Направленные цитотоксические аналоги антрациклина, композиция и способ лечения рака у млекопитающего.














Формула / Реферат
1. Соединение формулы
Q14-O-R-P (I)
где Q имеет химическую структуру

где -R- означает одинарную связь или -С(O)-(СН2)n-С(O)- и n равняется 0-7, R' выбран из группы, состоящей из NH2, ароматического или гидрированного 5- или 6-членного гетероцикла, имеющего, по крайней мере, один азот в кольце и такой гетероцикл, имеющий бутадиеновую часть, присоединенную к соседним углеродным атомам вышеупомянутого кольца, образует бициклическую систему, а Р означает Н или пептид, при условии, что когда R' означает NH2, тогда R-P означает другое, чем Н, и когда R-P означает Н, тогда R' означает другое, чем NH2.
2. Соединение по п.1, отличающееся тем, что R' выбран из группы, состоящей из NH2, пирролидин-1-ила, изоиндолин-2-ила, 3-пирролин-1-ила, 3-пирролидон-1-ила, 2-пирролин-1-ила, 3-пиперидон-1-ила, 1,3-тетрагидропиридин-1-ила, и Р означает Р1, Р2 и Р3,
где Р1 выбран из группы, включающей аналог LH-RH формулы
Aaa-Bbb-Ccc-Ser-Tyr-D-Lys(Xxx)-Leu-Arg-Pro-Ddd,
где (Ххх) означает водород, A2Bu или А2Рr, причем,
если Ааа означает Glp, тогда Bbb означает His, Ccc означает Тrр и Ddd означает Gly-NH2,
если Ааа означает Ac-D-Nal(2), тогда Bbb означает D-Phe(4Cl), Ccc означает D-Pal(3), D-Trp и Ddd означает D-Ala-NH2; и
если Ааа-Вbb-Ссс означают Ас, Ddd означает -NН-СН2-СН3,
при этом группа Q14-O-R- образует карбоксамидную связь со свободной аминогруппой остатка D-Lys или, по крайней мере, с одной из свободных аминогрупп A2Bu или А2Рr, когда они находятся в (Ххх),
Р2 означает аналог соматостатина формулы

причем если Ааа означает D-Phe, тогда Bbb означает Туr, Ccc означает Val и Ddd означает Тhr или Тrр,
если Ааа означает D-Trp, тогда Bbb означает Рhе, Ccc и Ddd являются Тhr,
при этом группа Q14-O-R- образует карбоксамидную связь с концевой аминогруппой остатка Ааа,
Р3 означает аналог антагониста бомбезина формулы
Aaa-Gln-Trp-Ala-Val-Gly-His-Leu-Bbb-NH2
где Ааа означает ничто или D-Phe, Bbb означает (CH2-NH)Leu, (CH2-Nh)Phe или (CH2-NH)Trp, или (СН2-N)Tас, причем группа Q14-O-R- образует карбоксамидную связь с концевой аминогруппой остатка Ааа, если он присутствует, или с Gln, если Ааа отсутствует.
3. Соединение по п.2, отличающееся тем, что n=3.
4. Соединение по п.3, отличающееся тем, что R' означает NН2.
5. Соединение по п.3, отличающееся тем, что R' означает 2-пирролин-1-ил.
6. Соединение по п.4, отличающееся тем, что Р означает Р1.
7. Соединение по п.5, отличающееся тем, что Р означает Р1.
8. Соединение по п.4, отличающееся тем, что Р означает P2.
9. Соединение по п.5, отличающееся тем, что Р означает Р2.
10. Соединение по п.4, отличающееся тем, что Р означает P3.
11. Соединение по п.5, отличающееся тем, что Р означает P3.
12. Соединение по п.1, отличающееся тем, что -R-P означает -Н и R' означает другое, чем NH2.
13. Соединение по п.12, отличающееся тем, что -R' означает пирролидин-1-ил.
14. Соединение по п.12, отличающееся тем, что -R' означает изоиндолин-2-ил.
15. Соединение по п.12, отличающееся тем, что -R' означает 3-пирролин-1-ил.
16. Соединение по п.12, отличающееся тем, что -R' означает 3-пирролидон-1-ил.
17. Соединение по п.12, отличающееся тем, что -R' означает 2-пирролин-1-ил.
18. Соединение по п.1, отличающееся тем, что является соединением формулы Glp-His-Trp-Ser-Tyr-D-Lys(Q114-O-glt)-Arg-Leu-Pro-Gly-NH2,
где Q114 означает доксорубицин-14-ил.
19. Соединение по п.1, отличающееся тем, что является соединением формулы Glp-His-Trp-Ser-Tyr-D-Lys(Q614-O-glt)-Arg-Leu-Pro-Gly-NH2,
где Q614 означает 3'-деамино-3'-(2"-пирролин-1"-ил)-доксорубицин-14-ил.
20. Соединение по п.1, отличающееся тем, что является соединением формулы

где Q114 означает доксорубицин-14-ил.
21. Соединение по п.1, отличающееся тем, что является соединением формулы

где Q614 означает 3'-деамино-3'-(2"-пирролин-1"-ил)-доксорубицин-14-ил.
22. Соединение по п.1, отличающееся тем, что является соединением формулы

где Q114 означает доксорубицин-14-ил.
23. Соединение по п.1, отличающееся тем, что является соединением формулы

где Q614 означает 3'-деамино-3'-(2"-пирролин-1"-ил)-доксорубицин-14-ил.
24. Соединение по п.1, отличающееся тем, что является соединением формулы Q114-O-glt-Gln-Trp-Ala-Val-Gly-His-Leu(CH2-NH)Leu-NH2,
где Q114 означает доксорубицин-14-ил.25. Соединение по п.1, отличающееся тем, что является соединением формулы Q614-O-glt-Gln-Trp-Ala-Val-Gly-His-Leu(CH2-NH)Leu-NH2,
где Q614 означает 3'-деамино-3'-(2"-пирролин-1"-ил)-доксорубицин-14-ил.
26. Композиция, включающая соединение по п.1 и фармацевтически приемлемый носитель.
27. Способ лечения рака у млекопитающего, включающий введение млекопитающему, в случае необходимости вышеупомянутого лечения, эффективного количества соединения по п.1.
Текст
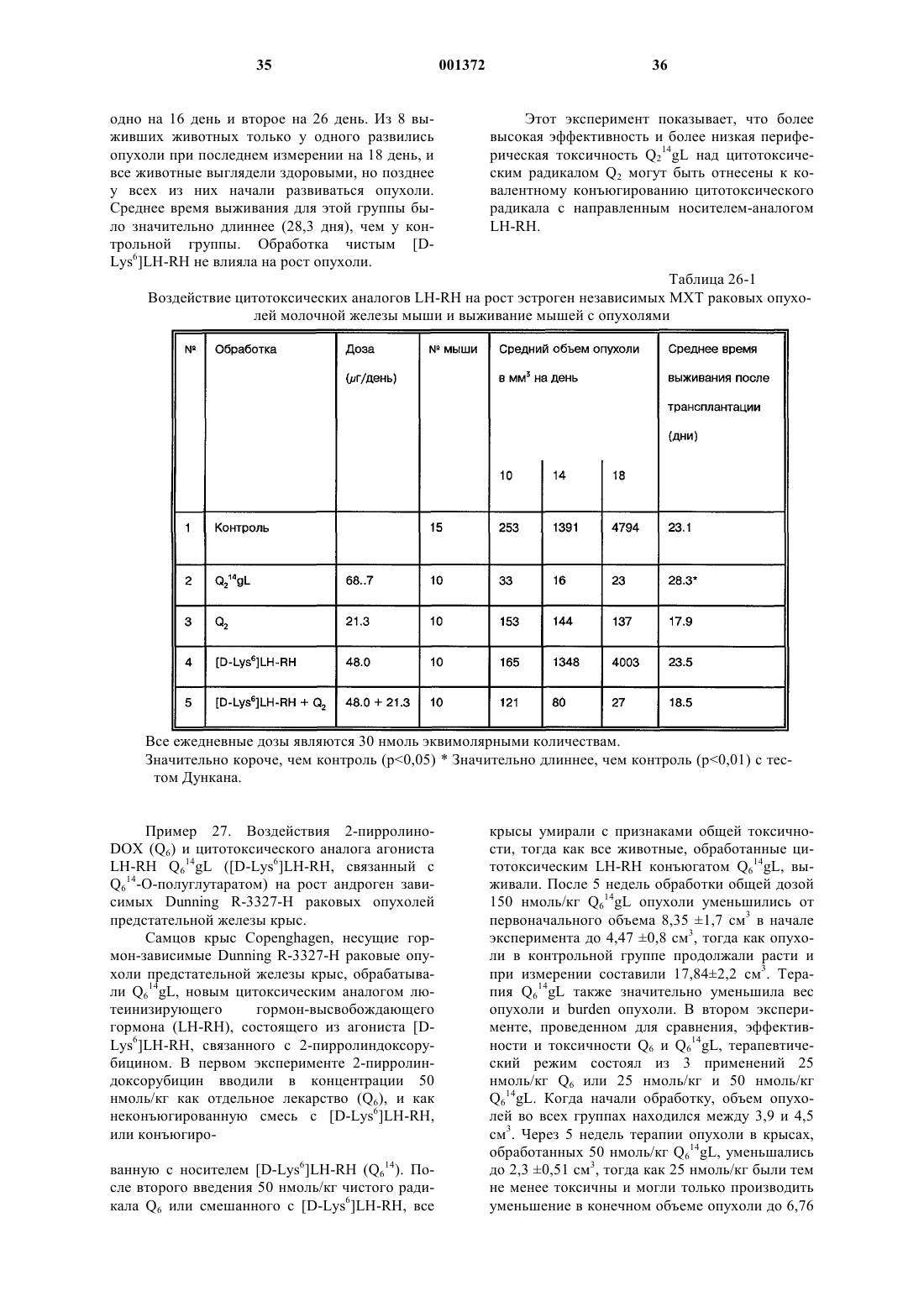
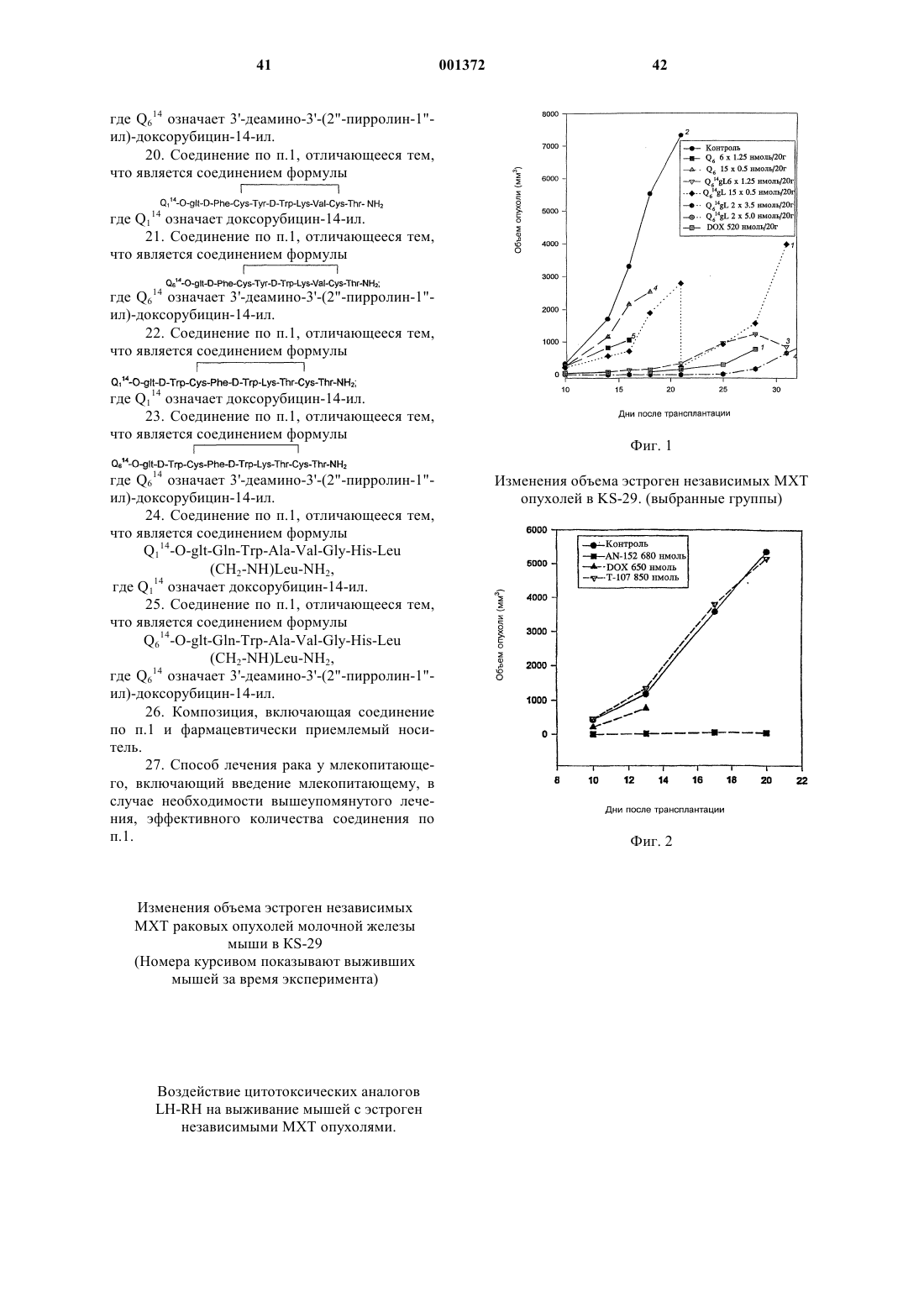
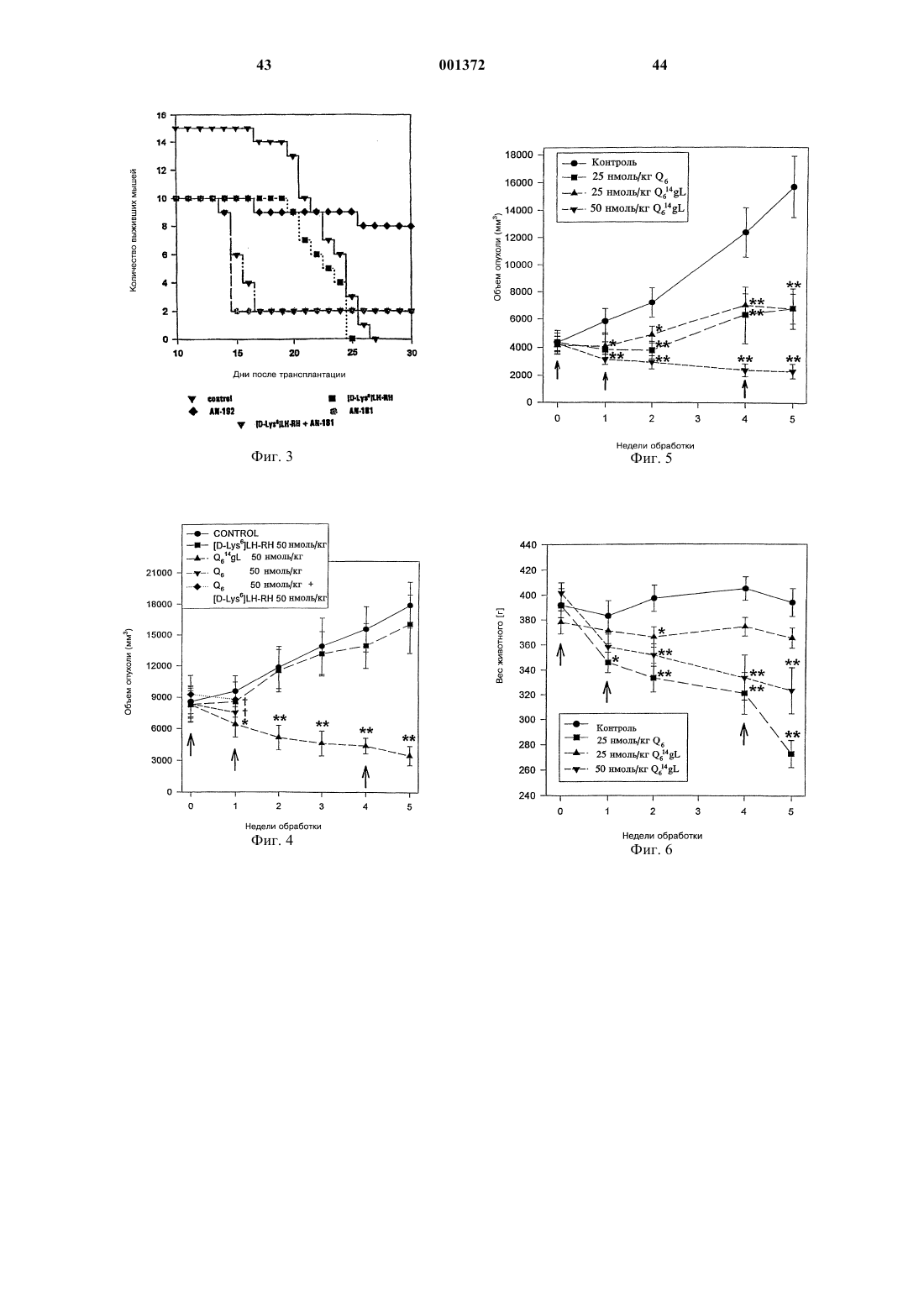
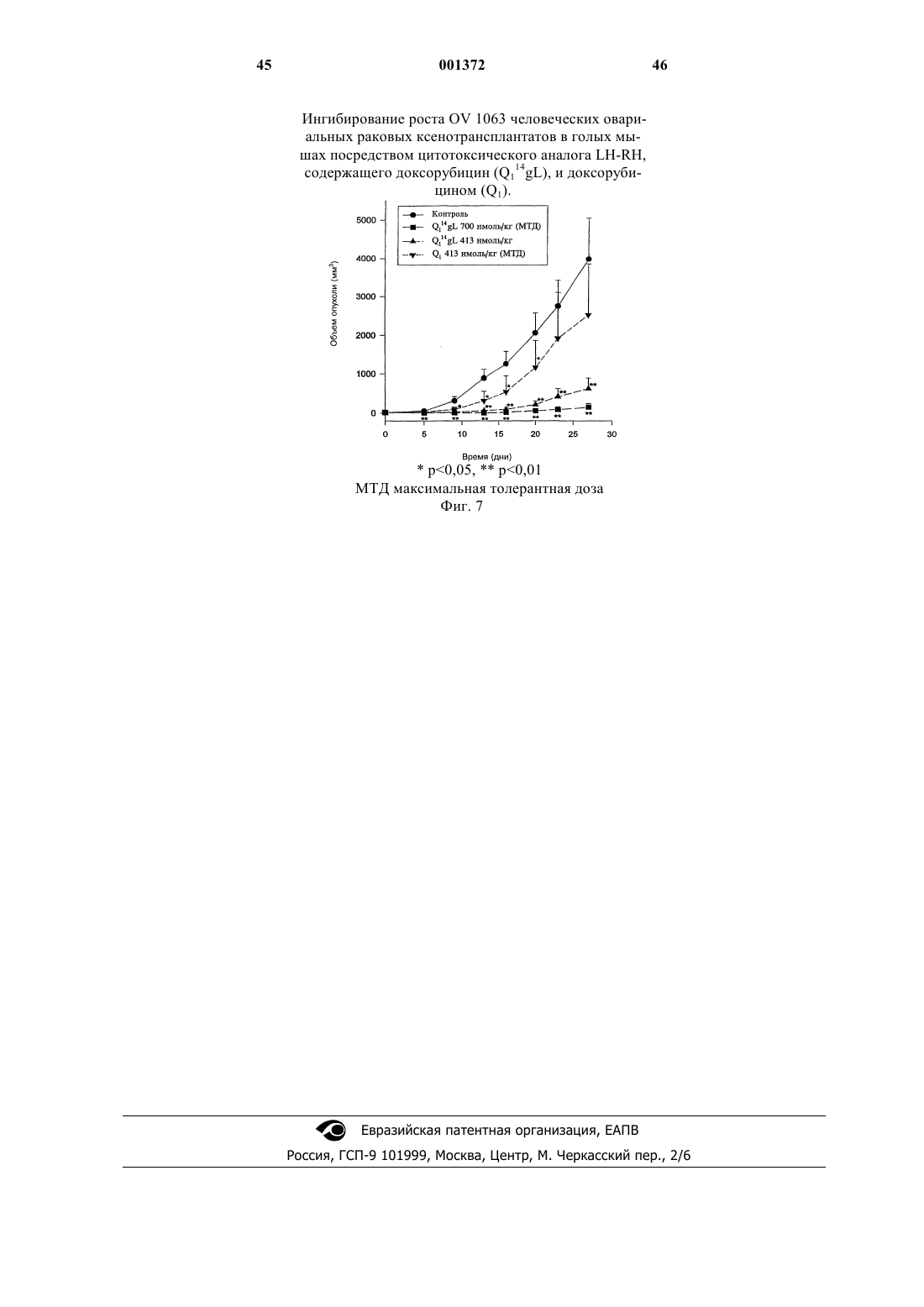
Это изобретение частично было сделано при поддержке правительства. Правительство имеет определенные права на эту заявку. Область техники, к которой относится изобретение Это изобретение относится к области химии направленных противораковых производных антрациклина. Более детально, оно касается доксорубицина (DOX) или его производных, модифицированных даунозамином (DM-DOX), ковалентно связанных с аналогами пептидных гормонов, таких как LH-RH, бомбезин и соматостатин. Эти ковалентные конъюгаты направляются к различным опухолям, имеющим рецепторы для этих аналогов пептидных гормонов. Уровень техникиLH-RH аналоги, которые имеют цитотоксические части в шестом положении, представлены в Schally, Janaky and Bajusz, EP 0450461 В 1, в принятой во внимание публикации от 6 сентября,1995 г.GnRH (LH-RH) аналоги для разрушения гонадотрофов описаны в Nett and Glode, WO 90/09799, опубликованной 7 сентября 1990 г. Эта заявка описывает токсины, подобные рицину,связанные с аналогами LH-RH, для разрушения гонадотрофов и лечения таким образом злокачественных опухолей, зависимых от половых гормонов. Также упомянуто производное LH-RH доксорубицина без подробностей, касающихся химии связывания. Цитотоксические аналоги соматостатина описаны Schally и соавт. в заявке на патент US,поданной 6 апреля 1990 г. и зарегистрированной 15 июля 1993 г. под серийным номером 08/076,846. Обзор A.V. Schally в Anti-Cancer Drugs 5,115-130 (1994) предоставляет подробности о присутствии на клеточных мембранах широкого ряда опухолей рецепторов для аналогов LH-RH,бомбезина или соматостатина.G. Weckbecker в своем обзоре в Farm. Ther. 60, 245-264 (1993) приводит несколько ссылок,которые показывают присутствие рецепторов и рецепторных подтипов для аналогов соматостатина на некоторых нормальных и опухолевых тканях. Бомбезинподобные пептиды и присутствие рецепторов бомбезина/GRP на различных нормальных и опухолевых тканях обсуждаются в обзоре N. Bunnett в Gut Peptides: Biochemistry and(Academic Press). В то же время доксорубицин (DOX) является наиболее широко используемым и очень сильнодействующим противораковым агентом. Однако определенные опухоли совсем не реагируют на него, и также его использование ограничено мультилекарственной резистентностью тропенией, которые являются результатами хронического лечения. Чтобы преодолеть эти недостатки и для дальнейшего использования этого огромного потенциала в уничтожении опухолевых клеток, присущего структуре антрациклиновых антибиотиков, описываются тысячи синтетических производных, включая их направленные аналоги, присоединенные к различным макромолекулам-носителям. Большая часть истории DOX и его аналогов описана в "Adriamycin", David W. Henry, ACSPress, (1981). Высокоактивный, алкилирующий, не обладающий перекрестной резистентностью 3'деамино-3'-(3"-циано-4"-морфолинил)-DОХ и производные из него, которые имеют противоопухолевую активность, описаны Mosher и соавт. в патенте US 4464529 от 7 августа 1984 г. Синтез и биологическая оценка этих "чрезвычайно сильнодействующих морфолинил антрациклинов" представлены в J. Med. Chem. 1984, 27, 638645. В Proc. Natl. Acad. Sci. USA Vol. 88, pp. 4845-4849, June 1991 Gao и соавт. описывают алкилирование формальдегидом последовательности ДНК посредством производного даунорубицина. Антрациклиновые аналоги, содержащие латентные алкилирующие заместители, описаны вJ. Med. Chem. 35, 3208-3214 (1992). Использование ,-дииодсоединения для алкилирования даунозаминового азота в DOX и получение таким образом нового морфолинилN-Трифторацетиладриамицин 14-О-полуглутарати-полуадипат описаны как аналоги Nтрифторацетиладриамицин 14-О-валерата (AD-32) с улучшенной растворимостью в воде Israel и соавт. в патенте US 4299822 от 10 ноября 1981 г.Horton и Priebe (J. Antibiotics, XXXVI, 12111215) описывают некоторые 14-O-эфиры различных антрациклиновых аналогов без сильных изменений в противораковой активности по сравнению с исходными 14-ОН аналогами. В области конструирования направленных хемотерапевтических агентов стремятся к следующим целям: стабильная связь между молекулойносителем и хемотерапевтическим агентом до тех пор, пока цель не достигнута; сохраняемые биологические характеристики молекулы-носителя внутри конъюгата, такие как сохраняемые связывающие свойства; сохраняемая фармакологическая активность хемотерапевтического агента внутри конъюгата,такая как сохраняемая цитотоксическая активность; получение в результате конъюгации аналогов с более интенсивной активностью и/или более низкой периферической токсичностью по сравнению с неконъюгированными частями. Конъюгация DOX посредством NalO4 окисления даунозаминовой части DOX, за которым следует восстановительное алкилирование,включающее первичный амин молекулы носителя, описана Sela и соавт. в патенте US 4263279 от 21 апреля 1981 г. Спейсер из цисаконитовой кислоты использовался для присоединения даунозаминового азота к макромолекулярным носителям рНчувствительной связью, как описано в Biochem.Biophys. Res. Commun. 1981, 102, 1048-1054. Образование сложноэфирных связей и C-N связей между 14-бромдаунорубицин и белками или поли-L-аминокислотами описано Zunino и соавт. в (1981) Tumori 67, 521-524 и в (1984) Eur.J. Cancer Clin. Oncol. 20, 421-425. Морфолино-DOX (высокоактивный, модифицированный даунозамином аналог DOX) конъюгировали с антителом через гидролизируемую(лизосомотропную,рН-чувствительную) гидразоновую связь, используя С-13 оксо-функцию цитотоксического агента, как описано в Bioconjugate Chemistry 1990 1(5), 325330. Чувствительность карбоксиамидной связи лейцинового остатка к ферментативной деградации удачно использовали в конъюгатах DOX,содержащих пептид в качестве "спейсерного плеча", предпочтительно Ala-Leu-Ala-Leu, где карбоксиконцевой Leu ацилирует дауназоминовый азот в DOX, и аминоконцевой Ala связывается с носителем через спейсер из дикарбоновой кислоты, как описано в Proc. Natl. Acad. Sci. USA 1982, 79, 626-629. Даунозаминовый азот в DOX ацилировали спейсером из глутаровой кислоты и связывали сLH-RH аналогами с сильной потерей цитотоксической активности, как описано в Proc. Natl.Acad. Sci. USA 1992, 89, 972-976. Следующие ссылки связаны с использованием соединений согласно настоящему изобретению для лечения различных человеческих опухолей: 1. Schally et. al. (1996) in Treatment withof Pancreatology 16, 277-280. 23. Halmos et.al. (1995) Cancer Res. 55, 280287. 24. Halmos et.al. (1994) Cancer Letters 85,111-118. 25. Oin et.al. (1994) J. Cancer Res. Clin. Oncol. 120, 519-528. 26. Oin et.al. (1994) Cancer Res. 54, 10351041. 27. Qin et.al. (1995) Int. J. Cancer 63, 257262. 28. Reile et.al. (1994) The Prostate 25, 29-38. 29. Pinski et.al (1994) Int. J. Cancer 57, 574580. 30. Radulovic et.al. (1992) P.S.E.B.M. 200,394-401. 31. Radulovic et.al. (1994) Acta Oncologica 33(6) 693-701. 32. Pinski et.al. (1993) Cancer Letters 71, 189196. 33. Pinski et.al. (1994) Br. J. of Cancer 70,886-892. 34. Pinski et.al. (1994) Cancer Res. 54, 58955901. Все упоминаемые цитирования приведены здесь как ссылки. Сущность изобретения Соединения этого изобретения являются новыми, направленными цитотоксическими пептидными гормонами, содержащими антрациклиновый цитотоксический агент, такой как DOX или DM-DOX, конъюгированный с пептидным гормоном, таким как аналоги LH-RH, бомбезина или соматостатина. Эти цитотоксические конъюгаты пептидных гормонов сконструированы для лечения опухолей, несущих специфические рецепторы для этого конъюгата, таких как рак груди, овариальный рак, эндометриальный рак, рак предстательной железы, рак поджелудочной железы, рак ободочной кишки, рак желудка и рак легких. Некоторые из этих (неконъюгированных) антрациклиновых цитотоксических агентов, использованных здесь, являются сами по себе новыми и очень сильнодействующими, однако, их уровень токсичности слишком высокий, чтобы использовать их в неконъюгированной форме. Модифицированные даунозамином аналогиDOX, представленные в этом изобретении, получали в ходе поиска новых, высокоактивных, не перекрещивающихся устойчивых аналогов DOX,пригодных для образования ковалентных конъюгатов с пептидными носителями. Этого образования стабильных, ковалентно связанных конъюгатов с полностью сохраняемыми биологическими активностями их компонентов достигали при использовании спейсера из дикарбоновой кислоты, подобной глутаровой кислоте. Одна карбоксильная группа спейсера образует сложноэфирную связь с 14-ОН группойDOX или DM-DOX, и другая карбоксильная группа спейсера образует карбоксамидную связь с хорошо выбранной свободной аминогруппой пептидного носителя. Соединения этого изобретения представлены общей формулой I-R- означает одинарную связь или -С(O)(СН 2)n-С(O)- и n = 0-7;R' означает NH2 или ароматические или гидрированные 5- или 6-членные гетероциклические соединения, имеющие, по крайней мере,один азот в кольце и необязательно имеющие бутадиеновую часть, присоединенную к соседним углеродным атомам вышеупомянутого кольца для образования бициклической системы; Р означает Н или пептидную часть, соответственно аналог LH-RH, соматостатина или бомбезина, но не исключая других физиологически активных пептидов. В частности, желатель 6 ны те LH-RH аналоги, которые имеют сродство к рецепторам неопластических клеток, особенно аналоги, имеющие D-Lys часть в 6 положении, а также укороченные аналоги соматостатина и бомбезина. Однако когда R' означает NH2, тогдаR-P означают другое, чем Н. Когда R-P означают Н, тогда R' означает другое, чем NH2. Новая синтетическая реакция была открыта в ходе этой работы. Не только было обнаружено,что доксорубицин и его производные могут быть связаны через дикарбоновую часть в 14 положении для получения новых фармакологически эффективных конъюгатов, но и разработали новый путь для получения частично насыщенных гетероциклических частей из вицинальных и дизъюнктных, т.е. ,- или ,-гидрокси первичных аминов. Специфическим применением в настоящем изобретении было образование 2"пирролинил и 1",3"-тетра-гидропиридинил частей на даунозаминовом сахаре. Однако эта реакция имеет более широкое применение. 5- и 6 членные частично насыщенные гетероциклические части могут быть образованы, когда вицинальный или дизъюнктивный гидроксиамин реагирует с галозамещенным альдегидом, имеющим 2 или 3 части между альдегидным углеродом и углеродным атомом, имеющим галоидгруппу. Эти части могут быть все метиленами, или может включаться гетероатом, такой как кислород. Эта реакция проводится в три стадии. Очень большой избыток галоальдегида реагирует с кислой солью гидроксиамина соответственно в полярном инертном безводном органическом растворителе. Таким образом формируется пятичленное оксазолидиновое кольцо (или шестичленное 1,3-тетрагидрооксазиновое кольцо) путем конденсации альдегидной группы с гидроксильной или аминогруппами. Этот продукт обрабатывают органическим основанием, соответственно четвертичным амином, посредством чего элементы галоводородной кислоты удаляются между галоидной частью первичного галоальдегида и вторичной аминогруппой оксазолидина или 1,3-тетрагидрооксазинового кольца, чтобы получить объединенную кольцевую структуру добавлением 5- или 6-членного кольца. Это основание затем нейтрализуют слабой кислотой,соответственно органической кислотой, такой как ледяная уксусная кислота. Обработка водной кислотой, соответственно органической кислотой,открывает оксазолидин или 1,3 тетрагидрооксазиновую часть объединенного кольца. Специалистам в этой области вероятно понятно, что в зависимости от исходного альдегида конечное азотсодержащее кольцо может содержать, по крайней мере, один дополнительный гетероатом, как упомянуто выше. Основная реакция может быть проиллюстрирована следующим образом где X' означает галоид, соответственно бромо или иодо, предпочтительно иодо,Y означает СH2, ОСН 2, CH2-CH2,Z ничего не означает или означает CH2. Когда Z ничего не означает, альдегидная часть образует 5-членное оксазолидиновое кольцо на первой стадии реакции. Когда Z означаетCH2, альдегидная часть образует 6-членное 1,3 тетрагидрооксазиновое кольцо. Тогда как такие кольцевые образования хорошо известны, в комбинации с закрытием кольца, производимым галоалкановой боковой цепью в основной среде,такой как третичный амин в безводной среде, эта реакция является новой и удивительной. Перечень чертежей Фиг. 1 является графиком объемных изменений эстроген независимых МХТ раковых опухолей молочной железы мыши при различных уровнях дозировки соединений настоящего изобретения и DOX; фиг. 2 - графиком объемных изменений эстроген независимых МХТ раковых опухолей молочной железы мыши при различных уровнях дозировки определенного соединения настоящего изобретения, соединения предшествующего уровня, DOX и контроля,фиг. 3 - графиком влияния некоторых цитотоксических LH-RH аналогов на выживание мышей с эстроген независимыми МХТ раковыми опухолями молочной железы мыши; фиг. 4 - графиком объема опухоли у самцовCopenhagen крыс, несущих крысиные Dunning K3327-H трансплантаты карциномы предстательной железы, в течение обработки агонистом предшествующего уровня и определенным соединением настоящего изобретения; фиг. 5 - графиком, показывающим влияние обработки определенным соединением настоящего изобретения и соответствующим цитотоксическим LH-RH аналогом на объем опухоли в крысах с Dunning К-3327-Н раком предстательной железы; фиг. 6 - графиком, показывающим влияние обработки определенным соединением настоящего изобретения и соответствующим цитотоксическим LH-RH аналогом на вес тела Copenhagen крыс, несущих Dunning K-3327-H рак предстательной железы; фиг. 7 - графиком, показывающим ингибирование роста опухоли, достигаемого обработкой определенным соединением настоящего изобретения и DOX. Сведения, подтверждающие возможность осуществления изобретения Часть Q, когда замещена в R' определенными предпочтительными группами, имеет обозначение подчастей от Q1 до Q8, из которых от Q2 доQ8 являются новыми цитотоксическими частями.R' имеет следующие предпочтительные значения, приводящие к желаемым Qx частям,перечисленным в скобках: NH2 (Q1), пирролидин-1-ил (Q2), изоиндолин-2-ил (Q3), 3 пирролин-1-ил (Q4), 3-пирролидон-1-ил (Q5), 2 пирролин-1-ил (Q6), 3-пиперидон-1-ил (Q7) или 1,3-тетрагидропиридин-1-ил (Q8). Таким образом, если R-P означает Н и -R' означает -NH2, Q1 означает DOX, если R-P означает Н и -R' означает пирролидин-1-ил, Q2 означает 3'-деамино-3'-(пирролидин-1"-ил)-доксорубицин (Q2); если R-P означает Н и -R' означает изоиндолин-2-ил, Q3 означает 3'-деамино-3'(изоиндолин-2"-ил)-доксорубицин (Q3); если R-P означает Н и -R' означает 3-пирролин-1-ил, Q4 означает 3'-деамино-3'-(3"-пирролин-1"-ил)доксорубицин (Q4); если R-P означает Н и -R' означает 3-пирролидон-1-ил, Q5 означает 3'деамино-3'-(3"-пирролидон-1"-ил)-доксорубицин (Q5); если R-P означает H и -R' означает 2 пирролин-1-ил, Q6 означает 3'-деамино-3'-(2"пирролин-1"-ил)-доксорубицин (Q6); если R-P означает H и -R' означает 3-пиперидон-1-ил, Q7 означает 3'-деамино-3'-(3"-пиперидон-1"-ил)доксорубицин (Q7); если R-P означает Н и -R' означает 1,3-тетрагидропиридин-1-ил, Q8 означает 3'-деамино-3'-(1",3"-тетрагидропиридин-1"ил)-доксорубицин (Q8). Соединения, содержащие даунозаминовый азот в пятичленном кольце, с алкилирующей функцией являются в 10-50 раз более активнымиin vitro, чем их гомологичные аналоги, содержащие даунозаминовый азот в шестичленном кольце. (Такими парами являются Q5 и Q7, а также Q6 и Q7.) В предпочтительных воплощениях настоящего изобретения в веществе формулы Q14-O-RP, R-P не являются водородом. Где Р является другим, чем водород, там он является Р 1, P2 и P3,соответственно где Р 1 означает LH-RH агонистноситель, LH-RH антагонист-носитель или укороченный LH-RH аналог-носитель, Р 2 означает укороченный аналог соматостатина и Р 3 означает антагонист бомбезина. Соответственно Р 1 означаетAaa-Bbb-Ccc-Ser-Tyr-D-Lys(Xxx)-Leu-Arg-Pro-Ddd, где (Ххх) является водородом или диаминозаместителем, таким как A2Bu или А 2 Рr,где, если Ааа означает Glp, тогда Bbb означаетPhe, Ccc означает D-Pal(3) и D-Trp и Ddd означает D-Ala-NH2; и где Aaa-Bbb-Ccc означают Ас,там Ddd означает -NН-СН 2-СН 3; Р 2 является где, если Ааа означает D-Phe, тогда Bbb означает Туr, Ccc означает Val и Ddd означает Тhr или Тrр; и если Ааа означает D-Trp, тогда Bbb означает Рhе и Ccc и Ddd являются Тhr; и P3 означаетAaa-Gln-Trp-Ala-Val-Gly-His-Leu-Bbb-NH2,где Ааа является ничем или D-Phe и Bbb является (CH2-NH)Leu, (СН 2-NH)Рhе, (СН 2NН)Тrр, (СН 2-N)Тас или (CH2-N)DMTac. В новых соединениях настоящего изобретения, содержащих аналоги LH-RH, цитотоксический радикал Q присоединен к D-Lys боковой цепи на LH-RH аналогах или к (Ххх) группе,присоединенной туда через спейсер из дикарбоновой кислоты как представлено в формуле VIIAaa-Bbb-Ccc-Ser-Tyr-D-Lys(Xxx)m(Q14-O-R)n-Leu-Arg-Pro-Ddd, где m означает 1 или 0 и n означает 1 или 2, при условии, что когда m является 1, т.е. (Ххх) означает A2Bu или A2Pr, n означает 1 или 2, когда m означает 0, т.е. является Н, n означает 1. В новых соединениях настоящего изобретения, содержащих аналоги соматостатина, цитотоксический радикал Q присоединяют к аминоконцу аналогов соматостатина через спейсер из дикарбоновой кислоты, как представлено в формуле VIII В новых соединениях настоящего изобретения, содержащих аналоги антагонистов бомбезина, цитотоксический радикал Q связан с аминоконцом антагонистов бомбезина как представлено в формуле IX Особенно предпочтительными воплощениями этого изобретения являются те пептидные конъюгаты, которые содержат Q1 и Q6 в качестве цитотоксических аналогов и глутаровую кислоту(n=3) в качестве спейсера из дикарбоновой кислоты, образующего 14-O-эфирную связь с Q1(2 пирролинодоксорубицин) и карбоксамидную связь с пептидным носителем. Наиболее предпочтительными воплощениями этого изобретения являются цитотоксические LH-RH аналоги со следующими формулами: 1. Glp-His-Trp-Ser-Tyr-D-Lys(Q114-O-glt)-Leu-ArgPro-Gly-NH2; 2. Glp-His-Trp-Ser-Tyr-D-Lys(Q614-O-glt)-Leu-ArgPro-Gly-NH2; цитотоксические аналоги соматостатина со следующими формулами В этом новом способе получения частично насыщенного гетероциклического кольца с азотом из вицинального или дизъюнктивного, т.е.,- или ,-гидроксиамина первая стадия реакции проводится в безводном инертном органическом полярном негидроксильном (апротонном) растворителе, соответственно диметилформамиде, при использовании значительного избытка,соответственно 30-кратного избытка галоальдегида, особенно эффективными являются 4 иодбутиральдегид и 5-иодвалеральдегид. Хотя изобретение не ограничивается этим, однако,бром может использоваться вместо иода. Эта реакция, а также последующие стадии могут проводиться при температуре окружающей среды. Стадию обработки основанием проводят в избытке, соответственно в 2-4-кратном избытке органического основания. Третичные амины,такие как триалкиламины, пригодны для этой цели. Таким образом образованное бициклическое кольцо раскрывают, чтобы освободить вицинальную или дизъюнктивную группу обработкой в органической кислоте в присутствии воды. Разбавленная водная трифторуксусная кислота соответственно в инертном органическом растворителе, таком как ацетонитрил, может использоваться. Этот продукт очищают удалением летучих соединений при пониженном давлении,избыток галосоединения экстрагируют гексаном и остаток очищают ВЭЖХ. Сокращения Для описания пептидов и их производных этого изобретения используются стандартные сокращения для аминокислот, как главным образом принято в области пептидной химии и как рекомендовано IUPAC-IUB Комиссией по биохимической номенклатуре (Europen J. Biochem.,138, 9-37 (1984. Сокращения для индивидуальных аминокислотных остатков основаны на тривиальном названии аминокислоты, например, Glp означает пироглутаминовую кислоту, His означает гистидин, Тrр означает триптофан и т.д. Сокращения указывают L-изомерную форму, кроме случаев выраженных особо, например, Ser означает Lсерин и D-Lys означает D-лизин. Сокращения необычных аминокислот в этом изобретении следующие:D-4-хлорфенилаланин. Пептидные последовательности пишутся в соответствии с принятым соглашением, когда Nконцевая аминокислота находится слева и Сконцевая аминокислота расположена справа,например, Glp-His-Trp. Формула Leu(CH2-NH)Leu-NH2 описывает восстановленную пептидную связь между лейцином и лейциновым амидным остатком на Сконце пептидной последовательности. Другие использованные сокращения:Tpi - 2,3,4,9-тетрагидро-1 Н-пиридо[3,4b]индол-3-карбоновая кислота. Аналитическая ВЭЖХ система Beckman оборудована моделью 168 диодного матричного детектора, и использовалась система "Золотое хроматографическое программное обеспечение"(Beckman) для контроля химических реакций и для проверки чистоты соединений этого изобретения. Использовалась колонка Dynamax С-18(250 х 4,6 мм; размер пор: 300 А; размер частиц: 12 (м). Система растворителей состояла из двух компонентов:(i) 0,1% TFA в воде и (ii) 0,1% TFA в 70%ном водном ацетонитриле, и использовалась в линейном градиентном режиме с увеличением на 1% (ii) в 1 мин для контроля за химическими реакциями. Эта система использовалась в изократическом режиме для контроля за чистотой.Beckman модель 342 полупрепаративной ВЭЖХ системы использовалась для выделения и очистки соединений этого изобретения. Использовалась колонка Aquapore Octyl (250 х 10 мм; размер пор: 300 А; размер частиц: 15 (м). Система растворителей была такой же, как описанная выше для аналитической ВЭЖХ. Анализ Для структурной идентификации производных доксорубицина использовались BrukerARX300 ЯМР спектрометр (300 МГц 1 Н частота,75 МГц 13 С частота) и масс-спектрометр с электрораспылением Funnigan-Mat TSQ 7000. Синтез пептидных носителей Пептиды этого изобретения часто вводят в форме фармацевтически приемлемых, нетоксических солей, таких как кислотно-аддитивные соли. Иллюстративными такими кислотноаддитивными солями являются гидрохлорид,гидробромид, сульфат, фосфат, фумарат, глюконат, таннат, малеат, ацетат, трифторацетат, цитрат, бензоат, сукцинат, альгинат, памоат, соль яблочной кислоты, аскорбат, тартрат и т.п. Если активный ингредиент должен вводиться в твердой лекарственной форме, эта таблетка может содержать фармацевтически приемлемый разбавитель, который включает связывающее вещество, такой как трагакант, кукурузный крахмал или желатин, дезинтегрирующий агент, такой как альгиновая кислота, и смазочный материал, такой как стеарат магния. Если введение желательно проводить в жидкой форме, подсластитель и/или ароматическое вещество могут использоваться как часть фармацевтически приемлемого разбавителя, может быть эффективным внутривенное введение в изотоническом солевом, фосфатно-буферном растворах или т.п. Фармацевтические композиции могут обычно содержать пептид в конъюгации соcтандартным, фармацевтически пригодным носителем. Обычно, эта дозировка может быть примерно от 1 до 100 мкг пептида на килограмм веса тела хозяина при внутривенном введении,оральные дозировки могут быть намного выше. В общем и целом, лечение субъектов этими пептидами в основном проводится таким же образом, как клиническое лечение, использующее другие аналоги LH-RH, соматостатина и аналоги доксорубицина. Эти пептиды могут вводиться млекопитающим внутривенно, подкожно, внутримышечно, орально, внутриназально или внутривагинально, чтобы добиться биологических гормональных эффектов через связывание со специфическими рецепторами. В случае LH-RH аналогов эти эффекты могут включать обратимую супрессию гонадной активности, и, в случае аналогов соматостатина, ингибирование гастроинтестинальной функции. Эффективные дозировки могут различаться в зависимости от формы введения и особых видов млекопитающих, которые обрабатываются. Примером одной типичной формы дозировки является физиологический солевой раствор, содержащий пептид, который вводится так, чтобы обеспечить дозу в пределах от примерно 0,1 до 2,5 мг/кг веса тела. Оральное введение пептида может производиться либо в твердой форме, либо в жидкой форме. Синтез пептидных носителей настоящего изобретения может быть преобразован любыми методиками, которые известны специалистам в области пептидной химии. Краткое изложение подходящих методик можно найти в М. Bodanszky, Principles of Peptide Synthesis, SpringerVerlag, Heidelberg, 1984. Методики для твердофазного пептидного синтеза могут быть обнаружены в учебнике J.M. Stewart and J.D. Yong, SolidUSA 85, 1637-1641 (1988) и 86, 6318-6322 (1989) и Janaky и соавт., Рrос. Natl. Acad. Sci. USA, 89,1023-1027 и 972-976 (1992). Синтез носителей-аналогов соматостатина,использованных в этом изобретении, представлен детально в примерах патента US 4650787, от 17 марта 1987 г., Andrew V. Schally и Ren Z. Cai. Описание этого синтеза может быть также обнаружено в статьях Cai и соавт., Рrос. Natl. Acad.Acad. Sci. USA 91, 12664-12668 (1994). Синтез производных доксорубицина, использованных в этом изобретении, и получение их конъюгатов с различными пептидными носителями представлен детально в следующих примерах, которые приведены как иллюстративные и не являются ограничивающими. Пример 1. Получение и выделение N-FmocDOX14-O-полуглутаратa. 50 мг (86 моль) соли DOX HCl растворяли в 1 мл DMF и добавляли 30 мг (90 моль) FmocOsu, после чего добавляли 31 мл (180 моль)DIPEA. После перемешивания в течение 3 ч реакция завершалась, что определяли аналитической ВЭЖХ. Растворитель выпаривали до состояния сухости в испарителе с высоким вакуумом Speed Vac и остаток кристаллизовали растиранием с 0,1% TFA в Н 2 О. Кристаллы фильтровали и один раз промывали холодным эфиром для удаления следов избытка Fmoc-Osu. После высушивания в эксикаторе получали 62 мг NFmoc-DOX 98% чистоты. Выход 94%. Этот промежуточный продукт реагировал ночь с 11,4 мг (100 моль) Glt2 О в 1 мл безводного DMF в присутствии 26,1 л (150 моль)DIPEA. Растворитель выпаривали в Speed Vac и оставшееся масло делали отверждали растиранием с 0,1% TFA (объем/объем). Сырье, получен 14 ное таким образом, содержит 70% N-FmосDОХ 14-О-полуглутарата, 20% непрореагировавшего N-Fmoc-DOX и 10% других примесей, что определяли аналитической ВЭЖХ. Этот сырой продукт может быть использован для получения пептидных DOX конъюгатов без дальнейшей очистки. Когда это сырье растворяли в 20 мл 60%-ного водного ацетонитрила, содержащего 0,1% TFA, и наносили на полупрепаративную ВЭЖХ, получали 45,7 мг конечного продукта NFmос-DОХ 14-О-полуглутарата 98% чистоты. Выход 64%. Пример 2. Получение и выделение 3'деамино-3'-(пирролидин-1"-ил)-доксорубицин ТFА соли (Q2) и ее TFA соль 14-O-полуглутарата(AN-193). 50 мг (86 моль) соли DOX HCl растворяли в 1 мл DMF и добавляли 171 л (1,3 ммоль) 15 кратного избытка 1,4-иодбутана, после чего добавляли 45 л (260 моль) 3-кратного избыткаDIPEA. Реакционную смесь перемешивали в течение ночи при комнатной температуре. После 16 ч реакция завершалась, что определяли аналитической ВЭЖХ. Растворитель выпаривали вSpeed Vac и оставшееся масло растворяли в 3 мл 0,1% TFA в Н 2 О и экстрагировали эфиром для удаления избытка 1,4-дииодбутана. Водный экстракт затем наносили на ВЭЖХ и получали 41,6 мг DOX производного 98% чистоты. Выход 68%. 41,6 мг (58 моль) полученной таким образом 3'-деамино-3'-(пирролидин-1"-ил)-доксорубицин TFA соли (Q2) подвергали реакции с 1,2 эквивалентом Glt20 в сухом DMF точно так, как описано в примере 1. Выход составлял 35% (16,9 мг) и чистота равнялась 98%. Пример 3. Получение и выделение 3'деамино-3'-(изоиндолин-2"-ил)-доксорубицин ТFА соли (Q3). 50 мг (86 моль) соли DOX HCl растворяли в 1 мл DMF и добавляли 226 мг (1,3 ммоль) 15 кратного избытка ,'-дихлор-орто-ксилена, после чего добавляли 45 л (260 моль) 3-кратного избытка DIPEA и каталитическое количествоTFA и экстрагировали 3 мл эфира для удаления избытка галогенсодержащего соединения. Сырье, полученное таким образом, наносили на ВЭЖХ. После очистки получали 36 мг конечного продукта 98% чистоты. Выход 55%. Пример 4. Получение и выделение 3'деамино-3'-(3"-пирролин-1"-ил)-доксорубицинTFA соли (Q4). 50 мг (86 моль) соли DOX HCl растворяли в 1 мл DMF и добавляли 136,8 л (1,3 ммоль) 15 кратного избытка цис-1,4-дихлор-2-бутена (Aldrich), после чего добавляли 45 л (260 молей) 3 кратного избытка DIPEA. Через 16 ч растворители удаляли Speed Vac и остаток растворяли в 3 мл 0,1%-ного водного TFA и экстрагировали 3 мл гексана для удаления избытка галогенсодер 15 жащего соединения. Полученное таким образом сырье наносили на ВЭЖХ. После очистки получали 22,6 мг конечного продукта 98% чистоты. Выход 37%. Пример 5. Получение и выделение 1-хлор 4-бром-2-бутанона (C4H6ClBrO) и 1-хлор-5-бром 2-пентанона (C5H8ClBrO). 100,8 л (1 ммоль) 3-бромпропионилхлорида (Aldrich) подвергали реакции с избытком диазометана в эфире. Через 1 ч эфирный раствор элюировали и каплю тестировали на ТСХ. Алюминиевые пластинки для тонкослойной хроматографии с силикагелем 60 F254 MerckArt5554 использовались как неподвижная фаза, и CHCL3:MeOH 95:5 (объем/объем) использовались как подвижная фаза. Для идентификации соединения по пятну реагент 2,4 динитрофенилгидразин (Vоgеl: А tехtbооk оfPractical Organic Chemistry, p. 1061, Third Edition,Longmans, New York) разбрызгивался на ТСХ пластинку после элюции. Таким образом полученное диазометилкетоновое производное проявлялось как желтое пятно с Rf 0.3. Этот эфирный раствор затем подвергали реакции с безводным HCl в эфире, превращая диазометилкетон в желаемый конечный продукт 1-хлор-4-бром-2 бутанон. Этот продукт проявлялся как желтое пятно,характерное для оксосоединений, с Rf 0.8 в той же системе растворителей и с реагентом для идентификации соединения по пятну, описанными выше. После выпаривания растворителя сырье наносили на колонку (15 см длиной, 2,5 см в диаметре), набитую 15 г силикагеля, Merck, марка 9385, меш 230-400, размер пор 60 А. Жидкой подвижной фазой был чистый СНСl3. Фракции,содержащие желаемый конечный продукт (охарактеризованный идентификацией по пятну, детально описанной выше), перемешивали и выпаривали до сухого состояния. Получали прозрачное масло массой 1,5 г. Выход 80%. 1-хлор-5-бром-2-пентанон получали из 4 бромбутирилхлорида точно таким же способом,как описано для 1-хлор-4-бром-2-бутанона, за исключением того, что использовали 4 бромбутирилхлорид вместо 3-бромпропионилхлорида. Получали 1,6 г прозрачного масла. Выход 80%. Пример 6. Получение и выделение 3'деамино-3'-(3"-пирролидон-1"-ил)-доксорубицинTFA соли (Q5). 50 мг (86 моль) соли DOX HCl растворяли в 1 мл DMF и добавляли 241 мг (1,3 ммоль) 15 кратного избытка 1-хлор-4-бром-2-бутанона,после чего добавляли 45 л (260 молей) 3 кратного избытка DIPEA. Через 16 ч растворители удаляли Speed Vac и остаток растворяли в 3 мл 0,1%-ного водного TFA и экстрагировали 3 мл гексана для удаления избытка галогенсодержащего соединения. Полученное таким образом сырье наносили на ВЭЖХ. После очистки полу 16 чали 20,6 мг конечного продукта 98% чистоты. Выход 33%. Пример 7. Получение и выделение 3'деамино-3'-(3"-пиперидон-1"-ил)-доксорубицинTFA соли (Q7). 50 мг (86 моль) соли DOX HCl растворяли в 1 мл DMF и добавляли 260 мг (1,3 ммоль) 15 кратного избытка 1-хлор-5-бром-2-пентанона,после чего добавляли 45 л (260 молей) 3 кратного избытка DIPEA. Через 16 ч растворители удаляли Speed Vac и остаток растворяли в 3 мл 0,1%-ного водного TFA и экстрагировали 3 мл гексана для удаления избытка галогенсодержащего соединения. Полученное таким образом сырье наносили на ВЭЖХ. После очистки получали 18 мг конечного продукта 95% чистоты. Выход 28%. Пример 8. Получение и выделение 4 иодбутиральдегида и 5-иодвалеральдегида. 1,3 мл (10 ммоль) 2-(3-хлорпропил)-1,3 диоксалана (4-хлор-n-бутиральдегид этилен ацеталь) (Fluka) растворяли в 200 мл ацетона, содержащего 30 г (200 ммоль, 20-кратный избыток)Nal. Этот раствор нагревали с обратным холодильником в течение 24 ч, после чего выпаривали до сухого состояния. Использовали 100 мл эфира для экстракции органического материала из неорганического твердого остатка. Этот эфирный раствор затем промывали 50 мл H2O, 50 мл 5%-ного водного Nа 2S2 О 3 раствора и 3 раза 50 мл H2O. Эфир удаляли в вакууме и оставшееся масло растворяли в 3 мл 50%-ной водной уксусной кислоты. Через 1 ч к этому раствору добавляли 100 мл эфира и уксусную кислоту, а также этиленгликоль удаляли, промывая 50 мл Н 2 О 3 раза. Главный продукт элюировался при Rf 0.8 на ТСХ в чистом СНСl3. Идентификацию соединения по пятну, которую использовали для альдегидной функции, проводили таким же образом,как описано для кетонов в примере 5. Затем удаляли эфир и черное масло наносили на колонку(15 см длиной, 2,5 см в диаметре), набитую 15 г силикагеля, Merck, марка 9385, меш 230-400,размер пор 60 А. Жидкой подвижной фазой был чистый СНСl3. Фракции, содержащие желаемый конечный продукт (охарактеризованный идентификацией по пятну, детально описанной выше), перемешивали и выпаривали до сухого состояния. Получали желтое масло массой 1,6 г. Выход 80%. 5-иодвалеральдегид получали точно таким же способом, начиная с 2-(4-хлорбутил)-1,3 диоксолана (5-хлор-n-валеральдегид этилен ацеталя) (Fluka). Получали 1,65 г желтого масла. Выход 80%. Пример 9. Получение и выделение 3'деамино-3'-(2"-пирролин-1"-ил)-доксорубицин чего добавляли 45 л (260 молей, 3-кратный избыток) DIPEA. Через 1 ч добавляли 100 л ледяной уксусной кислоты к этой реакционной смеси, в которую затем по каплям добавляли 5 мл 0,1% TFA в 70%-ном водном ацетонитриле (растворитель ii ВЭЖХ системы). Этот раствор разбавляли 2 мл 0,1%-ного водного раствора TFA,затем удаляли ацетонитрил в Speed Vac. Полученный раствор экстрагировали гексаном, чтобы удалить избыток галогенсодержащего соединения. Полученный таким образом материал наносили на ВЭЖХ. После очистки получали 52 мг конечного продукта 98% чистоты. Выход 85%. Пример 10. Получение и выделение 3'деамино-3'-(1",3"-тетрагидропиридин-1"-ил)доксорубицин ТFА соли (Q8). 50 мг (86 моль) соли DOX HCI растворяли в 1 мл DMF и добавляли 552 мг (2,6 ммоль) 30 кратного избытка 4-иодвалеральдегида, после чего добавляли 45 л (260 молей) 3-кратного избытка DIPEA. Через 1 ч добавляли 100 л ледяной уксусной кислоты к этой реакционной смеси, в которую затем по каплям добавляли 5 мл 0,1% TFA в 70%-ном водном ацетонитриле(растворитель ii ВЭЖХ системы). Этот раствор разбавляли 2 мл 0,1%-ного водного раствораTFA, затем удаляли ацетонитрил в Speed Vac. Полученный раствор экстрагировали гексаном,чтобы удалить избыток галогенсодержащего соединения. Полученный таким образом материал наносили на ВЭЖХ. После очистки получали 46 мг конечного продукта 98% чистоты. Выход 75%. Пример 11. Получение и выделение цитотоксического аналога агониста LH-RH, содержащего DOX. ([D-Lys6(DOX14-O-glt)]LH-RH,Q114gL). 60 мг (37,5 моль) [D-Lys6]LH-RH и 52 мг(Aldrich), 13,5 мг (100 моль) HOBt, а также 52 л (300 моль) DIPEA. После 1 ч перемешивания при комнатной температуре реакция завершалась. Растворители выпаривали и оставшееся масло кристаллизовали 3 мл этилацетата и затем дважды промывали 3 мл этилацетата. Затем растворяли 90 мг этого сырого твердого материала в 3 мл DMF и добавляли 300 мл пиперидина. Через 5 мин реакцию ставили в ледяную баню и подкисляли, добавляя смесь 300 л TFA, 700 л пиридина и 2 мл DMF. После выпаривания растворителей оставшееся масло отверждали этилацетатом. Полученное таким образом твердое сырье растворяли в 1 мл 70%-ного водного ацетонитрила, содержащего 0,1% TFA (i), разбавляли 3 мл 0,1%-ного водного TFA (ii) и наносили на полупрепаративную ВЭЖХ. Получали 40 мг Пример 12. Получение и выделение цитотоксического аналога агониста LH-RH, содержащего 2-пирролин-DОХ. ([D-Lys6(2-пирролинDOX14-O-glt)]LH-RH,Q614gL). 11,2 мг (5 моль) Q114gl (см. пример 11) растворяли в 200 л DMF и добавляли 30 мг (150 моль,30-кратный избыток) 4 иодбутиральдегида (пример 8), затем добавляли 3 л (17 моль) DIPEA. Через 1 ч реакция завершалась (см. пример 9) и 10 л ледяной уксусной кислоты добавляли к реакционной смеси, к которой затем добавляли по каплям 1 мл 0,1% TFA в 70%-ном водном ацетонитриле. Этот раствор затем разбавляли 1 мл 0,1 %-ного водного TFA и ацетонитрил удаляли в вакууме. Затем оставшийся водный раствор экстрагировали 1 мл гексана и наносили на ВЭЖХ. Получали 7,6 мг конечного продукта 99% чистоты. Выход 66%. Пример 13. Получение и выделение цитотоксического аналога соматостатина, содержащего DOX.DIPEA. После перемешивания в течение 1 ч при комнатной температуре реакция завершалась. После удаления растворителей в вакууме остаток кристаллизовали этилацетатом. Затем этот твердый материал растворяли в 1 мл DMF и добавляли 100 л пиперидина. Через 7 мин реакцию ставили в ледяную баню и подкисляли, добавляя смесь 100 л TFA, 300 л пиридина и 2 мл DMF. После выпаривания растворителей оставшееся масло отверждали этилацетатом. Таким образом полученное твердое сырье растворяли в 1 мл 70%-ного водного ацетонитрила, содержащего 0,1% TFA (i), разбавляли 3 мл 0,1%-ного водногоTFA (ii) и наносили на полупрепаративную ВЭЖХ. Получали 9,7 мг (5,1 моль) конечного продукта 95% чистоты. Выход 35%. Пример 14. Получение и выделение цитотоксического аналога соматостатина, содержащего 2-пирролин-DОХ. 6,4 мг (5 моль) растворяли в 100 л DMF и добавляли 2 пиppoлин-DOX14-O-пoлyглyтapaт (4,1 мг, 5 моль), после чего добавляли реагент ВОР (4,4 мг, 10 моль), HOBt (100 моль) и DIPEA (50 моль). После перемешивания в течение 2 ч реакционную смесь подкисляли 20 л АсОН и раз 19DОХ 14-О-полуглутарат получали из NFmос-DОХ 14-О-полуглутарата при отщеплении защитной группы Fmoc, как описано в примере 11. Выход 40%. Пример 15. Получение и выделение цитотоксического антагониста бомбезина, содержащего DOX (DOX14-O-glt-Gln-Trp-Ala-Val-GlyHis-Leu (CH2-NH)Leu-NH2, Q114gB). 20 мг (15,8 моль)HOBt, а также 17 л (100 моль) DIPEA. После перемешивания в течение 1 ч при комнатной температуре реакция завершалась. После удаления растворителей в вакууме остаток кристаллизовали этилацетатом. Затем этот твердый материал растворяли в 1 мл DMF и добавляли 100 л пиперидина. Через 5 мин реакцию ставили в ледяную баню и подкисляли, добавляя смесь 100 л TFA, 300 л пиридина и 2 мл DMF. После выпаривания растворителей оставшееся масло отверждали этилацетатом. Таким образом полученное твердое сырье растворяли в 1 мл 70%ного водного ацетонитрила, содержащего 0,1%(ii) и наносили на полупрепаративную ВЭЖХ. Получали 13,5 мг (7,1 моль) конечного продукта 98% чистоты. Выход 45%. Пример 16. Получение и выделение цитотоксического антагонистического аналога бомбезина,содержащего 2-пирролин-DОХ 2-пиppoлинDOX14-O-glt-Gln-Trp-Ala-Val-Gly-His-Leu(CH2NH)Leu-NH2, Q614gB. 9,5 мг (5 моль) Q114gB (см. пример 15) растворяли в 200 л DMF и добавляли 30 мг (150 моль,30-кратный избыток) 4 иодбутиральдегида (пример 8), затем добавляли 3 л (17 моль) DIPEA. Через 1 ч реакция завершалась (см. пример 9) и 10 л ледяной уксусной кислоты добавляли к реакционной смеси, к которой затем добавляли по каплям 1 мл 0,1% TFA в 70%-ном водном ацетонитриле. Этот раствор затем разбавляли 1 мл 0,1%-ного водного TFA и ацетонитрил удаляли в вакууме. Затем оставшийся водный раствор экстрагировали 1 мл гексана и наносили на ВЭЖХ. Получали 6 мг конечного продукта 98% чистоты. Выход 60%. Определение цитотоксической активности Клеточную линию МХТ эстроген независимой карциномы молочной железы мыши получили от Dr. Gunter Berndhardt, University of Regensburg, Германия. Все другие клеточные линии, использованные для определения антипролиферативной активности соединений этого изобретения, получали из American Type CultureCollection (ATCC). Для оценки активности аналогов использовали колориметрический цитотоксический анализ в планшетах для микротитрования, основанный на определении количества биомассы окрашиванием клеток кристаллическим фиолетовым,которое очень хорошо коррелирует с определением количества клеток. (Reile et al; Anal. Biochem. 187, 262-267, 1990; Bernhardt G. et al, Cancer Res. Clin. Oncol. (1992), 118, 35-43; Spruss Th.et al, J. Cancer Res. Clin. Oncol. 117, 435-443,1991; Gillies, R.J., Anal. Biochem. 159, 109-113,1986; Kueng, W. et al; Anal. Biochem., 182, 16-19,1989.) Протокол анализа Через один или два дня после посева клеток в 96-луночные планшеты культуральная среда заменяется на свежую среду, содержащую соединения, которые тестируются, и пустую свежую среду в случае контрольных культур. После различного времени инкубации клетки фиксируются глутаровым диальдегидом и сохраняются под фетальной бычьей сывороткой (FBS) при 4 С до конца эксперимента. Клетки окрашиваются кристаллическим фиолетовым, и связанная краска экстрагируется 70%-ным водным ЕtOН. Оптическая плотность измеряется EIA Reader(Bio-Tek Instruments) или Biomek 1000 (Beckman) на 590 нм или 600 нм соответственно. Каждая точка данных является средним значением из восьми культуральных лунок. Т/С значения высчитываются как Т/С=(Т-Со)/(С-Со),где Т=оптической плотности обработанных культур,С=оптической плотности контрольных (необработанных) культур, Со=оптической плотности культур в начале инкубации (t=0). Пример 17. Цитотоксическая активность invitro производных DOX, модифицированных даунозамином. Табл. 17-1 демонстрирует воздействие доксорубицина и его производных, модифицированных даунозамином, на MCF-7 клеточную линию карциномы молочной железы человека in vitro. Цитотоксические радикалы, содержащие даунозаминовый N, включенный в пятичленное кольцо с реакционной функцией, являлись в 5-50 раз более активными, чем их гомологичный аналог c шестичленным кольцом, как, например, 3 пирролидон-DOX (Q5) и 3-пиперидон-DOX (Q7),а также 2-пирролино-DОХ (Q6) и 1,3 тетрагидропиридин-DОХ (Q8). Воздействия доксорубицина и его производных, модифицированных даунозамином, на MCF-7 клеточную линию карциномы молочной железы человека in vitro Т/С значение при, М Соединение Время инкубации, ч 310-10 10-9 310-9 10-8 310-8 10-7 Доксорубицин 70 98 82 54-9 1,3-Тетрагидро 70 96 88 69 пиридин-DOX (Q8) 120 99 93 62 благодаря обработке. Следует отметить, 75 означает 75% выживших клеток по сравнению со 100% контроля или 25% ингибирования. Клетки инкубировали в IМЕМ среде, соПример 18. Полное сохранение цитотоксидержащей 5% HI-DCC-FBS (нагретая инактивической активности DOX в конъюгате Q114gL с рованная нагруженная декстраном угольная пыль, обработанная фетальной бычьей сыворотпептидом-агонистом LH-RH и сверхактивного кой), на 96-луночных планшетах. Относитель 2-пирролин-DОХ (Q6) в конъюгате Q614gL с пепное количество клеток в обработанных и контидом-агонистом LH-RH in vitro. трольных планшетах определяли методом окТаблица демонстрирует воздействие докрашивания кристаллическим фиолетовым и высорубицина и его производного, модифицироражали в значениях Т/С, где Т/С=(Т-Со/Сванного даунозамином,2-пирролиндоксоСо)х 100 [Т=поглощению обработанных кульрубицина (Q6) в сравнении с их конъюгатами с тур, С=поглощению контрольных культур,LH-RH агонистическим аналогом, [D-Lys6]LHСо=поглощению культур в начале инкубаRH (Q114gL и Q614gL соответственно) на ростMCF-7 клеточной линии карциномы молочной железы человека и клеточной линии МХТ эстции (t=0). Измеренное поглощение пропорциороген независимой карциномы молочной желенально количеству клеток.] зы мыши in vitro. Более низкие значения Т/С показывают уменьшение жизнеспособности раковых клеток Таблица 18-1 Соединение Т/С значение для MCF-7 клеточной линии при концентрации, М 310-11 Т/С значение для МХТ клеточной линии при концентрации, М 10-10 310-10 10-9 310-9 10-8 310-8 310-11 85MCF-7 клетки инкубировали в IMEM среде, содержащей 5% HI-DCC-FBS, в 96-луночных планшетах. МХТ клетки инкубировали вin vitro цитотоксическая активность аналогов соматостатина этого изобретения, содержащих Таблица 19-1 Воздействие цитотоксических аналогов соматостатина, содержащих доксорубицин, на рост клеточной линии MIIA РаСа-2 раковой опухоли поджелудочной железы человека in vitro Соединение Пример 20. Воздействие цитотоксических аналогов антагонистов бомбезина, содержащих доксорубицин, на рост клеток CFPAC-1 раковой опухоли поджелудочной железы человека invitro. Табл. 20-1 демонстрирует, что in vitro цитотоксическая активность антагонистических аналогов бомбезина этого изобретения, содержащих DOX, полностью сохраняется. Сохраненные связывающие свойства производных гормонов. Пример 21. Гормональные активности и рецепторные связывающие возможности цитотоксических аналогов-агонистовIC50 значение для гипофи- IC50 значение для резарных рецепторов крыс,цепторов раковой опухоли нМ грудной железы, НМ[D-Lys6]LH-RH 2,26 1,80 Ингибирование человеческого гормона роста - высвобождающегося гормона. В таблице 21-1LH отклики для аналогов Высвобождение гормона роста, вызванное определяли в диспергированных клетках гипо(hGH-RH(1-29)NH2), из опрыскиваемых крысифиза крысы, как описано S. Vigh и A.V. Schally в Peptides 5, 241-247 (1984). ных гипофизарных клеток аналогами сомато Аффинность связывания аналогов с гистатина S-98-I пофизарными рецепторами LH-RH крыс и рецепторами раковой опухоли грудной железы человека определяли в экспериментах конкурентного связывания, используя [125l] мечени S-121 ный [D-Trp6]LH-RH в качестве лиганда, как описано В. Szoke и соавт. В Peptides, 15(2), 359366 (1994). Аффинность связывания выражали в значениях IC50, концентрации немеченного в сравнении с их цитотоксическим производаналога, требуемой для ингибирования 50% ным,Q114gS98-1(DOX14-O-glt-S-98-1) и 121 14 специфического связывания радиоактивногоQ1 gS (DOX14-O-glt-S-121) соответственно. лиганда. В крысиную гипофизарную систему для Пример 22. Аналоги соматостатина ингивливания аналоги соматостатина вводили в тебируют секрецию гормона роста (GR) из опрычение 3 мин в виде 1 нМ дозы одновременно с 1 скиваемого крысиного гипофиза, как описано нМ hGH-RH(1-29)NH2. Настаивание с соматоCarlson и соавт. в Thyrotropin-releasing статиновыми аналогами проводили в течениеhypophyses, Endocrinolgy, 94, 1709-(1974). Сооттечение опрыскивания соматостатиновыми анаветственно этот метод использовали для сравлогами (0 мин) и через 30, 60 и 90 мин после нения гормональных активностей цитотоксичепрекращения введения. Данные представлены в ских аналогов соматостатина настоящего изотабл. 22-1. бретения с исходными молекулами носителя. Таблица 22-1GH высвобождение, вызванное 3 мин введением 1 Аналоги нМ hGH-RH(1-29)NH2 в различных временных точках соматостатина после настаивания с аналогами соматостатина 0 мин 30 мин 60 мин 90 мин Выражается как процент высвобождения GH, вызванного 3 мин введением 1 нМ hGH-RH(129)NH2 дo введения аналогов соматостатина. Пример 23. Изучение рецепторного связывания цитотоксических антагонистов бомбезина. Радиоактивное иодирование [Tyr4]BNRadio iodination kit, и выделение моноиодированных [l125-Tyr4]BN проводили как описано ранее (1). Связывание меченого [Tyr4]BN и замещение цитотоксическим бомбезиновым антагонистическим аналогом, Q614gВ проводили, используя сливающиеся Swiss 3T3 клетки(2) метода Kris и соавт. (3). Через три-пять дней после посева сливающиеся клетки промывали дважды солевым сбалансированным раствором Хэнка (HBSS) и инкубировали в течение 30 мин при 37 С с 50 рМ [l125-Tyr4]BN в отсутствие или присутствие нескольких концентраций немеченных конкурентов (Q614gB или BN) в общем объеме 0,5 мл связывающего буфера (DMEM с 50 мМ HEPES, 0,1% бычьего сывороточного альбумина (BSA), 5 мМ MgCl2 и 100 (г/мл бацитрацина, рН 7,4). Неспецифическое связывание определяли в присутствии 1 М немеченного лиганда. После трех промываний охлажденным во льду HBSS, содержащим 0,1% BSA (рН 7,4) клетки отделяли 0,05% трипсин/0,53 мМ ЭДТА раствором и переносили в пробирки. Радиоактивность измеряли гамма-счетчиком (Micromedic Systems Inc, Huntsville, AL). Данные связывания оценивали, используя программы McPherson анализа связывания радиоактивных лигандов(5). 1. Halmoss, et al., Cancer Letters, 85, 111118 (1994). 2. Cai, et al., Proc. Natl. Acad. Sci., USA 91:12664-12668, (1994). 3. Kris, et al., L. Biol. Chem, 262: 1121511220, (1987). 4. McPherson, G.A., J. Pharmaco Methods,14: 213-228, (1985). 5. Cheng and Prusoff, Biochem. Pharmacol. 22:3099-3108, (1973). Таблица 23-1 Характеристика специфического связывания цитотоксического антагониста бомбезинаQ614gB(2-пиppoлин-DOX14-O-glt-Gln-Trp-AlaVal-Gly-His-Leu-(-(CH2-N)Leu-NH2 с рецепторами бомбезина на клеточной линии Swiss 3T3 в сравнении с бомбезином Соединение Сравнительная эффективность и токсичность конъюгатов гормонов против цитотоксического свободного радикала. Пример 24. Обработка 2-пирролин-DОХ(Q6), цитотоксическим аналогом агониста LHRH Q614gL([D-Lys6]LH-RH, связанный с Q614-Ополуглутаратом) и (DOX) эстроген независимых МХТ раковых опухолей молочной железы мыши (KS-49). Для того чтобы сравнить опухолевую ингибиторную активность цитотоксического производного доксорубицина, Q6, и его направленного цитотоксического пептидного конъюгата, Q614gL, а также хорошо известного противонеопластического агента, DOX, и чтобы определить оптимальный путь введения и нетоксические дозы, наносили LH-RH рецептор положительной МХТ (3.2) ovex опухоли(1 мм 3) имплантировали подкожно в самок мышей B6D2F1. Через день после трансплантации мышей произвольно разделили на группы по пять животных и начали обработку. Соединения растворяли в 0,1% трифторуксусной кислоте (рН 2) и вводили внутрибрюшинно. Группы, расписания обработки и дозы, а также средние времена выживания представлены в табл. 24-1. Результаты суммированы в табл. 24-2 и фиг. 1. Табл. 24-2 показывает воздействие обработки Q6 и цитотоксическим аналогом LH-RHQ614gL на объемы опухолей и выживание мышей с эстроген независимыми раковыми опухолями молочной железы. Как показано в табл. 242, 1,25 нмоль Q6, введенного на 1, 2, 7, 8, 14 и 15 дней, вызывало сильную токсичность, охарактеризованную средним временем выживания 17,4 дня, которое значительно короче, чем у необработанных групп. Для сравнения, такая же доза Q614gL приводила к среднему времени выживания 30,8 дней, которое значительно длиннее, чем у необработанной контрольной группы. Более высокая эффективность Q614gL, чем у Q6,может также быть продемонстрирована при сравнении средних конечных объемов опухолей в группе 2 (1065 мм 3 на 16 день) и в группе 6(863 мм 3 на 31 день). Подобные результаты могут быть продемонстрированы при сравнении Q6 и Q614gL в различном расписании обработки, где 0,5 нмоль лекарств вводились пять дней в неделю в течение трех последовательных недель. Доксорубицин в токсической дозе (общее количество: 1560 нмоль, среднее время выживания: 20 дней) не мог уничтожать опухоль, тогда как обработка Q614gL в нетоксической дозе (общее количество: 7 нмоль, среднее время выжи 29 С 9 по 12 день доза возросла до 2,5 нмоль. С 9 по 12 день доза возросла до 5,0 нмоль. Выживание значительно дольше (р 0.01) Выживание значительно короче (р 0.01) или (р 0.05) при сравнении с контрольными данными при использовании теста Дункана. Пример 25. Воздействие одиночной обработки DOX, цитотоксических LH-RH аналогов Т-107 и Q114gL на эстроген независимые МХТ раковые опухоли молочной железы мыши (КS55). Тестовые соединения:DOX Анализ проводили следующим образом: Чтобы определить максимальные переносимые дозы и сравнить воздействия, кусочки МХТ (3.2) ovex опухоли (1 мм 3) имплантировали подкожно в самок мышей B6D2F1. Через день после трансплантации мышей произвольно разбивали на группы по пять животных и обраба тывали их одиночной инъекцией внутрибрюшинно. Группы и дозы представлены в табл. 251. Таблица также показывает число мышей, которые имели опухоли, когда измерялся объем и средние времена выживания для групп. Изменения объемов опухолей представлены на фиг 2. Соединения растворяли в 0,1% TFA (рН 2,0). Объем опухоли измеряли на 10, 13, 17 и 20 дни. Как показано в табл. 25-1 и на фиг. 2, Т 107, ([D-Lys6]LH-RH, связанный с N-глутарилDOX), совершенно неэффективен в ингибировании роста этой опухоли при дозе 850 нмоль/ 20 г мыши. Напротив, Q114gL, ([D-Lys6]LH-RH,связанный с 14-O-глутарил-DОХ) проявляет сильную супрессию роста опухоли (фиг.) при нетоксической дозе 650/20 г мыши. DОХ сам по себе был высокотоксичен (среднее время выживания: 13,6 дней) при одиночной дозе 650 нмоль/20 г мыши и значительно менее эффективен, чем Q114gL (фиг. 2). Время выживания значительно короче (р 0,01), чем в контролях.Время выживания значительно длиннее (р 0,01) или(р 0,05) по сравнению с контролем (одна мышь, которая случайно умерла на 2 день, выпала из этих двух групп). Пример 26. Воздействие цитотоксическихLH-RH аналогов на эстроген независимые МХТ раковые опухоли молочной железы мыши (KS47). Вещества, использованные для обработки. В более раннем эксперименте Q2 при ежедневной дозе 20 нмоль в течение 17 дней обнаруживал только среднее ингибиторное воздействие на рост опухоли, и он был токсичным при дозе 40 нмоль (среднее время выживания составляло 14,6 дней). Для настоящего эксперимента выбрали ежедневную дозу 30 нмоль, и сравнивали эффективность и токсичность[D-Lys6]LH-RH + Q2. Кусочки МХТ (3.2) ovex опухоли (1 мм 3) трансплантировали в самку мыши B6D2F1. Обработку начали через день после трансплантации и проводили в течение 12 дней посредством внутрибрюшинных инъекций один раз в день. Все группы получали эквимолярное количе ство соединений как показано в табл. 26-1. Опухоли измеряли на 10, 14 и 18 дни и рассчитывали объем опухоли. Данные представлены в табл. 26-1 и фиг. 3. Обработка ежедневной дозой 30 нмоль даунозамином, модифицированным доксорубициновым аналогом Q2 (пирролидин-DOX) приводила к сильному ингибиторному воздействию на рост опухоли (объем опухоли: 144 мм 3 на 14 день против 1391 мм 3 для контрольной группы),но вызывала сильную токсичность, убивая всех животных до конца эксперимента (среднее время выживания 17,9 дней). Подобным образом,Q2 комбинированный (смесь) с [D-Lys6]LH-RH,приводил к сильному ингибиторному воздействию на опухоль (объем опухоли: 80 мм 3 на 14 день), но среднее время выживания (18,5 дней) было значительно короче, чем у необработанной контрольной группы (23,1 день). В результате обработки Q214gL (Q2, ковалентно связанный с [D-Lys6]LH-RH) два животных умерли, 35 одно на 16 день и второе на 26 день. Из 8 выживших животных только у одного развились опухоли при последнем измерении на 18 день, и все животные выглядели здоровыми, но позднее у всех из них начали развиваться опухоли. Среднее время выживания для этой группы было значительно длиннее (28,3 дня), чем у контрольной группы. Обработка чистым [DLys6]LH-RH не влияла на рост опухоли. 36 Этот эксперимент показывает, что более высокая эффективность и более низкая периферическая токсичность Q214gL над цитотоксическим радикалом Q2 могут быть отнесены к ковалентному конъюгированию цитотоксического радикала с направленным носителем-аналогом Таблица 26-1 Воздействие цитотоксических аналогов LH-RH на рост эстроген независимых МХТ раковых опухолей молочной железы мыши и выживание мышей с опухолями Все ежедневные дозы являются 30 нмоль эквимолярными количествам. Значительно короче, чем контроль (р 0,05)Значительно длиннее, чем контроль (р 0,01) с тестом Дункана. Пример 27. Воздействия 2-пирролиноDОХ (Q6) и цитотоксического аналога агонистаQ614-О-полуглутаратом) на рост андроген зависимых Dunning R-3327-H раковых опухолей предстательной железы крыс. Самцов крыс Copenghagen, несущие гормон-зависимые Dunning R-3327-H раковые опухоли предстательной железы крыс, обрабатывали Q614gL, новым цитоксическим аналогом лютеинизирующего гормон-высвобождающего гормона (LH-RH), состоящего из агониста [DLys6]LH-RH, связанного с 2-пирролиндоксорубицином. В первом эксперименте 2-пирролиндоксорубицин вводили в концентрации 50 нмоль/кг как отдельное лекарство (Q6), и как неконъюгированную смесь с [D-Lys6]LH-RH,или конъюгированную с носителем [D-Lys6]LH-RH (Q614). После второго введения 50 нмоль/кг чистого радикала Q6 или смешанного с [D-Lys6]LH-RH, все крысы умирали с признаками общей токсичности, тогда как все животные, обработанные цитотоксическим LH-RH конъюгатом Q614gL, выживали. После 5 недель обработки общей дозой 150 нмоль/кг Q614gL опухоли уменьшились от первоначального объема 8,35 1,7 см 3 в начале эксперимента до 4,47 0,8 см 3, тогда как опухоли в контрольной группе продолжали расти и при измерении составили 17,842,2 см 3. Терапия Q614gL также значительно уменьшила вес опухоли и burden опухоли. В втором эксперименте, проведенном для сравнения, эффективности и токсичности Q6 и Q614gL, терапевтический режим состоял из 3 применений 25 нмоль/кг Q6 или 25 нмоль/кг и 50 нмоль/кгQ614gL. Когда начали обработку, объем опухолей во всех группах находился между 3,9 и 4,5 см 3. Через 5 недель терапии опухоли в крысах,обработанных 50 нмоль/кг Q614gL, уменьшались до 2,3 0,51 см 3, тогда как 25 нмоль/кг были тем не менее токсичны и могли только производить уменьшение в конечном объеме опухоли до 6,76 371,4 см 3, подобно тому, что получено для 25 нмоль/кг Q614gL (6,741 см 3), при сравнении с 15,6 2,2 см 3 для необработанных животных. Гистологическая оценка образцов показала значительное уменьшение митотических клеток только в Q614gL обработанных группах. LH-RH рецепторы с высокой связывающей емкостью были обнаружены на мембранах необработанных Dunning опухолевых образцов, но после обработки Q614gL никаких связывающих сайтов для LH-RH не смогли обнаружить. Ингибирование роста опухоли посредством AN-201 иQ614gL также связано со значительным уменьшением связывающей емкости EGF рецепторов. Как показано фиг. 4-6, направленный цитотоксический LH-RH аналог Q614gL является эффективным противоопухолевым агентом, вызывая регрессию Dunning R-3327-H раковых опухолей предстательной железы у крыс. Наши исследования также показывают, что цитотоксическийLH-RH аналог Q614gL является гораздо менее токсичным, чем введенный противонеопластический радикал (Q6), и значительно более активным в ингибировании роста опухоли. Подписи к фиг. для примера 27: Фиг. 4. Эксперимент I: Объем опухоли у самцов крыс Copenghagen, несущих трансплантаты крысиной Dunning R-3327-H раковой опухоли предстательной железы, в течение обработки, состоящей из 3 применений 50 нмоль/кг агониста [D-Lys6]LH-RH и 50 нмоль/кг цитотоксического LH-RH аналога Q614gL. Вертикальные линии показывают SEM. р 0,05; р 0,01 против контроля посредством нового, со множеством пределов теста Дункана. Обработка, показанная стрелками, применялась на 1, 8 и 29 дни. Животные, обработанные Q6, как отдельным лекарством, или неконъюгированной смесью с [D-Lys6]LH-RH, умерли на вторую неделю. В этих двух группах показан объем опухолей, зафиксированный на 8 день. Фиг. 5. Эксперимент II: Воздействие обработки 25 нмоль/кг 2-пирролиндоксо-рубицина (Q6), 25 нмоль/кг и 50 нмоль/кг цитотоксического LHRH аналога Q614gL на объем опухоли в крысах сDunning R-3327-H раком предстательной железы. Вертикальные линии показывают SEM. р 0,05; р 0,01 против контроля. Обработка,показанная стрелками, применялась 3 раза, на 1,8 и 29 дни. Фиг. 6. Эксперимент II: Воздействие обработки 25 нмоль/кг 2-пирролиндоксо-рубицина(Q6), 25 нмоль/кг и 50 нмоль/кг цитотоксического LH-RH аналога Q614gL на массу тела крысCopenghagen, несущих Dunning R-3327-H раковые опухоли предстательной железы. Вертикальные линии показывают SEM. р 0,05,р 0,01 против контроля. Обработка, показанная стрелками, применялась 3 раза, на 1, 8 и 29 дни. Пример 28. Сравнительное исследование воздействий доксорубицина (DOX) и направ 001372 38 ленного цитотоксического аналога агониста LHRH Q114gL ([D-Lys6]LH-RH, связанный с DOX14O-полуглутаратом) на рост OV-1063 человеческой овариальной раковой опухоли в голых мышах. Человеческую эпителиальную овариальную раковую клеточную линию OV-1063 получали из метастазирующей сосочковой цистаденокарциномы из яичника 57-летней пожилой женщины (Horowitz et.al. (1985) Oncology 42,332-337). Десять миллионов клеток OV-1063 вводили подкожно трем голым мышам для роста опухолей. Кусочки размером 1 мм 3 из этих опухолей трансплантировали в 60 животных для изучения ингибирования роста in vivo. Цель эксперимента заключалась в демонстрации того,что в результате присутствия рецепторов дляLH-RH был более эффективен и менее токсичен,чем DOX, который содержал цитотоксический радикал. Таким образом, воздействия цитотоксического LH-RH конъюгата сравнивали с воздействиями DOX, смесью DOX с молекулойносителем, чистым носителем и необработанными контрольными группами. Все инъекции вводились внутрибрюшинно. Соединения растворяли в 0,9% хлориде натрия в воде (солевой раствор). Мыши со средним размером опухоли приблизительно 15 мм 3 делились на шесть групп по девять животных и получали следующую обработку через 7 дней после трансплантации опухоли: группа 1, солевой раствор; группа 2,Q114gL дозой 700 нмоль/20 г животного; группа 3, Q114gL дозой 413 нмоль/20 г животного (максимальная толерантная доза, МТД для DOX); группа 4, DOX дозой 413 нмоль/20 г животного(МТД); группа 5, смесью 700 нмоль/20 г DOX и 700 нмоль/20 г [D-Lys6]LH-RH; группа 6, носитель - агонистический аналог [D-Lys6]LH-RH дозой 700 нмоль/20 г животного. Рецепторный анализ OV-1063 показал присутствие высокоаффинных связывающих сайтов для LH-RH. Результаты: как показано на фиг. 7, сильное ингибирование роста опухоли достигалось при обработке Q114gL дозой 413 нмоль/20 г(группа 3). Животные на проявляли признаков сильной токсичности. Для сравнения, обработкаDOX, вводившимся в той же дозе 413 нмоль/20 г (12 мг/кг, МТД, группа 4) не привела к значительному ингибированию роста опухоли в трех животных, выживших к концу эксперимента. Три животных умерли на пятый день, и шесть животных умерли на девятый день из-за токсичности. При более высокой дозе (700 нмоль/20 г, группа 2) Q114gL показывал очень сильное ингибирование роста опухоли (фиг. 7). Два из девяти животных умерли вследствие токсичности и одно животное погибло случайно. Шесть выживших животных оправлялись от примерно 20% потери веса в конце эксперимен 39 та. В группе 6, такая же высокая доза (700 нмоль/20 г) DOX смешивалась с 700 нмолями[D-Lys6]LH-RH. На 5 день все животные в этой группе умерли в результате сильной токсичности. Выводы: Наши результаты явно демонстрируют, что, благодаря присутствию рецепторов для LH-RH на клетках эпителиального овариального рака OV-1063, направленный цитотоксический LH-RH конъюгат Q114gL показывает более низкую токсичность и более высокую противоопухолевую активность, чем доксорубицин (Q1), который содержит цитотоксический радикал. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I где -R- означает одинарную связь или -С(O)(СН 2)n-С(O)- и n равняется 0-7, R' выбран из группы, состоящей из NH2, ароматического или гидрированного 5- или 6-членного гетероцикла,имеющего, по крайней мере, один азот в кольце и такой гетероцикл, имеющий бутадиеновую часть, присоединенную к соседним углеродным атомам вышеупомянутого кольца, образует бициклическую систему, а Р означает Н или пептид, при условии, что когда R' означает NH2,тогда R-P означает другое, чем Н, и когда R-P означает Н, тогда R' означает другое, чем NH2. 2. Соединение по п.1, отличающееся тем,что R' выбран из группы, состоящей из NH2,пирролидин-1-ила,изоиндолин-2-ила,3 пирролин-1-ила,3-пирролидон-1-ила,2 пирролин-1-ила,3-пиперидон-1-ила,1,3 тетрагидропиридин-1-ила, и Р означает Р 1, Р 2 и Р 3,где Р 1 выбран из группы, включающей аналогHis, Ccc означает Тrр и Ddd означает Gly-NH2,если Ааа означает Ac-D-Nal(2), тогда Bbb означает D-Phe(4Cl), Ccc означает D-Pal(3), DTrp и Ddd означает D-Ala-NH2; и если Ааа-Вbb-Ссс означают Ас, там Ddd означает -NН-СН 2-СН 3,при этом группа Q14-O-R- образует карбоксамидную связь со свободной аминогруппой остатка D-Lys или, по крайней мере, с одной из 40 свободных аминогрупп A2Bu или А 2 Рr, когда они находятся в (Ххх),Р 2 означает аналог соматостатина формулы причем если Ааа означает D-Phe, тогда Bbb означает Туr, Ccc означает Val и Ddd означает Тhr или Тrр,если Ааа означает D-Trp, тогда Bbb означает Рhе, Ccc и Ddd являются Тhr,при этом группа Q14-O-R- образует карбоксамидную связь с концевой аминогруппой остатка Ааа,Р 3 означает аналог антагониста бомбезина формулыAaa-Gln-Trp-Ala-Val-Gly-His-Leu-Bbb-NH2,где Ааа означает ничто или D-Phe, Bbb означает (CH2-NH)Leu, (CH2-Nh)Phe или (CH2NH)Trp, или (СН 2-N)Tас, причем группа Q14-OR- образует карбоксамидную связь с концевой аминогруппой остатка Ааа, если он присутствует, или с Gln, если Ааа отсутствует. 3. Соединение по п.2, отличающееся тем,что n=3. 4. Соединение по п.3, отличающееся тем,что R означает NН 2. 5. Соединение по п.3, отличающееся тем,что R' означает 2-пирролин-1-ил. 6. Соединение по п.4, отличающееся тем,что Р означает Р 1. 7. Соединение по п.5, отличающееся тем,что Р означает Р 1. 8. Соединение по п.4, отличающееся тем,что Р означает P2. 9. Соединение по п.5, отличающееся тем,что Р означает Р 2. 10. Соединение по п.4, отличающееся тем,что Р означает P3. 11. Соединение по п.5, отличающееся тем,что Р означает P3. 12. Соединение по п.1, отличающееся тем,что -R-P означает -Н и R' означает другое, чемGlp-His-Trp-Ser-Tyr-D-Lys(Q114-O-glt)-Arg-LeuPro-Gly-NH2,где Q114 означает доксорубицин-14-ил. 19. Соединение по п.1, отличающееся тем, что является соединением формулы где Q614 означает 3'-деамино-3'-(2"-пирролин-1"ил)-доксорубицин-14-ил. 20. Соединение по п.1, отличающееся тем,что является соединением формулы где Q114 означает доксорубицин-14-ил. 21. Соединение по п.1, отличающееся тем,что является соединением формулы где Q614 означает 3'-деамино-3'-(2"-пирролин-1"ил)-доксорубицин-14-ил. 22. Соединение по п.1, отличающееся тем,что является соединением формулы где Q114 означает доксорубицин-14-ил. 23. Соединение по п.1, отличающееся тем,что является соединением формулы где Q614 означает 3'-деамино-3'-(2"-пирролин-1"ил)-доксорубицин-14-ил. 24. Соединение по п.1, отличающееся тем,что является соединением формулы(CH2-NH)Leu-NH2,где Q614 означает 3'-деамино-3'-(2"-пирролин-1"ил)-доксорубицин-14-ил. 26. Композиция, включающая соединение по п.1 и фармацевтически приемлемый носитель. 27. Способ лечения рака у млекопитающего, включающий введение млекопитающему, в случае необходимости вышеупомянутого лечения, эффективного количества соединения по п.1. Изменения объема эстроген независимых МХТ раковых опухолей молочной железы мыши в КS-29(Номера курсивом показывают выживших мышей за время эксперимента)LH-RH на выживание мышей с эстроген независимыми МХТ опухолями. Фиг. 1 Изменения объема эстроген независимых МХТ опухолей в KS-29. (выбранные группы) Ингибирование роста OV 1063 человеческих овариальных раковых ксенотрансплантатов в голых мышах посредством цитотоксического аналога LH-RH,содержащего доксорубицин (Q114gL), и доксорубицином (Q1). р 0,05,р 0,01 МТД максимальная толерантная доза Фиг. 7
МПК / Метки
МПК: C07K 7/23, A61P 35/04, A61K 47/48
Метки: лечения, антрациклина, цитотоксические, направленные, композиция, аналоги, млекопитающего, способ, рака
Код ссылки
<a href="https://eas.patents.su/24-1372-napravlennye-citotoksicheskie-analogi-antraciklina-kompoziciya-i-sposob-lecheniya-raka-u-mlekopitayushhego.html" rel="bookmark" title="База патентов Евразийского Союза">Направленные цитотоксические аналоги антрациклина, композиция и способ лечения рака у млекопитающего.</a>
Производные пиридо[2,3-d]пиримидина и их фармацевтически приемлемые соли, фармацевтическая композиция, обладающая ингибирующей протеин-тирозин-киназу активностью, способ лечения заболеваний, опосредованных клеточной пролиферацией, и способ лечения рака, атеросклероза, псориаза или рестеноза

Номер патента: 897
Опубликовано: 26.06.2000
Авторы: Пэнек Роберт, Блэнкли Клифтон Джон, Босчелли Дайянэ Хэррис, Доэрти Эннет Мэриэн, Хэмби Джэймс Марино, Клачко Сильвестер
МПК: A61K 31/519, C07D 471/04, A61P 35/00...
Метки: ингибирующей, пиридо[2,3-d]пиримидина, лечения, пролиферацией, приемлемые, активностью, композиция, атеросклероза, опосредованных, фармацевтически, способ, протеин-тирозин-киназу, обладающая, соли, фармацевтическая, рестеноза, рака, клеточной, производные, псориаза, заболеваний
Формула / Реферат:
1. Производные пиридо[2,3-d]пиримидина общей формулы (I) где X означает иминогруппу, N-ацил, кислород или серу; R1 означает группы NR3R4S(O)mR3, где m является 0,1 или 2, или ОR3; R2, R3 и R4 независимо означают водород, группу (СН2)nPh, где Ph является фенилом, не замещенным или замещенным 1-3 остатками из группы, включающей галоген, гидроксил, карбоксил, алкил с 1-6 атомами углерода, не замещенный или замещенный гидроксилом,...
Липосомальная вакцинная композиция, применение липосомальной вакцинной композиции, способ приготовления липосомальной вакцинной композиции и способ лечения млекопитающего.

Номер патента: 839
Опубликовано: 24.04.2000
Авторы: Гарсон Натали Мари-Жозеф Клод, Фрид Мартин
МПК: A61K 39/39
Метки: способ, вакцинной, млекопитающего, применение, приготовления, липосомальной, вакцинная, лечения, липосомальная, композиция, композиции
Формула / Реферат:
1. Липосомальная вакцинная композиция, содержащая антиген или антигенную композицию, иммунологически активную сапониновую фракцию и стерин, характеризующаяся тем, что отношение сапониновая фракция : стерин составляет от 1:1 до 1:100 (по массе). 2. Вакцинная композиция по п.1, отличающаяся тем, что отношение сапониновая фракция : стерин составляет от 1:1 до 1:5 (по массе). 3. Вакцинная композиция по п.1, отличающаяся тем, что отношение...
Способ нормализации липопротеидного профиля плазмы крови у млекопитающего и фармацевтическая композиция для его осуществления

Номер патента: 514
Опубликовано: 28.10.1999
Автор: Бокен Томас
МПК: A61K 31/40
Метки: способ, нормализации, осуществления, плазмы, композиция, профиля, крови, млекопитающего, фармацевтическая, липопротеидного
Формула / Реферат:
1. Способ нормализации липопротеидного профиля плазмы крови у млекопитающего, отличающийся тем, что млекопитающему вводят фармацевтическую композицию, содержащую терапевтически эффективные количества ингибитора ацил-КоА-холестерин-О-ацилтрансферазы (АКоА ХАТ) и ингибитора 3-гидрокси-3-метил-глутарил-КоА-редуктазы (ГМГ-КоА-редуктазы). 2. Способ по п.1, отличающийся тем, что ингибитор АКоА ХАТ представляет собой...
Применение олигосахарида и аспирина для лечения тромбоэболических заболеваний, фармацевтическая композиция, способ лечения

Номер патента: 48
Опубликовано: 30.04.1998
Авторы: Стикема Якобус, Кариу Роже
МПК: A61K 31/725
Метки: применение, композиция, лечения, аспирина, заболеваний, тромбоэболических, олигосахарида, фармацевтическая, способ
Формула / Реферат:
1. Применение синтетического олигосахарида, который представляет собой селективный ингибитор фактора Ха, действующий через антитромбин III, в комбинации с аспирином для получения лекарственных средств, предназначенных для предупреждения или лечения тромбоэмболических заболеваний, имеющих место у млекопитающего, подвергающегося чрезкожной внутрипросветной ангиопластике. 2. Применение по п.1, отличающееся тем, что олигосахарид представляет собой...
Компонент в, используемый в качестве рубцующего агента, содержащая его фармацевтическая композиция и способ лечения

Номер патента: 1077
Опубликовано: 30.10.2000
Авторы: Мартелли Фабрицио, Донини Сильвия, Мастранджели Ренато, Боррелли Франческо
МПК: A61P 17/02, A61K 38/17
Метки: лечения, агента, качестве, композиция, содержащая, рубцующего, фармацевтическая, используемый, компонент, способ
Формула / Реферат:
1. Применение Компонента В, представляющего собой белок со следующей аминокислотной последовательностью SEQ ID NO:1: для получения лекарственных препаратов, пригодных в качестве рубцующих агентов. 2. Применение по п.1, отличающееся тем, что лекарственный препарат применяется в лечении ран, язв и других травматических повреждений любых тканей тела. 3. Фармацевтическая композиция в подходящей форме как рубцующий агент, содержащий Компонент...
Предыдущий патент: Система для защиты программного обеспечения на компакт-диске.
Следующий патент: Микропористый, содержащий карбамидные группы полиуретановый эластомер
Случайный патент: Ингибитор регулирующей апоптотические сигналы киназы