Фармацевтические композиции, содержащие белок плазмы
Номер патента: 4700
Опубликовано: 24.06.2004
Авторы: Петё Габор, Пааль Кристина, Кремпельш Кристина, Хегедюш Лайош















Формула / Реферат
1. Растворимый в воде фармацевтический препарат в твердом состоянии или в виде истинного водного раствора, содержащий
a) терапевтически активное соединение, обладающее низкой растворимостью в воде, менее 1x10-4 М/л, и существенным сродством связывания с белками плазмы (в дальнейшем "активное соединение"), означающим, что более чем 90% активного соединения связано с белком плазмы в водной среде при спонтанном равновесии и при комнатной температуре;
b) белковую фракцию плазмы в контролируемом агрегационном состоянии, исключающем нежелательную агрегацию белка,
c) причем активное соединение и белковая фракция связаны друг с другом нековалентными связями, и
d) где упомянутый водный раствор не включает какого-либо органического растворителя, и
e) в котором молярное соотношение активного соединения и белковой фракции находится в диапазоне от 1:0,05 до 1:100 и где концентрация указанного активного соединения в водном растворе соответствует терапевтически эффективной дозе.
2. Препарат по п.1, где белковая фракция плазмы представляет собой сывороточный альбумин человека, сывороточный альбумин животного, рекомбинантный сывороточный альбумин человека, рекомбинантный сывороточный альбумин животного, g-глобулин и рекомбинантный g-глобулин.
3. Препарат по п.1 или 2, где активным соединением является цитостатик, антибиотик, витамин, противовоспалительное, анальгетик, антивирусное средство, противосудорожное средство, иммуносупрессор, антиэпилептическое, анксиолитик, снотворное, противогрибковое средство, антикоагулянт, липидный ингибитор пероксидаз, коронарный вазодилятор, антиаритмическое средство, кардиотоническое, мочегонное, антитромботическое средство, стероидный гормон (прогестерон, андроген, тестоген) и фотосенсибилизатор, и где концентрация указанного активного соединения в водном растворе соответствует терапевтически эффективной дозе.
4. Препарат по любому из пп.1-3, где по меньшей мере одно или более одного активное соединение выбрано из группы, содержащей амфотерицин B, аналог адриамицина, апазон, азатиоприн, бромазепам, кампототецин, карбамазепин, клоназепам, циклоспорин A, диазепам, дикумарол, дигитоксин, дипиридамол, дизопирамид, флунитразепам, гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовая кислота, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус, тамоксифен, таксоноид, тестостерон, тирилазад, триоксален, вальпроевая кислота, варфарин и где белковая фракция выбрана из группы, состоящей из сывороточного альбумина человека, сывороточного альбумина животного, рекомбинантного сывороточного альбумина человека, рекомбинантного сывороточного альбумина животного, g-глобулина и рекомбинантного g-глобулина, в котором молярное соотношение активного соединения и белковой фракции находится в диапазоне от 1:0,05 до 1:100 и где концентрация заявленного активного соединения в водном растворе соответствует терапевтически эффективной дозе.
5. Препарат по любому из пп.1-4, содержащий в качестве терапевтически активного вещества таксоноид общей формулы

где R1 означает третичный амид бутилоксикарбоксикислоты или амид бензоила,
R2 означает водород, ацил или ацетильную группу, предпочтительно паклитаксел, и где белковая фракция выбрана из группы, состоящей из сывороточного альбумина человека, сывороточного альбумина животного, рекомбинантного сывороточного альбумина человека, рекомбинантного сывороточного альбумина животного, g-глобулина и рекомбинантного g-глобулина, в которых молярное соотношение активного соединения и белковой фракции находится в диапазоне от 1:0,05 до 1:100 и где концентрация терапевтически активного соединения в водном растворе соответствует терапевтически эффективной дозе.
6. Гомогенный, твердый препарат, состоящий преимущественно по меньшей мере из одного терапевтически активного соединения, имеющего низкую растворимость в воде, менее 1x10-4 М/л, выбранного из группы, содержащей амфотерицин B, аналог адриамицина, апазон, азатиоприн, бромазепам, кампототецин, карбамазепин, клоназепам, циклоспорин A, диазепам, дикумарол, дигитоксин, дипиридамол, дизопирамид, флунитразепам, гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовую кислоту, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус, тамоксифен, таксоноид, тестостерон, тирилазад, триоксален, вальпроевую кислоту, варфарин, и по меньшей мере одной белковой фракции, выбранной из группы, состоящей из сывороточного альбумина, g-глобулина, рекомбинантного сывороточного альбумина, рекомбинантного g-глобулина, и где указанное терапевтически активное соединение и указанная белковая фракция связаны друг с другом нековалентными связями и где молярное соотношение заявленного терапевтически активного соединения и белковой фракции находятся в диапазоне от 1:0,05 до 1:100, а растворимость в воде препарата достаточна для обеспечения концентрации, соответствующей терапевтически эффективной дозе активного соединения.
7. Гомогенный твердый препарат, состоящий преимущественно по меньшей мере из одного терапевтически активного соединения с растворимостью в воде менее чем 1x10-4 М/л, выбранного из группы, содержащей амфотерицин B, аналог адриамицина, апазон, азатиоприн, бромазепам, кампототецин, карбамазепин, клоназепам, циклоспорин A, диазепам, дикумарол, дигитоксин, дипиридамол, дизопирамид, флунитразепам, гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовую кислоту, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус, тамоксифен, таксоноид, тестостерон, тирилазад, триоксален, вальпроевую кислоту, варфарин, и белковой фракции, выбранной из группы, состоящей из гликопротеина, интерферона, интерлейкина и рекомбинантного гликопротеина, интерферона, интерлейкина, где терапевтически активные соединения и указанная белковая фракция связаны друг с другом нековалентными связями и где молярное соотношение терапевтически активного соединения и указанной белковой фракции находится в диапазоне от 1:0,05 до 1:100, а растворимость в воде препарата достаточна для обеспечения концентрации, соответствующей терапевтически эффективной дозе активного соединения.
8. Гомогенный, твердый, водорастворимый препарат по п.6 или 7, в котором молярное соотношение терапевтически активного соединения и указанной белковой фракции находится в диапазоне от 1,0:0,1 до 1:50.
9. Гомогенный, твердый, водорастворимый препарат по любому из пп.6-8, в котором указанное терапевтически активное соединение представляет собой таксоноид формулы

где R1 означает третичный амид бутилоксикарбоксикислоты или амид бензоила,
R2 означает водород, ацил или ацетильную группу, предпочтительно паклитаксел, и где белковая фракция выбрана из группы, состоящей из сывороточного альбумина человека, сывороточного альбумина животного, рекомбинантного сывороточного альбумина человека, рекомбинантного сывороточного альбумина животного, g-глобулина и рекомбинантного g-глобулина, причем молярное соотношение активного соединения и белковой фракции находится в диапазоне от 1:0,05 до 1:100 и концентрация активного соединения в водном растворе соответствует терапевтически эффективной дозе.
10. Препарат по любому из пп.1-9, в котором молярное соотношение терапевтически активного соединения и белковой фракции находится в диапазоне от 1:0,1 до 1:50.
11. Способ получения водорастворимого препарата в твердом состоянии, как указано в любом из пп.1-10, растворимость в воде которого достаточна для обеспечения концентрации соответствующей терапевтически эффективной дозе активного соединения, включающий
a) полное растворение терапевтически активного соединения в смешивающемся с водой фармацевтически приемлемом органическом растворителе,
b) объединение указанного раствора в органическом растворителе с водным раствором фракции белков плазмы в контролируемом агрегационном состоянии, с получением истинного раствора, содержащего заявленное активное соединение и указанную белковую фракцию, связанные вместе нековалентными связями,
c) удаление органического растворителя и лиофилизацию раствора или его концентрата.
12. Способ получения истинных водных растворов без примеси органических растворителей препарата по любому из пп.1-10, содержащих водонерастворимое терапевтически активное соединение, включающий
a) полное растворение терапевтически активного соединения в смешивающемся с водой фармацевтически приемлемым органическом растворителе,
b) объединение указанного раствора в органическом растворителе с водным раствором фракции белков плазмы в контролируемом агрегационном состоянии,
с получением истинного раствора, содержащего заявленное активное соединение и указанную белковую фракцию, связанные вместе нековалентными связями,
c) удаление органического растворителя и лиофилизацию раствора или его концентрата,
d) растворение полученного в твердом состоянии препарата в воде, не содержащей органического растворителя, с получением истинного водного раствора, свободного от органических растворителей, в котором концентрация заявленного активного соединения соответствует терапевтически эффективной дозе, годной для терапевтического введения.
13. Способ по п.12, включающий доведение раствора до конечного препарата в виде дозированной формы для парентерального введения.
14. Способ по любому из пп.11-13, в котором на стадии a) используют органический растворитель, имеющий следующие свойства:
a) в смеси с водой он способен полностью растворять активное соединение и
b) в смеси с водой, где вода составляет менее 50%, он не денатурирует белок.
15. Способ по п.14, в котором органический растворитель выбирают из группы, состоящей из алифатического C(2-4) моноспирта или полиспирта, 70-100% этанола, диметилформамида, метилформамида.
16. Способ по любому из пп.11-15, в котором на стадии b) в качестве белков плазмы используют белки, выбранные из группы, включающей сывороточный альбумин, иммуноглобулин, гликопротеин, интерферон, интерлейкин, рекомбинантный гликопротеин, рекомбинантный интерферон и рекомбинантный интерлейкин.
17. Способ по любому из пп.11-16, в котором на стадии a) используют по меньшей мере одно или более одного терапевтически активное соединение, выбранное из группы, включающей амфотерицин B, аналог адриамицина, апазон, азатиоприн, бромазепам, кампототецин, карбамазепин, клоназепам, циклоспорин A, диазепам, дикумарол, дигитоксин, дипиридамол, дизопирамид, флунитразепам, гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовую кислоту, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус, тамоксифен, таксоноид, тестостерон, тирилазад, триоксален, вальпроевую кислоту, варфарин.
18. Способ по любому из пп.11-17, в котором на стадии a) в качестве терапевтически активного соединения используют паклитаксел.
19. Способ лечения людей или ветеринарных пациентов терапевтически активным соединением, имеющим низкую растворимость в воде, менее 1x10-4 М/л, и обладающим существенным сродством к белкам плазмы, включающий введение пациенту, нуждающемуся в лечении указанным активным соединением, водных растворов препарата по любому из пп.1-10, при следующем диапазоне доз (в пересчете на активное соединение): паклитаксел/альбумин - 70-280 мг/введение, пропофол/альбумин - 6-10 мг/кг/ч, камптотецин/альбумин, гемфиброзил/альбумин, циклоспорин A/альбумин - по 3-5 мг/кг/день, амфотерицин B/альбумин - до 1,5 мг/кг/день, причем те же самые диапазоны доз применяются для соединений, содержащих соответствующие рекомбинантные белки.
20. Способ лечения людей или ветеринарных пациентов по п.19, включающий парентеральное введение указанных водных растворов.
Текст
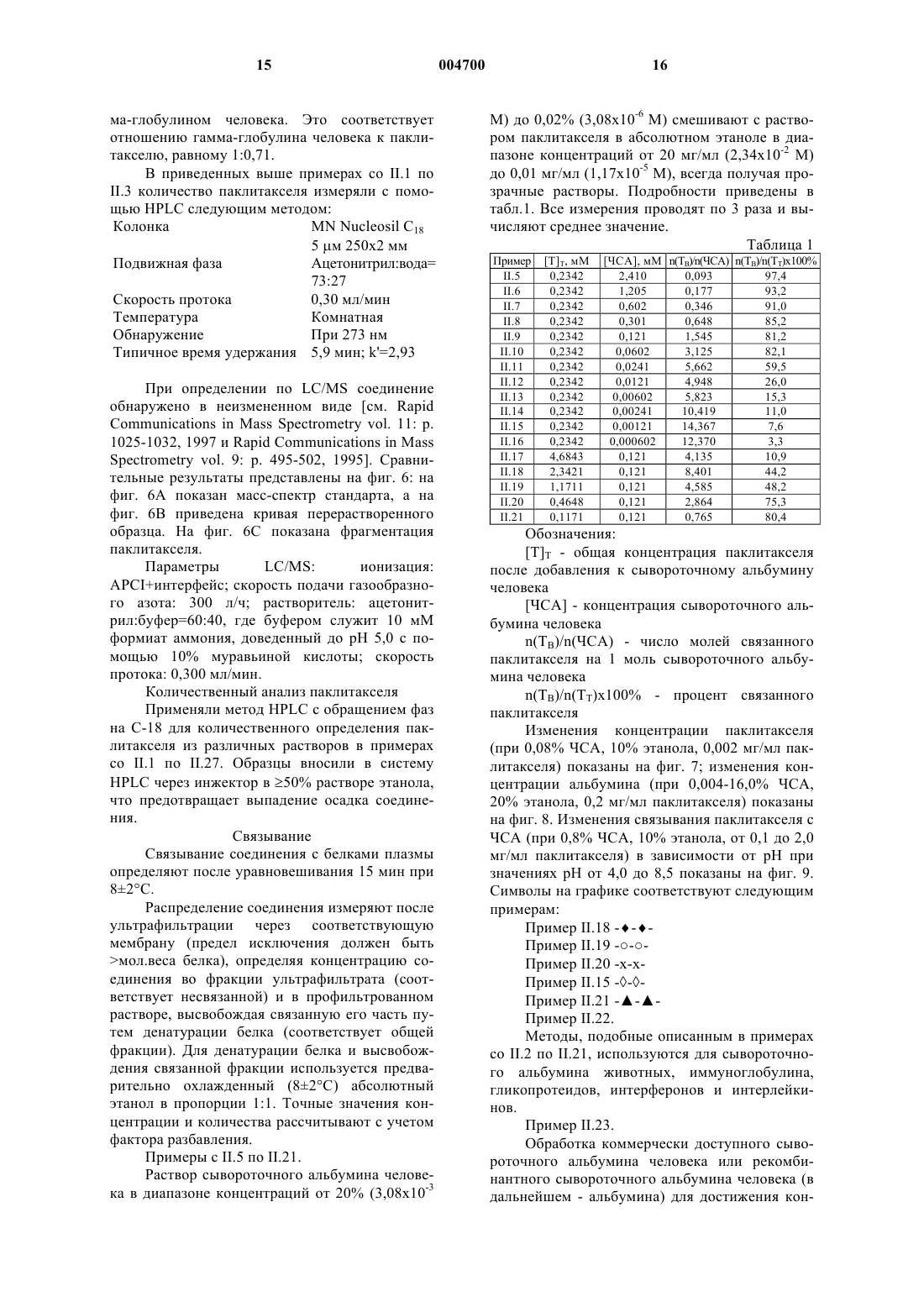
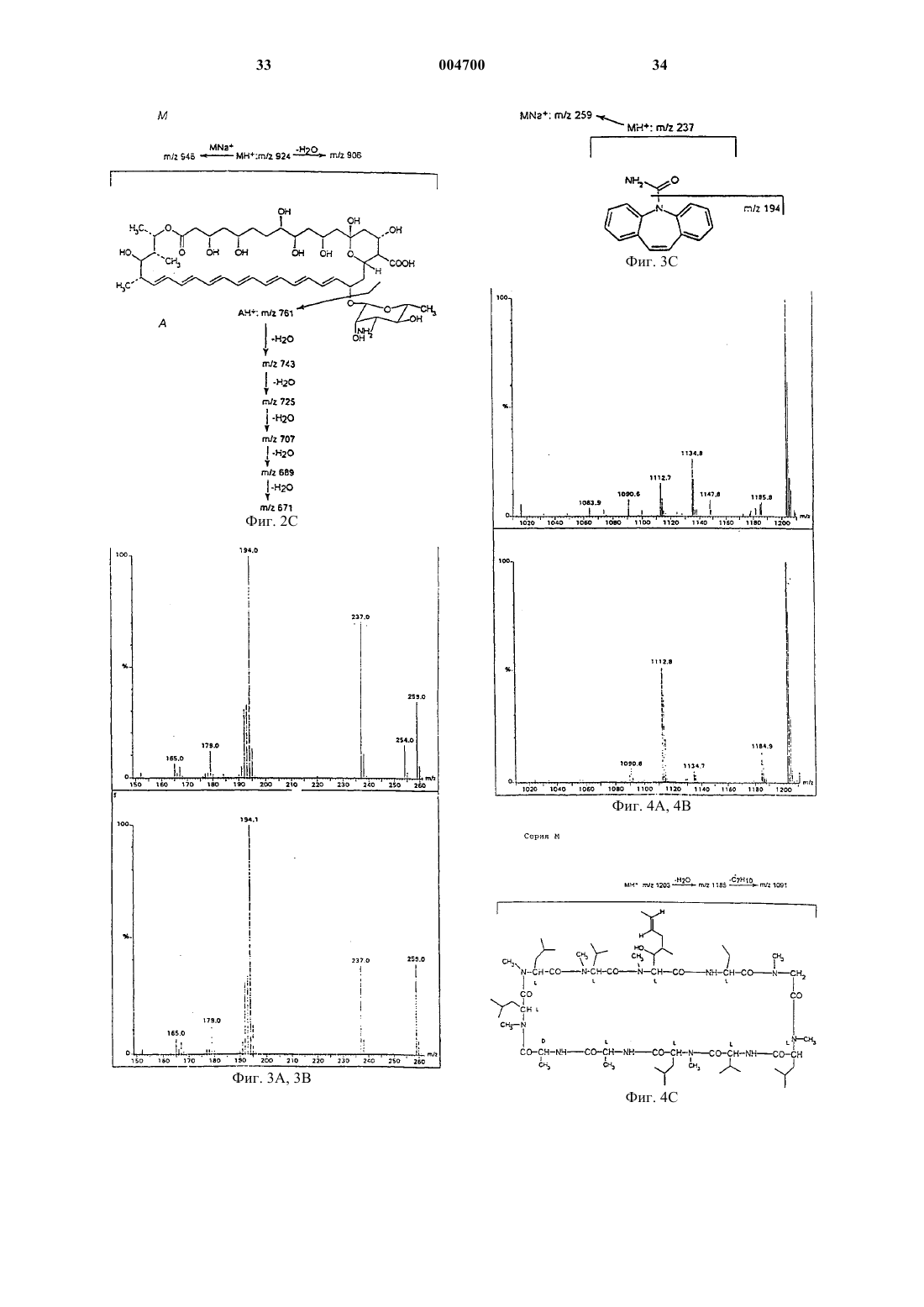
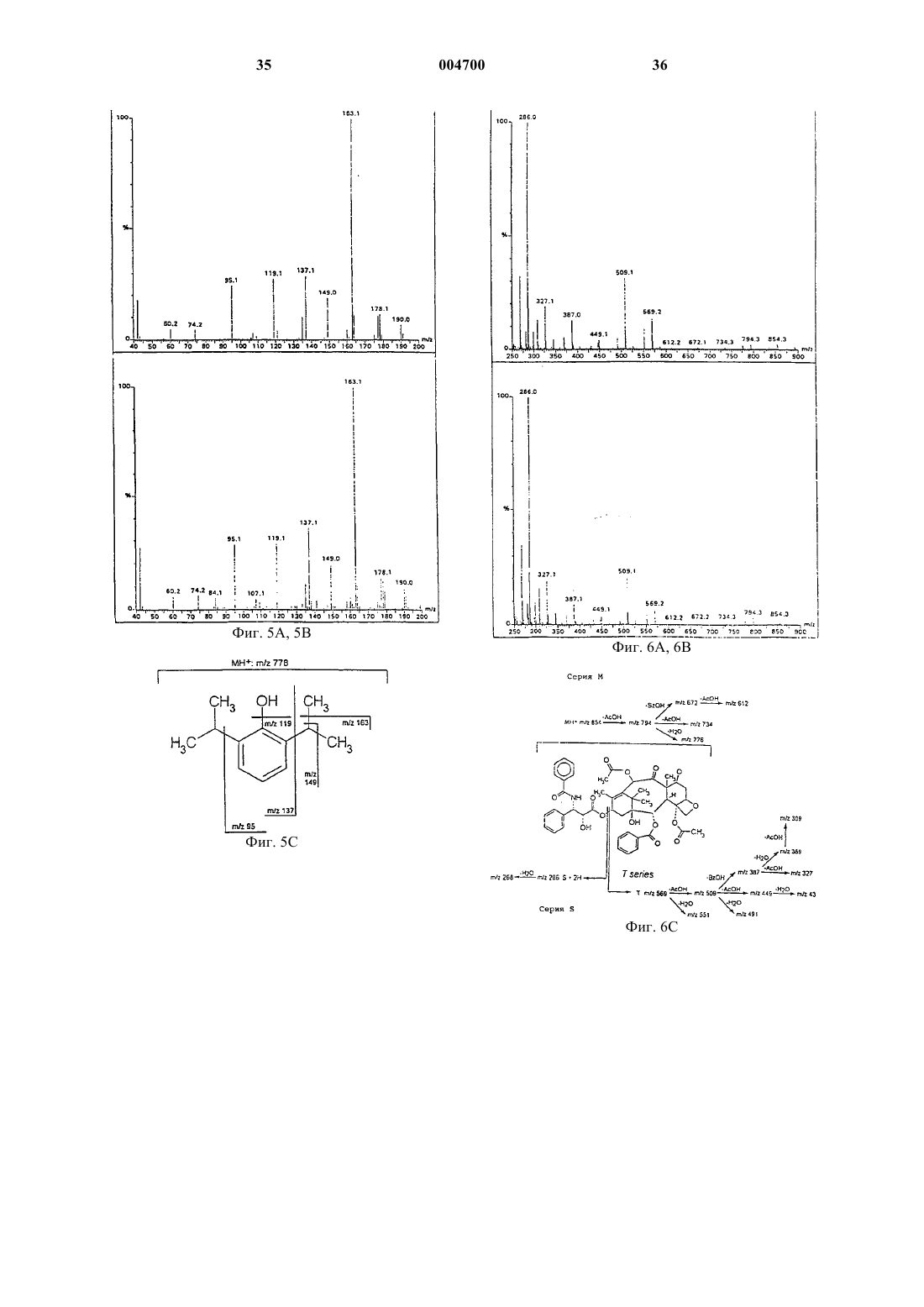
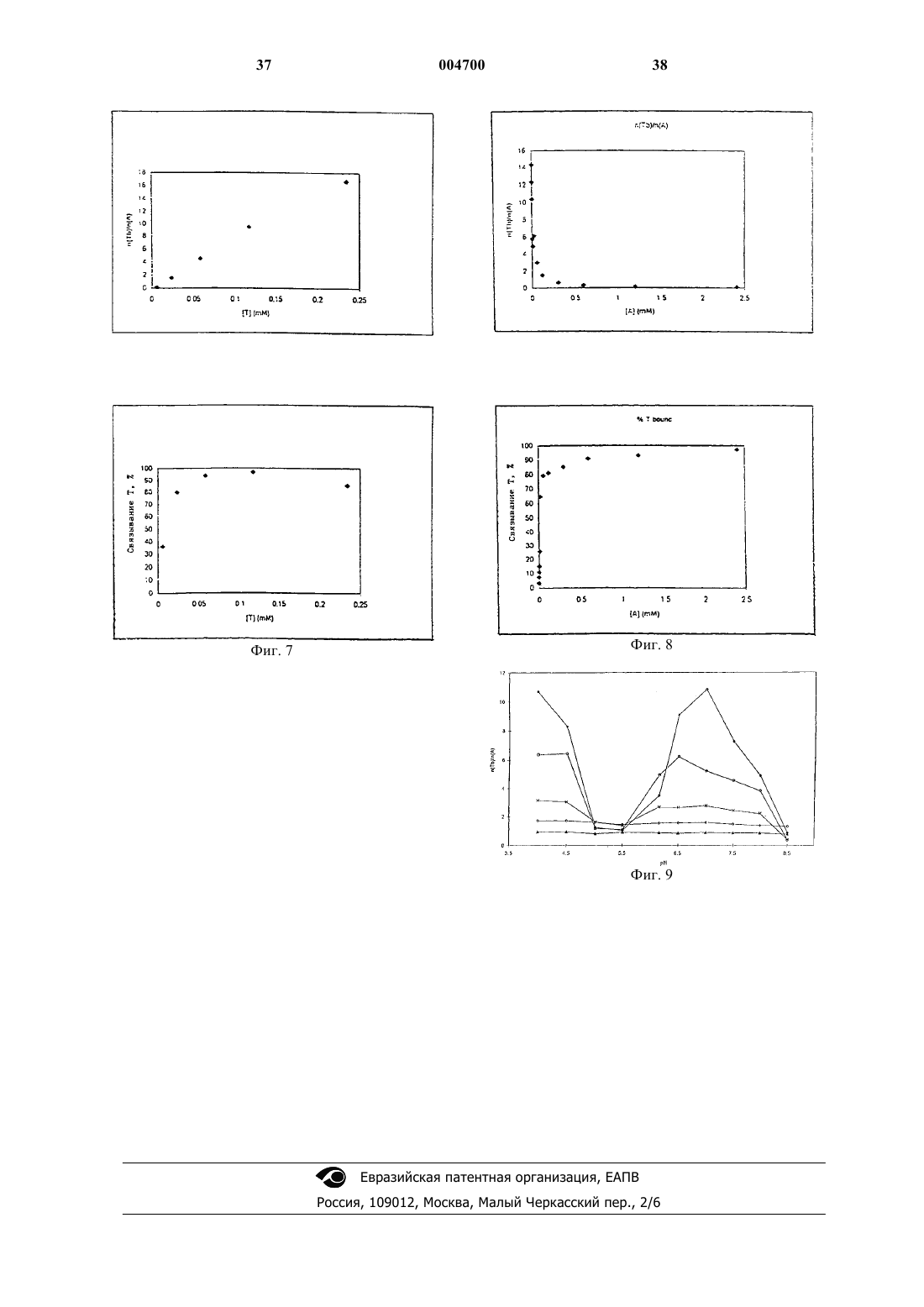
1 Настоящее изобретение касается нового метода, препаратов и композиций, применяющихся в терапии для введения терапевтически активных соединений, обладающих плохой растворимостью в воде и существенным сродством связывания с белками плазмы, и способов получения таких препаратов и композиций. Более конкретно, предметами изобретения в первую очередь являются препараты и фармацевтические композиции в твердой или жидкой форме, в основном для парентерального применения, состоящие из или содержащиеa) терапевтически активное соединение,имеющее низкую растворимость в воде и существенное сродство связывания с белками плазмы (в дальнейшем именуемое "активным соединением") иb) белковую фракцию плазмы в контролируемом агрегационном состоянии,вследствие чего указанное активное соединение и указанная белковая фракция связаны друг с другом нековалентными связями,c) а также, но необязательно, и другие фармацевтически приемлемые и, в основном,применяющиеся парентерально композиционные добавки - такие как вода, стабилизаторы,средства, подавляющие агрегацию белка. Гомогенные препараты, полученные по этому изобретению в твердом состоянии и состоящие из указанного белка и указанного соединения, растворимы в воде, а их водные растворы могут применяться парентерально или могут использоваться для получения парентеральных фармацевтических препаратов. Из предшествующего уровня техники известно, что биологически активные соединения обладают сильной терапевтической активностью, но их полезные свойства не могут проявиться вследствие плохой растворимости в жидких средах. Некоторые из них никогда не разрабатывались в качестве препаратов, тогда как небольшое число достигало лишь стадии"фазы I" при клинической разработке. Некоторые из них входят в состав "плохо биосовместимых" композиций, обладающих относительно высокой токсичностью, вызванной материалами, использованными для композиции. Типичный пример этого представляют препараты из группы таксонов, а конкретно - паклитаксел,являющийся сильным цитостатиком, применение которого, однако, ограничено вследствие токсичности его известной композиции в Клюцеле:твине 80 или в Клюцеле и растворителе 12,являющемся смесью 1:1 Кремафора EL:этанола.[Cancer Chemotherapy and Pharmacology (1994) 34: 465-471; Journal of the National Cancer Institute (1990) 1247-1259]. Кремафор EL (полиоксиэтилированное касторовое масло) обладает токсическими свойствами, вызывающими вазодиляцию, летаргию, гипотензию и т.д. Для снижения токсических побочных эффектов растворителя и адъюванта предлагались специальные 2 методы: введение очень маленьких доз в течение длительного периода времени, премедикация перед введением и др. (USP 5665761; USP 5621001; USP 5670537 и др.) Дальнейшее предложение состояло в сочетании активного соединения с диспергирующим средством, содержащимся в оболочке с белковыми стенками (USP 5 560 933), образованными путем реакции белка с маслом типа соевого масла - такие композиции предлагались для паклитакселя и амфотерицина. Однако, даже в новейшей литературе содержатся предостережения насчет курса применения паклитакселя (напр., см. "Guidance for Industry,изданное Департаментом Здравоохранения и Службы Человека CDER, сентябрь 1997, OGDL-8), в котором - вследствие реакции гиперчувствительности - всем пациентам, получающим паклитаксел, следует назначать премедикацию кортикостероидами, дифенгидрамином и Н 2 антагонистами. В дальнейшем было предложено получение парентеральных композиций определенных нерастворимых в воде дигидропиридинов путем растворения их в органическом растворителе или в смеси органического растворителя с водой и добавления водного раствора сывороточных белков человека к указанному раствору,чтобы свести к минимуму кристаллизацию нерастворимого активного соединения (венгерский патент 198381; DE Appl. 3702105). Однако полученная жидкость все-таки не была прозрачным раствором и содержала органический растворитель. Согласно публикации JP, документ 58216126 (см. ЕРО Patent Abstracts of Japan),некоторые производные карбоксикислот, несущие сильно защищенную ароматическую функциональную группу и растворимые в воде примерно до 0,1 мг/мл, растворялись при внесении их в разбавленные водные растворы сывороточного альбумина человека. Известно также, что оральные фармацевтические композиции были получены путем перемешивания активного ингредиента с яичным альбумином при высокой скорости от 5000 до 40000 об/мин в водном растворе и последующей возгонки растворителя (ЕРА 326618). В результате получили водную суспензию для орального применения, причем устранялись некоторые побочные эффекты активного ингредиента. Также известно, что некоторые нерастворимые в воде активные соединения обладают значительным сродством к белкам или сывороточным белкам. Приводим некоторые ссылки для паклитакселя [Cancer Chem. and Pharm. 3 Согласно новой литературе [The Lancet vol. 352(1998): 540-542] препарат Таксол вызывал слипание эритроцитов в "столбики", такой же эффект вызывало полиоксиэтилированное касторовое масло, служащее растворителем указанного препарата. Некоторые нерастворимые в воде лекарства были составлены с использованием токсичного Кремафора (циклоспорин, тенипозид, паклитаксел, амфотерицин В). Насколько нам известно, целый ряд высокоактивных, но нерастворимых в воде лекарств до сих пор отсутствует в продаже в лекарственных формах для парентерального и внутривенного применения, напр., ритонавит, карбамазепин,камфотетин, азатиопин, миконазол, флуконазол и др. Таким образом, существует потребность в разрешении проблемы, чтобы терапевтически ценные нерастворимые в воде вещества можно было вводить в водорастворимом виде, преимущественно парентерально, пациентам, требующим лечения указанными активными ингредиентами. Целью настоящего изобретения является удовлетворение этой потребности в отношении практически нерастворимых в воде активных ингредиентов, обладающих значительным сродством связывания с белками плазмы. Настоящее изобретение основано на понимании того, что связывание активных соединений с надлежащими белками посредством нековалентных связей перед введением представляет собой новую систему доставки с широкими возможностями для введения активных ингредиентов, плохо растворимых в воде. Согласно изобретению, образуются гомогенные твердые препараты, которые затем растворяют в воде и при этом получают биосовместимые, прозрачные водные растворы, пригодные для парентерального введения. Таким образом, изобретение представляет собой способ введения требуемых нерастворимых в воде активных ингредиентов без привлечения токсических элементов и в некоторых случаях в значительно более эффективной дозе, чем ранее. Термины, применяемые в настоящей заявке, которые в дальнейшем употребляются без расшифровки:R1: означает третичный амид бутилоксикарбоксикислоты или амид бензоила.R2: означает водородную или любую ацильную группу, преимущественно ацетил. Низкая растворимость в воде означает, что растворимость в воде при комнатной температуре 1 х 10-4 М. Существенное сродство связывания с белками плазмы означает, что 90% вещества связывается с белками в водной среде при спонтанном равновесии при комнатной температуре. ЧСА: сывороточный альбумин человека. ВДИ: вода для инъекций. 4 Один из предметов изобретения - это водорастворимая фармацевтическая композиция для человека, в основном для парентерального применения, содержащая терапевтически активное соединение с низкой растворимостью в воде и существенным сродством связывания с белками плазмы или белковой фракцией плазмы человека в контролируемом агрегационном состоянии. Другими предметами изобретения являются водорастворимые ветеринарные фармацевтические композиции, в основном для парентерального применения, содержащие терапевтически активное соединение с низкой растворимостью в воде и существенным сродством связывания с белками плазмы животного в контролируемом агрегационном состоянии. Плазма человека или животных, которая может присутствовать в препаратах и фармацевтических композициях согласно изобретению, и соответственно применяться в методах получения препаратов и композиций, может быть представлена любым из естественно присутствующих в плазме белков или фракций плазмы, таких как сывороточный альбумин,иммуноглобулин, гликопротеин, интерферон и/или интерлейкин, а также их рекомбинантными аналогами. В соединениях и композициях,предназначенных для лечения человека, отдается предпочтение натуральной сыворотке человека и рекомбинантным сывороточным белкам человека. Практически нерастворимые в воде активные ингредиенты, согласно изобретению, включают широкий спектр соединений, причем единственным ограничением является то, что они должны иметь существенное сродство к белку плазмы, выбранному для применения. Примеры таких активных ингредиентов включают следующие группы лекарственных средств цитостатики типа таксоноида, антибиотики,витамины, противовоспалительные средства,анальгетики, антиконвульсанты, иммуносупрессоры, антиэпилептические средства, анксиолитики, снотворные, противогрибковые средства,антикоагулянты, ингибиторы пероксидации липидов, коронарные вазодиляторы, антиаритмические средства, кардиотонические, мочегонные, антитромботические средства, стероидные гормоны (прогестерон, андроген, тестоген) и/или фотосенсибилизаторы. Можно одновременно использовать несколько активных ингредиентов после тщательного анализа и адаптации терапевтических доз с учетом сродства связывания к выбранным белкам, которые должны соответствовать пересмотренным требованиям. Для практического применения изобретения предложены препараты и фармацевтические композиции, содержащие, согласно вышесказанному, как минимум одно из следующих активных соединений: амфотерицин В, аналог адриамицина, апазон, азатиоприн, бромазепам, 5 камптотецин, карбамазепин, клоназепам, циклоспорин А, диазепам, дикумарол, дигитоксин,дипиридамол, дизопирамид, флунитразепам,гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовая кислота, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус,тамоксифен, таксоноид, тестостерон, тирилазад,триоксален, вальпроевая кислота и/или варфарин. Практическое применение изобретения составляют преимущественно препараты или композиции, подобные вышеописанным и содержащие таксоноиды, описываемые общей формулой I. Другое практическое применение, согласно изобретению, составляют паклитаксел и сывороточный альбумин человека, иммуноглобулин, гликопротеин, интерферон и/или интерлейкин или некоторые другие белковые фракции плазмы человека. Следующими особенно важными предметами изобретения являются гомогенные, твердые, водорастворимые препараты, содержащие как минимум одно активное соединение из группы: амфотерицин В, аналог адриамицина,апазон, азатиоприн, бромазепам, камптотецин,карбамазепин, клоназепам, циклоспорин А, диазепам, дикумарол, дигитоксин, дипиримадол,дизопирамид, флунитразепам, гемфиброзил,кетохлорин, кетоконазол, миконазол, нифлумовая кислота, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус, тамоксифен, таксоноид, тестостерон, тирилазад, триоксален, вальпроевая кислота и/или варфарин, и также содержащие как минимум один белок из группы: сывороточный альбумин человека, иммуноглобулин, гликопротеин, интерферон и/или интерлейкин или другую природную или рекомбинантную фракцию белков плазмы человека, в которых указанное активное соединение и указанная белковая фракция связаны друг с другом нековалентными связями, и в которых молярное соотношение указанного активного соединения и указанной белковой фракции находится в пределах от 1:0,05 до 1:100, предпочтительно от 1:0,1 до 1:50. Лучшими представителями вышеописанных являются следующие гомогенные, твердые,водорастворимые препараты, состоящие из следующих пар активных соединений и белков: таксоноиды общей формулы I - в этой формуле R1 означает третичный амид бутилоксикарбоксиловой кислоты или амид бензоила, аR2 означает водородную или ацильную группу,преимущественно ацетил- и белковая фракция плазмы; паклитаксел и сывороточный альбумин человека, рекомбинантный альбумин плазмы человека и/или -глобулин; 6 амфотерицин В и сывороточный альбумин человека, рекомбинантный альбумин плазмы человека и/или -глобулин; камптотецин и сывороточный альбумин человека, рекомбинантный альбумин плазмы человека и/или -глобулин; карбамазепин и сывороточный альбумин человека, рекомбинантный альбумин плазмы человека и/или -глобулин; циклоспорин А и сывороточный альбумин человека, рекомбинантный альбумин плазмы человека и/или -глобулин; пропофол и сывороточный альбумин человека, рекомбинантный альбумин плазмы человека и/или -глобулин. Из предыдущих объяснений ясно, что изобретение охватывает фармацевтические композиции, подобные вышеописанным, как в твердом состоянии, так и в виде водных растворов. По своей природной структуре - более конкретно, по химическому составу - молекулы белка имеют тенденцию к агрегации через свои специфические участки связывания. Степень агрегации зависит от параметров (температура,состав, относительная и абсолютная концентрация компонентов, затем рН, ионная сила) раствора, в котором присутствует белок. При использовании белков плазмы в соответствии с изобретением они, главным образом,находились в стабилизированном состоянии или состоянии контролируемой агрегации. Цель состоит в том, чтобы избежать такой агрегации белков, которая может ингибировать оптимальное связывание используемого на практике активного ингредиента. Нежелательную агрегацию белков можно контролировать присутствием других молекул, способных занимать некоторые или все участки связывания на макромолекулах, участвующих в агрегации, и тем самым избежать множественных связей белок-белок. Некоторые белки имеются в продаже в контролируемом агрегационном состоянии: они содержат стабилизаторы для предупреждения агрегации. Такое состояние, однако, не всегда является оптимальным для связывания с активным соединением, которое мы намерены использовать в соответствии с изобретением. Согласно изобретению термин "контролируемое агрегационное состояние" представляет собой наилучшее состояние связывания, когда белок способен связывать активное соединение точно таким образом, который желателен для намеченной задачи. Это не обязательно то состояние, когда максимальное количество молекул активного соединения связывается с белком, но есть случаи, когда желательна наибольшая степень связывания. Это означает, что в некоторых случаях необходимо удалить другие наполнители, напр.,из коммерчески доступной фракции сывороточного альбумина, такие как стабилизаторы, ион 7 ные компоненты и т.д. Это может быть необходимой начальной стадией процесса, в котором применяется метод согласно изобретению. Требуемые условия для установления надлежащего состояния агрегации строго зависят от конкретного активного соединения и соответствующей белковой фракции. Примеры, приведенные ниже, показывают(напр., паклитаксел и циклоспорин А), что они проявляют более сильное связывание с белковой фракцией плазмы в отсутствие других наполнителей (таких, как стабилизаторы, ионные компоненты, соли и др). Однако есть и такие активные соединения (напр., амфотерицин В и пропофол), связыванию которых не мешают,напр., стабилизаторы. Таким образом, следует определить надлежащее состояние агрегации используемого белка для каждой пары активного соединения/белка, применяемой согласно настоящему изобретению. При использовании пары паклитаксел и ЧСА важно устранить все стабилизаторы, входящие в состав коммерческих препаратов ЧСА,такие как N-ацетил-D,L-триптофан, каприлаты щелочных металлов, которые использовались для стабилизации белка во время пастеризации при 60 С. Амфотерицин В или пропофол может связываться с ЧСА также в присутствии этих стабилизаторов. В определенных случаях, когда желаемого состояния агрегации можно достичь с помощью воды, другие компоненты нужно удалить, напр., согласно процедуре, изложенной ниже в одном из примеров. Следует учитывать следующие аспекты для оптимального сочетания конкретных соединений с конкретными белковыми фракциями плазмы согласно изобретению:a) характеристики участка связывания, занимаемого соединением на белке;b) возможное присутствие в растворе других компонентов, занимающих тот же участок связывание или конкурирующих за него;c) физико-химические условия для конформации данного участка связывания и последствия связывания;d) известные терапевтические аспекты,напр.,i) паклитаксел на ЧСА имеет уникальные транспортные характеристики,ii) паклитаксел на интерлейкинах с известной терапевтической активностью переносчика;iii) циклоспорин А на гаммаиммуноглобулине с известной терапевтической активностью переносчика;iv) ритонавир на гамма-иммуноглобулине с известной терапевтической активностью переносчика; стабильность композиции. Одним из простейших средств контроля агрегации является вода. Используя надлежащее количество воды, можно подавить нежелатель 004700 8 ную агрегацию и подготовить белок для применения согласно изобретению - он перейдет в"контролируемое агрегационное состояние". При практическом применении изобретения композиции и соединения могут содержать в качестве добавок средства контроля агрегации белков и/или вспомогательные добавки, стабилизирующие раствор. Примерами таких добавок являются: вода, хлористый натрий, буфер, полиспирт типа глицерина, водорастворимые производные сахаров, главным образом, маннитол,сорбитол и/или дульцитол и другие. Следующим предметом настоящего изобретения является способ получения новых препаратов и фармацевтических композиций в соответствии с изобретением. Этот способ состоит из следующих стадий: а) растворение терапевтически активного соединения, имеющего низкую растворимость в воде и существенное сродство связывания с белками плазмы ("активного соединения"), в водорастворимом, фармацевтически приемлемом органическом растворителе,b) объединение этого раствора с водным раствором белковой фракции плазмы в контролируемом агрегационном состоянии,c) а также, но необязательно, с другим фармацевтически приемлемым вспомогательным компонентом - таким как средство контроля агрегации белка и/или стабилизатор - в результате чего получается настоящий раствор,содержащий указанное активное соединение и указанную белковую фракцию, связанные посредством нековалентных связей;d) удаление органического растворителя,предпочтительно путем ультрафильтрации, диализа, диафильтрации или лиофилизации раствора или его концентрата, или сочетанием этих обработок, в результате чего получается гомогенный водорастворимый жидкий или твердый препарат или фармацевтическая композиция,содержащая активное соединение и белковую фракцию плазмы;e) а также, но необязательно, растворение или разбавление твердого вещества или жидкости водой, в результате чего получается прозрачная, жидкая композицию, пригодная для терапевтического введения,f) а также, но необязательно, получение конечного препарата в виде парентеральной композиции (дозовой формы) для непосредственного применения. При получении новых гомогенных твердых препаратов, состоящих из активных соединений и белков, связанных нековалентными связями в соответствии с изобретением, предпочтительно применять процесс, состоящий из следующих шагов согласно изобретению:a) растворение терапевтически активного соединения в водорастворимом, фармацевтически приемлемом органическом растворителе;b) смешивание указанного раствора с водным раствором выбранной белковой фракции плазмы в контролируемом агрегационном состоянии, в результате чего получается настоящий раствор, содержащий указанное активное соединение и указанную белковую фракцию,связанные вместе нековалентными связями;c) удаление органического растворителя и лиофилизация раствора или его концентрата. Надлежащий способ наилучшего удаления органического растворителя зависит от активного соединения и соответственного белка. Из природы активного продукта (пары, состоящей из активного соединения и белка) следует, что применяемые методы должны обеспечивать мягкие условия. Лиофилизация приводит к получению гомогенных, твердых, водорастворимых препаратов, которые после растворения в воде можно вводить внутрибрюшинно. Может оказаться выгодным комбинирование вышеуказанных стадий, напр., процесс станет более экономичным, если вначале приготовить концентрат пары активное соединение/белок, а затем подвергнуть указанный концентрат лиофилизации. Некоторые пары активных соединений/белков (напр., пара амфотерицин В/сывороточный альбумин) можно успешно сконцентрировать путем ультрафильтрации или диализа. Некоторые другие пары (напр., паклитаксел/ЧСА) предпочтительнее обрабатывать посредством лиофилизации. Некоторые пары следует сначала подвергнуть ультрафильтрации и полученный концентрат затем подвергнуть лиофилизации. Специалисту в этой области понятно, что в ходе получения парентеральных фармацевтических средств разбавление водой включает и разбавление такими водными растворами, которые содержат другие добавки, пригодные для парентерального введения, напр., хлористый натрий. Надлежащий растворитель, применяемый согласно изобретению для растворения активного ингредиента на вышеуказанной стадии а),должен иметь следующие свойства: он должен обладать способностью к полному растворению активного ингредиента в его смеси с водой и его смесь с более 50% воды не должна денатурировать используемый белок. Перед началом проведения процесса согласно изобретению, используя активный ингредиент и выбранный белок, нужно определить адекватный растворитель на основе вышесказанного. Для этого подходят растворители, смеси которых, содержащие 50% воды, все еще способны растворять активный ингредиент. Предпочтительными растворителями, которые можно использовать на стадии а) вышеназванного процесса, например, могут быть любые представители группы, в которую входят алифатические С(2-4) моноспирты или полиоли, 004700 10 70-100% этанол, диметилформамид, метилформамид. При получении раствора, содержащего белок, можно добавлять средства контроля агрегации и/или стабилизаторы раствора. Подобные добавки включают дополнительное или оптимальное количество воды. Они также включают средства, способные частично заполнять некоторые из участков связывания белка для избежания агрегации, напр., любое из следующих средств: хлористый натрий, буфер, поли-спирт типа глицерина и/или водорастворимые производные сахаров, предпочтительно маннитол,сорбитол, дульцитол. При выборе оптимальных условий в случае любого активного ингредиента необходимо предварительно определить оптимальное сродство связывания и соответствующие агрегационные свойства. В нижеследующих примерах мы раскрываем полный метод подобных определений. Согласно предложенному практическому применению изобретения на стадии а) используются соединения: паклитаксел и компонент натуральной плазмы, такой как сывороточный альбумин, иммуноглобулин, гликопротеин, интерферон и/или интерлейкин или их рекомбинантные формы. Другие практические применения изобретения включают применение в качестве активных соединений нерастворимых в воде цитостатиков типа таксоноида, антибиотики, витамины, противовоспалительные, анальгетики, антиконвульсанты, иммуносупрессоры,антилептические средства, анксиолитики, снотворные, противогрибковые средства, антикоагулянты, ингибиторы пероксидации липидов,коронарные вазодиляторы, антиаритмические средства, кардиотонические, мочегонные, антитромботические средства, стероидные гормоны(прогестоген, андроген, тестоген) и/или фотосенсибилизаторы. Предпочтительно применение следующих активных соединений для процесса согласно изобретению: амфотерицин В, аналог адриамицина, апазон, азатиоприн, бромазепам,камптотецин, карбамазепин, клоназепам, циклоспорин А, диазепам, дикумарол, дигитоксин,дипиридамол, дизопирамид, флунитразепам,гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовая кислота, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус,тамоксифен, таксоноид, тестостерон, тирилазад,триоксален, вальпроевая кислота и/или варфарин. Предложенное практическое применение изобретения заключается в получении гомогенного, твердого, водорастворимого препарата,состоящего из паклитакселя и сывороточного альбумина человека, в котором активный ингредиент и белковая фракция плазмы могут образовывать нековалентные связи. Другое предложенное практическое применение изобрете 11 ния заключается в получении гомогенного,твердого, водорастворимого препарата, состоящего из таксоноида основной формулы I и белковой фракции плазмы, в котором активный ингредиент и белковая фракция плазмы образуют нековалентные связи. Из предыдущего объяснения ясно, что настоящее изобретение не ограничивается только активными соединениями или белками, перечисленными выше. Следующий предмет изобретения заключается в методе использования препаратов и композиций в соответствии с изобретением для лечения больных людей или животных. Метод состоит во ведении нуждающемуся в активном ингредиенте пациенту эффективной дозы композиции, полученной согласно изобретению. Вводимые дозы зависят как от активного ингредиента, так и от используемого белка. Можно вводить дозы, обеспечивающие, по крайней мере, те же уровни в крови, которые известны как эффективные при применении конкретных активных соединений при других способах введения. Представляем предложенный метод парентерального лечения больных людей и животных нерастворимыми в воде терапевтически активными соединениями, обладающими значительным сродством для связывания с белками плазмы, путем парентерального введения пациентам, нуждающимся в лечении указанным активным ингредиентом, эффективной дозы следующих препаратов, предпочтительно в следующих дозовых диапазонах (в пересчете на активное соединение),соответственно: паклитаксел/альбумин: 70-280 мг/прием; пропофол/альбумин: 6-10 мг/кг/ч; камптотецин/альбумин,гемфиброзил/альбумин, циклоспорин А/альбумин: 3-5 мг/кг/день; амфотерицин В/альбумин до 1,5 мг/кг/день, причем применяются те же дозовые диапазоны для соединений, содержащих соответствующие рекомбинантные белки. Соединения, композиции и методы данного изобретения дают следующие преимущества: становится возможным избежать использования биологически несовместимых растворителей, уменьшить или полностью избежать побочных эффектов, ограничивающих дозу,связанных с такими компонентами, как токсические растворители, поверхностно-активные средства, эмульсификаторы и т.п. применение белковых фракций плазмы при растворении медикаментов не оказывает дополнительных токсических эффектов - наоборот, они могут улучшить переносимость у пациентов, напр., в случае химиотерапии; в требуемых случаях можно повышать вводимую дозу по сравнению с препаратами,производимыми в настоящее время, тем самым давая возможность улучшить общий исход терапии. 12 Настоящее изобретение более подробно иллюстрируют следующие примеры, которыми оно не ограничивается. ПримерыI. Препаративные методы, анализы. Применялись следующие методы для определения связывания конкретного активного ингредиента (соединения) с белком:a) Ультрафильтрация. Образец в 1 мл прозрачного раствора, образованного смешиванием водного раствора,содержащего белок в контролируемом агрегационном состоянии, и раствора активного ингредиента в подходящем растворителе, фильтруют через ультрафильтрационную мембрану(предел исключения 30000 Да) и определяют активный ингредиент в ультрафильтрационной фракции. При измерении концентрации активного ингредиента в неотфильтрованном растворе общее количество (90%) обнаруживается в неизмененном виде.b) Лиофилизация. 1 мл вышеуказанного раствора лиофилизируют. После лиофилизации твердый остаток растворяют примерно в 1,00 мл дистиллированной воды, получая прозрачный раствор. При измерении концентрации активного ингредиента в этом растворе активный ингредиент не обнаруживается в водной фазе, но 100% его обнаруживается в белковой фракции.c) Анализ активного ингредиента. Количественное определение активного ингредиента проводится методом HPLC с УФдетекцией. Анализ методом HPLC можно проводить,напр., в системе HPLC Waters Millenium(Waters, MA, USA). Она состоит из следующих компонентов: насос Waters 616; контроллерWaters 717 plus, термостатируемый при +5 С; диодный детектор УФ/ВИЗ Waters 996. Эта система управляется и сбор данных осуществляется программой Waters Millenium v.2.02.0, работающей с персонального компьютера DigitalP486/166 (Digital Equipments, Irvin, UK). Условия необходимо оптимизировать индивидуально для каждого соединения, как показано в нижеследующих примерах для некоторых препаратов.d) Доказательство химической структуры. Применяется метод LC/MS для подтверждения того, что химическая структура выделенного из связанной фракции соединения осталась неизменной. Измерения LC/MS проводились на масс-спектрометре Finnigan Navigator(Finnigan,Manchester,UK) для одноквадрупольной LC/MS, используя режим ионизации ES или APCI, с помощью системы обработки данных MassLab v.2.0, работающей с персонального компьютера Digital Venturis FX/166(Digital Equipments, Irvin, UK). Рабочие параметры необходимо оптимизировать индивиду 13 ально для каждого конкретного соединения с помощью стандартов, как показано на следующих примерах.e) Подготовка образцов. Представлен следующий типичный метод подготовки образцов, который применяется для определения общей концентрации/количества соединения в образце методом HPLC и/илиLS/MS. Твердое содержимое лиофилизационного флакона реконституируют водой, раствор смешивают с абсолютным этанолом при соотношении объемов 1:1, при этом осаждаются белки плазмы, а вещество растворяется. После короткого центрифугирования раствор пригоден дляHPLC и/или LS/MS. При LS/MS его анализируют путем прямого введения образца, а приHPLC - путем отделения компонентов друг от друга. Оба метода дают ценную информацию о химической структуре родительского соединения и/или о возможных продуктах распада, что проиллюстрировано более подробно ниже для некоторых препаратов. Хроматографические и массспектроскопические данные из анализов HPLC и LS/MS могут подтвердить химическую эквивалентность между известным биологически активным соединением, использованным в качестве исходного материала, и соединением,выделенным после связывания с белковой фракцией согласно изобретению.f) Использованные материалы. Все использованные активные соединения по чистоте соответствовали USP XXIII. В экспериментах использовали следующие белковые фракции плазмы (=чистота по Евр. Фарм.): Альбумин человека,HUMAN Rt,20% р-рII. Получение, химические и физические анализы. В следующих примерах соотношения белок плазмы:субстрат для связывания в среднем попадают в интервал между 1:0,1-100. Пропорции соединение:ЧСА для связывания расчитывали, принимая мол. вес ЧСА=66 500, и мол. вес гамма-глобулина человека=150000 [см. 11 Science, vol. 244, р. 1195-1198, 1989, Vox Sang., 70: р. 203-209, 1996]. Пример II.1. 20% раствор (3,08x10-3 M) сывороточного альбумина человека в контролируемом агрега 004700 14 ционном состоянии и раствор паклитакселя 1 мг/мл (1,17 х 10-3 M) в абсолютном этаноле смешивают в пропорции 4:1 и перемешивают до получения прозрачного раствора. Раствор лиофилизируют, твердый остаток снова растворяют в количестве воды, достаточном для получения прозрачного раствора с концентрацией 20% сывороточного альбумина человека. Связывание определяют по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 99% паклитакселя связано с сывороточным альбумином человека. Это соответствует отношению сывороточного альбумина человека к паклитакселю, равному 1.0,1. Пример II.2. 4,44% раствор (6,67 х 10-4 М) сывороточного альбумина человека в контролируемом агрегационном состоянии и раствор паклитакселя 2,0 мг/мл (2,34 х 10-3 M) (м.в. 853,92) в абсолютном этаноле смешивают в пропорции 9:1 и перемешивают до получения прозрачного раствора. Раствор далее обрабатывают так, как описано в примере II.1. Связывание определяют по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 99% паклитакселя связано с сывороточным альбумином человека. Это соответствует отношению сывороточного альбумина человека к паклитакселю, равному 1:0,39. Пример II.3. 4,44% раствор (6,67 х 10-4 М) рекомбинантного сывороточного альбумина человека в контролируемом агрегационном состоянии и раствор паклитакселя 2,0 мг/мл (1,40 х 10-3 М) в абсолютном этаноле смешивают в пропорции 9:1 и перемешивают до получения прозрачного раствора. Раствор лиофилизируют, твердый остаток снова растворяют в количестве воды, достаточном для получения прозрачного раствора с концентрацией 20% рекомбинантного сывороточного альбумина человека. Связывание определяют по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 99% паклитакселя связано с рекомбинантным сывороточным альбумином человека. Это соответствует отношению рекомбинантного сывороточного альбумина человека к паклитакселю, равному 1:0,24. Пример II.4. 2,25% раствор (1,5x10-4 М) гаммаглобулинa человека, в контролируемом агрегационном состоянии и раствор паклитакселя 0,1 мг/мл (1,171 х 10-4 М) в абсолютном этаноле смешивают в пропорции 9:1 и перемешивают до получения прозрачного раствора. Раствор лиофилизируют, твердый остаток снова растворяют в количестве воды, достаточном для получения концентрации 16% гаммаглобулина человека в прозрачном растворе. Связывание определяют по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 98% паклитакселя связано с гам 15 ма-глобулином человека. Это соответствует отношению гамма-глобулина человека к паклитакселю, равному 1:0,71. В приведенных выше примерах со II.1 поII.3 количество паклитакселя измеряли с помощью HPLC следующим методом: КолонкаMN Nucleosil C18 5 м 250 х 2 мм Подвижная фаза Ацетонитрил:вода= 73:27 Скорость протока 0,30 мл/мин Температура Комнатная Обнаружение При 273 нм Типичное время удержания 5,9 мин; k'=2,93 При определении по LC/MS соединение обнаружено в неизмененном виде [см. RapidSpectrometry vol. 9: p. 495-502, 1995]. Сравнительные результаты представлены на фиг. 6: на фиг. 6 А показан масс-спектр стандарта, а на фиг. 6 В приведена кривая перерастворенного образца. На фиг. 6 С показана фрагментация паклитакселя. ПараметрыAPCI+интерфейс; скорость подачи газообразного азота: 300 л/ч; растворитель: ацетонитрил:буфер=60:40, где буфером служит 10 мМ формиат аммония, доведенный до рН 5,0 с помощью 10% муравьиной кислоты; скорость протока: 0,300 мл/мин. Количественный анализ паклитакселя Применяли метод HPLC с обращением фаз на С-18 для количественного определения паклитакселя из различных растворов в примерах со II.1 по II.27. Образцы вносили в системуHPLC через инжектор в 50% растворе этанола,что предотвращает выпадение осадка соединения. Связывание Связывание соединения с белками плазмы определяют после уравновешивания 15 мин при 82 С. Распределение соединения измеряют после ультрафильтрации через соответствующую мембрану (предел исключения должен быть мол.веса белка), определяя концентрацию соединения во фракции ультрафильтрата (соответствует несвязанной) и в профильтрованном растворе, высвобождая связанную его часть путем денатурации белка (соответствует общей фракции). Для денатурации белка и высвобождения связанной фракции используется предварительно охлажденный (82 С) абсолютный этанол в пропорции 1:1. Точные значения концентрации и количества рассчитывают с учетом фактора разбавления. Примеры с II.5 по II.21. Раствор сывороточного альбумина человека в диапазоне концентраций от 20% (3,08 х 10-3 16 М) до 0,02% (3,08x10-6 M) смешивают с раствором паклитакселя в абсолютном этаноле в диапазоне концентраций от 20 мг/мл (2,34 х 10-2 М) до 0,01 мг/мл (1,17 х 10-5 М), всегда получая прозрачные растворы. Подробности приведены в табл.1. Все измерения проводят по 3 раза и вычисляют среднее значение. Таблица 1 Пример[Т]T - общая концентрация паклитакселя после добавления к сывороточному альбумину человека[ЧСА] - концентрация сывороточного альбумина человекаn(ТB)/n(ЧСА) - число молей связанного паклитакселя на 1 моль сывороточного альбумина человекаn(ТB)/n(ТT)x100% - процент связанного паклитакселя Изменения концентрации паклитакселя(при 0,08% ЧСА, 10% этанола, 0,002 мг/мл паклитакселя) показаны на фиг. 7; изменения концентрации альбумина (при 0,004-16,0% ЧСА,20% этанола, 0,2 мг/мл паклитакселя) показаны на фиг. 8. Изменения связывания паклитакселя с ЧСА (при 0,8% ЧСА, 10% этанола, от 0,1 до 2,0 мг/мл паклитакселя) в зависимости от рН при значениях рН от 4,0 до 8,5 показаны на фиг. 9. Символы на графике соответствуют следующим примерам: Пример II.18 Пример II.19 Пример II.20 -х-хПример II.15 Пример II.21 Пример II.22. Методы, подобные описанным в примерах со II.2 по II.21, используются для сывороточного альбумина животных, иммуноглобулина,гликопротеидов, интерферонов и интерлейкинов. Пример II.23. Обработка коммерчески доступного сывороточного альбумина человека или рекомбинантного сывороточного альбумина человека (в дальнейшем - альбумина) для достижения кон 17 тролируемого агрегационного состояния с наилучшими условиями связывания молекул включает удаление стабилизаторов, таких как каприлат натрия, N-ацетил-D,L-триптофан и другие ионные компоненты и соли. а) Метод ультрафильтрации. Доводят раствор, содержащий 10% альбумин, до рН 3,0 с помощью хлорной кислоты и разводят до 5% по содержанию белка бидистиллированной водой. Концентрируют раствор до 10% по содержанию белка с помощью ультрафильтрации (предел исключения мембраны 30000 кД). Разводят раствор опять до 5% по содержанию белка в 1,0 мМ соляной кислоте. Концентрируют раствор до 10% по содержанию белка с помощью ультрафильтрации (предел исключения мембраны 30000 кД). Повторяют процедуру 12 раз, затем доводят до рН 6,9 с помощью 2,0 М водного раствора гидроокиси натрия и разбавляют раствор бидистиллированной водой до концентрации 5% по белку. Опять концентрируют раствор до 10% по содержанию белка с помощью ультрафильтрации (предел исключения мембраны 30 000 кД). Раствор снова разбавляют бидистиллированной водой до 5% по содержанию белка. Концентрируют раствор до 10% по содержанию белка с помощью ультрафильтрации (предел исключения мембраны 30 000 кД). Повторив процедуру 10 раз, получают чистую белковую фракцию, достаточно свободную от других наполнителей. К этому времени проводимость ультрафильтрата приближается к таковой бидистиллированной воды,использованной для разбавления. Этот белок пригоден к употреблению для связывания, напр., паклитакселя или циклоспорина.b) Использование диализа вместо ультрафильтрации дает сходные результаты. Обработка занимает примерно 48 ч. Пример II.24. 0,8% раствор (1,203x10-4 М) ЧСА и раствор 4,0 мг/мл амфотерицина В (4,33x10-3 М) (м.в. 924,09) в DMF смешивали в пропорции 9:1 и перемешивали до получения прозрачного раствора. Раствор лиофилизировали, твердый остаток снова растворяли в количестве воды, достаточном для обеспечения концентрации 20% ЧСА, получая прозрачный раствор. Связывание определяли по фракциям ультрафильтрата и осадка на фильтре,показывающeм, что 99,7% амфотерицина В связано с ЧСА. Это соответствует отношению ЧСА:амфотерицин В, равному 1:4. Пример II.25. 0,8% раствор (1,203x10-4 М) рекомбинантного сывороточного альбумина человека и раствор 40,0 мг/мл амфотерицина В (4,33x10-2 М) вDMF+НСl смешивают в пропорции 9:1 и перемешивают до получения прозрачного раствора. Раствор лиофилизуют, твердый остаток снова растворяют в количестве воды, достаточ 004700 18 ном для обеспечения конечной концентрации 20% рекомбинантного ЧСА, получая прозрачный раствор. Связывание определяют по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 99,5% амфотерицина В связано с ЧСА. Это соответствует отношению рекомбинантного ЧСА к амфотерицину В, равному 1:40. Амфотерицин В измеряли с помощьюMN Nucleosil C18 5 м 250 х 2 мм Подвижная фаза Ацетонитрил:буфер= 1:1 (буфер: 0,2% муравьиная кислота,доведенная до рН 4,0 триэтиламином) Скорость протока 0,30 мл/мин Температура Комнатная Обнаружение При 365 нм Типичное время удержания 5,3 мин; k'=1,41 При определении по LC/MS соединение обнаружено в неизмененном виде. Сравнительные результаты представлены на фиг. 2: на фиг. 2 А показан масс-спектр стандарта, на фиг. 2 В приведена кривая вновь растворенного образца. Фиг. 2 С показывает фрагментацию амфотерицина В. ПараметрыESI+интерфейс; скорость подачи газообразного азота: 300 л/ч; растворитель: 20 мМ формиат аммония, доведенный до рН 4,0 10% муравьиной кислотой; скорость протока 0,300 мл/мин. Пример II.26. 0,4% раствор (6,015x10-5 М) ЧСА в контролируемом агрегационном состоянии и раствор камптотецина 0,14 мг/мл (4,02x10-4 М)(м.в.=348,36) в абс. этаноле смешивали в пропорции 4:1 и перемешивали до получения прозрачного раствора. Раствор лиофилизировали,твердый остаток снова растворяли в количестве воды, достаточном для обеспечения конечной концентрации 20% ЧСА, получая прозрачный раствор. Связывание определяли по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 98% камптотецина связано с ЧСА. Это соответствует отношению ЧСА:кaмптoтeцин В, pавнoмy 1:5,34. Пример II.27. 0,4% раствор (6,015 х 10-5 М) рекомбинантного ЧСА в контролируемом агрегационном состоянии и раствор камптотецина 0,14 мг/мл(4,02 х 10-4 М) в абс. этаноле смешивали в пропорции 4:1 и перемешивали до получения прозрачного раствора. Раствор лиофилизировали, твердый остаток снова растворяли в количестве воды, достаточном для обеспечения конечной концентрации 20% рекомбинантного ЧСА, получая прозрачный раствор. Связывание определяли по фракциям ультрафильтрата и осадка на фильтре, 19 показывающим, что 98% камптотецина связано с ЧСА. Это соответствует отношению рекомбинантного ЧСА к камптотецину, равному 1:5,34. Мы измеряли камптотецин с помощьюMN Nucleosil C18 5 м 250x2 мм Подвижная фаза Ацетонитрил:буфер= 33:67 Скорость протока 0,33 мл/мин Температура Комнатная Обнаружение При 356 нм Типичное время удержания 6,9 мин; k'=2,45 При определении по LC/MS соединение обнаружено в неизмененном виде [Cancer Research, vol. 56: р.3689-3694, 1996]. Пример II.29. 0,4% раствор (6,015 х 10-5 М) рекомбинантного ЧСА в контролируемом агрегационном состоянии и раствор карбамазепина 8,0 мг/мл(3,39x10-2 М) (м.в. 236,27) в абс.этаноле смешивали в пропорции 19:1 и перемешивали до получения прозрачного раствора. Раствор лиофилизировали, твердый остаток снова растворяли в количестве воды, достаточном для обеспечения конечной концентрации 20% ЧСА, получая прозрачный раствор. Связывание определяли по фракциям ультрафильтрата и осадка на фильтре,показывающим, что 98% карбамазепина связано с ЧСА. Это соответствует отношению ЧСА:карбамазепин, равному 1:2,8. Мы измеряли карбамазепин с помощьюAцетонитрил:буфер= 1:1 (буфер: 0,2% муравьиная кислота,доведенная до рН 7,0 триэтиламином) Скорость протока 0,25 мл/мин Температура Комнатная Обнаружение При 285 нм Типичное время удержания 5,3 мин; k'=2,45 При определении по LC/MS соединение обнаружено в неизмененном виде [Eur J. Clin.Chem. Clin. Biochem, vol. 35(10): p.755-759,1997]. Сравнительные результаты представлены на фиг. 3: на фиг. 3 А показан масс-спектр стандарта, на фиг. 3 В приведена кривая вновь растворенного образца. Фиг. 3 С показывает фрагментацию карбамазепина. ПараметрыESI+интерфейс; скорость подачи газообразного азота: 300 л/ч; растворитель: 2 мМ формиат аммония, доведенный 10% муравьиной кислотой до рН 4,0; скорость протока: 0,250 мл/мин. Пример II.30. 4,0% раствор (6,015 х 10-4 М) ЧСА в контролируемом агрегационном состоянии и рас 004700 20 твор циклоспорина А 1,0 мг/мл (8,33 х 10-4 М) в абс.этаноле смешивали в пропорции 9:1 и перемешивали до получения прозрачного раствора. Раствор лиофилизировали, твердый остаток снова растворяли в количестве воды, достаточном для обеспечения концентрации 20% ЧСА, получая прозрачный раствор. Связывание определяли по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 97% циклоспорина А связано с ЧСА. Это соответствует отношению ЧСА:циклоспорин А, равному 1:0,14. Пример II.31. 2,0% раствор (3,008 х 10-4 М) рекомбинантного ЧСА в контролируемом агрегационном состоянии и раствор циклоспорина А 1,0 мг/мл(8,33x10-4 М) в абсолютном этаноле смешивали в пропорции 9:1 и перемешивали до получения прозрачного раствора. Раствор лиофилизировали, твердый остаток снова растворяли в количестве воды, достаточном для обеспечения концентрации 20% рекомбинантного ЧСА, получая прозрачный раствор. Связывание определяли по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 98% циклоспорина А связано с ЧСА. Это соответствует отношению рекомбинантного ЧСА к циклоспорину А, равному 1:0,29. Пример II.32. 2,25% раствор (1,50x10-4 M) раствора гамма-глобулина человека в контролируемом агрегационном состоянии и раствор циклоспорина А 8,0 мг/мл (3,39x10-2 M) в абсолютном этаноле смешивали в пропорции 9:1 и перемешивали до получения прозрачного раствора. Раствор лиофилизировали, твердый остаток снова растворяли в количестве воды, достаточном для обеспечения конечной концентрации 16% гамма-глобулина человека, получая при этом прозрачный раствор. Связывание определяли по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 98% циклоспорина А связано с гамма-глобулином человека. Это соответствует отношению рекомбинантного гамма-глобулина человека к циклоспорину А,равному 1:0,56. Циклоспорин А измеряли с помощьюMN Nucleosil C18 5 м 250 х 2 мм Подвижная фаза Ацетонитрил:вода: метанол:фосфорная кислота=700:260:40:0,05 Скорость протока 0,25 мл/мин Температура 80 С термостат Обнаружение При 205 нм Типичное время удержания 7,5 мин; k'=2,95 При определении по LC/MS соединение обнаружено в неизмененном виде [1]. Сравнительные результаты представлены на фиг. 4: на 21 фиг. 4 А показан масс-спектр стандарта, на фиг. 4 В приведена кривая вновь растворенного образца. Фиг. 4 С показывает фрагментацию циклоспорина А. Параметры LC/MS: ионизация: ESI+интерфейс; скорость подачи газообразного азота: 300 л/ч; растворитель: ацетонитрил/вода=60/40; скорость протока растворителя: 0,350 мл/мин. Пример II.33. 0,4% раствор (6,015 х 10-5 М) ЧСА и раствор пропофола 2,0 мг/мл (1,12 х 10-2 М) (м.в. 178,27) в абсолютном этаноле смешивали в пропорции 9:1 и перемешивали до получения прозрачного раствора. Раствор лиофилизировали, твердый остаток снова растворяли в количестве воды, достаточном для обеспечения концентрации 20% ЧСА, получая прозрачный раствор. Связывание определяли по фракциям ультрафильтрата и осадка на фильтре, показывающим, что 99% пропофола связано с ЧСА. Это соответствует отношению ЧСА:пропофол, равному 1:18,3. Пример II.34. 0,4% раствор (6,015 х 10-5 М) рекомбинантного ЧСА и раствор пропофола 2,0 мг/мл(1,12 х 10-2 М) в абсолютном этаноле смешивали в пропорции 9:1 и перемешивали до получения прозрачного раствора. Раствор лиофилизировали, твердый остаток снова растворяли в количестве воды, достаточном для обеспечения конечной концентрации 20% рекомбинантного ЧСА, получая прозрачный раствор. Связывание определяли по фракциям ультрафильтрата и осадка на фильтре,показывающим, что 99% пропофола связано с рекомбинантным ЧСА. Это соответствует отношению рекомбинантного ЧСА к пропофолу,равному 1:18,3. Пропофол измеряли с помощью HPLC следующим образом: КолонкаMN Nucleosil C18 5 м 250 х 2 мм Подвижная фаза Ацетонитрил:вода= 73:27 Скорость протока 0,30 мл/мин Температура Комнатная Обнаружение При 273 нм Типичное время удержания 6,1 мин; k'=1,77 При определении по LC/MS соединение обнаружено в неизмененном виде [J. of Chromatography В, 669: р. 358-365, 1995]. Сравнительные результаты представлены на фиг. 5: на фиг. 5 А показан масс-спектр стандарта, на фиг. 5 В приведена кривая вновь растворенного образца. Фиг. 5 С показывает фрагментацию пропофола. Параметры(1,213 х 10-4 М) ЧСА и 1,0 мл раствора амфотерицина В 4,0 мг/мл (4,33 х 10-3 М) в диметилформамиде до получения прозрачного раствора. Этот раствор диализировали против 2,0 л воды(ВДИ) при 4 С в течение 20 ч, защищая от света. Используя метод определения в примереII.24, нашли, что связывание составляет 99,6%,соответствуя отношению ЧСА:амфотерицин В,равному 1:3,5. При повторении процедуры диализа 5 раз концентрация DMF в растворе снижалась ниже предела определения (2 х 10-9 М).III. Дозировочные формы. Примеры с III.1 по III.6. Следуя способу получения с помощью лиофилизации, как описано выше, получают надлежащую фармацевтическую композицию. Вновь растворяя твердое вещество в соответствующем объеме ВДИ с тем, чтобы получить концентрацию 20% ЧСА, раствор доводят до концентрации, пригодной для терапевтического применения, как суммировано ниже для некоторых активных соединений. Пример Название Конц., мг/млIII.6 Пропофол 10,0 Вышеуказанные дозировочные формы можно затем довести до конечной формы во флаконах для инъекций и инфузий.IV. Биологические примеры. Исследования по биологической эквивалентности Биологическую эквивалентность определяли, сравнивая новые композиции, полученные по изобретению, с известными композициями,применяющимися при лечении и содержащими то же самое активное соединение с низкой растворимостью в воде. Такие известные композиции готовили в полиоксиэтилированном касторовом масле (Кремофор EL) и абсолютном этаноле. Использованные материалы Паклитаксел, растворенный в смеси полиоксиэтилированного касторового масла (Кремофор EL) с абсолютным этанолом в пропорции 1:1, сравнивали с водным раствором паклитаксел/ЧСА, полученным по изобретению согласно примеру II.2. Пример IV.1. Исследования in vitro. Проведены сравнительные исследования invitro для определения антипролиферативной и цитотоксической активности на клеточных линиях опухоли человека. Композиции паклитакселя в Кремофоре EL/абсолютном этаноле и в ЧСА сравнивали на клеточных линиях миело 23 идной лейкемии человека К 562, карциномы молочной железы MCF-7 и MDA-231 и яичниковOVCAR-5 [Anticancer Research, vol. 16: р. 24692478, 1996]. Метод Оценка ингибирования роста колоний. Монослойные культуры клеточных линий обрабатывали препаратом в двух вышеуказанных композициях при восьми различных концентрациях, а также в ДМСО/солевом растворе в качестве контроля. Культуры инкубировали 24, 48,72, 96 и 120 ч, соотв. Колонии окрашивали кристалл-виолетом и рассчитывали выживание обработанных клеток в виде процента от колоний,образованных необработанными клетками. В табл. с 2 А по 4B представлены результаты, полученные на различных клеточных линиях. В каждом исследовании показано выживание обработанных клеток, рассчитанное как процент от колоний, образованных необработанными клетками. Все значения являются средними из трех экспериментов. Таблица 2 А Клеточная линия: карцинома молочной железы Таблица 4 А Клеточная линия: миелоидная лейкемия человека К 562 Композиция: паклитаксел/Кремофор 25 Таблица 4 В Клеточная линия: миелоидная лейкемия человека К 562 Образец: паклитаксел/ЧСАin vivo. С терапевтической точки зрения, биоэквивалентность можно установить, демонстрируя эквивалентность таких фармакокинетических характеристик, как AUC (площадь под кривой),константы элиминации, период полужизни в плазме после введения одинаковой дозы тем же самым видам. Такой эксперимент был проведен на крысах для двух композиций, как в примереAUC означает площадь под кривой на графике концентрации в плазме от времени. Ее можно получить, измеряя концентрацию введенного соединения в плазме в различные периоды времени. Фармакокинетическое исследование на крысах Метод. Вводили болусом дозу паклитакселя 2,5 мг/кг в объеме 1,0 мл внутривенно крысам CR (Wi) BR (вес тела от 380 до 420 г), и отбирали образцы крови 1,0 мл в гепаринизованные пробирки от трех животных в каждый период времени, как указано ниже: Фракцию плазмы отделяли кратким центрифугированием при +5 С и хранили замороженной при -70 С до проведения аналитических измерений. Подготовка образцов. Замороженные образцы плазмы нагревали до +8 С, центрифугировали 5 мин при 5 000 об./мин. Отбирали 0,300-0,500 мл прозрачного раствора плазмы и наносили на экстракционный картридж Oasis 26 образца плазмы картридж промывали 1 мл метанола, затем 1 мл воды для прекондиционирования. Содержащийся в образце паклитаксел абсорбировался в картридже SPE, тогда как остальные компоненты вымывались 1 мл воды и 1 мл 30% раствора ацетонитрил/вода. Картридж высушивали продуванием воздуха. Паклитаксел элюировали из картриджа SPE с помощью 1 мл абсолютного этанола. Образец упаривали досуха азотом, хранили при -20 С до анализа. Остаток растворяли в 0,200 мл абсолютного этанола и вводили через инжектор для анализа HPLC. Условия HPLC были те же, что применялись для идентификации вещества. Результаты Полученные точки являлись средними из трех измерений образцов от трех индивидуальных животных. Вследствие этого различия между двумя кривыми, полученными в результате фармакокинетического исследования равных доз из двух различных композиций, оказались в пределах отклонения для индивидуальных образцов. Такая же форма кривой получается при нанесении на график всех индивидуальных данных, что указывает на отсутствие различий или минимальные различия фармакокинетических характеристик двух композиций. Пример IV.3. Исследования по оценке антипролиферативной и цитостатической активности in vivo показывают, что новые композиции оказывают положительное действие против раковых ксенотрансплантантов человека СН 1 и СН 1taх у голых мышей. Пример IV.4. Тесты на гиперчувствительность. Примерно у 45% больных, получавших паклитаксел, проявлялись реакции гиперчувствительности. Было доказано, что эти побочные эффекты связаны с одной добавкой в композиции, Кремофором EL, и наблюдались также с другими фармацевтическими препаратами, содержащими этот компонент. Эта реакция гиперчувствительности определяется как анафилактическая токсичность, проявляющаяся вследствие выброса гистамина, вызванного Кремофором EL. Наше исследование проводилось на самцах крыс CRL (WI) BR весом 130-150 г [14]. Вводимая доза паклитакселя была рассчитана примерно в 7,0 мг/кг, при внутривенном введении в общем объеме 1,0 мл. Для каждой временной точки и дозы была группа из 5 животных. Образцы крови собирали в содержащие гепарин пробирки через 2, 5 и 10 мин после введения. Плазму отделяли кратким центрифугированием. Образцы хранили при -70 С. Гистамин, содержащийся в образце, подвергали С 14-метилированию специфическим ферментом гистамин-N-метил-трансферазой. Уровень гистамина в образцах плазмы опреде 27 ляли путем измерения радиоактивности С 14 в образцах. Полученные данные показали, что Кремофор EL и содержащая его композиция вызывали значительный выброс гистамина, тогда как ЧСА и содержащая его композиция и сам паклитаксел не проявляли подобного эффекта. Пример IV.5. Такое же явление, как в примере IV.4, было обнаружено у человека в экспериментах invitro на образцах крови человека при количественной оценке активации хроматина лимфоцитов крови [Метод: Analytical and QuantitativeCytology and Histology, vol. 8: p.1, 1986]. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Растворимый в воде фармацевтический препарат в твердом состоянии или в виде истинного водного раствора, содержащийa) терапевтически активное соединение,обладающее низкой растворимостью в воде,менее 1 х 10-4 М/л, и существенным сродством связывания с белками плазмы (в дальнейшем"активное соединение"), означающим, что более чем 90% активного соединения связано с белком плазмы в водной среде при спонтанном равновесии и при комнатной температуре;b) белковую фракцию плазмы в контролируемом агрегационном состоянии, исключающем нежелательную агрегацию белка,c) причем активное соединение и белковая фракция связаны друг с другом нековалентными связями, иd) где упомянутый водный раствор не включает какого-либо органического растворителя, иe) в котором молярное соотношение активного соединения и белковой фракции находится в диапазоне от 1:0,05 до 1:100 и где концентрация указанного активного соединения в водном растворе соответствует терапевтически эффективной дозе. 2. Препарат по п.1, где белковая фракция плазмы представляет собой сывороточный альбумин человека, сывороточный альбумин животного, рекомбинантный сывороточный альбумин человека, рекомбинантный сывороточный альбумин животного, -глобулин и рекомбинантный -глобулин. 3. Препарат по п.1 или 2, где активным соединением является цитостатик, антибиотик,витамин, противовоспалительное, анальгетик,антивирусное средство, противосудорожное средство, иммуносупрессор, антиэпилептическое, анксиолитик, снотворное, противогрибковое средство, антикоагулянт, липидный ингибитор пероксидаз, коронарный вазодилятор, антиаритмическое средство, кардиотоническое, мочегонное, антитромботическое средство, стероидный гормон (прогестерон, андроген, тестоген) и фотосенсибилизатор, и где концентрация ука 004700 28 занного активного соединения в водном растворе соответствует терапевтически эффективной дозе. 4. Препарат по любому из пп.1-3, где по меньшей мере одно или более одного активное соединение выбрано из группы, содержащей амфотерицин В, аналог адриамицина, апазон,азатиоприн, бромазепам, кампототецин, карбамазепин, клоназепам, циклоспорин А, диазепам,дикумарол, дигитоксин, дипиридамол, дизопирамид, флунитразепам, гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовая кислота, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус, тамоксифен, таксоноид, тестостерон, тирилазад, триоксален,вальпроевая кислота, варфарин и где белковая фракция выбрана из группы, состоящей из сывороточного альбумина человека, сывороточного альбумина животного, рекомбинантного сывороточного альбумина человека, рекомбинантного сывороточного альбумина животного, глобулина и рекомбинантного -глобулина, в котором молярное соотношение активного соединения и белковой фракции находится в диапазоне от 1:0,05 до 1:100 и где концентрация заявленного активного соединения в водном растворе соответствует терапевтически эффективной дозе. 5. Препарат по любому из пп.1-4, содержащий в качестве терапевтически активного вещества таксоноид общей формулы где R1 означает третичный амид бутилоксикарбоксикислоты или амид бензоила,R2 означает водород, ацил или ацетильную группу, предпочтительно паклитаксел, и где белковая фракция выбрана из группы, состоящей из сывороточного альбумина человека, сывороточного альбумина животного, рекомбинантного сывороточного альбумина человека,рекомбинантного сывороточного альбумина животного, -глобулина и рекомбинантного глобулина, в которых молярное соотношение активного соединения и белковой фракции находится в диапазоне от 1:0,05 до 1:100 и где концентрация терапевтически активного соединения в водном растворе соответствует терапевтически эффективной дозе. 6. Гомогенный, твердый препарат, состоящий преимущественно по меньшей мере из одного терапевтически активного соединения,имеющего низкую растворимость в воде, менее 29 1 х 10-4 М/л, выбранного из группы, содержащей амфотерицин В, аналог адриамицина, апазон,азатиоприн, бромазепам, кампототецин, карбамазепин, клоназепам, циклоспорин А, диазепам,дикумарол, дигитоксин, дипиридамол, дизопирамид, флунитразепам, гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовую кислоту, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус, тамоксифен, таксоноид, тестостерон, тирилазад, триоксален,вальпроевую кислоту, варфарин, и по меньшей мере одной белковой фракции, выбранной из группы, состоящей из сывороточного альбумина, -глобулина, рекомбинантного сывороточного альбумина, рекомбинантного -глобулина,и где указанное терапевтически активное соединение и указанная белковая фракция связаны друг с другом нековалентными связями и где молярное соотношение заявленного терапевтически активного соединения и белковой фракции находятся в диапазоне от 1:0,05 до 1:100, а растворимость в воде препарата достаточна для обеспечения концентрации, соответствующей терапевтически эффективной дозе активного соединения. 7. Гомогенный твердый препарат, состоящий преимущественно по меньшей мере из одного терапевтически активного соединения с растворимостью в воде менее чем 1x10-4 М/л,выбранного из группы, содержащей амфотерицин В, аналог адриамицина, апазон, азатиоприн,бромазепам, кампототецин, карбамазепин, клоназепам, циклоспорин А, диазепам, дикумарол,дигитоксин, дипиридамол, дизопирамид, флунитразепам, гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовую кислоту, оксазепам, фенобарбитал, фенитоин, прогестерон,пропофол, ритонавир, сульфинпиразон, супрофен, такролимус, тамоксифен, таксоноид, тестостерон, тирилазад, триоксален, вальпроевую кислоту, варфарин, и белковой фракции, выбранной из группы, состоящей из гликопротеина, интерферона, интерлейкина и рекомбинантного гликопротеина, интерферона, интерлейкина, где терапевтически активные соединения и указанная белковая фракция связаны друг с другом нековалентными связями и где молярное соотношение терапевтически активного соединения и указанной белковой фракции находится в диапазоне от 1:0,05 до 1:100, а растворимость в воде препарата достаточна для обеспечения концентрации, соответствующей терапевтически эффективной дозе активного соединения. 8. Гомогенный, твердый, водорастворимый препарат по п.6 или 7, в котором молярное соотношение терапевтически активного соединения и указанной белковой фракции находится в диапазоне от 1,0:0,1 до 1:50. 9. Гомогенный, твердый, водорастворимый препарат по любому из пп.6-8, в котором ука 004700 30 занное терапевтически активное соединение представляет собой таксоноид формулы где R1 означает третичный амид бутилоксикарбоксикислоты или амид бензоила,R2 означает водород, ацил или ацетильную группу, предпочтительно паклитаксел, и где белковая фракция выбрана из группы, состоящей из сывороточного альбумина человека, сывороточного альбумина животного, рекомбинантного сывороточного альбумина человека,рекомбинантного сывороточного альбумина животного, -глобулина и рекомбинантного глобулина, причем молярное соотношение активного соединения и белковой фракции находится в диапазоне от 1:0,05 до 1:100 и концентрация активного соединения в водном растворе соответствует терапевтически эффективной дозе. 10. Препарат по любому из пп.1-9, в котором молярное соотношение терапевтически активного соединения и белковой фракции находится в диапазоне от 1:0,1 до 1:50. 11. Способ получения водорастворимого препарата в твердом состоянии, как указано в любом из пп.1-10, растворимость в воде которого достаточна для обеспечения концентрации соответствующей терапевтически эффективной дозе активного соединения, включающийa) полное растворение терапевтически активного соединения в смешивающемся с водой фармацевтически приемлемом органическом растворителе,b) объединение указанного раствора в органическом растворителе с водным раствором фракции белков плазмы в контролируемом агрегационном состоянии, с получением истинного раствора, содержащего заявленное активное соединение и указанную белковую фракцию,связанные вместе нековалентными связями,c) удаление органического растворителя и лиофилизацию раствора или его концентрата. 12. Способ получения истинных водных растворов без примеси органических растворителей препарата по любому из пп.1-10, содержащих водонерастворимое терапевтически активное соединение, включающийa) полное растворение терапевтически активного соединения в смешивающемся с водой фармацевтически приемлемым органическом растворителе,b) объединение указанного раствора в органическом растворителе с водным раствором 31 фракции белков плазмы в контролируемом агрегационном состоянии,с получением истинного раствора, содержащего заявленное активное соединение и указанную белковую фракцию, связанные вместе нековалентными связями,с) удаление органического растворителя и лиофилизацию раствора или его концентрата,d) растворение полученного в твердом состоянии препарата в воде, не содержащей органического растворителя, с получением истинного водного раствора, свободного от органических растворителей, в котором концентрация заявленного активного соединения соответствует терапевтически эффективной дозе, годной для терапевтического введения. 13. Способ по п.12, включающий доведение раствора до конечного препарата в виде дозированной формы для парентерального введения. 14. Способ по любому из пп.11-13, в котором на стадии а) используют органический растворитель, имеющий следующие свойства:a) в смеси с водой он способен полностью растворять активное соединение иb) в смеси с водой, где вода составляет менее 50%, он не денатурирует белок. 15. Способ по п.14, в котором органический растворитель выбирают из группы, состоящей из алифатического С(2-4) моноспирта или полиспирта, 70-100% этанола, диметилформамида, метилформамида. 16. Способ по любому из пп.11-15, в котором на стадии b) в качестве белков плазмы используют белки, выбранные из группы, включающей сывороточный альбумин, иммуноглобулин, гликопротеин, интерферон, интерлейкин,рекомбинантный гликопротеин, рекомбинантный интерферон и рекомбинантный интерлейкин. 17. Способ по любому из пп.11-16, в котором на стадии а) используют по меньшей мере одно или более одного терапевтически активное соединение, выбранное из группы, включающей амфотерицин В, аналог адриамицина, апазон,азатиоприн, бромазепам, кампототецин, карбамазепин, клоназепам, циклоспорин А, диазепам,дикумарол, дигитоксин, дипиридамол, дизопирамид, флунитразепам, гемфиброзил, кетохлорин, кетоконазол, миконазол, нифлумовую кислоту, оксазепам, фенобарбитал, фенитоин, прогестерон, пропофол, ритонавир, сульфинпиразон, супрофен, такролимус, тамоксифен, таксоноид, тестостерон, тирилазад, триоксален,вальпроевую кислоту, варфарин. 18. Способ по любому из пп.11-17, в котором на стадии а) в качестве терапевтически активного соединения используют паклитаксел. 19. Способ лечения людей или ветеринарных пациентов терапевтически активным соединением, имеющим низкую растворимость в воде, менее 1 х 10-4 М/л, и обладающим сущест 004700 32 венным сродством к белкам плазмы, включающий введение пациенту, нуждающемуся в лечении указанным активным соединением, водных растворов препарата по любому из пп.1-10, при следующем диапазоне доз (в пересчете на активное соединение): паклитаксел/альбумин - 70280 мг/введение, пропофол/альбумин - 6-10 мг/кг/ч, камптотецин/альбумин, гемфиброзил/альбумин, циклоспорин А/альбумин - по 3-5 мг/кг/день, амфотерицин В/альбумин - до 1,5 мг/кг/день, причем те же самые диапазоны доз применяются для соединений, содержащих соответствующие рекомбинантные белки. 20. Способ лечения людей или ветеринарных пациентов по п.19, включающий парентеральное введение указанных водных растворов.
МПК / Метки
МПК: A61K 47/42, A61P 35/00
Метки: содержащие, белок, плазмы, композиции, фармацевтические
Код ссылки
<a href="https://eas.patents.su/20-4700-farmacevticheskie-kompozicii-soderzhashhie-belok-plazmy.html" rel="bookmark" title="База патентов Евразийского Союза">Фармацевтические композиции, содержащие белок плазмы</a>
Ингибиторы металлопротеиназы, фармацевтические композиции, их содержащие, и их фармацевтические применения

Номер патента: 3294
Опубликовано: 24.04.2003
Авторы: Бендер Стивен Л., Мелник Майкл Дж., Дагнино Раймонд Джр., Жук Скотт Е., Дизон Майкл Е.
МПК: A61K 31/535, A61K 31/54, A61K 31/495...
Метки: применения, композиции, содержащие, металлопротеиназы, ингибиторы, фармацевтические
Формула / Реферат:
1. Соединение с формулой 1 где Z является O или S; Ar является моноциклической арильной или гетероарильной группой, выбранной из фенильной группы, или пяти- или шестичленной азот- или кислородсодержащей гетероарильной группы, незамещенной или замещенной одним или более заместителями, выбранными из галогрупп и цианогруппы; X выбран из S, S=O, O, N-R3 и N+(O-)-R4, где R3 является атомом водорода или C1-C6-алкильной группой, где указанная...
Мутантный il-6 человека и его внутренний фрагмент, кодирующие их последовательности днк, способы их получения, содержащие их фармацевтические композиции, содержащие их векторы, линии клеток- хозяев испособ лечения il-6 опосредованных заболеваний

Номер патента: 852
Опубликовано: 26.06.2000
Авторы: Гротзингер Йоахим, Розе-Йон Штефан, Элерс Марк
МПК: A61P 19/10, A61P 35/00, A61K 38/20...
Метки: мутантный, векторы, днк, содержащие, получения, опосредованных, композиции, человека, способы, лечения, кодирующие, фрагмент, испособ, последовательности, клеток, внутренний, хозяев, заболеваний, линии, фармацевтические
Формула / Реферат:
1. Мутантный интерлейкин-6 (IL-6) человека, имеющий аминокислотную последовательность содержащую следующие точечные мутации по сравнению с природным IL-6 человека: Pro 54, Glu 159, Pro 162, Leu 170 и Аrg 176. 2. Внутренний фрагмент мутантного IL-6 человека по п.1 формулы, обладающий аналогичной биологической активностью. 3. Последовательность ДНК, кодирующая мутантный IL-6 человека по п.1 формулы. 4. Последовательность ДНК, кодирующая...
Олигосахариды,способ их получения и содержащие их фармацевтические композиции

Номер патента: 4046
Опубликовано: 25.12.2003
Авторы: Штутцманн Жан-Мари, Мурье Пьер, Висков Кристиан, Валь Флоранс, Перрэн Элизабет
МПК: C07H 19/01, A61P 25/02, A61K 31/70...
Метки: композиции, содержащие, получения, олигосахариды,способ, фармацевтические
Формула / Реферат:
1. Олигосахариды формулы в которой n является целым числом от 0 до 25; R1, R3, R4 и R5, одинаковые или разные, обозначают атом водорода или радикал SO3M; R2 и R6, одинаковые или разные, обозначают атом водорода или радикал SO3M или COCH3 и M обозначает натрий, кальций, магний или калий, смесь этих олигосахаридов и их диастереоизомеры. 2. Олигосахариды формулы I по п.1, у которых R4 обозначает атом водорода, смесь этих олигосахаридов и их...
Фармацевтические композиции, содержащие ламивудин и зидовудин.

Номер патента: 2437
Опубликовано: 25.04.2002
Авторы: Форд Кэтрин Джиннетт, Гудсон Гэри Уэйн, Вуд Аллен Уэйн
МПК: A61K 31/7068, A61P 37/00
Метки: ламивудин, композиции, зидовудин, содержащие, фармацевтические
Формула / Реферат:
1. Способ получения фармацевтической композиции, содержащей 1) безопасное и терапевтически эффективное количество ламивудина или его фармацевтически приемлемого производного; 2) безопасное и терапевтически эффективное количество зидовудина или его фармацевтически приемлемого производного; и 3) фармацевтически приемлемый глидант; где фармацевтически приемлемый глидант присутствует в количестве от 0,05 до приблизительно 10,0% по массе, при котором...
Новые таксоиды,их получение и содержащие их фармацевтические композиции

Номер патента: 1533
Опубликовано: 23.04.2001
Авторы: Коммерсон Алан, Бушар Эрве
МПК: A61P 35/00, A61K 31/335, C07D 305/14...
Метки: композиции, таксоиды,их, получение, содержащие, новые, фармацевтические
Формула / Реферат:
1. Новые таксоиды общей формулы в которой Z обозначает радикал общей формулы в которой R1 обозначает бензоильный радикал или радикал R2-О-СО-, в котором R2 представляет собой алкильный радикал с 1-8 атомами углерода и R3 обозначает фенил или a - или b -нафтил; и либо R4 обозначает атом водорода, R6 и R7 вместе образуют кетонную функцию и R и R5 вместе образуют связь, либо R4 обозначает атом водорода или радикал алкокси с 1-6 атомами...
Предыдущий патент: Способ дозирования армирующих волокон для производства дисперсно-армированного бетона (варианты) и цепная тара для осуществления этого способа
Следующий патент: Амиды антраниловой кислоты и их применение в качестве лекарственных средств
Случайный патент: Повторно закрываемая упаковка и способ ее изготовления