как мыши, и статистическую значимость и максимальную эффективность относительно положительного контроля имипрамина применяли для оценки ответа на лечение. Соединение согласно примеру 1, активность которого измерили посредством этого анализа, проведенного, в основном, как описано выше, вызывало выраженные эффекты, подобные антидепрессивным,у мышей (F4,39=9,9, p0,0001). ED60 составляет 10,5 мг/кг, а максимальный эффект составляет 61% от времени в неподвижном состоянии, соответствующем носителю, при дозе 10 мг/кг относительно крыс контрольной группы, получавших носитель. В четырех независимых повторах этого исследования среднее значение ( среднеквадратическое отклонение) ED60 составляет 6,12,5 мг/кг, и максимальный эффект составляет 49,511,5% от времени в неподвижном состоянии, соответствующем носителю. По сравнению с этими результатами эффект антидепрессанта имипрамина, определенный с помощью многократных исследований, составляет 8,61,2 мг/кг (ED60) и 391,2% от времени в неподвижном состоянии, соответствующем носителю. У крыс соединение согласно примеру 2, активность которого измерили посредством этого анализа, проведенного, в основном, как описано выше, значительно уменьшало время,проведенное в неподвижном состоянии [F(4,29)=14,72, р 0,0001], вследствие введения препаратов в дозировках 4,1 и 13,8 мг/кг. Максимальный эффект соединения согласно примеру 2 заключается в уменьшении времени неподвижного состояния до 68% от времени для крыс, обработанных носителем. По сравнению с этими результатами введение 30 мг/кг имипрамина уменьшает время неподвижного состояния до 70% от времени для крыс контрольной группы, обработанных носителем. Эти результаты демонстрируют, что соединения согласно настоящему изобретению подходят в качестве лекарственных средств для лечения депрессии. Скрининг способности ингибировать взаимодействие транспортера PepT1 с глицилсаркозином(GlySar) in vitro и определение IC50 Анализы активности транспортера PepT1 осуществляли для проверки способности соединенийпролекарств на основе аминокислот взаимодействовать с кишечным транспортером PepT1, обеспечивающим абсорбцию. Клетки HeLa, полученные из кишечника человека (American Type Culture Collection), выращивали в среде Hyclone (Invitrogen,по каталогу SH30243), содержащей 10% фетальной телячьей сыворотки(FBS), 0,1 мМ не являющихся незаменимыми аминокислот (NEAA) и 100 ед./мл пенициллина со 100 мкг/мл стрептомицина, при 37 С во влажной атмосфере, содержащей 5% СО 2. Клеточную линию пересевали до 40 раз и затем выводили из эксперимента. Замороженные в ампулах на 1 мл клетки размораживали в водяной бане в течение 1-2 мин и добавляли к 5 мл клеточной среды при 37 С. В каждый флакон для тканевых культур (Т-флакон) помещали 8,5 мл свежей среды и 1,5 мл клеточного стока. Клетки пересевали дважды в течение недели. Эту процедуру выполняли путем промывания флаконов 10 мл фосфатно-солевого буфера с этилендиаминтетрауксусной кислотой (ФСБ-ЭДТУ), добавления 2 мл раствора трипсина на 2-5 мин для отделения клеток и добавления 8 мл свежей среды для ингибирования лишней активности трипсина. В каждый новый флакон помещали смесь 8,5 мл свежей среды и 1,5 мл клеточного стока с целью получения разведения клеток 1:6. Клетки инкубировали при 37 С до готовности для исследования поглощения. Клетки, достигшие состояния 70-80% конфлюентного монослоя в Т-флаконах, пересевали в планшеты за 1 день до процедуры трансфекции. Флакон с клеточным стоком обрабатывали ФСБ-ЭДТУ и трипсином для отделения клеток и, начиная с этого момента, применяли среду для трансфекции. Среда для трансфекции состоит из модифицированной по способу Дульбекко среды Игла с добавлениемNEAA. В каждую лунку добавляли 0,5 мл клеточной смеси (целевая концентрация клеток составляет 1,3105) и клетки инкубировали при 37 С в течение ночи. За 24 ч до анализа клетки трансфицировали геном РЕРТ 1. Трансфекционную смесь получали путем смешивания 600 мкл среды для трансфекции, не содержащей сыворотки, 18 мкл трансфицирующего реагента FuGene6 (Roche Diagnostics) и 11 мкг ДНК,кодирующей транспортер PepT1. Комплекс трансфицирующий реагент-ДНК инкубировали в течение 20 мин и добавляли в каждую лунку 24 мкл комплекса реагента с ДНК. Ингибирование опосредованной транспортером РЕРТ 1 активности, отвечающей за поглощение[глицил-1-2-14 С]глицилсаркозина (GlySar), измеряли в клетках, культивируемых в 24-луночных планшетах, через 24 ч после трансфекции, как опубликовано ранее (Zhang et al. 2004. J. Pharm. Exper Ther. 310: 437-445). Для измерения способности соединения согласно настоящему изобретению ингибировать поглощение [14 С]Gly-Sar, соединения-пролекарства в концентрации 5 мМ инкубировали с временно трансфицированными геном транспортера PepT1 клетками HeLa, достигшими состояния 80-90% конфлюентного монослоя, в среде для анализа поглощения с рН 6,0 в присутствии 5 мкМ [14 С]Gly-Sar (Moravek Biochemicals) и 20 мкМ немеченого Gly-Sar. Среда для анализа поглощения состоит из 140 мМ NaCl, 5,4 мМKCl, 1,8 мМ CaCl2, 0,8 мМ MgSO4, 5 мМ глюкозы, 25 мМ буфера на основе трис(гидрокси- 21020229 метил)аминометана (ТРИС). После приготовления раствора его рН доводили до 6,0 добавлением 2-(Nморфолино)этансульфоновой кислоты. Инкубацию проводили в объеме 500 мкл при комнатной температуре в течение 3 мин. Для остановки поглощения в конце времени инкубации среду для анализа поглощения аспирировали из лунок с монослоем клеток и добавляли в лунки по 500 мкл ледяного ФСБ. Клетки промывают 3 раза 500 мкл ФСБ комнатной температуры, не содержащим ионов Са 2+ и Mg2+. Затем клетки лизировали с помощью 300 мкл 1%-го водного раствора Тритона Х 100. Отбирали аликвоту объемом 200 мкл и определяли радиоактивность посредством жидкостного сцинтилляционного подсчета для измерения количества [14C]Gly-Sar, присутствующего в каждой из инкубационных лунок. Обеспечивали контроль, не содержащий ингибитора, и процент ингибирования каждым пролекарством вычисляли относительно этого контроля. Действие отрицательного контроля (глицина) и двух положительных контролей (цефадроксила и цефалексина) исследовали параллельно с каждым экспериментом для доказательства устойчивости тест-системы. Соединения-пролекарства, ингибирующие поглощение GlySar на том же уровне или лучше, чем цефалексин, считали приемлемыми. Средние значениястандартное отклонение составляли 10,19,5% (n=19) для глицина, 53,213,2% (n=19) для цефадроксила и 37,514,7%[14C]Gly-Sar и 20 мкМ немеченого Gly-Sar. Процедуры инкубации и забора образцов были в точности такими же, как и для отбора ингибиторов транспортера PepT1, описанного выше. Данные по поглощению [14 С]Gly-Sar оценивали для каждой из концентраций соединений-пролекарств и вычисляли значенияIC50. Соединения согласно примерам 2, 3, 4 и 5, как измерили посредством этого анализа, проведенного,в основном, как описано выше, в концентрации 5 мМ вызывали ингибирование поглощения [3 Н]Gly-Sar,опосредованного транспортером PepT1 человека, на 38% (n=3, станд. откл. = 18,4), 51% (n=1), 32% (n=1) и 44% (n=1), соответственно. Соединения согласно примерам 2 и 3, как измерили посредством этого анализа, проведенного, в основном, как описано выше, имели значения IC50 ингибирования поглощения[3H]Gly-Sar, опосредованного транспортером PepT1 человека, равные 1,84 мМ (среднеквадр. погр. = 0,42) и 5,36 мМ (среднеквадр. погр. = 1,69), соответственно. Эти результаты демонстрируют, что соединения согласно настоящему изобретению всасываются при пероральном введении за счет транспортераPepT1. Анализ кишечного гидролиза пролекарств in vitro Замороженные гомогенаты двенадцатиперстной кишки человека (с соотношением ткань:буфер 1:2,полученные с применением 100 мМ трис-фосфатного буфера, рН 7,4) не содержали и фенилметилсульфонилфторид (ФМСФ), и ЭДТА, приобретали в Celsius In Vitro Technologies (Baltimore, MD). Каждую партию двенадцатиперстной кишки человека получали от индивидуального донора, кишку разрезали и доли замораживали отдельно. Все оригинальные коллекции тканей создавали при 4 С и немедленно замораживали при -70 С. Кишечные гомогенаты человека размораживали и разбавляли в 100 мМ ФСБ, рН 7,4 до конечной концентрации белка, равной 0,5 мг/мл, непосредственно перед инкубациями. Инкубации проводили в 96-луночных планшетах, и все соединения-пролекарства исследовали в двух повторностях каждый день. Стоковые растворы соединений-пролекарств с концентрацией 1 мМ готовили в воде. Аликвоту кишечного гомогената с концентрацией белка 0,5 мг/мл объемом 200 мкл и 196 мкл 100 мМ ФСБ помещали в 96-луночный планшет в водяной бане с температурой 37 С. С помощью пипеточного дозатора на 96 лунок переносили 4 мкл 1 мМ раствора соединения-пролекарства в лунки с гомогенатом. Непосредственно после добавления соединения-пролекарства (нулевое время) и после инкубации в течение 1 ч 50 мкл образцов инкубационной смеси отбирали с помощью автоматического разового пипеточного дозатора для одновременного дозирования в 96 лунок и добавляли прямо к 200 мкл метанольного раствора для остановки реакции, содержащего 100 нг/мл внутреннего стандарта. Затем образцы центрифугировали при 3500 об/мин в течение 5 мин при 10 С. Супернатант (200 мкл) переносили в заключительный 96-луночный планшет для ПЦР и закрывали для ЖХ/МС/МС-анализа (жидкостной хроматографии с тандемной масс-спектрометрией). Концентрации гидролизуемых соединений согласно настоящему изобретению в инкубационных смесях определяли методом ЖХ/МС/МС с применением квадрупольного масс-спектрометра Sciex API 4000 с программным обеспечением Analyst версия 1.4.2, ионизацией путем электрораспыления с помощью пневматической системы и контролем селективных реакций (SRM). Применяли колонку для ВЭЖХWaters Atlantis T3 (202,1 мм, 5 мкм) при комнатной температуре со скоростью потока 1,0 мл/мин и градиентом подвижной фазы от 0,1% подвижной фазы А до 99% подвижной фазы А. Подвижная фаза А представляет собой смесь 1000:5 вода:гептафтормасляная кислота, а подвижная фаза В представляет собой смесь 1:1 метанол:ледяная уксусная кислота. Концентрации гидролизуемых соединений согласно настоящему изобретению в инкубационных смесях, содержащих гомогенат кишечника, определяли по стандартным кривым, получаемым путем по- 22020229 вторного двукратного разведения, начиная с концентрации 10 мкМ в 100 мМ ФСБ рН 7,4 и последующего добавления раствора метанол-внутренний стандарт для остановки реакции идентично опытным образцам. Средние значения и стандартные отклонения вычисляли с применением программы MicrosoftOffice Excel 2007. Степень гидролиза определяли как молярное процентное содержание образуемого соединения относительно концентрации добавленного соединения-пролекарства. Гидролиз положительного контроля, внутреннего соединения-пролекарства А, до внутреннего соединения-лекарственного средства А, проводимый в каждой серии опытов, составлял в среднем 75,3% (n=20). Итоговые значения затем нормализовали относительно образования внутреннего соединения-лекарственного средства А. Соединения согласно примерам 2-6, как было определено посредством данного анализа, проведенного, в основном, как описано выше, подвергались гидролизу в гомогенате двенадцатиперстной кишки человека относительно внутреннего соединения-пролекарства А на 61% (n=3, станд. откл. = 11,1), 53%(n=3, станд. откл. = 11,9), 45% (n=1), 35% (n=2; 34,1, 35,2) и 2% (n=1), соответственно. Эти результаты демонстрируют, что соединения согласно настоящему изобретению подвергаются гидролизу в кишечнике человека. Анализ гилролиза в присутствии гомогената фракции S9 печени человека in vitro Фракции S9 печени приобретали в Xenotech LLC (Lenexa, МО). Партия фракций состоит из пула от двух доноров, одного мужчины и одной женщины. Фракцию S9 печени получали и разбавляли с помощью буфера для гомогенизации, состоящего из 50 мМ Трис, рН 7,4 при 4 С и 150 мМ хлорида калия без ЭДТУ. Соединения-пролекарства инкубировали с гомогенатом печени в течение 2 ч при 37 С, после чего определяли концентрацию соединения посредством ЖХ/МС/МС. Гидролиз клопидогрела до производного карбоновой кислоты применяли в качестве положительного контроля анализа. Инкубацию проводили в формате 96-луночных планшетов, и все соединения-пролекарства исследовали в двух повторностях каждый день. Стоковые растворы соединений-пролекарств с концентрацией 1 мМ готовили в воде. Фракцию S9 печени человека разбавляли до конечной концентрации белка, равной 0,5 мг/мл, в 100 мМ ФСБ, рН 7,4. Аликвоту гомогената фракции S9 печени человека с концентрацией белка 0,5 мг/мл объемом 200 мкл и 196 мкл 100 мМ ФСБ помещали в 96-луночный планшет в водяной бане с температурой 37 С. С помощью пипеточного дозатора на 96 лунок переносили 4 мкл 1 мМ раствора пролекарства в лунки с гомогенатом. Для гарантии того, что гидролиз происходит не вследствие химической нестабильности,соединения-пролекарства также инкубировали только с ФСБ без фракции S9 печени. Непосредственно после добавления соединения-пролекарства (нулевое время) и после инкубации в течение 1 ч 50 мкл образцы инкубационной смеси отбирали с помощью автоматического разового пипеточного дозатора для одновременного дозирования в 96 лунок и добавляли прямо к 200 мкл метанольного раствора для остановки реакции, содержащего 100 нг/мл внутреннего стандарта. Затем образцы центрифугировали при 3500 об/мин в течение 5 мин при 10 С. Супернатант (200 мкл) переносили в заключительный 96 луночный планшет для ПЦР и закрывали для ЖХ/МС/МС-анализа. Количественное определение соединений, образованных во время инкубации, посредством ЖХ/МС/МС выполняли на приборе Sciex API 4000 с программным обеспечением Analyst версия 1.4.2,путем электрораспыления (положительная ионизация) с помощью пневматической системы и контролем селективных реакций (SRM). Применяли колонку для ВЭЖХ Waters Atlantis Т 3 (202,1 мм, 5 мкм) при температуре окружающей среды со скоростью потока подвижной фазы 1,0 мл/мин. Подвижная фаза А представляет собой смесь 1000:5 вода:гептафтормасляная кислота, а подвижная фаза В представляет собой смесь 1:1 метанол:ледяная уксусная кислота. Применяли градиент подвижной фазы, начиная с соотношения подвижных фаз А/В, равного 99,9/0,1, и заканчивая соотношением 1/99. Концентрации гидролизуемого соединения в инкубационных смесях определяли по стандартным кривым, получаемым путем повторного двукратного разведения, начиная с концентрации 10 мкМ в 100 мМ ФСБ рН 7,4 и последующего добавления раствора метанол-внутренний стандарт для остановки реакции идентично опытным образцам. Средние значения и стандартные отклонения вычисляли с применением программы Microsoft Office Excel 2007. Итоговые значения представляли как молярное процентное содержание образуемого соединения относительно концентрации добавленного соединенияпролекарства. Гидролиз клопидогрела до производного карбоновой кислоты, применяемый в качестве положительного контроля, составляет в среднем 73,0% (n=27). Соединения согласно примерам 2, 3, 5, 6 и 7, как измерили посредством этого анализа, проведенного, в основном, как описано выше, подвергались гидролизу в присутствии гомогената фракции S9 печени человека на 1% (n=1), 8% (n=1), 0,4% (n=1), 9% (n=2; 4,4, 13,6) и 29% (n=1), соответственно. Эти результаты демонстрируют, что соединения-пролекарства согласно настоящему изобретению подвергаются гидролизу в печени человека. Данные демонстрируют, что соединения-пролекарства на основе аминокислот согласно настоящему изобретению ингибируют поглощение субстрата GlySar транспортера PepT1 на том же уровне или лучше, по сравнению с цефадроксилом и цефалексином (Zhang et al., 2004. JPET 310: 437-445), что является прогностическим для всасывания при пероральном введении человеку, опосредованного транспортеромPepT1. Кроме всаывания соединения-пролекарства при попадании его в организм, необходим гидролиз соединения-пролекарства для получения активного соединения. Настоящие исследования гидролиза invitro демонстрируют, что соединения-пролекарства на основе аминокислот согласно настоящему изобретению могут подвергаться гидролизу в кишечнике человека. Гидролиз соединений-пролекарств с двумя сложноэфирными группами происходит в гомогенатах печени человека, демонстрируя, что соединенияпролекарства с двумя сложноэфирными группами согласно настоящему изобретению подвергаются гидролизу в организме человека после перорального введения. Вместе эти данные позволяют предположить,что соединения-пролекарства на основе аминокислот и соединения-пролекарства с двумя сложноэфирными группами согласно настоящему изобретению подвергаются гидролизу в организме человека. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы где R1 представляет собой водород, R2 представляет собой водород и R3 представляет собой водород;R1 представляет собой бензил, R2 представляет собой водород и R3 представляет собой бензил; илиR1 представляет собой (2-фторфенил)метил, R2 представляет собой водород и R3 представляет собой (2-фторфенил)метил; или его фармацевтически приемлемая соль. 2. Соединение по п.1, представляющее собой (1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль. 3. Соединение по п.2, представляющее собой (1R,2S,4R,5R,6R)-2-амино-4-(4 Н-1,2,4-триазол-3 илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту. 4. Соединение по п.1, представляющее собой (1R,2S,4R,5R,6R)-2-(2S)-2-аминопропаноил]амино]4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль. 5. Соединение по п.4, представляющее собой гидрохлорид (1R,2S,4R,5R,6R)-2-(2S)-2 аминопропаноил]амино]-4-(1H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты. 6. Соединение по п.1,представляющее собой(1R,2S,4R,5R,6R)-2-(2S)-2-амино-4 метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6 дикарбоновую кислоту или ее фармацевтически приемлемую соль. 7. Соединение по п.6, представляющее собой гидрохлорид (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4 метилсульфанилбутаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6 дикарбоновой кислоты. 8. Соединение по п.1,представляющее собой(1R,2S,4R,5R,6R)-2-(2S)-2-амино-4 метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль. 9. Соединение по п.8, представляющее собой гидрохлорид (1R,2S,4R,5R,6R)-2-(2S)-2-амино-4 метилпентаноил]амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты. 10. Соединение по п.1, представляющее собой (1R,2S,4R,5R,6R)-2-[(2-аминоацетил)амино]-4-(4H1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновую кислоту или ее фармацевтически приемлемую соль. 11. Соединение по п.10, представляющее собой гидрохлорид (1R,2S,4R,5R,6R)-2-[(2-амино- 24020229 ацетил)амино]-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты. 12. Соединение по п.1, представляющее собой дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0] гексан-2,6-дикарбоксилат или его фармацевтически приемлемую соль. 13. Соединение по п.12, представляющее собой дибензил(1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4 триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат. 14. Соединение по п.1, представляющее собой бис[(2-фторфенил)метил]-(1R,2S,4R,5R,6R)-2-амино 4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат или его фармацевтически приемлемую соль. 15. Соединение по п.14, представляющее собой гидрохлорид бис[(2-фторфенил)метил](1R,2S,4R,5R,6R)-2-амино-4-(4H-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилата. 16. Соединение, представляющее собой моногидрат моноаммониевой соли (1R,2S,4R,5R,6R)-2(2S)-2-амино-1-оксопропил]амино]-4-(4H-1,2,4-триазол-3-илтио)бицикло[3.1.0]гексан-2,6 дикарбоновой кислоты. 17. Соединение по п.16, представляющее собой моногидрат моноаммониевой соли(1R,2S,4R,5R,6R)-2-(2S)-2-амино-1-оксопропил]амино]-4-(4H-1,2,4-триазол-3-илтио)бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты в кристаллической форме, характеризующейся порошковой рентгеновской дифрактограммой, имеющей дифракционные пики по шкале 20,2, соответствующие 18,61 и 21,07. 18. Фармацевтическая композиция, содержащая соединение или соль по любому из пп.1-17 и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество. 19. Применение соединения или соли по любому из пп.1-17 в терапии. 20. Применение соединения или соли по любому из пп.1-17 для лечения психического расстройства, выбранного из группы, состоящей из биполярного расстройства, шизофрении, депрессии и генерализированного тревожного расстройства. 21. Применение соединения или соли по п.20, где психическое расстройство представляет собой биполярное расстройство. 22. Применение соединения или соли по п.20, где психическое расстройство представляет собой шизофрению. 23. Применение соединения или соли по п.20, где психическое расстройство представляет собой депрессию. 24. Применение соединения или соли по п.20, где психическое расстройство представляет собой генерализированное тревожное расстройство.
МПК / Метки
МПК: C07D 249/10, A61K 31/4196, A61P 25/00
Код ссылки
<a href="https://eas.patents.su/26-20229-agonisty-mglu2.html" rel="bookmark" title="База патентов Евразийского Союза">Агонисты mglu2</a>
Агонисты рецептора меланокортина

Номер патента: 19146
Опубликовано: 30.01.2014
Авторы: Ли Хиун Хо, Ли Хиун Мин, Ли Коо, Ахн Ин Ае, Шим Донг Суп, Чои Сунг Пил, Ли Санг -Дае, Чунг Соо Йонг, Моон Санг Пил
МПК: C07D 403/14
Метки: агонисты, рецептора, меланокортина
Формула / Реферат:
1. Соединение следующей формулы 1где R1 представляет собой водород или представляет собой C1-С10алкил, С3-С7циклоалкил, С6-С10арил, гетероцикл или гетероарил, каждый из которых является незамещенным или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из галогена, амино, С1-С4алкила, трифторметила, С1-С4алкокси, циано и оксо;R2 представляет собой фенил или шестичленный гетероарил, каждый из которых является незамещенным...
Мускариновые агонисты

Номер патента: 6853
Опубликовано: 28.04.2006
Авторы: Тернер Уилльям Уилсон Мл., Хитчкок Стефен Эндрю, Аллен Дженнифер Ребекка, Лиу Бин, Джеймисон Джеймс Эндрю
МПК: C07C 251/00
Метки: агонисты, мускариновые
Формула / Реферат:
1. Соединение формулы где Q, X, Y и Z независимо выбраны из группы, состоящей из CR1 и N, при условии, что не более чем два из Q, X, Y и Z представляют собой N, и по меньшей мере два из Q, X, Y и Z представляют собой СН; или Y представляет собой СН, Z представляет собой СН и группа Q=X представляет собой S, с образованием тиофенового кольца; R1, в каждом случае независимо, выбран из группы, состоящей из водорода, галогена, С1-С4-алкокси и...
Агонисты fxr

Номер патента: 15632
Опубликовано: 31.10.2011
Авторы: Варшавски Алан М., Стельзер Линдсэй Скотт, Ландер Питер Амброз, Очоада Джейсон Мэттью, Цюй Фучэн, Генин Майкл Джеймс, Доти Роберт Энтони, Ститс Райан Эдвард, Ма Тяньвэй, Белл Майкл Грегори, Маннинен Питер Рудольф
МПК: A61P 3/06, C07D 231/12, C07D 401/12...
Метки: агонисты
Формула / Реферат:
1. Соединение формулыгде р равен 0, или 1, или 2;X1 представляет собой С или N и Х2 представляет собой С или N; при условии, что X1 и Х2 одновременно не являются N;R1 и R2независимо выбраны из группы, состоящей из атома водорода, C1-С6алкила, С1-С6галогеналкила, C1-С6алкокси, C1-С6галогеналкокси, атома галогена, -SC1-С6алкила и -S-C1-С3галогеналкила;каждый R3независимо выбран из группы, состоящей из атома водорода, С1-С6алкила,...
Селективные β3-адренергические агонисты.

Номер патента: 1217
Опубликовано: 25.12.2000
Авторы: Шакер Энтони Дж., Винтер Марк А., Мэттьюз Дональд П., Макдональд Джон Х., Эврард Дебора А., Рито Кристофер Дж., Кровелл Томас А., Белл Майкл Г.
МПК: C07D 257/04, A61K 31/41
Метки: агонисты, beta;3-адренергические, селективные
Формула / Реферат:
1. Соединение формулы где R1 представляет ОН, галоген, SO2NHR2, CO2R2, CONHR2, NHCOR2, -NH (необязательно замещенный арил), СF3 или CF2H, причем термин "арил" обозначает необязательно замещенный фенил или нафтил; R1' представляет Н, галоген, C1-C4 алкил, ОН, SO2NHR2, СO2R2, CONHR2, NHCOR2, СF3 или CF2H; R2 представляет Н, C1-C4 алкил или арил, где арил определен как указано выше; R3 представляет Н или C1-C4 алкил; R4 представляет остаток,...
Селективные бетта 3-адренергические агонисты

Номер патента: 2778
Опубликовано: 29.08.2002
Авторы: Рито Кристофер Дж., Винтер Марк А., Шукер Энтони Дж., Дрост Кристин А., Белл Майкл Г., Мэттьюс Дональд П., Макдональд Джон Х.III, Джесудейсон Синтия Д., Нил Дэвид А., Кроуелл Томас А.
МПК: C07D 209/04, A61K 31/416
Метки: агонисты, бетта, селективные, 3-адренергические
Формула / Реферат:
1. Соединение формулы I где Х1 представляет -О-, -ОСН2-; R2 представляет Н, (C1-C4)алкил или фенил; R3 представляет водород; R4 является необязательно замещенным гетероциклом или группой, выбранной из группы, состоящей из X2 представляет связь или линейный или разветвленный алкилен с 1-5 атомами углерода; R5 представляет Н или (C1-C4)алкил; R6 представляет Н или (C1-C4)алкил; или R5 и R6 вместе с атомом углерода, к которому они присоединены,...
Предыдущий патент: Конденсированные тиазоло- и оксазолопиримидиноны
Следующий патент: Рекомбинантный модифицированный вирус осповакцины анкара (mva), экспрессирующий гомологичные последовательности, встроенные в поксвирусный геном, и его применение
Случайный патент: Способ изготовления контейнера для пищевых продуктов и напитков, содержащего теплообменник (варианты)