Соединения имидазолидинониламинопиримидина для лечения рака
Номер патента: 16177
Опубликовано: 28.02.2012
Авторы: Генри Джеймс Роберт, Ван Янь, Крич Джойс З., Ли Хун-Юй, Слейтер Мелисса Кейт, Брукс Харольд Бернс







Формула / Реферат
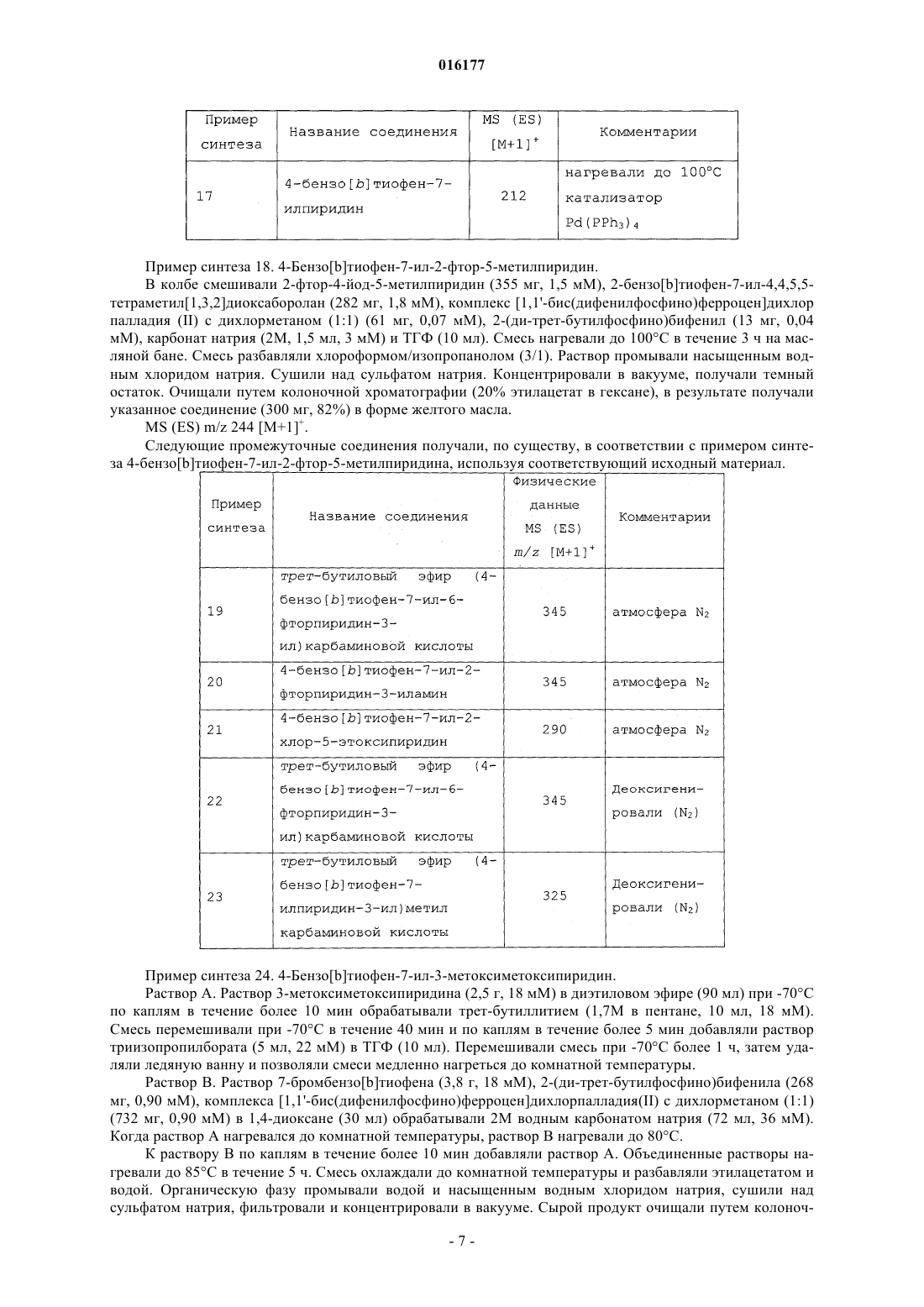
1. Соединение формулы

где R1 представляет собой водород, гидрокси, гидроксиметил, галоген, метил, фторметил, С1-С2алкокси, амино или метиламино;
R2 представляет собой водород, галоген или циано;
R3 представляет собой водород или галоген;
при условии, что по меньшей мере один из R1, R2 и R3 представляет собой водород; и
R4 представляет собой водород, галоген или метил; или
фармацевтически приемлемая соль такого соединения.
2. Соединение по п.1, отличающееся тем, что
R1 представляет собой водород, гидрокси, гидроксиметил, хлор, фтор, метил, фторметил, С1-С2алкокси, амино или метиламино;
R2 представляет собой водород, фтор или циано;
R3 представляет собой водород, хлор или фтор;
при условии, что по меньшей мере один из R1, R2 и R3 представляет собой водород; и
R4 представляет собой водород, хлор, фтор или метил; или
фармацевтически приемлемая соль такого соединения.
3. Соединение по п.1, отличающееся тем, что
R1 представляет собой водород или метил, фторметил;
R2 представляет собой водород или фтор;
R3 представляет собой водород, хлор или фтор;
при условии, что по меньшей мере один из R1, R2 и R3 представляет собой водород; и
R4 представляет собой водород, хлор, фтор или метил; или
фармацевтически приемлемая соль такого соединения.
4. Соединение по п.1, отличающееся тем, что R1 представляет собой галоген, R2 представляет собой водород, R3 представляет собой галоген и R4 представляет собой галоген, или фармацевтически приемлемая соль такого соединения.
5. Соединение, выбранное из группы, состоящей из
1-(2-{4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{5-фтор-4-[7-(2-фтор-5-метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(2-фтор-5-метилпиридин-4-ил)бензо[b]тиофен-2-ил]-5-метилпиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(2-фтор-5-метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-{2-[5-метил-4-(7-пиридин-4-илбензо[b]тиофен-2-ил)пиримидин-2-иламино]этил}имидазолидин-2-она,
1-(2-{5-хлор-4-[7-(2-фтор-5-метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-{2-[5-фтор-4-(7-пиридин-4-илбензо[b]тиофен-2-ил)пиримидин-2-иламино]этил}имидазолидин-2-она,
1-(2-{4-[7-(3-этоксипиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторпиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-(5-фтор-4-(7-(3-гидроксипиридин-4-ил)бензо[b]тиофен-2-ил)пиримидин-2-иламино)этил)имидазолидин-2-она,
1-(2-{4-[7-(2-хлор-5-этоксипиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{5-фтор-4-[7-(3-метоксипиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-(5-хлор-4-(7-(пиридин-4-ил)бензо[b]тиофен-2-ил)пиримидин-2-иламино)этил)имидазолидин-2-она,
1-(2-{5-хлор-4-[7-(5-хлор-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{5-хлор-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(5-хлор-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(2-хлор-5-фторпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(3-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{5-фтор-4-[7-(3-метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(5-хлор-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]-5-метилпиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(2,5-дихлорпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{5-фтор-4-[7-(2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(5-хлор-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
4-(2-(2-(2-(2-оксоимидазолидин-1-ил)этиламино)пиримидин-4-ил)бензо[b]тиофен-7-ил)пиколинонитрила,
1-(2-{4-[7-(3-аминопиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(3-метиламинопиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(3-аминопиридин-4-ил)бензо[b]тиофен-2-ил]-5-метилпиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(3-аминопиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(5-амино-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{4-[7-(3-амино-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил)имидазолидин-2-она,
1-(2-{5-фтор-4-[7-(2-фтор-5-гидроксиметилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил) имидазолидин-2-она и
1-(2-{5-фтор-4-[7-(2-фтор-5-(фторметил)пиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламино}этил) имидазолидин-2-она; или
фармацевтически приемлемая соль.
6. Фармацевтическая композиция, содержащая соединение по пп.1-5 или фармацевтически приемлемую соль указанного соединения в комбинации с фармацевтически приемлемым носителем, разбавителем или наполнителем.
7. Применение соединения по любому из пп.1-5 или фармацевтически приемлемой соли такого соединения для использования при изготовлении лекарственного средства.
Текст
СОЕДИНЕНИЯ ИМИДАЗОЛИДИНОНИЛАМИНОПИРИМИДИНА ДЛЯ ЛЕЧЕНИЯ РАКА Настоящее изобретение обеспечивает новые соединения имидазолидинониламинопиримидина,которые, как предполагают, можно применять в клинике для лечения рака посредством ингибирования Plk1, где R1 - водород, гидрокси, галоген, метил, C1-С 2 алкокси, амино или метиламино; R2 - водород, галоген или циано; R3 - водород или галоген; R4 - водород, галоген или метил; при условии, что по меньшей мере два из R1, R2, R3 и R4 являются водородами; R5 - водород,галоген или метил, или фармацевтически приемлемые соли.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US)Plk1 принадлежит к небольшому семейству протеинкиназ, характеризующихся связывающим фосфосерин/треонин доменом, известным как домен polo box. Plk1 играет ключевую роль в регуляции клеточного цикла. Предполагают, что помимо других функций Plk1 регулирует инициацию, прохождение и завершение митоза - стадии, на которой происходит деление раковых клеток. Следовательно, блокирование Plk1 в раковых клетках предотвращает их деление или митоз. Были обнаружены эффективные агенты, такие как алкалоиды барвинка (NAVELBINE), таксоидыVELCADE является противоопухолевым препаратом, который ингибирует протеосому 26S. Однако эти лекарственные средства вызывают значительные побочные эффекты в нормальных неделящихся клетках. Ингибиторы Plk действуют направленно на делящиеся клетки, что может позволить избежать нежелательной токсичности. Ингибиторы Plk1 известны в данной области техники. См., например, WO 06/066172. Далее в WO 06/021548 раскрыты определенные аналоги дигидроптеридинона (например, BI-2536) в качестве ингибиторов Plk1. В настоящее время BI-2536 находится на второй фазе клинических испытаний, но имеет высокий клиренс (CL1000 мл/мин), и его доза для человека ограничена миелосупрессией. До настоящего времени существует потребность в других соединениях, которые ингибируют Plk1 и обладают повышенной эффективностью или фармакокинетическими свойствами. Также желательно было бы получить ингибитор Plk1, подходящий для перорального введения. Настоящее изобретение обеспечивает новые соединения имидазолидинониламинопиримидина, которые, как предполагают, можно будет применять в клинике для лечения рака посредством ингибирования Plk1. Считают, что некоторые из этих соединений обладают повышенной эффективностью по сравнению с соединениями, раскрытыми в WO 06/066172. Кроме того, предполагают, что некоторые из этих соединений обладают улучшенными фармакокинетическими свойствами по сравнению с BI-2536. Дополнительно, благодаря тому что соединения согласно настоящему изобретению обладают биодоступностью при пероральном введении, предполагают, что некоторые из этих соединений можно вводить перорально. Краткое описание изобретения Настоящее изобретение обеспечивает соединения формулы IR3 представляет собой водород или галоген; при условии, что по меньшей мере один из R1, R2 и R3 представляет собой водород; иR4 представляет собой водород, галоген или метил; или фармацевтически приемлемые соли указанных соединений. Настоящее изобретение обеспечивает способ лечения ракового заболевания, выбранного из группы,состоящей из немелкоклеточного рака легких, рака полости рта и глотки, рака пищевода, рака желудка,меланомы, плоскоклеточного (эпидермоидного) рака кожи, рака молочной железы, рака яичника, рака эндометрия, рака прямой и толстой кишки, нейроглиомы, глиобластомы, карциномы щитовидной железы, рака шейки матки, рака поджелудочной железы, рака предстательной железы, гепатобластомы и неходжкинских лимфом, у млекопитающего, включающий введение нуждающемуся в этом млекопитающему эффективного количества соединения формулы I или фармацевтически приемлемой соли такого соединения. Настоящее изобретение также обеспечивает фармацевтическую композицию, включающую соединение формулы I или фармацевтически приемлемую соль указанного соединения, в комбинации с фармацевтически приемлемым носителем, растворителем или наполнителем. Настоящее изобретение обеспечивает также соединение формулы I или фармацевтически приемлемую соль указанного соединения для применения в изготовлении лекарственного средства. Дополнительно изобретение обеспечивает соединение формулы I или фармацевтически приемлемую соль такого соединения для применения в изготовлении лекарственного средства для лечения ракового заболевания,выбранного из группы, состоящей из немелкоклеточного рака легких, рака полости рта и глотки, рака-1 016177 пищевода, рака желудка, меланомы, плоскоклеточного (эпидермоидного) рака кожи, рака молочной железы, рака яичника, рака эндометрия, рака прямой и толстой кишки, нейроглиомы, глиобластомы, карциномы щитовидной железы, рака шейки матки, рака поджелудочной железы, рака предстательной железы, гепатобластомы и неходжкинских лимфом, у млекопитающего. Кроме того, настоящее изобретение обеспечивает фармацевтическую композицию, предназначенную для лечения ракового заболевания, выбранного из группы, состоящей из немелкоклеточного рака легких, рака полости рта и глотки, рака пищевода, рака желудка, меланомы, плоскоклеточного (эпидермоидного) рака кожи, рака молочной железы, рака яичника, рака эндометрия, рака прямой и толстой кишки, нейроглиомы, глиобластомы, карциномы щитовидной железы, рака шейки матки, рака поджелудочной железы, рака предстательной железы,гепатобластомы и неходжкинских лимфом, у млекопитающего и включающую соединение формулы I или фармацевтически приемлемую соль такого соединения в комбинации с фармацевтически приемлемым носителем, растворителем или наполнителем. Настоящее изобретение также обеспечивает соединения формулыR3 представляет собой водород или галоген;R4 представляет собой водород, галоген или метил; при условии, что по меньшей мере два из R1, R2, R3 и R4 представляют собой водород;R5 представляет собой водород, галоген или метил; или фармацевтически приемлемую соль указанного соединения. Подробное описание изобретения Общие химические термины, используемые в формулах выше, имеют свои обычные значения. Например, термин (C1-C2)алкокси означает метокси и этокси. Термин галоген означает фтор, хлор,бром и йод. Для специалиста в данной области очевидно, что большинство или все соединения согласно настоящему изобретению могут образовывать соли. Соединения согласно настоящему изобретению являются аминами и, соответственно, реагируют с любыми из множества неорганических и органических кислот с образованием фармацевтически приемлемых солей присоединения кислот. Такие фармацевтически приемлемые соли присоединения кислот и общая методология для их получения хорошо известны в данной области. См., например, P. Stahl, et al., HANDBOOK OF PHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE (VCHA/Wiley-VCH, 2002); S.M. Berge, et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January, 1977. Предпочтительными являются соединения формулы I, гдеa) R1 представляет собой водород или метил;c) R3 представляет собой водород или галоген;i) R1 представляет собой метил, R2 представляет собой водород, R3 представляет собой фтор, и R4 представляет собой метил. Схемы Специалисту в данной области понятно, что не все заместители в соединениях настоящего изобретения будут устойчивы к определенным условиям реакции, применяемым для синтеза соединений. Эти группы можно вводить на подходящем этапе синтеза или их можно защищать с последующим снятием защитных групп, если это необходимо или желательно. Специалисту в данной области понятно, что защитную группу можно удалить на любой удобной стадии синтеза соединений согласно настоящему изо-2 016177 бретению. Способы введения и удаления защитных групп для азота и кислорода хорошо известны в данной области: см., например, Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wileyand Sons, New York, Chapter 7 (1999). Кроме того, специалисту в данной области понятно, что во многих случаях порядок, в котором вводят группы, не является критичным. Конкретный порядок необходимых для получения соединений согласно настоящему изобретению стадий может зависеть от конкретного синтезируемого соединения, исходного соединения и относительной лабильности заместителей. Соединения согласно настоящему изобретению можно получить путем осуществления по меньшей мере двух описанных ниже вариантов. В схемах ниже все заместители, если не указано иное, совпадают с определенными ранее, а подходящие реагенты хорошо известны и признаны в данной области. На схеме 2 Y представляет собой галоген, a Z представляет собой бороновую кислоту. Схема 1(2) с образованием соединение формулы (5) в реакции нуклеофильного замещения. Такие реакции проводят в подходящем растворителе, таком как н-бутанол, диоксан, н-метилпиролидин-2-он (НМЛ) и тому подобное. Обычно реакции проводят при температурах примерно от 120 до 150 С с использованием масляной бани или микроволнового реактора. Типичная стехиометрия для этой реакции основана на использовании соединения формулы (3) и примерно 2 экв. 2-(аминоэтил)-1,3-дигидроимидазолона. Можно использовать аминные основания, такие как триэтиламин, диизопропилэтиламин и тому подобное. Схема 2 Соединение формулы (3) подвергают взаимодействию с соединением формулы (4) в реакции Сузуки с использованием пригодного палладиевого катализатора, такого как тетракис(трифенилфосфин)палладия(0), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) или подобный, в присутствии основания, такого как карбонат натрия, карбонат калия или подобный. Такие реакции проводят в подходящем растворителе, таком как ТГФ, диоксан, вода и тому подобное. Обычно реакции проводят при температурах примерно от 100 до 150 С с использованием масляной бани или микроволнового реактора. В случае необходимости получают фармацевтически приемлемую соль соединения согласно настоящему изобретению. Образование таких солей хорошо известно и принято в данной области техники. Понятно, что соединения формул (1) и (3) легко можно получить способами, подобными описанным выше процедурам, которые хорошо известны и общеприняты в данной области техники. Например,соединения формулы (1) получают путем осуществления связывания необязательно замещенного соединения пиридинила с необязательно замещенным соединением бензотиофенила по методу связывания Сузуки, как было описано выше. Полученный аддукт Сузуки боронилируют хорошо известными в данной области методами и затем подвергают реакции связывания с необязательно замещенным галогенидом по методу связывания Сузуки, как было описано выше. Соединения формулы (3) получают путем боронилирования необязательно замещенного соединения бензотиофенила хорошо известными в данной области методами с последующим добавлением 2-(аминоэтил)-1,3-дигидроимидазолона (2) к полученной бороновой кислоте/эфиру по механизму ароматического нуклеофильного замещения. Также следует понимать, что необходимые для получения соединений формулы (1) или (3) стадии можно выполнять в любом порядке, включая осуществление реакции фрагмента соединения формулы (1) или (3) с соединением формулы (2) и/или формулы (4), таким образом, чтобы осуществляемые позже связывание углеродуглеродными связями, реакция связывания и тому подобное обеспечили получение соединения согласно настоящему изобретению. Настоящее изобретение дополнительно проиллюстрировано следующими примерами соединений и-3 016177 синтеза. Эти примеры соединений и синтеза являются только иллюстрацией и никоим образом не ограничивают изобретение. Используемые в примерах и вариантах получения термины имеют обычные значения, если не указано иное. Названия соединениям из приведенных ниже примеров присваивали с использованием ChemDraw, Version 10. Получение 1. 2-Бензо[b]тиофен-7-ил-4,4,5,5-тетраметил[1,3,2]диоксаборолан. В колбе соединяли 7-бромбензо[b]тиофен (426 мг, 2 мМ), бис(пинаколато)дибор (756 мг, 3 мМ),комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (1:1) (81 мг, 0,1 мМ) и ацетат калия (294 мг, 3 мМ) в диметилсульфоксиде (ДМСО) (10 мл). Через смесь в течение 5 мин пропускали азот. Колбу закрывали и нагревали на масляной бане при 100 С в течение 4 ч. Смесь разбавляли хлороформом/изопропанолом (3/1). Раствор промывали насыщенным водным хлоридом натрия. Раствор сушили над сульфатом натрия. Раствор концентрировали в вакууме, в результате получали темный остаток. Очищали путем колоночной хроматографии (от гексана до 20% этилацетата в гексане), в результате получали указанное соединение (342 мг, 66%) в форме бесцветного твердого вещества.MS (ES) m/z 261 [M+l]+. Пример синтеза 2. Бензо[b]тиофен-7-бороновая кислота. В колбе Мортона на 12 л, снабженной механической мешалкой, соединяли 7-бромбензо[b]тиофен(300 г, 1,41 мМ) и триизопропилборат (403,6 г, 2,15 мМ) в безводном тетрагидрофуране (ТГФ) (4000 мл) и охлаждали смесь в атмосфере азота в ванне со смесью сухой лед/ацетон до -70 С. По каплям добавляли н-бутиллитий (1,6 М в гексане, 714 г, 1,68 мМ) с такой скоростью, чтобы внутренняя температура смеси не поднималась выше -67,5 С. После завершения добавления реакционную смесь оставляли перемешиваться при этой температуре в течение 1 ч. Охлаждающую ванну удаляли и медленно добавляли 4 л воды. Добавляли концентрированную HCl (75 мл) до достижения рН раствора примерно равного 2. Смесь оставляли для перемешивания в течение 1 ч. Добавляли достаточное количество 5N водного NaOH и доводили рН смеси примерно до рН 12. Фракции разделяли и сохраняли водную фракцию. Органическую фракцию разбавляли 4 л метил-трет-бутилового эфира и экстрагировали 1 л 5N водного NaOH. Фракции разделяли. Водную фракцию соединяли с полученным ранее водным экстрактом. Водную фракцию промывали дополнительным количеством метил-трет-бутилового эфира (4 л). Фракции разделяли и переносили водные фракции в 3-горлую круглодонную колбу на 12 л, оснащенную механической мешалкой. Раствор охлаждали до +5 С в ледяной ванне. Медленно добавляли концентрированную HCl до тех пор,пока рН раствора не достигал примерно рН 2. Смесь перемешивали в течение 30 мин и отфильтровывали полученное твердое вещество. Твердое вещество дважды промывали на воронке 2 л воды и оставляли сушиться на воздухе в течение 30 мин. Вещество помещали в вакуумную печь при 50 С и сушили в вакууме в течение ночи. Желтый цвет убирали путем суспендирования сухого твердого вещества в 2 л нгептана в течение 30 мин. Снова отфильтровывали твердое вещество, сушили на воздухе в течение 30 мин и сушили в вакууме при 40 С в течение ночи, в результате получали указанное соединение (188,8 г,75%) в форме белого твердого вещества. 1 Н ЯМР (400 МГц, CD3OD)7,86 (д, J=8 Гц, 1 Н), 7,49-7,57 (м, 2 Н), 7,30-7,39 (м, 2 Н). Пример синтеза 3. трет-Бутиловый эфир (6-фторпиридин-3-ил)карбаминовой кислоты. Круглодонную 3-горлую колбу на 100 мл оснащали магнитной мешалкой, термопарой с контролем нагревательной рубашки, холодильником и атмосферой азота. Колбу заполняли 5-амино-2 фторпиридином (5 г, 44,6 мМ), ТГФ (50 мл), 4-диметиламинопиридином (549 мг, 4,5 мМ, 10% мкМ) и ди-трет-бутилдикарбонатом (10,7 г, 49 мМ). Смесь нагревали до 50 С в течение 4 ч. Охлаждали и концентрировали под вакуумом. Остаток растворяли в дихлорметане/воде и фильтровали. Фильтрат переносили на делительную воронку и отделяли дихлорметановую фракцию. Дихлорметан сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. При хроматографии на силикагеле элюирование осуществляли изократической смесью 10% изопропанола/90% дихлорметана, в результате получали указанное соединение (1,64 г, 17%) в форме желто-коричневого прозрачного масла, которое затвердевает после вакуумной сушки.MS (EI) m/z 261 М+. Пример синтеза 4. трет-Бутиловый эфир (2-фторпиридин-3-ил)карбаминовой кислоты. Указанное соединение получали, по существу, в соответствии с примером синтеза трет-бутилового эфира (6-фторпиридин-3-ил)карбаминовой кислоты, используя соответствующий исходный материал.GCMS (EI) m/z 212 М+. Пример синтеза 5. N-(4-Йодпиридин-3-ил)-2,2-диметилпропионамид. Круглодонную 3-горлую колбу на 250 мл оснащали магнитной мешалкой, термопарой, баней из сухого льда/ацетона, атмосферой азота и воронкой для добавления. Заполняли колбу 2,2-диметил-Nпиридин-3-илпропионамидом (3,0 г, 16,8 мМ), диэтиловым эфиром (67 мл), тетраметилендиамином (4,68 г, 6,08 мл, 40,3 мМ). Реакционную смесь охлаждали до -78 С. Медленно в течение более 10 мин через стеклянный шприц добавляли н-бутиллитий (2,5 М раствор в гексане, 16,2 мл, 40,3 мМ). Реакционной смеси позволяли нагреться до -13 С в течение более 2 ч. Реакционную смесь охлаждали до -78 С. В смесь через воронку для добавления добавляли раствор йода (8,5 г, 33,6 мМ в 20 мл ТГФ)-4 016177 и перемешивали в течение 2,5 ч при -68 С. Реакцию гасили путем добавления насыщенного водного раствора NH4Cl (40 мл). Экстрагировали этилацетатом (100 мл) и удаляли водную фазу. Органическую фракцию промывали насыщенным водным раствором тиосульфата натрия (100 мл) и насыщенным водным хлоридом натрия. Органическую фазу сушили над сульфатом натрия и фильтровали. Концентрировали в вакууме, в результате получали продукт в форме коричневого масла. Проводили хроматографию на силикагеле (80 г), элюировали градиентом от 100% дихлорметана до 70% этилацетата/30% дихлорметана, в результате получали указанное соединение (1,19 г, 23%).MS (ES) m/z 305 [M+1]+. Следующие соединения получали, по существу, в соответствии с примером синтеза н-(4 йодпиридин-3-ил)-2,2-диметилпропионамида, используя соответствующий исходный материал. Пример синтеза 8. 3-Метоксиметоксипиридин. В ТГФ (20,6 мл) растворяли 3-гидроксипиридин (7 г, 74 мМ) и диметилформамид (34,4 мл) и охлаждали до -15 С. Добавляли трет-бутоксид калия (8,3 г, 74 мМ) и перемешивали при -15 С в течение 30 мин. К смеси по каплям на протяжении более 40 мин добавляли хлорметилметиловый эфир (5,81 мл, 77 мМ). После завершения добавления смесь перемешивали при -15 С в течение еще 1 ч. Ледяную ванну удаляли и оставляли смесь медленно нагреваться до 15 С. Смесь переливали в насыщенный водный хлорид натрия и интенсивно перемешивали в течение 10 мин. Полученный раствор экстрагировали тремя частями этилацетата. Органические экстракты объединяли и промывали насыщенным водным хлоридом натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Полученный продукт использовали без дальнейшей очистки. 1H ЯМР (400 МГц, CDCl3)8,42 (д, J=3 Гц, 1 Н), 8,28 (д, J=5 Гц, 1 Н), 7,37-7,42 (м, 1 Н), 7,21-7,27 (м,1 Н), 5,20 (с, 2 Н), 3,49 (с, 3 Н). Пример синтеза 9. 2-Хлор-5-метоксиметоксипиридин. В суспензию гидрида натрия (3,7 г, 93 мМ) в ДМФ (50 мл) по каплям более 45 мин добавляли раствор 2-хлор-5-гидроксипиридина (10 г, 77 мМ) в ДМФ (20 мл). Полученный раствор перемешивали при комнатной температуре в течение 1,5 ч. По каплям в течение более 45 мин добавляли хлорметилметиловый эфир (6,6 мл, 86 мМ). Полученную смесь перемешивали при комнатной температуре в течение 12 ч. Смесь разбавляли этилацетатом, водой и насыщенным водным хлоридом натрия. Органический раствор отделяли и промывали тремя частями воды, одной частью насыщенного водного хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Сырой продукт очищали путем колоночной хроматографии с 330 г силикагеля, проводили элюирование градиентом от гексана до 30% этилацетата в гексане более 20 мин и затем держали в 30% этилацетате в гексане в течение 30 мин, в результате получали указанное соединение (10,8 г, 81%) в форме прозрачного масла.MS (ES) m/z 174,0 [M+1]+. Следующие промежуточные соединения получали, используя, по существу, способы, сходные со способами, использованными для 2-хлор-5-метоксиметоксипиридина. Пример синтеза 11. 2-Хлор-4-йод-5-метоксиметоксипиридин. В раствор 2-хлор-5-метоксиметоксипиридина (10,8 г, 62 мМ) в ТГФ (300 мл) по каплям в течение более 10 мин добавляли трет-бутиллитий (1,7 М в пентане, 72 мл, 123 мМ) при -70 С. Полученный раствор перемешивали при -70 С в течение 30 мин. По каплям в течение более 30 мин добавляли раствор йода (23 г, 92 мМ) в ТГФ (150 мл). Полученный раствор перемешивали при -70 С в течение 1 ч. Ледяную ванну удаляли и позволяли реакционной смеси нагреться до комнатной температуры. Смесь разбавляли этилацетатом и водой и выделяли фазы. Водную фазу экстрагировали двумя частями этилацетата. Орга-5 016177 нические экстракты объединяли и промывали двумя частями водного тиосульфата натрия, одной частью воды, одной частью насыщенного водного хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Полученное твердое вещество растирали с гексаном. Твердое вещество выделяли путем фильтрации под вакуумом и промывали твердое вещество гексаном. Твердое вещество сушили в вакууме, в результате получали указанное соединение (10,8 г, 58%) в форме коричневого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО-d6)8,08 (с, 1 Н), 7,98 (с, 1 Н), 5,43 (с, 2 Н), 3,40 (с, 3 Н). Следующие промежуточные соединения получали, используя, по существу, способ для 2-хлор-4 йод-5-метоксиметоксипиридина.HCl (61 мл). Полученную смесь нагревали до 60 С в течение 3 ч. Смесь охлаждали до комнатной температуры и доводили рН до 7 путем медленного добавления насыщенного водного раствора бикарбоната натрия. Смесь экстрагировали тремя частями этилацетата. Органические экстракты объединяли и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, в результате получали указанное соединение (6,8 г, 98%) в форме коричневого твердого вещества, которое использовали без дальнейшей очистки. 1 Н ЯМР (400 МГц, ДМСО-d6)11,04 (с, 1 Н), 7,81-7,87 (м, 2 Н). Следующие промежуточные соединения получали, используя, по существу, способ для 6-хлор-4 йодпиридин-3-ола. Пример синтеза 15. 2-Хлор-5-этокси-4-йодпиридин. Раствор 6-хлор-4-йодпиридин-3-ола (4,9 г, 19 мМ) и карбоната калия (8,0 г, 58 мМ) в диметилформамиде (50 мл) обрабатывали этилиодидом (4,7 мл, 58 мМ). Нагревали до 60 С в течение 3 ч. Смесь охлаждали до комнатной температуры и фильтровали. Смесь разбавляли этилацетатом и промывали 10% водным раствором лимонной кислоты. Водные растворы объединяли и экстрагировали двумя дополнительными частями этилацетата. Органические экстракты объединяли и промывали тремя частями воды,одной частью насыщенного водного хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, в результате получали указанное соединение (5,1 г, 93%) в форме коричневого твердого вещества, которое использовали без дальнейшей очистки. 1 Н ЯМР (400 МГц, ДМСО-d6)8,00 (с, 1 Н), 7,93 (с, 1 Н), 4,18 (кв., J=7 Гц, 2 Н), 1,35 (т, J=7 Гц, 3 Н). Пример синтеза 16. 4-Бензо[b]тиофен-7-ил-2-хлорпиридин. В колбе смешивали 7-бромбензо[b]тиофен (1,7 г, 12 мМ), 2-хлор-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиридин (1,6 г, 7 мМ), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (1:1) (285 мг, 0,3 мМ), 2-(ди-трет-бутилфосфино)бифенил (63 мг, 0,2 мМ), карбонат натрия (2 М, 8 мл, 16 мМ) и ТГФ (20 мл). Смесь нагревали до 100 С в течение 3 ч. Смесь разбавляли хлороформом/изопропанолом (3/1). Раствор промывали насыщенным водным хлоридом натрия. Сушили над сульфатом натрия. Раствор концентрировали в вакууме, получали темный остаток. Его очищали путем колоночной хроматографии (от дихлорметана до 20% ТГФ в дихлорметане), в результате получали указанное соединение (1,14 г, 66%) в форме желтого твердого вещества.MS (ES) m/z 246 [M+1]+. Следующие соединения получали способами, сходными со способом, использованным для 4 бензо[b]тиофен-7-ил-2-хлорпиридина, с использованием ДМСО. Пример синтеза 18. 4-Бензо[b]тиофен-7-ил-2-фтор-5-метилпиридин. В колбе смешивали 2-фтор-4-йод-5-метилпиридин (355 мг, 1,5 мМ), 2-бензо[b]тиофен-7-ил-4,4,5,5 тетраметил[1,3,2]диоксаборолан (282 мг, 1,8 мМ), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлор палладия (II) с дихлорметаном (1:1) (61 мг, 0,07 мМ), 2-(ди-трет-бутилфосфино)бифенил (13 мг, 0,04 мМ), карбонат натрия (2 М, 1,5 мл, 3 мМ) и ТГФ (10 мл). Смесь нагревали до 100 С в течение 3 ч на масляной бане. Смесь разбавляли хлороформом/изопропанолом (3/1). Раствор промывали насыщенным водным хлоридом натрия. Сушили над сульфатом натрия. Концентрировали в вакууме, получали темный остаток. Очищали путем колоночной хроматографии (20% этилацетат в гексане), в результате получали указанное соединение (300 мг, 82%) в форме желтого масла.MS (ES) m/z 244 [M+1]+. Следующие промежуточные соединения получали, по существу, в соответствии с примером синтеза 4-бензо[b]тиофен-7-ил-2-фтор-5-метилпиридина, используя соответствующий исходный материал. Пример синтеза 24. 4-Бензо[b]тиофен-7-ил-3-метоксиметоксипиридин. Раствор А. Раствор 3-метоксиметоксипиридина (2,5 г, 18 мМ) в диэтиловом эфире (90 мл) при -70 С по каплям в течение более 10 мин обрабатывали трет-бутиллитием (1,7 М в пентане, 10 мл, 18 мМ). Смесь перемешивали при -70 С в течение 40 мин и по каплям в течение более 5 мин добавляли раствор триизопропилбората (5 мл, 22 мМ) в ТГФ (10 мл). Перемешивали смесь при -70 С более 1 ч, затем удаляли ледяную ванну и позволяли смеси медленно нагреться до комнатной температуры. Раствор В. Раствор 7-бромбензо[b]тиофена (3,8 г, 18 мМ), 2-(ди-трет-бутилфосфино)бифенила (268 мг, 0,90 мМ), комплекса [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (1:1)(732 мг, 0,90 мМ) в 1,4-диоксане (30 мл) обрабатывали 2 М водным карбонатом натрия (72 мл, 36 мМ). Когда раствор А нагревался до комнатной температуры, раствор В нагревали до 80 С. К раствору В по каплям в течение более 10 мин добавляли раствор А. Объединенные растворы нагревали до 85 С в течение 5 ч. Смесь охлаждали до комнатной температуры и разбавляли этилацетатом и водой. Органическую фазу промывали водой и насыщенным водным хлоридом натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Сырой продукт очищали путем колоноч-7 016177 ной хроматографии со 120 г силикагеля, элюировали градиентом от дихлорметана до этилацетата, в результате получали указанное соединение (3,8 г), содержащее небольшое количество исходного 3 метоксиметоксипиридина. Продукт использовали без дальнейшей очистки. 1 Н ЯМР (400 МГц, CDCl3)8,68 (с, 1 Н), 8,42 (д, J=4 Гц, 1 Н), 7,88 (д, J=8 Гц, 1 Н), 7,33-7,50 (м, 5 Н),5,12 (с, 2 Н), 3,36 (с, 3 Н). Пример синтеза 25. 2-Хлор-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин. В круглодонной колбе на 500 мл до -70 С в атмосфере азота охлаждали раствор 4-бензо[b]тиофен 7-ил-2-хлорпиридина (13 г, 53,1 мМ) и триизопропилбората (20 г, 106 мМ) в ТГФ (150 мл). К охлажденному раствору постепенно в течение более 30 мин добавляли диизопропиламид лития (2M B ТГФ, 53 мл,106 мМ). Смесь непрерывно перемешивали в течение еще 1 ч на охлаждающей бане. Смесь постепенно переносили в перемешиваемый раствор 2,4-дихлорпиримидина (12 г, 106 мМ), комплекса [1,1'бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (1:1) (2,2 г, 53 мМ) и карбоната натрия (35 мл, 3 М, 106 мМ) в ТГФ (150 мл) на период более 30 мин. Перемешивали с обратным холодильником в течение дополнительного 1 ч. Смесь охлаждали до комнатной температуры и разбавляли 500 мл смеси хлороформ/изопропанол (3/1) и 200 мл воды. Полученное твердое вещество собирали путем фильтрации и обрабатывали смесью хлороформ/изопропанол/вода. Твердое вещество промывали дихлорметаном и сушили в вакууме. Фракции разделяли с использованием смеси хлороформ/изопропанол/вода. Органическую фазу промывали водой и насыщенным водным хлоридом натрия,сушили над сульфатом натрия и концентрировали в вакууме, в результате получали коричневый остаток. Остаток очищали путем колоночной флэш-хроматографии (10% метанол в дихлорметане), в результате получали дополнительный продукт. Обе части объединяли и получали указанное соединение (13 г, 63%).MS (ES) m/z 358 [M+1]+. Следующие промежуточные соединения получали, по существу, в соответствии с примером синтеза 2-хлор-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидина, используя соответствующий исходный материал. Пример синтеза 39. 4-[2-(2-Хлор-5-фторпиримидин-4-ил)бензо[b]тиофен-7-ил]пиридин-3-ол. Раствор 2-хлор-5-фтор-4-[7-(3-метоксиметоксипиридин-4-ил)бензо[b]тиофен-2-ил]пиримидина (4 г,10 мМ) в ТГФ (10 мл) обрабатывали 5 Н HCl (3 мл). Смесь перемешивали при комнатной температуре в течение 6 ч. Реакционную смесь концентрировали в вакууме и разбавляли насыщенным водным бикарбонатом натрия и дихлорметаном. Фракции разделяли и фильтровали каждую фракцию. Твердое вещество из органической фазы промывали дихлорметаном, в результате получали указанное соединение (300 мг) в форме желто-коричневого твердого вещества. Твердое вещество из водной фракции промывали водой и сушили, в результате получали указанное соединение (300 мг) в форме желто-коричневого твердого вещества. Вещества смешивали, в результате получали указанное соединение (600 мг, 17%) в форме желто-коричневого твердого вещества.MS (ES) m/z 358 [M+1]+. Пример синтеза 40. 2-Хлор-4-[7-(3-этоксипиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторпиримидин. Раствор 4-[2-(2-хлор-5-фторпиримидин-4-ил)бензо[b]тиофен-7-ил]пиридин-3-ола (100 мг, 0,28 мМ) и карбоната цезия (100 мг, 0,28 мМ) в диметилформамиде (1 мл) обрабатывали этилйодидом (44 мг, 0,28 мМ). Смесь перемешивали при комнатной температуре в течение 12 ч. Смесь разбавляли этилацетатом и промывали раствор тремя частями воды и одной частью насыщенного водного хлорида натрия, сушили-9 016177 над сульфатом натрия, фильтровали и концентрировали в вакууме. Сырой продукт очищали путем колоночной хроматографии с 12 г силикагеля, элюировали градиентом от дихлорметана до этилацетата, в результате получали указанное соединение (48 мг, 45%) в форме коричневого твердого вещества.MS (ES) m/z 386 [M+1]+. Пример синтеза 41. 2-Хлор-5-фтор-4-[7-(3-метоксипиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин. Указанное соединение получали, по существу, в соответствии с примером синтеза 2-хлор-4-[7-(3 этоксипиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторпиримидина, используя соответствующий исходный материал.MS (ES) m/z 372 [M+1]+. Пример синтеза 42. 1-2-[4-(7-Бромбензо[b]тиофен-2-ил)-5-фторпиримидин-2-иламино]этилимидазолидин-2-он. В 1,4-диоксане (650 мл) соединяли 1-(2-аминоэтил)-2-имидазолон (100 г, 774 мМ) и 4-(7 бромбензо[b]тиофен-2-ил)-2-хлор-5-фторпиримидин (90 г, 2 62 мМ), смесь нагревали в атмосфере азота до 90 С при перемешивании в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры. Твердое вещество отфильтровывали и промывали водой (3500 мл) и диэтиловым эфиром (500 мл). Сушили в вакууме при 50 С, в результате получали указанное соединение (59,2 г, 52%) в форме желтого твердого вещества.MS (ES) m/z 436 [M+1]+. Следующие промежуточные соединения получали, по существу, в соответствии с примером синтеза 1-2-[4-(7-бромбензо[b]тиофен-2-ил)-5-фторпиримидин-2-иламино]этилимидазолидин-2-она, используя соответствующий исходный материал. Пример синтеза 46. 5-Бромметил-2-фтор-4-йодпиридин. В колбе объединяли 2-фтор-4-йодпиколин (10,0 г, 42,19 мМ), н-бромсукцинимид (9,76 г, 54,85 мМ),2,2'-азобисизобутиронитрил (3,46 г, 21,10 мМ) и сушили над CCl4 (100 мл). Нагревали в течение 16 ч при 70 С в атмосфере азота. Охлаждали до комнатной температуры. Разбавляли дихлорметаном и промывали водой и насыщенным водным хлоридом натрия. Фракции разделяли и сушили органическую фракцию над сульфатом магния. Концентрировали в вакууме, в результате получали сырой продукт. Очищали путем колоночной хроматографии (от 1 до 15% этилацетата в гексане), в результате получали указанное соединение (8,27 г, 62%).MS (EI) m/z 315 М+. Пример синтеза 47. 1-(2-5-Фтор-4-[7-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-он. В колбе объединяли 1-2-[4-(7-бромбензо[b]тиофен-2-ил)-5-фторпиримидин-2-иламино]этилимидазолидин-2-он (5,5 г, 12,6 мМ), бис(пинаколато)дибор (3,84 г, 15,3 мМ), (1,1'-бис(дифенилфосфино) ферроцен)дихлорпалладия(II) (1,0 г, 1,3 мМ) и ацетат калия (2,5 г, 25 мМ) в ДМСО (80 мл). Через смесь в течение 10 мин пропускали азот. Колбу закрывали и ставили в масляную баню, где нагревали до 85 С в течение суток. Смесь разбавляли смесью хлороформ/изопропиловый спирт (3/1). Раствор промывали насыщенным водным хлоридом натрия. Сушили ее над сульфатом натрия. Раствор концентрировали в вакууме, в результате получали темный остаток. Очищали остаток путем колоночной хроматографии(гексан 20% этилацетат в гексане 10% метанол в дихлорметане), в результате получали продукт в форме коричневого твердого вещества (5 г, 82%).MS (ES) m/z 484 [M+1]+. Пример синтеза 48. (6-Фтор-4-йодпиридин-3-ил)метанол. В круглодонной колбе соединяли 5-бромметил-2-фтор-4-йодпиридин (0,9 г, 2,85 мМ), нитрометан(15 мл, 2 78 мМ), тетрафторборат серебра (721 мг, 3,7 мМ) и диметилформамид (5 мл). Смесь перемешивали в течение суток при комнатной температуре. В смесь добавляли карбонат натрия (1,81 г, 17,1 мМ) и метанол (10 мл). Перемешивали при комнатной температуре в течение еще 4 ч. Реакционную смесь разбавляли хлороформом и промывали водой и насыщенным водным хлоридом натрия. Органическую фракцию отделяли от водной фракции и сушили над MgSO4. После фильтрации органический растворитель выпаривали в вакууме, в результате получали сырой продукт. Сырой продукт очищали путем колоночной флэш-хроматографии (элюировали 10% метанолом в дихлорметане), в результате получали желаемый продукт (0,6 г, 83%).MS (ES) m/z 254 [M+1]+. Пример синтеза 49. 2-Фтор-5-фторметил-4-йодпиридин. Трифторид диэтиламиносеры по каплям добавляли (892 мг, 4 мМ) в раствор (6-фтор-4-йодпиридин 3-ил)метанола в дихлорметане (25 мл) в круглодонной колбе в атмосфере азота и затем добавляли этанол(0,3 мл) при 0-5 С. Смесь перемешивали в течение 3 ч. Реакционную смесь переливали в насыщенный раствор бикарбоната натрия. Продукт экстрагировали хлороформом и промывали водой и насыщенным водным хлоридом натрия. Органическую фракцию отделяли от водной фракции и сушили над MgSO4. После фильтрации органический растворитель выпаривали в вакууме, в результате получали сырой продукт. Очищали сырой продукт путем колоночной флэш-хроматографии (10% метанол в дихлорметане), в результате получали указанное соединение (0,32 г, 53%). В выдерживающем давление сосуде соединяли 2-хлор-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2 ил]пиримидин (9 г, 25,1 мМ) и 2- (аминоэтил)-1,3-дигидроимидазолон (6,4 г, 50,2 мМ) в н-бутаноле (200 мл). Смесь нагревали на масляной бане до 120 С в течение 5 ч. Смесь разбавляли хлороформом/изопропанолом (3/1). Промывали раствор насыщенным водным хлоридом натрия. Сушили над сульфатом натрия. Раствор концентрировали в вакууме, получали темный остаток. Очищали путем колоночной хроматографии (от дихлорметана до 10% метанола в дихлорметане), в результате получали указанное соединение (9 г, 93%) в форме желтого твердого вещества.(36,8 мг, 0,3 мМ) и бикарбонат натрия (18,1 мг, 0,2 мМ). Добавляли тетракис(трифенилфосфин)палладия(0) (10,4 мг, 0,009 мМ). Смесь облучали при 150 С в течение 15 мин при перемешивании магнитной мешалкой. Сырую реакционную смесь переливали на колонки с сильной катионообменной смолой (KOC) (10 г). Желаемый продукт элюировали 2 Н аммиаком в метаноле (40 мл) и концентрировали при пониженном давлении. Очищали путем обратно-фазовой хроматографии (градиент от 30 до 90% при 80 мл/мин в течение 11 мин на колонках 30100 мм, 5 мм, C18 MS Xterra, растворитель А: вода с 0,01 М бикарбонатом аммония, растворитель В: ацетонитрил), в результате получали указанное соединение (20,4 мг, 25,1%). В микроволновом флаконе в ТГФ (3 мл) и воде (1,5 мл) соединяли 1-2-[4-(7-бромбензо[b]тиофен 2-ил)-5-хлорпиримидин-2-иламино]этилимидазолидин-2-он (500 мг, 1,1 мМ), 5-хлор-2-фторпиридин-4 бороновую кислоту (578 мг, 3,3 мМ), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (1:1) (90 мг, 0,11 мМ), 2-(ди-трет-бутилфосфино)бифенил (20 мг, 0,066 мМ) и карбонат натрия (350 мг, 3,3 мМ). В течение 5 мин через смесь пропускали азот. Смесь нагревали до 100 С в течение 10 мин. Органическую фракцию концентрировали и сушили в вакууме. Полученное из суспензии твердое вещество растворяли в дихлорметане/метаноле и очищали путем колоночной хроматографии (от 1% раствор 2N аммиака/метанола в дихлорметане до 10% раствора 2N аммиака/метанола в дихлорметане), в результате получали указанное соединение. Для дальнейшей очистки продукт растворяли в ДМСО и очищали путем обратно-фазовой колоночной хроматографии (от 50% ацетонитрила в воде (с 0,03%HCl) до 95% ацетонитрила в воде (с 0,03% HCl, в результате получали указанное соединение (146 мг,26%).MS (ES) m/z 503 [M+1]+. Следующие примеры получали, по существу, в соответствии с примером синтеза 1-(2-5-хлор-4-[7(5-хлор-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она, используя соответствующий исходный материал. В ДМСО (6 мл) соединяли 1-(2-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2 иламиноэтил)имидазолидин-2-он (100 мг, 0,22 мМ), цианид цинка (51 мг, 0,44 мМ),трис(дибензилиденацетон)дипалладия(0) (10 мг, 0,01 мМ) и 1,1'-бис(дифенилфосфино)ферроцен (6 мг,0,01 мМ). Смесь нагревали до 100 С в течение 4 ч. Смесь охлаждали до комнатной температуры и загружали на колонки с силикагелем. Элюировали на колонках 10% метанолом в дихлорметане, в результате получали указанное соединение (0,7 г, 72%) в форме желтого масла. В закрытом флаконе соединяли и нагревали до 85 С в течение 4 ч н-4-[2-(2-хлор-5 фторпиримидин-4-ил)бензо[b]тиофен-7-ил]пиридин-3-ил-2,2-диметилпропионамид (330 мг, 0,75 мМ), 2(аминоэтил)-1,3-дигидроимидазолон (386 мг, 3,0 мМ) и 1,4-диоксан (6 мл). Концентрировали в вакууме.- 16016177 Смесь разбавляли дихлорметаном и водой. Органический раствор промывали водой. Органический раствор сушили над сульфатом натрия. Раствор фильтровали и концентрировали в вакууме, в результате получали темный остаток. Очищали путем колоночной хроматографии (от дихлорметана до 7% метанола в дихлорметане), в результате получали N-[4-(2-5-фтор-2-[2-(2-оксоимидазолидин-1-ил)этиламино]пиримидин-4-илбензо[b]тиофен-7-ил)пиридин-3-ил]-2,2-диметилпропионамид. Промежуточное амидное соединение переносили в закрытый мембраной флакон на 40 мл. Добавляли магнит для перемешивания на мешалке и заполняли флакон водой (20 мл) и концентрированнойH2SO4 (5 мл). Флакон нагревали до 90 С на масляной бане в течение 5 ч. Реакционную смесь охлаждали до комнатной температуры и проводили через колонку с SCX (10 г). Элюировали смесью вода/метанол 1:1, затем 100% метанолом, затем смесью 1:1 дихлорметан/метанол и, наконец, элюировали продукт 10% раствором 2 М аммиака в метаноле/90% дихлорметана. Концентрировали в вакууме. При хроматографии на силикагеле (80 г) элюировали градиентом от 0 до 10% раствора 2 М аммиака/метанола в дихлорметане. Сушили в вакуумной печи при 42 С в течение 2 ч, в результате получали указанное соединение (192,6 мг, 48%) в форме твердого вещества золотистого цвета.MS (ES) m/z 450 [M+1]+. Следующие примеры получали, по существу, в соответствии с примером синтеза 1-(2-4-[7-(3 аминопиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторпиримидин-2-иламиноэтил)имидазолидин-2-она, используя соответствующий исходный материал. В закрытом флаконе соединяли и нагревали до 70 С в течение 15 ч трет-бутиловый эфир 4-[2-(2 хлорпиримидин-4-ил)бензо[b]тиофен-7-ил]-6-фторпиридин-3-илкарбаминовой кислоты (813 мг, 1,77- 17016177 мМ), 2-(аминоэтил)-1,3-дигидроимидазолон (919 мг, 7,11 мМ) и 1,4-диоксан (22 мл). Концентрировали в вакууме. Смесь разбавляли дихлорметаном и водой. Органический раствор промывали водой. Органический раствор сушили над сульфатом натрия. Фильтровали и концентрировали раствор в вакууме, получали темный остаток. Очищали путем колоночной хроматографии (дихлорметан и этилацетат), в результате получали трет-бутиловый эфир [6-фтор-4-(2-2-[2-(2-оксоимидазолидин-1-ил)этиламино]пиримидин-4-илбензо[b]тиофен-7-ил)пиридин-3-ил]карбаминовой кислоты. трет-Бутиловый эфир[6-фтор-4-(2-2-[2-(2-оксоимидазолидин-1-ил)этиламино]пиримидин-4 илбензо[b]тиофен-7-ил)пиридин-3-ил]карбаминовой кислоты растворяли в дихлорметане и адсорбировали на силикагеле (10 г) путем концентрирования под вакуумом. Сушили под глубоким вакуумом в течение 24 ч. Силикагель помещали в круглодонную колбу и нагревали в масляной бане до контролируемой температуры 98-99 С при глубоком вакууме в течение 2 ч. Охлаждали до комнатной температуры. Продукт экстрагировали на силикагеле 10% 7N аммиака в метаноле/90% дихлорметан. Концентрировали в вакууме. Хроматографировали на силикагеле, элюировали градиентом от 100% дихлорметана до 7% 2N аммиака в метаноле/93% дихлорметана, в результате получали указанное соединение (65,2 мг, 8,2%). Указанное соединение получали, по существу, в соответствии с примером синтеза 1-(2-4-[7-(5 амино-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она, используя соответствующий исходный материал. В закрытой выдерживающей давление пробирке в 5 мл диоксана соединяли 1-(2-5-фтор-4-[7(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-он (120 мг, 0,25 мМ), (6-фтор-4-йодпиридин-3-ил)метанол (100 мг; 0,32 мМ), хлорид (1,1'бис(дифенилфосфино)ферроцен)палладия(II) (10,14 мг; 0,01 мМ), 2-(ди-трет-бутилфосфино)бифенил (2 мг, 0,01 мМ) и карбонат натрия (2 М, 0,2 мл, 0,4 мМ). Смесь нагревали до 100 С в течение суток в масляной бане. Смесь охлаждали до комнатной температуры, разбавляли смесью хлороформ-изопропиловый спирт (3/1). Органическую фазу промывали насыщенным водным хлоридом натрия, сушили над сульфатом натрия и концентрировали до маслянистого остатка. Сырой продукт очищали путем колоночной флэш-хроматографии (10% метанол в дихлорметане), в результате получали указанное соединение (25 мг, 21%). В закрытой выдерживающей давление пробирке в 5 мл диоксана соединяли 1-(2-5-фтор-4-[7(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-он (120 мг, 248,26 мкМ), 2-фтор-5-фторметил-4-йодпиридин (100 мг, 392,15 мкМ), трис(дибензилиденацетон)дипалладий(0) (11,37 мг, 12,41 мкМ), трициклогексилфосфин (2,09 мг, 7,45 мкМ),фосфат калия (105,39 мг, 496,51 мкМ). Смесь нагревали до 100 С в течение 3 ч в масляной бане. ЖХ-МС показала пик при 485. Реакционную смесь охлаждали до комнатной температуры и разбавляли смесью хлороформ-изопропиловый спирт (3/1). Органический раствор промывали насыщенным водным хлоридом натрия, сушили над сульфатом натрия и концентрировали, в результате получали сырой продукт. Сырой продукт очищали путем флэшхроматографии (10% метанол в дихлорметане), в результате получали желаемый продукт (70 мг, 58,2%).MS (ES) m/z 485 [M+1]+. Тесты Показано, что Plk1 экспрессия повышена во многих опухолях человека, таких как немелкоклеточный рак легких, рак ротовой полости и носа, рак пищевода, рак желудка, меланома, рак молочной железы, рак яичника, рак эндометрия, рак прямой и толстой кишки, глиобластома, сосочковая карцинома, рак поджелудочной железы, предстательной железы, гепатобластома и неходжкинские лимфомы. Кроме того, экспрессия Plk1 имеет прогностическое значение для немелкоклеточного рака легких, рака носоглотки, пищевода, меланомы, гепатобластомы, прямой и толстой кишки и неходжкинских лимфомPlk1 регулируют прохождение митоза путем координирования созревания центросом, входа в митоз,разделения сестринских хроматид и цитокинеза (Eckerdt и Strebhardt 2006; Strebhardt and Ullrich 2006; vande Weerdt, B.C. и R.H. Medema (2006). Cell Cycle 5(8): 853-64). Ингибирование функции Plk1 путем введения антител, экспрессия доминантной негативной Plk1 и снижение количества антисмысловой мРНК приводит к возникновению однополярных веретен деления и задержке анафазы, что приводит к смерти митотической клетки в линиях опухолевых клеток, но к обратимой задержке G2 в нормальных нетрансформированных первичных линиях клеток. Кроме того, существуют сообщения о том, что Plk можно использовать при лечении рабдоидных опухолей (Morozov A., et al., Clinical Cancer Research. 13 (16):4721-30, (Aug 15, 2007. Активность соединения BI-2536 была продемонстрирована в доклинических моделях с использованием ксенографтных опухолей мыши НСТ 116, А 549 и NCIH460 (Baum, A., P. Garin-Chesa, et al. (2006). С 191 In vivo activity of BI 2536, a potent and selective inhibitor of mitotic kinase PLK1, in a range of cancerxenografts. Международная конференция AACR-NCI-EORTC "Molecular Targets and Cancer Therapeutics",Philidelphia, PA). Результаты следующих тестов указывают на то, что соединения согласно настоящему изобретению можно применять в качестве противораковых средств. Экспрессия и очистка Plk1. КДНК Plk1 человека, которую можно получить из множества источников, таких как Incyte (номер доступа: НМ 005030), можно напрямую соединить одним концом с полинуклеотидной последовательностью, экспрессирующей метку His6, такой как С-концевая метка FLAG-His6, встроить в подходящий вектор экспрессии, такой как вектор pFastBac (Invitrogen), и трансфецировать в подходящую систему, такую как бакуловирус, подобный описанному в Yue-Wei Qian, et al., Science 282, 1701 (1998) для xPlkk1. В случае использования вирусной системы экспрессии вирус (например, бакуловирус, несущий полинуклеотидный конструкт Plk1-Flag-His6 метка), вводили в культуру подходящих клеток-хозяев, таких как клетки Sf9. Когда экспрессировалось достаточное количество белка слияния Plk1-Flag-His6, например,примерно через 46 ч после инфицирования, культуру обрабатывали окадаевой кислотой (0,1 мкМ) в течение достаточного периода времени (например, 3 ч). Белки слияния Plk1-Flag-His6 очищали от остатков клеток, используя металлоаффинную смолу, такую как TALON (Clontech, Catalog 635503), с помощью хорошо известных в данной области техники методов. Очищенные тэги слияния Plk1-Flag-His6 хранили до использования в подходящей среде, такой как 10 мМ HEPES, 150 мМ NaCl, 0,01% TRITON X100, 1 мМ дитиотрейтол (DTT), 10% глицерин, рН 7,5, при -80 С в небольших аликвотах. Очищенный белок слияния Plk1-Flag-His6 идентифицировали методом MALDI (матрично-активированная лазерная десорбция/ионизация).- 19016177 Экспрессия и очистка GST-Cdc25C(1-206). Экспрессию кДНК гена Cdc25C человека, которую можно получить из множества источников, таких как Incyte (номер доступа: AY497474), можно осуществить в любой удобной системе экспрессии,после чего произвести очистку посредством хорошо известных методов, аналогичных тем, которые описаны в Bin Ouyang et al., Oncogene, 18, 6029-6036 (1999). Одна подходящая система включает выращивание в течение ночи при 18 С E.coli BL21, трансформированной вектором pGEX-2T (Amersham), в который встроена кДНК гена Cds25C человека, для индукции экспрессии использовали 1 мМ изопропилбета-D-тиогалактопиранозид. Экспрессированный белок GST-Cdc25C(1-206), являющийся субстратом для Plk1, можно очистить с помощью GLUTATHIONE SEPHAROSE 4B и хранить в подходящем растворе, таком как 10 мМHEPES, 100 мМ NaCl, рН 7,5, в небольших аликвотах при -80 С. Тест на ингибирование Plk1. Реакционная смесь с киназой Plk1 содержит фермент слияния Plk1-Flag-His6 (0,2 нг/мкл) в буфере,содержащем 50 мМ HEPES, рН 7,3, 1,0 мМ дитиотреитола, 5,0 мкМ АТФ, 10 мМ MgCl2, 0,01%TRITON X-100, 0,4 мкКюри 33 Р-АТФ и 0,06 мкг/мкл пептида GST-Cdc25c (1-206). Использовали соединения в форме 10 мМ исходных растворов в ДМСО. Соединения серийно разбавляли 1:3 в 20% ДМСО для построения кривой концентрация-ответ по 10 точкам, а затем разбавляли 1:5 (от 20 мкМ до итоговых 0,001 мкМ при итоговой концентрации ДМСО 4%) в реакционной смеси для определения активности соединения. Реакцию проводили при комнатной температуре в течение 60 мин и затем прекращали путем добавления 60 мкл 10,0% H3PO4. Реакционную смесь (85 мкл) переносили в 96-луночный планшет с фосфоцеллюлозным фильтром, смоченным 30 мкл 10,0% Н 3 РО 4, инкубировали при комнатной температуре в течение 20-30 мин и затем 3 раза промывали 0,5% H3PO4. Лунки высушивали, а затем добавляли 40 мкл MicroScintTM20 (Packard) и считывали на приборе Wallac MICROBETAJet. Значения процентного ингибирования для 10 точек кривой концентрация-ответ затем анализировали, например, с помощью программы ACTIVITY BASE (IDBS), используя логистическое уравнение с 4 параметрами. Абсолютные значения IC50 рассчитали на основании кривой, построенной по экспериментальным точкам. Все приведенные соединения имеют IC50 менее 100 нМ с минимальным значимым отношением(МЗО) 3,6. Например, соединение из примера 13 имеет IC50 примерно 23 нМ. Тест на содержание pHH3(S10), клеток в митозе и ДНК. Клетки HeLa из Американской коллекции стандартных культур клеток (АТСС) помещали по 200 клеток/лунка в 96-луночные планшеты Beckman Dickinson BIOCOAt и инкубировали в MEM (минимальная поддерживающая среда, например, GIBCO, каталог 11095) с 10% ФБС (фетальная бычья сыворотка) при 37 С, 5% CO2 в течение 24 ч. Клетки обрабатывали путем добавления соединения (в 0,25% ДМСО) в среду, в 10 дозах дипазона от 0,5 до 0,0098 мкМ. После 23 ч выдержки с соединением клетки фиксировали, например, с помощью фиксатора PREFER [Anatech LTD., catalog414] в течение 30 мин,затем разрушали, используя 0,1% TRITON X100 в растворе фосфатно-солевого буфера (ФСБ) в течение 15 мин. Клетки 3 раза промывали ФСБ, затем расщепляли РНКазой в концентрации 50 мкг/мл. К клеткам добавляли первичное антитело к фосфорилированному гистону Н 3 (Upstate Cat06-570), 1:500 в ФСБ с 1% бычьим сывороточным альбумином (БСА) и оставляли на ночь при 4 С. После 3-кратной промывки ФСБ, клетки инкубировали с меченым А 1 еха 488 вторичным антителом (Invitrogen catA11008) в течение 1 ч при комнатной температуре. Снова 3 раза промывали ФСБ и затем на 30 мин добавляли 15 мкМ пропидия йодид (Molecular Probes catР 3566) для окрашивания ядер. Флюоресценцию планшетов считывали с помощью ACUMEN EXPLORER [лазерный сканирующий флюоресценцию цитометр для микропланшетов (состоит из 488 нм ионного аргонового возбуждающего лазера и фотоумножающей трубки детекции), производится ТТР LABTECH LTD.], для определения фосфорилированного гистона Н 3, содержания ДНК и доли клеток в митозе по результата определения уплотнения ДНК. Анализ изображения основан на сигналах флюоресценции от клеток для идентификации клеток в разных субпопуляциях. pHH3(S10)-положительные клетки идентифицировали по средней интенсивности на 500-530 нм выше порога. Для идентификации индивидуальных клеток (клетки с содержанием ДНК от 2 до 4N) и субпопуляций клеточного цикла (клетки 2N, клетки 4N) использовали общую интенсивность от пропидия иодида/ДНК на 655-705 нм. Максимальную интенсивность на 575-640 нм использовали для идентификации уплотнения ДНК, которую использовали в качестве маркера определения клеток в митозе среди клеток 4N. Результаты теста выражаются как процентная доля каждой идентифицированной субпопуляции, % рННЗ, % 2N, % 4N, % клеток в митозе и общее число клеток. Значение ЕС 50 определяли по кривой, аппроксимированной аппроксимации каждого набора результатов 4-параметрической логистической кривой с использованием ACTIVITY BASE. Полученные ЕС 50 Для PHH3(s10), содержания ДНК и митоза имеют МЗО 2,6, 2,4 и 2,5 соответственно. Например, соединение из примера 13 имеет ЕС 50pHH3(s10) = 42 нМ (n=2), ЕС 50 содержание ДНК = 40 нМ (n=2) и ЕС 50 митотическое = 45 нМ (n=1). Тест на антипролиферативное действие. Влияние соединений на пролиферацию клеток можно определить, используя клетки и методы пролиферации клеток, известные в данной области (Robert С. Squatrito et al., Gynecological Oncology, 58, 101- 20016177 105, (1995. Например, клетки HCT116, которые можно получить из АТСС, высевали в концентрации 2000 клеток/лунка в 96-луночный планшет и оставляли в течение суток во влажном CO2-инкубаторе при 37 С. После 20-24 ч инкубации добавляли серийно разбавленные (полулогарифмическое разбавление) соединения и возвращали планшеты в инкубатор. После выдержки подходящей продолжительности(например, 72 ч) определяли пролиферацию клеток, используя хорошо известные методы. В одном методе в планшет с клетками добавляли 10 мкл соли тетразолия, такой как Alamar Blue. После соответствующей экспозиции в красителе определяли флюоресценцию (возбуждение на 530 нм, испускание на 580 нм). Полученное значение IC50 имеет МЗО 3,1. Например, соединение из примера 13 имеет IC50 11 нМ(n=3). Соединения согласно настоящему изобретению предпочтительно имеют форму фармацевтических композиций, вводимых рядом различных путей. Наиболее предпочтительны композиции для перорального введения. Подобные фармацевтические композиции и способы их получения хорошо известны в данной области. См., например, REMINGTON: SCIENCE AND PRACTICE OF PHARMACY (A. Gennaro,et al., eds., 19th ed., Mack Publishing Co., 1995). Соединения формулы I в целом являются эффективными в широком диапазоне дозировок. Например, дозировка на день обычно находится в пределах от примерно 0,01 до примерно 20 мг/кг массы тела,более предпочтительно от 0,1 до 20 мг/кг массы тела. В некоторых случаях более подходящим может быть уровень дозы, находящийся ниже нижнего предела указанного дипазона, в то время как в других случаях можно применять большие дозы при отсутствии каких-либо нежелательных побочных эффектов,и следовательно указанный диапазон дозировок никоим образом не ограничивает объем настоящего изобретения. Понятно, что количество фактически вводимого соединения будет определено врачом в зависимости от значимых факторов, включая состояние, которое нужно лечить, выбранный путь введения,конкретное вводимое соединение или соединения, возраст, вес и реакцию конкретного пациента и тяжесть симптомов заболевания данного пациента. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулыR3 представляет собой водород или галоген; при условии, что по меньшей мере один из R1, R2 и R3 представляет собой водород; иR4 представляет собой водород, галоген или метил; или фармацевтически приемлемая соль такого соединения. 2. Соединение по п.1, отличающееся тем, чтоR3 представляет собой водород, хлор или фтор; при условии, что по меньшей мере один из R1, R2 и R3 представляет собой водород; иR4 представляет собой водород, хлор, фтор или метил; или фармацевтически приемлемая соль такого соединения. 3. Соединение по п.1, отличающееся тем, чтоR1 представляет собой водород или метил, фторметил;R2 представляет собой водород или фтор;R3 представляет собой водород, хлор или фтор; при условии, что по меньшей мере один из R1, R2 и R3 представляет собой водород; иR4 представляет собой водород, хлор, фтор или метил; или фармацевтически приемлемая соль такого соединения. 4. Соединение по п.1, отличающееся тем, что R1 представляет собой галоген, R2 представляет собой водород, R3 представляет собой галоген и R4 представляет собой галоген, или фармацевтически прием- 21016177 лемая соль такого соединения. 5. Соединение, выбранное из группы, состоящей из 1-(2-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2 она,1-(2-5-фтор-4-[7-(2-фтор-5-метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил) имидазолидин-2-она,1-(2-4-[7-(2-фтор-5-метилпиридин-4-ил)бензо[b]тиофен-2-ил]-5-метилпиримидин-2-иламиноэтил) имидазолидин-2-она,1-(2-4-[7-(2-фтор-5-метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она,1-2-[5-метил-4-(7-пиридин-4-илбензо[b]тиофен-2-ил)пиримидин-2-иламино]этилимидазолидин-2 она,1-(2-5-хлор-4-[7-(2-фтор-5-метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил) имидазолидин-2-она,1-2-[5-фтор-4-(7-пиридин-4-илбензо[b]тиофен-2-ил)пиримидин-2-иламино]этилимидазолидин-2 она,1-(2-4-[7-(3-этоксипиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторпиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-(5-фтор-4-(7-(3-гидроксипиридин-4-ил)бензо[b]тиофен-2-ил)пиримидин-2-иламино)этил)имидазолидин-2-она,1-(2-4-[7-(2-хлор-5-этоксипиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламино этил)имидазолидин-2-она,1-(2-5-фтор-4-[7-(3-метоксипиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-(5-хлор-4-(7-(пиридин-4-ил)бензо[b]тиофен-2-ил)пиримидин-2-иламино)этил)имидазолидин-2 она,1-(2-5-хлор-4-[7-(5-хлор-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил) имидазолидин-2-она,1-(2-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-5-хлор-4-[7-(2-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-4-[7-(5-хлор-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламиноэтил) имидазолидин-2-она,1-(2-4-[7-(2-хлор-5-фторпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламиноэтил) имидазолидин-2-она,1-(2-4-[7-(3-хлорпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-5-фтор-4-[7-(3-метилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-4-[7-(5-хлор-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]-5-метилпиримидин-2-иламиноэтил) имидазолидин-2-она,1-(2-4-[7-(2,5-дихлорпиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламиноэтил) имидазолидин-2-она,1-(2-5-фтор-4-[7-(2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-4-[7-(5-хлор-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она,4-(2-(2-(2-(2-оксоимидазолидин-1-ил)этиламино)пиримидин-4-ил)бензо[b]тиофен-7-ил)пиколинонитрила,1-(2-4-[7-(3-аминопиридин-4-ил)бензо[b]тиофен-2-ил]-5-фторопиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-4-[7-(3-метиламинопиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-4-[7-(3-аминопиридин-4-ил)бензо[b]тиофен-2-ил]-5-метилпиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-4-[7-(3-аминопиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин 2-она,1-(2-4-[7-(5-амино-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она,1-(2-4-[7-(3-амино-2-фторпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2-иламиноэтил)имидазолидин-2-она,- 22016177 1-(2-5-фтор-4-[7-(2-фтор-5-гидроксиметилпиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2 иламиноэтил)имидазолидин-2-она и 1-(2-5-фтор-4-[7-(2-фтор-5-(фторметил)пиридин-4-ил)бензо[b]тиофен-2-ил]пиримидин-2 иламиноэтил)имидазолидин-2-она; или фармацевтически приемлемая соль. 6. Фармацевтическая композиция, содержащая соединение по пп.1-5 или фармацевтически приемлемую соль указанного соединения в комбинации с фармацевтически приемлемым носителем, разбавителем или наполнителем. 7. Применение соединения по любому из пп.1-5 или фармацевтически приемлемой соли такого соединения для использования при изготовлении лекарственного средства.
МПК / Метки
МПК: A61K 31/381, A61K 31/496, A61K 31/4436, A61K 31/444, A61K 31/4439, A61P 35/00, C07D 409/14
Метки: имидазолидинониламинопиримидина, рака, лечения, соединения
Код ссылки
<a href="https://eas.patents.su/24-16177-soedineniya-imidazolidinonilaminopirimidina-dlya-lecheniya-raka.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения имидазолидинониламинопиримидина для лечения рака</a>
N-α-(бензилоксикарбонил)-l-γ-глутамил-3-[[2-[[бис[бис(2-хлорэтил)амино]фосфонил]окси]этил]сульфонил]-l-аланил-2(r)-фенилглицин или его соль, фармацевтическая композиция, содержащая это соединение, применение этого соединения для лечения рака и способ лечения рака с помощью этого соединения

Номер патента: 16052
Опубликовано: 30.01.2012
Авторы: Гевель Ронан И., Поломски Роберт Э., Шоу Стивен Р., Колиер Стивен Дж., Жичкин Павел Э., Иди Деннис Л., Истам Стивен А., Кьэрсгорд Ханс Й., Меклер Харолд, Херр Джейсон Р., Боуланджер Уилльям А., Эрнандес-Абад Педро Э.
МПК: A61P 35/00
Метки: композиция, лечения, соединение, соль, соединения, содержащая, это, этого, помощью, применение, фармацевтическая, n-α-(бензилоксикарбонил)-l-γ-глутамил-3-[[2-[[бис[бис(2-хлорэтил)амино]фосфонил]окси]этил]сульфонил]-l-аланил-2(r)-фенилглицин, способ, рака
Формула / Реферат:
1. Соединение формулыили его соль.2. Фармацевтическая композиция, включающая соединение по п.1 или его соль.3. Применение соединения по п.1 или его соли для приготовления лекарственного средства, предназначенного для лечения рака.4. Способ лечения рака у млекопитающего, включающий введение млекопитающему соединения по п.1 или его...
Соединения и способы для лечения и диагностики рака легкого

Номер патента: 5140
Опубликовано: 30.12.2004
Авторы: Ванг Тонгтонг, Фэн Ликвун
МПК: A61K 39/395, A61P 35/00, C07K 14/47...
Метки: соединения, рака, способы, легкого, лечения, диагностики
Формула / Реферат:
1. Изолированный полинуклеотид, включающий последовательность, выбранную из группы, которая состоит из (a) последовательности, представленной в SEQ ID NO:347; (b) комплементов последовательности, представленной в SEQ ID NO:347; (c) полинуклеотида, включающего нуклеотидную последовательность SEQ ID NO:347 или ее часть; и (d) дегенеративных вариантов последовательности, представленной в SEQ ID NO:347. 2. Изолированный полипептид, включающий...
Замещённые 5-фенилпиримидины в терапии рака, фармацевтическая композиция на их основе и способ лечения рака у животных

Номер патента: 14098
Опубликовано: 30.08.2010
Авторы: Гроте Томас, Швеглер Анья, Шивек Франк, Мюллер Бернд, Блеттнер Карстен, Ябс Торстен, Наве Барбара, Райнхаймер Йоахим
МПК: A61K 31/505, C07D 239/42, A61K 31/506...
Метки: замещённые, способ, рака, животных, фармацевтическая, лечения, 5-фенилпиримидины, композиция, терапии, основе
Формула / Реферат:
1. Применение замещенных 5-фенилпиримидинов формулы I и их фармацевтически приемлемых солей в терапии ракагде X означает группу формулы NR1R2, в которойR1 означает C1-C6-алкил, C2-C6-алкенил, C1-C8-галоалкил или C3-C8-циклоалкил, которые могут быть замещены C1-C6-алкилом,R2 означает водород, C1-C6-алкил или C2-C6-алкенил,R1 и R2вместе с атомом азота, к которому они присоединены, образуют пирролидиниловое, 3,6-дигидро-2Н-пиридин-1-иловое,...
Производные бороновой кислоты (варианты), способ их получения, промежуточные соединения, композиция для ингибирования активности протеасом, способы ингибирования активности протеасомы, фактора транскрипции nf-kb и деградации белка и способ лечения рака

Номер патента: 10804
Опубликовано: 30.12.2008
Авторы: Мента Эрнесто, Икбаль Мохамед, Бернардини Раффаэлла, Д`аразмо Джермано, Мессина Маклафлин Патриция, Чаттерджи Санкар, Феррети Эдмондо, Бернареджи Альберто, Де Мунари Серджо, Кассара Паоло Дж., Олива Амброджио
МПК: A61K 31/69, C07F 5/02, A61P 35/00...
Метки: фактора, бороновой, способ, белка, производные, варианты, лечения, соединения, промежуточные, протеасомы, транскрипции, протеасом, получения, активности, кислоты, nf-kb, способы, ингибирования, рака, композиция, деградации
Формула / Реферат:
1. Производные бороновой кислоты общей формулы (I) или их фармацевтически приемлемые соли, стереоизомерные и таутомерные формы, где R1 означает C1-C8алкил, C2-C8алкенил, C2-C8алкинил или C3-C7циклоалкил; R2 означает Н, -(CH2)aCH2NHC(=NR4)NH-Y, -(CH2)bCH2CONR5R6, -(CH2)cCH2N(R4)CONH2, -(CH2)dCH(R7)NR9R10 или -(CH2)eCH(R7)ZR8; a, b, и с, каждый независимо, равны 0, 1, 2, 3, 4, 5 или 6; d и е, каждый независимо, равны 0, 1, 2, 3 или 4; R4...
Замещенные имидазопиримидины для предупреждения и лечения рака

Номер патента: 9038
Опубликовано: 26.10.2007
Авторы: Фернандес Гарсия Андрес, Катена Руис Хуан Лоренсо, Фернандес Серрат Анна, Бальса Лопес Долорс, Серра Комас Кармен, Фарреронс Гальеми Карлес, Лагунас Арналь Кармен, Сальседо Рока Каролина
МПК: A61K 31/505, A61P 35/00, C07D 235/00...
Метки: замещенные, имидазопиримидины, рака, лечения, предупреждения
Формула / Реферат:
1. Соединение общей формулы (I) его стереоизомеры и их смеси, его полиморфы и их смеси, и всех их фармацевтически приемлемые сольваты и аддитивные соли, где А1, А2, A3, А4, А5, В1, В2, B3, В4 и В5 означают радикалы, независимо выбираемые из группы, состоящей из Н, (С1-С4)алкила, (С3-С7)циклоалкила, CF3, OCF3, CN, (CH2)nOR1, (CH2)nNR1R2, CONR1R2, F, Cl, Br, I, NR1R2, NR2COR1, OR1, COR1, COOR1, COSR1, OCOR1, SR1, SOR1, S(O)OH, SO2R1, SO2NR2R3,...
Предыдущий патент: Композиция с замедленным высвобождением и способ ее получения
Следующий патент: Соединения имидазолидинониламинопиримидина
Случайный патент: Способ получения меламина