N, n-дизамещенные диазоциклоалканы, используемые для лечения заболеваний центральной нервной системы (цнс), вызываемых серотонинергической дисфункцией
Номер патента: 7503
Опубликовано: 27.10.2006
Авторы: Теста Родольфо, Леонарди Амедео, Рива Карло, Мотта Джанни





















Формула / Реферат
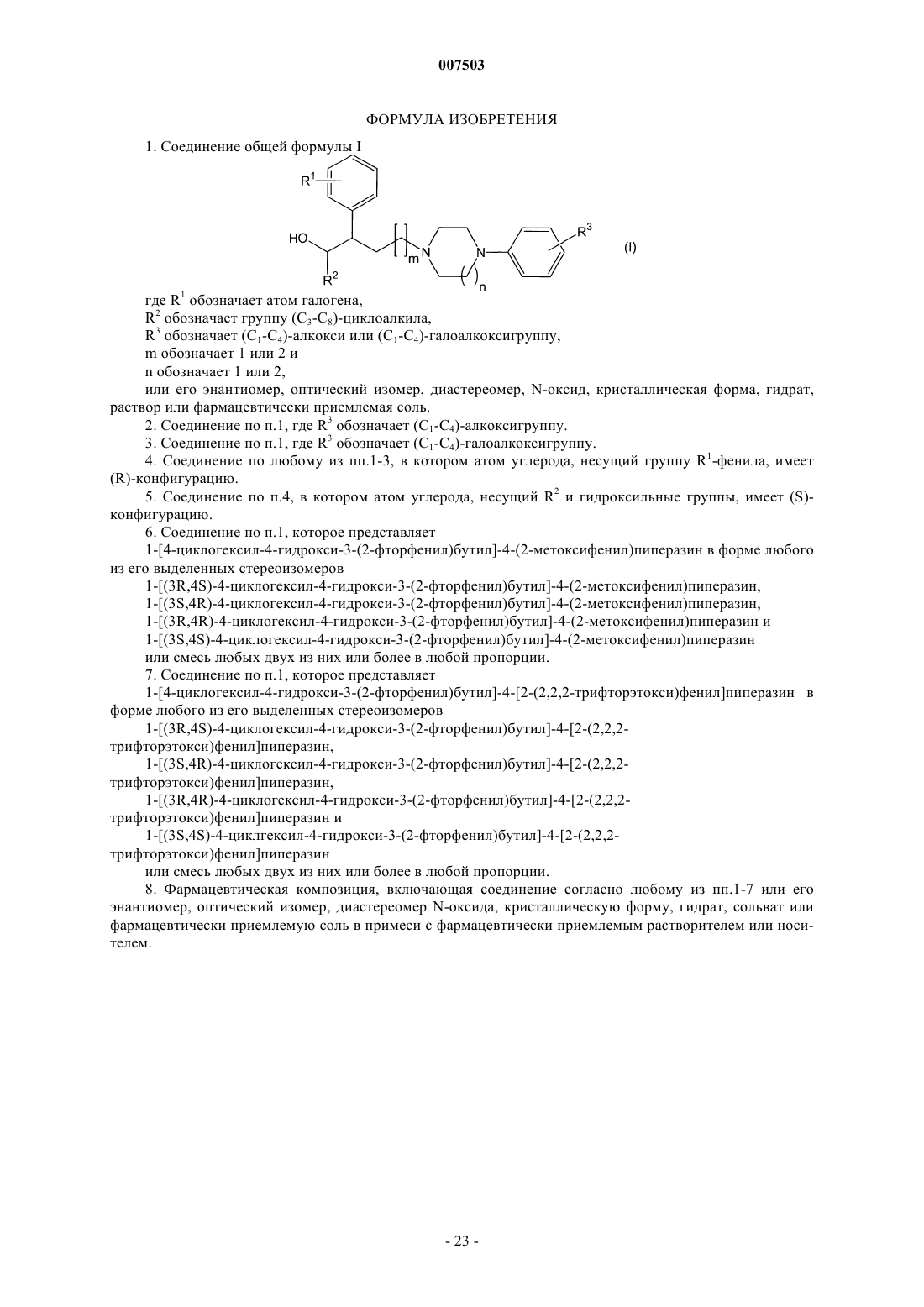
1. Соединение общей формулы I

где R1 обозначает атом галогена,
R2 обозначает группу (С3-С8)-циклоалкила,
R3 обозначает (С1-С4)-алкокси или (С1-С4)-галоалкоксигруппу,
m обозначает 1 или 2 и
n обозначает 1 или 2,
или его энантиомер, оптический изомер, диастереомер, N-оксид, кристаллическая форма, гидрат, раствор или фармацевтически приемлемая соль.
2. Соединение по п.1, где R3 обозначает (C1-C4)-алкоксигруппу.
3. Соединение по п.1, где R3 обозначает (С1-С4)-галоалкоксигруппу.
4. Соединение по любому из пп.1-3, в котором атом углерода, несущий группу R1-фенила, имеет (R)-конфигурацию.
5. Соединение по п.4, в котором атом углерода, несущий R2 и гидроксильные группы, имеет (S)-конфигурацию.
6. Соединение по п.1, которое представляет
1-[4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин в форме любого из его выделенных стереоизомеров
1-[(3R,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин,
1-[(3S,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин,
1-[(3R,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин и
1-[(3S,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин
или смесь любых двух из них или более в любой пропорции.
7. Соединение по п.1, которое представляет
1-[4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин в форме любого из его выделенных стереоизомеров
1-[(3R,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин,
1-[(3S,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин,
1-[(3R,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин и
1-[(3S,4S)-4-циклгексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин
или смесь любых двух из них или более в любой пропорции.
8. Фармацевтическая композиция, включающая соединение согласно любому из пп.1-7 или его энантиомер, оптический изомер, диастереомер N-оксида, кристаллическую форму, гидрат, сольват или фармацевтически приемлемую соль в примеси с фармацевтически приемлемым растворителем или носителем.
Текст
007503 Изобретение касается N,N-диземещенных диазоциклоалканов, имеющих сродство для серотонинергических рецепторов, содержащих их фармацевтических соединений, а также использования таких соединений и композиций. У млекопитающих мочеиспускание является сложным процессом, который требует объединенного действия мочевого пузыря, его внутренних и внешних сфинктеров, мускулатуры диафрагмы таза, и неврологического контроля над этими мышцами на трех уровнях (в стенке мочевого пузыря или сфинктере непосредственно, в автономных центрах спинного мозга и в центральной нервной системе на уровне центра мочеиспускания в варолиевом мосту (ЦМВМ) в стволе мозга (мост) под контролем коры головного мозга) (De Groat, Neurobiology of Incotinence, Ciba Foundation Symposium 151:27, 1990). Мочеиспускание происходит в результате сокращения непроизвольной мускулатуры, которая состоит из чередующихся гладкомышечных волокон, под контролем парасимпатической автономной системы, начинающейся из крестцового отдела спинного мозга. Простой рефлекс опорожнения запускается нервами, чувствительными к боли, температуре и растяжению, которые проходят от мочевого пузыря к крестцовому отделу спинного мозга. Однако чувствительные пути от мочевого пузыря также достигают ЦМВМ, генерируя нервные импульсы, которые обычно подавляют торможение крестцового отдела спинного мозга кортикальным ингибированием рефлекторной дуги, и расслаблением мышцы диафрагмы таза и внешнего сфинктера. Наконец происходят сокращения непроизвольной мускулатуры и опорожнение. Отклонения в сторону снижения функции мочевых путей, например, дизурия, недержание и энурез, являются обычными среди населения в целом. Дизурия включает частые мочеиспускания, ночную полиурию и позывы на мочеиспускание, и может быть вызвана циститом (включая промежуточный цистит), простатитом или доброкачественной гиперплазией предстательной железы (ДГПЖ) (которые поражают приблизительно 70% пожилых мужчин), или неврологическими расстройствами. Синдромы недержания мочи включают недержание мочи при напряжении, недержание при позывах на мочеиспускание, недержание вследствие переполнения мочевого пузыря и смешанное недержание. Энурез относится к ненамеренному выделению мочи ночью или во время сна. Как правило, лечение нервно-мышечной дисфункции при снижении функции мочевых путей включало введение соединений, непосредственно воздействующих на мышцы мочевого пузыря, например флавоксатов, спазмолитических препаратов (Ruffman, J. Int. МЭД. Res. .16:317, 1988), которые также воздействуют на ЦМВМ (Guarneri et al., Drugs of Today ,30:91, 1994), или антихолинергических соединений, например оксобутинина (Andersson, Drugs 36:477, 1988) и толтеродина (Nilvebrant, Life Sci. 68 (2223): 2549, 2001). Известно также применение антагонистов 1-адренергических рецепторов для лечения ДГПЖ, но оно основано на другом механизме действия (Lepor, Urology, 42:483, 1993). Однако лечение,включающее прямое торможение тазовой мускулатуры (включая непроизвольную мускулатуру), может иметь нежелательные побочные эффекты, например неполное опорожнение или аккомодационный паралич, тахикардию и сухость во рту (Andersson, Drugs 35:477, 1988). Таким образом, было бы предпочтительно использовать соединения, которые действуют через центральную нервную систему для воздействия, например, на рефлексы крестцового отдела спинного мозга и/или ингибирование проводящих путей ЦМВМ таким образом, чтобы восстанавливалось нормальное функционирование механизма мочеиспускания. В патенте США 5346896 описаны 5-HT1A связующие вещества, которые можно использовать при лечении таких заболеваний центральной нервной системы (ЦНС) как, например, беспокойства. В патенте ЕР 0924205 описаны соединения арилпиперазинов, которые связываются с 5-HT1A рецепторами. Настоящее изобретение касается соединений общей формулы In обозначает 1 или 2,а также их изомеры, оптические изомеры, диастереомеры, N-оксиды (например, N-пиперазин оксиды), кристаллические формы, гидраты, сольваты или фармацевтически приемлемые соли.-1 007503 Соединения формулы I могут существовать как четыре стереоизомера, которые могут присутствовать в рацемических смесях или в любой другой комбинации. Рацемические смеси могут быть подвергнуты энантиомерному обогащению для получения композиций, обогащенных в специфическом энантиомере, или растворенных в отдельных энантиомерах и в композициях, включающих отдельные энантиомеры. Энантиомерное обогащение может быть выражено как ее (энантиомерный избыток) как указано ниже. Предпочтительными являются соединения формулы I, где атом углерода, несущий группу R1 фенила, имеет (R) конфигурацию. Наиболее предпочтительны соединения, где атом углерода, несущий группу R1-фенила, имеет (R) конфигурацию, а смежный атом углерода, несущий R2 и гидроксильные группы, одновременно имеет (S) конфигурацию. Изобретение также включает метаболиты предшествующих соединений формулы I, обладающие тем же самым типом активности, в дальнейшем называемые активными метаболитами. Настоящее изобретение также рассматривает пролекарства, которые преобразуются в организме в ходе обмена веществ, для получения любого из предшествующих соединений. Согласно другому варианту осуществления изобретение касается фармацевтических композиций,включающих соединения формулы I, энантиомеры, диастереомеры, N-оксиды, кристаллические формы,гидраты, сольваты или фармацевтически приемлемые соли таких соединений формулы I, в примеси с фармацевтически приемлемыми растворителями или носителями. Согласно следующим вариантам осуществления, изобретение касается способа понижения частоты сокращений мочевого пузыря вследствие растяжения мочевого пузыря у нуждающихся в этом млекопитающих (например человека) путем назначения эффективного количества по меньшей мере одного соединения настоящего изобретения для понижения частоты сокращений мочевого пузыря вследствие растяжения мочевого пузыря у млекопитающих. Согласно другим вариантам осуществления, изобретение касается способа повышения объема мочевого пузыря у нуждающихся в этом млекопитающих (например человека) путем введения эффективного количества по меньшей мере одного соединения согласно настоящему изобретению для повышения объема мочевого пузыря у млекопитающих. Еще одним вариантом осуществления изобретения является способ лечения нарушений мочевого тракта у нуждающихся в этом млекопитающих (например человека) путем введения эффективного количества по меньшей мере одного соединения согласно настоящему изобретению для улучшения по меньшей мере одного из следующих состояний: позывы к мочеипусканию, повышенная активность мочевого пузыря, повышенная частота мочеиспусканий, пониженная растяжимость мочевого пузыря (пониженная емкость мочевого пузыря), цистит (включая промежуточный цистит), недержание мочи, истечение мочи,энурез, дизурия, слабость мочевого пузыря и затруднения при опорожнении мочевого пузыря. Для лечения вышеуказанных заболеваний соединения согласно изобретению можно вводить в комбинации с другими веществами такими, как, например, антимускариновые лекарственные средства, 1 адренергические антагонисты, ингибиторы циклооксигеназных ферментов, которые могут ингибировать СОХ 1 и СОХ 2 изозимы или которые, кроме того, могут быть избирательными для СОХ 2 изозима, и производных их NO доноров. Согласно другому варианту осуществления, настоящее изобретение касается способа лечения млекопитающих, страдающих нарушениями центральной нервной системы (ЦНС), вызываемых серотонинергической дисфункцией, путем введения эффективного количества по меньшей мере одного соединения настоящего изобретения для лечения нарушения ЦНС. Такие дисфункции включают тревогу, депрессию, гипертензию, нарушения цикла сон/бодрствование, нарушения питания, поведения, половой функции и когнитивных способностей у млекопитающих (в частности, у человека), вызываемых ударом,ранением, слабоумием, и вызываемых развитием неврологических состояний, вызываемых гиперактивностью, связанной с дефицитом внимания (ADHD), привыканием к чрезмерному употреблению лекарственных средств, прекращением приема препарата, синдромом раздражения кишечника, но не ограничены ими. Лечение можно проводить путем поставки соединения согласно изобретению к среде 5-HT1A серотонинергического рецептора, например, к экстрацеллюлярной внеклеточной питательной среде (системно или локально введением млекопитающему, обладающему таким 5-HT1A рецептором) количества соединения согласно изобретению, эффективного для лечения вышеуказанных нарушений. В предпочтительном варианте осуществления изобретение касается способа лечения млекопитающих (включая человека), страдающего от нарушения мочевого тракта, путем добавления по меньшей мере одного соединения согласно изобретению к среде 5-HT1A рецептора в количестве, эффективном для повышения длительности неподвижности мочевого пузыря при отсутствии сокращений. Наиболее предпочтительным является то, когда повышение длительности неподвижности мочевого пузыря достигают при небольшом воздействии (например, понижении или повышении) на давление при мочеиспускании или отсутствии такового эффекта. Предпочтительными группами циклоалкила R2 являются те, которые имеют от 3 до 6 атомов углерода, например, группы циклопропила, циклобутила, циклопентила и циклогексила. Термин "галоген" обозначает фтор, хлор, бром и йод.-2 007503 Термин "галоалкокси" обозначает моногалоалкоксигруппы, которые являются алкоксигруппами,замещенными одним атомом галогена, и полигалоалкоксигруппами, которые являются алкоксигруппами,замещенными по меньшей мере 2 атомами галогена. Предпочтительной галоалкоксигруппой является 2,2,2-трифторэтокси."Метаболит" описанного здесь соединения обозначает производное соединения, которое образуется, когда соединение метаболизируется. Термин "активный метаболит" обозначает биологически активное производное соединения, которое образуется, когда соединение метаболизируется. Термин "метаболизируется" обозначает совокупность процессов, при которых специфическое вещество изменяется в живом организме. На все соединения, присутствующие в организме, воздействуют ферменты в пределах организма для получения энергии и/или удаления их из организма. Специфические ферменты производят определенные структурные изменения в соединении. Цитохром Р 450, например, катализирует ряд окислительных и восстановительных реакций. Уридин дифосфат глюкуронилтрансферазы, например, катализирует передачу активизированных молекул глюкуроновой кислоты к ароматическим спиртам, алифатическим спиртам, карбоновым кислотам, аминам и свободным сульфгидрильным группам. Дальнейшая информация о метаболизме описана в The Pharmacological Basis of therapeutics, 9th Edition, McGraw - Hill(1996), pages 11-17. Метаболиты описанных здесь соединений могут быть идентифицированы либо путем введения соединений хозяину и анализа образцов ткани хозяина, либо инкубацией соединений с печеночными клетками или другими системами in vitro, такими, как цитохромы или микросомы, и анализом полученных соединений. Оба способа известны в данной области техники. Используемый здесь термин "стереоизомер" обозначает соединение, полученное из тех же самых атомов, связанных теми же самыми атомами, но имеющими различные трехмерные структуры, которые не взаимозаменяются. Трехмерные структуры называют конфигурациями. Используемый здесь термин"энантиомер" обозначает два стереоизомера, молекулы которых являются несуперналагаемыми зеркальными отображениями друг друга. Используемый здесь термин "оптический изомер" эквивалентен термину "энантиомер". Соединения, которые являются стереоизомерами друг друга, но не являются энантиомерами друг друга, называются диастереомерами. Термины "рацемат" или "рацемическая смесь" обозначают смеси равных частей энантиомеров. Термин "хиральный центр" обозначает атом углерода, к которому присоединены четыре различных группы. Используемый здесь термин "энантиомерное обогащение" относится к повышению количества одного энантиомера по сравнению с другим. Удобным способом выражения полученного энантиомерного обогащения является понятие энантиомерного избытка где Е 1 - количество первого энантиомера, и Е 2 - количество второго энантиомера. Таким образом, если начальное отношение двух энантиомеров составляет 50:50, как присутствует в рацемической смеси, и достигается энантиомерное обогащение, достаточное для получения конечного соотношения 50:30, то энантиомерное обогащение ее в отношении первого энантиомера составляет 25%. Однако, если конечное соотношение составляет 90:10, то ее в отношении первого энантиомера составляет 80%. Согласно одному варианту осуществления изобретения, предпочтительно энантиомерное обогащение ее более 90%, ее более 95% наиболее предпочтительно, а особенно наиболее предпочтительно ее более 99%. Энантиомерное обогащение может определить специалист в данной области техники, используя стандартные методы и процедуры, такие, как высокоэффективная жидкостная хроматография с хиральной колонкой. Выбор адекватной хиральной колонки, элюента и условий, необходимых для эффекта разделения энантиомерной пары находится в пределах знаний специалиста в данной области техники. Кроме того, энантиомеры соединений формулы I могут быть растворены специалистом в данной области техники, используя стандартные методы, известные области техники, например описанные J.Jacques, et al., "Enantiomers, Racemates, and Resolutions ", John Wiley and Sons, Inc, 1981. Примеры обратного растворения включают методы перекристаллизации или хиральную хроматографию. Диастереомеры отличаются как по физическим свойствам, так и по химической реакционной способности. Смесь диастереомеров может быть разделена на энантиомерные пары, основанные на растворимости, фракционной кристаллизции или хроматографических свойствах, например, с помощью хроматографа с тонким слоем, колоночной хроматографии или HPLC (жидкостной хроматографии высокого давления). Очистку сложных смесей диастереомеров в энантиомерах обычно проводят в две стадии. В первой стадии смесь диастереомеров растворяют в энантиомерных парах, как описано выше. На второй стадии далее энантиомерные пары очищают в композициях, обогащенных для одного или другого энантиомера или, более предпочтительно, растворяют в композиции, включающей чистые энантиомеры. Обратное растворение изомеров обычно требует реакции или межмолекулярного взаимодействия с хиральным агентом, например, растворителем или колоночной матрицей. Обратное растворение энантиомеров можно проводить, например, преобразованием смеси изомеров, например, рацемической смеси, в смесь диа-3 007503 стереомеров реакцией с чистым энантиомером второго агента, то есть растворяющегося агента. Два полученных диастереомерных продукта затем могут быть разделены. Разделенные диастереомеры затем повторно преобразуют в чистые энантиомеры обращением начального химического превращения. Обратное растворение энантиомеров можно также проводить с помощью различий в их нековалентной связи с хиральным веществом, например, хроматографией на гомохиральных абсорбентах. Нековалентная связь между энантиомерами и хроматографическим адсорбатом созданных диастереомерных комплексов приводит к разному распределению подвижных и связанных состояний в хроматографической системе. Поэтому два энантиомера проводят через хроматографическую систему, например колонку, с различными скоростями, что приводит к их разделению. Хиральные колонки растворения известны в области техники и выпускаются серийно (например,MetaChem Technologies Inc, подразделением ANSYS Technologies, Inc, Lake Forest, CA). Энантиомеры могут быть проанализированы и очищены при использовании, например, хиральных стационарных фаз(ХСФ) для HPLC. Хиральные HPLC колонки обычно содержат одну форму энантиомерного соединения,иммобилизированного к поверхности наполнителя диоксида кремния. Для осуществления хирального разделения должны быть, по меньшей мере, три точки одновременного взаимодействия между ХСФ и одним определяемым при анализе энантиомером вещества, с одним или большим количеством этих взаимодействий, являющихся стереохимически зависимыми.D-фенилглицин и L-лейцин являются ХСФ Типа 1 и используют комбинации р-р взаимодействия,водородные связи, диполь - дипольные взаимодействия, и пространственные взаимодействия для осуществления хирального распознавания. Для растворения на колонке Типа 1 определяемый энантиомер должен содержать функциональную группу, комплементарную к этому ХСФ таким образом, чтобы анализируемое вещество подвергалось эфирным взаимодействиям с ХСФ. Образец должен предпочтительно содержать один из следующих функциональных групп: р-кислота или р-основание, донор водородной связи и/или акцептор, или амид диполь. Получение производных иногда используют для добавления интерактивных сайтов к недостающим соединениям. Наиболее встречающиеся производные включают получение амидов из аминов и карбоновых кислот.MetaChiral ODM является ХСФ типа II. Первичные механизмы получения комплексов растворенного вещества-ХСФ осуществляют с помощью взаимных притяжений, но комплексы включения также играют важную роль. Образование водородной связи, пи - пи, и межплоскостные взаимодействия диполей важны для хирального обратного растворения на MetaChiral ODM. Получение производных часто необходимо, когда молекула растворенного вещества не содержит группы, необходимые для взаимодействий растворенного вещества и колононки. Получение производных, обычное для бензиламидов, также необходимо для некоторых сильно полярных молекул подобно аминам и карбоновым кислотам, которые иначе взаимодействовали бы слишком сильно со стационарной фазой посредством нестереоспецифических взаимодействий. Согласно предпочтительному варианту осуществления изобретения R1 формулы I обозначает атом фтора. Также предпочтительно, если R1 обозначает атом фтора в 2-положении кольца фенила. Предпочтительной группой, которую обозначает R2, является незамещенная группа циклогексила. Предпочтительным заместителем, который обозначает R3, является алкокси, более предпочтительно метокси, и наиболее предпочтительно метоксигруппу в положении 2 кольца фенила. Предпочтительным соединением согласно настоящему изобретению является 1-[4-циклогексил-4 гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин формулы II где Z1 и Z2 представляют хиральные центры. Соединения формулы II могут существовать в виде одного из четырех стереоизомеров: Эти соединения можно назвать следующим образом: 1-[(3R,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин,1-[(3S,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин,1-[(3R,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин и 1-[(3S,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин. Соединения формулы II могут быть разделены на диастереомерные пары, например, разделением на тонкослойной хроматографии. Эти диастереомерные пары упомянуты здесь как диастереомер с верхней тонкослойной хроматографией Rf; и диастереомер с пониженной тонкослойной хроматографией Rf. Диастереомеры могут быть дополнительно обогащены для специфических энантиомеров или растворены в отдельных энантиомерах, с помощью способов, известных в данной области техники, например описанных здесь. Согласно другому предпочтительному варианту осуществления, изобретение касается соединения 1-[4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазина, которое может существовать в четырех стереоизомерах: 1-[(3R,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин,1-[(3S,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин,1-[(3R,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин, и 1-[(3S,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин,и их энантиомеры, оптические изомеры, диастереомеры, N-оксиды (например, N-пиперазин оксиды), кристаллические формы, гидраты, сольваты и фармацевтически приемлемые соли. Предпочтительны 3R, 4S и 3R, 4R. Наиболее предпочтительны 3R, 4S. Комбинированное лечение В некоторых примерах осуществления изобретения нарушений мочевых путей лечат путем введения соединения формулы I в комбинации с дополнительным 5-НТ 1 Аантагонистом или антагонистом одного или более дополнительных классов рецепторов. В предпочтительных вариантах осуществления соединение формулы I вводят в комбинации с антагонистом 1 - адренергических, или мускариновых рецепторов. В следующих примерах осуществления изобретения заболевания пониженной функции мочевых путей лечат введением соединения формулы I в комбинации с одним или более ингибиторами циклооксигеназного фермента, который может ингибировать как СОХ 1, так и СОХ 2 изозимы или который также может быть селективным для СОХ 2 изозима, и NO донора их производных. Примеры антимускариновых лекарств для введения в комбинации с соединением формулы I включают оксобутинин, толтеродин, дарифенацин, и темиверин. Соединение формулы I можно вводить в комбинации с 1 - адренергическими антагонистами для терапии симптомов пониженной функции мочевых путей, независимо от того, связаны ли они с ВРН. Предпочтительные 1 - адренергические антагонисты, подходящие для введения в комбинации с соединением формулы I, включают, например, празосин, доксазосин, теразосин, алфузосин, и тамсулосин. До-5 007503 полнительные 1 - адренергические антагонисты, подходящие для введения в комбинации с соединением формулы I, описаны в патентах США 5,990,114; 6,306,861; 6,365,591; 6,387,909; и 6,403,594. Примеры 5 - HT1A антагонистов, которые можно вводить в комбинации с соединением формулы I,представлены у Leonardi et al., J. Pharmacol. Exp. Ther. 299: 1027 - 1037, 2001 (например, Rec 15/3079),патенте США 6,071,920, а другие производные фенилпиперазина описаны в международной заявкеWO 99/06383 и в находящеся на рассмотрении заявке на патент США под серийным 10/266,088 и 10/266,104, поданной 7 октября 2002. Дополнительные 5 - HT1A антагонисты включают DU - 125530 и относящиеся к ним соединения, описанные в патенте США 5,462,942, а также и робалзотан и относящиеся к ним соединения, описанные в международной заявке WO 95/11891. Примеры селективных СОХ 2 ингибиторов, которые можно вводить в комбинации с соединением формулы I, включают без ограничения, нимесулид, мелоксикам, рофекоксиб, целекоксиб, парекоксиб и вальдекоксиб. Дополнительные примеры селективных СОХ 2 ингибиторов описаны, без ограничения, в патенте США 6,440,963. Примеры не-селективных СОХ 1 - СОХ 2 ингибиторов включают без ограничения, ацетилсалициловую кислоту, нифлумовую кислоту, флуфенамовую кислоту, энфенамовую кислоту,меклофенамовую кислоту, толфенамовую кислоту, тиапрокарболовую кислоту, ибупрофен, напроксен,кетопрофен, флурбипрофен, фурпрофен, индометацин, ацетметацин, проглуметацин, кеторолак, дихлофенак, этодолак, сулиндак, фентиазак, теноксикам, лорноксикам, цинноксикам, ибупроксам, набуметон,толметин, амтолметин. Соответственно, каждое из вышеуказанных веществ представляет собой неограниченные примеры СОХ ингибиторов, которые можно вводить в комбинации с соединением формулы I. Примеры производных СОХ ингибиторов, которые могут вводить в комбинации с соединением формулы I, включают производные СОХ ингибиторов, несущие нитратные (нитроокси) или нитритные группы, как представленные, например, в международной заявке WO 98/09948, способные высвобождатьNO in vivo. Фармацевтические композиции Далее изобретение касается фармацевтических композиций, включающих соединение формулы I или его энантиомеры, диастереомеры, N - оксиды, кристаллические формы, гидраты, сольваты, активные метаболиты или фармацевтически приемлемые соли. Фармацевтическая композиция может также включать дополнительные добавки, например фармацевтически приемлемый носитель или растворитель,ароматизатор, подслащивающее вещество, консервант, краситель, связующее вещество, суспендирующий агент, диспергирующий агент, пигмент, дезинтегратор, наполнитель, растворитель, смазку, поглотительный усилитель, бактерицид и т.п., стабилизатор, мягчитель, пищевое масло, или любую комбинацию двух или более из указанных добавок. Подходящие фармацевтически приемлемые носители или растворители включают без ограничения,этанол, воду, глицерол, гель алоэ вера, аллантоин, глицерин, масла витамина А и Е, минеральное масло,физиологический раствор с фосфатным буфером, PPG2 пропионат миристила, карбонат натрия, фосфат калия, растительное масло, жидкий животный жир и солкеталь. Подходящие связующие компоненты включают без ограничения, крахмал, желатин, природные сахара, например глюкозу, сахарозу и лактозу, гранулированные подслащивающие вещества, натуральные и синтетические смолы, например камедь, трагакант, растительную смолу, альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воска и т.п. Подходящие дезинтеграторы включают без ограничения, крахмал, например кукурузный крахмал,метилцеллюлозу, агар, бентонит, ксантановую смолу и т.п. Подходящие смазки включают без ограничения, олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Подходящие суспендирующие агенты включают без ограничения, бентонит. Подходящие диспергирующие и суспендирующие агенты включают без ограничения, синтетические и натуральные смолы, например камедь, трагакант, акацию, альгинат, декстран, карбоксиметилцеллюлозу натрия, метилцеллюлозу, поливинилпирролидон и желатин. Примеры дополнительных добавок включают без ограничения, сорбит, тальк, стеариновую кислоту и фосфат дикальция. Лекарственные формы Фармацевтическая композиция может быть получена в виде таких лекарственных форм, как таблетки, пилюли, капсулы, болюсы, порошки, гранулы, стерильные парентеральные растворы, стерильные парентеральные суспензии, стерильные парентеральные эмульсии, элексиры, тинктуры, дозируемые аэрозоли или жидкие распылители, капли, ампулы, автоинъекционные устройства или суппозитории. Лекарственные формы можно использовать для перорального, парентерального, внутриносового, подъязычного или ректального введения, либо для введения ингаляцией или вдуванием, в виде трансдермальных бляшек, а также лиофилизированных композиций. В целом, можно использовать любые формы доставки активных ингредиентов, которые обеспечивают к системное наличие таких ингредиентов. Предпочтительной лекарственной формой является пероральная форма, наиболее предпочтительно - твердая пероральная форма; поэтому предпочтительные лекарственные формы включают таблетки, пилюли и капсулы. Однако также предпочтительны парентеральные препараты.-6 007503 Твердые лекарственные формы можно получать смешиванием активных реагентов согласно изобретению с фармацевтически приемлемым носителем и любыми другими желательными добавками, как описано выше. Смесь обычно перемешивают до получения гомогенной смеси активных реагентов согласно настоящему изобретению и образования носителя и любых других желательных добавок, то есть активные реагенты распределены равномерно по всей композиции. В этом случае композицию можно получать в виде сухих или влажных гранул. Лекарственные формы можно получать, например, в виде формы "немедленного высвобождения". Лекарственные формы "немедленного высвобождения" обычно получают в виде таблеток, которые высвобождают по меньшей мере 60-90% активного ингредиента в течение 30-60 мин при проведении тестов на растворимость лекарственного средства, например, по стандарту Фармакопеи США 711. В предпочтительном варианте осуществления лекарственные формы немедленного действия высвобождают около 75% активного ингредиента в течение приблизительно 45 мин. Могут также быть получены такие лекарственные формы как, например, формы с контролируемым высвобождением. Лекарственные формы с контролируемым, непрерывным, долговременным или замедленным высвобождением обозначают эквивалентные термины, которые описывают вид доставки активного реагента, что происходит, когда активный реагент высвобождается из носителя при установленной и управляемой скорости в течение времени, которое составляет в целом порядка минут, часов или дней, обычно в пределах приблизительно от 60 мин до 3 дней, вместо того, в отличие от немедленного рассеивания перед попаданием в пищеварительный тракт или после контакта с желудочной жидкостью. Контролируемая скорость высвобождения может изменяться как функция разнообразных факторов. Факторы, влияющие на расход в контролируемом высвобождении, включают размер, состав, пористость, структуру заряда, и степень гидратации поставляющего носителя активного ингредиента(ов), кислотность среды ( внутренней либо внешней на носитель для поставки), а также растворимость активного реагента в физиологической среде, то есть специфическое местоположение в пищеварительном тракте. Типичные параметры для теста на растворимость контролируемых лекарственных форм указаны в стандарте Фармакопеи США 724. Можно также получать лекарственные формы для доставки активного реагента в многофазных стадиях, при которых первая фракция активного ингредиента высвобождается при первой скорости, а по меньшей мере вторые фракции активного ингредиента высвобождаются при второй скорости. В предпочтительном варианте осуществления, лекарственная форма может быть получена для доставки активного реагента двухфазным способом, включающим первую фазу "немедленного высвобождения", во время которой фракция активного ингредиента доставляется при скорости, указанной выше для лекарственных форм немедленного высвобождения, а также вторую фазу "контролируемого высвобождения",при которой остаток активного ингредиента высвобождается под контролем, как описано выше для лекарственных форм дозировки с контролируемым высвобождением. Таблетки или пилюли могут иметь покрытие или могут быть изготовлены иначе для получения лекарственной формы с задержанным и/или непрерывным действием, например, с контролируемым высвобождением и с задержанным высвобождением. Например, таблетка или пилюля могут включать компонент внутренней дозировки и внешней дозировки, причем последний находится в виде слоя или защитного покрытия над первым. Эти два компонента могут быть разделены тонкокишечным слоем, который служит для противодействия разложению в желудке и позволяет внутреннему компоненту проходить неповрежденным в двенадцатиперстную кишку или обеспечивает его задержанное высвобождение. Разлагаемые микроорганизмами полимеры для контроля высвобождения активных реагентов включают полимолочную кислоту, полиэпсилон капролактон, полигидроксимасляную кислоту, полиортоэфиры, полиацетали, полидигидропираны, полицианакрилаты и сшитые или амфипатические блоксополимеры гидрогелей, но не ограничены ими. Для жидких лекарственных форм дозировки, активные вещества или их физиологически приемлемые соли растворяют, суспендируют или эмульгируют, произвольно с обычно используемыми веществами, такими, как солюбилизаторы, эмульгаторы или другие добавки. Растворители для активных комбинаций и соответствующих физиологически приемлемых солей могут включать воду, растворы физиологических солей или спиртов, например этанол, пропандиол или глицерол. Дополнительно, могут быть использованы растворы сахаров, например растворы глюкозы или манитола. В настоящем изобретении можно также использовать смесь различных упомянутых растворителей. Трансдермальные лекарственные формы также рассматриваются в соответствии с настоящим изобретением. Трансдермальной формой может быть диффузионная трансдермальная система (трансдермальные бляшки), в которой используют либо жидкий резервуар, либо систему матрицы препарата в связующем веществе. Другие трансдермальные лекарственные формы включают, без ограничения гели для наружного применения, лосьоны, мази, трансмукозальные системы и устройства, а также ионофоретические (путем электрической диффузии) системы доставки. Трансдермальные лекарственные формы можно использовать для задержанного высвобождения и длительного высвобождения активных реагентов настоящего изобретения.-7 007503 Фармацевтические композиции и лекарственные формы согласно настоящему изобретению для парентерального введения, и в частности, инъекций, обычно включают фармацевтически приемлемый носитель, как описано выше. Предпочтительным жидким носителем является растительное масло. Инъекция может быть, например, внутривенной, эпидуральной, интратекальной, внутримышечной, внутрипросветной, интратрахеальной или подкожной. Активные вещества можно также вводить в форме липосомной системы поставки, например в виде малых однослойных везикул, больших однослойных везикул и мультичешуйчатых везикул. Липосомы можно получать из ряда фосфолипидов, например таких, как холестерин, стеариламин или фосфатидилхолины. Активные вещества согласно настоящему изобретению можно также соединять с растворимыми полимерами, например наводимыми носителями лекарственных средств. Такие полимеры включают без ограничения, поливинилпирролидон, сополимеры пирана, полигидроксипропилметакриламидофенол,полигидроксиэтиласпартамидофенол, и полиэтиленоксиполилизин, замещенный остатками пальмитоила. Введение Фармацевтическую композицию или лекарственные формы согласно настоящему изобретению можно вводить рядом способов, такими, как оральный и тонкокишечный, внутривенный, внутримышечный, подкожный, трансдермальный, трансмукозальный (включая ректальный и трансбуккальный), а также ингаляцией. Предпочтительными являются оральный или трансдермальный способы введения(например, твердые или жидкие составы или кожные бляшки, соответственно). Фармацевтическую композицию или лекарственные формы, включающие эффективное количество состава согласно настоящему изобретению, можно вводить животному, предпочтительно человеку, при необходимости лечения нервно-мышечной дисфункции при пониженной функции мочевых путей, как описано Е. J. McGuire в "Campbell's UROLOGY", 5th Ed., 616-638, 1986, W.B. Saunders Company, a также пациентам, страдающим от любой физиологической дисфункции, вызванной недостаточной функцией рецептора 5-НТ 1 А. Такие дисфункции включают без ограничения такие заболевания центральной нервной системы, как депрессия, тревога, нарушение питания, половой функции, привыкание к препаратам и связанные с этим проблемы. Используемый здесь термин "эффективное количество" относится к количеству, которое приводит к измеримому улучшению по меньшей мере одного симптома или параметра определенного заболевания. В предпочтительном варианте осуществления изобретения с помощью соединения лечат такие заболевания мочевых путей, как, например, позывы на мочеиспускание, повышенная активность мочевого пузыря, повышенная частота мочеиспускания, пониженная растяжимость мочевого пузыря (пониженная емкость мочевого пузыря), цистит (включая промежуточный цистит), недержание, истечение мочи, энурез, дизурия, слабость мочевого пузыря и затруднения при опорожнении мочевого пузыря, либо заболевания центральной нервной системы, вызываемые серотонергической дисфункцией, например тревога,депрессия, гипертензия, нарушения цикла сон/бодрствование, нарушения пищевого поведения, половой функции и когнитивных способностей у млекопитающих (в частности, у человека), связанные с ударом,повреждением, слабоумием, и развитием неврологических нарушений, заболеваний, вызываемых гиперактивностью, связанной с дефицитом внимания (ADHD), привыканием к чрезмерному употреблению лекарственных средств, прекращением приема препарата, синдромом раздражения кишечника. Фармацевтическую композицию или лекарственную форму согласно настоящему изобретению можно вводить согласно дозировке и режиму введения, определяемому стандартным клиническим тестированием в свете вышеуказанных нормативов для получения оптимальной активности при снижении токсичности или побочных эффектов для специфического пациента. Однако такая тонкая регулировка терапевтического режима производится стандартным клиническим тестированием в свете представленных здесь нормативов. Дозировка активных соединений согласно настоящему изобретению может изменяться в зависимости от ряда факторов, например, таких, как причины возникновения болезни, состояние индивидуума,вес, пол и возраст, а также способ введения. Эффективное количество для лечения заболеваний может быть легко определено эмпирическими способами, известными обычным специалистам в данной области техники, например, путем определения матрицы дозировок и частоты введения, а также сравнением группы экспериментальных единиц или предметов в каждом пункте в матрице. Точное количество соединения, которое должно быть назначено пациенту, изменяется в зависимости от состояния и серьезности нарушения и физического состояния пациента. Измеримое улучшение любого симптома или параметра может быть определено специалистом в данной области техники или сообщено пациентом врачу. Следует понимать, что любое клинически или статистически значительное ослабление или улучшение любого симптома или параметра заболеваний мочевых путей находится в пределах области изобретения. Под клинически значительным ослаблением или улучшением следует понимать такое, которое заметно для пациента и/или врача. К примеру, один пациент может страдать от нескольких симптомов дизурии одновременно, например, позывов на мочеиспускание и чрезмерной частоты мочеиспускания, либо и того, и другого, и они могут быть снижены путем использования способа согласно настоящему изобретению. В случае недер-8 007503 жания, любое снижение частоты или объема нежелательного выхода мочи рассматривают как положительный эффект от данного способа лечения. Количество назначаемого вещества может колебаться приблизительно в диапазоне от 0,01 до 25 мг/кг/день, предпочтительно приблизительно между 0,1 и 10 мг/кг/день и наиболее предпочтительно между 0,2 и 5 мг/кг/день. Следует понимать, что фармацевтические композиции согласно настоящему изобретению не обязательно содержат полное количество вещества, которое является эффективным при лечении заболеваний, поскольку такое эффективное количество может быть обеспечено путем введения множества доз таких фармацевтических композиций. В предпочтительном варианте осуществления настоящего изобретения, соединения получают в капсулах или таблетках, предпочтительно содержащих 50-200 мг соединений согласно изобретению, и предпочтительно вводят пациенту при общей ежедневной дозе 50-400 мг, предпочтительно 150-250 мг и наиболее предпочтительно приблизительно 200 мг, в случае недержания мочи и дисфункциях при лечении с лигандом 5-HT1A рецептора. Полные ежедневные дозировки можно вводить многократно, например, 4 суб -дозы, в сутки. В предпочтительных вариантах осуществления полную ежедневную дозировку вводят в 1 или 2 дозировках в сутки. Фармацевтическая композиция для парентерального введения содержит приблизительно от 0.01 % до 100% по весу активных веществ согласно настоящему изобретению на 100% веса всей фармацевтической композиции. В целом, трансдермальные лекарственные формы содержат приблизительно от 0,01 до 100 % по весу активных веществ на 100% общего веса лекарственной формы. Фармацевтическую композицию или лекарственную форму можно вводить как однократную ежедневную дозу, либо полную ежедневную дозу, а также можно принимать в виде раздельных доз. Кроме того, желательным может быть совместное или последовательное введение другого соединения для лечения заболевания. Примеры такого комбинированного лечения описаны выше. При комбинированном лечении, когда соединения находятся в раздельных дозированных составах,соединения можно вводить одновременно, либо каждое из них можно вводить в разные периоды времени. Например, соединение согласно изобретению можно вводить утром, и антимускариновое соединение можно вводить вечером, либо наоборот. Дополнительные соединения можно также вводить в определенных интервалах. Порядок введения будет зависеть от ряда факторов, включая возраст, вес, пол и медицинское состояние пациента; серьезность и этиологию подлежащих лечению заболеваний, способа введения, функции почек и печени пациента, истории лечения пациента, а также чувствительности пациента. Определение порядка введения может быть отработано очень точно, и такая точная отработка осуществляется стандартными клиническими методами в соответствии с представленными здесь указаниями. Применение. Способы лечения Не вдаваясь в теорию, следует отметить, что введение антагониста 5 - HT1A рецептора предотвращает нежелательную активность крестцовых рефлексов и/или корковых механизмов, контролирующих мочеиспускание. Таким образом, предполагается, что, используя соединения согласно настоящему изобретению, можно лечить широкий диапазон нейро-мышечных дисфункций нижних мочевых путей, включая без ограничения дизурию, недержание и энурез (сверхактивный мочевой пузырь). Дизурия включает частые мочеиспускания, ночную полиурию, позывы на мочеиспускание, пониженную растяжимость мочевого пузыря (пониженная емкость мочевого пузыря), трудности при опорожнении мочевого пузыря, то есть объем мочи, удаляемой во время мочеиспускания, ниже оптимального. Синдромы недержания включают недержание мочи при напряжении, недержание при позывах на мочеиспускание и недержание при энурезе, а также смешанные формы недержания. Энурез относится к ненамеренному выходу мочи ночью либо во время сна. Соединения согласно настоящему изобретению можно также использовать для лечения заболеваний центральной нервной системы, вызываемых серотонергической дисфункцией. Синтез соединений по настоящему изобретению Соединения согласно изобретению в основном получают в соответствии со следующей схемой: Группы А и R являются такими же, как R2 и R1, как определено со ссылкой на общую формулу (I). Группа В эквивалентна группе R3-фенил, как определено со ссылкой на общую формулу (I). R4 представляет алкил. Исходное вещество (1) обрабатывают основанием, предпочтительно трет-бутилатом калия, затем производят алкилирование 2 - бромацеталдегид диалкилацеталем или другим карбонилом, защищенным 2 - галоацетальдегидом (например, Rа,алкильные группы могут также быть соединены в цикл для получения диоксолана или кольца диоксана). Другие альтернативные и соответствующие основания для проведения конденсации включают амид лития, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат калия, карбонат цезия и т.п. с добавлением катализаторов фазового переноса или без них. Реакцию предпочтительно проводят в растворителе, например диметил сульфоксиддиметиле или толуоле при температуре 0 С для дефлегмирования. Обработкой (2) кислотой, например соляной кислотой или р-толуол-сульфокислотой или трифторуксусной кислотой в соответствующем органическом растворителе получают альдегид (3). В целом реакцию проводят в протонном растворителе, например смеси водной кислоты и ацетона или тетрагидрофурана, при температуре приблизительно от 5 до 75 С, предпочтительно при температуре окружающей среды. Предпочтительный и подобный способ состоит из проведения реакции в смеси водной трифторуксусной кислоты в хлорированном растворителе при температуре окружающей среды. Альдегид (3) реагирует с желательным арил диазоциклоалканом (4) в соответствии с восстановительной процедурой аминирования с получением продукта (5). Реакцию предпочтительно проводят при температуре окружающей среды в нереакционноспособном растворителе, например дихлорэтане или хлористом метилене или хлороформе в присутствии натриевого триацетоксиборгидрида, и в основном завершают в течение от одного до 24 ч (см. например A. F. Abdel-Magid, et al., J. Org. Chem., 61, 3849(1996, либо ее могут проводить в протонном растворителе (например, метаноле) при помощи натриевого цианоборгидрида произвольно в присутствии молекулярных сит. Восстановление (5) до спирта (I) сразу проводят, используя восстановитель, например борогидрид натрия или гидрид диизобутилалюминия, либо другой алюминий или гидрид бора, либо другой способ восстановления для проведения конверсии кетона до спирта, хорошо известный специалистам в данной области техники для получения гидрокси соединения (I). Реакцию предпочтительно проводят в органическом растворителе, например метаноле или хлористом метилене или тетрагидрофуране при температуре приблизительно от - 20 С до температуры окружающей среды. Исходное вещество (1) либо выпускается серийно, либо может быть получено реакцией связывания подходящего амида Weinreb (6) [См. Nahm and Weinreb, Tetrahedron Lett., 22, 3815, (1981)] с соединением(7), как описано в схеме 2 выше, где М обозначает металлическую соль, например галид лития или магния. Реакцию предпочтительно проводили под инертной атмосферой предпочтительно под азотом, в апротонном растворителе, например тетрагидрофуране, при температуре окружающей среды или более низких температурах до -78 С. Кроме того, эфир со структурой АСООалкила можно обработать замещенным бензилмагний хлоридом или бензилмагний бромидом при стандартных условиях, известных в данной области техники для получения кетона структуры (1). Предпочтительный и подобный путь синтеза (1) представляет катализируемую палладием реакцию ацил галида с соединением (7), где М представляет галид цинка.- 10007503 В частности, соединения формулы (5) могут получать в ходе методики, описанной в схеме 3. Все заместители, если не обозначено иначе, как определены ранее. Реактивы и исходные вещества являются легко доступными специалисту в данной области техники. В схеме 3, например, на стадии А, циклогексанкарбонил хлорид добавляют к смеси соответствующего хлорида бензилцинка или бромида и соответствующего катализатора палладия, например, дихлоробистрифенилфосфин)палладия (II), перемешиваемой при 0 С в растворителе, например тетрагидрофуране. Затем перемешивание продолжают при температуре окружающей среды в течение 4-24 ч. Затем реакцию гасят, например, водным насыщенным раствором хлористого аммония. Кетон (8) получают экстракцией с помощью обычной методики исследования. Кетон (8) можно очищать методами, известными в данной области техники, например, флэш-хроматографией на силикагеле с соответствующим элюентом, например, этилацетат/гексан, для получения очищенного вещества. Кроме того, неочищенный кетон(8) можно переносить в стадию В. В схеме 3, стадия В, кетон (8) алкилируют с бромацетальдегид диэтил ацеталем при условиях, известных в данной области техники, для получения соединения структуры (9). Например, кетон (8) растворяют в соответствующем органическом растворителе, таком как диметил сульфоксид или толуол, и обрабатывают небольшим избытком соответствующего основания, например трет-бутилатом калия. Реакционную смесь перемешивают в течение приблизительно 15-30 мин при температуре между 0 С и температурой флегмы растворителя и добавляют к реакционной смеси по каплям бромацетальдегид диэтилацеталь. Специалист в данной области техники сразу может определить, что бромацетальдегид диметилацеталь, бромацетальдегид этиленацеталь и т.п. можно использовать вместо соответствующего диэтила ацеталя. В схеме 3, стадия С, соединение (9) гидролизуют при кислотных условиях для получения альдегида(10) способом, аналогичным методике, описанной в схеме I. В частности, например, соединение (9) растворяют в соответствующем органическом растворителе, например дихлорметане, и обрабатывают соответствующей кислотой, например водной трифторуксусной кислотой. Реакционную смесь перемешивают в течение приблизительно 1-6 ч при комнатной температуре. Затем реакционную смесь разбавляют тем же растворителем, промывают соляным раствором, органический слой отделяют, высушивают над безводным сульфатом натрия, фильтруют и концентрируют под вакуумом для получения альдегида (10). Альдегид (10) можно очищать методами, известными в данной области техники, например флэшхроматографией на силикагеле с соответствующим элюентом, таким как этилацетат/гексан. Кроме того,неочищенный альдегид (10) можно использовать непосредственно в стадии D. В схеме 3, стадия D, альдегид (10) восстановительно аминируют, при условиях, известных в данной области техники, с диазоциклоалканом (4), для получения кетона (5) способом, аналогичным методике,описанной в схеме I. В частности, например, альдегид (10) растворяют в соответствующем органическом растворителе, например метиленхлориде. К этому раствору добавляют приблизительно 1,05 или более эквивалентов диазоциклоалкана (4). Уксусную кислоту можно произвольно добавлять для облегчения растворения диазоциклоалкана (4). Затем добавляют приблизительно 1,4-1,5 эквивалентов триацетоксиборгидрида натрия, и реакционную смесь перемешивают при комнатной температуре в течение приблизительно 3-5 ч. Реакцию затем гасят добавлением соответствующего основания, такого как водный карбонат натрия или гидроксид для получения рН приблизительно 8-12. Затем погашенную реакционную смесь экстрагируют с соответствующим органическим растворителем, например метиленхлоридом. Органические экстракты соединяют, промывают соляным раствором, высушивают, фильтруют и концентрируют под вакуумом для получения соединения формулы (5). Затем это вещество можно очищать методами, известными в данной области техники, например флэш-хроматографией на силикагеле с соответствующим элюентом, таким как этилацетат/петролейный эфир или гексан. Кроме того, соединения структуры (5) можно получать согласно методике, описанной в схеме 4. Все заместители, если не отмечено иначе, такие, как определены ранее. Реактивы и исходные вещества доступны специалисту в данной области техники. В схеме 4, стадия А, альдегид (11) добавляют к соответствующему органометаллическому реактиву(12) при условиях, известных в данной области техники для получения спирта (13). Примеры подходящих органометаллических реактивов включают реактивы Гриньяра, алкиллитиевые реактивы, алкилцинковые реактивы, и т.п. Предпочтительными являются реактивы Гриньяра. Для примеров типичных реактивов Гриньяра и условий реакции см. J. March, "Advanced Organic Chemistry: Reactions, Mechanisms,and Structure", 2nd edition, McGraw - Hill, с. 836 - 841 (1977). В частности, альдегид (11) растворяют в соответствующем органическом растворителе, например тетрагидрофуране или толуоле, охлаждают приблизительно до -5 С и обрабатывают приблизительно 1,1-1,2 эквивалентами реактива Гриньяра формулы(12), где М - MgCl или MgBr. Реакционную смесь перемешивают приблизительно 0,5-6 ч, затем резко охлаждают и изолируют спирт (13) с помощью хорошо известной рабочей методики. В схеме 4, стадия В, спирт (13) окисляют при стандартных условиях, хорошо известных в данной области техники, описанными J. March, "Advanced Organic Chemistry: Reactions, Mechanisms, and Structure", 2nd edition, McGraw - Hill, с. 1082-1084 (1977), для получения кетона (1). Кетон (1) является исходным веществом, используемым в вышеуказанной схеме 1. Например, вышеупомянутое окисление можно также проводить, используя стандартные условия окисления Сверна, известные специалистам в данной области техники (Marx, Tidwell - J. Org. Chem. 49,788 (1984, или же спирт (13) растворяют в соответствующем органическом растворителе, например метиленхлориде, раствор охлаждают во влажной ледяной ванной ацетона и обрабатывают 2,5-3,0 эквивалентами диметилсульфоксида. После перемешивания в течение приблизительно 30 мин реакционную смесь затем обрабатывают приблизительно 1,8 эквивалентами Р 2 О 5. Реакционную смесь перемешивают в течение приблизительно 3 ч и затем предпочтительно обрабатывают около 30 мин приблизительно 3,5 эквивалентами подходящего амина, например триэтиламином. Затем охлаждающую ванну удаляют, а реакционную смесь перемешивают в течение приблизительно 8-16 ч. Затем кетон (1) изолируют стандартными методами экстракции, известными в данной области техники. В схеме 4, стадия С, кетон (1) обрабатывают подходящим основанием, затем добавляют алкен (15),где X обозначает соответствующую уходящую группу для получения соединения (14). Например, кетон(1) объединяют с избытком алкена (15) в соответствующем органическом растворителе, например тетрагидрофуране, и охлаждают влажной ледяной ванной ацетона. Примеры соответствующих уходящих групп включают Cl, Br, I, тозилат, мезилат, и т.п. Предпочтительные остаточные группы включают Cl иBr. Добавляют приблизительно 1,1 эквивалента соответствующего основания и перемешивают реакционную смесь в течение приблизительно 2 ч при комнатной температуре. Примеры соответствующих оснований включают трет-бутилат калия, гидрид натрия, NaN(Si(CH3)3)2, литийдиизопропиламид,KN(Si(CH3)3)2, NaNH2, этилат натрия, метилат натрия и т.п. Трет-бутилат калия является предпочтительным подходящим основанием. Затем реакцию гасят водной кислотой, а соединение (14) изолируют обычной рабочей методикой. В схеме 4, стадия D, соединение (14) обрабатывают соответствующим окислителем для получения альдегида (3). Альдегид (3) также получают в схеме 1. Примеры соответствующих окислителей включают озон, NaIO4/катализатор осмия, и т.п. Озон является предпочтительным окислителем. Примеры соответствующих окислительных реактивов и условий описаны J. March, "Advanced Organic Chemistry: Reactions, Mechanisms, and Strukture", 2nd edition,McGraw - Hill, с. 1090 -1096 (1977). Например, соединение (14) растворяют в соответствующем органическом растворителе, таком как метанол, добавляют небольшое количество Судан III и раствор охлаждают приблизительно до -20 С. Озон кипит в растворе в течение приблизительно 4 ч, до тех пор, пока розовый цвет не изменится на- 12007503 светло-желтый цвет. Затем добавляют восстановитель, например Me2S или трибутилфосфин. Концентрацией получают промежуточный диметил ацеталь альдегида (3). Этот диметил ацеталь сразу гидролизуют при стандартных кислотных условиях для получения альдегида (3). Кроме того, прямой кислотной обработкой неочищенной реакционной смеси получают альдегид (3). Помимо этого, альдегид (3) можно получать непосредственно озонолизом (14) в неацетальном полученном растворителе, например хлористом метилене. В схеме 4, стадия Е, альдегид (3) восстановительно аминируют при условиях, аналогичных выше описанным в схеме 3, стадия D, для получения соединения (5). Соединение 5 также получают в схеме I. На схеме 5 представлен альтернативный синтез получения кетона (5). Все заместители, если не отмечено иначе, такие, как указаны ранее. Реактивы и исходные вещества доступны специалисту в данной области техники. В схеме 5, стадия А, альдегид (3) конденсируют с диазоциклоалканом (4) при стандартных условиях, известных в данной области техники, для получения энамина (15). Например, приблизительно 1,05 эквивалента альдегида (3), растворенного в соответствующем органическом растворителе, например изопропиловом эфире уксусной кислоты или изопропаноле, добавляют к чистому диазоциклоалкану (4),свободному основанию. Дополнительный органический растворитель добавляют для получения жидкого раствора, и реакционную смесь перемешивают в течение приблизительно 1-2 ч. Затем изолируют энамин(15) стандартными методами, например улавливание фильтрацией. В схеме 5, стадия В, энамин (15) гидрируют при условиях, известных специалисту в данной области техники для получения соединения (5). Например, энамин (15) добавляют к соответствующему органическому растворителю, такому, как изопропиловый спирт, и каталитическим количеством 5% палладия на углероде в колбе Парра. Смесь помещают под давлением 50 фунтов на квадратный дюйм (344850 паскалей) водорода и взбалтывают в течение приблизительно 2 дней при комнатной температуре. Затем жидкий раствор фильтруют для удаления катализатора, а фильтрат концентрируют для получения соединения (5). Каким бы способом они не были бы получены, кетоинтермедиаты (5) могут быть преобразованы в соответствующие конечные соединения формулы I путем реакции с восстановителями, в частности теми,из которых получают анионы водорода, например борогидрид натрия или DIBAL-H. Такие кетоинтермедиаты (5), восстановленные без предыдущего растворения, в целом генерируют смесь диастереоизомеров, где пара (RS, SR) является количественно преобладающей над (RR, SS) парой. Затем (RS, SR) пару выделяют колоночной хроматографией на силикагели и растворяют в отдельных(RS) и (SR) энантиомерах, например, хроматографией на хиральной стационарной фазе или другими способами, известными специалистам в данной области техники. Кроме того, рацемические кетоны (5) можно растворять на его два энантиомера с помощью известных способов, a (R)-5 затем подвергают восстановлению, получая предпочтительно (S, R) энантиомер,который является предпочтительным согласно настоящему изобретению, и его можно легко очищать физическими способами. Стереохимия В схеме 1 соединения I получают в син /анти смеси диастереомеров при соотношении в зависимости от используемого условия реакции. Диастереомеры можно разделять обычными методами, известными специалистам в данной области техники, включая фракционную кристаллизацию оснований или их солей, либо хроматографическими методами, например жидкостной хроматографией (LC) или флэшхроматографией. Для обоих диастереомеров (+) энантиомер формулы Iа можно отделять от (-) энантиомера, используя технологии и методики, известные в данной области техники, как описано J. Jacques, etal., "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc, 1981. Например, хиральную хроматографию с соответствующим органическим растворителем, таким, как этанол/ацетонитрил и упаковка Chiralpak AD, 20 мкм можно также использовать для эффективного разделения энантиомеров. Свободные основания формулы I, их диастереомеры или энантиомеры можно преобразовывать в соответствующие фармацевтически приемлемые соли при стандартных условиях, известных в данной области техники. Например, свободное основание формулы I растворяют в соответствующем органическом растворителе, к примеру в метаноле, обрабатывают одним эквивалентом малеиновой или щавелевой кислоты, например, одним или двумя эквивалентами соляной кислоты или метаносульфоновой кислоты, и затем концентрируют под вакуумом для получения соответствующей фармацевтически прием- 13007503 лемой соли. Затем остаток можно очищать перекристаллизацией из соответствующего органического растворителя или смеси органического растворителя, например метанол/диэтилового эфира.N - оксиды соединений формулы I можно синтезировать в соответствии с обычными методиками окисления, известными специалистам в данной области техники. Методика окисления, описаннаяP.Brougham et al. (Synthesis, 1015 - 1017, 1987), позволяет дифференцировать два атома азота кольца пиперазина для получения N - оксида, а также и N, N'-диоксида. Следующие примеры представляют типичный синтез соединений формулы I, как в общем описано выше. Эти примеры только иллюстрируют, но никоим образом не ограничивают изобретение. Реактивы и исходные вещества легко доступны для обычного специалиста в данной области техники. Пример 1. 1-[(SR-RS)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (диастереомер с верхней тонкослойной хроматографией Rf) (Пр. 1) 1-[(3S,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (Пр. 1 Х) 1-[(3R,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (Пр. 1Y) Пример 2. 1-[(RR-SS)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (диастереомер с низкой тонкослойной хроматографией Rf) (Пр. 2) 1-[(3R,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (Пр. 2 Х) 1-[(3S,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (Пр. 2Y) Циклогексил 2-фторбензил кетон (Соединение 1 а) К смеси 36 мл 2-фторбензилцинк хлорида (0,5 М раствор в тетрагидрофуране) и 0,008 г дихлоробис(трифенилфосфин)палладия (II), перемешиваемой при 0 С, добавляли по каплям через шприц 2,14 мл циклогексанкарбонил хлорида. После этого реакционную смесь перемешивали при комнатной температуре в течение 4 ч, гасили водным насыщенным раствором хлорида аммония (25 мл), экстрагировали с 20 мл EtOAc, который высушивали (Na2SO4) и выпаривали до степени сухости в вакууме, получая 3,52 г указанного неочищенного соединения, которое можно использовать в следующей стадии без дальнейшей очистки. 1 Н - NMR (CDCl3, ): 1,10-2,05 (m, 10 Н), 2,47 (tt, 1 Н), 3,77 (s, 2 Н), 6,97 - 7,32 (m, 4 Н) 4-Циклогексил-4-оксо-3-(2-фторфенил)-бутиральдегид диэтилацеталь (Соединение 1b) Раствор 5,02 г соединения 1 а в 136 мл толуола нагревали с флегмой, восстанавливая 35 мл толуола перегонкой для удаления воды. Затем добавляли 3,18 г трет-бутоксида калия и продолжали перемешивание с флегмой в течение 30 мин; реакционную смесь охлаждали до 80 С и добавляли 4,27 мл 2 бромацетальдегид диэтил ацеталя. Через 18 ч при дефлегмировании реакционную смесь охлаждали при комнатной температуре, гасили водным насыщенным раствором хлорида аммония (30 мл) и экстрагировали с 30 мл EtOAc. Экстракты высушивали (Na2SO4) и выпаривали до степени сухости в вакууме, получая неочищенное вещество, которое очищали флэш-хроматографией (петролейный эфир - EtOAc 92,5:7,5), получая 2,97 г указанного чистого вещества. 1 Н - NMR (CDCl3, ): 1,00-2,10 (m, 17H), 2,20-2,52 (m, 2H), 3,30-3,72 (m, 4H), 4,25-4,45 (m, 2H), 6,907,35 (m, 4H) 4-Циклогексил-4-оксо-3-(2-фторфенил)бутиральдегид (Соединение 1 с) Смесь 1,12 г соединения 1b, 9 мл водной 50% трифторуксусной кислоты и 18 мл СН 2 Сl2 перемешивали в течение 2 ч при комнатной температуре, затем разбавляли 10 мл СН 2 Сl2. Органический слой отделяли, промывали соляным раствором (2 х 15 мл), высушивали (Na2SO4) и выпаривали до степени сухости в вакууме для получения неочищенного вещества (0.88 г), используя в следующей стадии без дальнейшей очистки. 1 Н - NMR (CDCl3, ): 0,90-2,10 (m, 10 Н), 2,25-2,70 (m, 2H), 3,12-3,52 (m, 1H), 4,60-4,80 (m, 1H), 6,957,40 (m, 4H), 9,75 (s, 1H) 1-[4-циклогексил-4-оксо-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (Соединение 1d) Смесь 0.88 г соединения 1 с, 0.84 г 1-(2-метоксифенил)пиперазин HCl, 1,06 г триацетоксиборгидрида натрия и 33 мл CH2Cl2 перемешивали при комнатной температуре в течение 1 ч, выдерживали в течение ночи, подщелачивали 20% водным Na2 СО 3 Органический слой отделяли, промывали соляным раствором (2 х 30 мл), высушивали (Na2SO4) и выпаривали до степени сухости в вакууме для получения неочищенного вещества (1,46 г), которое использовали в следующей стадии без дальнейшей очистки. Образец очищали флэш-хроматографией (петролейный эфир-EtOAc 6:4), получая чистый образец. 1 Н - NMR (CDCl3, ): 1,05-2,00 (m, 11H), 2,20-2,44 (m, 4H), 2,45-2,72 (m, 4H), 2,90-3,20 (m, 4H), 3,85(SR,RS)-1-Циклогексил-4-[4-(2-метоксифенил)пиперазин-1-ил]-2-(2-фторфенил)бутан-1-ол (диастереомер с верхней тонкослойной хроматографией Rf) и(RR,SS)-1-Циклогексил-4-[4-(2-метоксифенил)пиперазин 1-ил]-2-(2-фторфенил)бутан-1-ол (диастереомер с низкой тонкослойной хроматографией Rf) К раствору 1,46 г соединения 1d в 33 мл метанола, перемешиваемого при 0 С, добавляли 0,19 г борогидрида натрия и перемешивали смесь при комнатной температуре в течение 4 ч. Растворитель выпа- 14007503 ривали, а неочищенный продукт реакции поглощали водой и экстрагировали с EtOAc. Органический слой отделяли, промывали соляным раствором (2x15 мл), высушивали (Na2SO4) и выпаривали до степени сухости в вакууме для получения неочищенного вещества, которое очищали последовательной флэшхроматографией (петролейный эфир- EtOAc -2 N аммиак в метаноле 75:25:2; петролейный эфир-EtOAc2N аммиак в метаноле 80:20:2), получая 0.82 г соединения примера 1 (верхняя тонкослойная хроматография Rf; элюент: петролейный эфир-EtOAc-2N аммиак в метаноле 70:30:2), затем 0,062 г соединения примера 2 (нижняя тонкослойная хроматография Rf; тот же элюент). Пр. 1: 1 Н - NMR (CDCl3, ): 0,80-1,40 (m, 6H), 1,50-1,82 (m, 4H), 1,85-2,10 (m, 3 Н), 2,21-2,45 (m, 2H),2,52-2,85 (m, 4H), 2,98-3,26 (m, 4H), 3,28-3,42 (m, 1H), 3,50-3,60 (m, 1 Н), 3,85 (s, 3 Н), 6,80-7,30 (m, 7 Н),7,62-7,80 (m, 1 Н); О пик не обнаруживаем Пр. 2: 1 Н - NMR (CDCl3, ): 0,75-2,00 (m, 13 Н), 2,00-2,30 (m, 1 Н), 2,31-2,55 (m, 2 Н), 2,56-2,95 (m,4 Н), 3,00-3,30 (m, 4 Н и ОН), 3,60 (dd, 1H), 3,85 (s, 3 Н), 6,80-7,38 (m, 8 Н) 1-[(3S,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (Пример 1 Х) Это соединение получали хиральной колоночной хроматографией на соединении из примера 1, используя Chiralpak AD (0.46x25 см), элюируя с н-гексан-EtOH 95:5 (поток = 0.5 мл/мин; детектор УФ 247 нм). 1-[(3R,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (Пример 1Y) Это соединение получали хиральной колоночной хроматографией на соединении из примера 1, используя Chiralpak AD (0.46x25 см), элюируя с н-гексан-этанолом 95:5 (поток = 0.5 мл/мин; детектор УФ 247 нм). 1-[(3R,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (Пример 2 Х) Это соединение получали хиральной колоночной хроматографией на соединении из примера 2, используя Chiralpak AD (2x25 см), элюируя с н-гексан-этанолом 85:15 (поток = 8 мл/мин; детектор УФ 254 нм). 1-[(3S,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин (Пример 2Y) Это соединение получали хиральной колоночной хроматографией на соединении из примера 2, используя Chiralpak AD (2x25 см), элюируя с н-гексан-этанолом 85:15 (поток = 8 мл/мин; детектор УФ 254 нм). Абсолютная стереохимия соединений 1 Х и 2Y, в форме их солей с бромидом водорода, была определена дифракцией рентгеновских лучей монокристалла следующим образом. Эксперимент дифракции рентгеновских лучей монокристалла: Монокристалл в форме игл был выбран для анализа дифракции рентгеновских лучей и установлен на стекловолокне. Данные собирали на детекторе рентгеновских лучей фирмы Rigaku Rapid с видеоплатой в форме цилиндра с отверстием = 45.0 х 25.6 см. Все контролировали персональным компьютером на базе Windows 2000, с программным обеспечением Rapid Auto, версия 1.06 (Rigaku, 2000), при низкой температуре (-120 К), с Micromax-002 микро- софокусными зеркалами СuК излучение [(СuК) =1.5405]. Индексацию проводили от трех 3 рамок колебаний, которые были выставлены на 360 с. Все отражения измеряли в пяти группах изображения с шестью рамками в каждой группе; время экспозиции составляло 160 с на градус. Среди них пять групп изображений были под углами phi = 0, 90, 180, 270 с chi=50 и phi=0 с chi=0 всех рамок были дельта омега = 30, и которые приводили к 2 mах = 136.3. Интервал образец/детектора составлял 12,74 см. Программа восстановления данных, Rapid Auto версия 1.06 (Rigaku, 2000), определила, что Laue группой была - 1, а общее количество 7,986 отражений было объединено для структуры раствора и очисток. Результаты по монокристаллу Структуру растворяли прямыми способами, используя SIR92 (Altomare et al. 1994). Все вычисления проводили, используя CrystalStructure 3,0 (MSC/Rigaku, 2002; Watkin et al., 1996, Carruthers и Watkin,1979) кристаллографический пакет программ. В испытуемом растворе было 38 неводородных атомов в асимметричной единице. Очистка наименьших квадратов включала координаты всех неводородных атомов и анизотропные тепловые параметры. Заключительный цикл полных - матричных квадратов наименьшей очистки на F основывался на 6,297 отражениях с l 3(I), конвергентных с факторами совпадения: R=0,071, S=2,224, Rw=0,073. Абсолютную конфигурацию определяли, используя рассчитанный Flack x параметр, который составлял 0,00 сesd = 0,04. Ожидаемые значения составляют 0,0 (в пределах 3 esd's) для правильной и +1,0 для инвертированной абсолютной структуры.Watkin, D.J., Prout, C.K. Carruthers, J.R. and Betteridge, P.W., CRYSTALS Issue 10, Chemical Crystallography Laboratory, Oxford, UK. Пример 3. 1-[(RS,SR)-4-Циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин (диастереомер с верхней тонкослойной хроматографией Rf) Пример 4. 1-[(RR,SS)-4-Циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин (диастереомер с низкой тонкослойной хроматографией Rf) 1-[4-Циклогексил-4-оксо-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин (Соединение 3 а) Указанное соединение получали, следуя методике, описанной для соединения 1d, но используя 1(2,2,2-трифторэтоксифенил)пиперазин HCl вместо 1-(2-метоксифенил)пиперазин HCl. После очистки флэш-хроматографией (петролейный эфир - EtOAc 7:3), получали указанное в заголовке соединение(51%). 1 Н - NMR (CDCl3, ): 1,00-1,85 (m, 10 Н), 1,86-2,05 (m, 1H), 2,20-2,44 (m, 4H), 2,45-2,70 (m, 4H), 2,953,18 (m, 4H), 4,25-4,60 (m, 3 Н), 6,850-7,30 (m, 8H) 1-[(RS,SR)-4-Циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин (диастереомер с верхней тонкослойной хроматографией Rf) и 1-[(RR,SS)-4-Циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин (диастереомер с низкой тонкослойной хроматографией Rf) Вышеуказанные соединения синтезировали, используя методику, описанную для соединения из примеров 1 и 2, но используя соединение 3 а как исходное вещество вместо соединения 1d. После очистки флэш-хроматографией (петролейный эфир - EtOAc - 2N аммиак в метаноле 60:40:2) получали соединение из примера 3 в виде диастереомера, имеющего верхний Rf (79%); фракции, содержащие соединение из примера 4 в виде диастереомера, имеющего низкий Rf, проходили повторную очистку флэшхроматографией (петролейный эфир - EtOAc - 2N аммиак в метаноле 75:25:1), для получения чистого вещества (35%). Пр. 3: 1 Н - NMR (CDCl3, ): 0,80-1,40 (m, 6 Н), 1,45-1,80 (m, 4 Н), 1,85-2,10 (m, 3 Н), 2,21-2,45 (m, 2 Н),2,50-2,75 (m, 4 Н), 2,95-3,26 (m, 4 Н), 3,30-3,42 (m, 1 Н), 3,50-3,60 (m, 1 Н), 3,70-4,30 (br, 1 Н, О), 4,38 (q, 2 Н),6,85-7,30 (m, 7 Н), 7,65-7,75 (m, 1 Н) Пр. 4: 1 Н - NMR (CDCl3, ): 0,75-1,95 (m, 12 Н), 2,05-2,90 (m, 7 Н и ОН), 3,00-3,30 (m, 5 Н), 3,65 (d,1 Н), 4,38 (q, 2 Н), 6,85-7,40 (m, 8 Н) Пример 5 (Сравнительный). 1-[(RS,SR)-4-Циклогексил-4-гидрокси-3-фенилбутил]-4-(2-метоксифенил)пиперазин Бензилциклогексил кетон (Соединение 5 а) К раствору 11 мл 0,5 М раствора бромида цинка бензила в безводном тетрагидрофуране добавляли при 0 С 5 мг бис-трифенилфосфинпалладий дихлорида и 0,66 мл циклогексанкарбонил хлорида. Смесь перемешивали при комнатной температуре в течение 1,5 ч, гасили насыщенным раствором хлористого аммония и экстрагировали с этилацетатом. Собранные органические слои промывали водой, высушивали(Na2SO4), а растворитель выпаривали под вакуумом. Неочищенное вещество очищали флэшхроматографией, элюируя с петролейным эфиром - EtOAc 95:5, для получения 0,78 г (52%) указанного в заголовке соединения. 1 Н - NMR (CDCl3, ): 1,09-1,92 (m, 10 Н), 2,32-2,53 (m, 1H), 3,72 (s, 2H), 7,12-7,39 (m, 5H) 4-Циклогексил-4-оксо-3-фенилбутиральдегид диэтилацеталь (Соединение 5b) К раствору 1,78 г соединения 5 а в 30 мл безводного диметилформамида добавляли при комнатной температуре 0.37 г дисперсии масла 60% гидрида натрия и смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 1.46 мл 2-бромацетальдегид диэтилацеталя, и смесь перемешивали при комнатной температуре в течение 1 ч и при 80 С в течение 1 ч, охлаждали до комнатной температуры,гасили водой и экстрагировали EtOAc. Собранные органические слои промывали водой, высушивали(Na2SO4), и растворитель выпаривали под вакуумом. Неочищенное вещество очищали флэш-хроматографией, элюируя петролейным эфиром - EtOAc 95:5 для получения 1.17 г (42 %) вышеуказанного соединения. Н - NMR (CDCl3, ): 1,01-1,98 (m, 17H), 2,28-2,48 (m, 2H), 3,30-3,71 (m, 4H), 3,98 (t, 1H), 4,25 (t,1H), 7,12-7,39 (m, 5H) 4-Циклогексил-4-оксо-3-фенилбутиральдегид (Соединение 5 с) Раствор 1.17 г соединения 5b в 10 мл ацетона и 22,1 мл 2 Н HCl перемешивали при комнатной температуре в течение 5 ч. После выдерживания смеси в течение ночи, водный слой экстрагировали EtOAc. Собранные органические слои промывали водой, высушивали (Na2SO4), а растворитель выпаривали под вакуумом для получения 0.84 г (100%) вышеуказанного соединения, сразу используемого без дальнейшей очистки. 1-(4-Циклогексил-4-оксо-3-фенилбутил)-4-(2-метоксифенил)пиперазин (Соединение 5d) К раствору 0,84 г соединения 5 с и 1.19 г 1-(2-метоксифенил)пиперазина в 30 мл дихлорметана добавляли 1,48 г триацетоксиборгидрида натрия и 0,98 мл уксусной кислоты и перемешивали полученную смесь при комнатной температуре в течение 5 ч. После выдерживания смеси в течение ночи, органический слой промывали избытком 1 М NaOH, затем - водой, высушивали (Na2SO4), и выпаривали растворитель под вакуумом. Неочищенное вещество очищали флэш-хроматографией, элюируя петролейным эфиром-EtOAc 7:3 для получения 1.45 г (99%) указанного в заголовке соединения. 1 Н - NMR (CDCl3, ): 1,01-2,05 (m, 11H), 2,20-3,30 (m, 12H), 3,82 (s, 3H), 3,91-4,02 (m, 1H), 6,75-7,08(m, 4H), 7,12-7,39 (m, 5H) 1-[(RS,SR)-4-Циклогексил-4-гидрокси-3-фенилбутил]-4-(2-метоксифенил)пиперазин В раствор 1,21 г соединения 5d в 60 мл дихлорметана при -78 С по каплям добавляли 11,5 мл 1 М раствора диизобутилалюминий гидрида (DIBAL-H) в толуоле. Смесь перемешивали при -78 С в течение 1 ч, гасили при -60 С насыщенным водным раствором NH4Cl и экстрагировали хлороформом. Собранные органические слои промывали водой, высушивали (Na2SO4), и выпаривали растворитель под вакуумом. Неочищенное вещество очищали флэш-хроматографией, элюируя петролейным эфиром - EtOAc-2H аммиак в метаноле 30:70:2, для получения 1,06 г (80%) указанного в заголовке соединения. 1 Н - NMR (CDCl3, ):1,01-1,38 (m, 6H), 1,51-1,78 (m, 4H), 1,86-2,02 (m, 3H), 2,22-2,38 (m, 3 Н), 2,522,78 (m, 4H), 2,75-2,95 (m, 1H), 3,02-3,20 (m, 4H), 3,48 (t, 1H), 3,82 (s, ЗН), 6,80-7,03 (m, 4H), 7,10-7,38 (m,5H). Пример 6: связывание радиолиганда с различными рецепторами 6 А. Рекомбинантные 5-НТ 1A-рецепторы человека Способ: Геномный клон, кодирующий 5 НТ 1 А - серотонергический рецептор человека, устойчиво трансфектировали в линии клеток человека (HeLa). HeLa клетки выращивали как мономолекулярные слои в модифицированной по способу Дульбекко среде Игла, (DMEM), содержащей 10% эмбриональную бычью сыворотку, гентамицин и 5% диоксид углерода (0,1 мг/мл), при 37 С. Клетки отделяли из колбы для роста при 95% слиянии соскобленных клеток и лизировали в холодном 5 мм Трисе и 5 мМ буфере этилендиаминтетрауксусной кислоты (рН 7,4). Гомогенаты центрифугировали при 40000 х г х 20 мин, и гранулы ресуспендировали в небольшом объеме холодного 5 мМ Триса и 5 мМ буфера этилендиаминтетрауксусной кислоты (рН 7,4), немедленно замораживали и сохраняли при -70 С до использования.[3H]8-ОН-DРАТ связывание: в день эксперимента мембраны клеток ресуспендировали в инкубационном буфере: 50 мМ Трис HCl (рН 7.4), 2.5 мМ MgCl2, 10 мМ паргилина (Fargin et al., Nature 335, 358360, 1988). Мембраны инкубировали в конечном объеме 1 мл в течение 30 мин при 30 С с 1 нм [3 Н] 8 ОН-DPAT, при отсутствии или присутствии тестируемых соединений. Неспецифическое связывание определяли в присутствии 10 мкМ 5 - НТ. Инкубацию останавливали добавлением холодного Трис - HCl буфера и быстрой фильтрацией через -Whatman - GF/B, предварительно обработанный 0.2 % - полиэтиленимином, или фильтр SchleicherSchuell - GF52. Сродство тестируемых соединений оценивали как ингибирование специфического связывания радиолиганда с 5 - HT1A рецепторами (IC50), используя нелинейную программу для вычерчивания эмпирической кривой (De Lean et al., Am. J. Physiol. 235, E97 - E102 (1978). Значение IC50 преобразовывали к константе сродства (Ki) уравнением Cheng et al., Biochem. Pharmacol. 22, 3099 - 3108 (1973.[35S]GTPS связывание: в день эксперимента мембраны клеток от клеток HeLa, трансфектируемых с клонированными 5-НТ 1Aрецепторами человека, повторно суспензировали в буфере рН 7.4, содержащем 20 мм HEPES, 3 мМ MgCl2 и 120 мм NaCl (Stanton, J. A.; Beer, M. С. Eur. J. Pharmacol. 320, 267-275,1997). Мембраны инкубировали с 10 мкМ GDP и понижающимися концентрациями тестируемого лекарства (от 100 мкМ до 0,1 нм) или понижающимися концентрациями 5-НТ (от 100 мкМ до 0.1 нм, соответствующая кривая) в течение 20 мин при 30C в окончательном объеме около 0,25 мл. [35S]GTPS (200250 пМ в 10 мкл), добавляли к образцам и инкубировали в течение следующих 30 мин при 30 С. Неспецифическое связывание определяли в присутствии 10 мкМ GTPS. Инкубацию прекращали добавлением ледяного буфера HEPES и быстрой фильтрацией на Unifilter GF/C фильтрах, используя сборщик клетокFiltermate (Packard). Фильтры промывали четыре раза общим количеством 1,2 мл того же самого буфера. Радиоактивность считали с помощью жидкостной сцинцилляционной спектрометрии с эффективностью 90% (TopCount Packard). Стимуляцию [35S]GTPS связывания, индуцируемого тестируемыми соедине- 17007503 ниями, выражали как % увеличение связывания вышеописанного основного значения, наблюдаемую с 5 НТ максимальную стимуляцию принимали за 100%. Кривую концентрация-ответ агонистической активности анализировали в соответствии с нелинейной эмпирической программой (De Lean et al., Am. J.Physiol. 235, E97-E102, 1978). Стимуляция [35S]GTPS связывания представляет функциональную корреляцию связывания агониста соединения с 5-HT1A рецепторами. Стимуляцию, индуцированную эндогенным лигандом 5-НТ, считают максимально достигаемой стимуляцией. Соединения, стимулирующие [35S]GTPS связывание на более низком уровне, считают частичными агонистами. Соединения, которые не стимулируют[35S]GTPS связывание, считают нейтральными антагонистами. Результаты Результаты, представленные в табл. 1, показывают, что соединения тестируемого изобретения имеют высокое сродство в 5-HT1A рецепторам. Соединения пр. 1, пр. 1Y, пр. 2 и пр. 2 Х были более эффективными, чем пр. 5 (статистическое значение р 0.01). По отношению к [35S]GTPS связывания, соединения пр. 1 Х и 2Y были частичными агонистами, вызывающими стимуляцию [35S]GTPS связывания. Другие соединения не стимулировали [35S]GTPS связывание, проявляя себя, как нейтральные антагонисты. Таблица 1. Связывание с 5-HT1A рецепторами 6 В. Рекомбинантные подтипы 1-адренорецепторов человека Способ: Связывание с клонированными подтипами 1-адренорецепторами человека проводили в мембранах из СНО клеток (клетки яичника китайского хомяка), трансфектируемых электропорацией с ДНК экспрессией гена, кодирующего каждый подтип 1-адренорецептора. Клонирование и стабильную экспрессию 1-адренорецептора человека проводили, как было описано ранее (Testa et al. Pharmacol. Comm., 6: 79-86, 1995).CHO клетки выращивали во взвешенном состоянии в модифицированной по способу Дульбекко среде Игла, добавляли 10% сыворотку плода жеребенка и гентамицин (50 мкг/мл), при 37 С в 7% СО 2 в увлажняемом инкубаторе. Клетки собирали центрифугированием, растворяли в 5 мМ ледяного Трис и 5 мМ буфера ЭДТА (этилендиаминтетрауксусная кислота) (рН 7.4) и мягко гомогенизировали. Гомогенаты центрифугировали при 40000 х г х 20 мин, и гранулы повторно суспендировали в небольшом объеме 5 мМ ледяного Трис и 5 мМ буфера ЭДТА (этилендиаминтетрауксусная кислота) (рН 7.4) и сразу замораживали и хранили при -80 С до использования. Мембраны СНО клеток повторно суспендировали и инкубировали в 50 мм Трис, рН 7.4, с 0.2 нм[3 Н] празосина, в окончательном объеме 1,02 мл в течение 30 мин при 25 С, при отсутствии или присутствии конкурирующего лекарства. Неспецифическое связывание определяли в присутствии 10 мкМ фентоламина. Инкубацию останавливали быстрой фильтрацией через обработанные 0,2% полиэтиленимином фильтры Schleicher и Schuell GF52, используя сборщик клеток Tomtec (PerkinElmer). Фильтры промывали 3 х 1 мл ледяного Трис буфера, высушивали и измеряли радиоактивность в жидкостном сцинтилляционном счетчике Betaplate (Wallac). Ингибирование неспецифического связывания у соединений анализировали, чтобы оценить значение IC50, используя нелинейную программу Allfit для вычерчивания эмпирической кривой (De Lean et al.,Am. J. Physiol. 235, E97-E102 (1978). Значение IC50 преобразовывали к константе сродства (Ki) уравнением Cheng и Prusoff (Biochem. Pharmacol. 22, 3099 - 3108 (1973.- 18007503 Результаты Результаты, представленные в табл. 2, показывают, что тестируемые соединения согласно изобретению имеют различное сродство для подтипов 1-адренорецепторов, но особенно эффективны в 1dподтипе. Селективность (определенная как отношение между значениями Ki в подтипах 1 адренорецептора и значением Ki в 5-HT1A рецепторе) соединений согласно изобретению была в целом лучше, чем у соединения, описанного в известном уровне техники (Пр. 5). Таблица 2. Сродство к связыванию (Ki, нм) для подтипов 1-адренорецепторов и селективности против 5-HT1A рецепторов 6 С. Рекомбинанты D3 рецепторов дофамина крыс Способ: Использовали клонированные D3 рецепторы дофамина крыс, постоянно экспрессированные в СНО клетках (Chio et al. Mol. Pharmacol. 45, 51-60, 1994). Мембраны получали механическим разрушением гранул клеток в 50 мМ ледяного Трис, 5 мМ ЭДТА, 5 мМ EGTA, рН 7,4, затем центрифугировали при низкой (1000g), средней (20000g) и высокой (80000g) степенях скорости. Использовали конкурирующий меченный лиганд связывания экспериментов 11 концентраций лекарственного средства, управляемых в двойном экземпляре в формате анализа эффекта близости сцинцилляций (SPA). Использовали меченный лиганд [3 Н]-7-ОН-DPAT (154 Кю/ммоль). Неспецифическое связывание (75-95% общего количества) определяли с помощью холодного галоперидола, добавленного в избытке (3 мкМ). Общее связывание определяли с помощью буфера: 20 мМ HEPES, 10 мМ MgSO4 150 Мм NaCl, 1 мМ ЭДТА (рН 7.4). Связывающие смеси получали в гибких, 96-клеточных, пластинах Wallac Micro-Beta путем добавления 11 мкл растворенного лекарственного средства, 11 мкл меченного лиганда, и 178 мкл суспензии мембран/шариков SPA (100 мг покрытых WGA SPA шариков, инкубированных с 5-15 мкг белок/пластина в 10 мл связывающего буфера в течение 30 мин при комнатной температуре, далее проводили медленное центрифугирование и повторное суспензирование в 2 мл связывающего буфера). После запечатывания и инкубации при комнатной температуре в течение 1 ч пластины считали в сцинтилляционном счетчикеIC50 значения из обоих способов анализа оценивали, подгоняя данные к модели конкуренции с одним сайтом: Y = T/(1 + 10log(Х) - log(IC50, где Y обозначает специфическую СРМ связь при концентрации X и T обозначает специфическую СРМ связь в отсутствии конкурента. Константы ингибирования (Ki) рассчитывали, используя уравнение Cheng-Prushoff (Biochem. Pharmacol. 22: 3099-3108, 1973). Результаты Результаты, представленные в табл. 3, показывают, что соединения, тестируемые согласно изобретению, имеют сродство для D3 подтипа дофаминергического рецептора. Селективность (вычисленная как отношение между значениями Ki в D3-дофаминергическом подтипе и значение Ki в 5-HT1A рецепторе) соединений согласно изобретению была лучшей, чем селективность соединения, описанного в известном уровне техники (Пр. 5). Таблица 3. Сродство к связыванию для D3 рецепторов дофамина и селективность против 5-HT1A рецепторов Пример 67. Воздействия на ритмичные сокращения при опорожнении мочевого пузыря, вызываемые наполнением мочевого пузыря у анестезированных крыс А. Способ: Использовали самок крыс линии Sprague - Dawley весом 225 - 275 г (Crl: CD (SD) IGS BR, CharlesRiver Italia). Животных размещали со свободным доступом к пище и воде и содержали при искусственном 12-часовом чередовании светлого и темного циклов при 22-24 С в течение по меньшей мере одной недели, помимо времени эксперимента. Активность сокращений, вызываемых при опорожнении мочевого пузыря, оценивали согласно способу Dray (Dray J., Pharmacol. Methods, 13:157, 1985), с некоторыми модификациями по Guarneri (Guarneri, Pharmacol. Res. 27:173, 1993). Вкратце, крыс анестезировали подкожной инъекцией 1,25 г/кг (5 мл/кг) уретана, затем в мочевой пузырь вводили катетер через уретру, используя тюбинг из РЕ 50 полиэтилена, заполненный физиологическим раствором. Катетер закрепляли в месте с лигатурой вокруг внешнего мочеиспускательного отверстия и связывали с обычными датчиками- 19007503 давления (Statham P23 ID/P23 XL). Внутрипузырное давление непрерывно отображалось на регистрирующем устройстве (Battaglia Rangoni KB 135 с DCI/TI усилителем). Затем мочевой пузырь заполняли через регистрирующий катетер возрастающими объемами теплого (37 С) физиологического раствора до наступления рефлекторных сокращений, опорожняющих мочевой пузырь (обычно 0,8-1,5 мл). Для внутривенной инъекции биоактивных соединений в яремную вену вставляли тюбинг из полиэтилена, заполненный физиологическим раствором. Из цистометрограммы оценивали количество сокращений, зарегистрированных за 15 мин до (основные значения) и после лечения, а также среднюю амплитуду этих сокращений (средняя высота пиков в мм рт.ст.). Поскольку большинство соединений быстро оказывали действие в начале и приводили к полному прекращению сокращений мочевого пузыря, то биоактивность оценивали, измеряя продолжительность покоя мочевого пузыря (то есть, продолжительность времени, в течение которого не происходили сокращения). Было также зарегистрировано число тестируемых животных, у которых наблюдали снижение количества сокращений более чем на 30% от наблюдаемых в основной период. Для сравнения эффективности тестируемых соединений для ингибирования сокращений, опорожняющих мочевой пузырь, вычисляли равномерные эффективные дозы, приводящие к исчезновению сокращений на 10 мин, ЭД 10 мин рассчитывали посредством линейной регрессии, используя метод наименьших квадратов. Экстраполируемые дозы, которые вызвали снижение числа сокращений более 30% у 50% крыс, подвергшихся лечению (ЭД 50), оценивали способом по Bliss (Bliss С. I., Quart J. Pharm. Pharmacol. 11, 192 - 216, 1938). В. Результаты Быстрое растяжение мочевого пузыря у анестезированных уретаном крыс вызывало ряд ритмичных сокращений, опорожняющих мочевой пузырь, характеристики которых описаны (Maggi et al., J. Pharmacol. Exp. Ther., 380:83, 1986; Maggi et al., J. Pharmacol. Exp. Ther., 230: 500, 1984). Частота этих сокращений зависит от сенсорной центростремительной силы рефлекса мочеиспускания и целостности центра мочеиспускания, в то время как их амплитуда зависит от функции рефлекса центробежной силы. В этой модели системы те соединения, которые воздействуют главным образом на центральную нервную систему (например, морфин) вызывают блокирование опорожняющих сокращений, в то время как лекарства, которые действуют на уровне непроизвольной мускулатуры, например оксобутинин, снижают амплитуду сокращений мочевого пузыря. Результаты, полученные после введения соединений известного уровня техники и соединений согласно изобретению, представлены в табл. 4. Соединения согласно изобретению превосходили исходные стандарты в блокировании индуцированных объемом ритмичных сокращений мочевого пузыря. Пр. 1Y был более эффективным, чем пр. 5,будучи его экстраполируемой дозой, вызывающей 10 мин исчезновения сокращений по крайней мере 2 нижних слоев. Кроме того, после введения 0,3 мг/кг пр. 1Y, сокращения мочевого пузыря исчезали в течение 24 мин, тогда как после введения 0,3 мг/кг пр. 5 это время составляло 14 мин. По сравнению с соединениями согласно изобретению, оксобутинин имел эффект снижения амплитуды сокращений в зависимости от дозы, со значением ЭД 50 (экстраполируемая доза, вызывающая 30% снижение амплитуды сокращений у 50% крыс, подвергшихся лечению) 240 мкг/кг. В этой дозировке,оксобутинин не вызывал блокаду сокращений мочевого пузыря, и это происходило вследствие определенного (антимускариновое) механизма действия, который отличается от такового у соединений согласно изобретению. Таблица 4. Воздействия на ритмичные сокращения, опорожняющие мочевой пузырь, после внутривенного введения Данные представляют значения ЭД 10min (экстраполируемая доза, вызывающая 10 мин исчезновения сокращений), ЭД 50(частота) значения (экстраполируемые дозы, вызывающие сокращение количества сокращений 30% у 50% крыс, подвергшихся лечению), и ЭД 50 (амплитуда) значения (экстраполируемые дозы, вызывающие 30% снижение амплитуды сокращений у 50% крыс, подвергшихся лечению). н.а. = не активный; нет значительного снижения высоты пиков- 20007503 Пример 8. Воздействие на цистометрические параметры у находящихся в сознании крыс после орального введения А. Способ: Использовали самцов крыс линии Sprague - Dawley [Crl: CD (SD) IGS BR] 300-400 г, поставляемых Charles River Italia. Животных размещали со свободным доступом к пище и воде и содержали при искусственном 12-часовом чередовании светлого/темного циклов при температуре 22-24 С, помимо времени эксперимента. Для определения количественных уродинамических параметров у находящихся в сознании крыс цистометрографические исследования проводили согласно методике, описанной (Guarneriet al., Pharmacol. Res. 24: 175, 1991). Крыс анестезировали внутрибрюшинным введением 3 мл/кг раствора Эквитензина (пентобарбитал 30 мг/кг и хлораль гидрат 125 мг/кг) и помещали в лежачее положение. В побритой и очищенной брюшной стенке делали разрез срединной линии длиной приблизительно 10 мм. Мочевой пузырь осторожно освобождали от прилегающих тканей, опорожняли и затем вводили канюлю через разрез в стенке мочевого пузыря, используя канюлю из полиэтилена (внутренний диаметр 0,58 мм, наружный диаметр 0,96 мм), которая была постоянно пришита шелковой нитью. Канюлю выводили через подкожный проход в залопаточную область, где ее связывали с пластмассовым переходным устройством для исключения риска удаления ее животным. Для тестирования лекарств использовали крыс спустя один день после имплантации. В день эксперимента крыс помещали в модифицированные клетки по Bollman, то есть ограничивающие клетки, которые были достаточно велики, чтобы позволить крысам принимать нормальное положение согнувшись, но и достаточно узкие, чтобы предотвратить переворачивание. После периода стабилизации приблизительно до 20 мин, свободный конец канюли мочевого пузыря связывали через Т образную трубу с датчиком давления (Statham P23XL) и с перистальтическим насосом (Gilson minipuls 2) для продолжительной инфузии теплого (37 С) физиологического раствора в мочевой пузырь, при постоянной скорости 0.1 мл/мин. Сигнал интралюминарного давления во время инфузии физиологического раствора в мочевой пузырь непрерывно регистрировали на многоканальном самописце (Rectigraph - 8 КБSan-ei с ВМ 614/2 усилителем от Biomedica Mangoni). Использовали цистометрограмму для оценки уродинамических параметров емкости мочевого пузыря (ЕМП) и давление мочеиспускания (ДМ). ЕМП (в мл) определяли как объем вводимого в мочевой пузырь физиологического раствора, необходимый, чтобы вызвать сокращение непроизвольной мускулатуры, сопровождаемого мочеиспусканием. ДМ (мм рт.ст.) определяли как максимальное внутрипузырное давление, вызванное сокращением в течение мочеиспускания. Оценивали основные значения ЕМП и ДМ как среднее значение, наблюдаемое в цистометрограммах, зарегистрированных в начальный период 30-60 мин. Во время определения основных ЕМП и ДМ инфузию прерывали, и тестируемые соединения вводили перорально через трубку в желудке. Инфузию мочевого пузыря возобновляли, и изменения в ЕМП и ДМ оценивали из средних значений,полученных в цистометрограммах, наблюдаемых в течение спустя 1, 2, 3, 4 и 5 ч после лечения. Соединения вводили в объеме 2 мл/кг, а группы контрольных животных получали то же количество носителя(0,5% метилцеллюлоза в воде) перорально. Результаты представлены на соответствующих чертежах, на которых фиг. 1 - изменения во времени ЕМП и ДМ у крыс после перорального введения носителя (круги) или 10.0 мг/кг соединения (1-циклогексил-4-[4-(2-метоксифенил)-1-пиперазинил]-2-(2-фторфенил)бутан 1-ола; верхняя тонкослойная хроматография Rf) примера 1 (квадраты). Данные представляют % изменения по сравнению с основным значением в разное время лечения, "n" = количество крыс/группа. Значение, показанное как Р(между курсами лечения: ANOVA CONTRAST VARIABLES) указывает, что наблюдалось различие между тенденцией в контрольной (носитель) группе и группе, получающей лечение. Звездочки ( = р 0.05,= р 0.01 и= р 0.001) указывают статистическую значимость между наблюдаемым значением в указанное время и базовым значением (во время лечения). Фиг. 2 - изменения во времени ЕМП и ДМ у крыс после перорального введения носителя (круги) или 3,0 мг/кг оксобутинина (квадраты). Данные представлены, как на фиг. 1. Статистический анализ Данные были выражены как среднее значениестандартная ошибка. Процентные изменения ЕМП и ДМ по сравнению с основными значениями, а такжезначения (различие в мл или мм рт.ст.) ЕМП и ДМ (ЕМП или ДМ во время "х" минус основного значения) также оценивали для каждой крысы/времени. На фигурах данные представлены как % изменения по сравнению с основным значением. Статистический анализ значений ЕМП и ДМ, а такжезначений проводили с помощью программного обеспечения S.A.S./STAT, версия 6.12. Наблюдаемые различия между носителем (контроль), и тестируемыми вариантами лечения оценивали назначения ЕМП и ДМ, тогда как различия между значениями в разное время по сравнению с основными значениями анализировали на первичных данных ЕМП и ДМ.- 21007503 Пример 9. Ингибирование стереотипии (ритмичных непроизвольных движений передних лап), вызываемое 8 - ОН - DPAT у крыс (постсинаптический антагонизм)A. Способ: Ингибирующее действие антагониста 5-НТ 1A-рецептора на стереотипию передних лап, вызванное у крысах подкожной инъекцией 8-ОН-DPAT, оценивали способом по Tricklebank (Tricklebank et al., Eur. J.Pharmacol.,117: 15, 1985) с незначительными модификациями, как описано ниже. Использовали самцов крыс линии Sprague - Dawley [Crl: CD (SD) IGS BR] весом 150 - 175 г отCharles River Italia. Животных размещали со свободным доступом к пище и воде и содержали при искусственном чередовании 12 - часовых светлого и темного циклов при температуре 22-24 С. В день эксперимента крыс помещали отдельно в прозрачные пластмассовые контейнеры за 10-15 мин до введения носителя или тестируемых соединений. Для оценки антагонистической активности после перорального приема соединения вводили за 1 и 4 ч до индукции стереотипии 8 - ОН - DPAT (1 мг/кг подкожно). Сеансы наблюдения длились 30 с и начинались спустя 3 мин после лечения 8 - ОН - DPAT и повторялись каждые 3 мин в течение 15 мин. Отмечали появление симптома, вызванного постсинаптической стимуляцией 5-HT1A рецепторов, и измеряли интенсивность, используя шкалу интенсивности, где: 0 = отсутствует, 1 = сомнительная, 2 = присутствует и 3 = интенсивная. Поведенческие шкалы для крыс, получивших лечение, собирали в течение времени наблюдения (5 периодов наблюдения) и выражали как среднее значение 4 крыс/доз. Использовали изменение средних значений у животных, получивших лечение, по сравнению с контрольной(носитель) группой, выраженное как процент ингибирования, для определения количественной антагонистической активности.B. Результаты: Результаты представлены в табл. 5. Эти результаты демонстрируют, что введение соединений согласно изобретению приводит к значительной и длительной постсинаптической активности антагониста 5-НТ 1 А-рецептора. Чтобы оценить эффективность соединений согласно изобретению по ингибированию стереотипии (ритмичные движения передних лап), вызванной 8-OH-DPAT, значение ЭД 75 (эквиэффективная доза, ингибирующая движение передних лап на 75%) оценивали посредством линейной регрессии, используя метод наименьших квадратов. После внутривенного введения соединения Пр. 2 Х, Пр. 1Y и Пр. 5 были фактически эквиэффективными. После перорального введения, Пр. 1Y было более эффективным, чем Пр. 5, в частности, на четыре часа после введения. Таблица 5. Ингибирование ритмичных непроизвольных движений передних лап, вызванное 8-OH-DPAT у крыс (постсинаптический антагонизм)- 22007503 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение общей формулы In обозначает 1 или 2,или его энантиомер, оптический изомер, диастереомер, N-оксид, кристаллическая форма, гидрат,раствор или фармацевтически приемлемая соль. 2. Соединение по п.1, где R3 обозначает (C1-C4)-алкоксигруппу. 3. Соединение по п.1, где R3 обозначает (С 1-С 4)-галоалкоксигруппу. 4. Соединение по любому из пп.1-3, в котором атом углерода, несущий группу R1-фенила, имеет(R)-конфигурацию. 5. Соединение по п.4, в котором атом углерода, несущий R2 и гидроксильные группы, имеет (S)конфигурацию. 6. Соединение по п.1, которое представляет 1-[4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин в форме любого из его выделенных стереоизомеров 1-[(3R,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин,1-[(3S,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин,1-[(3R,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин и 1-[(3S,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-(2-метоксифенил)пиперазин или смесь любых двух из них или более в любой пропорции. 7. Соединение по п.1, которое представляет 1-[4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2-трифторэтокси)фенил]пиперазин в форме любого из его выделенных стереоизомеров 1-[(3R,4S)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2 трифторэтокси)фенил]пиперазин,1-[(3S,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2 трифторэтокси)фенил]пиперазин,1-[(3R,4R)-4-циклогексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2 трифторэтокси)фенил]пиперазин и 1-[(3S,4S)-4-циклгексил-4-гидрокси-3-(2-фторфенил)бутил]-4-[2-(2,2,2 трифторэтокси)фенил]пиперазин или смесь любых двух из них или более в любой пропорции. 8. Фармацевтическая композиция, включающая соединение согласно любому из пп.1-7 или его энантиомер, оптический изомер, диастереомер N-оксида, кристаллическую форму, гидрат, сольват или фармацевтически приемлемую соль в примеси с фармацевтически приемлемым растворителем или носителем.
МПК / Метки
МПК: A61P 13/02, A61K 31/495, C07D 243/08, A61K 31/551, C07D 295/096, A61P 25/00, A61P 13/10
Метки: n-дизамещенные, дисфункцией, нервной, цнс, используемые, диазоциклоалканы, заболеваний, серотонинергической, центральной, вызываемых, лечения, системы
Код ссылки
<a href="https://eas.patents.su/25-7503-n-n-dizameshhennye-diazocikloalkany-ispolzuemye-dlya-lecheniya-zabolevanijj-centralnojj-nervnojj-sistemy-cns-vyzyvaemyh-serotoninergicheskojj-disfunkciejj.html" rel="bookmark" title="База патентов Евразийского Союза">N, n-дизамещенные диазоциклоалканы, используемые для лечения заболеваний центральной нервной системы (цнс), вызываемых серотонинергической дисфункцией</a>
4-,5-, 6-и 7-индольные производные, полезные при лечении заболеваний центральной нервной системы

Номер патента: 6155
Опубликовано: 27.10.2005
Авторы: Банг-Андерсен Бенни, Келер Ян
МПК: A61P 25/06, A61K 31/4045, C07D 401/14...
Метки: полезные, 7-индольные, нервной, 4-,5, производные, центральной, заболеваний, системы, лечении
Формула / Реферат:
1. 4-, 5-, 6- или 7-Метилензамещенное индолильное производное формулы I где заместитель R представляет собой арил или гетероарил, где указанная арильная или гетероарильная группа может быть замещена один или несколько раз заместителями, выбранными из атома галогена, циано-, нитрогруппы, C1-6-алкила, C2-6-алкенила, C2-6-алкинила, C3-8-циклоалкила, C3-8-циклоалкил-C1-6-алкила, C1-6-алкокси-, C1-6-алкилтио-, гидроксигруппы, гидрокси-C1-6-алкила,...
Замещенные 4-(6-фтор-(1н)-индол-3-ил)-1,2,3,6-тетрагидропиридин для лечения нарушений центральной нервной системы

Номер патента: 1469
Опубликовано: 23.04.2001
Автор: Фэйрхерст Джон
МПК: C07D 513/06, A61K 31/475, A61P 25/24...
Метки: 4-(6-фтор-(1н)-индол-3-ил)-1,2,3,6-тетрагидропиридин, системы, лечения, нервной, нарушений, замещенные, центральной
Формула / Реферат:
1. Соединение формулы где R1 представляет водород, C1-4-алкил, C1-4-алкокси или галоген и R2 представляет водород, C1-4-алкил или C1-4-алкокси, или его соль. 2. Соединение по п.1, где как R1, так и R2 представляют водород или либо R1, либо R2 представляет водород. 3. Соединение по пп.1 или 2, где R1 представляет водород, метил, метокси или фтор и R2 представляет водород или метил. 4. 2,2-Диоксид...
Лечение нарушений центральной нервной системы селективными модуляторами рецепторов эстрогена

Номер патента: 2360
Опубликовано: 25.04.2002
Авторы: Брайант Генри Ульман, Пол Стивен Марк, Бэйлес Келли Рени, Нэдлер Мэри Патриция
МПК: A61P 25/24, C07D 333/52, A61K 31/381...
Метки: центральной, селективными, нарушений, модуляторами, рецепторов, системы, лечение, нервной, эстрогена
Формула / Реферат:
1. Способ лечения у пациента, нуждающегося в таком лечении, нарушения центральной нервной системы, выбранного из депрессии, смены настроений и болезни Альцгеймера, включающий введение терапевтически эффективного количества соединения, имеющего структуру или его фармацевтически приемлемой соли или пролекарства R1 и R2 независимо выбирают из группы, состоящей из гидрокси и алкокси, содержащего от одного до четырех атомов углерода ; и R3 и R4...
Лечение нарушений центральной нервной системы с использованием антагонистов оксидазы d-аминокислот и d-аспартатоксидазы

Номер патента: 6654
Опубликовано: 24.02.2006
Авторы: Коэн Даниель, Чумаков Илья
МПК: A61P 25/24, A61K 31/198, A61P 25/18...
Метки: нервной, оксидазы, d-аминокислот, d-аспартатоксидазы, антагонистов, системы, нарушений, использованием, лечение, центральной
Формула / Реферат:
1. Способ идентификации молекулы-кандидата для лечения нарушения ЦНС, включающий: a) контактирование полипептида оксидазы D-аминокислот (DAO) или D-аспартатоксидазы (DDO) или его биологически активного фрагмента с тест-соединением; b) определение, (i) связывается ли указанное соединение с указанным полипептидом, или (ii) ингибирует активность указанного полипептида; и c) если указанное соединение связывается с указанным полипептидом или...
Устройство для пользователей с последствиями поражения центральной нервной системы и /или повреждением опорно-двигательного аппарата тела

Номер патента: 3800
Опубликовано: 30.10.2003
Авторы: Чугунов Виталий Викторович, Семенова Ксения Александровна, Аверьянов Андрей Игоревич
Метки: последствиями, системы, повреждением, пользователей, аппарата, тела, устройство, нервной, поражения, опорно-двигательного, или, центральной
Формула / Реферат:
1. Устройство для пользователей с последствиями поражения центральной нервной системы и/или повреждением опорно-двигательного аппарата тела, содержащее реклинатор (1), расположенный в верхней области тела пользователя и приспособленный для разведения надплечий и приведения лопаток к позвоночнику; средство коррекции средней области тела пользователя, расположенное в области поясницы; по меньшей мере одно средство (16) коррекции бедра и голени...
Предыдущий патент: Производные никотин-или изоникотинбензотиазола
Следующий патент: Синергическая комбинация лиганда альфа-2-дельта и ингибитора pde5 для применения при лечении боли
Случайный патент: Способ получения 3-изопропил-1н-2,1,3-бензотиадиазин-4(3н)-он-2,2-диоксида