Способ получения пирролидиноксимов






Формула / Реферат
1. Способ получения соединения формулы (I)

в которой А обозначает карбонильную группу -(С=O)-;
В выбран из группы, включающей оксадиазольное кольцо, амидогруппу формулы -(C=O)-NR3R4 и
-(CH2)n-X-R8;
в которой оксадиазольное кольцо описывается любой из формул


R1 обозначает Н или C1-C6-алкил;
R2 выбран из группы, включающей арил, гетероарил и насыщенный или ненасыщенный 3-8-членный циклоалкил;
R3 и R4 независимо выбраны из группы, включающей водород, C1-C6-алкил, С2-C6-алкенил, C2-C6-алкинил, алкоксигруппу, сульфанил, ацил, алкоксикарбонил, аминокарбонил, насыщенный или ненасыщенный 3-8-членный циклоалкил, который может содержать 1-3 гетероатома, выбранных из группы, включающей N, О, S, арил, гетероарил, C1-C6-алкиларил и C1-C6-алкилгетероарил;
X обозначает О или NR9;
R8 выбран из группы, включающей водород, C1-C6-алкил, C1-C6-алкиларил, гетероарил, C1-C6-алкилгетероарил, C2-C6-алкенил, C2-C6-алкениларил, C2-C6-алкенилгетероарил, C2-C6-алкинил, C2-C6-алкиниларил, C2-C6-алкинилгетероарил, C3-C8-циклоалкил, гетероциклоалкил, C1-C6-алкилциклоалкил, C1-C6-алкилгетероциклоалкил, C1-C6-алкилкарбоксигруппу, ацил, C1-C6-алкилацил, C1-C6-алкилацилоксигруппу, C1-C6-алкилалкоксигруппу, алкоксикарбонил, C1-C6-алкилалкоксикарбонил, аминокарбонил, C1-C6-алкиламинокарбонил, C1-C6-алкилациламиногруппу, C1-C6-алкилуреидогруппу, аминогруппу, C1-C6-алкиламиногруппу, сульфонилоксигруппу, C1-C6-алкилсульфонилоксигруппу, сульфонил, C1-C6-алкилсульфонил, сульфинил, C1-C6-алкилсульфинил, C1-C6-алкилсульфанил и C1-C6-алкилсульфониламиногруппу;
R7 выбран из группы, включающей водород, сульфонил, аминогруппу, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, где указанные алкильные, алкенильные, алкинильные цепи необязательно включают гетероатом, выбранный из группы, включающей N, О и S, арил, гетероарил, насыщенный или ненасыщенный 3-8-членный циклоалкил, гетероциклоалкил, где указанные циклоалкильные, гетероциклоалкильные, арильные или гетероарильные группы необязательно сконденсированы еще с 1-2 циклоалкильными, гетероциклоалкильными, арильными или гетероарильными группами, ацильный фрагмент, C1-C6-алкиларил, C1-C6-алкилгетероарил, C1-C6-алкениларил, C1-C6-алкенилгетероарил, C1-C6-алкиниларил, C1-C6-алкинилгетероарил, C1-C6-алкилциклоалкил, C1-C6-алкилгетероциклоалкил, C1-C6-алкенилциклоалкил, C1-C6-алкенилгетероциклоалкил, C1-C6-алкинилциклоалкил, C1-C6-алкинилгетероциклоалкил, алкоксикарбонил, аминокарбонил, C1-C6-алкилкарбоксигруппу, C1-C6-алкилацил, C1-C6-алкилацилоксигруппу, C1-C6-алкилалкоксигруппу, C1-C6-алкилалкоксикарбонил, C1-C6-алкиламинокарбонил, C1-C6-алкилациламиногруппу, C1-C6-алкилуреидогруппу, C1-C6-алкиламиногруппу, C1-C6-алкиламмониевую группу, C1-C6-алкилсульфонилоксигруппу, C1-C6-алкилсульфонил, C1-C6-алкилсульфинил, С1-C6-алкилсульфанил, C1-C6-алкилсульфониламиногруппу, C1-C6-алкиламиносульфонил, гидроксигруппу, галоген и цианогруппу;
R9 выбран из группы, включающей водород, C1-C6-алкил, C1-C6-алкиларил, С1-C6-алкилгетероарил, арил и гетероарил;
R8 и R9 совместно с атомом N, с которым они связаны, могут образовать пяти-восьмичленное насыщенное или ненасыщенное гетероциклоалкильное кольцо; и
n является целым числом, равным от 1 до 3;
где "арил" означает ненасыщенную ароматическую карбоциклическую группу, содержащую от 6 до 14 атомов углерода, включающую одно кольцо или несколько конденсированных колец,
"гетероарил" означает моноциклическую гетероароматическую группу или бициклическую или трициклическую гетероароматическую группу, содержащую конденсированные кольца,
"гетероциклоалкил" означает C3-C8-циклоалкильную группу, в которой вплоть до 3 атомов углерода заменены гетероатомами, выбранными из группы, включающей О, S, NR, где R обозначает водород или метил,
"ацил" означает группу -C(O)R, в которой R означает "C1-C6-алкил", "арил", "гетероарил", "C3-C8-циклоалкил", "гетероциклоалкил", "C1-C6-алкиларил" или "C1-C6-алкилгетероарил",
указанный способ включает следующие стадии:
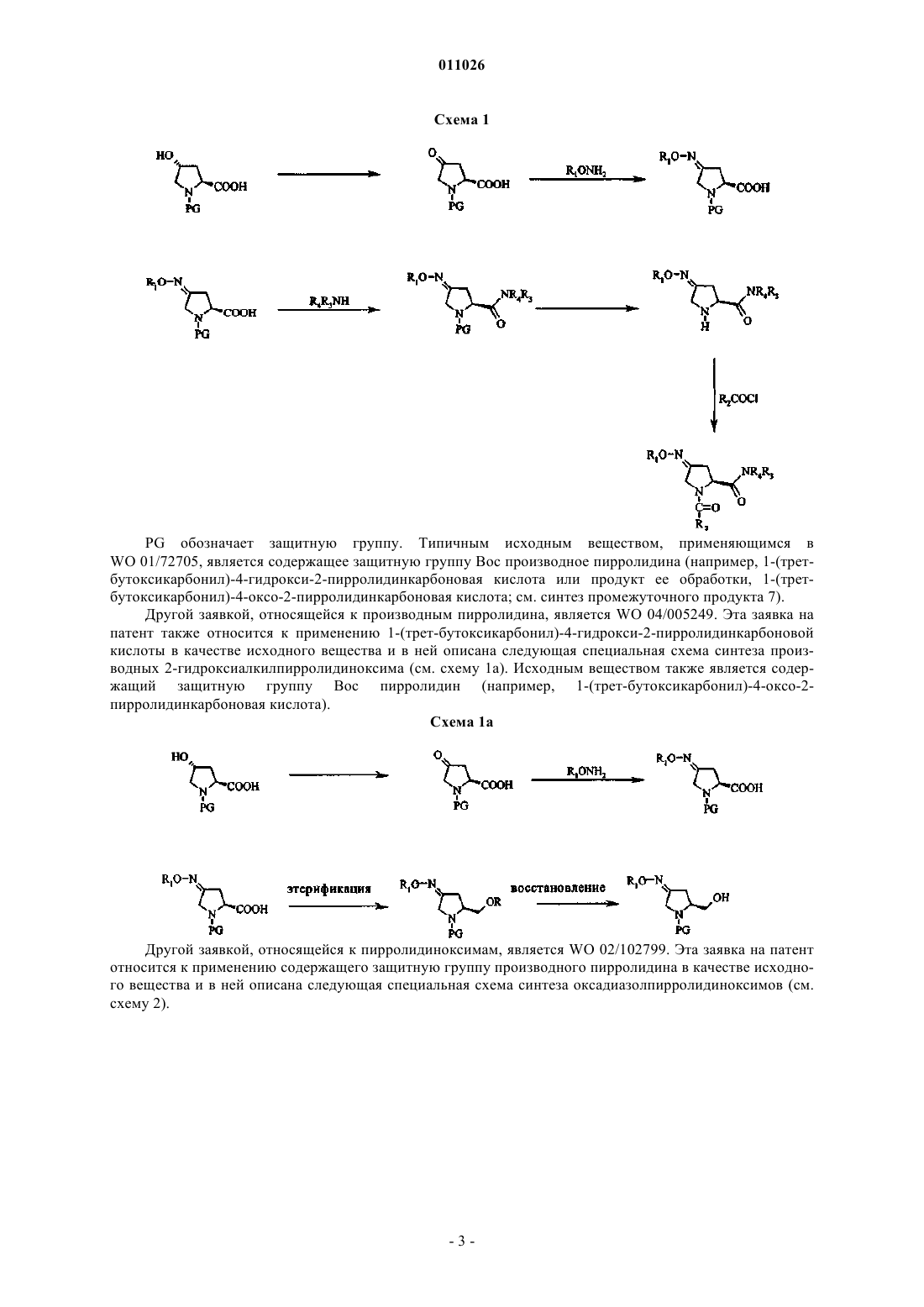
стадия 1: превращение пирролидина формулы (II) в ацилпроизводное формулы (IV) с использованием ацилирующего реагента (III)

стадия 2: окисление ацилпроизводного (IV) окислительным реагентом с получением пирролидона формулы (V)

стадия 3: превращение пирролидона формулы (V) в соединение (VII) с использованием подходящего алкоксиламина, арилоксиламина или гидроксиламина общей формулы (VI)

стадия 4: превращение соединения (VII) с помощью амина общей формулы (VIII) или N-гидроксиамидина общей формулы (IX) с получением соединений (Ia) и (Ib) или превращение соединения (VII) сначала в нитрил (VIIa), который затем превращают в гидроксиамидин (VIIb), который затем вводят в реакцию с карбоновой кислотой R7-СООН и получают соединение (Ic), или сначала проведение этерификации и последующего восстановления соединения (VII) с использованием подходящего этерифицирующего или восстановительного реагента соответственно с получением соединения (Id)

2. Способ получения соединения формулы (I) по п.1

в которой А обозначает карбонильную группу -(С=O)-;
В обозначает амидогруппу формулы -(C=O)-NR3R4 или оксадиазольное кольцо, описываемое любой из формул


R7 выбран из группы, включающей водород, сульфонил, аминогруппу, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, где указанные алкильные, алкенильные, алкинильные цепи необязательно включают гетероатом, выбранный из группы, включающей N, О и S, арил, гетероарил, насыщенный или ненасыщенный 3-8-членный циклоалкил, гетероциклоалкил, уфх указанные циклоалкильные, гетероциклоалкильные, арильные или гетероарильные группы необязательно сконденсированы еще с 1-2 циклоалкильными, гетероциклоалкильными, арильными или гетероарильными группами, ацильный фрагмент, C1-C6-алкиларил, C1-C6-алкилгетероарил, C1-C6-алкениларил, C1-C6-алкенилгетероарил, C1-C6-алкиниларил, C1-C6-алкинилгетероарил, C1-C6-алкилциклоалкил, C1-C6-алкилгетероциклоалкил, C1-C6-алкенилциклоалкил, C1-C6-алкенилгетероциклоалкил, C1-C6-алкинилциклоалкил, C1-C6-алкинилгетероциклоалкил, алкоксикарбонил, аминокарбонил, C1-C6-алкилкарбоксигруппу, C1-C6-алкилацил, C1-C6-алкилацилоксигруппу, C1-C6-алкилалкоксигруппу, C1-C6-алкилалкоксигруппу-карбонил, C1-C6-алкиламинокарбонил, C1-C6-алкилациламиногруппу, C1-C6-алкилуреидогруппу, C1-C6-алкиламиногруппу, C1-C6-алкиламмониевую группу, C1-C6-алкилсульфонилоксигруппу, C1-C6-алкилсульфонил, C1-C6-алкилсульфинил, С1-C6-алкилсульфанил, C1-C6-алкилсульфониламиногруппу, C1-C6-алкиламиносульфонил, гидроксигруппу, галоген и цианогруппу;
R1 обозначает Н или C1-C6-алкил;
R2 выбран из группы, включающей арил, гетероарил и насыщенный или ненасыщенный 3-8-членный циклоалкил;
R3 и R4 независимо выбраны из группы, включающей водород, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, алкоксигруппу, сульфанил, ацил, алкоксикарбонил, аминокарбонил, насыщенный или ненасыщенный 3-8-членный циклоалкил, который может содержать 1-3 гетероатома, выбранных из группы, включающей N, О, S, арил, гетероарил, C1-C6-алкиларил и C1-C6-алкилгетероарил;
указанный способ включает следующие стадии:
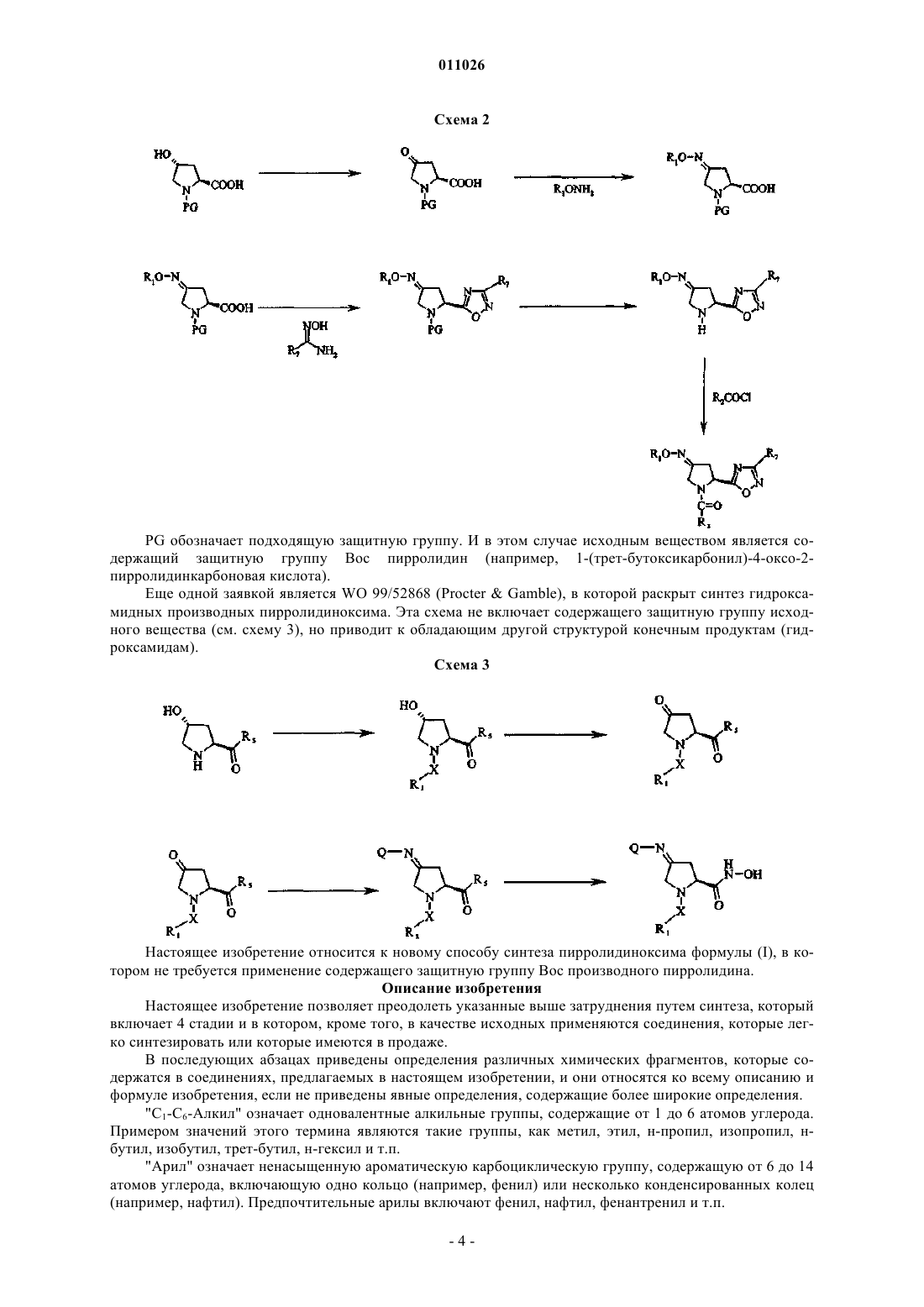
стадия 1: превращение пирролидина формулы (II) в ацилпроизводное формулы (IV) с использованием ацилирующего реагента (III)

стадия 2: окисление ацилпроизводного (IV) окислительным реагентом с получением пирролидона формулы (V)

стадия 3: превращение пирролидона формулы (V) в соединение (VII) с использованием подходящего алкоксиламина, арилоксиламина или гидроксиламина общей формулы (VI)

стадия 4: превращение соединения (VII) с помощью амина общей формулы (VIII) или N-гидроксиамидина общей формулы (IX) с получением соединений (Ia) и (Ib), или превращение соединения (VII) сначала в нитрил (VIIa), который затем превращают в гидроксиамидин (VIIb), который затем вводят в реакцию с карбоновой кислотой R7-СООН и получают соединение (Ic)

3. Способ по п.1 или 2, в котором ацилхлорид, применяющийся на стадии 1, представляет собой 1'1-бифенил-4-карбонилхлорид или 2'-метил-1'1-бифенил-4-карбонилхлорид.
4. Способ по любому из пп.1-3, в котором окислительный реагент, применяющийся на стадии 2, представляет собой комплекс пиридин-триоксид серы (Ру-SO3) в комбинации с ДМСО.
5. Способ по любому из пп.2-4, в котором реакцию проводят в присутствии триэтиламина.
6. Способ по любому из пп.1-5, в котором алкоксиламин, применяющийся на стадии 3, представляет собой О-метилгидроксиламингидрохлорид.
7. Способ по любому из пп.1-6, в котором R1 обозначает метильную группу, R2 обозначает бифенил.
8. Способ по любому из пп.1-7, в котором В обозначает амидогруппу формулы -(C=O)NHR5, где R5 обозначает C1-C6-алкиларильную группу.
9. Способ по п.8, в котором R5 обозначает фенилэтильную группу, которая замещена аминогруппой или гидроксигруппой.
10. Способ по любому из пп.1-7, в котором В обозначает 1,2,4-оксадиазольный заместитель


где R7 обозначает C1-C6-алкил или циклоалкил, необязательно содержащий 1 или 2 гетероатома.
11. Способ по любому из пп.1, 3, 4 или 6, 7, в котором В обозначает -(CH2)n-X-R8, где X обозначает О, R8 обозначает водород и n равно 1.
12. Способ по любому из пп.1-11, в котором соединение выбрано из группы, включающей
(2S,4E и 4Z)-N-[(2S)-2-гидрокси-2-фенилэтил]-4-(метоксиимино)-1-[(2'-метил[1,1'-бифенил]-4-ил) карбонил]-2-пирролидинкарбоксамид,
(3E,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-[3-(2-гидроксиэтил)-1,2,4-оксадиазол-5-ил]-3-пирролидинон-O-метилоксим,
(3Z,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-[3-(2-гидроксиэтил)-1,2,4-оксадиазол-5-ил]-3-пирролидинон-O-метилоксим,
(3E,5S)-5-[3-(2-гидроксиэтил)-1,2,4-оксадиазол-5-ил]-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-он-O-метилоксим,
(3E,5S)-5-[3-(2-гидроксиэтил)-1,2,4-оксадиазол-5-ил]-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-он-O-метилоксим,
(3EZ,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-{5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-3-пирролидинон-О-метилоксим,
(3Z,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-{5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил}-3-пирролидинон-О-метилоксим,
(3E,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-{5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил}-3-пирролидинон-О-метилоксим,
(3EZ,5S)-5-{5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил}-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-он-O-метилоксим,
(3Z,5S)-5-{5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил}-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-он-О-метилоксим,
(3E,5S)-5-{5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил}-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-он-O-метилоксим и
(3Z/E,5S)-1-(бифенил-4-илкарбонил)-(5-гидроксиметил)пирролидин-3-он-О-метилоксим.
Текст
011026 Краткое содержание изобретения Настоящее изобретение относится к новому способу синтеза пирролидиноксимов общей формулы(I). Соединения применимы при лечении и/или предупреждении преждевременных родов, рождения недоношенного ребенка и дисменореи. Область техники, к которой относится изобретение Настоящее изобретение относится к новому способу синтеза пирролидиноксимов общей формулы где А обозначает карбонильную группу -(С=O)-; В выбран из группы, включающей замещенное и незамещенное оксадиазольное кольцо, амидогруппу формул -(C=O)-NR3R4 и -(CH2)n-X-R8;R2 выбран из группы, включающей или содержащей незамещенный или замещенный арил, незамещенный или замещенный гетероарил, незамещенный или замещенный насыщенный или ненасыщенный 3-8-членный циклоалкил. Более предпочтительной является арильная, в особенности фенильная группа,которая необязательно замещена, например, другой фенильной группой (с образованием бифенильного фрагмента);R3 и R4 независимо выбраны из группы, включающей или содержащей водород, незамещенный или замещенный C1-C6-алкил, незамещенный или замещенный C2-C6-алкенил, незамещенный или замещенный C2-C6-алкинил, незамещенную или замещенную алкоксигруппу, незамещенный или замещенный сульфанил, ацил, алкоксикарбонил, аминокарбонил, незамещенный или замещенный насыщенный или ненасыщенный 3-8-членный циклоалкил, который может содержать 1-3 гетероатома, выбранных из группы, включающей N, О, S, незамещенный или замещенный арил, незамещенный или замещенный гетероарил, незамещенный или замещенный C1-C6-алкиларил, незамещенный или замещенный C1-C6 алкилгетероарил;R8 и R9 совместно с атомом N, с которым они связаны, могут образовать 5-8-членное насыщенное или ненасыщенное гетероциклоалкильное кольцо;n является целым числом, равным от 1 до 3. Предпочтительными производными пирролидина являются такие соединения формулы (I), в которых R1 обозначает метильную группу, R2 обозначает замещенный или незамещенный бифенил. В контексте одного предпочтительного варианта осуществления В обозначает амидогруппу формулы -(C=O)NHR5, в которой R5 обозначает незамещенную или замещенную C1-C6-алкиларильную группу,например, фенилэтильную группу, которая необязательно замещена гидрофильными фрагментами,включая аминогруппу и гидроксигруппу. В контексте другого предпочтительного варианта осуществления заместитель В обозначает 1,2,4 оксадиазольный заместитель, который может быть присоединен к пирролидиновому кольцу по приведенным ниже схемам (Ха) или (Xb) В указанных формулах (Ха) и (Xb) R7 выбран из группы, включающей или содержащей водород,сульфонил, аминогруппу, незамещенный или замещенный C1-C6-алкил, незамещенный или замещенныйC2-C6-алкенил, незамещенный или замещенный C2-C6-алкинил, где указанные алкильные, алкенильные,алкинильные цепи могут включать гетероатом, выбранный из группы, включающей N, О и S, незамещенный или замещенный арил, незамещенный или замещенный гетероарил, незамещенный или замещенный насыщенный или ненасыщенный 3-8-членный циклоалкил, незамещенный или замещенный гетероциклоалкил, где указанные циклоалкильные, гетероциклоалкильные, арильные или гетероарильные группы могут быть сконденсированы еще с 1-2 циклоалкильными, гетероциклоалкильными, арильными или гетероарильными группами, ацильный фрагмент, незамещенный или замещенный C1-C6-алкиларил,незамещенный или замещенный C1-C6-алкилгетероарил, незамещенный или замещенный C1-C6 алкениларил, незамещенный или замещенный C1-C6-алкенилгетероарил, незамещенный или замещенныйC1-C6-алкиниларил, незамещенный или замещенный C1-C6-алкинилгетероарил, незамещенный или замещенный C1-C6-алкилциклоалкил, незамещенный или замещенный C1-C6-алкилгетероциклоалкил, незамещенный или замещенный C1-C6-алкенилциклоалкил, незамещенный или замещенный C1-C6 алкенилгетероциклоалкил, незамещенный или замещенный C1-C6-алкинилциклоалкил, незамещенный или замещенный C1-C6-алкинилгетероциклоалкил, замещенный или незамещенный алкоксикарбонил,замещенный или незамещенный аминокарбонил, замещенную или незамещенную C1-C6 алкилкарбоксигруппу, замещенный или незамещенный С 1-C6-алкилацил, незамещенную или замещенную C1-C6-алкилацилоксигруппу, незамещенную или замещенную C1-C6-алкилалкоксигруппу, незамещенный или замещенный C1-C6-алкилалкоксикарбонил, незамещенный или замещенный С 1-C6 алкиламинокарбонил, незамещенную или замещенную C1-C6-алкилациламиногруппу, незамещенную или замещенную C1-C6-алкилуреидогруппу, незамещенную или замещенную C1-C6-алкиламиногруппу, незамещенную или замещенную C1-C6-алкиламмониевую группу, незамещенную или замещенную C1-C6 алкилсульфонилоксигруппу, незамещенный или замещенный C1-C6-алкилсульфонил, незамещенный или замещенный C1-C6-алкилсульфинил, незамещенный или замещенный C1-C6-алкилсульфанил, незамещенную или замещенную C1-C6-алкилсульфониламиногруппу, незамещенный или замещенный C1-C6 алкиламиносульфонил, гидроксигруппу, галоген, цианогруппу. В предпочтительном варианте осуществления R7 обозначает незамещенную или замещенную C1-C6 алкильную группу, например, метильную или этильную группу, которая необязательно может быть замещена гидрофильными фрагментами, включая аминогруппу и гидроксигруппу, или R7 обозначает 38-членный циклоалкил, необязательно содержащий 1 или 2 гетероатома, например, пирролидин, фуранил, тиенил, пиперидин, морфолин или пиперазин. В контексте другого предпочтительного варианта осуществления заместитель В обозначает группу формулы -(CH2)n-X-R8, в которой X обозначает О, R8 обозначает водород и n равно 1. В способе используются имеющиеся в продаже или легко получаемые исходные соединения. Уровень техники Схема синтеза пирролидиноксимов формулы (I) хорошо известна. В нескольких документах описан синтез таких соединений. Например, в WO 01/72705 раскрыт синтез амидопроизводного пирролидиноксима, представленный ниже (схема 1).WO 01/72705, является содержащее защитную группу Boc производное пирролидина (например, 1-(третбутоксикарбонил)-4-гидрокси-2-пирролидинкарбоновая кислота или продукт ее обработки, 1-(третбутоксикарбонил)-4-оксо-2-пирролидинкарбоновая кислота; см. синтез промежуточного продукта 7). Другой заявкой, относящейся к производным пирролидина, является WO 04/005249. Эта заявка на патент также относится к применению 1-(трет-бутоксикарбонил)-4-гидрокси-2-пирролидинкарбоновой кислоты в качестве исходного вещества и в ней описана следующая специальная схема синтеза производных 2-гидроксиалкилпирролидиноксима (см. схему 1 а). Исходным веществом также является содержащий защитную группу Boc пирролидин (например, 1-(трет-бутоксикарбонил)-4-оксо-2 пирролидинкарбоновая кислота). Схема 1 а Другой заявкой, относящейся к пирролидиноксимам, является WO 02/102799. Эта заявка на патент относится к применению содержащего защитную группу производного пирролидина в качестве исходного вещества и в ней описана следующая специальная схема синтеза оксадиазолпирролидиноксимов (см. схему 2).PG обозначает подходящую защитную группу. И в этом случае исходным веществом является содержащий защитную группу Boc пирролидин (например, 1-(трет-бутоксикарбонил)-4-оксо-2 пирролидинкарбоновая кислота). Еще одной заявкой является WO 99/52868 (ProcterGamble), в которой раскрыт синтез гидроксамидных производных пирролидиноксима. Эта схема не включает содержащего защитную группу исходного вещества (см. схему 3), но приводит к обладающим другой структурой конечным продуктам (гидроксамидам). Схема 3 Настоящее изобретение относится к новому способу синтеза пирролидиноксима формулы (I), в котором не требуется применение содержащего защитную группу Boc производного пирролидина. Описание изобретения Настоящее изобретение позволяет преодолеть указанные выше затруднения путем синтеза, который включает 4 стадии и в котором, кроме того, в качестве исходных применяются соединения, которые легко синтезировать или которые имеются в продаже. В последующих абзацах приведены определения различных химических фрагментов, которые содержатся в соединениях, предлагаемых в настоящем изобретении, и они относятся ко всему описанию и формуле изобретения, если не приведены явные определения, содержащие более широкие определения."C1-C6-Алкил" означает одновалентные алкильные группы, содержащие от 1 до 6 атомов углерода. Примером значений этого термина являются такие группы, как метил, этил, н-пропил, изопропил, нбутил, изобутил, трет-бутил, н-гексил и т.п."Арил" означает ненасыщенную ароматическую карбоциклическую группу, содержащую от 6 до 14 атомов углерода, включающую одно кольцо (например, фенил) или несколько конденсированных колец(например, нафтил). Предпочтительные арилы включают фенил, нафтил, фенантренил и т.п."Гетероарил" означает моноциклическую гетероароматическую или бициклическую или трициклическую содержащую конденсированные кольца гетероароматическую группу. Конкретные примеры гетероароматических групп включают необязательно замещенный пиридил, пирролил, фурил, тиенил,имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиразолил, 1,2,3-триазолил, 1,2,4-триазолил,1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, 1,3,4-триазинил, 1,2,3 триазинил, бензофурил, [2,3-дигидро]бензофурил, изобензофурил, бензотиенил, бензотриазолил, изобензотиенил, индолил, изоиндолил, 3H-индолил, бензимидазолил, имидазо[1,2-а]пиридил, бензотиазолил,бензоксазолил, хинолизинил, хиназолинил, фталазинил, хиноксалинил, циннолинил, нафтиридинил, пиридо[3,4-b]пиридил, пиридо[3,2-b]пиридил, пиридо[4,3-b]пиридил, хинолил, изохинолил, тетразолил,5,6,7,8-тетрагидрохинолил, 5,6,7,8-тетрагидроизохинолил, пуринил, птеридинил, карбазолил, ксантенил и бензохинолил."C3-C8-Циклоалкил" означает насыщенную карбоциклическую группу, содержащую от 3 до 8 атомов углерода, включающую одно кольцо (например, циклогексил) или несколько конденсированных колец (например, норборнил). Предпочтительные циклоалкилы включают циклопентил, циклогексил,норборнил и т.п."Гетероциклоалкил" означает C3-C8-циклоалкильную группу, соответствующую приведенному выше определению, в которой вплоть до 3 атомов углерода заменены гетероатомами, выбранными из группы, включающей О, S, NR, R определяется, как водород или метил. Предпочтительные гетероциклоалкилы включают пирролидин, пиперидин, пиперазин, 1-метилпиперазин, морфолин и т.п."C2-C6-Алкенил" означает алкенильные группы, предпочтительно содержащие от 2 до 6 атомов углерода и включающие одну или большее количество алкенильных ненасыщенных фрагментов. Предпочтительные алкенильные группы включают этенил (-СН=CH2), н-2-пропенил (аллил, -СН 2 СН=СН 2) и т.п."C2-C6-Алкинил" означает алкинильные группы, предпочтительно содержащие от 2 до 6 атомов углерода и включающие одну или большее количество алкинильных ненасыщенных фрагментов. Предпочтительные алкинильные группы включают этинил (-ССН), пропинил (-CH2CСН) и т.п."C1-C6-Алкиламинокарбонил" означает группу -C(O)NRR', в которой все R, R' независимо обозначают водород или "C1-C6-алкил"."C1-C6-Алкилациламиногруппа" означает группу -NR(CO)R', в которой все R, R' независимо обозначают водород или "C1-C6-алкил"."Сульфонил" означает группу "-SO2-R", в которой R выбран из группы, включающей Н, "C1-C6 алкил", "C1-C6-алкил", необязательно замещенный атомами галогена, такую как, например, группа -SO2CF3, "арил", "гетероарил", "C1-C6-алкиларил" или "C1-C6-алкилгетероарил"."Сульфоксигруппа" означает группу "-S(O)-R", в которой R выбран из группы, включающей Н,"C1-C6-алкил", "C1-C6-алкил", необязательно замещенный атомами галогена, такую как, например, группа"Сульфинил" означает группу "-SO-R'R", в которой R выбран из группы, включающей Н, "C1-C6 алкил", "C1-C6-алкил", необязательно замещенный атомами галогена, такую как, например, группа -SOCF3, "арил", "гетероарил", "C1-C6-алкиларил" или "C1-C6-алкилгетероарил"."Аминогруппа" означает группу -NRR', в которой все R, R' независимо обозначают водород, "C1-C6 алкил", "C2-C6-алкенил", "C2-C6-алкинил", "С 3-C8-циклоалкил", "гетероциклоалкил", "арил", "гетероарил", "C1-C6-алкиларил" или "C1-C6-алкилгетероарил", "C2-C6-алкениларил", "C2-C6-алкенилгетероарил","С 2-C6-алкиниларил", "C2-C6-алкинилгетероарил", "C1-C6-алкилциклоалкил", "C1-C6-алкилгетероциклоалкил", и в которой R и R' совместно с атомом азота, с которым они связаны, необязательно могут образовать 3-8-членное гетероциклоалкильное кольцо."Уреидогруппа" означает группу -NRC(O)NR'R", в которой все R, R', R" независимо обозначают водород, "C1-C6-алкил", "C2-C6-алкенил", "C2-C6-алкинил", "С 3-С 8-циклоалкил", "гетероциклоалкил","арил", "гетероарил", "C1-C6-алкиларил" или "C1-C6-алкилгетероарил", "C2-C6-алкениларил", "C2-C6 алкенилгетероарил", "C2-C6-алкиниларил", "C2-C6-алкинилгетероарил", "C1-C6-алкилциклоалкил", "C1-C6 алкилгетероциклоалкил" и где R' и R" совместно с атомом N, с которым они связаны, необязательно могут образовать 3-8-членное гетероциклоалкильное кольцо."Замещенный или незамещенный". Если иное ограничение не налагает определение конкретного заместителя, перечисленные в указанном выше наборе группы, такие как "алкил", "арил" и "гетероарил" и т.п. необязательно могут содержать от 1 до 5 заместителей, выбранных из группы, включающей "C1-C6 алкил", "аминогруппу", "арил", "гетероарил", "сульфинил", "сульфонил", "алкоксигруппу", "сульфанил",-5 011026"галоген", "карбоксигруппу", цианогруппу, гидроксигруппу, меркапогруппу, нитрогруппу и т.п. Способ, предлагаемый в настоящем изобретении, включает следующие 4 стадии. В контексте настоящего изобретения соединения формулы (I) получают с использованием в качестве исходного вещества не содержащую защитную группу 4-гидроксипирролидинкарбоновую кислоту формулы (II). Соединение (II) имеется в продаже или его можно получить по известным методикам. Стадия 1. На первой стадии (см. схему 4) пирролидин формулы (II) превращают в ацилпроизводное формулы (IV) с помощью подходящего ацилирующего реагента (III), например, ацилхлорида, ангидрида,карбоновой кислоты или сложного эфира. Предпочтительным ацилирующим реагентом является 1,1'бифенил-4-карбонилхлорид или 2'-метил-1,1'-бифенил-4-карбонилхлорид. Получение таких соединений раскрыто, например, в WO 01/72705. Схема 4 Реакцию предпочтительно проводят в присутствии основания, например гидроксида натрия или гидроксида калия (условия Шоттена-Баумана), или с использованием органического основания, включая триэтиламин, N,N-диизопропилэтиламин или пиридин. Стадия 2. Затем ацилпроизводное (IV) окисляют подходящим окислительным реагентом и получают пирролидон формулы (V). Одним подходящим окислительным реагентом является комплекс пиридин-триоксид серы (Ру-SO3), с которым используется ДМСО (диметилсульфоксид) в качестве растворителя. Реакцию предпочтительно проводят в присутствии триэтиламина. Дополнительные примеры подходящих окислительных реагентов включают, например, оксалилхлорид/ДМСО, ангидрид трифторуксусной кислоты/ДМСО, дициклогексилкарбодиимид/ДМСО, пиридинийдихромат, пиридинийхлорхромат, окисление по Джону и периодинан Десса-Мартина, 1,1,1 трис(ацетокси)-1-5,2-бензйодоксол-3(1 Н)-он. Схема 5 Стадия 3. Затем соединение формулы (V) превращают в соединение (VII) с использованием подходящего алкоксиламина, арилоксиламина или гидроксиламина общей формулы (VI), например, Ометилгидроксиламингидрохлорида (такое соединение имеется в продаже) в присутствии органического основания, такого как триэтиламин или N,N-диизопропилэтиламин. Схема 6 Стадия 4. Затем соединение (VII) превращают в соединения (Ia) или (Ib) с использованием амина общей формулы (VIII) или N-гидроксиамидоксима общей формулы (IX). Получение Nгидроксиамидоксима общей формулы (IX) раскрыто, например, в WO 02/102799. В случае, если необходимо получить конечный продукт (Ic), стадию 4 необходимо изменить, так чтобы сначала получить N-гидроксиамидоксим (VIIb) путем превращения соединения (VII) в нитрил(VIIa) (например, непосредственно из кислоты (это известно из литературы) или через амид), который затем дополнительно вводят в реакцию с карбоновой кислотой формулы R7-COOH или, например, соответствующим ацилхлоридом и в заключение получают соединение (Ic) после нагревания промежуточного продукта, например, с избытком пиридина. Для реакции амидоксима (VIIb) с карбоновой кислотой предпочтительно использовать реагенты реакции сочетания, например N-(3-диметиламинопропил)-N'этилкарбодиимидгидрохлорид, карбонилдиимидазол, дициклогексилкарбодиимид, пивалоилхлорид, изобутилхлорформиат (или любой другой из обычных реагентов, применяющихся при образовании пептидной связи). В случае, если необходимо получить конечный продукт (Id), в котором R6 обозначает водород, для превращения карбоксигруппы в гидроксиалкильную группу можно использовать различные хорошо известные этерифицирующие и восстановительные реагенты. Примерами этерифицирующих реагентов являются диметилсульфат, метилйодид, метилтозилат, производные диазометана, такие как триметилсилилдиазометан, которые все являются этерифицирующими реагентами, действующими в слабощелочной или нейтральной среде. Примерами восстановительных реагентов являются борогидрид лития, алюмогидрид лития, натрий-бис-(2-метоксиэтокси) алюмогидрид лития, (Red-A1), диизобутилалюминийгидрид(ДИБАЛ) и т.п. Конечные продукты формул (Ia), (Ib), (Ic) и (Id) можно дополнительно превратить, в частности, по фрагменту R1, R2, R7 и R8. Таким образом, в конечном продукте (Ic), в котором R7 содержит функциональную группу, указанный фрагмент можно превратить в другой фрагмент с помощью подходящих средств, включая гидролиз, этерификацию, омыление, алкилирование и т.п. Кроме того, соединения,предлагаемые в настоящем изобретении, можно подвергнуть дополнительным стадиям очистки, включая хроматографию и перекристаллизацию. Новый способ синтеза для получения соединения формулы (I) не включает применение относительно дорогостоящего содержащего защитную группу Boc пирролидин, а в нем используется недорогой и легко доступный 3-гидроксипролин. Другое преимущество нового способа синтеза относится к получению соединений, содержащих по-7 011026 лярные фрагменты, присоединенные в положения 2 карбоксамида или оксадиазола, например, если R3,R4, R7 являются фрагментом (например, алкилом или арилом), который содержит, например, гидроксильный или аминный заместитель, включая циклический амин. Предлагаемый новый способ исключает конечную стадию образования N-концевой группы (как показано на схеме 2), включающую использование нуклеофила (например, ацилхлорида), который может взаимодействовать как с пирролидинамином, так и с указанным вторым полярным фрагментом, например гидроксильным или аминным заместителем. В одном варианте осуществления новый способ синтеза можно применять для промышленного изготовления соединений формулы (I). Настоящее изобретение иллюстрируется приведенными ниже примерами. Пример 1. Получение (2S,4 Е и 4Z)-N-[(2S)-2-гидрокси-2-фенилэтил]-4-(метоксиимино)-1-[(2'метил[1,1'-бифенил]-4-ил)карбонил]-2-пирролидинкарбоксамида. Стадия 1. Получение (4R)-4-гидрокси-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролина (соединение (IV) на схеме 4). 4-Гидрокси-L-пролин (0,625 мас.ч.) и воду (3,3 об.ч.) помещали в колбу с отводом объемом 20 л. К содержимому по каплям прибавляли триэтиламин (2,42 об.ч.), так чтобы температура поддерживалась в диапазоне от 10 до 20 С. Прибавляли тетрагидрофуран (5,0 об.ч.) и реакционную смесь охлаждали до температуры, равной от 0 до 5 С. Хлорангидрид 2'-метил-1,1'-бифенил-4-карбоновой кислоты(1,0 мас.ч.) и тетрагидрофуран (5,0 об.ч.) помещали в отдельную колбу, перемешивали в течение от 5 до 10 мин и затем прибавляли к реакционной смеси, так чтобы температура поддерживалась в диапазоне от 0 до 10 С. Реакционную смесь нагревали при температуре, равной от 15 до 25 С, в течение 60-120 мин и поддерживали при температуре, равной от 15 до 25 С, пока по данным анализа с помощью ТСХ (тонкослойная хроматография) реакция не завершалась. Полученную смесь концентрировали в вакууме при температуре, равной от 35 до 40 С, к остатку прибавляли воду (10,0 об.ч.) и этилацетат (5,0 об.ч.) и содержимое перемешивали в течение от 5 до 10 мин. Слои разделяли, водную фазу подкисляли до pH 1 водным раствором хлористо-водородной кислоты (6 М, примерно 3,0 об.ч.) и полученную взвесь охлаждали и выдерживали при температуре, равной от 0 до 10 С, в течение от 25 до 40 мин. Осадок собирали фильтрованием, отделенное твердое вещество переносили в подходящую колбу с отводом и суспендировали в теплой (от 35 до 60 С) воде(5,0 об.ч.) в течение от 10 до 25 мин. Твердое вещество собирали фильтрованием и повторяли суспендирование в горячей воде, описанное выше. После второго суспендирования твердое вещество подвергали азеотропной сушке с помощью толуола (25,0 об.ч.) при температуре, равной от 40 до 50 С. К остатку прибавляли этилацетат (2,5 об.ч.) и гептаны (2,5 об.ч.), полученную взвесь охлаждали и выдерживали при температуре, равной от 0 до 5 С, в течение от 30 до 40 мин, фильтровали, собранное твердое вещество промывали предварительно охлажденной смесью (от 0 до 5 С) этилацетат:гептаны (1:1, 2,0 об.ч.) и сушили в вакууме при температуре, равной от 30 до 40 С, до постоянной массы и получали (4R)-4 гидрокси-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-1-пролин в виде белого твердого вещества. Выход: 85,9%. Стадия 2. Получение 1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-4-оксо-1-пролина (соединение (V) на схеме 5).(4R)-4-Гидрокси-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролин (продукт, полученный на стадии 1, 1,0 мас.ч.) и диметилсульфоксид (2,5 об.ч.) помещали в колбу с отводом объемом 20 л. Содержимое нагревали до температуры, равной от 35 до 40 С, и поддерживали при этой температуре до полного растворения. Раствор охлаждали до температуры, равной от 5 до 10 С, в атмосфере азота и прибавляли триэтиламин (3,0 об.ч.), так чтобы температура поддерживалась в диапазоне от 5 до 20 С. Комплекс пиридин-триоксид серы (1,47 мас.ч.) и диметилсульфоксид (4,9 об.ч.) помещали в отдельную колбу, перемешивали в течение от 5 до 10 мин и затем прибавляли к реакционной смеси, так чтобы температура поддерживалась в диапазоне от 15 до 25 С. Реакционную смесь перемешивали при температуре, равной от 15 до 25 С, пока по данным анализа с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) реакция не завершалась (обычно от 1 до 3 ч). Содержимое сосуда охлаждали до температуры, равной от 0 до 10 С, и реакцию останавливали водным раствором хлористо-водородной кислоты (3 М,8 об.ч.), поддерживая температуру ниже 30 С. Затем прибавляли тетрагидрофуран (5,0 об.ч.) и гептаны(1,0 об.ч.), слои разделяли, водную фазу экстрагировали тетрагидрофураном (25,0 об.ч.) и объединенные органические фазы промывали водным раствором хлористо-водородной кислоты (1 М, 22,0 об.ч.) и насыщенным рассолом (22,0 об.ч.). Водные промывочные растворы объединяли и подвергали обратной экстракции тетрагидрофураном (21,0 об.ч.), органические фазы объединяли, сушили над сульфатом магния (3 мас.ч.) и фильтровали. Осадок на фильтре промывали тетрагидрофураном (1,0 об.ч.) и фильтраты концентрировали в вакууме при температуре, равной от 40 до 45 С, и получали бледно-коричневое вспененное вещество. К остатку прибавляли этилацетат (10,0 об.ч.), содержимое перемешивали в течение от 5 до 10 мин и растворитель удаляли в вакууме при температуре, равной от 40 до 45 С. Остаток переносили в колбу, прибавляли этилацетат (8,0 об.ч.) и содержимое кипятили с обратным холодильни-8 011026 ком. Прибавляли взвесь активированного угля (0,14 мас.ч.) в этилацетате (5,0 об.ч.) и кипячение с обратным холодильником возобновляли и продолжали в течение от 20 до 30 мин. Содержимое охлаждали до температуры, равной от 40 до 45 С, фильтровали, осадок на фильтре промывали этилацетатом (2,5 об.ч.) и фильтраты концентрировали до 2,5-3,0 об.ч. в вакууме при температуре, равной от 40 до 45 С. Взвесь разбавляли этилацетатом (0,5 об.ч.) и кипятили с обратным холодильником. Прибавляли гептан (3,0 об.ч.) и содержимому давали охладиться до температуры, равной от 15 до 25 С, в течение от 1 до 2 ч. Взвесь дополнительно охлаждали до температуры, равной от 0 до 5 С, в течение от 2 до 3 ч, фильтровали и осадок на фильтре промывали смесью этилацетат:гептан [(1:1), 1,0 об.ч.], предварительно охлажденной до температуры, равной от 0 до 5 С, затем гептаном (5,0 об.ч.). Выделенное твердое вещество сушили в вакууме при температуре, равной от 40 до 45 С, и получали 1-[(2'-метил-1,1'-бифенил-4 ил)карбонил]-4-оксо-L-пролина в виде почти белого твердого вещества. Выход: 60,3%. Стадия 3. Получение 4-метоксиимино-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролина (соединение (VII) на схеме 6). 1-[(2'-Метил-1,1'-бифенил-4-ил)карбонил]-4-оксо-1-пролин (полученный на стадии 2, 1,0 мас.ч.), Ометилгидроксиламингидрохлорид (0,285 мас.ч.) и дихлорметан (20 об.ч.) помещали в колбу с отводом объемом 20 л и охлаждали до температуры, равной от 0 до 5 С. В колбу прибавляли триэтиламин(0,91 об.ч.), так чтобы температура поддерживалась в диапазоне от 0 до 10 С, реакционную смесь нагревали до температуры, равной от 15 до 25 С, и поддерживали в этом температурном диапазоне в течение от 16 до 20 ч. Реакционную смесь концентрировали в вакууме при температуре, равной от 40 до 45 С,остаток растворяли в этилацетате (10,0 об.ч.) и промывали водным раствором хлористо-водородной кислоты (1 М, 25,0 об.ч.). Водные промывочные растворы объединяли и подвергали обратной экстракции этилацетатом (5,0 об.ч.), органические экстракты объединяли и промывали насыщенным рассолом(10,0 об.ч.), сушили над сульфатом магния (0,5 мас.ч.), фильтровали и осадок на фильтре промывали этилацетатом (5,0 об.ч.). Фильтраты концентрировали в вакууме при температуре, равной от 40 до 45 С,и получали 4-метоксиимино-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролин в виде ожидаемой смесиE:Z. Выход: 95,6%. Стадия 4. Получение N-[2-гидрокси-2-фенилэтил]-4-(метоксиимино)-1-[(2'-метил[1,1'-бифенил]-4 ил)карбонил]-2-пирролидинкарбоксамида (соединение (Ia) на схеме 7). 4-Метоксиимино-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролин (полученный на стадии 3,1,0 мас.ч.) и дихлорметан (10,0 об.ч.) помещали в колбу с отводом объемом 20 л и охлаждали до температуры, равной от 0 до 5 С, в атмосфере азота. N-Метилморфолин (0,78 об.ч.) прибавляли при температуре, равной от 0 до 5 С, а затем пивалоилхлорид (0,37 об.ч.) при температуре, равной от 0 до 5 С. Содержимое сосуда перемешивали при температуре, равной от 0 до 5 С, пока не завершалось образование смешанного ангидрида (обычно от 30 до 60 мин). В отдельную колбу с отводом объемом 20 л помещали(S)-2-амино-1-фенилэтанол (0,47 мас.ч., 1,2 экв.) и дихлорметан (3,0 об.ч.) и полученную смесь перемешивали в течение от 5 до 25 мин. Затем раствор охлаждали до температуры, равной от 10 до 15 С, и прибавляли смешанный ангидрид, так чтобы температура поддерживалась в диапазоне от 5 до 15 С. Реакционную смесь нагревали до температуры, равной от 15 до 25 С, и поддерживали в этом температурном диапазоне, пока по данным анализа с помощью ВЭЖХ реакция не завершалась. Полученную смесь концентрировали в вакууме при температуре, равной от 35 до 45 С, остаток подвергали распределению между трет-бутилметиловым эфиром (ТБМЭ, 10,0 об.ч.) и водным раствором лимонной кислоты (0,1 М,5,0 об.ч.), слои разделяли и органическую фазу дополнительно промывали водным раствором лимонной кислоты (0,1 М, 25,0 об.ч.), насыщенным водным раствором гидрокарбоната натрия (25,0 об.ч.) и насыщенным рассолом (5,0 об.ч.). Органическую фазу сушили над сульфатом магния (1 мас.ч.), фильтровали и осадок на фильтре промывали с помощью ТБМЭ (2,0 об.ч.). Фильтраты концентрировали в вакууме при температуре, равной от 35 до 45 С, и получали коричневое полужидкое вещество. К остатку прибавляли дихлорметан (5,0 об.ч.) и содержимое концентрировали в вакууме при температуре, равной от 35 до 45 С, и получали смолу. Процедуру повторяли со второй порцией дихлорметана (1,0 об.ч.) и неочищенный конечный продукт получали в виде ожидаемой смеси E:Z. Выход: 84,4%. Пример 2. (3E,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-[3-(2-гидроксиэтил)-1,2,4-оксадиазол-5-ил]-3 пирролидинон-O-метилоксим;(3,30 л, 3,3 об.ч.) помещали в колбу с отводом объемом 20 л. По каплям прибавляли триэтиламин(2,570 л, 2,57 об.ч.), так чтобы температура поддерживалась в диапазоне от 10 до 15 С, и полученную смесь охлаждали до температуры, равной от 0 до 5 С. 1,1'-Бифенил-4-карбонилхлорид (1,00 кг,3,78 моль, 1,0 мас.ч.) и тетрагидрофуран (5,00 л, 5,0 об.ч.) помещали в отдельную колбу, перемешивали в виде взвеси в течение от 5 до 10 мин и прибавляли к реакционной смеси в течение от 40 до 50 мин, так чтобы температура поддерживалась в диапазоне от 0 до 10 С. Реакционную смесь нагревали от 15 до-9 011026 25 С, в течение от 60 до 120 мин и поддерживали при температуре, равной от 15 до 25 С, пока по данным анализа с помощью ТСХ реакция не завершалась (дихлорметан:метанол:уксусная кислота 90:10:1; визуализация УФ-излучением; Rf продукта равен 0,13). Реакционную смесь концентрировали при пониженном давлении при температуре, равной от 35 до 40 С, к остатку прибавляли воду (8,00 л, 8,0 об.ч.) и этилацетат (5,00 л, 5,0 об.ч.) и содержимое перемешивали в течение от 5 до 10 мин. Слои разделяли, водную фазу подкисляли до pH 1 путем быстрого прибавления водного раствора хлористо-водородной кислоты (6 М, примерно 900 мл, 0,9 об.ч.) и полученную взвесь охлаждали до температуры, равной от 0 до 10 С, в течение от 40 до 50 мин. Осадок собирали фильтрованием, отделенное твердое вещество суспендировали в теплой воде (от 35 до 60 С, 5,00 л, 5,0 об.ч.) в течение от 10 до 25 мин и твердое вещество собирали фильтрованием. Суспендирование в воде повторяли, как это описано выше. Собранные твердые вещества объединяли с полученным из такой же порции, помещали в колбу с отводом объемом 20 л,прибавляли ацетон (10,00 л, 5,0 об.ч.) и реакционную смесь нагревали и продолжали кипячение с обратным холодильником возобновляли (примерно 65 С) в течение от 10 до 20 мин. Полученной смеси давали охладиться до температуры, равной от 15 до 25 С, перемешивали при температуре, равной от 15 до 25 С, в течение от 12 до 18 ч, и дополнительно охлаждали и выдерживали при температуре, равной от 0 до 5 С, в течение 60 мин. Осадок собирали фильтрованием и промывали смесью этилацетат:ацетон (1:1,4,00 л, 2 об.ч.). Твердое вещество сушили досуха на фильтре и дополнительно сушили в вакууме при температуре, равной от 40 до 45 С, до постоянной массы и получали (2S,4R)-1-(бифенил-4-илкарбонил)4-гидроксипирролидин-2-карбоновую кислоту в виде бежевого твердого вещества. Фильтраты концентрировали примерно до 3,00 л при пониженном давлении и получали вторую порцию материала, который собирали фильтрованием, промывали смесью этилацетат:гептаны (1:1, 24,00 л, 22 об.ч.) и высушивали на фильтре. Сушка в вакууме при температуре, равной от 40 до 45 С, до постоянной массы давала искомое соединение в виде бежевого твердого вещества. Полное количество полученного вещества: 2,616 кг. Выход: 91,9%. Стадия 2. Получение (2S)-1-(бифенил-4-илкарбонил)-4-оксо-пирролидин-2-карбоновой кислоты(2S,4R)-1-(Бифенил-4-илкарбонил)-4-гидроксипирролидин-2-карбоновую кислоту (0,806 кг,1,0 мас.ч.) и диметилсульфоксид (5,00 л, 6,25 об.ч.) помещали в колбу с отводом объемом 20 л и перемешивали в атмосфере азота до полного растворения. Раствор охлаждали до температуры, равной от 10 до 15 С, и прибавляли триэтиламин (2,40 л, 3,0 об.ч.), так чтобы внутренняя температура поддерживалась в диапазоне от 10 до 20 С. К реакционной смеси порциями прибавляли комплекс пиридин-триоксид серы(1,224 кг, 1,53 мас.ч.), так чтобы внутренняя температура поддерживалась в диапазоне от 10 до 25 С. Перемешивание при температуре, равной от 15 до 25 С продолжали, пока по данным анализа с помощью ТСХ реакция не завершалась (дихлорметан:метанол:уксусная кислота 90:10:1; Rf продукта равен 0,28),обычно в течение от 1 до 3 ч. Реакционную смесь охлаждали до температуры, равной от 0 до 10 С, и реакцию останавливали водным раствором хлористо-водородной кислоты (3 М, 6,460 л, 8,0 об.ч.) поддерживая температуру ниже 30 С. Прибавляли тетрагидрофуран (2,00 л, 2,5 об.ч.) и этилацетат (2,00 л,2,5 об.ч.), слои разделяли, водную фазу экстрагировали тетрагидрофураном:этилацетат (1:1, 4,00 л,5,0 об.ч.) и объединенные экстракты промывали водным раствором хлористо-водородной кислоты (1 М,21,60 л, 22,0 об.ч.) и насыщенным рассолом (1,60 л, 2,0 об.ч.). К органической фазе прибавляли активированный уголь (160 г, 0,2 мас.ч.) и полученную взвесь нагревали и продолжали кипячение с обратным холодильником (от 65 до 70 С) в течение 0,5 ч. Реакционную смесь охлаждали до температуры,равной от 20 до 30 С, прибавляли сульфат магния (375 г, 0,5 мас.ч.), перемешивание продолжали в течение 10 мин и смесь фильтровали через целит. Собранные твердые вещества промывали этилацетатом(20,800 л, 21,0 об.ч.) и объединенные фильтраты концентрировали при пониженном давлений при температуре, равной от 40 до 45 С, и получали искомое соединение, (2S)-1-(бифенил-4-илкарбонил)-4 оксо-пирролидин-2-карбоновую кислоту в виде вязкого оранжевого масла (0,769 кг, Выход: 96,0%). Вещество использовали на следующей стадии без дополнительной очистки. Стадия 3. Получение (2S)-1-(бифенил-4-илкарбонил)-4-(метоксиимино)пирролидин-2-карбоновой кислоты (соединение (VII) на схеме 6). Неочищенную (2S)-1-(бифенил-4-илкарбонил)-4-оксопирролидин-2-карбоновую кислоту (1,550 кг,5,01 моль, 1,0 мас.ч.), О-метилгидроксиламингидрохлорид (0,620 кг, 7,42 моль, 0,40 мас.ч.) и дихлорметан (12,40 л, 8,0 об.ч.) помещали в колбу с отводом объемом 20 л и охлаждали до температуры, равной от 0 до 5 С. К реакционной смеси в течение от 45 до 60 мин прибавляли триэтиламин (1,752 л, 1,13 об.ч.),так чтобы внутренняя температура поддерживалась в диапазоне от 0 до 10 С. Реакционную смесь нагревали до температуры, равной от 15 до 25 С, и поддерживали в этом температурном диапазоне (обычно от 12 до 18 ч), пока по данным анализа с помощью ТСХ реакция не завершалась (дихлорметан:метанол:уксусная кислота 90:10:1, визуализация УФ-излучением; Rf продукта равен 0,27, 0,35 Z, Е). Реакционную смесь концентрировали при пониженном давлении при температуре, равной от 40 до 45 С,остаток растворяли в этилацетате (12,40 л, 8,0 об.ч.) и промывали водным раствором хлористоводородной кислоты (2 М, 24,650 л, 23,0 об.ч.). Водные промывочные растворы объединяли и подвер- 10011026 гали обратной экстракции этилацетатом (4,650 л, 3,0 об.ч.). Органические экстракты объединяли, промывали насыщенным рассолом (4,650 л, 3,0 об.ч.), сушили над сульфатом магния (770 г, 0,5 мас.ч.), фильтровали и осадок на фильтре промывали этилацетатом (4,650 л, 3,0 об.ч.). Фильтраты концентрировали при пониженном давлений при температуре, равной от 40 до 45 С, и получали бежевое твердое вещество. Неочищенный продукт суспендировали в этилацетате (3,10 л, 2,0 об.ч.) при температуре, равной от 15 до 20 С, в течение 15 мин прибавляли циклогексан (12,40 л, 8,0 об.ч.) и полученную взвесь охлаждали и выдерживали при температуре, равной от 0 до 5 С, в течение 1 ч. Осадок собирали фильтрованием, промывали смесью этилацетат:циклогексан (1:2; 4,650 л, 3,0 об.ч.) и сушили в вакууме при температуре,равной от 40 до 45 С, до постоянной массы и получали искомый продукт в виде бежевого твердого вещества (1,132 кг, Выход: 66,8%). Выделенные фильтраты (из 9 циклов указанной выше реакции) объединяли и концентрировали при пониженном давлении при температуре, равной от 40 до 45 С. Остаток (примерно 1,00 кг) суспендировали в горячем виде (от 70 до 75 С) в этилацетате (7,00 л), охлаждали и выдерживали при температуре,равной от 0 до 5 С, в течение 2 ч, фильтровали и собранные твердые вещества сушили в вакууме при температуре, равной от 40 до 45 С, до постоянной массы и получали вторую порцию (2S)-1-(бифенил-4 илкарбонил)-4-(метоксиимино)пирролидин-2-карбоновой кислоты (0,732 кг, 4,9% от теоретического выхода). Стадия 4 а. Получение(2S)-1-(Бифенил-4-илкарбонил)-4-(метоксиимино)пирролидин-2-карбоновую кислоту (0,560 кг,1,0 мас.ч.) и тетрагидрофуран (8,40 л, 15,0 об.ч.) помещали в колбу с отводом объемом 20 л и охлаждали до температуры, равной от 0 до 5 С. Порциями прибавляли карбонилдиимидазол (0,280 кг, 0,5 мас.ч.),так чтобы внутренняя температура поддерживалась в диапазоне от 0 до 10 С. Реакционную смесь нагревали и перемешивали при температуре, равной от 15 до 20 С (от 1 до 2 ч), пока по данным анализа с помощью ТСХ реакция не завершалась (этилацетат, визуализация УФ-излучением). Затем одной порцией прибавляли N-гидрокси-3-триэтилсиланилоксипропионамидин (0,381 кг, 0,68 мас.ч., 1,0 экв., скорректировано на содержание силанола) в виде раствора в тетрагидрофуране (2,80 л, 5,0 об.ч.) и перемешивание продолжали при температуре, равной от 15 до 25 С, и за протеканием реакций следили на основании анализа с помощью ТСХ (этилацетат, визуализация УФ-излучением). Завершение реакции отмечено через 1 ч. Реакционную смесь концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, и остаток объединяли с двумя порциями, полученными при таком же количестве исходных веществ. К объединенному веществу прибавляли пиридин (5,040 л, 3 об.ч.) и полученную смесь нагревали и поддерживали при температуре, равной от 85 до 90 С, пока по данным анализа с помощью ВЭЖХ циклизация не завершалась. Реакционную смесь концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, темный маслообразный остаток обрабатывали этилацетатом (16,80 л,10 об.ч.) и промывали 25% водным раствором лимонной кислоты (35,00 л, 33,0 об.ч.). Водные экстракты объединяли и проводили обратную экстракцию этилацетатом (5,00 л, 3 об.ч.), объединенные органические фазы промывали рассолом (5,00 л, 3,0 об.ч.), сушили над сульфатом магния (1,680 кг,1 мас.ч.), фильтровали и осадок на фильтре промывали этилацетатом (1,70 л). Объединенные фильтраты концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, и получали неочищенный (2S)-1-(бифенил-4-карбонил)-5-[3-(2-триэтилсиланилоксиэтил)-1,2,4-оксадиазол-5-ил]пирролидин-3-он-O-метилоксим в виде коричневого масла, которое использовали без дополнительной очистки(2,796 кг, 108%). Стадия 4b. Получение (2S)-1-(бифенил-4-карбонил)-5-[3-(2-гидроксиэтил)-1,2,4-оксадиазол-5 ил]пирролидин-3-он-O-метилоксима. Неочищенный (2S)-1-(бифенил-4-карбонил)-5-[3-(2-триэтилсиланилоксиэтил)-1,2,4-оксадиазол-5 ил]пирролидин-3-он-O-метилоксим (1,398 кг, 1,0 мас.ч.) в виде раствора в тетрагидрофуране (6,990 л,5,0 об.ч.) обрабатывали 1% водным раствором трифторуксусной кислоты (3,495 л, 2,5 об.ч.). Анализ с помощью ТСХ (этилацетат; визуализация УФ-излучением; Rf продукта равен 0,35, 0,48 Z, Е) указывал на завершение реакции через 30 мин. Значение pH реакционной смеси доводили до pH 7 насыщенным водным раствором гидрокарбоната натрия (1,00 л, 0,72 об.ч.) и прибавляли этилацетат (6,990 л, 5 об.ч.). Слои разделяли, органическую фазу промывали насыщенным водным раствором гидрокарбоната натрия(2,796 л, 2,0 об.ч.), водные промывочные растворы объединяли и проводили обратную экстракцию этилацетатом (2,796 л, 2,0 об.ч.). Органические фазы объединяли, промывали рассолом (4,794 л, 3 об.ч.), сушили над сульфатом магния (1,164 кг, 0,75 мас.ч.), фильтровали и осадок на фильтре промывали этилацетатом (20,699 л, 20,5 об.ч.). Объединенные фильтраты концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, и получали маслообразный остаток, который объединяли с остатком от второй порции, полученной при таком же количестве исходных веществ. Полное количество неочищенного вещества: 2,592 кг. Неочищенное вещество растворяли в ацетонитриле (2,592 л, 1 об.ч.),прибавляли гептаны (26,00 л, 10 об.ч.) и полученную смесь нагревали и поддерживали при температуре,равной от 45 до 55 С, в течение 30 мин. Нижнюю ацетонитрильную фазу отделяли, при энергичном пе- 11011026 ремешивании прибавляли к трет-бутилметиловому эфиру (56,00 л, 22 об.ч.), смесь охлаждали и выдерживали при температуре, равной от 0 до 5 С, в течение от 1 до 2 ч, фильтровали и концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, и получали искомое соединение в виде бледно-желтого твердого вещества (2,037 кг, 93,3%). Пример 2 а. (3E,5S)-5-[3-(2-гидроксиэтил)-1,2,4-оксадиазол-5-ил]-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-он-O-метилоксим; (3Z,5S)-5-[3-(2-гидроксиэтил)-1,2,4-оксадиазол-5-ил]-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-он-О-метилоксим. Стадия 1. Получение (4R)-4-гидрокси-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролина (соединение (IV) на схеме 4). 4-Гидрокси-L-пролин (0,625 мас.ч.) и воду (3,3 об.ч.) помещали в колбу с отводом объемом 20 л. К содержимому по каплям прибавляли триэтиламин (2,42 об.ч.), так чтобы температура поддерживалась в диапазоне от 10 до 20 С. Прибавляли тетрагидрофуран (5,0 об.ч.) и реакционную смесь охлаждали до температуры, равной от 0 до 5 С. Хлорангидрид 2'-метил-1,1'-бифенил-4-карбоновой кислоты (1,0 мас.ч.) и тетрагидрофуран (5,0 об.ч.) помещали в отдельную колбу, перемешивали в течение от 5 до 10 мин и затем прибавляли к реакционной смеси, так чтобы температура поддерживалась в диапазоне от 0 до 10 С. Реакционную смесь нагревали при температуре, равной от 15 до 25 С, в течение 60-120 мин и поддерживали при температуре, равной от 15 до 25 С, пока по данным анализа с помощью ТСХ реакция не завершалась. Полученную смесь концентрировали в вакууме при температуре, равной от 35 до 40 С, к остатку прибавляли воду (10,0 об.ч.) и этилацетат (5,0 об.ч.) и содержимое перемешивали в течение от 5 до 10 мин. Слои разделяли, водную фазу подкисляли до pH 1 водным раствором хлористо-водородной кислоты (6 М, примерно 3,0 об.ч.) и полученную взвесь охлаждали и выдерживали при температуре,равной от 0 до 10 С, в течение от 25 до 40 мин. Осадок собирали фильтрованием, отделенное твердое вещество переносили в подходящую колбу с отводом и суспендировали в теплой (от 35 до 60 С) воде(5,0 об.ч.) в течение от 10 до 25 мин. Твердое вещество собирали фильтрованием и повторяли суспендирование в горячей воде, описанное выше. После второго суспендирования твердое вещество подвергали азеотропной сушке с помощью толуола (25,0 об.ч.) при температуре, равной от 40 до 50 С. К остатку прибавляли этилацетат (2,5 об.ч.) и гептаны (2,5 об.ч.), полученную взвесь охлаждали и выдерживали при температуре, равной от 0 до 5 С, в течение от 30 до 40 мин, фильтровали, собранное твердое вещество промывали предварительно охлажденной смесью (от 0 до 5 С) этилацетат:гептаны (1:1, 2,0 об.ч.) и сушили в вакууме при температуре, равной от 30 до 40 С, до постоянной массы и получали (4R)-4 гидрокси-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-1-пролин в виде белого твердого вещества. Выход: 85,9%. Стадия 2. Получение 1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-4-оксо-L-пролина (соединение (V) на схеме 5).(4R)-4-Гидрокси-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролин (продукт, полученный на стадии 1, 1,0 мас.ч.) и диметилсульфоксид (2,5 об.ч.) помещали в колбу с отводом объемом 20 л. Содержимое нагревали до температуры, равной от 35 до 40 С, и поддерживали при этой температуре до полного растворения. Раствор охлаждали до температуры, равной от 5 до 10 С, в атмосфере азота и прибавляли триэтиламин (3,0 об.ч.), так чтобы температура поддерживалась в диапазоне от 5 до 20 С. Комплекс пиридин-триоксид серы (1,47 мас.ч.) и диметилсульфоксид (4,9 об.ч.) помещали в отдельную колбу, перемешивали в течение от 5 до 10 мин и затем прибавляли к реакционной смеси, так чтобы температура поддерживалась в диапазоне от 15 до 25 С. Реакционную смесь перемешивали при температуре, равной от 15 до 25 С, пока по данным анализа с помощью ВЭЖХ реакция не завершалась (обычно от 1 до 3 ч). Содержимое сосуда охлаждали до температуры, равной от 0 до 10 С и реакцию останавливали водным раствором хлористо-водородной кислоты (3 М, 8 об.ч.), поддерживая температуру ниже 30 С. Затем прибавляли тетрагидрофуран (5,0 об.ч.) и гептаны (1,0 об.ч.), слои разделяли, водную фазу экстрагировали тетрагидрофураном (25,0 об.ч.) и объединенные органические фазы промывали водным раствором хлористо-водородной кислоты (1 М, 22,0 об.ч.) и насыщенным рассолом (22,0 об.ч.). Водные промывочные растворы объединяли и подвергали обратной экстракции тетрагидрофураном (21,0 об.ч.), органические фазы объединяли, сушили над сульфатом магния (3 мас.ч.) и фильтровали. Осадок на фильтре промывали тетрагидрофураном (1,0 об.ч.) и фильтраты концентрировали в вакууме при температуре,равной от 40 до 45 С, и получали бледно-коричневое вспененное вещество. К остатку прибавляли этилацетат (10,0 об.ч.), содержимое перемешивали в течение от 5 до 10 мин и растворитель удаляли в вакууме при температуре, равной от 40 до 45 С. Остаток переносили в колбу, этилацетат (8,0 об.ч.) прибавляли и содержимое кипятили с обратным холодильником, взвесь активированного угля (0,14 мас.ч.) в этилацетате (5,0 об.ч.) прибавляли и кипячение с обратным холодильником возобновляли и продолжали в течение от 20 до 30 мин. Содержимое охлаждали до температуры, равной от 40 до 45 С, фильтровали,осадок на фильтре промывали этилацетатом (2,5 об.ч.) и фильтраты концентрировали до 2,5-3,0 об.ч. в вакууме при температуре, равной от 40 до 45 С. Взвесь разбавляли этилацетатом (0,5 об.ч.) и кипятили с обратным холодильником. Прибавляли гептан (3,0 об.ч.) и содержимому давали охладиться до температуры, равной от 15 до 25 С, в течение от 1 до 2 ч. Взвесь дополнительно охлаждали до температуры,- 12011026 равной от 0 до 5 С, в течение от 2 до 3 ч, фильтровали и осадок на фильтре промывали смесью этилацетат:гептан [(1:1), 1,0 об. частей] предварительно охлажденной до температуры, равной от 0 до 5 С, затем гептаном (5,0 об.ч.). Выделенное твердое вещество сушили в вакууме при температуре, равной от 40 до 45 С, и получали 1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-4-оксо-L-пролин в виде почти белого твердого вещества. Выход: 60,3%. Стадия 3. Получение 4-метоксиимино-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролина (соединение (VII) на схеме 6). 1-[(2'-Метил-1,1'-бифенил-4-ил)карбонил]-4-оксо-L-пролин (полученный на стадии 2, 1,0 мас.ч.), Oметилгидроксиламингидрохлорид (0,285 мас.ч.) и дихлорметан (20 об.ч.) помещали в колбу с отводом объемом 20 л и охлаждали до температуры, равной от 0 до 5 С. Триэтиламин (0,91 об.ч.) в колбу прибавляли так, чтобы температура поддерживалась в диапазоне от 0 до 10 С, реакционную смесь нагревали до температуры, равной от 15 до 25 С, и поддерживали в этом температурном диапазоне в течение от 16 до 20 ч. Реакционную смесь концентрировали в вакууме при температуре, равной от 40 до 45 С, остаток растворяли в этилацетате (10,0 об.ч.) и промывали водным раствором хлористо-водородной кислоты(1 М, 25,0 об.ч.). Водные промывочные растворы объединяли и подвергали обратной экстракции этилацетатом (5,0 об.ч.), органические экстракты объединяли и промывали насыщенным рассолом(10,0 об.ч.), сушили над сульфатом магния (0,5 мас.ч.), фильтровали и осадок на фильтре промывали этилацетатом (5,0 об.ч.). Фильтраты концентрировали в вакууме при температуре, равной от 40 до 45 С,и получали 4-метоксиимино-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролин в виде ожидаемой смесиE:Z. Стадия 4 а. Получение (3EZ,5S)-1-[1-[(2'-метилбифенил-4-ил)карбонил]-5-3-[2-(триэтилсилил)окси]этил-1,2,4-оксадиазол-5-ил]пирролидин-3-он-O-метилоксима (соединение (Ib) на схеме 7). Раствор 4-метоксиимино-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролина (80,0 г, 227,02 ммоль,1,00 экв.) в тетрагидрофуране (1,00 л) охлаждали до внутренней температуры, равной от 0 до 5 С. Порциями прибавляли карбонилдиимидазол (38,65 г, 238,37 ммоль, 1,05 экв.), так чтобы температура поддерживалась в диапазоне от 0 до 5 С. Реакционную смесь нагревали и перемешивали при температуре,равной от 20 до 25 С, пока по данным анализа с помощью ВЭЖХ реакция не завершалась (от 2 до 3 ч)(реакцию останавливали 2,0 М аммиаком в метаноле). Затем по каплям прибавляли N-гидрокси-3 триэтилсиланилоксипропионамидин (81,25 г, 238,37 ммоль, 1,05 экв., скорректировано на содержание силанола) в виде раствора в тетрагидрофуране (330 мл), так чтобы внутренняя температура поддерживалась в диапазоне от 20 до 25 С, и перемешивание продолжали при температуре, равной от 20 до 25 С, и за протеканием реакции следили на основании анализа с помощью ВЭЖХ. Завершение реакции отмечено через 18 ч. Реакционную смесь концентрировали при пониженном давлении при температуре, равной от 40 до 45 С. К веществу прибавляли пиридин (500 мл) и полученный раствор нагревали и поддерживали при температуре, равной от 85 до 90 С, пока по данным анализа с помощью ВЭЖХ циклизация не завершалась (от 2 до 3 ч). Реакционную смесь концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, темный маслообразный остаток обрабатывали этилацетатом (1,00 л) и промывали 25% водным раствором лимонной кислоты (3400 мл). Водные экстракты объединяли и проводили обратную экстракцию этилацетатом (250 мл), объединенные органические фазы промывали рассолом (1,00 л), сушили над сульфатом магния, фильтровали и осадок на фильтре промывали этилацетатом. Объединенные фильтраты концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, и получали неочищенный (3EZ,5S)-1-1-[(2'-метилбифенил-4-ил)карбонил]-5-(3-[2-(триэтилсилил)окси]этил)-1,2,4-оксадиазол-5-илпирролидин-3-он-O-метилоксим в виде коричневого масла, которое использовали без какой-либо дополнительной очистки (126,04 г, 104%). Стадия 4b. Получение (3Z,5S)-5-[3-(2-гидроксиэтил)-1,2,4-оксадиазол-5-ил]-1-[(2'-метилбифенил-4 ил)карбонил]-3-пирролидинон-O-метилоксима. Неочищенный (3EZ,5S)-1-[1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-5-3-[2'-(триэтилсилил)окси]этил-1,2,4-оксадиазол-5-ил]пирролидин-3-он-O-метилоксим (126,04 г) объединяли с другой порцией,полученной при таком же количестве исходных веществ (полное количество: 257,8 г, 482,11 ммоль,1,0 экв.). Прибавляли ацетонитрил (1,29 л, 5,0 об.ч.) и полученный раствор обрабатывали 5% водным раствором трифторуксусной кислоты (1,065 л). Анализ с помощью ЖХМС (жидкостная хроматография масс-спектроскопия) указывал на завершение реакции после перемешивания в течение ночи. Прибавляли гидрокарбонат натрия (48,5 г, 1,2 экв.) и реакционную смесь перемешивали в течение 5 мин. Продукт экстрагировали этилацетатом (3500 мл), объединенные экстракты промывали разбавленным вдвое насыщенным рассолом (3300 мл), сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, и получали двухфазный маслообразный/твердый остаток. Остаток повторно растворяли в ацетонитриле (1,00 л), промывали гептаном(3200 мл), разделяли и концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, и получали искомое соединение в виде коричневого масла (183,9 г, 90,7% от теоретического выхода). Очистку продукта E/Z проводили на колонке (Novasep, с использованием диоксида кремния,- 13011026 40-63 мкм; EtOAc/циклогексан = 2:3, затем чистый этилацетат), а затем с помощью дополнительной хроматографии (Novasep, с использованием диоксида кремния, 15-25 мкм; EtOAc/циклогексан = 1:1). Эти две очистки позволили удалить большую часть побочных продуктов и получить бледно-желтое масло. Третью очистку проводили при таких же условиях, как и вторую хроматографию, и получали чистый Z изомер в виде бесцветного масла, содержащий 5-10% соответствующего кетона. Растворение в смеси ТГФ/ДХМ (тетрагидрофуран/дихлорметан)=1:4 (всего: 7 об.ч.), обработка связанным с полимером трисамином (1 г на 4,5 г Z изомера) в течение от 24 до 48 ч, фильтрование и концентрировали при пониженном давлении при температуре, равной от 40 до 45 С, давали (3Z,5S)-5-[3-(2-гидроксиэтил)-1,2,4 оксадиазол-5-ил]-1-[(2'-метилбифенил-4-ил)карбонил]-3-пирролидинон-O-метилоксим в виде почти белого твердого вещества (выход в диапазоне: 30-35%). Пример 3. (3EZ,5S)-1-([1,1'-Бифенил]-4-илкарбонил)-5-5-[(диметиламино)метил]-1,2,4-оксадиазол 3-ил-3-пирролидинон-O-метилоксим; (3Z,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-3-пирролидинон-О-метилоксим; (3E,5S)-1-([1,1'-бифенил]-4-илкарбонил)5-5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-3-пирролидинон-О-метилоксим. В этом примере стадии 1, 2 и 3 являются такими же, как и в примере 2. Стадия 4 а. Получение (2S,4Z)-1-([1,1'-бифенил]-4-илкарбонил)-4-(метоксиимино)-2-пирролидинкарбонитрила (соединение (VIIa) на схеме 7). Трехгорлую колбу объемом 6 л в атмосфере азота, содержащую (2S)-1-(бифенил-4-илкарбонил)-4(метоксиимино)пирролидин-2-карбоновую кислоту (151,95 г; 449,39 ммоль; 1,00 экв.) в сухом ТГФ(2500,00 мл), охлаждали до -20 С и затем прибавляли триэтиламин (62,46 мл; 449,39 ммоль; 1,00 экв.)-35 С. К раствору в течение 10 мин прибавляли этилхлорформиат (42,78 мл; 449,39 ммоль; 1,00 экв.),поддерживая температуру равной -35 С. Реакционную смесь перемешивали в течение 2 ч, давая температуре повыситься вплоть до -20 С. В течение 5 мин по каплям прибавляли еще 4 мл этилхлорформиата и реакционную смесь перемешивали при -20 С, в течение 30 мин. Насыщенный раствор аммиака в ТГФ получали пропусканием аммиака через 500 мл сухого ТГФ в течение 20 мин при -60 С в атмосфере азота в трехгорлой колбе объемом 2 л. Раствор аммиака прибавляли в колбу для проведения реакций через капельную воронку, поддерживая температуру ниже -25 С. Раствору давали нагреться до комнатной температуры в течение 3 ч и реакционную смесь перемешивали при в течение ночи. Реакционную смесь охлаждали до 10 С и в течение 10 мин при -60 С по каплям прибавляли еще 250 мл насыщенного раствора аммиака в ТГФ. Затем реакционную смесь перемешивали, давая температуре повыситься до комнатной. Непосредственно через реакционную смесь при 15 С пропускали аммиак в течение 10 мин, а затем перемешивали в течение 3 ч. Реакционную смесь концентрировали в вакууме до объема, равного 1 л. Полученную взвесь фильтровали и полученный остаток промывали с помощью 0,1 н. NaOH. Твердое вещество промывали водой и сушили и получали (2S,4Z)-1-([1,1'-бифенил]-4-илкарбонил)-4-(метоксиимино)-2 пирролидинкарбоксамид (102,10 г; 67,34%). В трехгорлую колбу объемом 3 л помещали (2S,4Z)-1-([1,1'бифенил]-4-илкарбонил)-4-(метоксиимино)-2-пирролидинкарбоксамид (102,10 г; 302,63 ммоль; 1,00 экв.) и толуол-4-сульфонилхлорид (86,54 г; 453,94 ммоль; 1,50 экв.) в пиридине (1 500,00 мл) и перемешивали при 80 С в течение ночи до завершения реакции. Летучие компоненты удаляли в вакууме и остаток растворяли в ДХМ (1 л). Органическую фазу промывали с помощью 1 н. HCl (2500 мл), затем насыщенным раствором NaHCO3 (1500 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали и получали черный остаток (масса=178 г). Этот остаток растворяли в ДХМ 350 мл полученную суспензию фильтровали и получали порошок кремового цвета. Для очистки фильтрат вводили в хроматографическую колонку (Novasep) (дихлорметан). Необходимые фракции объединяли и концентрировали и получали коричневый остаток, который объединяли с выделенным ранее твердым веществом (порошок кремового цвета). Объединенные твердые вещества разбавляли метил-трет-бутиловым эфиром (500 мл), суспензию фильтровали и промывали метил-трет-бутиловым эфиром и получали(2S,4Z)-1-([1,1'-бифенил]-4-илкарбонил)-4-(метоксиимино)-2-пирролидинкарбонитрил (60,00 г; 62,08%). Стадия 4b. Получение (3EZ,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-5-[(диметиламино)метил]-1,2,4 оксадиазол-3-ил-3-пирролидинон-O-метилоксима; (3Z,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-3-пирролидинон-O-метилоксима; (3E,5S)-1-([1,1'-бифенил]-4 илкарбонил)-5-5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-3-пирролидинон-О-метилоксима. В двухгорлую колбу объемом 2 л, содержащую (2S,4Z)-1-([1,1'-бифенил]-4-илкарбонил)-4(метоксиимино)-2-пирролидинкарбонитрил (59,10 г; 185,06 ммоль; 1,00 экв.) и гидроксиламингидрохлорид (15,43 г; 222,07 ммоль; 1,20 экв.) в EtOH (1 200,00 мл) при комнатной температуре в течение 5 мин по каплям прибавляли триэтиламин (30,87 мл; 222,07 ммоль; 1,20 экв.). Затем реакционную смесь перемешивали при 80 С в течение ночи, пока не обнаруживалось завершение реакции. Смеси давали охладиться до комнатной температуры и EtOH удаляли в вакууме. Прибавляли воду (1 л) и суспензию фильтровали. Для удаления всех побочных продуктов твердое вещество дважды промывали ацетонитрилом(2100 мл), затем диэтиловым эфиром (1100 мл) и получали продукт чистоты 75%. После сушки в вакууме при комнатной температуре получали (2S,4Z)-1-(бифенил-4-карбонил)-N'-гидрокси-4- 14011026(2S,4Z)-1-(бифенил-4-карбонил)-N'-гидрокси-4-(метоксиимино)пирролидин-2 карбоксамида (11,5 г; 32,63 ммоль; 1,00 экв.), 4-диметиламинопиридина (4,78 г; 39,16 ммоль; 1,20 экв.),N,N-диметилглицина (=R7-COOH; 4,04 г; 39,16 ммоль; 1,20 экв.) в 1000 мл смеси ДХМ/ДМФ (диметилформамид) (1:1) прибавляли N-(3-диметиламинопропил)-N'-этилкарбодиимидгидрохлорид (6,88 г; 35,90 ммоль; 1,10 экв.). Полученную бежевую суспензию перемешивали при комнатной температуре. Перемешивание продолжали в течение ночи. Растворитель удаляли при пониженном давлении, оставшийся коричневый маслообразный остаток растворяли в дихлорметане, дважды промывали 5% лимонной кислотой (для разрушения эмульсии требовалось прибавление рассола) и дважды насыщенным раствором NaHCO3, органический слой сушили над MgSO4 и концентрировали при пониженном давлений и получали 12,45 г желто-коричневатого твердого вещества. Указанное твердое вещество разделяли на 3 одинаковые порции (4,15 г), каждую порцию растворяли в 500 мл пиридина и полученные растворы нагревали при внешней температуре, равной 120 С, до завершения реакции. Порции объединяли, пиридин удаляли в вакууме, полученный остаток растворяли в ДХМ, дважды промывали 5% лимонной кислотой (вследствие образования эмульсии разделение фаз происходило только после прибавления рассола), сушили над MgSO4 и выпаривали при пониженном давлении и получали 12,9 г черного масла. Неочищенный продукт предварительно очищали путем фильтрования через слой (диоксид кремния; дихлорметан/МеОН = 95:5) и получали 10,67 г коричневого масла. Очистку E/Z продукта проводили на колонке (с использованием обычного диоксида кремния;EtOAc/циклогексан=9:1). Первая очистка позволила полностью удалить все побочные продукты и выделить продукт в виде почти белого твердого вещества (масса=6,73 г). Вторая очистка, проведенная при таких же условиях, дала чистый Z изомер: (3Z,5S)-1-([1,1'-бифенил]-4-илкарбонил)-5-5[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-3-пирролидинон-О-метилоксим (4,937 г; 36%). Пример 3 а. (3EZ,5S)-5-5-[(Диметиламино)метил]-1,2,4-оксадиазол-3-ил-1-[(2'-метилбифенил-4 ил)карбонил]-пирролидин-3-он-O-метилоксим; (3Z,5S)-5-5-[(диметиламино)метил]-1,2,4-оксадиазол-3 ил-1-[(2'-метилбифенил-4-ил)карбонил]-пирролидин-3-он-O-метилоксим;(3E,5S)-5-5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-1-[(2'-метилбифенил-4-ил)карбонил]-пирролидин-3-он-Oметилоксим. В этом примере стадии 1, 2 и 3 являются такими же, как и в примере 2 а. Стадия 4 а. Получение (2S,4EZ)-4-(метоксиимино)-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин 2-карбонитрила (соединение (VIIa) на схеме 7). Трехгорлую колбу объемом 6 л в атмосфере азота, содержащую 4-метоксиимино-1-[(2'-метил-1,1'бифенил-4-ил)карбонил]-1-пролин (150,00 г; 425,66 ммоль; 1,00 экв.) в сухом ТГФ (2,5 л), охлаждали до-25 С и затем прибавляли триэтиламин (80,11 мл; 576,30 ммоль; 1,63 экв.) (температура повышалась до-23 С). Раствор перемешивали в течение 10 мин и температуру доводили до -40 С. К раствору в течение 30 мин прибавляли этилхлорформиат (54,86 мл; 576,30 ммоль; 1,63 экв.), поддерживая температуру ниже-35 С. Реакционную смесь перемешивали в течение 2,5 ч, давая температуре повыситься вплоть до-19 С. Получали оранжевую суспензию. Насыщенный раствор аммиака в ТГФ получали пропусканием аммиака через 500 мл сухого ТГФ в течение 20 мин при -40 С в атмосфере азота в трехгорлой колбе объемом 1 л. Раствор аммиака (400 мл) прибавляли в колбу для проведения реакции через капельную воронку, поддерживая температуру ниже -25 С. Полученному раствору давали нагреться до -20 С в течение 1 ч, после чего обнаружено завершение реакции и смеси давали нагреться до комнатной температуры в течение ночи. Реакционную смесь концентрировали в вакууме до объема, равного 200 мл, и полученный остаток разбавляли с помощью 600 мл МТБЭ (метил-трет-бутиловый эфир). Полученную суспензию фильтровали, осадок на фильтре промывали с помощью МТБЭ (2200 мл), объединенные фильтраты дополнительно разбавляли этилацетатом (400 мл) и промывали водой (2500 мл). Водную фазу подвергали обратной экстракции этилацетатом (300 мл), объединенные органические фазы сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении и получали (2S,4EZ)-4(метоксиимино)-1-[(2'-метилбифенил-4-ил)карбонил]-L-пролинамид (163,64; 109,4% от теоретического выхода). Затем продукт использовали без очистки. В трехгорлую колбу объемом 3 л помещали (2S,4EZ)4-(метоксиимино)-1-[(2'-метилбифенил-4-ил)карбонил]-L-пролинамид (149,56 г; 425,61 ммоль; 1,00 экв.,расчет основан на равном 100% выходе для предыдущей стадии) и толуол-4-сульфонилхлорид (121,71 г; 638,41 ммоль; 1,50 экв.) в пиридине (1,5 л) и перемешивали при 80 С до завершения реакции (4,5 ч). Летучие компоненты удаляли в вакууме при температуре, равной от 40 до 45 С, и остаток растворяли в ДХМ (1 л). Органическую фазу промывали с помощью 1 н. HCl (2500 мл), затем насыщенным раствором NaHCO3 (1 500 мл). Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали и получали черный остаток. Этот остаток растворяли в ДХМ (350 мл) и для очистки вводили в хроматографическую колонку (Novasep) (дихлорметан). Необходимые фракции объединяли и концентрировали и получали коричневый остаток, который использовали без какой-либо дополнительной очистки. Выход:(3EZ,5S)-5-5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-1-(2'метилбифенил-4-ил)карбонилпирролидин-3-он-O-метилоксима; (3Z,5S)-5-5-[(диметиламино)метил]1,2,4-оксадиазол-3-ил-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-он-O-метилоксима; (3E,5S)-55-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-онO-метилоксима. В двухгорлую колбу объемом 2 л, содержащую (2S,4EZ)-4-(метоксиимино)-1-[(2'-метилбифенил-4 ил)карбонил]пирролидин-2-карбонитрил (136,38 г; 278,70 ммоль; 1,00 экв.) и гидроксиламингидрохлорид (27,11 г; 390,18 ммоль; 1,40 экв.) в этаноле (1,5 л), при комнатной температуре в течение 5 мин по каплям прибавляли триэтиламин (54,23 мл; 390,18 ммоль; 1,40 экв.). Затем реакционную смесь перемешивали при 80 С в течение ночи, пока не обнаруживалось завершение реакции. Смеси давали охладиться до комнатной температуры и этанол удаляли в вакууме. Прибавляли воду (1 л) и суспензию фильтровали. Оставшееся твердое вещество дважды промывали ацетонитрилом (2150 мл) и сушили в вакууме при комнатной температуре и получали (2S,4EZ)-N'-гидрокси-4-(метоксиимино)-1-[(2'-метилбифенил-4 ил)карбонил]пирролидин-2-карбоксамид (52,00 г; 50,92%). К суспензии(2S,4EZ)-N'-гидрокси-4-(метоксиимино)-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-2-карбоксамида (19,00 г; 51,85 ммоль; 1,00 экв.), 4-диметиламинопиридина (7,60 г; 62,22 ммоль; 1,20 экв.), N,N-диметилглицина (=R7-COOH; 6,42 г; 62,22 ммоль; 1,20 экв.) в 1,8 л смеси ДХМ/ДМФ (1:1) прибавляли N-(3-диметиламинопропил)-N'-этилкарбодиимидгидрохлорид (10,93 г; 57,04 ммоль; 1,10 экв.). Полученную суспензию перемешивали при комнатной температуре в течение ночи, после чего образовывался раствор. Растворитель удаляли при пониженном давлении, полученный остаток растворяли в дихлорметане, дважды промывали 5% лимонной кислотой и дважды насыщенным раствором NaHCO3, органический слой сушили над сульфатом магния и концентрировали при пониженном давлении и получали 24,57 г коричневатого аморфного твердого вещества. Указанное твердое вещество разделяли на 5 одинаковых порций (4,91 г), каждую порцию растворяли в 200 мл пиридина и полученные растворы нагревали при внешней температуре, равной 120 С , до завершения реакции. Порции объединяли, пиридин удаляли в вакууме и полученный остаток предварительно очищали с помощью хроматографии (Novasep, 100% этилацетат) и получали коричневое масло (масса=10,28 г). Очистка с помощью флэш-хроматографии, проведенная при таких же условиях, дала желтое масло (масса=2,62 г),которое несколько раз очищали при таких же условиях и получали искомое соединение в виде чистого Z изомера: (3Z,5S)-5-5-[(диметиламино)метил]-1,2,4-оксадиазол-3-ил-1-[(2'-метилбифенил-4-ил)карбонил]пирролидин-3-он-O-метилоксим (3,54 г; 15%). Пример 4. (3Z/E,5S)-1-(Бифенил-4-илкарбонил)-(5-гидроксиметил)пирролидин-3-он-О-метилоксим. В этом примере стадии 1, 2 и 3 являются такими же, как и в примере 1. Стадия 4. (4Z/E,2S) Метил-1-(бифенил-4-илкарбонил)-(4-метоксиимино)пирролидин-2-карбоксилат(этерификация на схеме 7). 4-Метоксиимино-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]-L-пролин (1 мас.ч.), ацетон (10 об.ч.) и карбонат калия (1 мас.ч.) в атмосфере азота помещали в колбу подходящего объема. Содержимое охлаждали до температуры, равной от 0 до 10 С, и прибавляли диметилсульфат, поддерживая температуру ниже 10 С. Реакционную смесь нагревали до температуры, равной от 16 до 25 С, и поддерживали в этом температурном диапазоне до завершения реакции (ожидаемое время: от 1 до 2 ч). Содержимое концентрировали в вакууме при температуре, равной от 40 до 45 С. К остатку прибавляли этилацетат (8 об.ч.) и воду (8 об.ч.) и слои разделяли. Органическую фазу промывали насыщенным рассолом (8 об.ч.) и затем сушили над сульфатом натрия (2 мас.ч.). Содержимое фильтровали, осадок на фильтре промывали этилацетатом (1 об.ч.) и фильтраты концентрировали в вакууме при температуре, равной от 40 до 45 С. Остаток растворяли в дихлорметане (1 об.ч.) и полученный раствор разделяли на 2 порции для проведения хроматографии. Каждый раствор очищали с помощью флэш-хроматографии на "сухой" колонке с использованием диоксида кремния (1,8 мас.ч.) при элюировании с помощью 25 об./об.% этилацетата в гептанах (12 об.ч.), а затем с помощью 50 об./об.% этилацетат/гептаны (12 об.ч.) для удаления содержащихся в небольшом количестве примесей. Фракции, содержащие продукт, полученный из 2 колонок, объединяли и концентрировали в вакууме при температуре, равной от 40 до 45 С. Остаток растворяли в ТГФ(2,5 об.ч.) и повторно концентрировали в вакууме при температуре, равной от 40 до 45 С, и получали искомый продукт (от 80 до 100%, от 83 до 104 мас./мас.%). Стадия 5. Получение (3Z/E,5S)-1-(бифенил-4-илкарбоннл)-(5-гидроксиметил)пирролидин-3-он-Oметилоксима (восстановление в соединение (Id) на схеме 7).(4Z/E,2S) Метил-1-(бифенил-4-илкарбонил)-(4-метоксиимино)пирролидин-2-карбоксилат (1 мас.ч.),ТГФ (4,7 об.ч.) и метанол (4,7 об.ч.) помещали в колбу подходящего объема. Раствор охлаждали до температуры, равной от 0 до 10 С, в атмосфере азота и порциями прибавляли борогидрид лития (0,1 мас.ч.),поддерживая температуру ниже 20 С. Реакционную смесь перемешивали при температуре, равной от 16 до 25 С, до завершения реакции по данным ТСХ (ожидаемое время: от 2 до 3 ч). Реакцию останавливали путем прибавления воды (0,8 об.ч.) и концентрировали в вакууме при температуре, равной от 40 до 45 С. К остатку прибавляли этилацетат (10 об.ч.) и воду (5 об.ч.) и слои разделяли. Водную фазу подвергали обратной экстракции этилацетатом (2 об.ч.). Органические фазы объединяли и промывали с помощью(5 об.ч.). Органический раствор сушили над сульфатом магния (2 мас.ч.). Содержимое фильтровали и фильтрат концентрировали в вакууме при температуре, равной от 40 до 45 С, и получали искомый продукт (от 80 до 100%, от 74 до 92 мас./мас.%). Затем неочищенный продукт очищали. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы (I) в которой А обозначает карбонильную группу -(С=O)-; В выбран из группы, включающей оксадиазольное кольцо, амидогруппу формулы -(C=O)-NR3R4 и-(CH2)n-X-R8; в которой оксадиазольное кольцо описывается любой из формулR2 выбран из группы, включающей арил, гетероарил и насыщенный или ненасыщенный 3-8 членный циклоалкил;R3 и R4 независимо выбраны из группы, включающей водород, C1-C6-алкил, С 2-C6-алкенил, C2-C6 алкинил, алкоксигруппу, сульфанил, ацил, алкоксикарбонил, аминокарбонил, насыщенный или ненасыщенный 3-8-членный циклоалкил, который может содержать 1-3 гетероатома, выбранных из группы,включающей N, О, S, арил, гетероарил, C1-C6-алкиларил и C1-C6-алкилгетероарил;R7 выбран из группы, включающей водород, сульфонил, аминогруппу, C1-C6-алкил, C2-C6-алкенил,C2-C6-алкинил, где указанные алкильные, алкенильные, алкинильные цепи необязательно включают гетероатом, выбранный из группы, включающей N, О и S, арил, гетероарил, насыщенный или ненасыщенный 3-8-членный циклоалкил, гетероциклоалкил, где указанные циклоалкильные, гетероциклоалкильные, арильные или гетероарильные группы необязательно сконденсированы еще с 1-2 циклоалкильными,гетероциклоалкильными, арильными или гетероарильными группами, ацильный фрагмент, C1-C6 алкиларил, C1-C6-алкилгетероарил, C1-C6-алкениларил, C1-C6-алкенилгетероарил, C1-C6-алкиниларил, C1C6-алкинилгетероарил, C1-C6-алкилциклоалкил, C1-C6-алкилгетероциклоалкил, C1-C6-алкенилциклоалкил, C1-C6-алкенилгетероциклоалкил, C1-C6-алкинилциклоалкил, C1-C6-алкинилгетероциклоалкил, алC1-C6-алкилацил,C1-C6-алкилкоксикарбонил,аминокарбонил,C1-C6-алкилкарбоксигруппу,ацилоксигруппу, C1-C6-алкилалкоксигруппу, C1-C6-алкилалкоксикарбонил, C1-C6-алкиламинокарбонил,C1-C6-алкилациламиногруппу, C1-C6-алкилуреидогруппу, C1-C6-алкиламиногруппу, C1-C6-алкиламмониевую группу, C1-C6-алкилсульфонилоксигруппу, C1-C6-алкилсульфонил, C1-C6-алкилсульфинил, С 1-C6 алкилсульфанил, C1-C6-алкилсульфониламиногруппу, C1-C6-алкиламиносульфонил, гидроксигруппу,галоген и цианогруппу;R8 и R9 совместно с атомом N, с которым они связаны, могут образовать пяти-восьмичленное насыщенное или ненасыщенное гетероциклоалкильное кольцо; иn является целым числом, равным от 1 до 3; где "арил" означает ненасыщенную ароматическую карбоциклическую группу, содержащую от 6 до 14 атомов углерода, включающую одно кольцо или несколько конденсированных колец,"гетероарил" означает моноциклическую гетероароматическую группу или бициклическую или трициклическую гетероароматическую группу, содержащую конденсированные кольца,"гетероциклоалкил" означает C3-C8-циклоалкильную группу, в которой вплоть до 3 атомов углерода заменены гетероатомами, выбранными из группы, включающей О, S, NR, где R обозначает водород или метил,"ацил" означает группу -C(O)R, в которой R означает "C1-C6-алкил", "арил", "гетероарил", "C3-C8 циклоалкил", "гетероциклоалкил", "C1-C6-алкиларил" или "C1-C6-алкилгетероарил",указанный способ включает следующие стадии: стадия 1: превращение пирролидина формулы (II) в ацилпроизводное формулы (IV) с использованием ацилирующего реагента (III) стадия 2: окисление ацилпроизводного (IV) окислительным реагентом с получением пирролидона формулы (V) стадия 3: превращение пирролидона формулы (V) в соединение (VII) с использованием подходящего алкоксиламина, арилоксиламина или гидроксиламина общей формулы (VI) стадия 4: превращение соединения (VII) с помощью амина общей формулы (VIII) или Nгидроксиамидина общей формулы (IX) с получением соединений (Ia) и (Ib) или превращение соединения(VII) сначала в нитрил (VIIa), который затем превращают в гидроксиамидин (VIIb), который затем вводят в реакцию с карбоновой кислотой R7-СООН и получают соединение (Ic), или сначала проведение этерификации и последующего восстановления соединения (VII) с использованием подходящего этерифицирующего или восстановительного реагента соответственно с получением соединения (Id) 2. Способ получения соединения формулы (I) по п.1 в которой А обозначает карбонильную группу -(С=O)-; В обозначает амидогруппу формулы -(C=O)-NR3R4 или оксадиазольное кольцо, описываемое любой из формулR7 выбран из группы, включающей водород, сульфонил, аминогруппу, C1-C6-алкил, C2-C6-алкенил,C2-C6-алкинил, где указанные алкильные, алкенильные, алкинильные цепи необязательно включают гетероатом, выбранный из группы, включающей N, О и S, арил, гетероарил, насыщенный или ненасыщенный 3-8-членный циклоалкил, гетероциклоалкил, где указанные циклоалкильные, гетероциклоалкильные, арильные или гетероарильные группы необязательно сконденсированы еще с 1-2 циклоалкильными,гетероциклоалкильными, арильными или гетероарильными группами, ацильный фрагмент, C1-C6 алкиларил, C1-C6-алкилгетероарил, C1-C6-алкениларил, C1-C6-алкенилгетероарил, C1-C6-алкиниларил, C1C6-алкинилгетероарил, C1-C6-алкилциклоалкил, C1-C6-алкилгетероциклоалкил, C1-C6-алкенилциклоалкил, C1-C6-алкенилгетероциклоалкил, C1-C6-алкинилциклоалкил, C1-C6-алкинилгетероциклоалкил, алкоксикарбонил, аминокарбонил, C1-C6-алкилкарбоксигруппу, C1-C6-алкилацил, C1-C6-алкилацилоксигруппу, C1-C6-алкилалкоксигруппу, C1-C6-алкилалкоксигруппу-карбонил, C1-C6-алкиламинокарбонил,C1-C6-алкилациламиногруппу,C1-C6-алкилуреидогруппу,C1-C6-алкиламиногруппу,C1-C6-алкиламмониевую группу, C1-C6-алкилсульфонилоксигруппу, C1-C6-алкилсульфонил, C1-C6-алкилсульфинил,С 1-C6-алкилсульфанил, C1-C6-алкилсульфониламиногруппу, C1-C6-алкиламиносульфонил, гидроксигруппу, галоген и цианогруппу;R2 выбран из группы, включающей арил, гетероарил и насыщенный или ненасыщенный 3-8 членный циклоалкил;R3 и R4 независимо выбраны из группы, включающей водород, C1-C6-алкил, C2-C6-алкенил, C2-C6 алкинил, алкоксигруппу, сульфанил, ацил, алкоксикарбонил, аминокарбонил, насыщенный или ненасыщенный 3-8-членный циклоалкил, который может содержать 1-3 гетероатома, выбранных из группы,включающей N, О, S, арил, гетероарил, C1-C6-алкиларил и C1-C6-алкилгетероарил;- 19011026 указанный способ включает следующие стадии: стадия 1: превращение пирролидина формулы (II) в ацилпроизводное формулы (IV) с использованием ацилирующего реагента (III) стадия 2: окисление ацилпроизводного (IV) окислительным реагентом с получением пирролидона формулы (V) стадия 3: превращение пирролидона формулы (V) в соединение (VII) с использованием подходящего алкоксиламина, арилоксиламина или гидроксиламина общей формулы (VI) стадия 4: превращение соединения (VII) с помощью амина общей формулы (VIII) или Nгидроксиамидина общей формулы (IX) с получением соединений (Ia) и (Ib), или превращение соединения (VII) сначала в нитрил (VIIa), который затем превращают в гидроксиамидин (VIIb), который затем вводят в реакцию с карбоновой кислотой R7-СООН и получают соединение (Ic) 3. Способ по п.1 или 2, в котором ацилхлорид, применяющийся на стадии 1, представляет собой 1'1 бифенил-4-карбонилхлорид или 2'-метил-1'1-бифенил-4-карбонилхлорид. 4. Способ по любому из пп.1-3, в котором окислительный реагент, применяющийся на стадии 2,представляет собой комплекс пиридин-триоксид серы (Ру-SO3) в комбинации с ДМСО. 5. Способ по любому из пп.2-4, в котором реакцию проводят в присутствии триэтиламина. 6. Способ по любому из пп.1-5, в котором алкоксиламин, применяющийся на стадии 3, представляет собой О-метилгидроксиламингидрохлорид. 7. Способ по любому из пп.1-6, в котором R1 обозначает метильную группу, R2 обозначает бифенил. 8. Способ по любому из пп.1-7, в котором В обозначает амидогруппу формулы -(C=O)NHR5, где R5 обозначает C1-C6-алкиларильную группу. 9. Способ по п.8, в котором R5 обозначает фенилэтильную группу, которая замещена аминогруппой или гидроксигруппой. 10. Способ по любому из пп.1-7, в котором В обозначает 1,2,4-оксадиазольный заместитель где R7 обозначает C1-C6-алкил или циклоалкил, необязательно содержащий 1 или 2 гетероатома. 11. Способ по любому из пп.1, 3, 4 или 6, 7, в котором В обозначает -(CH2)n-X-R8, где X обозначает О, R8 обозначает водород и n равно 1. 12. Способ по любому из пп.1-11, в котором соединение выбрано из группы, включающей
МПК / Метки
МПК: C07D 207/22, C07D 413/04
Метки: пирролидиноксимов, получения, способ
Код ссылки
<a href="https://eas.patents.su/22-11026-sposob-polucheniya-pirrolidinoksimov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения пирролидиноксимов</a>
Производные сложных эфиров о-диметилтрамадола, способ их получения и применение для получения лекарственного препарата

Номер патента: 3742
Опубликовано: 28.08.2003
Авторы: Чалаукс Фрейкса Мария, Дель Кастильо Нието Хуан Карлос, Угуэт Клотет Хоан, Де Рамон Амат Элизабет, Моурелье Мансини Марисабель
МПК: A61P 25/04, A61K 31/24, C07C 219/26...
Метки: производные, препарата, сложных, о-диметилтрамадола, применение, эфиров, способ, получения, лекарственного
Формула / Реферат:
1. Производные сложных эфиров O-диметилтрамадола общей формулы (I) где R1 или R2 - OH-группа; R3 - атом водорода; или R2 и R3 вместе образуют двойную связь; R4 - атом водорода или C1-C3алкилгруппа; R5 - атом водорода, NH2, NH-R11 или O-R11-группа; R6 - атом водорода, CO-R11, O-R11-группы или атом галогена; R7 - атом водорода, C1-C5-алкил-, C2-C5-O-алкенил-, фенилгруппы; или R6 и R7 - CH=CR12-CR13=CH-группа, образующая возможно...
Новый способ получения алендроновой кислоты

Номер патента: 3861
Опубликовано: 30.10.2003
Авторы: Лидор-Хадас Рами, Лифшиц Ревитал
МПК: C07F 9/38
Метки: алендроновой, новый, кислоты, получения, способ
Формула / Реферат:
1. Способ получения алендроновой кислоты, включающий стадии а) взаимодействия соединения формулы I с H3PO3 где R - имидогруппа и R1 выбирают из группы, которая состоит из атома хлора, брома, йода или фтора, гидрокси-, амино-, -OR2 или -OC(O)R2-группы, где R2 - C1-C12алкил-, C1-C12циклоалкил- или C1-C12арилгруппа, б) взаимодействия продукта стадии (а) с агентом, снимающим защитную группу, и в) выделения алендроновой кислоты. 2. Способ по п.1,...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Ларкин Джон Патрик, Крок Вероник, Руссель Патрик, Колладан Колетт
МПК: C07D 487/04
Метки: 10-диоксо-6н-пиридазино, октагидро-6, способ, получения, применение, соединений, диазепин-1-карбоновой, активных, терапевтически, производные, 1,2, 1,2-а, кислоты
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Мазюри Алан, Бонне Алан, Дельтиль Мишель
МПК: C07H 17/08
Метки: биологически, 5-0-дезозаминил-6-0-метилэритронолида, применение, получения, способ, активных, продуктов, производные
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Способ получения производного тетразола

Номер патента: 4633
Опубликовано: 24.06.2004
Авторы: Балло Илдико, Фишер Янош, Немеш Андраш, Надьне Багди Юдит, Веркне Папп Эва, Хегедюш Иштван, Цибула Ласло, Дойчне Юхас Ида, Петеньи Эндрене, Крейдл Янош, Фаркаш Йенёме
МПК: C07D 403/10, A61K 31/41, A61P 9/12...
Метки: производного, способ, получения, тетразола
Формула / Реферат:
1. Способ синтеза лозартана калия формулы (I) имеющего химическое название 2-н-бутил-4-хлор-1-[(2'-(тетразол-5-ил)-1,1'-бифенил-4-ил)метил]имидазол-5-метанол калия, с использованием в качестве исходного соединения 2-н-бутил-4-хлор-1-[(2'-(2-трифенилметил-2H-тетразол-5-ил)-1,1'-бифенил-4-ил)метил]-1H-имидазол-5-метанол формулы (III) отличающийся тем, что проводят реакцию соединения формулы (III) в среде спирта формулы R-OH (VI), где R...
Предыдущий патент: Новые соединения
Следующий патент: Фанера и способ ее изготовления
Случайный патент: Способ обновления и использования баз географических данных и пространственно-распределенная гибридная навигационная система для его реализации