Новый способ получения каберголина
Номер патента: 10689
Опубликовано: 30.10.2008
Авторы: Галамбош Янош, Деметер Адам, Кашшай Ференцне, Балинт Шандорне, Игначне Сендрей Дьёрдьи, Шебок Ференц, Цибуля Ласло










Формула / Реферат
1. Способ получения каберголина (I)

включающий следующие стадии:
(а) взаимодействие соединения формулы (XIII)

где R1 представляет собой С1-4алкильную группу, в присутствии катализатора
(i) с соединением формулы (XIV)
![]()
где R2 представляет собой необязательно замещенную линейную или разветвленную C1-6алкильную группу и X представляет атом брома или хлора, или
(ii) с соединением формулы (XV)
![]()
где R2 представляет собой группу, имеющую значения, указанные выше для формулы (XIV);
(b) взаимодействие полученного карбаматного производного формулы (XVI)

где R1 и R2 представляют собой группы, имеющие значения, указанные выше, с 3-(диметиламино)пропиламином в присутствии катализатора;
(с) взаимодействие полученного эрголин-8b-карбоксамида формулы (XVII)

где R2 представляет собой группу, имеющую значения, указанные выше, с этилизоцианатом в присутствии лиганда(ов) и соли металла группы Ib и IIb в качестве катализатора,
(d) взаимодействие полученного содержащего защитные группы производного N-ацилмочевины формулы (XVIII)

где R2 представляет собой группу, имеющую значения, указанные выше, с сильной водной неорганической кислотой;
(е) взаимодействие полученного вторичного амина формулы (XIX)

с электрофильным производным аллилового спирта в присутствии палладий- или никельсодержащего катализатора и, необязательно, в присутствии лиганда(ов) с образованием каберголина (I).
2. Способ по п.1, где R1 представляет собой метил и R2 представляет собой трет-бутил.
3. Способ по любому из пп.1 или 2, где стадию (а) осуществляют при температуре от 0 до 50шС в присутствии катализатора 4-диметиламинопиридина в углеводородном галогенсодержащем растворителе.
4. Способ по любому из пп.1 или 2, где стадию (b) осуществляют при температуре от 50 до 70шС в растворителе С1-4алифатическом спирте в присутствии катализатора 2-гидроксипиридина.
5. Способ по любому из пп.1 или 2, где стадию (с) осуществляют в углеводородном галогенсодержащем растворителе в присутствии катализаторов хлорида меди(I), и/или хлорида меди(II), и/или бромида меди(I), и/или иодида меди(I) и лиганда трифенилфосфина или три-п-толилфосфина при температуре от 30 до 50шС.
6. Способ по любому из пп.1 или 2, где стадию (d) осуществляют при температуре от 40 до 80шС в водной хлористо-водородной кислоте.
7. Способ по любому из пп.1 или 2, где на стадии (е) электрофильное производное аллилового спирта представляет собой аллилацетат, катализатором является тетракис(трифенилфосфин)палладий(0) и взаимодействие осуществляют в ароматическом углеводородном растворителе при температуре от 20 до 50шС.
8. Соединение формулы (XVI)

где R1 представляет С1-4алкильную группу и R2 представляет необязательно замещенную C1-6алкильную группу.
9. Соединение по п.8, где R1 представляет собой метил и R2 представляет собой трет-бутил.
10. Соединение формулы (XVII)

где R2 представляет необязательно замещенную C1-6алкильную группу.
11. Соединение по п.10, где R2 представляет собой трет-бутил.
12. Соединение формулы (XVIII)

где R2 представляет необязательно замещенную C1-6алкильную группу.
13. Соединение по п.12, где R2 представляет собой трет-бутил.
14. Соединение формулы (XIX)

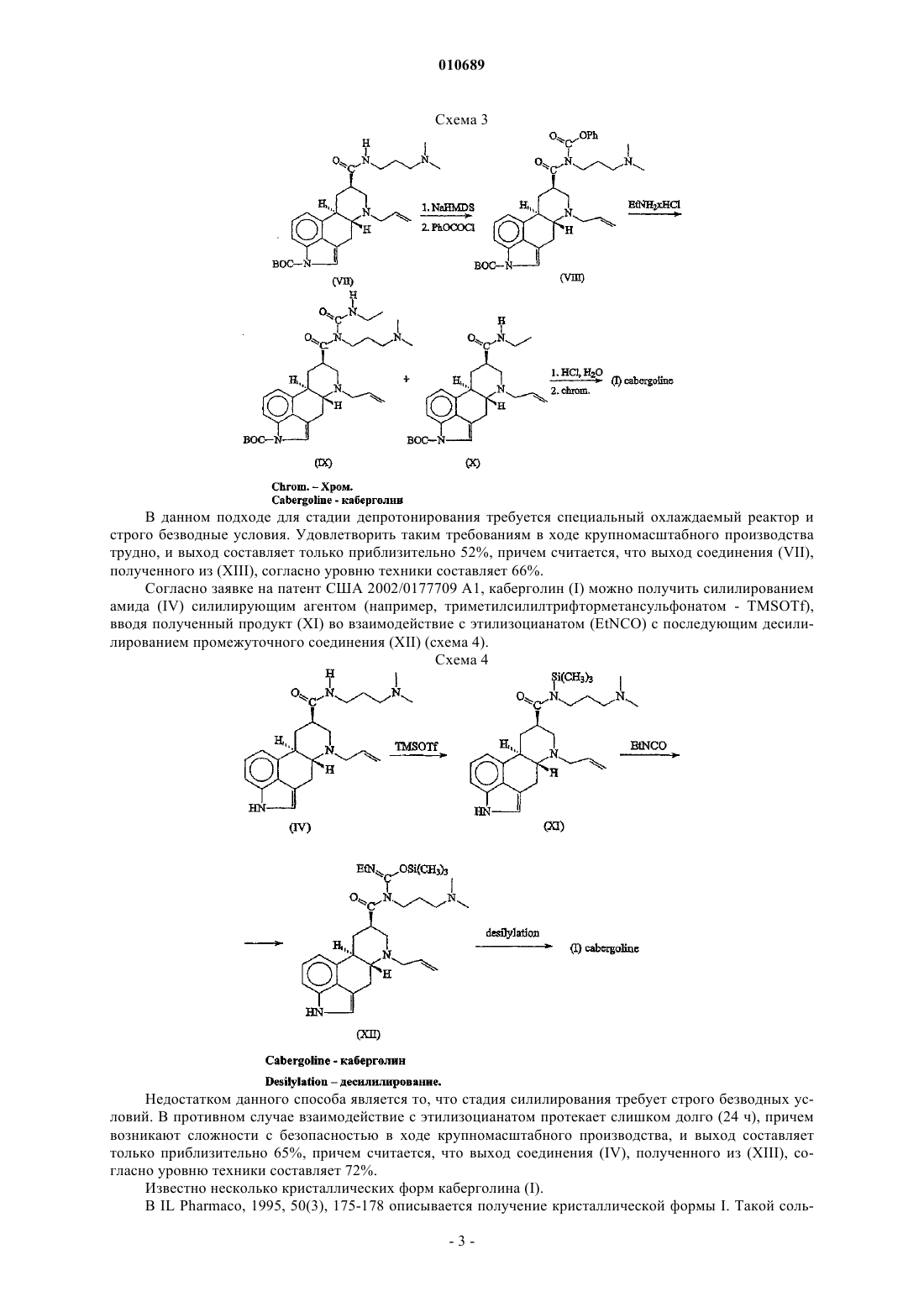
15. Полиморфная аморфная форма каберголина (I), характеризующаяся инфракрасным (ИК) спектром, показанным на фиг. 3, и дополнительно характеризующаяся картиной дифракции рентгеновских лучей (XRD) в порошке, показанной на фиг. 1.
16. Способ получения полиморфной аморфной формы каберголина (I), где очищенный хроматографически каберголин (I) в форме масла растворяют в подходящем органическом растворителе и из полученного раствора несколько раз частично удаляют растворитель в вакууме при температуре от 0 до 30шС до тех пор, пока не получат не маслянистый, а твердый продукт.
17. Способ по п.16, где растворителем является ацетон, метилацетат или дихлорметан.
Текст
010689 Область техники, к которой относится изобретение Данное изобретение относится к новому способу получения каберголина формулы (I), к новым промежуточным соединениям, используемым в указанном способе, к полиморфной аморфной форме каберголина (I) и ее получению. Уровень техники 6-Аллил-N-[3-(диметиламино)пропил]-N-[(этиламино)карбонил]эрголин-8-карбоксамид - международное непатентованное название каберголин - формулы (I) является мощным допаминовым агонистом и применим в качестве лекарственного средства против болезни Паркинсона и в качестве ингибитора пролактина (Eur. J. Med. Chem., 1989, 24,421-426 и патент США 5382669). Каберголин (I) получали, прежде всего, согласно патенту США 4526892 взаимодействием 6 аллилэрголин-8-карбоновой кислоты (II) с 1-[3-(диметиламино)пропил]-3-этилкарбодиимидом (EDC) В таком случае получают оба регизомера (I) и (III), и выход выделенного каберголина (I) составляет только приблизительно 21% вследствие трудностей выделения, причем считается, что выход соединения(II), полученного из (XIII), согласно уровню техники составляет 70%. В Eur. J. Med. Chem., 1989, 24, 421-426, описывается другой способ получения каберголина (I), который основан на прямом взаимодействии 6-аллил-N-[3-(диметиламино)пропил]эрголин-8 карбоксамида (IV) с этилизоцианатом (EtNCO) (схема 2). Так как такое взаимодействие ведет к равновесию, требуется использование большого избытка этилизоцианата (до 40 экв.) для разумной конверсии, и реакцию необходимо проводить при температуре выше 100 С в толуоле в течение нескольких часов. Использование большого количества токсичного этилизоцианата в жестких условиях реакции представляет серьезную опасность в случае получения каберголина (I) в крупном масштабе. Кроме того, конверсия в (I) неполная, и происходит конкурентное ацилирование индольного азота с образованием соединений (V) и (VI). Такая побочная реакция осложняет очистку продукта и снижает выход, который составляет только приблизительно 58%, причем считается,что выход соединения (IV), полученного из (XIII), согласно уровню техники составляет 72%. Способ, описанный в патенте США 5382669 и в Syn. Lett., 1995, 605-606, показал, что катализ солями меди в присутствии фосфиновых лигандов позволяет осуществлять взаимодействие с этилзоцианатом при комнатной температуре только с 3 экв. этилзоцианата. Однако, несмотря на умеренные условия реакции, конверсия и соотношение каберголина (I) и побочных продуктов (V и VI) не сильно отличаются от термического взаимодействия без катализатора. Выход составляет только 48 и 57%, причем считается,что выход соединения (IV), полученного из (XIII), согласно уровню техники составляет 72%. В J. Org. Chem., 2002, 67, 7147-7150 описывается способ получения каберголина (I) без использования этилизоцианата, что решает также проблему полноты ацилирования индольного азота. Первой стадией является защита индольного азота амида (IV) предпочтительно в виде третбутилкарбамата (VII). Удлинение амидной боковой цепи совершается депротонированием соединения (VII) гексаметилдисилазидом натрия (NaHMDS) с последующим захватом аниона фенилхлорформиатом (PhOCOCl) с образованием фенилкарбамата (VIII). Взаимодействие соединения (VIII) с гидрохлоридом этиламина (EtNH2HCl) дает ВОС-каберголин(IX), но также образуется этиламид (X). Удаление защитной группы осуществляют из смеси (IX) и (X) 1N соляной кислотой. Затем очищенный каберголин (I) извлекают подщелачиванием с последующей хроматографией на диоксиде кремния (схема 3). В данном подходе для стадии депротонирования требуется специальный охлаждаемый реактор и строго безводные условия. Удовлетворить таким требованиям в ходе крупномасштабного производства трудно, и выход составляет только приблизительно 52%, причем считается, что выход соединения (VII),полученного из (XIII), согласно уровню техники составляет 66%. Согласно заявке на патент США 2002/0177709 А 1, каберголин (I) можно получить силилированием амида (IV) силилирующим агентом (например, триметилсилилтрифторметансульфонатом - TMSOTf),вводя полученный продукт (XI) во взаимодействие с этилизоцианатом (EtNCO) с последующим десилилированием промежуточного соединения (XII) (схема 4). Схема 4 Недостатком данного способа является то, что стадия силилирования требует строго безводных условий. В противном случае взаимодействие с этилизоцианатом протекает слишком долго (24 ч), причем возникают сложности с безопасностью в ходе крупномасштабного производства, и выход составляет только приблизительно 65%, причем считается, что выход соединения (IV), полученного из (XIII), согласно уровню техники составляет 72%. Известно несколько кристаллических форм каберголина (I). В IL Pharmaco, 1995, 50(3), 175-178 описывается получение кристаллической формы I. Такой соль-3 010689 ватированный безводный продукт кристаллизуется из диэтилового эфира. В заявке на патент WO 01/70740 А 1 описывается новый способ получения кристаллической формыI из новой кристаллической формы V. Форму V, представляющую собой сольват с толуолом, получают из смеси очищенного каберголина (I) с толуолом и диэтиловым эфиром длительным сложным способом при низкой температуре реакции и с выходом только 45%. Кристаллическую форму I получают сушкой формы V в вакууме. В заявке на патент WO 01/72746 А 1 описывается получение кристаллической формы VII из кристаллической формы I. Согласно такому способу суспензию формы I в н-гептане или 1,4-диоксане перемешивают в течение 48 ч и затем суспензию фильтруют и получают кристаллическую форму VII. Выход составляет 45,2%. В заявке на патент WO 01/72747 А 1 описывается кристаллическая форма II и способ ее получения с выходом приблизительно 70% перемешиванием каберголина (I) в течение нескольких суток в органическом растворителе (например, диэтиловом эфире) при низкой температуре. Раскрытие изобретения Неожиданно обнаружилось, что получение каберголина (I) способом через новые промежуточные соединения по настоящему изобретению коммерчески более выгодно, чем раскрытыми ранее известными способами, из-за высокого выхода (приблизительно 78%), более умеренных условий взаимодействия и более короткого времени взаимодействия. Другим преимуществом применения указанных промежуточных соединений является то, что они имеют высокую гидрофобность по сравнению с известными промежуточными соединениями, содержащими две основные функциональные группы, так что их очистка хроматографией с нормальной фазой (при необходимости) является высокоэффективной из-за большего различия во времени удерживания между ними и побочными продуктами. Настоящее изобретение относится к способу получения каберголина (I) из эрголин-8-карбоновой кислоты и C1-4 алкилэфиров через новые промежуточные соединения и полиморфной аморфной формы каберголина (I). Способ включает защиту функциональной вторичной аминогруппы и индольного азота C1-4 алкилэфиров эрголин-8-карбоновой кислоты в виде карбаматных производных, амидирование полученного соединения с защитными группами 3-(диметиламино)пропиламином, взаимодействие амида с этилизоцианантом, удаление защитных групп и взаимодействие полученного вторичного амина без защитных групп с электрофильным производным аллилового спирта с образованием каберголина (I). Настоящее изобретение также относится к новым промежуточным соединениям, используемым в указанном способе. Изобретение также относится к новой аморфной форме каберголина (I) и ее получению. Осуществление изобретения Изобретение относится к способу получения каберголина (I), включающему следующие стадии:(а) взаимодействие соединения формулы (XIII), где R1 представляет C1-4 алкильную группу, в присутствии катализатора(i) с соединением формулы (XIV) где R2 представляет необязательно замещенную линейную или разветвленную C1-6 алкильную группу, иX представляет атом брома или хлора, или(ii) с соединением формулы (XV) где R2 представляет собой группу, имеющую значения, указанные выше для формулы (XIV);(b) взаимодействие полученного карбаматного производного формулы (XVI), где R1 и R2 представляют собой группы, имеющие значения, указанные выше, с 3-(диметиламино)пропиламином (DMAPA) в присутствии катализатора;R2 представляет собой группу, имеющую значения, указанные выше, с этилизоцианатом (EtNCO) в присутствии лиганда(ов) и соли металла группы Ib и IIb в качестве катализатора,(d) взаимодействие полученного содержащего защитные группы производного N-ацилмочевины формулы (XVIII), где R2 представляет собой группу, имеющую значения, указанные выше, с сильной водной неорганической кислотой (водн./кислота);(e) взаимодействие полученного вторичного амина формулы (XIX) с электрофильным производным аллилового спирта в присутствии палладий- или никельсодержащего катализатора и, необязательно, в присутствии лиганда(ов) с образованием каберголина (I). Процедура осуществления взаимодействий показана на схеме 5.(XIII) с соединением формулы (XIV) или (XV). Исходные вещества эфиры эрголин-8-карбоновой кислоты формулы (XIII) можно получить любым способом, известным в технике, например описанным в патенте US 4166182 или в Collect. Czech.Chem. Commun., 1983, 48(5), 1483-1489. Способы, служащие для защиты как вторичных аминогрупп, так и индольного азота, хорошо известны в технике. Так, указанные функциональные группы можно защитить, например, в виде Nалкильных, N-арильных, N-силильных, N-сульфонильных производных или в виде карбаматов. Был проверен ряд защитных групп, и обнаружено, что лучше всего защищать исходные вещества формулы (XIII) в виде карбаматов формулы (XVI). Соединения формул (XIV) и (XV) коммерчески доступны или их можно получить хорошо известными процедурами или процедурами, аналогичными описанным в литературе. Соединения формул (XIV) и (XV) можно использовать в 2-10-кратном молярном количестве, предпочтительно 2-5-кратном молярном количестве, относительно количества эфира эрголин-8-карбоновой кислоты формулы (XIII). Взаимодействие можно осуществить при температуре от -50 С до температуры образования флегмы реакционной смеси в подходящем апротонном растворителе в присутствии органического или неорганического основания как катализатора. Предпочтительно реакционную стадию (а) осуществляют при температуре от 0 до 50 С в присутствии катализатора 4-диметиламинопиридина в углеводородном галогенсодержащем растворителе. Предпочтительно R1 представляет собой метил и R2 представляет собой трет-бутил. Стадия (b) способа относится к взаимодействию карбаматного производного формулы (XVI), полученного на стадии (а), с 3-(диметиламино)пропиламином. Амидирование 3-(диметиламино)пропиламином производных метилового эфира эрголин-8 карбоновой кислоты, содержащих основной азот в положении 6 эрголинового скелета, можно осуществить известными способами. Взаимодействие можно осуществить с большим избытком амина(i) кипячением реакционной смеси с обратным холодильником в течение 10-12 ч без растворителя(приблизительно при 135 С) в присутствии катализатора уксусной кислоты;(ii) нагреванием реагентов в течение 18 ч при 100 С в этиленгликоле в качестве растворителя с каталитическим количеством 2-гидроксипиридина. Найдено, что отсутствие основного азота в D-цикле эрголинового скелета оказывает существенное действие на время реакции и температуру, требуемые при реакции амидирования.-5 010689 Так, взаимодействие карбаматных производных формулы (XIV) с 3-(диметиламино)пропиламином,осуществляемое известными способами, можно завершить при температуре ниже 70 С за несколько часов. Из-за пониженной температуры полученное производное эрголин-8-карбоновой кислоты формулы(XVII) имеет высокую чистоту и выход свыше 95%. Амидирование можно осуществить при температуре от 40 до 70 С. Катализаторы ускоряют реакцию амидирования. Все известные катализаторы можно использовать,при условии, что они не оказывают вредного воздействия на трет-бутоксикарбонильные защитные группы. Примерами катализаторов являются органические и неорганические основания, такие как гидроксиды или карбонаты щелочных металлов, алкоголяты щелочных или щелочно-земельных металлов, пиридин или его производные, третичные амины, и т.д.; соли, такие как хлорид аммония, ацетат меди(II),хлорид магния; и другие катализаторы, такие как трибромид бора, амиды диметилалюминия, смешанные амиды олова (II) или их смеси. Амидирование можно осуществлять в отсутствие растворителя или в присутствии подходящего растворителя. Предпочтительно стадию (b) осуществляют при температуре от 50 до 70 С в растворителе C1-4 алифатическом спирте в присутствии катализатора 2-гидроксипиридина. Полученный амид формулы (XVII) можно использовать на следующей стадии после выделения из реакционной массы, следуя обычным процедурам, или можно подвергнуть воздействию на следующей стадии без выделения. Стадия (с) способа относится к взаимодействию производного эрголин-8-карбоновой кислоты формулы (XVII), полученного на стадии (b), с этилизоцианатом (EtNCO). Этилизоцианат можно использовать в 1-4-кратном молярном количестве, предпочтительно 2-3 кратном молярном количестве, относительно количества амида (XVII). Необязательно, взаимодействие (XVII) с этилизоцианатом можно ускорить катализом металлами в присутствии координационного(ых) соединения(ий). Подходящими металлсодержащими катализаторами являются соли металлов групп Ib и IIb, предпочтительно соли меди(I) и меди(II). Наиболее предпочтительными солями являются хлорид меди(I), хлорид меди(II), бромид меди(I) и иодид меди(I). Лиганды в координационном(ых) соединении(ях) с металлами Ib и IIb предпочтительно содержат атомы фосфора, азота и/или кислорода. Примерами лигандов являются триарилфосфины, третичные амины, нитрилы и соединения типа простых эфиров. Предпочтительными лигандами являются триарилфосфины, наиболее предпочтительными лигандами являются трифенилфосфин и три-п-толилфосфин. Взаимодействие осуществляют в присутствии подходящего апротонного органического растворителя при температуре от 0 С до температуры образования флегмы реакционной смеси. Предпочтительно стадию (с) осуществляют в углеводородном галогенсодержащем растворителе в присутствии катализаторов хлорида меди(I), и/или хлорида меди(II), и/или бромида меди(I), и/или иодида меди(I) и лиганда трифенилфосфина или три-п-толилфосфина при температуре от 30 до 50 С. Продукт реакции можно выделить последующими обычными процедурами. Так как полученные содержащие защитные группы производные N-ацилмочевины формулы (XVIII) имеют высокую гидрофобность (по сравнению с известными промежуточными соединениями, содержащими две основные функциональные группы), их очистка хроматографией с нормальной фазой (при необходимости) является высокоэффективной из-за увеличенного различия во времени удерживания между нужными соединениями и побочными продуктами. Стадия (d) способа относится к взаимодействию содержащего защитные группы производного Nацилмочевины формулы (XVIII), полученного на стадии (с), с сильной водной неорганической кислотой с образованием N-[3-(диметиламино)пропил]-N-[(этиламино)карбонил]эрголин-8-карбоксамида(XIX). Способы отщепления защитных групп карбаматного типа от основного азота и индольного азота хорошо известны в технике, но не каждый способ подходит для обеих целей. В других случаях с некоторыми обычно используемыми способами (например, с муравьиной кислотой или трихлоруксусной кислотой) не получают значительной конверсии, в то время как другие способы (например, с трифторуксусной кислотой или трифторметансульфоновой кислотой) не выдерживает субстрат (XVIII) и/или продукт реакции (XIX). Обнаружено, что защитные группы карбаматного типа соединений (XVIII) можно легко удалить при прямом взаимодействии с сильными водными неорганическими кислотами. В данном случае термин"сильная кислота" обозначает кислоту с величиной pK в воде менее 2. Примерами сильных неорганических кислот являются хлористо-водородная кислота, бромисто-водородная кислота, серная кислота. Удаление защитной группы можно осуществить при температурах от 0 С до температуры образования флегмы реакционной смеси. Из-за наличия основных азотсодержащих функциональных групп с основным азотом как субстрат(XVIII), так и продукт реакции (XIX) полностью растворяются в водной кислой среде, так что органический растворитель для осуществления реакции удаления защитной группы не требуется. Предпочтительно стадию (d) осуществляют при температуре от 40 до 80 С в водной хлористо-6 010689 водородной кислоте. Полученное соединение формулы (XIX) можно использовать на следующей стадии после выделения из реакционной смеси, следуя обычным процедурам, или можно подвергнуть взаимодействию на следующей стадии без выделения. Стадия (е) способа относится к взаимодействию полученного 6-деаллилкаберголина формулы(XIX) с электрофильным производным аллилового спирта с образованием каберголина (I). Из-за образования значительного количества производных четвертичных аммониевых оснований аллилирование функциональной вторичной аминогруппы в (XIX) нельзя осуществить обычно используемыми аллилирующими агентами (например, аллилгалогенидами, аллиларилсульфонатами, аллилалкилсульфонатами). Однако четвертичных побочных продуктов не наблюдается, когда продукт реакции(I) получают реакцией нуклеофильного аллилзамещения. Так, взаимодействие 6-деаллилкаберголина(XIX) с электрофильным производным аллилового спирта в органическом растворителе в присутствии палладиевого или никелевого катализатора и лиганда(ов) дает каберголин (I) высокой чистоты. Примерами электрофильного производного аллилового спирта являются аллилкарбоксилаты, такие как аллилацетат или аллилбензоат, и аллилфениловый эфир. Каталитическая система может быть гомогенной или гетерогенной, предпочтительно каталитическая система является гомогенной. Примерами гетерогенного катализатора являются палладий на активированном угле или полистироле при наличии фосфорсодержащих лигандов, палладий, лигированный полистиролфосфинатом или фосфинированным диоксидом кремния. Примерами гомогенного катализатора являются тетракис(трифенилфосфин)палладий(0), тетракис(трифенилфосфин)никель, бис(циклоокта-1,5 диен)никель, [1,4-бис(дифенилфосфино)бутан]никель, димер хлорида аллилпалладия и цис,цис,цис 1,2,3,4-тетракис(дифенилфосфинометил)циклопентан. Взаимодействие можно осуществить в подходящем апротонном органическом растворителе при температуре от 0 С до температуры образования флегмы реакционной смеси. Предпочтительно на стадии (е) электрофильное производное аллилового спирта представляет собой аллилацетат, катализатором является тетракис(трифенилфосфин)палладий(0), и взаимодействие осуществляют в ароматических углеводородных растворителях при температуре от 20 до 50 С. Продукт реакции можно выделить и очистить, следуя обычным процедурам. Каберголин (I) можно превратить в фармацевтически приемлемые соли. Затем каберголин (I) или его фармацевтически приемлемые соли можно ввести в состав с фармацевтически приемлемым носителем или разбавителем и получить фармацевтическую композицию. Промежуточные соединения формул (XVI), (XVII), (XVIII) и (XIX) являются новыми соединениями. Неожиданно обнаружилось, что если очищенный хроматографически маслянистый каберголин (I) растворить в подходящем органическом растворителе и из полученного раствора растворитель частично удалять несколько раз в вакууме при температуре от 0 до 30 С до остатка, который является не маслянистым, а твердым продуктом, получают новую полиморфную аморфную форму каберголина (I), которая идентифицируется аналитическими методами рентгеновской дифракции (XRD), дифференциальной сканирующей калориметрии (DSC) и ИК. Химическая чистота полиморфной аморфной формы каберголина (I), полученной вышеуказанным способом, составляет 99,5% (ВЭЖХ). Преимуществом описанного способа получения каберголина (I) является более короткое технологическое время и высокий выход, который практически является количественным. Предпочтительно используемым растворителем является ацетон, метилацетат или дихлорметан. Согласно аналитическим и фармакологическим исследованиям авторов изобретения полиморфная аморфная форма каберголина (I) является весьма устойчивой, и скорость растворения и абсорбционные свойства аморфоной формы каберголина (I) являются благоприятными. Поэтому применение аморфоной формы каберголина (I) в фармацевтических композициях является более выгодным по сравнению с известными кристаллическими формами. Далее изобретение иллюстрируется приведенными примерами, не являющимися ограничительными. Пример 1. Синтез метилового эфира 1,6-ди(трет-бутоксикарбонил)эрголин-8-карбоновой кислоты(XVI, R1 = метил, R2 = трет-бутил). К раствору 13,05 г (48,2 ммоль) метилового эфира эрголин-8-карбоновой кислоты (XIII, R1 = метил) в 400 мл дихлорметана последовательно добавляют 20 мл триэтиламина, 1,0 г 4-диметиламинопиридина и 42,1 г (193,1 ммоль) ди-трет-бутилдикарбоната, и реакционную смесь перемешивают при 40 С в течение 5 ч. Смесь охлаждают до температуры окружающей среды и промывают 3100 мл раствора хлорида натрия. Органический слой сушат над безводным сульфатом натрия и концентрируют в вакууме. Кристаллизация из гексана дает 21,6 г (95,2%) названного в заголовке соединения. Н NMR (CDCl3, TMS, 500 MHz)1.46 (s, 9H, N(6)-COOC(CH3)3); 1.66 (s, 9H, N(1)-СООС(CH3)3); 188 (td, 1H, J=13.1Hz, 9.5Hz, H-9); 2.81 (ddd, 1H, J=13.1Hz, 8.1Hz, 3.7Hz3 H-9); 2.86-2.94 (m, 1H, H-8); 3.06 (ddd, 1H, J=14.9Hz, 11.6Hz, 2.1Hz, H-4); 3.14 (td, 1H, J=13.2Hz, 3.6Hz, H-10); 3.31 (dd, 1H, J=15.1Hz,4.0Hz, H-4); 3.65 (td, 1H, J=11.4Hz, 4.0Hz, H-5); 3.70 (dd, 1H, J=14.2Hz, 5.6Hz, H-7); 3.73 (s, 3H,COOCH3); 3.92 (dd, 1H, J=14.2Hz, 4.5Hz, H-7); 7.03 (d, 1H, J=7.2Hz, H-12); 7.20-7.30 (m, 2H, H-2, H-13); 7.80 (br d, 1H, H-14). Здесь и далее: NMR - ЯМР, TMS - TMC, MHz - МГц, Hz - Гц,Пример 2. Синтез N-[3-(диметиламино)пропил]-1,6-ди(трет-бутоксикарбонил)эрголин-8-карбоксамида (XVII, R2 = трет-бутил). Смесь 14,7 г (31,24 ммоль) метилового эфира 1,6-ди(трет-бутоксикарбонил)эрголин-8-карбоновой кислоты (XVI, R1 = метил, R2 = трет-бутил), 58,8 мл 3-(диметиламино)пропиламина, 29,4 мл 2-пропанола и 3,68 г 2-гидроксипиридина перемешивают при 70 С в течение 8 ч. Реакционную смесь охлаждают до температуры окружающей среды и добавляют 230 мл дихлорметана. Полученную смесь промывают 3120 мл раствора хлорида натрия. Органический слой сушат над безводным сульфатом натрия. Осушенный раствор или передают на последующую стадию без выделения продукта реакции, или его концентрируют в вакууме и получают 16,4 г (97,1%) названного в заголовке соединения. 1(ddd, 1H, J=13.4Hz, 8.2 Hz, 3.1Hz, H-9); 3.05 (ddd, 1H, J=14.2Hz, 11.8Hz, 2.1Hz, H-4); 3.13 (td, 1H,J=13.3Hz, 3.6Hz, Н-10); 3.28 (dd, 1H, J=14.8Hz, 3.9Hz, H-4); 3.28-3.42 (m, 2H, CONHCH2CH2CH2N(CH3)2); 3.60 (dd, 1H, J=14.8Hz, 6.0Hz, H-7); 3.65 (td, 1H, J=11.4Hz, 3.9Hz, H-5); 3.92 (dd, 1H, J=14.8Hz, 3.7Hz, H7); 7.04 (d, 1H, J=7.4Hz, H-12); 7.22-7.32 (m, 2H, H-2, H-13); 7.62 (t, 1H, J=5.0Hz,CONHCH2CH2CH2N(CH3)2); 7.79 (br d, 1H, H-14). Пример 3. Синтез N-[3-(диметиламино)пропил]-N-[(этиламино)карбонил]-1,6-ди(трет-бутоксикарбонил)эрголин-8-карбоксамида (XVIII, R2 = трет-бутил). К раствору 15,5 г (28,67 ммоль) N-[3-(диметиламино)пропил]-1,6-ди(трет-бутоксикарбонил)эрголин-8-карбоксамида (XVII, R2 = трет-бутил) в 350 мл дихлорметана последовательно добавляют 0,8 г трифенилфосфина, 0,3 г хлорида меди(I) и 6,8 мл (86 ммоль) этилизоцианата и реакционную смесь перемешивают при 35 С в течение 4 ч. Реакционную смесь концентрируют в вакууме, продукт реакции очищают на слое диоксида кремния и получают 16,9 г (96,4%) названного в заголовке соединения. 1(XIX). К 12,7 г (20,76 ммоль) N-[3-(диметиламино)пропил]-N-[(этиламино)карбонил]-1,6-ди(третбутоксикарбонил)эрголин-8-карбоксамида (XVIII, R2 = трет-бутил) добавляют 230 мл 4 М соляной кислоты и реакционную смесь перемешивают при 35 С в течение 2 ч. Реакционную смесь охлаждают до температуры окружающей среды, добавляют 200 мл дихлорметана и доводят pH до 11 концентрированным водным раствором аммиака. Органический слой отделяют и водный слой экстрагируют 260 мл дихлорметана. Объединенные органические слои сушат над безводным сульфатом натрия. Осушенный раствор или подвергают воздействию на последующей стадии без выделения продукта реакции, или его концентрируют в вакууме и получают 8,1 г (94,8%) названного в заголовке соединения. 1 Н NMR (CDCl3, TMS, 500 MHz)1.19 (t, 3H, J=7.3Hz, CONHCH2CH3); 1.80-1.92 (m, 3H, H-9,CONCH2CH2CH2N(CH3)2); 2.27 (s, 6H, CONCH2CH2CH2N(CH3)2); 2.39 (t, 2H, J=6.7Hz,CONHCH2CH2CH2N(CH3)2); 2.60 (br s, 1H, N(6)H); 2.68-2.90 (m, 4H, H-4, H-9, H-5, H-10); 2.97 (t, 1H,J=12.3Hz, H-7); 3.07 (dd, 1H, J=14.5Hz, 4.0Hz, H-4); 3.26-3.42 (m, 4H, H-7, H-8, CONHCH2CH3); 3.743.92 (m, 2H, CONCH2CH2CH2N(CH3)2); 6.84-6.90 (m, 2H, H-2, H-12); 7.10-7.20 (m, 2H, H-13, H-14); 8.16 (s,1H, N(1)H); 9.45 (br t, 1H, CONHCH2CH3). Пример 5. Синтез 6-аллил-N-[3-(диметиламино)пропил]-N-[(этиламино)карбонил]эрголин-8 карбоксамида (I) (каберголина). К суспензии 9,0 г (21,87 ммоль) N-[3-(диметиламино)пропил]-N-[(этиламино)карбонил]эрголин-8 карбоксамида (XIX) в 250 мл толуола добавляют 0,5 г тетракис(трифенилфосфин)палладия(0) и 5 мл аллилацетата и реакционную смесь перемешивают при температуре окружающей среды в течение 2 ч. Полученную смесь промывают 100 мл воды. Органический слой сушат над безводным сульфатом натрия.-8 010689 Осушенный раствор концентрируют в вакууме, продукт реакции очищают на слое диоксида кремния и получают 9,1 г (92,3%) названного в заголовке соединения. 1(d, 1H, J=7.0Hz, H-12); 6.97 (s, 1H, H-2); 7.01 (t, 1H, J=7.5Hz, H-13); 7.13 (d, 1H, J=8.0Hz, H-14); 9.04 (t, 1H,J=5.0Hz, CONHCH2CH3); 10.60 (s, 1H,N(1)H). Пример 6. Получение полиморфной аморфной формы каберголина (I).a) Растворяют 10 г очищенного хроматографически каберголина (I) в форме масла в 50 мл ацетона. Раствор концентрируют в вакууме при 25-30 С до приблизительно 15 г. Полученный маслянистый остаток растворяют в 40 мл ацетона и раствор концентрируют в вакууме при 25-30 С до приблизительно 12 г. Полученный маслянистый остаток снова растворяют в 30 мл ацетона и раствор концентрируют в вакууме при 25-30 С до 10 г. Полученный твердый каберголин (I) сушат в вакууме при 25-30 С до свободного от растворителя состояния и получают 9,8 г (98%) названного в заголовке соединения.b) Осуществляют то же самое, что в примере 6 а, но с использованием метилацетата в качестве растворителя, и получают 9,85 г (98,5%) названного в заголовке соединения. с) Осуществляют то же самое, что в примере 6 а, но с использованием дихлорметана в качестве растворителя, и получают 9,82 г (98,2%) названного в заголовке соединения. Согласно аналитическим исследованиям методами XRD, DSC и ИК кристаллическая форма продукта является аморфной. Дифракция рентгеновских лучей (XRD) в порошке.XRD осуществляют с использованием порошкового дифрактометра Philips PW 1840 Compact. Дифференциальная сканирующая калориметрия (DSC). Исследования методом DSC проводят на приборе Mettler Toledo DSC 821. Фиг. 1 показывает спектры XRD аморфной формы каберголина (I). Фиг. 2 показывает спектры DSC аморфной формы каберголина (I). Фиг. 3 показывает ИК-спектры аморфной формы каберголина (I). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения каберголина (I)(а) взаимодействие соединения формулы (XIII)-9 010689 где R2 представляет собой необязательно замещенную линейную или разветвленную C1-6 алкильную группу и X представляет атом брома или хлора, или(ii) с соединением формулы (XV) где R2 представляет собой группу, имеющую значения, указанные выше для формулы (XIV);(b) взаимодействие полученного карбаматного производного формулы (XVI) 3 где R2 представляет собой группу, имеющую значения, указанные выше, с этилизоцианатом в присутствии лиганда(ов) и соли металла группы Ib и IIb в качестве катализатора,(d) взаимодействие полученного содержащего защитные группы производного N-ацилмочевины формулы (XVIII) где R2 представляет собой группу, имеющую значения, указанные выше, с сильной водной неорганической кислотой;(е) взаимодействие полученного вторичного амина формулы (XIX)- 10010689 с электрофильным производным аллилового спирта в присутствии палладий- или никельсодержащего катализатора и, необязательно, в присутствии лиганда(ов) с образованием каберголина (I). 2. Способ по п.1, где R1 представляет собой метил и R2 представляет собой трет-бутил. 3. Способ по любому из пп.1 или 2, где стадию (а) осуществляют при температуре от 0 до 50 С в присутствии катализатора 4-диметиламинопиридина в углеводородном галогенсодержащем растворителе. 4. Способ по любому из пп.1 или 2, где стадию (b) осуществляют при температуре от 50 до 70 С в растворителе С 1-4 алифатическом спирте в присутствии катализатора 2-гидроксипиридина. 5. Способ по любому из пп.1 или 2, где стадию (с) осуществляют в углеводородном галогенсодержащем растворителе в присутствии катализаторов хлорида меди(I), и/или хлорида меди(II), и/или бромида меди(I), и/или иодида меди(I) и лиганда трифенилфосфина или три-п-толилфосфина при температуре от 30 до 50 С. 6. Способ по любому из пп.1 или 2, где стадию (d) осуществляют при температуре от 40 до 80 С в водной хлористо-водородной кислоте. 7. Способ по любому из пп.1 или 2, где на стадии (е) электрофильное производное аллилового спирта представляет собой аллилацетат, катализатором является тетракис(трифенилфосфин)палладий(0) и взаимодействие осуществляют в ароматическом углеводородном растворителе при температуре от 20 до 50 С. 8. Соединение формулы (XVI) где R1 представляет С 1-4 алкильную группу и R2 представляет необязательно замещенную C1-6 алкильную группу. 9. Соединение по п.8, где R1 представляет собой метил и R2 представляет собой трет-бутил. 10. Соединение формулы (XVII) где R2 представляет необязательно замещенную C1-6 алкильную группу. 11. Соединение по п.10, где R2 представляет собой трет-бутил. 12. Соединение формулы (XVIII) где R2 представляет необязательно замещенную C1-6 алкильную группу. 15. Полиморфная аморфная форма каберголина (I), характеризующаяся инфракрасным (ИК) спектром, показанным на фиг. 3, и дополнительно характеризующаяся картиной дифракции рентгеновских лучей (XRD) в порошке, показанной на фиг. 1. 16. Способ получения полиморфной аморфной формы каберголина (I), где очищенный хроматографически каберголин (I) в форме масла растворяют в подходящем органическом растворителе и из полученного раствора несколько раз частично удаляют растворитель в вакууме при температуре от 0 до 30 С до тех пор, пока не получат не маслянистый, а твердый продукт. 17. Способ по п.16, где растворителем является ацетон, метилацетат или дихлорметан.
МПК / Метки
МПК: C07D 457/06
Метки: кабэрголина, способ, новый, получения
Код ссылки
<a href="https://eas.patents.su/14-10689-novyjj-sposob-polucheniya-kabergolina.html" rel="bookmark" title="База патентов Евразийского Союза">Новый способ получения каберголина</a>
Способ получения кристаллической формы i каберголина

Номер патента: 5928
Опубликовано: 25.08.2005
Авторы: Палланца Джанфранко, Рамелла Джулиано, Мадженес Стефания, Унгари Марио, Томази Аттилио
МПК: C07D 457/04
Метки: формы, кристаллической, кабэрголина, получения, способ
Формула / Реферат:
1. Способ получения каберголина формы I, который включает кристаллизацию продукта из смеси толуола/диэтилового эфира, содержащей неочищенный каберголин, с последующим выделением и сушкой полученных кристаллов. 2. Способ по п.1, в котором кристаллизация включает растворение неочищенного каберголина в смеси толуола/диэтилового эфира, охлаждение полученного раствора, сбор полученного сольвата формы V каберголина, имеющего рентгенограмму порошковой...
Кристаллическая форма ii каберголина

Номер патента: 4376
Опубликовано: 29.04.2004
Авторы: Мадженес Стефания, Пандольфи Марко, Рамелла Джулиано, Томази Аттилио, Унгари Марио
МПК: C07D 457/06
Метки: кабэрголина, кристаллическая, форма
Формула / Реферат:
1. Кристаллическая форма II каберголина. 2. Кристаллическая форма II каберголина по п.1, которая является безводной, несольватированной и имеет процентную чистоту выше чем 85%. 3. Кристаллическая форма II каберголина по п.1, которая является безводной, несольватированной и имеет процентную чистоту выше чем 98%. 4. Кристаллическая форма II каберголина, имеющая порошковую рентгенограмму согласно фиг. 1. 5. Фармацевтическая композиция, содержащая...
Комбинированное лечение заболевания цнс, особенно болезни паркинсона, путём совместного введения каберголина и прамипексола

Номер патента: 4474
Опубликовано: 29.04.2004
Автор: Гомес-Мансилья Балтасар
МПК: A61P 25/00, A61K 31/48
Метки: лечение, прамипексола, комбинированное, путём, введения, паркинсона, особенно, заболевания, совместного, цнс, кабэрголина, болезни
Формула / Реферат:
1. Способ лечения пациентов, страдающих заболеванием ЦНС, включающий введение пациенту комбинации каберголина и прамипексола или их фармакологически приемлемых солей. 2. Способ по п.1 для лечения пациентов, страдающих болезнью Паркинсона. 3. Способ по п.2, при котором первоначальная доза каберголина вводится пациенту в количестве от 0,5 до 1 мг в день на пациента и увеличивается с недельными интервалами до терапевтической дозы 2, 4, 6, 8 или 10...
Новый способ получения изоиндолина

Номер патента: 3515
Опубликовано: 26.06.2003
Авторы: Фуже Клод, Лекове Жан-Пьер, Совие Жан-Клод
МПК: C07D 209/44
Метки: изоиндолина, новый, получения, способ
Формула / Реферат:
1. Способ синтеза изоиндолина, отличающийся тем, что раствор фталонитрила в тетрагидрофуране, в смеси тетрагидрофуран/вода или в диметоксиэтане подвергают воздействию водорода под давлением 100-180 бар при температуре 30-100шC в присутствии 5%-ной Pt/C. 2. Способ по п.1, отличающийся тем, что растворителем служит тетрагидрофуран. 3. Способ по п.1, отличающийся тем, что растворителем служит смесь тетрагидрофуран/вода, в которой содержание воды не...
Новый способ получения алендроновой кислоты

Номер патента: 3861
Опубликовано: 30.10.2003
Авторы: Лифшиц Ревитал, Лидор-Хадас Рами
МПК: C07F 9/38
Метки: алендроновой, получения, способ, новый, кислоты
Формула / Реферат:
1. Способ получения алендроновой кислоты, включающий стадии а) взаимодействия соединения формулы I с H3PO3 где R - имидогруппа и R1 выбирают из группы, которая состоит из атома хлора, брома, йода или фтора, гидрокси-, амино-, -OR2 или -OC(O)R2-группы, где R2 - C1-C12алкил-, C1-C12циклоалкил- или C1-C12арилгруппа, б) взаимодействия продукта стадии (а) с агентом, снимающим защитную группу, и в) выделения алендроновой кислоты. 2. Способ по п.1,...
Предыдущий патент: Одностадийный способ получения жевательной резинки
Следующий патент: Трёхзамещённые производные арилов и гетероарилов в качестве модуляторов метаболизма и профилактика и лечение связанных с ними нарушений
Случайный патент: Лекарственное средство и его применение для лечения воспалительных и/или обструктивных заболеваний дыхательных путей