Производные третичного амина в качестве ингибиторов фосфодиэстеразы-4
Номер патента: 18069
Опубликовано: 30.05.2013
Авторы: Рицци Андреа, Армани Элисабетта, Ла Порта Елена, Перетто Илария












Формула / Реферат
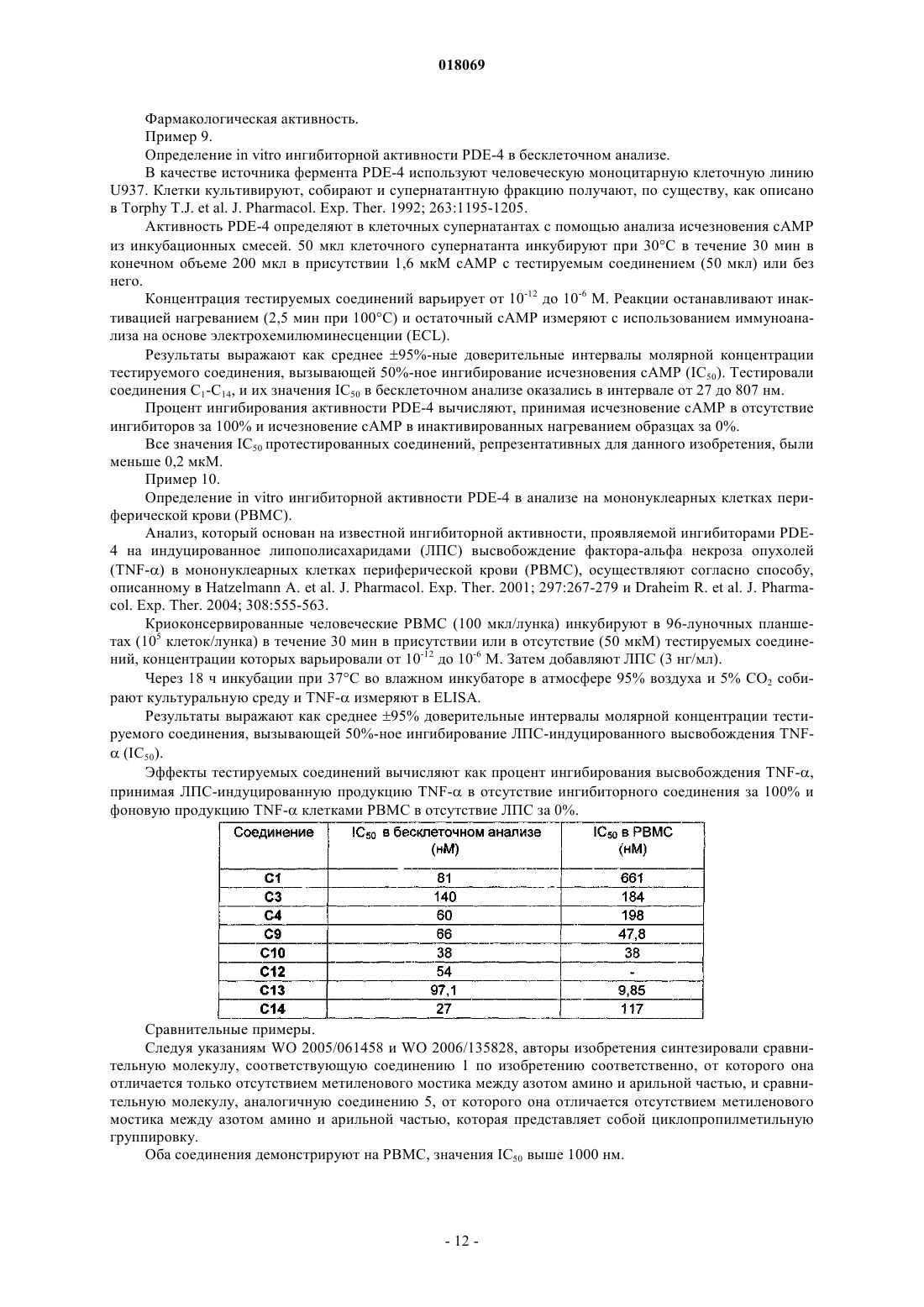
1. Соединение общей формулы (I)

где n=1 или 2;
R1 и R2 являются разными или одинаковыми и независимо выбраны из группы, состоящей из C1-C4-алкилокси, C3-C7-циклоалкилокси и (C3-C7)циклоалкил-(C1-C3)алкилокси;
по меньшей мере один из R1 и R2 представляет собой C1-C4-алкилокси;
А представляет собой арильную кольцевую систему, имеющую от 6 до 10 кольцевых атомов, или гетероарильную кольцевую систему, имеющую от 5 до 9 кольцевых атомов, в которой один кольцевой атом представляет собой гетероатом, выбранный из N, S или O, которые возможно замещены одним или более заместителями, независимо выбранными из группы, состоящей из C1-C6-алкила, OR3, где R3 выбран из группы, состоящей из Н, C1-C6-алкила, C3-C7-циклоалкила; атомов галогена, -CN, NO2, NR6R7, где R6 и R7 являются разными или одинаковыми и независимо выбраны из группы, состоящей из Н, C1-C6-алкила, HNSO2R9, где R9 представляет собой C1-C4-алкил, SO2R10, где R10 представляет собой C1-C4-алкил, ОН или NR6R7, где R6 и R7 являются Н, COOR13, где R13 представляет собой Н, C1-C4-алкил,
и его фармацевтически приемлемые соли.
2. Соединение по п.1, где А представляет собой возможно замещенный фенил.
3. Соединение по п.1, где А представляет собой возможно замещенное гетероарильное кольцо, выбранное из группы, состоящей из фурана или бензотиофена.
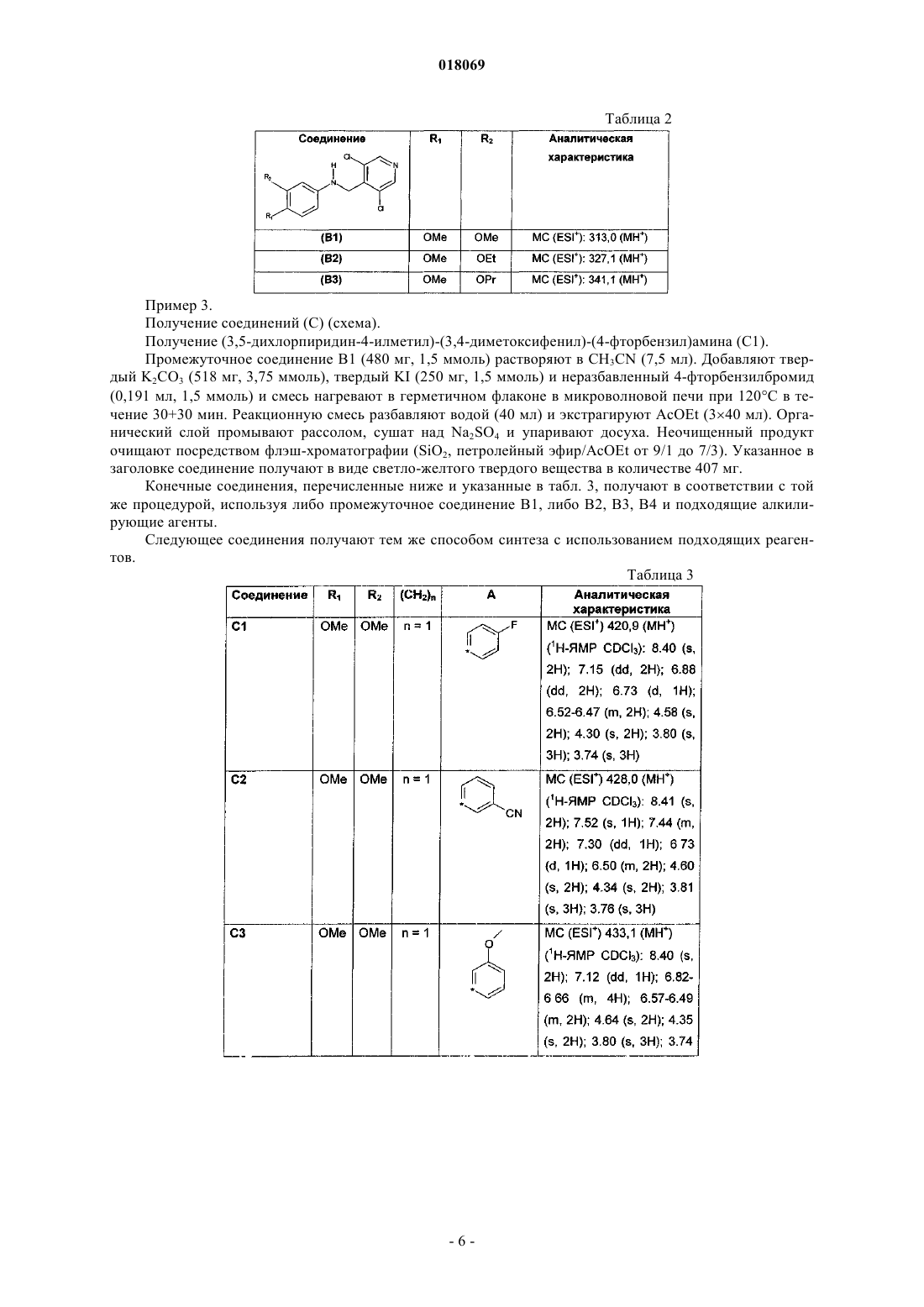
4. Соединение по п.1, выбранное из
(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)-(4-фторбензил)амина;
3-{[(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амино]метил}бензонитрила;
(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)-(3-метоксибензил)амина;
(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)-(4-метансульфонилбензил)амина;
бензил(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амина;
(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)фенэтиламина;
(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксибензил)-(3,4-диметоксифенил)амина;
(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)-(3-фтор-4-метоксибензил)амина;
(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)фуран-2-илметиламина;
4-{[(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амино]метил}бензолсульфонамида;
4-{[(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амино]метил}бензойной кислоты метилового эфира;
N-(3-{[(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амино]метил}фенил)метансульфонамида;
(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)-(4-метоксибензил)амина;
(3,5-дихлорпиридин-4-илметил)-(3-этокси-4-метоксифенил)-(4-метоксибензил)амина;
(3,5-дихлорпиридин-4-илметил)-(3-изопропокси-4-метоксифенил)-(4-метоксибензил)амина;
4-{[(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амино]метил}бензойной кислоты;
(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)тиофен-2-илметиламина;
бензо[b]тиофен-2-илметил(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амина;
3-{[(3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амино]метил}фенола.
5. Соединение по п.4, представляющее собой (3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)-(4-фторбензил)амин (соединение 1).
6. Соединение по п.4, представляющее собой (3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)фуран-2-илметиламин (соединение 9).
7. Соединение по п.4, представляющее собой (3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)-(4-метоксибензил)амин (соединение 13).
8. Фармацевтическая композиция, обладающая ингибиторной активностью в отношении фосфодиэстеразы-4 (PDE-4), содержащая соединение по любому из пп.1-7 в смеси с одним или более фармацевтически приемлемыми носителями и/или эксципиентами.
9. Применение соединения по любому из пп.1-7 в качестве лекарственного средства, обладающего ингибиторной активностью в отношении PDE-4.
10. Применение соединения по любому из пп.1-7 для изготовления лекарственного средства для предупреждения и/или лечения воспалительного заболевания, расстройства или состояния, характеризующегося или ассоциированного с нежелательным воспалительным иммунным ответом или индуцированного или ассоциированного с избыточной секрецией TNF-α и PDE-4.
Текст
ПРОИЗВОДНЫЕ ТРЕТИЧНОГО АМИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ-4 Изобретение относится к ингибиторам (I) фермента фосфодиэстеразы-4 (PDE-4). Более конкретно,изобретение относится к соединениям, которые представляют собой третичные амины, способам получения таких соединений, содержащим их композициям и их терапевтическому применению. Рицци Андреа, Армани Элисабетта,Перетто Илария, Ла Порта Елена (IT) Поликарпов А.В., Борисова Е.Н. (RU) Область изобретения Настоящее изобретение относится к ингибиторам фермента фосфодиэстеразы-4 (PDE-4). Более конкретно, это изобретение относится к производным третичного амина, способам получения таких соединений, содержащим их композициям и их терапевтическому применению. Предшествующий уровень техники Фосфодиэстеразы (PDE), специфичные к циклическим нуклеотидам, составляют семейство с 11 изоферментами, известными в настоящее время, которые катализируют гидролиз различных циклических нуклеозидмонофосфатов (включая cAMP и cGMP). Эти циклические нуклеотиды действуют в качестве вторичных мессенджеров в клетках и в качестве мессенджеров переносят импульсы от рецепторов на клеточной поверхности, связывающих различные гормоны и нейротрансмиттеры. PDE регулируют уровень циклических нуклеотидов в клетках и поддерживают гомеостаз циклических нуклеотидов, разлагая такие циклические мононуклеотиды, что приводит к завершению их роли в качестве мессенджеров. Эти изоферменты можно сгруппировать в соответствии с их специфичностью в отношении гидролиза cAMP или cGMP, их чувствительности к регуляции кальцием, кальмодулином или cGMP и их селективным ингибированием различными соединениями.PDE-4 является cAMP-специфичной, и ее ингибирование вызывает расслабление дыхательных путей, противовоспалительную, повышенную когнитивную и антидепрессантную активность. Поэтому ингибиторы изоферментов PDE-4 представляют собой терапевтические агенты, которые могут быть полезными в лечении заболеваний, вовлекающих воспаление, таких как астма или артрит, или заболеваний центральной нервной системы, таких как когнитивное ухудшение или потеря памяти. Известны различные химические классы ингибиторов PDE-4. В частности, ингибиторы PDE-4, относящиеся к классу третичных аминов, раскрыты вWO 2005/061458 и WO 2006/135828. Однако общеизвестно, что соединения, имеющие значения IC50 выше 1000 нм, могут демонстрировать неудовлетворительную терапевтическую активность. Как следствие, активность известных ингибиторов PDE-4, в частности, относящихся к классу третичных аминов, все еще требует улучшения. Поэтому предмет настоящего изобретения состоит в том, чтобы предложить соединения, относящиеся к классу третичных аминов, с улучшенной активностью относительно известных ингибиторовPDE-4. Краткое описание сущности изобретения Изобретение направлено на соединения, действующие в качестве ингибиторов фермента фосфодиэстеразы-4 (PDE-4), способы получения таких соединений, содержащие их композиции и их терапевтическое применение, в частности изобретение направлено на производные третичных аминов общей формулы (I)R1 и R2 являются разными или одинаковыми и независимо выбраны из группы, состоящей изC1-C4-алкилокси, C3-C7-циклоалкилокси и (C3-C7)циклоалкил-(C1-C3)алкилокси; по меньшей мере один из R1 и R2 представляет собой C1-C4-алкилокси; А представляет собой арильную или гетероарильную кольцевую систему, имеющую от 5 до 10 кольцевых атомов, в которой по меньшей мере один кольцевой атом представляет собой гетероатом (например, N, S или О) и которая возможно замещена одним или более заместителями, независимо выбранными из группы, состоящей из C1-C6-алкила, OR3, где R3 выбран из группы, состоящей из Н,C1-C6-алкила, C3-C7-циклоалкила; атомов галогена, CN, NO2, NR6R7, где R6 и R7 являются разными или одинаковыми и независимо выбраны из группы, состоящей из Н, C1-C6-алкила, HNSO2R9, где R9 представляет собой C1-C4-алкил, SO2R10, где R10 представляет собой C1-C4-алкил, ОН или NR6R7, где R6 и R7 являются такими, как определено выше; COOR13, где R13 представляет собой Н, C1-C4-алкил,и их фармацевтически приемлемые соли. В настоящем изобретении также предложены фармацевтические композиции соединений общей формулы (I) в отдельности или в комбинации с одним или более фармацевтически приемлемыми носителями. В другом аспекте в настоящем изобретении предложено применение соединений общей формулы(I) для изготовления лекарственного средства для предупреждения и/или лечения любого заболевания,где требуется ингибирование PDE-4. В настоящем изобретении также предложено применение соединений общей формулы (I) в изготовлении лекарственного средства. В дополнительном аспекте в настоящем изобретении предложено применение соединений общей формулы (I) для изготовления лекарственного средства для предупреждения и/или лечения воспалительного заболевания, расстройства или состояния, характеризующегося или ассоциированного с нежелательным воспалительным иммунным ответом или индуцированного или ассоциированного с избыточной секрецией TNF- и PDE-4. Определения Термин "атомы галогена", как использовано в данном описании, включает в себя фтор, хлор, бром и йод, предпочтительно хлор. Как использовано в данном описании, выражение "линейный или разветвленный C1-Cx-алкил", где х представляет собой целое число, большее 1, относится к алкильным группам с прямой и разветвленной цепью, где число атомов углерода находится в диапазоне от 1 до x. Особенно предпочтительными алкильными группами являются метил, этил, н-пропил, изопропил и трет-бутил. Возможно, один или более атомов водорода в указанных группах могут быть замещены атомами галогена, предпочтительно хлора или фтора. Производные выражения "C2-C6-алкенил" и "C2-C6-алкинил" следует понимать аналогичным образом. Как использовано в данном описании, выражение "C3-Cx-циклоалкил", где x представляет собой целое число, большее 3, относится к циклическим неароматическим углеводородным группам, содержащим от 3 до x кольцевых атомов углерода. Примеры включают в себя циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Возможно, один или более атомов водорода в указанных группах могут быть замещены атомами галогена, предпочтительно хлора или фтора. Производное выражение "С 5-Сх-циклоалкенил", где х представляет собой целое число, большее 5,следует понимать аналогичным образом. Как использовано в данном описании, выражение "кольцевая система" относится к моно- или бициклическим кольцевым системам, которые могут быть насыщенными, частично ненасыщенными или ненасыщенными, таким как арил, C3-C8-циклоалкил или гетероарил, имеющим от 5 до 10 кольцевых атомов, в которых по меньшей мере один кольцевой атом представляет собой гетероатом (например, N, S или О). Примеры подходящих моноциклических систем включают в себя тиофен, фенил и фуран. Примеры подходящих бициклических систем включают в себя нафтил и бензотиофен. Подробное описание изобретения Настоящее изобретение относится к производным третичного амина, в которых заместители представляют собой ароматическое кольцо, замещенное двумя группами алкилокси, арилметильной группой и пиридинилметильной группой. Более конкретно, настоящее изобретение относится к производным третичных аминов общей формулы (I) Фармацевтически приемлемые соли включают в себя соли, полученные путем взаимодействия соединения с неорганической или органической кислотой с образованием соли, например солей соляной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, камфорсульфоновой кислоты, щавелевой кислоты, малеиновой кислоты, янтарной кислоты и лимонной кислоты. Фармацевтически приемлемые соли также включают в себя соли, в которых кислотные функциональные группы, когда они присутствуют, подвергают взаимодействию с подходящим основанием с образованием, например, солей натрия, калия, кальция, магния, аммония и хлорида. Обнаружили, что, когда фенильная группа в А замещена циклоалкильной группировкой, активность падает, в частности, в клеточном анализе. Более того, из анализа результатов скрининга следует, что донор водородной связи или заместители акцептора на фенильном кольце в области А, по-видимому, являются предпочтительными, фактически они дают начало соединениям, демонстрирующим улучшенную ингибиторную активность в бесклеточном анализе. Также обнаружили, что соединения, в которых А непосредственно связан с азотом амино, демонстрируют активность выше 1000 нм в анализе IC50 на мононуклеарных клетках периферической крови(РВМС). В одном из предпочтительных воплощений R1 и R2 представляют собой C1-C4-алкилокси. В конкретном воплощении изобретения А представляет собой гетероарильное кольцо, выбранное из группы, состоящей из фурана или бензотиофена. В другом конкретном воплощении изобретения А представляет собой нафтил. В одном из предпочтительных воплощений изобретения А представляет собой фенил. В одном из предпочтительных воплощений возможный заместитель Rx кольцевой системы А выбран из группы, состоящей из C1-C6-алкила, атома галогена, предпочтительно фтора; SO2R10, где R10 представляет собой C1-C4-алкил, предпочтительно метил, или NH2; CN; ОН; COR8, где R8 представляет собой предпочтительно ОН; HNSO2R9, где R9 представляет собой C1-C4-алкил, предпочтительно метил. В одном из предпочтительных воплощений настоящего изобретения Rx представляет собой донор водородной связи или заместитель акцептора, выбранный из группы, состоящей из OR3, где R3 представляет собой C1-C6-алкил, предпочтительно метил, или NR6R7. В соответствии с предпочтительным воплощением в настоящем изобретении предложены следующие соединения. Соединения общей формулы (I) могут быть получены согласно общепринятым способам. Примеры способов, которые могут быть использованы, раскрыты ниже и указаны на схеме. Как указано на схеме, соединения общей формулы (I) получают согласно способу, который включает в себя следующие стадии, причем способ получения амина формулы (А) хорошо известен: 1-я стадия - функционализация амина формулы (А) путем восстановительного аминирования подходящим альдегидом (PhCl2)-CHO с получением вторичного амина общей формулы (В). Эта реакция может быть осуществлена, например, путем образования иминного промежуточного соединения в толуоле с молекулярными ситами с последующим выпариванием растворителя и последующим восстановлением иминного производного боргидридом натрия (NaBH4) в этаноле; 2-я стадия - дополнительная функционализация вторичного амина общей формулы (В) либо посредством восстановительного аминирования, либо посредством алкилирования первичными алкилирующими агентами с получением конечных соединений общей формулы (С). В настоящем изобретении также предложены фармацевтические композиции соединений общей формулы (I) в смеси с одним или более фармацевтически приемлемыми носителями, например, описанными в Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A. Введение соединений по настоящему изобретению можно осуществлять в соответствии с потребностями пациента, например перорально, интраназально, парентерально (подкожно, внутривенно, внутримышечно, интрастернально и посредством инфузии), посредством ингаляции, ректально, интравагинально, местно, локально, трансдермально и посредством глазного введения. Для введения соединений по изобретению могут быть использованы различные твердые пероральные лекарственные формы, в том числе такие твердые формы, включающие таблетки, желатиновые капсулы, капсулы, каплеты, гранулы,лепешки и нефасованные порошки. Соединения по настоящему изобретению можно вводить в отдельности или объединять с различными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмалы) и известными эксципиентами, включающими, но не ограничивающимися этим, суспендирующие агенты, солюбилизаторы, буферные агенты, связующие агенты, разрыхлители, консерванты, красители, ароматизаторы, смазывающие агенты и т.п. Предпочтительными также являются капсулы, таблетки и гели с высвобождением во времени. Для введения соединений по изобретению также могут быть использованы различные жидкие пероральные лекарственные формы, включающие водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие лекарственные формы также могут содержать подходящие инертные разбавители, известные в данной области, такие как вода, и подходящие эксципиенты, известные в данной области, такие как консерванты, увлажняющие агенты, подсластители, ароматизаторы, а также агенты для эмульгирования и/или суспендирования соединений по изобретению. Соединения по изобретению можно инъецировать, например внутривенно, в форме изотонического стерильного раствора. Также возможными являются другие препараты. Суппозитории для ректального введения соединений по настоящему изобретению могут быть приготовлены путем смешивания соединения с подходящим эксципиентом, таким как масло какао, салицилаты и полиэтиленгликоли. Препараты для вагинального введения могут быть в форме крема, геля, пасты, пены или спрея, содержащего, в дополнение к активному ингредиенту, подходящие известные носители. Для местного введения фармацевтическая композиция может быть в форме кремов, мазей, линиментов, лосьонов, эмульсий, суспензий, гелей, растворов, паст, порошков, спреев и капель, подходящих для введения в кожу, глаз, ухо или нос. Местное введение также может включать трансдермальное введение с помощью таких средств, как трансдермальные пластыри. Дозировки соединений по настоящему изобретению зависят от множества факторов, включая конкретное заболевание, подлежащее лечению, тяжесть симптомов, путь введения, частоту введения доз,конкретное используемое соединение, эффективность, токсикологический профиль и фармакокинетический профиль соединения. Преимущественно соединения общей формулы (1) можно вводить, например, в дозировке от 0,001 до 1000 мг/сутки, предпочтительно от 0,1 до 500 мг/сутки. Соединения общей формулы (I) можно вводить для предупреждения и/или лечения любого заболевания, где требуется ингибирование PDE-4. Указанное заболевание включает заболевания, вовлекающие воспаление, такие как астма и хроническое обструктивное заболевание легких (CODP), аллергические болезненные состояния, такие как атопический дерматит, крапивница, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, эозинофильная гранулема, псориаз, воспалительный артрит, ревматоидный артрит, септический шок, язвенный колит, болезнь Крона, поражение миокарда и головного мозга при реперфузии, хронический гломерулонефрит, эндотоксиновый шок, муковисцидоз,артериальный рестеноз, атеросклероз, кератоз, ревматоидный спондилит, остеоартрит, пирез, сахарный диабет, пневмокониоз, токсическая и аллергическая контактная экзема, атопическая экзема, себорейная экзема, простой лишай, солнечный ожог, зуд в аногенитальной области, очаговая алопеция, гипертрофированные рубцы, дискоидная красная волчанка, системная красная волчанка, фолликулярные и обширные пиодермии, эндогенные пурпура, нефрит, воспалительное заболевание кишечника, лейкоз, рассеянный склероз, желудочно-кишечные заболевания, аутоиммунные заболевания и т.п. Они также включают неврологические и психиатрические расстройства, такие как болезнь Альцгеймера, рассеянный склероз, боковой амиотрофический склероз (ALS), множественная системная атрофия (MSA), шизофрения, болезнь Паркинсона, болезнь Гентингтона, болезнь Пика, депрессия, инсульт и поражение спинного мозга. Настоящее изобретение теперь будет дополнительно описано с помощью следующих примеров. Пример 1. Получение промежуточных соединений (А) (схема). Получение (3-этокси-4-метоксифенил)амина (А 2). Стадия 1. Получение 2-этокси-1-метокси-4-нитробензола. 2-Метокси-5-нитрофенол (508 мг, 3 ммоль) растворяют в DMF (20 мл) в атмосфере азота. Добавляют K2CO3 (900 мг, 6,5 ммоль), KI (490 мг, 2,95 ммоль) и этилбромид (0,250 мл, 3,3 ммоль), и суспензию нагревают до 40C в течение 28 ч. Смесь разбавляют AcOEt (60 мл) и экстрагируют 1 н. NaOH (40 мл) и водой (40 мл). Органический слой сушат над Na2SO4 и упаривают досуха. Неочищенный продукт используют на следующей стадии без очистки. Стадия 2. Получение 3-этокси-4-метоксианилина. Неочищенный продукт, полученный на стадии 1, растворяют в этаноле (99%, 20 мл). ДобавляютPd/C (10%, 60 мг) и формиат аммония (1,71 г) и полученную смесь перемешивают при комнатной температуре в течение 1 ч. Катализатор удаляют фильтрацией и растворитель выпаривают при пониженном давлении. Неочищенный продукт очищают посредством флэш-хроматографии (SiO2, петролейный эфир/AcOEt от 7/3 до 5/5). Указанное в заголовке соединение получают в количестве 325 мг. Ту же процедуру используют для синтеза (А 2) (3-изопропокси-4-метоксифенил)амина с использованием подходящих реагентов. Таблица 1 Пример 2. Получение промежуточных соединений (В) (схема). Получение (3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амина (В 1). Имеющиеся в продаже 3,4-диметоксифениламин (1,74 г, 11,3 ммоль) и 3,5-дихлорпиридин-4 карбальдегид (2,0 г, 11,3 ммоль) растворяют в толуоле (40 мл) в атмосфере азота. Добавляют молекулярные сита (4 , 1 г) и смесь нагревают до температуры дефлегмации. Мониторинг реакции осуществляют с помощью ТСХ-анализа (петролейиый эфир/AcOEt 7/3): образование иминного промежуточного соединения завершается через 3 ч. Молекулярные сита удаляют фильтрацией и растворитель выпаривают. Остаток растворяют в этаноле (99%, 40 мл). Раствор охлаждают до 0C и добавляют NaBH4 (559 мг,14,7 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 18 ч, затем добавляют воду (50 мл) и смесь экстрагируют AcOEt (360 мл). Органический слой промывают рассолом, сушат над Na2SO4 и упаривают досуха. Неочищенный продукт очищают посредством флэш-хроматографии(SiO2, петролейный эфир/AcOEt от 9/1 до 7/3). Указанное в заголовке соединение получают в виде желтого твердого вещества (3,04 г). Следующие соединения получают, следуя той же процедуре синтеза, с использованием подходящих реагентов.(0,191 мл, 1,5 ммоль) и смесь нагревают в герметичном флаконе в микроволновой печи при 120C в течение 30+30 мин. Реакционную смесь разбавляют водой (40 мл) и экстрагируют AcOEt (340 мл). Органический слой промывают рассолом, сушат над Na2SO4 и упаривают досуха. Неочищенный продукт очищают посредством флэш-хроматографии (SiO2, петролейный эфир/AcOEt от 9/1 до 7/3). Указанное в заголовке соединение получают в виде светло-желтого твердого вещества в количестве 407 мг. Конечные соединения, перечисленные ниже и указанные в табл. 3, получают в соответствии с той же процедурой, используя либо промежуточное соединение В 1, либо В 2, В 3, В 4 и подходящие алкилирующие агенты. Следующее соединения получают тем же способом синтеза с использованием подходящих реагентов. Таблица 3 Следующие алкилирующие агенты (как перечислено в табл. 4), используемые для синтеза перечисленных выше конечных соединений, не доступны в продаже, и их синтезируют согласно примерам 4-6. Таблица 4 Пример 4. Получение алкилирующих агентов (K) (схема). Получение 4-бромметил-бензол-сульфонамида (K1). Стадия 1. 4-Гидроксиметил-бензол-сульфонамид.LiAlH4 (235 мг, 6,2 ммоль) суспендируют в сухом THF (10 мл) в атмосфере азота. Суспензию охлаждают до 0C и добавляют 4-сульфамоил-бензойную кислоту (500 мг, 2,48 ммоль, в виде суспензии в 10 мл сухого THF). Полученную смесь затем подвергают дефлегмации в течение 18 ч. Реакционную смесь гасят путем добавления 3 н. HCl при 0C. Гашеную смесь экстрагируют AcOEt, органический слой сушат над Na2SO4 и упаривают досуха. Неочищенный продукт очищают посредством флэш-9 018069 хроматографии (SiO2, петролейный эфир/AcOEt от 9/1 до 1/1) с получением указанного в заголовке соединения в количестве 105 мг. Стадия 2. 4-Бромметил-бензол-сульфонамид. 4-Гидроксиметил-бензол-сульфонамид (0,105 мг, 0,56 ммоль) растворяют в DCM (5 мл). Добавляют трифенилфосфин на полимерной подложке (294 мг, 2,4 ммоль/г, 1,12 ммоль) и смесь перемешивают с помощью шейкера при комнатной температуре в течение 10 мин. Затем добавляют CBr4 (557 мг,1,68 ммоль) и перемешивание продолжают в течение 3 ч. Реагент на подложке удаляют фильтрацией,растворитель упаривают, и неочищенный продукт очищают посредством флэш-хроматографии (SiO2,петролейный эфир/AcOEt 9/1) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (70 мг). Алкилирующие агенты K3 и K4 синтезируют, как описано на стадии 2 этого же примера 4, начиная с соответствующих имеющихся в продаже производных спиртов. Пример 5. Получение фуран-2-илметилового эфира метансульфоновой кислоты (K2). Фуран-2-илметанол (0,477 мл, 5,5 ммоль) растворяют в сухом DCM (10 мл). Раствор охлаждают до 0C и добавляют по каплям триэтиламин (1,16 мл, 8,25 ммоль) и хлорид метансульфоновой кислоты(0,554 мл, 7,15 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 3 ч. Суспендированное твердое вещество (триэтиламина гидрохлорид) удаляют фильтрацией, фильтрат упаривают досуха и неочищенный продукт используют на следующей стадии без очистки. Стадия 2. 4-Бромметил-3,5-дихлорпиридин.(3,5-Дихлорпиридин-4-ил)метанол (918 мг, 5,15 ммоль) растворяют в сухом DCM (25 мл). Добавляют трифенилфосфин (2,70 г, 10,3 ммоль) и смесь перемешивают при комнатной температуре в течение 10 мин. Затем раствор охлаждают до 0C и добавляют CBr4 (5,12 г, 15,4 ммоль). Смесь перемешивают при комнатной температуре в течение 30 мин, затем растворитель выпаривают и неочищенный продукт очищают посредством флэш-хроматографии (SiO2, петролейный эфир/AcOEt 95/5) с получением 850 мг указанного в заголовке соединения. Пример 6. Получение N-(3-хлорметилфенил)метансульфонамида (K5). Стадия 1. (3-Аминофенил)метанол. 3-Аминобензойную кислоту (1,05 г, 7,65 ммоль) растворяют в THF (30 мл) в атмосфере азота. Добавляют боран (BH3, 1 М раствор в THF, 24 мл, 24 ммоль) и полученный раствор перемешивают при комнатной температуре в течение 20 ч. Реакционную смесь затем вливают в насыщенный раствор NH4Cl(100 мл) и экстрагируют AcOEt (3100 мл). Органический слой сушат над Na2SO4 и упаривают досуха с получением 388 мг указанного в заголовке соединения, которое используют на следующей стадии без дополнительной очистки. Стадия 2. N-(3-хлорметилфенил)метансульфонамид. Раствор (3-аминофенил)метанола (388 мг, 3,15 ммоль), хлорида лития (270 мг, 6,3 ммоль) и 2,6-лутидина (0,821 мл, 6,93 ммоль) в DMF (10 мл) охлаждают при 0C в атмосфере азота. Добавляют по каплям метансульфонилхлорид (0,536 мл, 6,93 ммоль) и смесь перемешивают при комнатной температуре в течение 3 ч. Затем добавляют воду (30 мл) и смесь экстрагируют AcOEt (340 мл). Органический слой промывают рассолом, сушат над Na2SO4 и упаривают досуха. Неочищенный продукт очищают посредством флэш-хроматографии (SiO2, петролейный эфир/AcOEt от 10/0 до 8/2) с получением 330 мг указанного в заголовке соединения. Пример 7. 4-[(3,5-Дихлорпиридин-4-илметил)-(3,4-диметоксифенил)амино]метилбензойная кислота (С 16). Соединение С 11 (350 мг, 0,75 ммоль) растворяют в МеОН (14 мл). Добавляют 1 н. KOH (3 мл, 3 ммоль) и смесь подвергают дефлегмации в течение 1 ч. Растворитель выпаривают, остаток растворяют в воде (15 мл) и обрабатывают 3 н. HCl до pH 1. Затем раствор экстрагируют AcOEt (340 мл), органический слой сушат над Na2SO4 и упаривают досуха. Неочищенный продукт очищают посредством флэшхроматографии (SiO2, AcOEt/петролейный эфир от 1/1 до 1/0) с получением указанного в заголовке соединения в количестве 230 мг. Пример 8. Получение конечного соединения (С) (схема). Синтез (3,5-дихлорпиридин-4-илметил)-(3,4-диметоксифенил)тио-3-илметиламина. Промежуточное соединение В 1 (200 мг, 0,64 ммоль) растворяют в МеОН (10 мл). Добавляют молекулярные сита (4 , 200 мг) и затем тиофен-3-карбальдегид (0,056 мл, 0,64 ммоль). Смесь перемешивают при комнатной температуре в течение 2 ч, затем добавляют NaBH3CN (121 мг, 1,9 ммоль). Добавляют по каплям АсОН до pH 5. Через 48 ч молекулярные сита удаляют фильтрацией и добавляют воду (20 мл). Смесь экстрагируют AcOEt (350 мл), органический слой промывают рассолом, сушат над Na2SO4 и упаривают досуха. Неочищенный продукт очищают посредством флэш-хроматографии (SiO2, петролейный эфир/AcOEt от 9/1 до 7/3) с получением указанного в заголовке соединения в количестве 173 мг. Соединения С 17, С 18 и С 19 получают, следуя той же процедуре синтеза, используя промежуточное соединение В 1 и подходящие альдегиды с использованием подходящих реагентов. Таблица 6 В таблице приняты следующие обозначения:- ЯМР; Фармакологическая активность. Пример 9. Определение in vitro ингибиторной активности PDE-4 в бесклеточном анализе. В качестве источника фермента PDE-4 используют человеческую моноцитарную клеточную линиюU937. Клетки культивируют, собирают и супернатантную фракцию получают, по существу, как описано в Torphy T.J. et al. J. Pharmacol. Exp. Ther. 1992; 263:1195-1205. Активность PDE-4 определяют в клеточных супернатантах с помощью анализа исчезновения cAMP из инкубационных смесей. 50 мкл клеточного супернатанта инкубируют при 30C в течение 30 мин в конечном объеме 200 мкл в присутствии 1,6 мкМ cAMP с тестируемым соединением (50 мкл) или без него. Концентрация тестируемых соединений варьирует от 10-12 до 10-6 М. Реакции останавливают инактивацией нагреванием (2,5 мин при 100C) и остаточный cAMP измеряют с использованием иммуноанализа на основе электрохемилюминесценции (ECL). Результаты выражают как среднее 95%-ные доверительные интервалы молярной концентрации тестируемого соединения, вызывающей 50%-ное ингибирование исчезновения cAMP (IC50). Тестировали соединения C1-C14, и их значения IC50 в бесклеточном анализе оказались в интервале от 27 до 807 нм. Процент ингибирования активности PDE-4 вычисляют, принимая исчезновение cAMP в отсутствие ингибиторов за 100% и исчезновение cAMP в инактивированных нагреванием образцах за 0%. Все значения IC50 протестированных соединений, репрезентативных для данного изобретения, были меньше 0,2 мкМ. Пример 10. Определение in vitro ингибиторной активности PDE-4 в анализе на мононуклеарных клетках периферической крови (РВМС). Анализ, который основан на известной ингибиторной активности, проявляемой ингибиторами PDE4 на индуцированное липополисахаридами (ЛПС) высвобождение фактора-альфа некроза опухолей(TNF-) в мононуклеарных клетках периферической крови (РВМС), осуществляют согласно способу,описанному в Hatzelmann A. et al. J. Pharmacol. Exp. Ther. 2001; 297:267-279 и Draheim R. et al. J. Pharmacol. Exp. Ther. 2004; 308:555-563. Криоконсервированные человеческие РВМС (100 мкл/лунка) инкубируют в 96-луночных планшетах (105 клеток/лунка) в течение 30 мин в присутствии или в отсутствие (50 мкМ) тестируемых соединений, концентрации которых варьировали от 10-12 до 10-6 М. Затем добавляют ЛПС (3 нг/мл). Через 18 ч инкубации при 37C во влажном инкубаторе в атмосфере 95% воздуха и 5% СО 2 собирают культуральную среду и TNF- измеряют в ELISA. Результаты выражают как среднее 95% доверительные интервалы молярной концентрации тестируемого соединения, вызывающей 50%-ное ингибирование ЛПС-индуцированного высвобождения TNF (IC50). Эффекты тестируемых соединений вычисляют как процент ингибирования высвобождения TNF-,принимая ЛПС-индуцированную продукцию TNF- в отсутствие ингибиторного соединения за 100% и фоновую продукцию TNF- клетками РВМС в отсутствие ЛПС за 0%. Сравнительные примеры. Следуя указаниям WO 2005/061458 и WO 2006/135828, авторы изобретения синтезировали сравнительную молекулу, соответствующую соединению 1 по изобретению соответственно, от которого она отличается только отсутствием метиленового мостика между азотом амино и арильной частью, и сравнительную молекулу, аналогичную соединению 5, от которого она отличается отсутствием метиленового мостика между азотом амино и арильной частью, которая представляет собой циклопропилметильную группировку. Оба соединения демонстрируют на РВМС, значения IC50 выше 1000 нм. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение общей формулы (I)R1 и R2 являются разными или одинаковыми и независимо выбраны из группы, состоящей изC1-C4-алкилокси, C3-C7-циклоалкилокси и (C3-C7)циклоалкил-(C1-C3)алкилокси; по меньшей мере один из R1 и R2 представляет собой C1-C4-алкилокси; А представляет собой арильную кольцевую систему, имеющую от 6 до 10 кольцевых атомов, или гетероарильную кольцевую систему, имеющую от 5 до 9 кольцевых атомов, в которой один кольцевой атом представляет собой гетероатом, выбранный из N, S или O, которые возможно замещены одним или более заместителями, независимо выбранными из группы, состоящей из C1-C6-алкила, OR3, где R3 выбран из группы, состоящей из Н, C1-C6-алкила, C3-C7-циклоалкила; атомов галогена, -CN, NO2, NR6R7,где R6 и R7 являются разными или одинаковыми и независимо выбраны из группы, состоящей из Н,C1-C6-алкила, HNSO2R9, где R9 представляет собой C1-C4-алкил, SO2R10, где R10 представляет собойC1-C4-алкил, ОН или NR6R7, где R6 и R7 являются Н, COOR13, где R13 представляет собой Н, C1-C4-алкил,и его фармацевтически приемлемые соли. 2. Соединение по п.1, где А представляет собой возможно замещенный фенил. 3. Соединение по п.1, где А представляет собой возможно замещенное гетероарильное кольцо, выбранное из группы, состоящей из фурана или бензотиофена. 4. Соединение по п.1, выбранное из(3,5-дихлорпиридин-4-илметил)-(3,4 диметоксифенил)-(4-метоксибензил)амин (соединение 13). 8. Фармацевтическая композиция, обладающая ингибиторной активностью в отношении фосфодиэстеразы-4 (PDE-4), содержащая соединение по любому из пп.1-7 в смеси с одним или более фармацевтически приемлемыми носителями и/или эксципиентами. 9. Применение соединения по любому из пп.1-7 в качестве лекарственного средства, обладающего ингибиторной активностью в отношении PDE-4. 10. Применение соединения по любому из пп.1-7 для изготовления лекарственного средства для предупреждения и/или лечения воспалительного заболевания, расстройства или состояния, характеризующегося или ассоциированного с нежелательным воспалительным иммунным ответом или индуцированного или ассоциированного с избыточной секрецией TNF- и PDE-4. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2
МПК / Метки
МПК: A61P 25/28, A61P 25/00, A61P 29/00, A61K 31/44, A61P 25/16, C07D 401/12
Метки: третичного, ингибиторов, амина, качестве, фосфодиэстеразы-4, производные
Код ссылки
<a href="https://eas.patents.su/14-18069-proizvodnye-tretichnogo-amina-v-kachestve-ingibitorov-fosfodiesterazy-4.html" rel="bookmark" title="База патентов Евразийского Союза">Производные третичного амина в качестве ингибиторов фосфодиэстеразы-4</a>
Производные фталазина в качестве ингибиторов фосфодиэстеразы 4

Номер патента: 3702
Опубликовано: 28.08.2003
Авторы: Норчини Габриеле, Пеллачини Франко, Мораццони Габриеле, Гранчини Джанкарло, Наполетано Мауро
МПК: C07D 237/30, A61K 31/502
Метки: фталазина, ингибиторов, фосфодиэстеразы, качестве, производные
Формула / Реферат:
1. Соединение формулы I где ---- представляет одинарную или двойную связь; B представляет метилен; A представляет пиридин, замещенный одним заместителем или большим количеством заместителей; R представляет два атома водорода или C = O группу, когда ---- представляет одинарную связь, или, когда ---- представляет двойную связь, R представляет водород, необязательно замещенный арил или гетероцикл, (C1-8)алкил, (C2-8)алкенил или (C2-8)алкинил,...
Производные 6-фенилпиридил-2-амина, полезные в качестве ингибиторов nos

Номер патента: 2907
Опубликовано: 31.10.2002
Автор: Лоу Джон Адамс III
МПК: A61K 31/4418, A61P 11/06, C07D 213/73...
Метки: полезные, качестве, производные, ингибиторов, 6-фенилпиридил-2-амина
Формула / Реферат:
1. Соединение формулы где R1 и R2 независимо выбраны из водорода, гидрокси, метила и метокси; и G является группой формулы где n равно нулю или единице; Y представляет собой NR3R4, (С1-С6)алкил или аралкил, где арильная группировка указанного аралкила является фенилом или нафтилом, а алкильная группировка является нормальной или разветвленной и содержит от 1 до 6 атомов углерода и где указанный (С1-С6)алкил и арильная группировка указанного...
Новые спиротрициклические производные и их применение в качестве ингибиторов фосфодиэстеразы-7

Номер патента: 6815
Опубликовано: 28.04.2006
Авторы: Дюкро Пьер, Вернье Фабрис, Лортиуа Эдвиг, Бернарделли Патрик
МПК: C07D 239/70, A61P 29/00, A61K 31/527...
Метки: спиротрициклические, производные, новые, качестве, применение, фосфодиэстеразы-7, ингибиторов
Формула / Реферат:
1. Соединение, имеющее следующую формулу (I) где a) X1, Х2, Х3 и Х4 одинаковые или различные и выбраны из C-R1, где R1 выбран из Q1 или низшего алкила, низшего алкенила или низшего алкинила, причем эти группы не замещены или замещены одной или несколькими группами Q2; группы Х5-R5, где X5 выбран из одинарной связи; низшего алкилена, низшего алкенилена или низшего алкинилена, возможно прерванных 1 или 2 гетероатомами, выбранными из О, S, S(=O),...
Производные индазола и их использование в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и продуцирования фактора некроза опухоли (фно)

Номер патента: 2113
Опубликовано: 24.12.2001
Автор: Марфат Энтони
МПК: C07D 401/12, A61K 31/415, A61P 11/06...
Метки: производные, опухоли, продуцирования, индазола, типа, фосфодиэстеразы, фно, фдэ, ингибиторов, использование, фактора, некроза, качестве
Формула / Реферат:
1. Соединение формулы (I) или их фармацевтически приемлемые соли, в которых R является Н, C1-C9 алкилом, -(СН2)m (5-10 членным гетероциклилом), где m равно от 0 до 2, или (Z1)b(Z2)с(С6-С10 арилом), где b и с независимо равны от 0 до 1, Z1 является C1-С6 алкиленом или C2-C8 алкениленом и Z2 является О, S, SO2 или NR12; и где указанные R группы необязательно замещены от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген,...
Производные пирролидина в качестве ингибиторов фосфодиэстеразы, специфичной к циклическому амф

Номер патента: 5686
Опубликовано: 28.04.2005
Авторы: Джоунс Закари С., Шлахтер Стивен, Мартинс Тимоти Дж., Годино Джон Дж., Кесицки Эдвард А., Ньюхаус Брэдли Дж., Фаулер Керри У., Одинго Джошуа, Оливер Эйми, Бэрджесс Лоренс И.
МПК: A61K 31/40, C07D 295/20
Метки: качестве, специфичной, ингибиторов, фосфодиэстеразы, амф, производные, пирролидина, циклическому
Формула / Реферат:
1. Соединение, имеющее формулу где R1 выбран из группы, включающей водород, C1-6алкил, мостиковый алкил, выбранный из группы, включающей норборнил, адамантил, бицикло[2.2.2]октил, бицикло[3.2.1]гептил, бицикло[3.2.1]октил, бицикло[4.1.0]гептил, бицикло[3.1.0]гексил и декагидронафтил, замещенный или незамещенный арил, выбранный из группы включающей фенил, нафтил, бифенил, тетрагидронафтил, инданил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил,...