Применение производных индола в качестве активаторов nurr-1 при лечении болезни паркинсона
Номер патента: 18079
Опубликовано: 30.05.2013
Авторы: Маккрири Эндрю, Бубиа Бенаисса, Ван Донген Мария Йоханна Петронелла, Пупардин-Оливье Оливия, Ден Хартог Якобус Антониус Йозеф, Талландье Мирелль, Ван Влиет Бернар Йоханнес












Формула / Реферат
1. Производное индола формулы (I)

где R1 представляет собой галоген или трифторметильную группу;
R2 представляет собой атом водорода или C1-C4-алкильную группу;
R3 представляет собой изопропильную или трет-бутильную группу;
n равно 3 или 4,
или его фармацевтически приемлемые соли.
2. Соединение по п.1, где R3 представляет собой изопропильную группу.
3. Соединение по п.1, где R3 представляет собой трет-бутильную группу.
4. Соединение по любому из пп.1-3, где R2 представляет собой атом водорода.
5. Фармацевтическая композиция, содержащая по меньшей мере одно соединение по любому из пп.1-4 в качестве активного вещества и по меньшей мере один фармацевтически приемлемый эксципиент.
6. Применение производного индола по любому из пп.1-4 для изготовления лекарства, предназначенного для лечения или предупреждения заболеваний, при которых полезна активация рецепторов NURR-1.
7. Применение по п.6 для изготовления лекарства, предназначенного для лечения и предупреждения нейродегенеративных заболеваний.
8. Применение по п.7, где вышеупомянутое заболевание представляет собой болезнь Паркинсона.
Текст
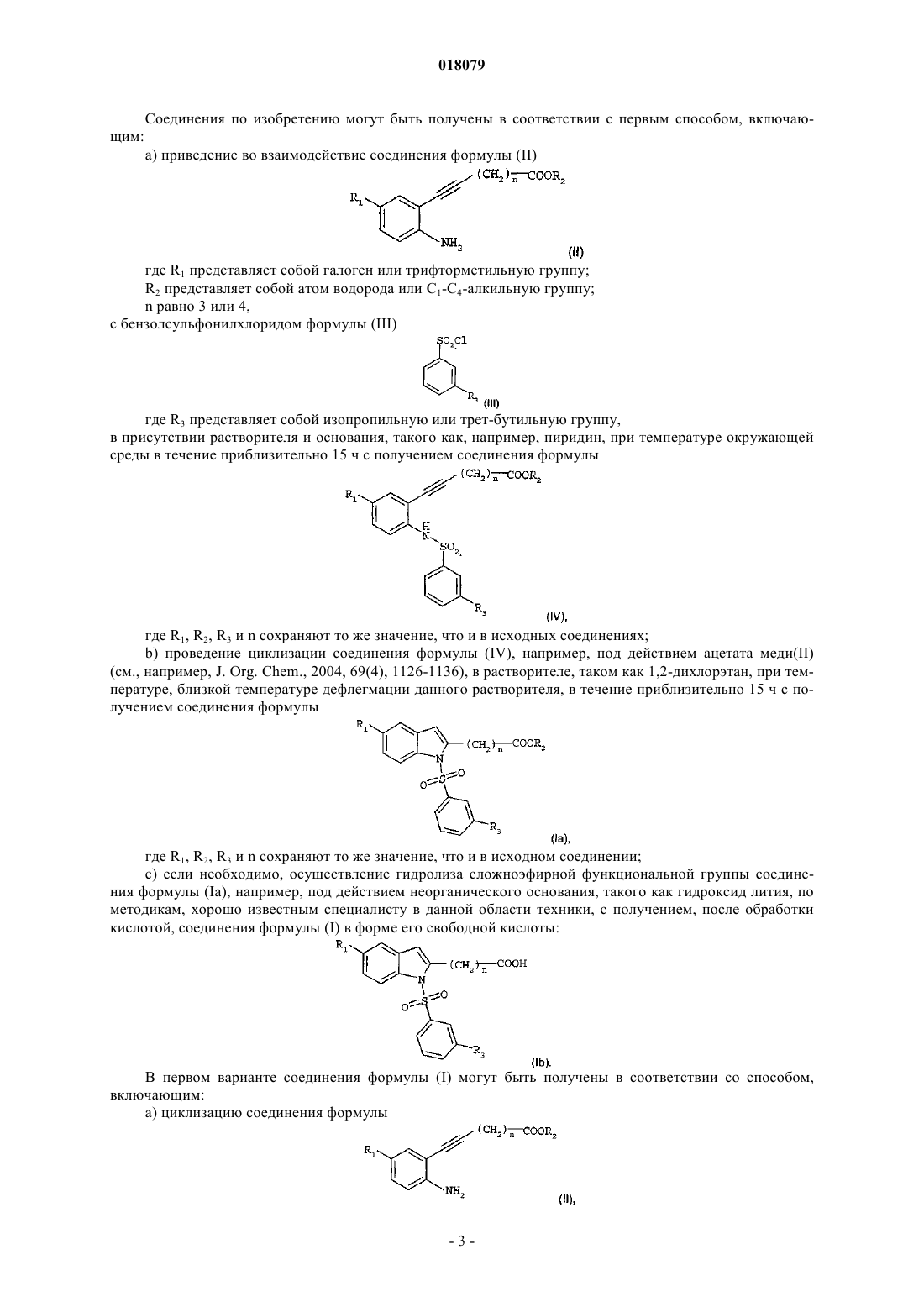
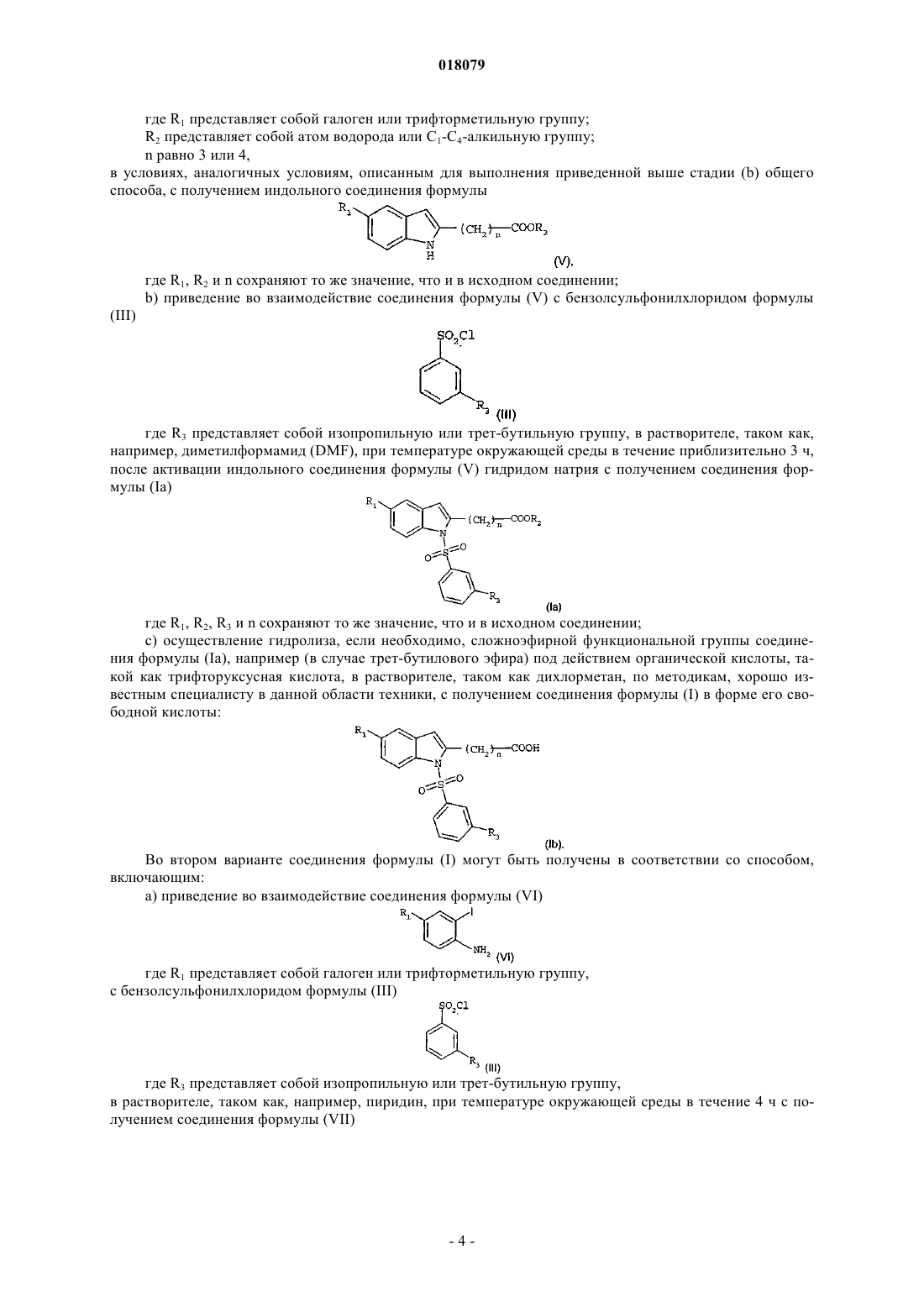
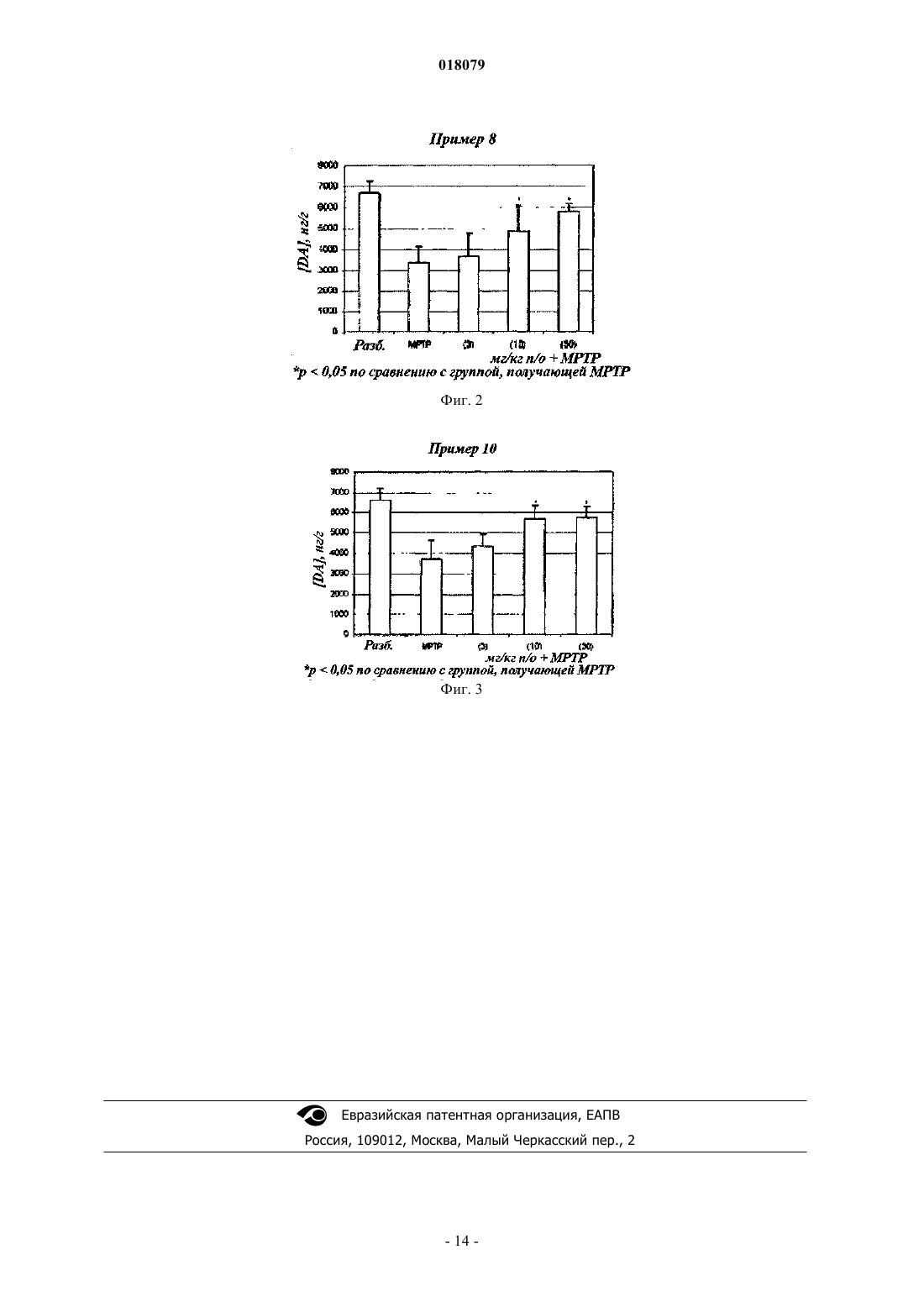
ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ИНДОЛА В КАЧЕСТВЕ АКТИВАТОРОВ NURR-1 ПРИ ЛЕЧЕНИИ БОЛЕЗНИ ПАРКИНСОНА Изобретение относится к производному индола формулы (I) Бубиа Бенаисса (FR), Ван Влиет Бернар Йоханнес, Ден Хартог Якобус Антониус Йозеф, Маккрири Эндрю(NL), Талландье Мирелль (FR), Ван Донген Мария Йоханна Петронелла в которой R1 представляет собой галоген или трифторметильную группу; R2 представляет собой атом водорода или C1-C4-алкильную группу; R3 представляет собой изопропильную группу или трет-бутильную группу и n равно 3 или 4, а также фармацевтически приемлемым солям указанного соединения формулы (I). Изобретение может быть использовано при лечении болезни Паркинсона. Настоящее изобретение относится к новому терапевтическому применению некоторых производных индола в лечении и/или предупреждении заболеваний, вовлекающих ядерные рецепторы NURR-1. Более конкретно, данное изобретение относится к применению этих соединений для изготовления лекарства для лечения и/или предупреждения болезни Паркинсона. Предшествующий уровень техники Нейродегенеративные заболевания определяют как заболевания, характеризующиеся прогрессирующей дисфункцией нервной системы. Зачастую их ассоциируют с атрофией структур центральной или периферической нервной системы. Они включают, среди прочего, такие заболевания, как болезнь Альцгеймера, болезнь Крейцфельдта-Якоба, болезнь Гентингтона, болезнь Паркинсона, "лизосомальные болезни", прогрессирующий супрануклеарный паралич, множественный склероз и амиотрофический боковой склероз. Среди нейродегенеративных заболеваний болезнь Паркинсона представляет собой заболевание, которое поражает около четырех миллионов людей по всему миру. Хотя поражению подвержены индивидуумы любого возраста, в большинстве случаев она встречается у пожилых людей (причем этой болезнью поражены 2% популяции людей старше 65 лет). Она характеризуется дегенерацией дофаминергических нейронов черной субстанции. Дофамин является нейротрансмиттером, который играет центральную роль в контроле произвольных движений, в когнитивных функциях и в проявлении поведения, ассоциированного с эмоциями. Современная терапевтическая стратегия лечения болезни Паркинсона заключается в ослаблении симптомов посредством компенсации дефицита дофамина благодаря введению метаболического предшественника, такого как L-DOPA (L-3,4-дигидроксифенилаланин). В настоящее время увеличение частоты возникновения этой патологии сделало необходимой разработку новых терапевтических агентов, которые играют существенную роль в дифференцировке и выживании нейронов. Данная разработка привела к идентификации соединений, способных активировать ядерные рецепторы, вовлеченные в патогенез болезни Паркинсона. Высокоэкспрессируемый в головном мозге транскрипционный фактор NURR-1, член суперсемейства орфановых ядерных рецепторов, идентифицирован как играющий существенную роль в развитии и сохранении дофаминергических нейронов среднего мозга (Zetterstrom, Solomin and al. 1997, Science. 1997Apr 11; 276(5310): 248-50). Ядерный рецептор NURR-1 участвует в сохранении дофаминергического фенотипа путем регуляции специфических генов дофаминергических (DA) нейронов. Он также способствует выживаемости DA нейронов посредством защиты их от воздействия токсических веществ. Таким образом, ядерный рецептор NURR-1 действует в качестве специфического транскрипционного фактора дофаминергических нейронов, активность которых можно регулировать путем модулирования дофаминергической нейропередачи в болезни Паркинсона. Этот рецептор связывается с ДНК в форме мономеров, гомодимеров или гетеродимеров с RXR (ретиноидным X рецептором), ядерным рецептором, который является гетеропартнером для многих других членов семейства ядерных рецепторов. RXR участвует во многих физиологических процессах, таких как метаболизм липидов, метаболизм глюкозы, развитие и дифференцировка. Так, NURR-1 взаимодействует с изоформамииRXR. Экспрессия RXR осуществляется повсеместно, тогда как экспрессия RXR сосредоточена, в первую очередь, в головном мозге и, более конкретно, в полосатом теле, гипоталамусе и гипофизе. Образованные комплексы NURR-1/RXR и NURR-1/RXR обладают способностью регулировать транскрипцию в ответ на лиганд RXR. Таким образом, RXR положительно модулирует способность к активации транскрипции NURR-1. Следовательно, идентификация соединений, способных индуцировать активность комплексовNURR-1/RXR и NURR-1/RXR, с вероятностью позволит сделать доступными новые направления для лечения болезни Паркинсона. В документе WO 2003/015780 описываются гетероциклические соединения, которые действенны в отношении лечения болезни Паркинсона. Кроме того, в документах WO 2004/072050, FR 2903105, FR 2903106 и FR 2903107 описываются соединения, представляющие собой активаторы рецептора NURR-1, а применение гетероциклических соединений, которые модулируют активность рецепторов семейства NGFI-B (членом которого являетсяNURR-1), описано в документе WO 2005/047268 И наконец, в документе WO 2005/056522 описываются производные индола, которые представляют собой активаторы ядерных рецепторов PPAR (рецепторов, активируемых пролифератором пероксисом) и находят применение в качестве активного начала лекарств для лечения некоторых заболеваний сердечнососудистой системы. В этом контексте обнаружено, и это является тем, что составляет основу настоящего изобретения,что некоторые соединения, являющиеся производными индола и охватывающиеся общей формулой,приведенной в документе WO 2005/056522, представляют собой селективные агонисты NURR-1/RXR иNURR-1/RXR, способные ингибировать дегенерацию нейронов, которая наблюдается при болезни Паркинсона. Таким образом, неожиданно обнаружено, что соединения по изобретению, в дополнение к их эффективности в качестве активатора PPAR, демонстрируют очень высокий потенциал к активации гетеродимеров NURR-1/RXR и NURR-1/RXR. Поэтому данные соединения, благодаря своим уникальным свойствам, представляют особенный интерес с точки зрения их применения в лечении или предупреждении заболеваний, вовлекающих рецептор NURR-1, в особенности нейродегенеративных заболеваний и,более конкретно, болезни Паркинсона. Во-первых, согласно настоящему изобретению в качестве новых продуктов предложены соединения, представляющие собой производные индола, выбранные из: 1) соединений формулы (I) где R1 представляет собой галоген или трифторметильную группу;R2 представляет собой атом водорода или C1-C4-алкильную группу;R3 представляет собой изопропильную (1-метилэтильную) группу или трет-бутильную (1,1 диметилэтильную) группу;n-равно 3 или 4; 2) фармацевтически приемлемых солей указанных соединений формулы (I). Обнаружено (и это является вкладом в уровень техники), что одновременное присутствие изопропильного заместителя или трет-бутильного заместителя в мета-положении на бензолсульфонильной группе и галогена или трифторметильной группы в положении 5 индола наделяет соединения по изобретению удивительной и совсем неожидаемой активностью в отношении рецепторов NURR-1. Таким образом, соединения по изобретению имеют химическую структуру, которая, несмотря на то, что в большинстве случаев охватывается общей формулой, описанной в документе WO 2005/056522,представляет собой результат выбора который специалист в данной области техники не смог бы осуществить в процессе исследования соединений, предназначенных для лечения болезни Паркинсона. Во-вторых, согласно данному изобретению предложены вышеупомянутые соединения для их применения в качестве фармакологически активных веществ, а также фармацевтические композиции, содержащие их. В-третьих, согласно данному изобретению предложено применение по меньшей мере одного соединения формулы (I) или одной из его фармацевтически приемлемых солей в качестве активного начала для изготовления лекарства, предназначенного для лечения заболеваний, вовлекающих рецептор NURR1, особенно нейродегенеративных заболеваний, таких как, более конкретно, болезнь Паркинсона. Подробное описание В настоящем описании C1-C4-алкильная группа представляет собой линейную или разветвленную насыщенную углеводородную цепь, имеющую 1-4 атома углерода, и, более конкретно, группу метил,этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил или 1,1-диметилэтил. Галоген представляет собой атом фтора или хлора. Соединения формулы (I), в которых R2 является атомом водорода, представляют собой карбоновые кислоты, которые могут быть использованы в форме свободной кислоты или в форме солей, причем указанные соли получают путем объединения данной кислоты с нетоксичным органическим или неорганическим основанием, которое предпочтительно является фармацевтически приемлемым. Неорганические основания, которые могут быть использованы, включают, например, гидроксид натрия, гидроксид калия,гидроксид магния или гидроксид кальция. Органические основания, которые могут быть использованы,включают, например, амины, аминоспирты, основные аминокислоты, такие как лизин или аргинин, или также соединения, которые несут функциональную группу четвертичного аммония, такие как, например,бетаин или холин. Соединения по изобретению могут быть получены в соответствии с первым способом, включающим: а) приведение во взаимодействие соединения формулы (II) где R1 представляет собой галоген или трифторметильную группу;R2 представляет собой атом водорода или C1-C4-алкильную группу; где R3 представляет собой изопропильную или трет-бутильную группу,в присутствии растворителя и основания, такого как, например, пиридин, при температуре окружающей среды в течение приблизительно 15 ч с получением соединения формулы где R1, R2, R3 и n сохраняют то же значение, что и в исходных соединениях;b) проведение циклизации соединения формулы (IV), например, под действием ацетата меди(II)(см., например, J. Org. Chem., 2004, 69(4), 1126-1136), в растворителе, таком как 1,2-дихлорэтан, при температуре, близкой температуре дефлегмации данного растворителя, в течение приблизительно 15 ч с получением соединения формулы где R1, R2, R3 и n сохраняют то же значение, что и в исходном соединении; с) если необходимо, осуществление гидролиза сложноэфирной функциональной группы соединения формулы (Ia), например, под действием неорганического основания, такого как гидроксид лития, по методикам, хорошо известным специалисту в данной области техники, с получением, после обработки кислотой, соединения формулы (I) в форме его свободной кислоты: В первом варианте соединения формулы (I) могут быть получены в соответствии со способом,включающим: а) циклизацию соединения формулы где R1 представляет собой галоген или трифторметильную группу;R2 представляет собой атом водорода или C1-C4-алкильную группу;n равно 3 или 4,в условиях, аналогичных условиям, описанным для выполнения приведенной выше стадии (b) общего способа, с получением индольного соединения формулы где R1, R2 и n сохраняют то же значение, что и в исходном соединении;b) приведение во взаимодействие соединения формулы (V) с бензолсульфонилхлоридом формулы где R3 представляет собой изопропильную или трет-бутильную группу, в растворителе, таком как,например, диметилформамид (DMF), при температуре окружающей среды в течение приблизительно 3 ч,после активации индольного соединения формулы (V) гидридом натрия с получением соединения формулы (Ia) где R1, R2, R3 и n сохраняют то же значение, что и в исходном соединении; с) осуществление гидролиза, если необходимо, сложноэфирной функциональной группы соединения формулы (Ia), например (в случае трет-бутилового эфира) под действием органической кислоты, такой как трифторуксусная кислота, в растворителе, таком как дихлорметан, по методикам, хорошо известным специалисту в данной области техники, с получением соединения формулы (I) в форме его свободной кислоты: Во втором варианте соединения формулы (I) могут быть получены в соответствии со способом,включающим: а) приведение во взаимодействие соединения формулы (VI) где R1 представляет собой галоген или трифторметильную группу,с бензолсульфонилхлоридом формулы (III) где R3 представляет собой изопропильную или трет-бутильную группу,в растворителе, таком как, например, пиридин, при температуре окружающей среды в течение 4 ч с получением соединения формулы (VII) где R1 и R3 сохраняют то же значение, что и в исходных соединениях;b) приведение во взаимодействие соединения формулы (VII) с производным ацетилена формулы где R2 представляет собой атом водорода или C1-C4-алкильную группу;n равно 3 или 4,в присутствии йодида одновалентной меди, катализатора на основе палладия, такого как, например, бис(трифенилфосфин)палладия хлорид, и органического основания, такого как, например, триэтиламин, в растворителе, таком как, например, диметилформамид (DMF), при температуре от температуры окружающей среды до 80C в течение 12 ч с получением соединения формулы где R1, R2, R3 и n сохраняют то же значение, что и в исходных соединениях; с) циклизацию приведенного выше соединения формулы (IV) в условиях, аналогичных условиям,описанным для выполнения приведенной выше стадии (b) общего способа, с получением индольного соединения формулы где R1, R2, R3 и n сохраняют то же значение, что и в исходном соединении;d) осуществление гидролиза, если необходимо, сложноэфирной функциональной группы соединения формулы (Ia), например (в случае трет-бутилового эфира) под действием органической кислоты, такой как трифторуксусная кислота, в растворителе, таком как дихлорметан, по методикам, хорошо известным специалисту в данной области техники, с получением соединения формулы (I) в форме его свободной кислоты: где R1, R2, R3 и n сохраняют то же значение, что и в исходном соединении. Необходимо отметить, что в некоторых условиях стадии (b) и (c) этого способа предпочтительно могут быть выполнены за одну операцию (так называемый способ в одном реакторе). Соединение формулы (II), где R1 представляет собой галоген или трифторметильную группу, R2 представляет собой C1-C4-алкильную группу, а n равно 3 или 4, может быть получено в результате взаимодействия ортойоданилина формулы со сложным эфиром алкиновой кислоты формулыn равно 3 или 4,в присутствии йодида одновалентной меди, катализатора на основе палладия, такого как, например, бис(трифенилфосфин)палладия хлорид, и органического основания, такого как, например, триэтиламин, в растворителе, таком как, например, диметилформамид (DMF), при температуре от температуры окружающей среды до 80C в течение 1-12 ч. Сложный эфир алкиновой кислоты формулыn равно 3 или 4,может быть получен, начиная с соответствующей алкиновой кислоты, в результате последовательного действия оксалилхлорида и затем алкилата металла формулы R2OM, где M представляет собой щелочной металл, такой как, например, натрий или калий. Соединения по изобретению в форме солей кислоты формулы (Ib) с органическим или неорганическим основанием можно получить традиционным способом, используя методы, хорошо известные специалисту в данной области техники, например путем смешивания стехиометрических количеств кислоты формулы (Ib) и основания в растворителе, таком как, например, вода или водно-спиртовая смесь, и затем посредством лиофилизации полученного раствора. На некоторых реакционных стадиях, описанных выше, возможно и предпочтительно заменить традиционные методы нагревания нагреванием микроволновым облучением, используя реакторы, подходящие для этого типа реакции. В этом случае специалисту в данной области техники будет понятно, что продолжительность нагревания будет существенно снижена по сравнению с продолжительностью, необходимой в случае общепринятого нагревания. Приведенные далее примеры получения соединений, соответствующих формуле (I), обеспечат лучшее понимание изобретения. В этих примерах, которые не ограничивают объем изобретения, примеры, озаглавленные как "подготовительный пример", представляют собой примеры, описывающие синтез промежуточных соединений, а примеры, озаглавленные как "примеры", описывают синтез соединений формулы (I) по изобретению. В описании использованы следующие сокращения: мМ - миллимоль(и),THF - тетрагидрофуран,DMF - диметилформамид,DCM - дихлорметан. Точки плавления измеряют на стенде Кофлера, а спектральные величины ядерного магнитного резонанса охарактеризовывают химическим сдвигом, рассчитанным относительно TMS (тетраметилсилана), по числу протонов, ассоциируемых с сигналом и по форме сигнала (s для синглета, d для дублета, t для триплета, q для квадруплета, m для мультиплета). Для каждого соединения указывают используемые рабочую частоту и растворитель. Температура окружающей среды составляет 205C. Подготовительный пример 1. 6-[2-[3-(1-Метилэтил)фенил)сульфонил)амино]-5-(трифторметил)фенил]-5-гексиновая кислота,сложный метиловый эфир. Готовили раствор 42,90 г (150,39 мМ) сложного метилового эфира 6-[2-амино-5(трифторметил)фенил]-5-гексиновой кислоты в 500 мл пиридина и добавляли 37,90 г (173,29 мМ) 3-(1 метилэтил)бензолсульфонилхлорида. Смесь перемешивали при температуре окружающей среды в течение 15 ч и затем выливали в смесь льда и соляной кислоты. Полученную кислотную смесь трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния и концентрировали при пониженном давлении. Оставшееся масло очищали хроматографией на силикагеле, элюируя смесью циклогексан/этилацетат (9/1; об./об.). Это позволило получить 29,09 г ожидаемого соединения в виде бледно-желтого масла (выход = 41%). 1 Н ЯМР (DMSO-d6, 250 МГц):1,12 (d, J=6,9 Гц, 6H), 1,76 (q, J=7,0 Гц, 2H), 2,40 (t, J=7,0 Гц, 2H),2,44 (t, J=7,0 Гц, 2H), 2,92 (q, J=6,9 Гц, 1H), 3,62 (s, 3H), 7,47-7,51 (m, 4H), 7,62-7,66 (m, 3H), 9,68 (s, 1H). Пример 1. 1-3-(1-Метилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-бутановая кислота, сложный метиловый эфир. Готовили раствор 28,12 г (60,15 мМ) сложного эфира, полученного согласно подготовительному примеру 1, в 250 мл 1,2-дихлорэтана и добавляли 12,49 г (62,55 мМ) ацетата (двухвалентной) меди моногидрата. Смесь помещали в атмосферу азота и подвергали кипячению с обратным холодильником при перемешивании в течение приблизительно 15 ч. Реакционную смесь фильтровали и твердый остаток после фильтрации промывали на фильтре, используя DCM. Объединенные фильтраты концентрировали при пониженном давлении. Это позволило получить 27,70 г ожидаемого соединения в виде бежевых кристаллов (выход = 99%). т.пл. = 115C. Пример 2. 1-3-(1-Метилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-бутановая кислота. 27,50 г (58,82 мМ) сложного эфира, полученного согласно примеру 1, смешивали с 450 мл THF и добавляли 4,23 г (176,47 мМ) гидроксида лития в 100 мл воды. Смесь перемешивали в течение приблизительно 15 ч при температуре окружающей среды и затем охлаждали до 0C. Затем постепенно добавляли 180 мл 1 н. соляной кислоты при тщательном перемешивании. Органическую фазу отделяли и половину растворителя выпаривали без нагревания при пониженном давлении. Остаток после выпаривания трижды экстрагировали дихлорметаном. Объединенные органические фазы сушили над сульфатом магния,фильтровали и концентрировали при пониженном давлении. Это позволило получить 26,22 г ожидаемого продукта в виде белого порошка (выход = 98%). т.пл.= 160 С. Пример 2 а. 1-3-(1-Метилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-бутановая кислота, натриевая соль. 68 мг (0,15 мМ) кислоты, полученной согласно примеру 2, в растворе в 4 мл тетрагидрофурана смешивали с 6 мг (0,15 мМ) гидроксида натрия в растворе в 3 мл воды. Смесь перемешивали при температуре окружающей среды в течение 6 ч и затем концентрировали при пониженном давлении. Это позволило получить 65 мг ожидаемой соли в виде белого кристаллического порошка (выход = 91%). т.пл. = 231C. Пример 2b. 1-3-(1-Метилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-бутановая кислота, соль пиперазина. 400 мг (0,88 мМ) кислоты, полученной согласно примеру 2, растворяли в 10 мл тетрагидрофурана и добавляли 76 мг (0,88 мМ) пиперазина. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды и затем концентрировали при пониженном давлении. Это позволило получить 400 мг ожидаемой соли в виде белого кристаллического порошка (выход = 46%). т.пл. = 147 С. Пример 2 с. 1-3-(1-Метилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-бутановая кислота, соль трис(гидроксиметил)аминометана. 400 мг (0,88 мМ) кислоты, полученной согласно примеру 2, растворяли в 10 мл тетрагидрофурана и добавляли 106,85 мг (0,88 мМ) трис-(гидроксиметил)аминометана. Добавляли 3 мл воды, получая раствор. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды и затем концентрировали при пониженном давлении. Остаток трижды переносили в метанол, каждый раз выпаривая растворитель при пониженном давлении. Это позволило получить 480 мг ожидаемой соли в виде белого кристаллического порошка (выход = 95%). т.пл. = 126 С. Подготовительный пример 2. 6-[5-Хлор-2-3-(1-метилэтил)фенил]сульфонил]амино]фенил]-5-гексиновая кислота, сложный метиловый эфир. Процедуру выполняли тем же способом, как и в подготовительном примере 1, начиная со сложного метилового эфира 6-(2-амино-5-хлорфенил)-5-гексиновой кислоты, ожидаемое соединение получали в виде коричневого масла (выход = 96%). 1 Н ЯМР (DMSO-d6, 300 МГц):1,13 (d, J=6,9 Гц, 6H) 1,71 (q, J=7,1 Гц, 2 Н), 2,33 (t, J=7,1 Гц, 2H),2,42 (t, J=7,4 Гц, 2H), 2,91 (q, J=6,9 Гц, 1H), 3,61 (s, 3H), 7,26 (d, J=7,3 Гц, 1H), 7,34-7,40 (m, 3H), 7,49-7,57(m, 2H), 7,76-7,78 (m, 1H), 9,68 (s, 1H). Пример 3. 1-3-(1-Метилэтил)фенил]сульфонил]-5-хлор-1H-индол-2-бутановая кислота, сложный метиловый эфир. Готовили раствор 0,3 г (0,69 мМ) сложного эфира, полученного согласно подготовительному примеру 2, в 13 мл 1,2-дихлорэтана и добавляли 0,21 г (1,05 мМ) ацетата двухвалентной меди моногидрата. Проводили облучение реакционной смеси в микроволновой печи при 120C в течение 15 мин, затем охлаждали и фильтровали. Остаток на фильтре промывали DCM и затем фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле, элюируя смесью циклогексан/этилацетат (9/1; об./об.). Это позволило получить 0,23 г ожидаемого соединения в виде бежевого твердого вещества (выход = 77%). т.пл. = 94-97 С. Н ЯМР (DMSO-d6, 250 МГц):1,11 (d, J=6,9 Гц, 6H), 1,95 (q, J=7,4 Гц, 2H), 2,42 (t, J=7,4 Гц, 2H),2,94 (q, J=7,4 Гц, 1H), 3,02 (t, J=7,4 Гц, 2H), 3,59 (s, 3H), 6,61 (s, 1H), 7,32 (dd, J=2,2 и 8,9 Гц, 1H), 7,47 (t,J=7,9 Гц, 1H), 7,56-7,63 (m, 4H), 8,06 (d, J=8,9 Гц, 1H). Пример 4. 1-3-(1-Метилэтил)фенил]сульфонил]-5-хлор-1H-индол-2-бутановая кислота. Процедуру выполняли способом, аналогичным таковому примера 2, начиная с соединения, полученного согласно примеру 3, с получением ожидаемого продукта в виде темно-бежевого твердого вещества (выход = 93%). т.пл. = 128C. Подготовительный пример 3. 6-Гептиновая кислота, сложный 1,1-диметилэтиловый эфир. 8,00 г (63,41 мМ) 6-гептиновой кислоты растворяли в смеси 137 мл безводного дихлорметана и 0,70 мл безводного диметилформамида. По каплям добавляли 16,10 г (126,83 мМ) оксалилхлорида. Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч в атмосфере азота и затем упаривали в атмосфере азота. Оставшийся продукт переносили в 137 мл тетрагидрофурана. Смесь охлаждали до 0C и смешивали порциями с 14,23 г (126,83 мМ) трет-бутилата калия. Реакционную смесь выдерживали при температуре окружающей среды при перемешивании в течение 1 ч. Затем добавляли 200 г льда и 200 мл воды. Смесь экстрагировали 3 раза, используя 200 мл эфира, затем объединенные органические фазы сушили над сульфатом магния и концентрировали при пониженном давлении. Это позволило получить 7,46 г ожидаемого соединения в виде коричневого масла (выход = 65%). 1 Н ЯМР (DMSO-d6, 250 МГц):1,40 (s, 9H), 1,40-1,45 (m, 4H), 2,13-2,22 (т, 4 Н), 2,75 (t, J=2,7 Гц,1H). Подготовительный пример 4. 7-[2-Амино-5-(трифторметил)фенил]-6-гептиновая кислота, сложный 1,1-диметилэтиловый эфир. Готовили раствор 9,78 г (34,07 мМ) 2-йод-4-(трифторметил)анилина и 7,45 г (40,89 мМ) сложного эфира 6-гептиновой кислоты, полученного согласно подготовительному примеру 3, в 136 мл триэтиламина. Добавляли 1,20 г (1,70 мМ) дихлор-бис-(трифенилфосфин)палладия и 0,3 г (1,70 мМ) йодида одновалентной меди. Реакционную смесь перемешивали, нагревали при температуре дефлегмации в атмосфере азота в течение 3 ч и затем концентрировали при пониженном давлении. Остаток после выпаривания переносили в этилацетат и промывали раствором гидрокарбоната натрия (приблизительно 1 М в воде), затем 1 н. соляной кислотой и окончательно дистиллированной водой. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Это позволило получить 12,38 г ожидаемого соединения в виде коричневого масла (выход = 71%). 1 Н ЯМР (DMSO-d6, 250 МГц):1,40 (s, 9H), 1,53-1,68 (m, 4H), 2,24 (t, J=8,4 Гц, 2H), 2,48 (t,J=8,1 Гц, 2H), 5,93 (s, 2H), 6,78 (d, J=10,2, 1H), 7,28-7,33 (m, 2H). Подготовительный пример 5. 5-Трифторметил-1H-индол-2-пентановая кислота, сложный 1,1-диметилэтиловый эфир. Готовили раствор 7,63 г (22,35 мМ) сложного трет-бутилового эфира 7-[2-амино-5(трифторметил)фенил]-6-гептиновой кислоты в 44,70 мл 1,2-дихлорэтана и добавляли 6,69 г (33,52 мМ) ацетата двухвалентной меди моногидрата. Смесь подвергали кипячению с обратным холодильником при перемешивании в течение 48 ч. Реакционную смесь фильтровали через полиамидный фильтр и затем фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле, элюируя смесью циклогексан/этилацетат (9/1; об./об.). Это позволило получить 3,42 г ожидаемого соединения в виде желтого порошка (выход = 45%). 1 Н ЯМР (DMSO-d6, 250 МГц):1,38 (s, 9H), 1,51-1,57 (m, 2H), 1,67-1,73 (т, 2 Н), 2,23 (t, J=8,4 Гц,2 Н), 2,75 (t, J=8,7 Гц, 2 Н), 6,31 (s, 1 Н), 7,28 (dd, J=2,1 и 10,2 Гц, 1H), 7,44 (d, J=10,2 Гц, 1H), 7,79 (s, 1H). Пример 5. 1-3-(1-Метилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-пентановая кислота, сложный 1,1-диметилэтиловый эфир. 46,87 мг (1,17 мМ) гидрида натрия (60%-ного в масле) добавляли к раствору 200,00 мг (0,59 мМ) сложного эфира, полученного согласно подготовительному примеру 5, в 0,5 мл DMF при 0C Эту смесь перемешивали в течение 5 мин и по-прежнему при 0C, добавляли раствор 192,20 мг (0,88 мМ) 3-(1-метилэтил)бензолсульфонилхлорида в 0,5 мл DMF. Смесь перемешивали при температуре окружающей среды в течение 3 ч и затем добавляли раствор хлорида аммония, чтобы нейтрализовать следовые количества гидрида натрия. Смесь экстрагировали дихлорметаном. Органическую фазу концентрировали при пониженном давлении и затем полученную реакционную смесь использовали в реакции на следующей стадии без очистки. 1 Пример 6. 1-3-(1-Метилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-пентановая кислота. Готовили раствор 200,00 мг (0,38 мМ) сложного эфира, полученного согласно примеру 5, в 1 млDCM и добавляли 1 мл трифторуксусной кислоты. Реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч, затем переносили в DCM и концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле, элюируя смесью циклогексан/этилацетат (6/4; об./об.). Это позволило получить 50,00 мг ожидаемого соединения в виде беловатого порошка (выход = 26%). т.пл. = 119C. Подготовительный пример 6. 3-(1,1-Диметилэтил)-N-[2-йод-4-(трифторметил)фенил]бензолсульфонамид. Готовили раствор 1,03 г (3,59 мМ) 2-йод-4-(трифторметил)анилина в 5 мл пиридина и добавляли 1,00 г (4,31 мМ) 3-(1,1-диметилэтил)бензолсульфонилхлорида. После этого реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч. Реакционную смесь промывали 1 н. соляной кислотой и дважды экстрагировали этилацетатом. Органическую фазу сушили над сульфатом магния и затем фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле, элюируя смесью циклогексан/этилацетат (градиент от 100/0 до 90/10; об./об.). Это позволило получить 730 мг ожидаемого соединения в виде белого кристаллического порошка (выход = 42%). т.пл. = 111C. Пример 7. 1-3-(1,1-Диметилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-бутановая кислота, сложный метиловый эфир. В атмосфере азота готовили смесь 250 мг (0,52 мМ) соединения, полученного согласно подготовительному примеру 6, 4,93 мг (0,03 мМ) йодида одновалентной меди, 9,08 мг (0,01 мМ) бис-(трифенилфосфин)дихлорпалладия и 3 мл триэтиламина. Реакционную смесь перемешивали при температуре окружающей среды в течение 10 мин. Добавляли 120,31 мг (0,95 мМ) сложного метилового эфира 5-гексиновой кислоты в растворе в 3 мл диметилформамида. Реакционную смесь нагревали при температуре дефлегмации в течение 3 ч, затем промывали водой и экстрагировали этилацетатом. Органическую фазу сушили над сульфатом магния и концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле, элюируя смесью циклогексан/этилацетат(95/5; об./об.). Это позволило получить 115 мг ожидаемого продукта в виде бежевого кристаллического порошка (выход = 46%). т.пл. = 84C. Пример 8. 1-3-(1,1-Диметилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-бутановая кислота. Процедуру выполняли тем же способом, как и для примера 2, начиная с соединения, полученного согласно примеру 7, с получением ожидаемого продукта в виде белого порошка (выход = 27%). т.пл. = 135-141C. Пример 9. 1-3-(1,1-Диметилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-пентановая кислота,сложный метиловый эфир. В атмосфере азота готовили смесь 57,93 г (119,87 мМ) соединения, полученного согласно подготовительному примеру 6, и 350 мл диметилформамида и перемешивали до полного растворения продукта. Затем последовательно добавляли 21,84 г (155,83 мМ) сложного метилового эфира 4-пентиновой кислоты, 1,14 г (5,99 мМ) йодида одновалентной меди и 1,68 г (2,40 мМ) бис(трифенилфосфин)дихлорпалладия. Эту смесь перемешивали при температуре окружающей среды в течение 15 мин и затем смешивали, добавляя по каплям 174 мл триэтиламина. Реакционную смесь нагревали в течение 14 ч при 80C, охлаждали, затем проводили гидролиз, используя 1 л воды, и экстрагировали этилацетатом. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный маслянистый продукт растворяли при 40C в изопропиловом эфире. Полученный раствор фильтровали и концентрировали при пониженном давлении. Полученный продукт перекристаллизовывали из смеси 140 мл изопропанола и 60 мл воды. Это позволило получить 46,51 г ожидаемого продукта в виде беловатого твердого вещества (выход = 78%). т.пл. = 77C. Пример 10. 1-3-(1,1-Диметилэтил)фенил]сульфонил]-5-(трифторметил)-1H-индол-2-пентановая кислота. Процедуру выполняли тем же способом, как и для примера 2, начиная с соединения, полученного согласно примеру 9, с получением ожидаемого продукта в виде беловатого твердого вещества (выход = 94%). т.пл. = 135C. Описанные выше соединения по изобретению представлены в табл. 1. Таблица 1 Фармакологическая активность. Соединения по изобретению подвергали биологическим тестам с целью оценки их потенциала на предмет лечения или предупреждения некоторых нейродегенеративных паталогий. Начнем с того, что с использованием анализа in vitro измеряли способность соединений по изобретению действовать в качестве активатора гетеродимеров, образуемых ядерным рецептором NURR-1 и ядерными рецепторами RXR. В качестве первичного скринингового теста использовали анализ трансактивации. Cos-7-клетки совместно трансфицировали плазмидой, экспрессирующей химерную структуру (человеческий рецепторNURR-1)-Gal4, плазмидой, экспрессирующей человеческий рецептор RXR (рецептор RXR или RXR),и репортерной плазмидой 5Gal4pGL3-TK-Luc. Трансфекцию осуществляли, используя химический агент(Jet PEI). Трансфицированные клетки распределяли в 384-луночные планшеты и отставляли на 24 ч. Через 24 ч культуральную среду заменяли. В культуральную среду добавляли тестируемые продукты (конечная концентрация от 10-4 до 30-10 М). После инкубации в течение ночи измеряли экспрессию люциферазы после добавления "SteadyGlo" в соответствии с инструкциями производителя (Promega). 4-6-Метил-2-фенил-5-(2-пропенил)-4-пиримидинил]амино]бензойную кислоту (агонист RXR, названный ХСТ 0135908) в концентрации 210-5 М использовали в качестве сравнения. Уровни индукции рассчитывали относительно базальной активности каждого гетеродимера. Результаты выражали в виде процента уровня индукции относительно уровня индукции, полученного для сравнения (уровень индукции соединения сравнения условно равен 100%). Соединения по изобретению демонстрируют уровень индукции до 104% (NURR-1/RXR) и 88%(NURR-1/RXR) и величины EC50 вплоть до 26 нМ (NURR-1/RXR) и 20 нМ (NURR-1/RXR). Некоторые соединения по изобретению имеют ЕС 50 менее 100 нМ, особенно в отношении гетеродимера NURR-1/RXR. В качестве примера для соединений по изобретению получены сравнительные результаты, приведенные в табл. 2, выраженные в виде процента относительно соединения сравнения - активатора NURR1/RXR (ХСТ 0135908). С целью сравнения исследование также было проведено для соединения из примера 76 заявкиWO 2007/026097, имеющего относительно близкую соединениям по изобретению структуру, и результаты для которого показывают, что концентрация, при которой данное соединение дает половину от максимальной эффективности (EC50), по меньшей мере в 10 раз больше концентрации соединений, описанных в изобретении. Первую серию тестов in vivo проводили, используя ряд соединений по изобретению, с целью определения их фармакокинетического профиля для головного мозга и плазмы на самцах мышей C57BL6 и,следовательно, подтверждения того, что эти соединения проходят через гематоэнцефалический барьер. Использовали следующий далее протокол. Для этого исследования использовали самцов C57BL6 мышей (25-30 г) от Janvier, Le Genest-St-Isle,France (12 мышей на одну дозу). Животных кормили стандартным кормом для грызунов (Purina Mills, St. Louis, МО), размещали в клетках и выдерживали в условиях 12 ч/12 ч цикла свет/темнота, поддерживая при этом в помещении температуру 222C и уровень влажности 5510%. Мышей не подвергали голоданию перед введением. На протяжении всего эксперимента воду предоставляли без ограничения. Тестируемое соединение вводили перорально (п/о) в концентрации 10 мг/кг. Для перорального введения в дозе 10 мг/кг животных кормили через желудочный зонд суспензией тестируемого соединения (10 мл/кг), приготовленной в 1%-ной метилцеллюлозе (с вязкостью 400 сП(400 мПас. Животных умерщвляли под анестезией в моменты времени 15, 30 мин, 1, 3, 6 и 8 ч после зондового кормления. В каждый момент времени и у каждого умерщвленного животного отбирали кровь и извлекали головной мозг. По 1 мл крови, собранной в 1,5 мл пробирки, содержащие по 20 мкл упаренного антикоагулянта(раствора гепарината натрия в концентрации 1000 международных единиц (IU)/мл), центрифугировали при 4500 g в течение 3 мин, получая приблизительно 400 мкл плазмы. Плазму разделяли на две аликвоты по 200 мкл, которые хранили при -20C до момента проведения экстракции путем осаждения белков с последующим анализом жидкостной хроматографией в сочетании с тандемной масс-спектрометрией(LC-MS/MS) для количественного определения тестируемого соединения. Сразу после изъятия образцы головного мозга погружали в жидкий азот и затем хранили при -20C для анализа. После этого образцы головного мозга измельчали в присутствии смеси водного/органического растворителя с получением гомогената. Затем эти гомогенаты центрифугировали, тестируемое соединение экстрагировали из полученного супернатанта жидкость-жидкостной экстракцией и далее проводили количественное определение с помощью LC-MS/MS. Фармакокинетические (ФК) параметры определяли, основываясь на некомпартментальном подходе в Excel. Площадь под кривой (AUC0-t) определяли методом трапеций с линейной аппроксимацией. В качестве примера представлены следующие результаты, полученные с использованием соединений примеров 2, 8 и 10 (табл. 3). Вторую серию тестов in vivo проводили, используя соединения по изобретению, с целью подтверждения того, что данные молекулы обладают ожидаемым нейропротективным эффектом. Соединение из примера 2 тестировали в модели мышей, обработанных 1-метил-4-фенил-1,2,3,6 тетрагидропиридином (МРТР), чтобы подтвердить его потенциальную активность. МРТР является нейротоксином, который усиливает необратимые симптомы болезни Паркинсона, разрушая некоторые нейроны в черной субстанции головного мозга. Ниже приведен использованный протокол. Самцов C57BL6/J мышей в возрасте 10-12 недель в начале исследований разделяли на группы по 8 животных. Соединение вводили перорально и два раза в сутки, в общей сложности в течение 11 суток. Введение начинали за 3 суток до обработки токсином МРТР в концентрации 20 или 25 мг/кг. МРТР вводили один раз в сутки внутрибрюшинной инъекцией в течение 5 суток. Введение тестируемого соединения продолжали в течение 3 суток после обработки МРТР. Одна группа мышей получала только разбавитель (0,5%-ный раствор метилцеллюлозы). Животных подвергали эвтаназии по окончании последнего зондового кормления и извлекали полосатое тело. Из полосатого тела экстрагировали дофамин и количество дофамина (DA), выраженное в нг на один 1 г полосатого тела (среднееSEM (стандартная ошибка среднего, измеряли высокоэффективной жидкостной хроматографией (HPLC) с применением электрохимической детекции. Полученные результаты приведены на фиг. 1-3. Эти результаты показывают, что введение МРТР усиливает характерное снижение уровня дофамина в полосатом теле и что соединения, соответствующие примерам 2, 8 и 10, ослабляют дозозависимым образом действие МРТР, токсина, который усиливает синдром паркинсонизма. Таким образом, значительный эффект наблюдается в дозах 10 и 30 мг/кг: соединения по изобретению, введенные перорально, обладают способностью восстановления дофаминергической активности,ингибированной МРТР, в головном мозге. Соединения этого типа, которые проходят через гематоэнцефалический барьер и оказывают благоприятное действие на коммуникацию между нейронами, могут быть предпочтительно использованы в качестве активного начала в лекарствах, предназначенных для лечения болезни Паркинсона. Эти результаты in vitro и in vivo показывают, что соединения по изобретению способны модифицировать механизмы данного заболевания в некоторых животных и клеточных моделях и приостанавливать дегенеративный процесс путем увеличения количества нейропротекторных агентов, противодействующих клеточной смерти дофаминергических нейронов. Таким образом, данные результаты подтверждают интерес к этим соединениям с точки зрения их применения в качестве активного начала в лекарствах,предназначенных для предупреждения или лечения нейродегенеративных заболеваний и, более конкретно, болезни Паркинсона. Кроме того, согласно изобретению предложена фармацевтическая композиция, содержащая в качестве активного начала по меньшей мере одно соединение формулы (I) или одну из его фармацевтически приемлемых солей. В другом аспекте настоящее изобретение ставит задачу применения фармацевтической композиции такого типа для предупреждения или лечения заболеваний, вовлекающих рецептор NURR-1, особенно нейродегенеративных заболеваний и, более конкретно, болезни Паркинсона. Эти фармацевтические композиции можно изготовить традиционным способом, используя фармацевтически приемлемые эксципиенты, с получением форм, которые можно вводить парентерально или предпочтительно перорально, таких как, например, таблетки или капсулы. В случае инъекционных форм соединения формулы (I) следует предпочтительно применять в виде солей, которые растворимы в водной среде. Как указано выше, такие соли предпочтительно образуются между соединением формулы (Ib) (кислотной) и фармакологически приемлемым нетоксичным основанием. Композиция может представлять собой либо раствор соединения в изотонической водной среде в присутствии растворимых эксципиентов, либо лиофилизат соединения, к которому экстемпорально добавляют растворитель. Эти соединения из подготовительных примеров можно вводить в перфузионной форме или в виде болюс-инъекции, в зависимости от нужд пациента. С практической точки зрения в случае парентерального введения соединения суточная доза для человека предпочтительно составляет 2-250 мг. Соединения из подготовительных примеров, которые могут быть введены перорально, предпочтительно будут представлены в виде капсулы или таблетки, содержащей тонкоизмельченное или, еще лучше, микронизированное соединение по изобретению, смешанное с эксципиентами, хорошо известными специалисту в данной области техники, такими как, например, лактоза, прежелатинизированный крахмал и стеарат магния. Например, смесь, составленная из 500 г соединения из примера 2, тонкоизмельченного, 500 г прежелатинизированного крахмала, 1250 г лактозы, 15 г лаурилсульфата натрия и 235 г поливинилпирролидона, подвергали грануляции. Затем эту гранулированную смесь добавляли к 20 г стеарата магния и 80 г микрокристаллической целлюлозы и полученную смесь распределяли, после размалывания и просеивания, по 260 мг в капсулы. Это позволило изготовить капсулы, каждая из которых содержит 50 мг активного начала. С практической точки зрения в случае перорального введения соединения суточная доза для человека предпочтительно составляет 5-500 мг. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Производное индола формулы (I) где R1 представляет собой галоген или трифторметильную группу;R2 представляет собой атом водорода или C1-C4-алкильную группу;R3 представляет собой изопропильную или трет-бутильную группу;n равно 3 или 4,или его фармацевтически приемлемые соли. 2. Соединение по п.1, где R3 представляет собой изопропильную группу. 3. Соединение по п.1, где R3 представляет собой трет-бутильную группу. 4. Соединение по любому из пп.1-3, где R2 представляет собой атом водорода. 5. Фармацевтическая композиция, содержащая по меньшей мере одно соединение по любому из пп.1-4 в качестве активного вещества и по меньшей мере один фармацевтически приемлемый эксципиент. 6. Применение производного индола по любому из пп.1-4 для изготовления лекарства, предназначенного для лечения или предупреждения заболеваний, при которых полезна активация рецепторовNURR-1. 7. Применение по п.6 для изготовления лекарства, предназначенного для лечения и предупреждения нейродегенеративных заболеваний. 8. Применение по п.7, где вышеупомянутое заболевание представляет собой болезнь Паркинсона.
МПК / Метки
МПК: C07D 209/18, A61K 31/405, A61P 25/16, C07D 209/12, A61P 25/28
Метки: производных, индола, применение, качестве, паркинсона, nurr-1, активаторов, болезни, лечении
Код ссылки
<a href="https://eas.patents.su/15-18079-primenenie-proizvodnyh-indola-v-kachestve-aktivatorov-nurr-1-pri-lechenii-bolezni-parkinsona.html" rel="bookmark" title="База патентов Евразийского Союза">Применение производных индола в качестве активаторов nurr-1 при лечении болезни паркинсона</a>
Применение замещённых 2 – аминотетралинов для профилактического лечения болезни паркинсона

Номер патента: 11387
Опубликовано: 27.02.2009
Авторы: Шеллер Дитер, Дрессен Франк
МПК: A61K 31/381, A61P 25/16
Метки: замещённых, применение, паркинсона, профилактического, лечения, аминотетралинов, болезни
Формула / Реферат:
1. Применение соединения общей формулы I где n равно от 1 до 5; R1 выбран из группы и где X выбран из S, О или NH; R2 представляет собой ОА, где А выбран из Н, С1-12алкила, С1-12алкоксиметила или группы где R6 и R7 независимо представляют собой С1-20алкил, фенил или метоксифенил; каждый из R3 и R4 представляет собой Н; R5 представляет собой С1-3алкил; где соединение формулы I представлено в виде рацемата или в виде чистого (R)- или...
Терапевтические средства, содержащие кислородные анион-радикалы и/или продукты их последующего превращения и расщепления, и их применение для лечения болезни паркинсона

Номер патента: 1107
Опубликовано: 30.10.2000
Автор: Гольдштайн Наум
МПК: A61K 33/40
Метки: последующего, терапевтические, применение, кислородные, болезни, превращения, расщепления, продукты, анион-радикалы, паркинсона, лечения, средства, содержащие
Формула / Реферат:
1. Фармацевтическое средство для лечения болезни Паркинсона или других заболеваний, связанных с тремором, отличающееся тем, что оно содержит кислородные анион-радикалы и/или продукты их последующего превращения или расщепления. 2. Фармацевтическое средство по п.1, отличающееся тем, что кислородными анион-радикалами и/или продуктами их последующего превращения или расщепления являются гидропероксидные радикалы, пероксид водорода, другие активные...
Применение производных 2н-[1,3]-оксазино [3,2-a] индола для лечения невропатической боли

Номер патента: 8927
Опубликовано: 31.08.2007
Авторы: Ализи Алессандра, Гульельмотти Анджело, Поленцани Лоренцо, Каццолла Никола
МПК: A61K 31/5365, A61P 25/02
Метки: невропатической, лечения, применение, индола, 2н-[1,3]-оксазино, производных, 3,2-a, боли
Формула / Реферат:
1. Применение соединения формулы I где R представляет Н, линейную или разветвленную алкильную цепь, имеющую от 1 до 12 атомов углерода, или арилалкильную группу, где алкильный фрагмент имеет от 1 до 4 атомов углерода, или циклогексилметил, и его кислотно-аддитивных солей с фармацевтически приемлемыми органическими или неорганическими кислотами для приготовления фармацевтической композиции, активной при лечении невропатической боли. 2....
Применение фенилацетильных производных для производства лекарственных средств для лечения болезни фон хиппеля-линдау (vhl)

Номер патента: 12908
Опубликовано: 26.02.2010
Автор: Бурзинский Станислав Р.
МПК: A61K 31/192, A61K 31/197, A61K 31/198...
Метки: применение, производных, vhl, лекарственных, фенилацетильных, фон, производства, средств, болезни, лечения, хиппеля-линдау
Формула / Реферат:
1. Применение фенилацетилглутамина (PG) или его соли; фенилацетилизоглутамина (isoPG) или его соли и фенилуксусной кислоты (PN) или ее соли для получения лекарственного средства, эффективного для лечения пациентов, страдающих от болезни фон Хиппеля-Линдау.2. Применение по п.1, в котором лекарственное средство включает в себя эффективное количество фенилацетилглутамината натрия, фенилацетилизоглутамината натрия и фенилацетата натрия.3. Применение...
Катехоламиновые производные, полезные для лечения болезни паркинсона

Номер патента: 18011
Опубликовано: 30.04.2013
Авторы: Мерк Нильс, Пюшль Аск, Банг-Андерсен Бенни, Йергенсен Мортен, Ларсен Дженнифер
МПК: A61P 25/00, C07D 491/04, A61K 31/4741...
Метки: паркинсона, полезные, катехоламиновые, производные, лечения, болезни
Формула / Реферат:
1. Соединение структуры Iгде n=1,R1 и R2 объединены и образуют метиленовую (CH2) группу иR3 выбирают из группы, состоящей из водорода, метила, этила, н-пропила, циклопропила, циклобутила, аллила, пропаргила, гидроксиэтила, 3-фторпропила и 2-фторэтила;или его фармацевтически приемлемая кислотно-аддитивная соль.2. Соединение по п.1, где R3 выбирают из группы, состоящей из водорода, метила, этила, н-пропила, аллила и пропаргила.3. Соединение по...
Следующий патент: Соединения, пригодные для лечения дегенеративных и воспалительных заболеваний
Случайный патент: Способ кондиционирования донора органов