Кристаллические формы 2-тиазолил-4-хинолинилоксипроизводного, активного ингибитора hcv
Номер патента: 18603
Опубликовано: 30.09.2013
Авторы: Беркенбуш Тило, Бьюсакка Карл Алан, Варсолона Ричард Дж., Йэгер Буркхард





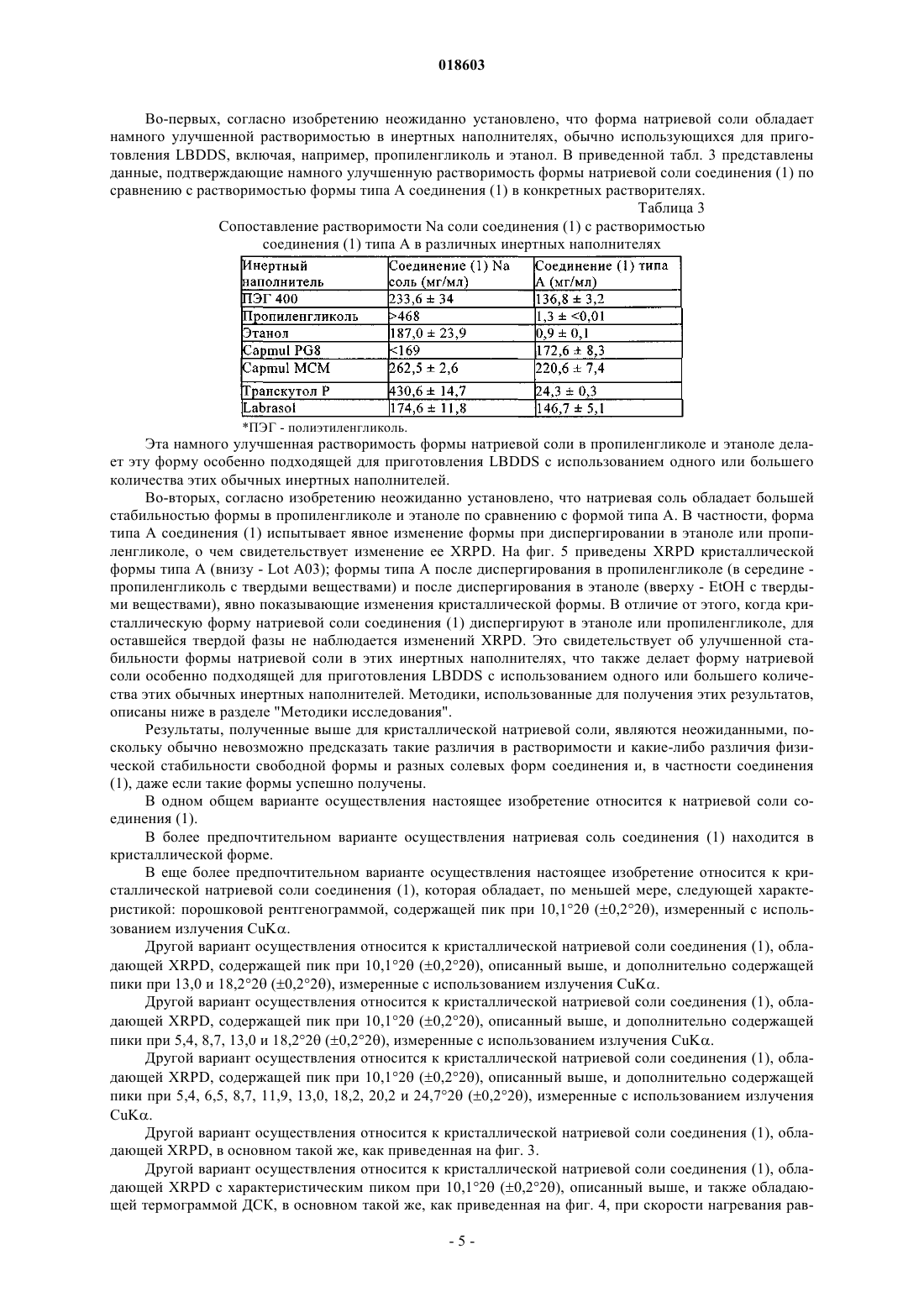


Формула / Реферат
1. Соединение формулы (1) в кристаллической форме

2. Кристаллическое соединение формулы (1) по п.1, обладающее порошковой рентгенограммой, содержащей пик при 9,6°2θ (±0,2°2θ), измеренный с использованием излучения CuKα.
3. Кристаллическое соединение по п.2, для которого порошковая рентгенограмма дополнительно содержит пик при 19,8°2θ (±0,2°2θ), измеренный с использованием излучения CuKα.
4. Кристаллическое соединение формулы (1) по п.1, обладающее порошковой рентгенограммой, содержащей пики при 4,8, 6,8, 9,6, 13,6, 17,3, 19,8 и 24,5°2θ (±0,2°2θ), измеренные с использованием излучения CuKα.
5. Натриевая соль соединения формулы (1)

6. Натриевая соль по п.5 в кристаллической форме.
7. Кристаллическая натриевая соль по п.6, обладающая порошковой рентгенограммой, содержащей пик при 10,1°2θ (±0,2°2θ), измеренный с использованием излучения CuKα.
8. Кристаллическая натриевая соль по п.7, для которой порошковая рентгенограмма дополнительно содержит пики при 13,0 и 18,2°2θ (±0,2°2θ), измеренные с использованием излучения CuKα.
9. Кристаллическая натриевая соль по п.8, для которой порошковая рентгенограмма дополнительно содержит пики при 5,4 и 8,7°2θ (±0,2°2θ), измеренные с использованием излучения CuKα.
10. Кристаллическая натриевая соль по п.6, обладающая порошковой рентгенограммой, содержащей пики при 5,4, 6,5, 8,7, 10,1, 11,9, 13,0, 18,2, 20,2 и 24,7°2θ (±0,2°2θ), измеренные с использованием излучения CuKα.
11. Кристаллическая натриевая соль по п.6, обладающая порошковой рентгенограммой, полученной с использованием излучения CuKα, которая в основном такая, как приведенная на фиг. 3.
12. Смесь соединения формулы (1) и его натриевой соли

где не менее 50% указанного соединения содержится в форме натриевой соли соединения по пп.5-10 или 11.
13. Фармацевтическая композиция, содержащая натриевую соль по пп.5-10 или 11 и фармацевтически приемлемый носитель или разбавитель.
14. Фармацевтическая композиция по п.13, в которой не менее 50% натриевой соли соединения формулы (1) в композиции содержится в форме кристаллического соединения по пп.6-10 или 11.
15. Применение кристаллического соединения формулы (1) по п.1 или натриевой соли соединения формулы (1) по п.5 или их смесей для приготовления фармацевтической композиции, предназначенной для лечения вирусной инфекции гепатита С у млекопитающего.
Текст
В изобретении приведены описания новых кристаллических форм соединения (1) и его натриевой соли и способов их получения, содержащих их фармацевтические композиции, и их применение для лечения вирусной инфекции гепатита С (HCV): Беркенбуш Тило (DE), Бьюсакка Карл Алан (US), Йэгер Буркхард (DE),Варсолона Ричард Дж. (US) Веселицкая И.А., Пивницкая Н.Н.,Кузенкова Н.В., Веселицкий М.Б.,Каксис Р.А., Комарова О.М., Белоусов Ю.В. (RU)(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) По заявке на данное изобретение испрашивается приоритет по следующим предварительным заявкам US: 61/097291, поданной 16 сентября 2008 г., и 61/150826, поданной 9 марта 2009 г. Область техники, к которой относится изобретение Настоящее изобретение относится к новым кристаллическим формам соединения (1) и натриевой соли соединения (1), описанным в настоящем изобретении, способам их получения, содержащим их фармацевтическим композициям и их применению для лечения вирусной инфекции гепатита C (HCV). Уровень техники Соединение (1) известно как селективный и активный ингибитор серинпротеазы HCV NS3 Соединение (1) входит в число серии ациклических пептидных ингибиторов HCV, раскрытых в патентах US 6323180 и 7514557 и в публикации заявки на патент US2005/0020503. Соединение (1), в частности, раскрыто как соединение 1055 в публикации заявки на патент US 2005/0020503 и как соединение 1008 в патенте US 7514557. Соединение (1) можно получить по общим методикам, приведенным в цитированных выше ссылках, которые включены в настоящее изобретение в качестве ссылки. Соединение (1) также может быть известно в виде приведенного ниже альтернативного изображения его химической структуры, которая эквивалентна описанной выше структуре:R2 обозначает При синтезе по общим методикам, приведенным в указанных выше ссылках, соединение (1) получают в виде аморфного твердого вещества, которое представляет собой форму, обычно менее подходящую для крупномасштабного фармацевтического производства. Таким образом, необходимо получить соединение (1) в кристаллической форме, чтобы приготовить препараты, соответствующие действующим фармацевтическим требованиям и спецификациям. Кроме того, способ, с помощью которого получают соединение (1), должен быть подходящим для крупномасштабного производства. Кроме того, желательно, чтобы продукт находился в форме, которую легко фильтровать и легко сушить. Наконец, с экономической точки зрения желательно, чтобы продукт был стабильным в течение длительных периодов времени и не требовались специальные условия хранения. Краткое изложение сущности изобретения Согласно изобретению неожиданно и непредвиденно впервые было установлено, что соединение(1) можно получить в кристаллической форме, а также в форме его натриевой соли и более предпочтительно натриевой соли в кристаллической форме. Таким образом, настоящее изобретение относится к соединению (1) в кристаллической форме, которое в одном варианте осуществления представляет собой новую кристаллическую полиморфную форму, обозначенную в настоящем изобретении, как тип A, а также новую кристаллическую натриевую соль соединения (1). Эти новые кристаллические формы позволяют преодолеть затруднения при фармацевтической обработке, характерные для использования аморфной формы и формы натриевой соли, в частности они обладают другими характеристиками, делающими их особенно подходящими для обработки фармацевтической композиции, как это подробно описано ниже. В одном варианте осуществления настоящее изобретение относится к соединению (1) в кристаллической форме. В более предпочтительном варианте осуществления авторы настоящего изобретения открыли новую кристаллическую полиморфную форму соединения (1), обозначенную ниже в настоящем изобретении как тип A. Тип A обладает характеристической порошковой рентгенограммой (XRPD) с характеристическими пиками, выраженными в 2 (0,22), при 4,8, 6,8, 9,6, 13,6, 17,3, 19,8 и 24,5, измеренными с использованием излучения CuK. Другой вариант осуществления относится к натриевой соли соединения (1), и эту натриевую соль можно получить в кристаллической форме. Кристаллическая натриевая соль соединения (1) обладает характеристической порошковой рентгенограммой (XRPD) с характеристическими пиками, выраженными в 2 (0,22), при 5,4, 6,5, 8,7, 10,1, 11,9, 13,0, 18,2, 20,2 и 24,7, измеренными с использованием излучения CuK. Еще один вариант осуществления относится к фармацевтической композиции, содержащей тип A или натриевую соль соединения (1) или их смеси и по меньшей мере один фармацевтически приемлемый носитель или разбавитель. Еще один вариант осуществления относится к способу лечения инфекции HCV у млекопитающего,включающему введение указанному млекопитающему типа A или натриевой соли соединения (1), или их смесей в терапевтически эффективном количестве. Краткое описание чертежей На фиг. 1 приведена характеристическая порошковая рентгенограмма (XRPD) типа A. На фиг. 2 приведена термограмма ДСК кристаллов типа A, причем ДСК проводят при скорости нагревания, равной 10C/мин, в закрытом тигле. На фиг. 3 приведена характеристическая порошковая рентгенограмма (XRPD) кристаллической натриевой соли соединения (1). На фиг. 4 приведена термограмма ДСК кристаллов кристаллической натриевой соли соединения(1), причем ДСК проводят при скорости нагревания, равной 10C/мин, в открытой чашке. На фиг. 5 приведены XRPD для кристаллической формы типа A соединения (1) (внизу); кристаллической формы типа A соединения (1) после диспергирования в пропиленгликоле (в середине) и кристаллической формы типа A соединения (1) после диспергирования в этаноле (вверху). Подробное описание изобретения Определения. Терминам, специально не определенным в настоящем изобретении, следует придавать значения,которые им должен придавать специалист в данной области техники в соответствии с раскрытием и контекстом. Однако если не указано иное, то при использовании в описании и прилагаемой формуле изобретения приведенные ниже термины обладают указанными значениями. Термин "тип A" означает кристаллическую полиморфную форму соединения (1), которая обладает порошковой рентгенограммой, содержащей, по меньшей мере, характеристический пик при 9,62(0,22), измеренный с использованием излучения CuK. Предполагается, что этот характеристический пик отличает тип A от других кристаллических форм соединения (1). Термин "примерно" означает с отклонением, составляющим не более 5% и более предпочтительно не более 1% от данного значения или диапазона. Например, "примерно 3,7%" означает от 3,5 до 3,9%,предпочтительно от 3,66 до 3,74%. Если термин "примерно" относится к диапазону значений, например,"примерно от X до Y%", то термин "примерно" указывает на изменение и верхнего (X) и нижнего (Y) значений указанного диапазона. Например, выражение "примерно от 20 до 40%" эквивалентно выражению "от примерно 20 до примерно 40%". Термин "фармацевтически приемлемый" применительно к веществу при использовании в настоящем изобретении означает, что вещество в соответствии с тщательной медицинской оценкой пригодно для использования при взаимодействии с тканям человека и низших животных без нежелательной токсичности, проявления раздражения, аллергической реакции и т.п., в соответствии с разумным отношением риск/польза и эффективно для предназначенного применения, когда вещество используется в фарма-2 018603 цевтической композиции. Термин "лечение" применительно к лечению патологического состояния пациента включает:(i) подавление или улучшение протекания патологического состояния у пациента, например остановку или замедление его развития; или(ii) облегчение патологического состояния у пациента, т.е. обеспечение ремиссии или излечения патологического состояния. В случае HCV лечение включает уменьшение нагрузки вирусом HCV у пациента. Кристаллическое соединение (1). Соединение (1) выделено в виде кристаллической полиморфной формы, обозначенной в настоящем изобретении как тип A. Обычно тип A обладает характеристической порошковой рентгенограммойXRPD типа A приведена на фиг. 1. Положения и относительные интенсивности характеристических пиков на XRPD, приведенной на фиг. 1, представлены в табл. 1. Таблица 1 На фиг. 2 приведена полученная с помощью дифференциальной сканирующей калориметрии (ДСК) термограмма кристаллов типа A, причем ДСК проводят при скорости нагревания 10C/мин в закрытом тигле. В одном общем варианте осуществления настоящее изобретение относится к соединению (1) в кристаллической форме. Другой более предпочтительный вариант осуществления относится к кристаллической полиморфной форме соединения (1), которая обладает, по меньшей мере, следующей характеристикой: порошковой рентгенограммой, содержащей пик при 9,62 (0,22), измеренный с использованием излученияCuK. Другой вариант осуществления относится к кристаллической полиморфной форме соединения (1),обладающей XRPD, содержащей пик при 9,62 (0,22), описанный выше, и дополнительно содержащей пик при 19,82 (0,22), измеренный с использованием излучения CuK. Другой вариант осуществления относится к кристаллической полиморфной форме соединения (1),обладающей XRPD, содержащей пик при 9,62 (0,22), описанный выше, и дополнительно содержащей пики при 4,8 и 19,82 (0,22), измеренные с использованием излучения CuK. Другой вариант осуществления относится к кристаллической полиморфной форме соединения (1),обладающей XRPD, содержащей пик при 9,62 (0,22), описанный выше, и дополнительно содержащей пики при 4,8, 6,8, 13,6, 17,3, 19,8 и 24,52 (0,22), измеренные с использованием излученияCuK. Другой вариант осуществления относится к кристаллической полиморфной форме соединения (1),обладающей XRPD, в основном такой же, как приведенная на фиг. 1. Другой вариант осуществления относится к кристаллической полиморфной форме соединения (1),обладающей XRPD, содержащей пик при 9,62 (0,22), описанный выше, и также обладающей термограммой ДСК, в основном такой же, как приведенная на фиг. 2, при скорости нагревания, равной 10C/мин, в закрытом тигле. Другой вариант осуществления относится к количеству соединения (1), в котором не менее 50%,предпочтительно не менее 75%, более предпочтительно не менее 95%, более предпочтительно не менее 99% указанного соединения содержится в кристаллической форме, например в форме типа A кристаллической полиморфной формы, охарактеризованной с помощью любого из описанных выше посредствомXRPD вариантов осуществления. Наличие таких количеств типов А в количестве соединения (1) обычно измеримо с помощью проводимого посредством XRPD анализа соединения. Другой вариант осуществления относится к фармацевтической композиции, содержащей соединение (1) и фармацевтически приемлемый носитель или разбавитель, в которой не менее 50%, предпочтительно не менее 75%, более предпочтительно не менее 95%, более предпочтительно не менее 99%, соединения (1) в композиции содержится в кристаллической форме, например в форме типа A кристаллической полиморфной формы, охарактеризованной с помощью любого из описанных выше посредством Настоящее изобретение относится к способу получения типа A, который включает кристаллизацию соединения (1) из раствора в растворителях при условиях, которые дают тип A. Точные условия, при которых образуется тип A, можно определить эмпирически и можно привести только методики, которые оказались подходящими при практическом применении. Согласно изобретению установлено, что тип A соединения (1) можно получить способом, включающим следующие стадии, который также является вариантом осуществления настоящего изобретения:(i) растворение соединения (1) в алифатическом спиртовом растворителе, необязательно содержащем воду в качестве сорастворителя, путем нагревания смеси до температуры примерно 65-75C, с получением раствора;(ii) добавление воды к раствору, полученному на стадии (i), с поддержанием температуры раствора примерно 70-75C, с получением взвеси;(iii) охлаждение взвеси, полученной на стадии (ii), с получением твердого вещества;(iv) сбор твердого вещества, полученного на стадии (iii), и сушка указанного вещества при температуре примерно 65-80C, с получением соединения (1)типа A. Алифатические спирты, которые можно использовать в этом способе, включают, например, этанол(например, денатурированный крепостью 100 или 100% чистоты), 1-пропанол, 2-пропанол, 1-бутанол,изобутиловый спирт и изопентиловый спирт, предпочтительно этанол. Полученные кристаллы типа A можно извлечь по любым обычным методикам, известным в данной области техники. На конечной стадии (iv) твердые вещества, полученные на стадии (iii), можно собрать и высушить при высокой температуре по обычным методикам сбора и высокотемпературной сушки, например фильтрованием и сушкой в вакуумном сушильном шкафу. В одном предпочтительном варианте осуществления аморфное соединение (1) растворяют в алифатическом спиртовом растворителе (например, этаноле), содержащем примерно до 10% об./об. воды в качестве сорастворителя, путем перемешивания и нагревания смеси до температуры, равной примерно 72-74C, пока соединение (1) полностью не растворится. Готовят отдельный добавочный водный раствор, содержащий воду и примерно до 10% об./об. алифатического спирта (например, этанола), и этот добавочный водный раствор добавляют примерно с линейной скоростью к раствору соединения (1), поддерживая температуру смеси равной примерно 72-74C. Тип A соединения (1) начинает кристаллизоваться во время добавления водного раствора. Полученную взвесь кристаллов охлаждают и перемешивают и затем кристаллы отфильтровывают, промывают и сушат при температуре, равной примерно 6575C, по обычным методикам. Разумеется, эти стадии способа можно облегчить с помощью обычных методик смешивания, например перемешивания и других обычных методик, для которых хорошо известно, что они облегчают проведение способа. Натриевая соль соединения (1). Установлено, что натриевая соль соединения формулы (1) является особенно подходящей для фармацевтической обработки, поскольку ее можно получить в виде стабильной кристаллической формы. Обычно кристаллическая натриевая соль соединения (1) обладает характеристической порошковой рентгенограммой (XRPD) с характеристическими пиками, выраженными в 2 (0,22), при 5,4, 6,5, 8,7,10,1, 11,9, 13,0, 18,2, 20,2 и 24,7.XRPD кристаллической натриевой соли соединения (1) приведена на фиг. 3. Положения и относительные интенсивности характеристических пиков на XRPD, приведенной на фиг. 3, представлены в табл. 2. Таблица 2 На фиг. 4 приведена полученная с помощью дифференциальной сканирующей калориметрии (ДСК) термограмма кристаллической натриевой соли соединения (1) кристаллов, причем ДСК проводят при скорости нагревания, равной 10C/мин, в открытом тигле. Согласно изобретению неожиданно установлено, что форма натриевой соли обладает особыми характеристиками, делающими ее особенно подходящей для обработки фармацевтической композиции. В частности, форма натриевой соли обладает некоторыми характеристиками, делающими ее особенно подходящей для приготовления системы доставки лекарственного средства на основе липидов (LBDDS). Во-первых, согласно изобретению неожиданно установлено, что форма натриевой соли обладает намного улучшенной растворимостью в инертных наполнителях, обычно использующихся для приготовления LBDDS, включая, например, пропиленгликоль и этанол. В приведенной табл. 3 представлены данные, подтверждающие намного улучшенную растворимость формы натриевой соли соединения (1) по сравнению с растворимостью формы типа A соединения (1) в конкретных растворителях. Таблица 3 Сопоставление растворимости Na соли соединения (1) с растворимостью соединения (1) типа A в различных инертных наполнителях Эта намного улучшенная растворимость формы натриевой соли в пропиленгликоле и этаноле делает эту форму особенно подходящей для приготовления LBDDS с использованием одного или большего количества этих обычных инертных наполнителей. Во-вторых, согласно изобретению неожиданно установлено, что натриевая соль обладает большей стабильностью формы в пропиленгликоле и этаноле по сравнению с формой типа A. В частности, форма типа A соединения (1) испытывает явное изменение формы при диспергировании в этаноле или пропиленгликоле, о чем свидетельствует изменение ее XRPD. На фиг. 5 приведены XRPD кристаллической формы типа A (внизу - Lot A03); формы типа A после диспергирования в пропиленгликоле (в середине пропиленгликоль с твердыми веществами) и после диспергирования в этаноле (вверху - EtOH с твердыми веществами), явно показывающие изменения кристаллической формы. В отличие от этого, когда кристаллическую форму натриевой соли соединения (1) диспергируют в этаноле или пропиленгликоле, для оставшейся твердой фазы не наблюдается изменений XRPD. Это свидетельствует об улучшенной стабильности формы натриевой соли в этих инертных наполнителях, что также делает форму натриевой соли особенно подходящей для приготовления LBDDS с использованием одного или большего количества этих обычных инертных наполнителей. Методики, использованные для получения этих результатов,описаны ниже в разделе "Методики исследования". Результаты, полученные выше для кристаллической натриевой соли, являются неожиданными, поскольку обычно невозможно предсказать такие различия в растворимости и какие-либо различия физической стабильности свободной формы и разных солевых форм соединения и, в частности соединения(1), даже если такие формы успешно получены. В одном общем варианте осуществления настоящее изобретение относится к натриевой соли соединения (1). В более предпочтительном варианте осуществления натриевая соль соединения (1) находится в кристаллической форме. В еще более предпочтительном варианте осуществления настоящее изобретение относится к кристаллической натриевой соли соединения (1), которая обладает, по меньшей мере, следующей характеристикой: порошковой рентгенограммой, содержащей пик при 10,12 (0,22), измеренный с использованием излучения CuK. Другой вариант осуществления относится к кристаллической натриевой соли соединения (1), обладающей XRPD, содержащей пик при 10,12 (0,22), описанный выше, и дополнительно содержащей пики при 13,0 и 18,22 (0,22), измеренные с использованием излучения CuK. Другой вариант осуществления относится к кристаллической натриевой соли соединения (1), обладающей XRPD, содержащей пик при 10,12 (0,22), описанный выше, и дополнительно содержащей пики при 5,4, 8,7, 13,0 и 18,22 (0,22), измеренные с использованием излучения CuK. Другой вариант осуществления относится к кристаллической натриевой соли соединения (1), обладающей XRPD, содержащей пик при 10,12 (0,22), описанный выше, и дополнительно содержащей пики при 5,4, 6,5, 8,7, 11,9, 13,0, 18,2, 20,2 и 24,72 (0,22), измеренные с использованием излученияCuK. Другой вариант осуществления относится к кристаллической натриевой соли соединения (1), обладающей XRPD, в основном такой же, как приведенная на фиг. 3. Другой вариант осуществления относится к кристаллической натриевой соли соединения (1), обладающей XRPD с характеристическим пиком при 10,12 (0,22), описанный выше, и также обладающей термограммой ДСК, в основном такой же, как приведенная на фиг. 4, при скорости нагревания рав-5 018603 ной 10C/мин, в открытом тигле. Другой вариант осуществления относится к количеству соединения (1), в котором не менее 50%,предпочтительно не менее 75%, более предпочтительно не менее 95%, более предпочтительно не менее 99% указанного соединения содержится в форме кристаллической натриевой соли соединения (1), которую можно охарактеризовать с помощью любого из описанных выше посредством XRPD вариантов осуществления. Наличие таких количеств кристаллической натриевой соли соединения (1) в количестве соединения (1) обычно измеримо с помощью проводимого посредством XRPD анализа соединения. Другой вариант осуществления относится к фармацевтической композиции, содержащей натриевую соль соединения (1) и фармацевтически приемлемый носитель или разбавитель. В более предпочтительном варианте осуществления не менее 50%, предпочтительно не менее 75%, более предпочтительно не менее 95%, более предпочтительно не менее 99% соединения (1) натриевой соли в композиции содержится в кристаллической форме, например в форме кристаллической натриевой соли соединения (1),которую можно охарактеризовать с помощью любого из описанных выше посредством XRPD вариантов осуществления. Настоящее изобретение относится к способу получения кристаллической натриевой соли соединения (1), который включает кристаллизацию соединения (1) из раствора в растворителях при условиях,которые дают кристаллическую натриевую соль. Точные условия, при которых образуется кристаллическая натриевая соль, можно определить эмпирически и можно привести только методики, которые оказались подходящими при практическом применении. Согласно изобретению установлено, что кристаллическую натриевую соль соединения (1) можно получить способом, включающим следующие стадии, который также является вариантом осуществления настоящего изобретения:(i) растворение соединения (1) в кетонах или ацетатах, необязательно содержащих воду в качестве сорастворителя, путем нагревания смеси в виде взвеси или путем получения истинного раствора;(ii) добавление воды к раствору, полученному на стадии (i), с поддержанием температуры раствора равной примерно 50-70C, с получением раствора или взвеси;(iii) внесение затравки кристаллической натриевой соли соединения (1);(iv) охлаждение взвеси, полученной на стадии (iii), с получением твердого вещества;(iv) сбор твердого вещества, полученного на стадии (iii), сушка указанного вещества при температуре, равной примерно 45-75C, и получение кристаллической натриевой соли соединения (1). Дополнительные альтернативные способы получения кристаллической натриевой соли соединения(1) приведены в разделе "Примеры", каждый из которых является дополнительным вариантом осуществления настоящего изобретения. Фармацевтические композиции и способы. С учетом показанной ингибирующей активности соединения (1) по отношению к серинпротеазеHCV NS3 указанные выше формы соединения (1), включая тип A и формы натриевой соли, применимы в качестве средств против HCV. Поэтому указанные формы применимы для лечения инфекции HCV у млекопитающего и их можно использовать для приготовления фармацевтической композиции, предназначенной для лечения инфекции HCV или облегчения одного или более его симптомов у пациента. Кроме того, с помощью клинических исследований показано, что форма натриевой соли соединения (1) эффективна для лечения пациентов, инфицированных HCV. Подходящие дозы и режимы лечения конкретного пациента можно определить по методикам, известным в данной области техники, и с учетом раскрытия в патенте US 6323180 В 1 и в публикации заявки на патент US2005/0020503. Обычно для лечения инфекции HCV у млекопитающего вводят терапевтически эффективное количество. В одном варианте осуществления взрослому человеку вводят примерно от 50 до 1000 мг, более предпочтительно от примерно 120 до примерно 480 мг в сутки в виде одной или нескольких доз. Разумеется, конкретные оптимальные дозы и режимы лечения любого конкретного пациента зависят от множества факторов, включая возраст, массу тела, общее состояние здоровья, пол, диету, время введения, скорость выведения, использующуюся комбинацию лекарственных средств, тяжесть и протекание инфекции, предрасположенность пациента к инфекции и решение лечащего врача. Обычно наиболее желательно вводить соединение при концентрации, которая обычно приводит к противовирусно эффективным результатам без проявления каких-либо опасных или вредных побочных эффектов. Эти кристаллические формы соединения (1) или его натриевой соли при выбранной дозе обычно вводят пациенту с помощью фармацевтической композиции. Различные типы композиций, которые можно использовать в настоящем изобретении, см., например, описание в патенте US 6323180 В 1 и в публикации заявки на патент US2005/0020503. Фармацевтическую композицию можно вводить перорально, парентерально или с помощью имплантированного резервуара. Термин "парентеральный" при использовании в настоящем изобретении методики подкожной, внутривенной, внутримышечной, внутрисуставной, внутрисиновиальной, надчревной, внутриоболочечной и проводимой внутрь пораженных тканей инъекции или вливания. Предпочтительны пероральное введение или введение путем инъекции. Фармацевтические композиции, предлагаемые в настоящем изобретении, могут содержать любые обычные нетоксичные фармацевтически приемлемые носители, разбавители, вспомогательные вещества,-6 018603 инертные наполнители или растворители. В некоторых случаях значение рН композиции можно отрегулировать фармацевтически приемлемыми кислотами, основаниями или буферами для увеличения стабильности приготовленной композиции соединения или формы его доставки. Фармацевтические композиции могут находиться в форме стерильного препарата для инъекции,например стерильного водного раствора или масляной суспензии для инъекции. Эту суспензию можно приготовить по методикам, известным в данной области техники, с использованием подходящих диспергирующих или смачивающих агентов (таких как, например. Tween 80) и суспендирующих агентов. Фармацевтические композиции также могут находиться в форме пероральной фармацевтической композиции, содержащей тип A или натриевую соль соединения (1) или их смеси и по меньшей мере один фармацевтически приемлемый носитель или разбавитель. Пероральные фармацевтические композиции можно вводить перорально в любой перорально приемлемой дозированной форме, включая, но не ограничиваясь только ими, таблетки, капсулы (например, капсулы из твердого или мягкого желатина),включая капсулы, заполненные жидкостью, и водные суспензии и растворы. В случае таблеток для перорального применения обычно использующиеся носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие агенты, такие как стеарат магния. Для перорального введения в форме капсул обычно использующиеся разбавители включают лактозу и сухой кукурузный крахмал. Примеры капсул из мягкого желатина, которые можно использовать, включают раскрытые в ЕР 649651 В 1 и патенте US 5985321. Если водную суспензию вводят перорально, то активные ингредиенты объединяют с эмульгирующими и суспендирующими агентами. При необходимости можно добавить некоторые подсластители и/или красители. Другие подходящие для указанных выше препаратов и композиций приведены в стандартных фармацевтических публикациях, например в "Remington's Pharmaceutical Sciences", 19 ed., Mack PublishingCompany, Easton, Penn., 1995. Обычно, когда кристаллическую натриевую соль готовят в жидком разбавителе, например в виде жидкого раствора или суспензии для перорального введения или введения путем инъекции, включая,например, заполненные жидкостью капсулы, натриевая соль теряет свой кристаллический характер. Тем не менее, конечная фармацевтическая композиция на жидкой основе содержит новую натриевую соль соединения (1), и поэтому ее следует считать отдельным вариантом осуществления настоящего изобретения. Только благодаря открытию способа приготовления натриевой соли в стабильной кристаллической форме авторы настоящего изобретения смогли обеспечить эффективную фармацевтическую обработку и приготовление фармацевтического препарата с использованием формы натриевой соли. Поэтому конечная фармацевтическая композиция, содержащая форму натриевой соли, полученную в соответствии с этим открытием, считается другим объектом и вариантом осуществления настоящего изобретения. Методики исследования. 1. Порошковая рентгенография. Анализы с помощью порошковой рентгенографии проводили на порошковом дифрактометре BrukerAXS X-Ray Powder Diffractometer Model D8 Discover, выпускающемся фирмой Bruker AXS, Inc. of Madison, WI, с использованием излучения CuK. Прибор снабжен длинной рентгеновской трубкой с тонкой фокусировкой. Для трубки использовали электропитание 40 кВ и 40 мА. Прибор работал в режиме параллельных пучков с зеркалом Gobel с использованием выходной щели 0,6 мм, фокусирующей щели Соллера 0,4, монохроматора дифрагированного луча на основе плоского кристалла LiF и сцинтилляционного детектора на основе NaI. Сканирование детектором проводили при угле трубки, равном 12. Сканирование проводили от 2 до 402 с шагом 0,05, длительность шага равна 4 с. Для проверки юстировки прибора использовали кварцевый эталонный стандарт. Образцы готовили для анализа путем закрепления в кварцевом держателе с нулевым фоном. 2. Исследование с помощью ДСК. Исследование с помощью ДСК проводили на приборе ТА DSC Q 1000. Термограмму дифференциальной сканирующей калориметрии получали для образца, нагреваемого со скоростью, равной 10C/мин,в закрытом тигле в токе азота. 3. Исследования растворимости и изменения формы. Растворимость соединения (1) в виде типа A или в виде формы натриевой соли исследовали в различных неводных растворителях. Растворы готовили путем добавления избытка соединения (1) к равному от 0,25 до 1,0 мл объему инертного наполнителя во флаконах коричневого стекла с винтовыми крышками, покрытыми тефлоном. Образцы вращали при комнатной температуре в течение до 4 дней. Образцы обрабатывали путем центрифугирования (14000 об/мин в настольной центрифуге Eppendorf model 5415C) и фильтровали через фильтр из поливинилиденфторида с отверстиями размером 0,45 мкм. Для определения растворимости фильтрат анализировали с помощью ВЭЖХ. Анализ с помощью ВЭЖХ проводили на приборе Agilent 1100 с использованием градиентного или изократического режима. В обеих методиках использовали смесь ацетонитрил/вода (с добавлением 0,1% трифторуксусной кислоты) и стационарную фазу АСЕ С-18 с нагреванием колонки при 40-45C. Длину волны детектирования устанавливали равной при 220 или 264 нм. Влажные твердые вещества собирали и анализировали для определе-7 018603 ния изменения формы (стабильности) с помощью XRPD. Анализ с помощью XRPD проводили для определения изменения формы на порошковом дифрактометре Bruker AXS X-Ray Powder Diffractometer Model D8 Discover или D8 Advance, выпускающемся фирмой Bruker AXS, Inc. of Madison, WI, с использованием излучения CuK. Для трубки использовали электропитание 40 кВ и 40 мА или 40 кВ и 30 мА. Прибор(ы) работал в режиме параллельных пучков с зеркалом Gobel с использованием выходной щели 0,6 мм с фокусирующей щелью Соллера 0,4 и монохроматора дифрагированного луча на основе плоского кристалла LiF или с использованием 1 мм ограничивающей щели с системой фокусирующих щелей Соллера 0,12 мм. Для некоторых анализов также использовали конфигурацию Брэгга-Брентано с D8 Advance с 1 мм ограничивающей щелью с системой фокусирующих щелей Соллера 0,12 мм. В каждой конфигурации/приборе использовали сцинтилляционный детектор на основе NaI. Сканирование детектором проводили при угле трубки, равном 12. Сканирование проводили от 2 до 35 или 402 с шагом 0,05, длительность шага равна 0,6 или 4 с. Для проверки юстировки прибора использовали кварцевый эталонный стандарт. Образцы готовили для анализа путем закрепления в кварцевом держателе с нулевым фоном или в держателе, покрытом с помощью Ni. Для более полного понимания настоящего изобретения ниже приведены примеры. Эти примеры приведены для иллюстрации вариантов осуществления настоящего изобретения и их не следует считать каким-либо образом ограничивающими объем настоящего изобретения. Реагенты, использованные в приведенных ниже примерах, можно получить так, как описано в настоящем изобретении, или, если это не описано в настоящем изобретении, то они имеются в продаже или их можно получить из имеющихся в продаже веществ по методикам, известным в данной области техники. Например, некоторые исходные вещества можно получить по методикам, описанным в заявках на международные патенты WO 00/09543,WO 00/09558, WO 00/59929, патентах US 6323180, 6608027 и 7514557 и в публикации заявки на патентUS2005/0020503. Если не указано иное, растворители, температуры, давления и другие параметры реакции легко может подобрать специалист с общей подготовкой в данной области техники. Обычно за протеканием реакции при необходимости можно следить с помощью высокоэффективной жидкостной хроматографии(ВЭЖХ) и промежуточные и конечные продукты можно очистить с помощью хроматографии на силикагеле и/или с помощью перекристаллизации. Примеры Пример 1. Получение исходного вещества, соединения 11 на основе хинолина. Стадия 1. Дианион амида 1 (получен точно так, как описано выше, из 1,00 г амида 1) охлаждали до -78C, затем шприцем в течение 5 мин по каплям добавляли 2,19 мл перфтороктилбромид (8,46 ммоль, 1,75 экв.). Затем темную реакционную смесь помещали в баню при -10C. Через 2 ч осторожно добавляли 10 мл 1 н.HCl и смесь экстрагировали с помощью EtOAc (225 мл), сушили (MgSO4) и растворители удаляли в вакууме. Затем остаток хроматографировали на силикагеле при элюировании смесью 4:1 гексан: EtOAc и получали 1,13 г бромамида 5 (81%) в виде бесцветного масла. 1(dd, J=1,3, 8,3 Гц, 1 Н), 3,87 (s, 3H), 1,33 (s, 9H). 13 С ЯМР (100 МГц, CDCl3) : 176,57 (s), 155,74 (s), 136,98 (s), 128,34 (d), 113,63 (d), 106,86 (d),103,07 (s), 56,26 (q), 40,20 (s), 27,45 (q). Стадия 2. 0,25 г бромамида 5 (0,87 ммоль, 1 экв.), 2,0 мл концентрированной HCl (24 ммоль, 28 экв.) и 1,0 мл диглима нагревали при 100C в течение 24 ч. Затем смесь охлаждали и отфильтровывали (продукт). Фильтрат выпаривали в вакууме с использованием H2O для азеотропного удаления всех растворителей. Остаток растирали с EtOAc для осаждения дополнительного количества продукта, который также отфильтровывали. Объединенные твердые вещества сушили и получали 0,16 г (77%) броманилина 6.HCl в виде светло-коричневого твердого вещества. 1HCl броманизидина (5,73 г, 24,0 ммоль), трихлорид алюминия (3,52 г) и хлорбензол (15,0 мл) при КТ (комнатная температура) помещали в высушенную в сушильном шкафу трехгорлую колбу объемом 100 мл (температура повышалась до 30C). Затем полученную смесь перемешивали в течение 10 мин,затем охлаждали до 0-5C и после этого медленно добавляли ацетонитрил (1,89 мл, 36,0 ммоль), а затем добавляли BCl3 (2,82 г), вводимый в реакционную смесь в виде газа (или жидкости), поддерживая температуру ниже 5C. Затем полученную смесь перемешивали при КТ в течение 20 мин, затем нагревали при 85-100C в течение 16 ч. ВЭЖХ показывала, что реакция завершилась (SM0,5% при 220 нм). Смесь охлаждали до 50C, затем добавляли толуол (15 мл) и после этого при 50C медленно добавляли ИПС (изопропиловый спирт) (11,1 мл), затем медленно добавляли воду (32 мл). Полученную смесь перемешивали в течение еще 2 ч при этой же температуре, затем добавляли 3 г целита и при перемешивании смесь охлаждали до КТ. Фильтровали, затем органическую фракцию промывали водой 115 мл, с помощью 215 м: 5% NaHCO3, 115 мл воды, затем концентрировали при пониженном давлении и получали 3,92-4,4 г искомого продукта с выходом выделенного вещества, равным 68-72%. 1H-ЯМР (400 МГц, CDCl3) : 7,72 (d, J=9,0 Гц, 1 Н), 7,1 (br s, 2H), 6,28 (d, J=9,1 Гц, 1 Н), 3,94 (s, 3H),2,55 (s, 3H). Стадия 4. Оксалилхлорид (8,15 мл) в течение 5 мин по каплям добавляли к холодной смеси (105C) тиазолкарбоновой кислоты 8 (20,18 г), растворенной в ТГФ (тетрагидрофуран) (300 мл) и ДМФ (диметилформамид) (300 мкл), поддерживая внутреннюю температуру равной 105C. Реакционная смесь становилась желтой и гомогенной. Охлаждающую баню удаляли и смеси давали достичь температуры окружающей среды в течение 30 мин. Наблюдалось выделение газа. Смесь перемешивали при температуре окружающей среды в течение от 30 мин до 1 ч. При 105C добавляли раствор анилина 7 (19,8 г), ДМАП(диметиламинопиридин) (140 мг) и ТГФ (35 мл). При 105C в течение 10 мин порциями добавляли Et3N(13,2 мл). Баню со льдом удаляли и смесь нагревали до 652C и перемешивали в течение ночи (18 ч). Смеси давали достичь температуры окружающей среды, разбавляли с помощью EtOAc (150 мл) и промывали водой (150 мл). К органической порции добавляли NaHCO3 (5%, 225 мл) и смесь перемешивали при температуре окружающей среды в течение 30 мин. Органическую порцию концентрировали при пониженном давлении примерно при 40C. К полученному веществу добавляли EtOAc (150 мл) и оставшуюся воду удаляли и смесь концентрировали при пониженном давлении примерно при 40C (для азеотропной отгонки воды). Добавляли EtOAc (94 мл) и полученную взвесь перемешивали в течение 2-6 ч и фильтровали. Твердое вещество промывали с помощью EtOAc (30 мл), затем гептаном (30 мл) и сушили воздухом в течение 1 ч и получали искомый продукт с выходом 70%. 1(d, 1H, J=8,7 Гц), 7,70 (d, 1H, J=8,7 Гц), 7,86 (s, 1H), 8,98 (bs, 1H), 10,13 (bs, 1H). Стадия 5. В колбу объемом 2 л помещали трет-бутоксид калия (112 г). При комнатной температуре добавляли сухой ДМЭ (диметиловый эфир) (экзотермическая реакция: температура повышалась до 35C). Полученный раствор нагревали примерно до 80C и 10 порциями медленно добавляли амид (88 г), так чтобы температура поддерживалась равной 80-85C. После завершения добавления реакционную смесь перемешивали при 85C в течение 2 ч. Во время протекания реакции осаждалось твердое вещество. Анализ с помощью ВЭЖХ показывал, что к этому времени реакция завершилась (степень превращения: 100%). Реакционную смесь охлаждали до комнатной температуры и затем до 10C с помощью охлаждающей бани. Для остановки реакции медленно добавляли водный раствор 2 н. HCl (примерно 500 мл), так чтобы температура поддерживалась ниже 25C. Значение pH доводили до 4-5. Добавляли примерно 100 мл воды (Примечание: количество воды можно изменить для облегчения фильтрования) и полученную суспензию перемешивали при комнатной температуре в течение 5-10 ч. Продукт выделяли фильтрованием,промывали с помощью ТГФ и сушили в вакууме. Выход: 81 г, выход 96%. 1 Н-ЯМР (400 МГц, ДМСО-d6 (ДМСО - диметилсульфоксид: 1,14 (6 Н, d, J=6,8 Гц, i-Pr), 2,48 (1 Н,гептет, J=6,8 Гц, i-Pr), 3,99 (3 Н, s, МеО), 6,75 (1H, s, H-3), 7,24 (1 Н, d, J=8,5 Гц, Н-6), 8,10 (1 Н, d, J=8,5 Гц,Н-5), 8,22 (1 Н, s, Н-5'), 9,87 (1 Н, s, ОН), 12,40 (1 Н, s, амидная группа NH). Стадия 6. В колбу объемом 100 мл помещали исходное вещество, хинолон (4,22 г) и диоксан (40 мл). Добавляли POCl3 (4,6 г) и смесь нагревали при 75C. Через 2 ч ВЭЖХ показывала, что реакция завершилась(степень превращения 99,7%). Реакционную смесь охлаждали до комнатной температуры и затем ее выливали в 100 мл насыщенного раствора NaHCO3 и 20 мл EtOAc. Полученную суспензию перемешивали в течение 3 ч. Продукт выделяли фильтрованием, промывали с помощью EtOAc и сушили в вакууме. Выход: 4,0 г, 90,9%. 1 В трехгорлую колбу объемом 250 мл, снабженную термопарой, патрубком для подачи азота и стержнем для магнитной мешали, помещали N-циклопентилоксикарбонил-трет-L-лейцин (20,0 г,82,2 ммоль, 1,0 экв.), 1-гидроксибензотриазол (12,73 г, 90,42 ммоль, 1,1 экв.) и 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (17,33 г, 90,42 ммоль, 1,1 экв.). Колбу продували азотом и начинали перемешивание. В колбу добавляли безводный ДМФ (62 мл) и смесь перемешивали в течение 20 мин при комнатной температуре (примерно 24C). Реакция являлась умеренно экзотермической, внутренняя температура повышалась до 29C. К реакционной смеси одной порцией добавляли твердый гидрохлорид метилового эфира транс-4-гидроксипролина (14,93 г, 82,2 ммоль, 1,0 экв.). К реакционной смеси по каплям в течение 25 мин шприцем добавляли диизопропилэтиламин (14,36 мл,82,2 ммоль, 1,0 экв.). Внутренняя температура повышалась от 29 до 34,5C. Реакционную смесь перемешивали в течение 1,75 ч и получали соединение 12. Затем реакцию останавливали с помощью 0,1 М HCl(100 мл), внутренняя температура повышалась до 34C. Реакционную смесь трижды экстрагировали с помощью 75 мл этилацетата и органические слои объединяли. Органический слой промывали с помощью 75 мл H2O и 275 мл насыщенного раствора NaHCO3. Органический слой (примерно 235 мл) переносили в колбу объемом 500 мл, снабженную механической мешалкой, короткой насадкой для перегонки, внутренней и внешней термопарами и в вакууме, создаваемом лабораторной вакуумной установкой(110 мм рт. ст.), перегоняли до минимального объема, допускающего перемешивание, при внутренней температуре ниже 35C и при температуре масляной бани, равной 40C. Затем к этой неочищенной смеси 12 добавляли тетрагидрофуран (150 мл) и ее перегоняли до минимального объема, допускающего перемешивание. В колбу добавляли тетрагидрофуран (100 мл) и повторно перегоняли до минимального объема, допускающего перемешивание. Насадку для перегонки заменяли на капельную воронку. В колбу добавляли тетрагидрофуран (100 мл) и метанол (50 мл) и раствор перемешивали в течение примерно 15 мин. В капельную воронку помещали 3,2 М раствор LiOH (77 мл, 246,6 ммоль, 3 экв.) и его добавляли в течение 45 мин. Температура повышалась от 22 до 29C и реакционная смесь становилась немного мутной. Смесь охлаждали в бане с холодной водой, затем реакцию останавливали путем медленного (в течение 45 мин) добавления 4 М HCl (58-65 мл) для доведения pH до 3,5, что вызывало небольшое повышение температуры до 27C. Колбу снабжали насадкой для перегонки и метанол и тетрагидрофуран удаляли путем перегонки при пониженном давлении при температуре бани, равной 40C, и при внутренней температуре ниже 30C. Смесь дважды экстрагировали с помощью 150 мл МТБЭ (метил-трет-бутиловый эфир). Раствор в МТБЭ концентрировали при пониженном давлении (350 мм рт. ст.) до минимального объема, допускающего перемешивание. Добавляли 50 мл МТБЭ, его удаляли с помощью перегонки при внутренней температуре ниже 35C. Реакционная смесь представляла собой прозрачную вязкую жидкость, добавляли 20 мл МТБЭ, смесь нагревали при 50C, раствор был прозрачным, масляную баню удаляли и раствор охлаждали до КТ, 24C, в течение 1,5 ч. Затем к полученной взвеси добавляли 60 мл МТБЭ, перемешивали в течение 2 ч, затем взвесь фильтровали с использованием 20 мл МТБЭ для переноса смеси. Затем твердое вещество сушили в вакууме при 35C до постоянной массы, 16,4 г (52%) и получали содержащий 1/3 МТБЭ сольват соединения 13 в виде бесцветного твердого вещества. Температура плавления 117-124C;H-ЯМР (400 МГц, ДМСО, главный описанный поворотный изомер) : 6,76 (d, J=9,3 Гц, 1 Н), 5,15 (s,1 Н), 4,92 (m, 1 Н), 4,31 (br s, 1H), 4,26 (t, J=8,3 Гц, 1 Н), 4,19 (d, J=9,3 Гц, 1 Н), 3,63 (m, 2H), 3,06 (s, 1H,(МТБЭ, 2,08 (m, 1H), 1,87-1,48 (m, 9H), 1,09 (s, 3 Н, (МТБЭ, 0,92 (s, 9H). Пример 3. Получение исходного вещества, соединения 16, карбоксипроизводного трипептида. В колбе объемом 25 мл соединение 14 растворяли в 3 мл ДМФ. В указанном порядке при комнатной температуре добавляли HOBt (149 мг, 1,1 ммоль), ЭДХ (N-(3-диметиламинопропил)-N'этилкарбодиимидгидрохлорид) (211 мг, 1,1 ммоль), 13 (290 мг, 1,0 ммоль) и i-Pr2NEt (129 мг, 1,0 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в 15 мл водного раствора NaHCO3 и экстрагировали этилацетатом (20 мл). Органический слой промывали с помощью HCl (0,5 н., 210 мл) и насыщенным водным раствором NaHCO3(10 мл). После удаления растворителя в роторном испарителе получали соединение 15 в виде белого твердого вещества, 0,46 г (выход 95%). 1(1H, dd, J=10,5, 2,0 Гц), 5,20 (1H, d, J=18,0 Гц), 5,20-5,25 (1H, m), 5,65-5,77 (1H, ddd, J=18,0, 10,5, 2,0 Гц),7,78 (1H, br) част./млн. 320 мг сложного эфира 15 (0,667 ммоль, 1 экв.) растворяли в 6,7 мл ТГФ + 3,4 мл МеОН при температуре окружающей среды в атмосфере N2. Затем к этому раствору по каплям в течение 5 мин добавляли 3,34 мл 1,6 М LiOH (5,34 ммоль, 8 экв.). Через 1,5 ч растворители удаляли в вакууме и остаток разбавляли с помощью 15 мл EtOAc + 10 мл насыщенного раствора NaCl, затем добавляли 1 н. HCl до установления pH 3,45. Фазы разделяли и водную фазу повторно экстрагировали с помощью 15 мл EtOAc. Объединенные слои, содержащие EtOAc, промывали с помощью H2O (150 мл), сушили (MgSO4) и растворители удаляли в вакууме и получали масло. Масло подвергали азеотропной перегонке с МТБЭ (115 мл) и остаток сушили в высоком вакууме и получали 320 мг соединения 16 (100%) в виде бесцветного вспененного вещества. Точная масса, рассчитано для C23H35N3O7: 465,25; найдено (ИЭ - (ионизация электрораспылением: 464,29;H-ЯМР (400 МГц, ДМСО, главный описанный поворотный изомер) : 12,40 (br s, 1H), 8,49 (s, 1H),6,77 (d, J=8,2 Гц, 1 Н), 5,71 (m, 1H), 5,22-4,85 (m, 4H), 4,36-4,10 (m, 3H), 3,80-3,21 (m, 4H), 2,00-1,42 (m,11H), 0,92 (s, 9H). Пример 4. Получение аморфного соединения (1) по реакции SNAr с использованием дипептида. Методика SNAr 1. В трехгорлую круглодонную колбу объемом 100 мл помещали 1,93 г соединения 13 (5,00 ммоль,1 экв.), затем ее откачивали/заполняли с помощью Ar (3), затем шприцем добавляли 17,0 мл ДМСО и получали прозрачный бесцветный раствор. Затем колбу повторно откачивали/заполняли с помощью Ar(3), затем одной порцией добавляли 2,53 г неразбавленного t-BuOK (22,5 ммоль, 4,5 экв.). Наблюдали повышение температуры максимально до 31,5C. Колбу откачивали/заполняли с помощью Ar (3), затем перемешивали в вакууме, создаваемом лабораторной вакуумной установкой (60 мм), в течение 1 ч и наблюдали небольшое вспенивание (-t-BuOH). Откачанную колбу заполняли с помощью Ar, затем одной порцией добавляли 2,20 г неразбавленного 11 (5,00 ммоль, 1 экв.). Наблюдали повышение температуры до 28,6C. Колбу откачивали/заполняли с помощью Ar (3), затем при температуре окружающей среды перемешивали в вакууме, создаваемом лабораторной вакуумной установкой, при защите от воздействия света. Через 6,5 ч откачанную колбу заполняли с помощью Ar и отбирали образец для ВЭЖХ, в котором обнаруживалось 2% непрореагировавшего соединения 11. Затем колбу охлаждали в бане с холодной водой до 18C и затем шприцем в течение 10 мин добавляли 1,72 мл ледяной НОАс (30 ммоль, 6 экв.). Наблюдали повышение температуры до 20,5C. Смесь перемешивали в течение 10 мин, затем в течение 15 мин по каплям добавляли во вторую колбу, содержащую тщательно перемешанный раствор 30 мл pH 3,5 Н 2 О (0,001 М HCl) при 18C, что сразу приводило к образованию осадка, и наблюдали повышение температуры до 21,0C. 2,0 мл ДМСО использовали для смывания остатка в водную смесь, затем промывали с помощью 5,0 мл 0,001 М HCl. Полученную суспензию перемешивали в течение 15 мин, затем добавляли 30 мл смеси 1:1 EtOAc:МТБЭ и смесь энергично перемешивали в течение 15 мин. Перемешивание останавливали и фазам давали разделиться. Наблюдали быстрое разделение фаз и образование 2 прозрачных фаз без образования загрязненного слоя. Затем нижнюю водную фазу повторно экстрагировали с помощью 30 мл смеси 1:1 EtOAc:МТБЭ (также быстрое разделение) и органические экстракты объединяли и сохраняли. Водную фазу отбрасывали, как отход. Затем органический раствор промывали с помощью Н 2 О (330 мл) и опять все повторные экстракции приводили к быстрому разделению фаз без образования загрязненного слоя, затем EtOAc отгоняли до минимального объема, допускающего перемешивание. Затем остаток подвергали азеотропной перегонке с 30 мл ТГФ (2), повторно перегоняли до минимального объема, допускающего перемешивание. Полученную взвесь неочищенного соединения 18 сразу использовали для реакции сочетания пептида. Точная масса, рассчитано для C34H42BrN5O8S: 759,19; найдено (МС - (масс-спектроскопия: 757,92. Методика SNAr 2. 1,00 г соединения 13 (2,59 ммоль, 1 экв.) и 1,35 г соединения 11 (2,59 ммоль, 1 экв.) помещали в сухую колбу. Затем колбу откачивали/заполняли с помощью Ar (3), затем шприцем добавляли 10 мл сухого ДМСО. Колбу повторно откачивали/заполняли с помощью Ar (3), затем охлаждали до 19C в бане с холодной водой. Затем к этой смеси по каплям в течение 30 мин добавляли 2 М растворKDMO/гептан (5,71 мл, 11,7 ммоль, 4,5 экв.). Через 6 ч ВЭЖХ показывала, что реакция завершилась. Реакцию останавливали с помощью 0,89 мл НОАс (6 экв.) и при перемешивании медленно добавляли H2O до 25 мл, что приводило к образованию осадка. Затем смесь экстрагировали с помощью ИПАЦ (изопропилацетат) (225 мл). Объединенные фазы, содержащие ИПАЦ, промывали с помощью H2O (125 мл),сушили (MgSO4) и растворители удаляли в вакууме и получали твердое вещество, которое подвергали азеотропной перегонке с MeCN (125 мл) и затем разбавляли гептаном и получали взвесь. Взвесь фильтровали и сушили и получали 1,80 г соединения 18 (91%). Методика сочетания пептида 1. К взвеси в ТГФ неочищенного соединения 18, полученного по методике SNAr 1 (5,00 ммоль, 1 экв.) в атмосфере Ar при температуре окружающей среды в колбе, защищенной от воздействия света, добавляли 1,72 г соединения 14 (5,5 ммоль, 1,1 экв.) и 25 мл ТГФ. Затем раствор охлаждали до 5C в атмосфере Ar, затем шприцем в течение 5 мин по каплям добавляли 0,958 мл ДИЭА (диизопропилэтиламин)(5,50 ммоль, 1,1 экв.). Через 5 мин после завершения добавления ДИЭА добавляли 0,85 г гидрата НОВТ(6,00 ммоль, 1,2 экв.) и затем одной порцией добавляли 1,05 г неразбавленного ЭДХ (5,50 ммоль,1,1 экв.). Затем колбу снимали с охлаждающей бани и затем полученную смесь перемешивали при температуре окружающей среды в атмосфере Ar в течение 4 ч. Отбирали образец для ВЭЖХ, в котором обнаруживалось 2% непрореагировавшего соединения 18. Смесь охлаждали до 5C, затем через капельную воронку в течение 5 мин по каплям добавляли 40 мл 0,1 н. HCl, затем 40 мл EtOAc. Смесь энергично перемешивали в течение 15 мин, затем перемешивание прекращали и фазам давали разделиться. Затем нижнюю водную фазу повторно экстрагировали с помощью 40 мл EtOAc и органические фазы объединяли и сохраняли. Водную фазу отбрасывали, как отход. Затем органический раствор промывали с помощью H2O (140 мл), насыщенным раствором NaHCO3 (240 мл) и повторно с помощью H2O (140 мл),затем перегоняли до минимального объема, допускающего перемешивание. Затем остаток подвергали азеотропной перегонке с МТБЭ (240 мл) и повторно перегоняли до минимального объема, допускающего перемешивание. Остаток сушили в высоком вакууме и получали 4,70 г неочищенного соединения 19 в виде оранжевого твердого вещества, по данным ВЭЖХ обладающего чистотой, составляющей 78,3%. Затем это вещество хроматографировали на силикагеле при элюировании смесью 2:1 EtOAc:гексан и получали 3,01 г (68% за 2 стадии) чистого соединения 19 в виде желтого порошкообразного вещества. Точная масса, рассчитано для C41H51BrN6O9S: 882,26, МС+: 883,30. 1H ЯМР (400 МГц, ДМСО, главный описанный поворотный изомер) : 12,32 (s, 1H), 8,69 (s, 1H),8,14 (d, J=9,2 Гц, 1 Н), 8,03 (s, 1 Н), 7,45 (s, 1H), 7,33 (d, J=9,4 Гц, 1 Н), 6,97 (d, J=8,6 Гц, 1 Н), 5,65 (m, 1 Н),5,40 (s, 1 Н), 5,20 (dd, J=1,5, 17 Гц, 1 Н), 5,06 (dd, J=1,6, 10,2 Гц, 1 Н), 5,56 (s, 1 Н), 4,46 (m, 1 Н), 4,37 (d,J=9 Гц, 1H), 4,08 (m, 1 Н), 3,99 (s, 3 Н), 3,90 (m, 1H), 3,56 (s, 3 Н), 2,81 (m, 1 Н), 2,51 (m, 1 Н), 2,25 (m, 1 Н),2,07 (m, 1H), 1,70-1,32 (m, 7 Н), 1,30 (m, 3 Н), 1,15 (d, J=8,1 Гц, 6 Н), 0,95 (s, 9H). Методика сочетания пептида 2. В 4-горлую круглодонную колбу объемом 5 л, снабженную механической мешалкой, капельной воронкой и термопарой, помещали 69,57 г соединения 14 (222 ммоль, 1,3 экв.), затем откачивали/заполняли с помощью Ar (3). Затем к этой смеси добавляли 200 мл ТГФ раствора соединения 18 (содержал 129,85 г, 171 ммоль, 1 экв.), затем добавляли 523 мл ТГФ для доведения конечного объема ТГФ до 1 л. Затем смесь охлаждали до 4,0C в атмосфере Ar. Затем через капельную воронку в течение 10 мин по каплям добавляли 38,67 мл ДИЭА (222 ммоль, 1,3 экв.), и внутренняя температура снижалась до 2,4C. Смесь выдерживали в течение 5 мин, затем добавляли 29,98 г НОВТ Н 2 О (222 ммоль, 1,3 экв.), затем 42,57 г ЭДХ (222 ммоль, 1,3 экв.). После этого внутренняя температура становилась равной 3,6C. Затем баню удаляли. Внутренняя температура повышалась до 20,5C за 90 мин. Через 4 ч после завершения добавления ЭДХ ВЭЖХ показывала, что реакция завершилась. Смесь охлаждали до 4,0C, затем в течение 30 мин через капельную воронку добавляли 750 мл 0,1 н. HCl, что приводило к повышению температуры до 9,5C. Затем к этой смеси добавляли 250 мл насыщенного раствора NaCl, затем 1 л ИПАЦ. После 5 мин энергичного перемешивания смесь помещали в делительную воронку и фазы разделяли. Затем нижнюю водную фазу повторно экстрагировали с помощью 500 мл ИПАЦ и фазы, содержащие ИПАЦ,объединяли. Затем их последовательно промывали с помощью H2O (11 л), насыщенным растворомNaHCO3 (11 л) и затем с помощью H2O (11 л). Затем смесь механически перемешивали в течение 12 ч для осаждения хинолона 7. Затем смесь фильтровали через воронку с фильтром средней пористости и фильтрат перегоняли до минимального объема, допускающего перемешивание. Затем остаток подвергали азеотропной перегонке с МТБЭ (2400 мл) и повторно перегоняли до минимального объема, допускающего перемешивание. Остаток сушили в высоком вакууме и получали 128 г соединения 19 в виде желтого твердого вещества, по данным ВЭЖХ обладающего чистотой, составляющей 89%. 140 мг соединения 19 (0,158 ммоль, 1 экв.) растворяли в 1,6 мл ТГФ + 0,80 мл МеОН при температуре окружающей среды в атмосфере N2. Затем к этому раствору по каплям в течение 5 мин добавляли 0,79 мл 1,6 М LiOH (1,27 ммоль, 8 экв.). Через 1,5 ч органические растворители удаляли в вакууме и остаток разбавляли с помощью 10 мл EtOAc + 10 мл насыщенного раствора NaCl. Затем значение pH доводили до 5,75 с помощью 1 н. HCl. Смесь энергично перемешивали в течение 1 ч, затем фазы разделяли. Водную фазу повторно экстрагировали с помощью 10 мл EtOAc. Затем объединенные фазы, содержащиеEtOAc, затем промывали с помощью H2O (225 мл), сушили (MgSO4) и растворители удаляли в вакууме и получали 125 мг соединения (1) (91%) в виде аморфного желтого порошкообразного вещества. Пример 5. Получение аморфного соединения (1) по реакции SNAr с использованием трипептида. В колбу помещали 233 мг карбоксипроизводного трипептида 16 (0,50 ммоль), затем колбу откачивали/заполняли с помощью Ar (3). Затем добавляли 1,7 мл ДМСО и колбу откачивали/заполняли с помощью Ar (3). Затем смесь охлаждали в бане с холодной водой, затем добавляли 317 мг t-BuOK(2,82 ммоль, 5,63 экв.). Колбу повторно откачивали/заполняли с помощью Ar (3), затем перемешивали в вакууме 60 мм в течение 1 ч. Затем добавляли 220 мг хинолина 11 (0,50 ммоль, 1 экв.) и колбу откачивали/заполняли с помощью Ar (3), затем перемешивали в вакууме 60 мм в темноте при температуре окружающей среды в течение 3 ч. Затем добавляли 0,30 мл НОАс, затем полученный раствор добавляли с помощью 25 мл 0,001 М HCl, что приводило к образованию осадка. Взвесь фильтровали, твердые вещества промывали с помощью 25 мл H2O. Твердое вещество сушили в атмосфере N2 в течение 2 ч, затем хроматографировали на силикагеле при элюировании с помощью EtOAc и получали 226 мг (52%) соединения (1) в виде аморфного желтого твердого вещества. Дополнительные методики получения аморфного соединения (1) приведены в патенте US 6323180 В 1 и в публикациях заявок на патенты US2005/0020503 и 2005/0267151, которые включены в настоящее изобретение в качестве ссылки. Пример 6. Получение соединения (1) типа A. Аморфное соединение (1) (партия 7, 13,80 г) помещали в трехгорлую колбу объемом 1000 мл. В колбу добавляли абсолютный этанол (248,9 г). При перемешивании содержимое колбы нагревали со скоростью 60C/ч до 74C (твердые вещества не растворяются при 74C). Затем к полученной взвеси при перемешивании и поддерживая температуру равной 74C с постоянной скоростью в течение 4 ч добавляли воду (257,4 г). После завершения добавления воды температуру с постоянной скоростью, равной 8C/ч, доводили до температуры окружающей среды, затем при перемешивании выдерживали при температуре окружающей среды в течение 6 ч. Полученное твердое вещество собирали фильтрованием и промывали с помощью 50 мл смеси 1/1 (мас./мас.) EtOH/вода. Влажные твердые вещества сушили в воронке в течение 30 мин, проводя отсасывание осадка на фильтре в токе N2. (Анализ этого образца с помощью XRPD показал, что рентгенограмма сходна с рентгенограммой сольвата с EtOH). Затем твердые вещества сушили при 65-70C в вакууме (Р=25 дюймов рт. ст.) и продували азотом в течение 1,5 ч. С помощью XRPD подтверждали, что полученные твердые вещества (12,6 г, скорректированный выход 95,5%) представляют собой соединение (1) типа A. Специфическая XRPD и термограмма ДСК соединения (1) типа A приведены на фиг. 1 и 2. Пример 7. Получение натриевой соли соединения (1) - методика 1. 2,1 г аморфной натриевой соли соединения (1) и 8,90 г ацетона помещали в колбу и перемешивали при температуре окружающей среды в течение 3 ч. Взвесь отфильтровывали от маточного раствора и полученное твердое вещество в течение 20 мин сушили в токе азота. Собирали 1,51 г кристаллической натриевой соли соединения (1) в виде твердого вещества. Пример 8. Получение натриевой соли соединения (1) - методика 2. 15,6 г соединения (1) типа A, 175 мл ацетона и 3,6 мл воды помещали в реактор объемом 250 мл и нагревали при 53C для растворения твердых веществ. В реактор добавляли 900 мкл 10 н. NaOH и в раствор вносили затравку типа A. Раствор с затравкой перемешивали при 53C в течение 10 мин. Добавляли вторую порцию 10 н. NaOH объемом 900 мкл и смесь перемешивали при 53C в течение 30 мин и за это время образовывалась взвесь. Взвесь охлаждали до 19C при скорости охлаждения, равной 15C/ч, и выдерживали в течение ночи при 19C. Конечную полученную взвесь фильтровали и влажные твердые вещества промывали с помощью 15 мл ацетона. Твердые вещества сушили в течение 1 ч при 52C в вакууме с током азота и затем в течение 1 ч твердые вещества выдерживали на воздухе в лаборатории. Собирали 12,1 г кристаллической натриевой соли соединения (1) в виде твердого вещества. Пример 9. Получение натриевой соли соединения (1) - методика 3. В реактор помещали 25,4 кг аморфного соединения (1), 228 л ТГФ и 11,1 кг 10 мас.% раствораNaOH (водного). Компоненты перемешивали при 25C для растворения всех твердых веществ. Полученный раствор фильтровали и реактор и фильтр промывали с помощью 23 л ТГФ. 180 л растворителя удаляли путем отгонки при атмосферном давлении при 65C. Добавляли 195 л МИБК (метилизобутилкетон) и 166 л растворителя удаляли с помощью перегонки в вакууме при 44C. В реактор добавляли 161 л МИБК и 0,41 кг воды и содержимое нагревали до 70C. Добавляли 255 г затравки натриевой соли соединения (1) при 70C и в течение 1,5 ч добавляли 1,42 л воды. После завершения добавления воды взвесь выдерживали при 70C в течение 45 мин и затем охлаждали до 45C в течение 1 ч. Полученную взвесь фильтровали и промывали с помощью 64 л МИБК, содержащего 0,8 мас.% воды. Влажный осадок на фильтре сушили при 55C и получали 25 кг кристаллической натриевой соли соединения (1). Пример 10. Получение натриевой соли соединения (1) - методика 4. В реактор помещали 2,00 г аморфного соединения (1), 9,96 г ТГФ и 0,11 г воды и перемешивали при температуре окружающей среды для растворения твердых веществ. При перемешивании по каплям добавляли 0,820 мл 21 мас.% раствора NaOEt в этаноле и получали раствор А. Во второй реактор помещали 15,9 г n-BuAc и 160 мкл воды и нагревали при 65C (раствор В). 2,56 г раствора А добавляли к раствору В при 65C и в полученную смесь вносили 40 мг затравки натриевой соли соединения (1). Смесь с затравкой выдерживали при 65C в течение 45 мин. К раствору А добавляли 2,56 г раствора В и выдерживали в течение 45 мин в течение 4 отдельных интервалов. После завершения добавления и выдерживания взвесь охлаждали до 50C в течение 1 ч и фильтровали. Влажный осадок на фильтре промывали с помощью 6 мл n-BuAc, содержащего 0,5 мас.% воды. Конечные твердые вещества сушили при 50C в вакууме при продувании азотом. Собирали кристаллическую натриевую соль соединения (1) в виде твердого вещества. Пример 11. Получение натриевой соли соединения (1) - методика 5. При комнатной температуре раствор этоксида натрия в этаноле (21 мас.%; 306 мл) при перемешивании добавляли к раствору соединения (1) (745 г) в ТГФ (2000 мл) и воде (76,5 мл). После перемешивания в течение 30 мин смесь фильтровали и фильтр промывали с помощью ТГФ (85 мл). Полученный раствор нагревали до 65C и в течение 30 мин обрабатывали профильтрованным бутилацетатом (6640 мл,необязательно предварительно нагретым до 65C). Добавляли затравочные кристаллы (0,50 г) и смесь перемешивали при 65C в течение 2 ч, причем кристаллизация начиналась примерно через 30 мин. Суспензию охлаждали до 50C в течение 1 ч и перемешивали при этой же температуре в течение еще 1 ч. Искомое соединение выделяли фильтрованием, промывали профильтрованным бутилацетатом (765 мл,необязательно предварительно нагретым до 50C) и сушили при 65C в течение примерно 16 ч и получали кристаллическую натриевую соль соединения (1) ( 725 г). Специфическая XRPD и термограмма ДСК кристаллической натриевой соли соединения (1) приведены на фиг. 3 и 4. 2. Кристаллическое соединение формулы (1) по п.1, обладающее порошковой рентгенограммой, содержащей пик при 9,62 (0,22), измеренный с использованием излучения CuK. 3. Кристаллическое соединение по п.2, для которого порошковая рентгенограмма дополнительно содержит пик при 19,82 (0,22), измеренный с использованием излучения CuK. 4. Кристаллическое соединение формулы (1) по п.1, обладающее порошковой рентгенограммой, содержащей пики при 4,8, 6,8, 9,6, 13,6, 17,3, 19,8 и 24,52 (0,22), измеренные с использованием излучения CuK. 5. Натриевая соль соединения формулы (1) 6. Натриевая соль по п.5 в кристаллической форме. 7. Кристаллическая натриевая соль по п.6, обладающая порошковой рентгенограммой, содержащей пик при 10,12 (0,22), измеренный с использованием излучения CuK. 8. Кристаллическая натриевая соль по п.7, для которой порошковая рентгенограмма дополнительно содержит пики при 13,0 и 18,22 (0,22), измеренные с использованием излучения CuK. 9. Кристаллическая натриевая соль по п.8, для которой порошковая рентгенограмма дополнительно содержит пики при 5,4 и 8,72 (0,22), измеренные с использованием излучения CuK. 10. Кристаллическая натриевая соль по п.6, обладающая порошковой рентгенограммой, содержащей пики при 5,4, 6,5, 8,7, 10,1, 11,9, 13,0, 18,2, 20,2 и 24,72 (0,22), измеренные с использованием излучения CuK. 11. Кристаллическая натриевая соль по п.6, обладающая порошковой рентгенограммой, полученной с использованием излучения CuK, которая в основном такая, как приведенная на фиг. 3. 12. Смесь соединения формулы (1) и его натриевой соли где не менее 50% указанного соединения содержится в форме натриевой соли соединения по пп.510 или 11. 13. Фармацевтическая композиция, содержащая натриевую соль по пп.5-10 или 11 и фармацевтически приемлемый носитель или разбавитель. 14. Фармацевтическая композиция по п.13, в которой не менее 50% натриевой соли соединения формулы (1) в композиции содержится в форме кристаллического соединения по пп.6-10 или 11. 15. Применение кристаллического соединения формулы (1) по п.1 или натриевой соли соединения формулы (1) по п.5 или их смесей для приготовления фармацевтической композиции, предназначенной для лечения вирусной инфекции гепатита С у млекопитающего.
МПК / Метки
МПК: A61P 31/12, C07D 417/14, A61K 31/4709
Метки: 2-тиазолил-4-хинолинилоксипроизводного, формы, ингибитора, кристаллические, активного
Код ссылки
<a href="https://eas.patents.su/20-18603-kristallicheskie-formy-2-tiazolil-4-hinoliniloksiproizvodnogo-aktivnogo-ingibitora-hcv.html" rel="bookmark" title="База патентов Евразийского Союза">Кристаллические формы 2-тиазолил-4-хинолинилоксипроизводного, активного ингибитора hcv</a>
Новые кристаллические формы периндоприл эрбумина

Номер патента: 8603
Опубликовано: 29.06.2007
Авторы: Штресслер Кристоф, Леллек Вит, Фесслер Роже
МПК: C07D 209/42
Метки: формы, периндоприл, кристаллические, новые, эрбумина
Формула / Реферат:
1. Кристаллическая форма d периндоприл эрбумина, обладающая следующими параметрами дифракции при порошковой рентгенографии с использованием облучения СuKa: 2. Кристаллическая форма e периндоприл эрбумина, обладающая следующими параметрами дифракции при порошковой рентгенографии с использованием облучения СuKa: 3. Лекарственное средство, содержащее кристаллическую форму периндоприл эрбумина по п.1 или 2. 4. Применение кристаллических форм...
Кристаллические формы eto2c-ch2-(r)cgl-aze-pab-oh

Номер патента: 3264
Опубликовано: 27.02.2003
Авторы: Эдвардссон Даниель, Хедстрём Лена, Петтерссон Урсула, Лундблад Анита
МПК: A61K 38/55, A61P 7/02, C07K 5/062...
Метки: формы, кристаллические, eto2c-ch2-(r)cgl-aze-pab-oh
Формула / Реферат:
1. По существу, кристаллическая форма EtO2C-CH2-(R)Cgl-Aze-Pab-OH или его фармацевтически приемлемой соли. 2. Соединение по п.1, которое находится в безводной форме. 3. Соединение по п.2, которое находится не в форме соли. 4. Соединение по любому из пп.1-3, которое содержит не более 2 маc.% воды. 5. Соединение по любому из пп.1-4, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5шC/мин в...
Кристаллические формы макролидных соединений, обладающих противовоспалительной активностью

Номер патента: 13082
Опубликовано: 26.02.2010
Авторы: Брешелло Роберто, Пеллачини Франко, Микьелетто Иван, Мораззони Габриеле, Наполетано Мауро, Массаччеси Франко, Котарка Ливиус, Вердзини Массимо, Мелотто Элиза, Ди Мария Алессандро, Мараньи Паоло, Рестелли Анджело, Браджа Дарио
МПК: A61K 31/7048, C07H 17/08, A61P 29/00...
Метки: соединений, активностью, формы, обладающих, противовоспалительной, кристаллические, макролидных
Формула / Реферат:
1. Кристаллическая форма I соединения формулы (I)характеризующаяся порошковой рентгенограммой, содержащей значения угла 2q, составляющие приблизительно 4,9; приблизительно 8,5; приблизительно 9,1; приблизительно 9,6; приблизительно 10,3; приблизительно 11,1; приблизительно 14,5; приблизительно 17,0; приблизительно 18,2; приблизительно 19,3.2. Кристаллическая форма по п.1, характеризующаяся порошковой рентгенограммой, в основном соответствующей...
Солевые формы ингибитора mtor

Номер патента: 18144
Опубликовано: 30.05.2013
Авторы: Вайзор Гари К., Малвихилл Кристен Мишель, Речка Йозеф А., Кастелано Арлиндо Л.
МПК: A61K 45/06, C07D 487/04
Метки: солевые, ингибитора, формы
Формула / Реферат:
1. Фармацевтически приемлемая соль транс-4-[4-амино-5-(7-метокси-1Н-индол-2-ил)имидазо[5,1-f][1,2,4]триазин-7-ил]циклогексанкарбоновой кислоты, в которой соль выбирают из L-аргининовой или трометаминовой соли.2. Соль по п.1, в которой солевая форма представляет собой L-аргининовую соль.3. Соль по п.1, в которой солевой формой является трометаминовая соль.4. Соль по п.1, которая представляет собой гидратированную или сольватированную форму...
Новые кристаллические формы гидробромида клопидогреля и способы их получения

Номер патента: 8972
Опубликовано: 26.10.2007
Авторы: Пигера Павел, Гайичек Йосеф, Степанкова Гана
МПК: C07D 495/04, C07D 221/00, C07D 333/00...
Метки: формы, получения, кристаллические, способы, новые, гидробромида, клопидогреля
Формула / Реферат:
1. Гидробромид клопидогреля в кристаллической форме I, отличающийся рентгенограммой с характеристическими межплоскостными расстояниями d, равными 4,01; 4,39 и 3,17 Е. 2. Гидробромид клопидогреля в кристаллической форме I по п.1, отличающийся межплоскостными расстояниями d, равными 3,12; 6,99; 5,5; 4,29 и 3,65 Е. 3. Гидробромид клопидогреля в кристаллической форме I по п.1 или 2, отличающийся наличием в инфракрасном спектре полос поглощения при...
Предыдущий патент: Способ и устройство для регенерации тканей полости рта
Следующий патент: Способ получения хлорсодержащих солей алюминия и технологическая линия для их получения
Случайный патент: Дробилка и способ дробления материала