Лекарственное средство, активное в отношении невропатической боли
Номер патента: 19405
Опубликовано: 31.03.2014
Авторы: Поленцани Лоренцо, Гульельмотти Анджело, Фурлотти Гвидо, Каццолла Никола, Ализи Мария Алессандра











Формула / Реферат
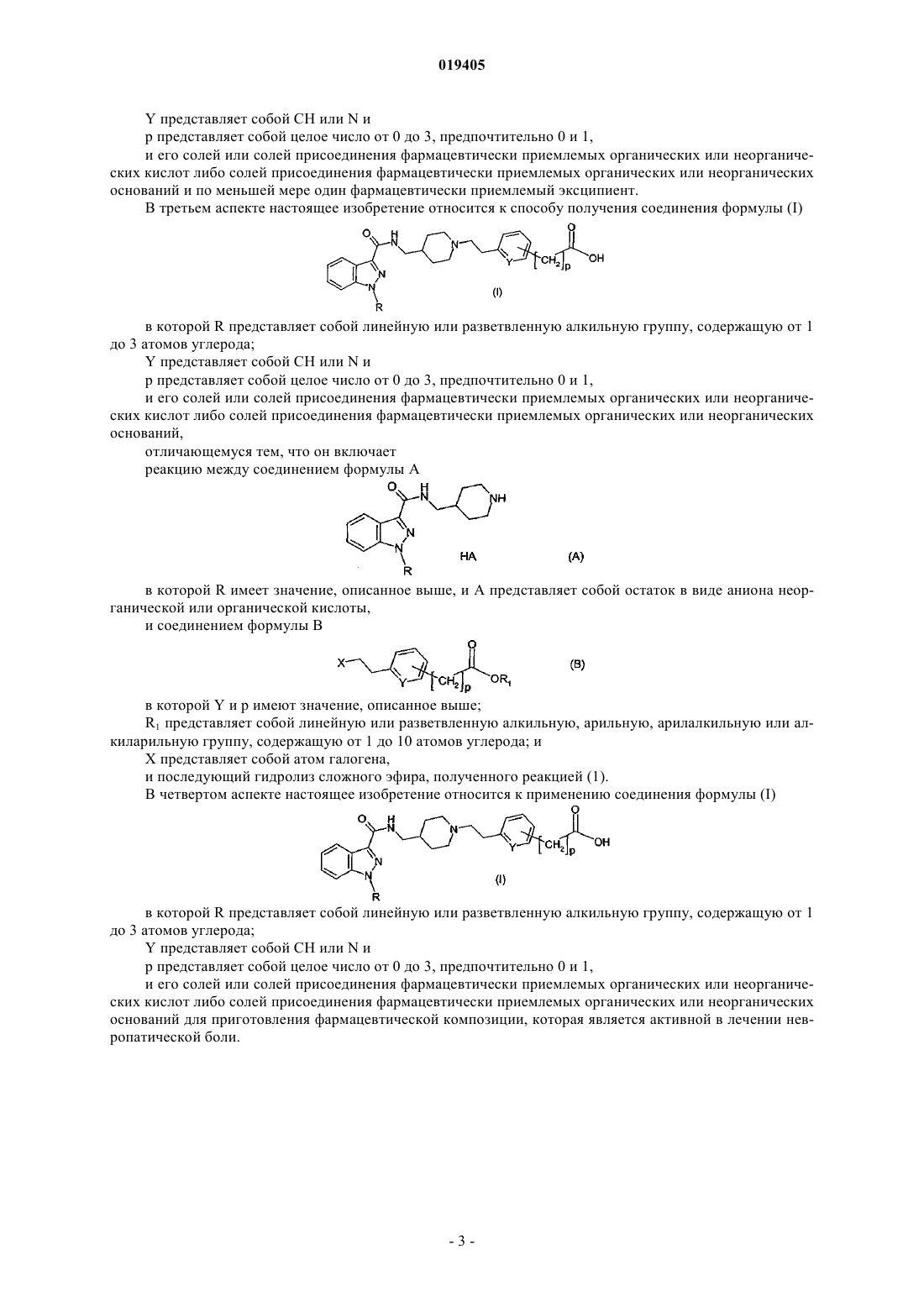
1. Соединение формулы (I)
в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;
Y представляет собой СН или N и
р представляет собой целое число от 0 до 3,
и его соли либо соли присоединения фармацевтически приемлемых органических или неорганических кислот или соли присоединения фармацевтически приемлемых органических или неорганических оснований.
2. Соединение по п.1, где R представляет собой линейную или разветвленную алкильную группу, содержащую от 2 до 3 атомов углерода.
3. Соединение по п.1, где Y представляет собой СН.
4. Соединение по п.1, где р представляет собой целое число от 0 до 1.
5. Соединение по п.1, где R, Y, р и положение группы
представлены в следующей таблице:
6. Соединение по любому одному из предшествующих пунктов, где упомянутую фармацевтически приемлемую органическую или неорганическую кислоту выбирают из группы, включающей щавелевую кислоту, малеиновую кислоту, метансульфокислоту, паратолуолсульфокислоту, янтарную кислоту, лимонную кислоту, винную кислоту, молочную кислоту, хлористо-водородную кислоту, фосфорную кислоту, серную кислоту, аспарагиновую кислоту и глутаминовую кислоту.
7. Соединение по любому одному из пп.1-5, где упомянутое фармацевтически приемлемое органическое или неорганическое основание выбирают из группы, включающей метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, пропиламин, дипропиламин, трипропиламин, этилендиамин, моноэтаноламин, диэтаноламин, триэтаноламин, гуанидин, морфолин, пиперидин, пирролидин, пиперазин, 1-бутилпиперидин, 1-этил-2-метилпиперидин, N-метилпиперазин, 1,4-диметилпиперазин, N-бензилфенилэтиламин, N-метилглюкозамин, трис-(гидроксиметил)аминометан, аргинин, лизин, аммиак, гидроксид натрия, гидроксид кальция, гидроксид калия, гидроксид алюминия, гидроксид железа, гидроксид магния и гидроксид цинка.
8. Фармацевтическая композиция, содержащая эффективное количество соединения формулы (I)
в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;
Y представляет собой СН или N и
р представляет собой целое число от 0 до 3,
и его солей либо солей присоединения фармацевтически приемлемых органических или неорганических кислот или солей присоединения фармацевтически приемлемых органических или неорганических оснований и по меньшей мере один фармацевтически приемлемый эксципиент.
9. Фармацевтическая композиция по п.8, в которой упомянутый по меньшей мере один фармацевтически приемлемый эксципиент выбирают из группы, включающей разделительные средства, связывающие вещества, разрыхлители, наполнители, разбавители, красители, флюидизирующие вещества, регуляторы сыпучести, скользящие вещества, консервирующие вещества, стабилизаторы, увлажнители, абсорбенты, поверхностно-активные вещества, буферные растворы, соли для регулирования осмотического давления, эмульгаторы, ароматизирующие вещества и подслащивающие вещества.
10. Фармацевтическая композиция по п.8, в которой соединение формулы (I) определено в соответствии с любым одним из пп.1-7.
11. Фармацевтическая композиция по любому одному из пп.8-10, где фармацевтическую композицию выбирают из группы, включающей таблетки, капсулы, таблетки, покрытые оболочкой, гранулы, растворы и сиропы для перорального введения, антисептические пластыри, растворы, пасты, кремы и мази для трансдермального введения, суппозитории для ректального введения, стерильные растворы для инъекционного или аэрозольного введения, либо в лекарственной форме с мгновенным высвобождением, либо в лекарственной форме с замедленным высвобождением.
12. Фармацевтическая композиция по любому одному из пп.8-11, где упомянутая фармацевтическая композиция содержит количество соединения формулы (I) или его соли присоединения кислоты либо соли присоединения основания, которое надежно обеспечивает уровень введения от 0,001 до 100 мг/кг/день соединения формулы (I), представленного в свободной форме.
13. Фармацевтическая композиция по п.12, в которой упомянутое количество будет надежно обеспечивать уровень введения от 0,05 до 50 мг/кг/день.
14. Фармацевтическая композиция по п.12, в которой упомянутое количество будет надежно обеспечивать уровень введения от 0,1 до 10 мг/кг/день.
15. Способ получения соединения формулы (I)
в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;
Y представляет собой СН или N и
р представляет собой целое число от 0 до 3,
и его солей или солей присоединения фармацевтически приемлемых органических или неорганических кислот либо солей присоединения фармацевтически приемлемых органических или неорганических оснований,
отличающийся тем, что он включает:
(1) реакцию между соединением формулы (А)
в которой R имеет значение, описанное выше, и А представляет собой остаток в виде аниона неорганической или органической кислоты,
и соединением формулы (В)
в которой Y и р имеют значение, описанное выше;
R1 представляет собой линейную или разветвленную алкильную, арильную, арилалкильную или алкиларильную группу, содержащую от 1 до 10 атомов углерода; и
X представляет собой атом галогена, и
(2) последующий гидролиз сложного эфира, полученного реакцией (1).
16. Способ получения по п.15, в котором упомянутый остаток А выбирают из группы, включающей галогенид, фосфат, сульфат, нитрат и органический карбоксилат.
17. Способ получения по п.16, в котором упомянутый остаток А выбирают из группы, включающей хлорид, бромид и йодид.
18. Способ получения по п.15, в котором упомянутый остаток X выбирают из группы, включающей хлорид, бромид и йодид.
19. Способ получения по п.15, в котором упомянутую группу R1 выбирают из группы, включающей линейный или разветвленный алкил, содержащий 1-6 атомов углерода, арил, арилалкил и алкиларил, содержащий 7-8 атомов углерода.
20. Способ получения по любому одному из пп.15-19, в котором упомянутую реакцию (1) проводят в органическом растворителе, выбранном из группы, включающей полярные апротонные растворители.
21. Способ получения по п.20, в котором упомянутый органический растворитель выбирают из группы, включающей ацетон, метилэтилкетон, тетрагидрофуран, диметилформамид, диметилсульфоксид, диоксан и ацетонитрил.
22. Способ получения по любому одному из пп.15-21, в котором упомянутую реакцию (1) проводят в присутствии основного органического или неорганического соединения.
23. Способ получения по любому одному из пп.15-22, в котором упомянутую реакцию (1) проводят в присутствии активирующего вещества, выбранного из группы, включающей йодид калия, йодид цезия, йодид тетрабутиламмония и йодид триметилфениламмония.
24. Применение соединения формулы (I)
в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;
Y представляет собой СН или N и
р представляет собой целое число от 0 до 3,
и его солей или солей присоединения фармацевтически приемлемых органических или неорганических кислот либо солей присоединения фармацевтически приемлемых органических или неорганических оснований для приготовления фармацевтической композиции для лечения невропатической боли.
25. Применение соединения по п.24 для приготовления фармацевтической композиции для лечения невропатической боли, вызванной диабетами, раком, иммунодефицитом, травмой, ишемией, рассеянным склерозом, ишиалгией, тригеминальной невралгией и постгерпетическим синдромом.
Текст
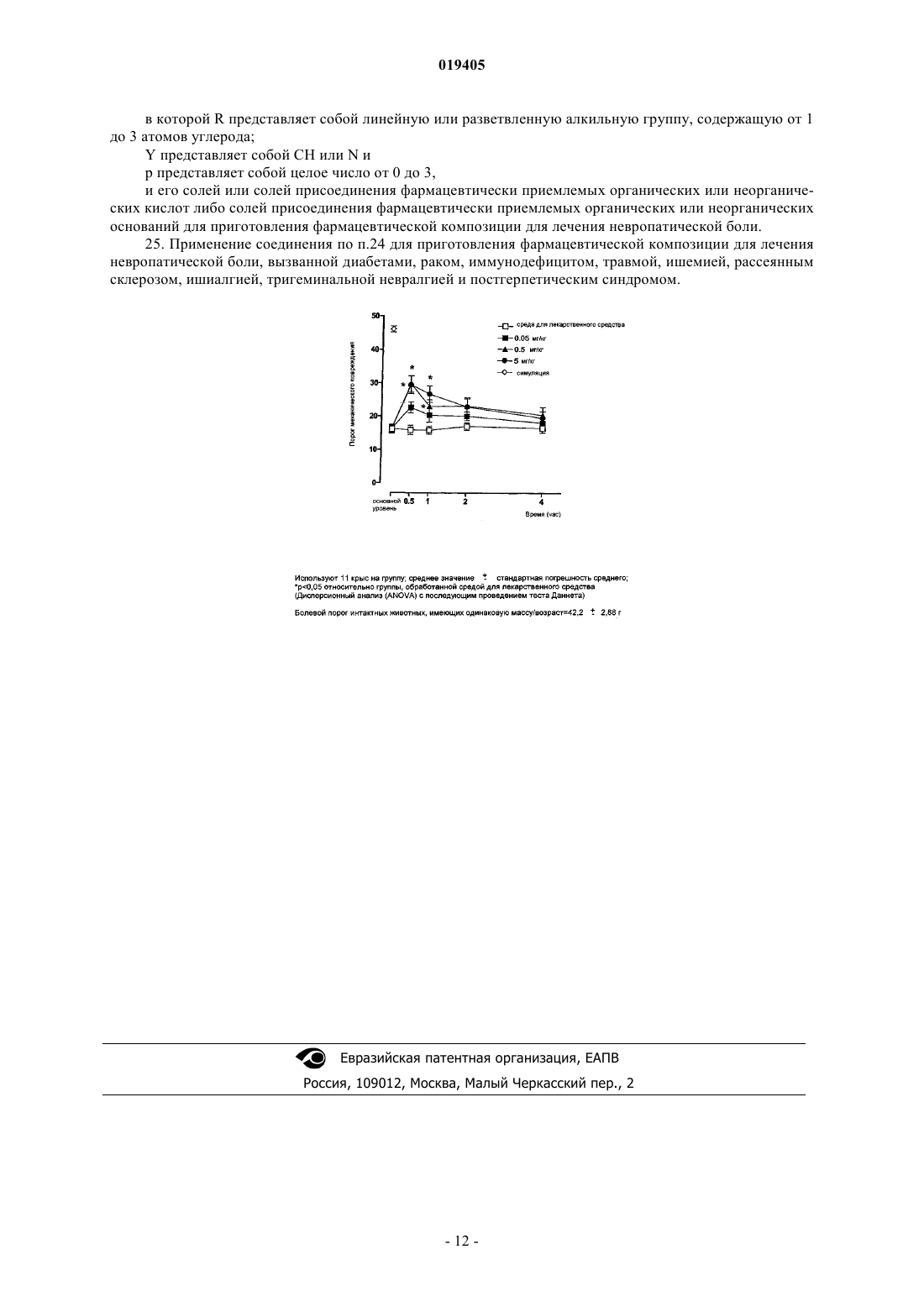
Изобретение относится к соединению формулы (I), в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода, Y представляет собой СН или N и р представляет собой целое число от 0 до 3 и предпочтительно 0 и 1,и к его солям, либо солям присоединения фармацевтически приемлемых органических или неорганических кислот, либо солям присоединения фармацевтически приемлемых органических или неорганических оснований. Изобретение также относится к способу получения соединения формулы (I) и к фармацевтической композиции, содержащей это соединение. Изобретение также относится к применению индазола для приготовления фармацевтической композиции, которая является активной в лечении невропатической боли.(71)(73) Заявитель и патентовладелец: АЦЬЕНДЕ КИМИКЕ РЬЮНИТЕ АНДЖЕЛИНИ ФРАНЧЕСКО Область техники, к которой относится изобретение Настоящее изобретение относится к новому индазолу, к способу получения его и к фармацевтической композиции, содержащей его. Изобретение также относится к применению индазола для получения фармацевтической композиции, которая является активной в лечении невропатической боли. Известный уровень техники Хроническая боль свидетельствует о ряде патологий, от которых страдает, в среднем, приблизительно 10-20% взрослого населения. Хроническая боль, как правило, сопутствует клиническим состояниям, свойственным хроническим и/или дегенеративным нарушениям. Хроническая боль отличается от острой боли в основном длительностью. Острая боль имеет продолжительность в несколько дней или недель, и она связана с восстановлением после события, которое вызвало эту боль (травма, ожоги, чрезмерное напряжение, хирургические и зуболечебные вмешательства и т.п.). С другой стороны, хроническая боль проявляется в течение месяцев и даже лет, вызывая мышечное напряжение, ограниченную подвижность, усталость, потерю аппетита и апатию. Характерными примерами патологий, охарактеризованных хронической болью, являются ревматоидный артрит, остеоартрит, фибромиалгия, невропатии и т.д. [Ashbum M.A., Staats P.S. Management ofchronic pain, Lancet, 1999, 353: 1865-69]. Хроническая боль, в частности невропатическая боль, часто является изнуряющей и вызывающей слабость и является причиной потери работоспособности и причиной плохого качества жизни. В связи с этим также наступает социально-экономический ущерб. Аналгетические лекарственные средства, используемые в настоящий момент в лечении невропатической боли, включают нестероидные противовоспалительные лекарственные средства (NSAID), антидепрессанты, опиоидные аналгетики и противосудорожные средства [Woolf C.J., Mannion R.J. Neuropathic pain: aetiology, symptoms, mechanism, and management, Lancet, 1999, 353: 1959-1964]. Однако общеизвестно, что хроническую боль, в частности невропатическую боль, трудно лечить лекарственными средствами, доступными в настоящий момент. Как следствие, разработка новых лекарственных средств всегда была одной из основных задач фармацевтической промышленности. Кроме того, несмотря на многочисленные исследовательские усилия, направленные на выявление подходящего аналгетического соединения, существует значительный ряд пациентов, чье болезненное состояние по-прежнему не имеет подходящего лечения [Scholz J., Woolf C.J. Can we conquer pain NatNeusci, 2002, 5: 1062-76]. Некоторые лекарственные средства, активные в отношении серотонинергической системы, известны по их способности контролировать хроническую боль, в частности невропатическую боль. Заявки на патент ЕР-А-0975623 и WO 98/46589 относятся к многочисленным соединениям формулы (1) включая те, у которых R6 представлен арилом, возможно замещенным гидроксильной или галогенной группой. В соответствии с вышеупомянутыми документами соединения формулы (А) имеют сродство в отношении 5 НТ 4-рецепторов и действуют в качестве частичных агонистов или антагонистов серотонина, было предложено использовать их в лечении расстройств моторики желудочно-кишечного тракта,недержания мочи, аритмии сердца и расстройств центральной нервной системы, таких как нарушения памяти и тревога. Заявки на патент WO 2005/013989 и ЕР 1646387 относятся к применению производных индазола для приготовления фармацевтической композиции, которая является активной в лечении невропатической боли, где производные индазола представлены формулой (2) ниже в которой X может представлять собой N или СН, в том случае, когда X представляет собой N, R является водородом, а в том случае, когда X представляет собой CH, R является атомом водорода, гидроксильной группой, алкилом с линейной или разветвленной цепью, содержащей от 1 до 3 атомов углерода, алкоксигруппой с линейной или разветвленной цепью, содержащей от 1 до 3 атомов углерода, или атомом галогена. Заявка на патент WO 2004/101548 относится к применению некоторых производных индазола для приготовления фармацевтической композиции, которая является активной в лечении хронической боли. Некоторые из производных индазола, раскрытые в заявке на патент WO 2004/101548, представлены формулой (3) ниже где Ra представляет собой Н, NH2C(О), CH3C(O)NH, CH3SO2, CH3SO2NH, линейный или разветвленный C1-С 3-алкил, линейную или разветвленную C1-C3-алкоксигруппу или галоген;Rb представляет собой Н, линейный или разветвленный C1-C6-алкил, арил(С 1-С 3)алкил, необязательно замещенный одним или двумя атомами галогена, C1-С 3-алкильной группой или C1-C3 алкоксигруппой;Rc представляет собой гидроксигруппу, аминогруппу, ди(С 1-С 3)алкиламиногруппу, три(C1 С 3)алкиламмониометил, нитрогруппу, трифторметил, нитрил, CH3C(O)NH, CH3SO2NH, CH3SO2, R'RNSO2, где R' и R являются Н, или линейным или разветвленным C1-C6-алкилом;Rd представляет собой Н, гидроксигруппу, аминогруппу, ди(C1-С 3)алкиламиногруппу, три(C1C3)алкиламмониометил, нитрогруппу, трифторметил, нитрил, CH3C(O)NH, CH3SO2NH, CH3SO2, R'RNSO2, где R' и R имеют значения, установленные выше, с условием, что в том случае, когда Ra и Rd являются, оба, водородом, a Rb является изопропилом, Rc не является гидроксигруппой. К сожалению, лекарственные средства, активные в отношении серотонинергической системы и которые в текущий момент используют, проявляют побочное и отрицательное действие, которое имеет место вследствие низкой селективности в отношении других серотонинергических рецепторов, что часто способствует приостановке лечения или сведению на нет лечения в случае некоторых типов пациентов,которые, помимо наличия хронической боли, имеют сопутствующие патологические состояния некоторых органов, таких как сердечно-сосудистая система. В сущности, взаимодействие с серотонинергической системой и даже в большей степени с конкретными субрецепторами, такими как 5HT1B или 5HT2A, может приводить к возникновению отрицательных воздействий на сердечно-сосудистую систему, включая воздействия на ритм сердца. Кроме того, были обнаружены взаимодействия с ионным каналом hERG (ген специфических калиевых каналов сердца), которые могут быть ответственными за отрицательные побочные реакции в сердечно-сосудистой системе. Описание изобретения Неожиданно было обнаружено, что в том случае, когда группа R формулы (2) представлена карбоксильной или алкенилкарбоксильной группой, соединения формулы (В) сохраняют свою активность в отношении невропатической боли с увеличением сродства к серотонинергическому 5 НТ 4-рецептору и повышением селективности в отношении других серотонинергических рецепторов и по отношению к каналу hERG. Таким образом, настоящее изобретение относится к соединению формулы (I) в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;Y представляет собой СН или N и р представляет собой целое число от 0 до 3 и предпочтительно 0 и 1,и к его солям или к солям присоединения фармацевтически приемлемых органических или неорганических кислот либо к солям присоединения фармацевтически приемлемых органических или неорганических оснований. Во втором аспекте настоящее изобретение относится к фармацевтической композиции, содержащей эффективное количество соединения формулы (I) в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;Y представляет собой CH или N и р представляет собой целое число от 0 до 3, предпочтительно 0 и 1,и его солей или солей присоединения фармацевтически приемлемых органических или неорганических кислот либо солей присоединения фармацевтически приемлемых органических или неорганических оснований и по меньшей мере один фармацевтически приемлемый эксципиент. В третьем аспекте настоящее изобретение относится к способу получения соединения формулы (I) в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;Y представляет собой СН или N и р представляет собой целое число от 0 до 3, предпочтительно 0 и 1,и его солей или солей присоединения фармацевтически приемлемых органических или неорганических кислот либо солей присоединения фармацевтически приемлемых органических или неорганических оснований,отличающемуся тем, что он включает реакцию между соединением формулы А в которой R имеет значение, описанное выше, и А представляет собой остаток в виде аниона неорганической или органической кислоты,и соединением формулы ВR1 представляет собой линейную или разветвленную алкильную, арильную, арилалкильную или алкиларильную группу, содержащую от 1 до 10 атомов углерода; иX представляет собой атом галогена,и последующий гидролиз сложного эфира, полученного реакцией (1). В четвертом аспекте настоящее изобретение относится к применению соединения формулы (I) в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;Y представляет собой СН или N и р представляет собой целое число от 0 до 3, предпочтительно 0 и 1,и его солей или солей присоединения фармацевтически приемлемых органических или неорганических кислот либо солей присоединения фармацевтически приемлемых органических или неорганических оснований для приготовления фармацевтической композиции, которая является активной в лечении невропатической боли. Примеры соединений, представленных вышеупомянутой формулой (I), проиллюстрированы в следующей табл. 1. Колонка "Положение" указывает положение карбоксильной или алкенилкарбоксильной группы в ариле относительно углерода, связанного с N-этилпиперидиногруппой. Таблица 1 Примеры фармацевтически приемлемых органических и неорганических кислот представляют собой щавелевую кислоту, малеиновую кислоту, метансульфокислоту, паратолуолсульфокислоту, янтарную кислоту, лимонную кислоту, винную кислоту, молочную кислоту, хлористо-водородную кислоту,фосфорную кислоту, серную кислоту. Аминокислоты, такие как аспарагиновая кислота и глутаминовая кислота, также могут быть использованы в качестве органических кислот. Примеры фармацевтически приемлемых органических и неорганических оснований представляют собой моно-, ди- и триалкиламины, например метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, пропиламин, дипропиламин, трипропиламин, этилендиамин, моно-, ди- и триалканоламины, например моноэтаноламин, диэтаноламин и триэтаноламин; гуанидин, морфолин, пиперидин, пирролидин, пиперазин, 1-бутилпиперидин, 1-этил-2-метилпиперидин, N-метилпиперазин, 1,4 диметилпиперазин, N-бензилфенилэтиламин, N-метилглюкозамин, трис-(гидроксиметил)аминометан,аммиак, гидроксид натрия, гидроксид кальция, гидроксид калия, гидроксид алюминия, гидроксид железа, гидроксид магния и гидроксид цинка. Аминокислоты, такие как аргинин и лизин, также могут быть использованы в качестве органических оснований. Примеры патологий, охарактеризованных невропатической болью, представляют собой диабеты,рак, иммунодефицит, травму, ишемию, множественный склероз, ишиалгию, тригеминальную невралгию и постгерпетический синдром. Предпочтительно фармацевтические композиции настоящего изобретения получают в форме подходящих лекарственных форм, включающих эффективную дозу по меньшей мере одного соединения (I) или его соли либо соли присоединения фармацевтически приемлемой органической или неорганической кислоты или соли присоединения фармацевтически приемлемого органического и неорганического основания и по меньшей мере один фармацевтически приемлемый эксципиент. Термин "фармацевтически приемлемый эксципиент" предназначен для обозначения без каких-либо особых ограничений материала, который подходит для получения фармацевтической композиции, которая должна быть введена живому существу. Такие материалы, известные в данной области, представляют собой, например, разделительные средства, связывающие вещества, разрыхлители, наполнители, разбавители, красители, флюидизирующие вещества, регуляторы сыпучести (вещества, способствующие скольжению), скользящие вещества,консервирующие вещества, стабилизаторы, увлажнители, абсорбенты, поверхностно-активные вещества,буферные растворы, соли для регулирования осмотического давления, эмульгаторы, ароматизирующие вещества и подслащивающие вещества. Полезными примерами фармацевтически приемлемых эксципиентов являются сахара, такие как лактоза, глюкоза или сахароза, крахмалы, такие как кукурузный крахмал и картофельный крахмал, целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы, трагакантовая камедь, ржаной солод, желатин, тальк, масло какао, парафины, масла, такие как арахисовое масло, хлопковое масло, подсолнечное масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло, гликоли, такие как пропиленгликоль, полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль, сложные эфиры, такие как этилолеат и этиллаурат, агар-агар, буферные препараты,такие как гидроксид магния и гидроксид алюминия, альгиновая кислота, вода, изотонические растворы,этанол, буферные растворы, сложные полиэфиры, поликарбонаты, полиангидриды и т.п. Примерами подходящих лекарственных форм являются таблетки, капсулы, таблетки, покрытые оболочкой, гранулы, растворы и сиропы для перорального введения; антисептические пластыри, растворы, пасты, кремы и мази для трансдермального введения; суппозитории для ректального введения и стерильные растворы для инъекционного или аэрозольного введения. Другие подходящие лекарственные формы представляют собой формы с замедленным высвобождением или формы на основе липосом либо для перорального, либо для инъекционного пути введения. При необходимости особых видов терапии фармацевтическая композиция настоящего изобретения может содержать другие фармакологически активные ингредиенты, одновременное введение которых является полезным. Количество соединения (I) или его соли присоединения кислоты либо его соли присоединения основания в фармацевтической композиции настоящего изобретения может варьироваться в пределах широкого диапазона в зависимости от известных факторов, например типа патологии, которой сопутствует невропатическая боль, которая должна быть вылечена, серьезности заболевания, массы пациента, лекарственной формы, выбранного пути введения, числа введений за сутки и эффективности выбранного соединения формулы (I). Тем не менее, оптимальное количество может быть легко определено обычным способом специалистом в данной области. Обычно количество соединения (I) или его соли присоединения кислоты либо соли присоединения основания в фармацевтической композиции настоящего изобретения берут таким, чтобы оно гарантированно обеспечивало уровень введения от 0,001 до 100 мг/кг/день соединения (I), находящегося в форме основания. Предпочтительно уровень введения будет находиться между 0,05 и 50 мг/кг/день и даже более предпочтительно между 0,1 и 10 мг/кг/день. Лекарственные формы фармацевтической композиции настоящего изобретения могут быть приготовлены в соответствии с методиками, хорошо известными химикам-фармацевтам, включая смешение,гранулирование, прессование, растворение, стерилизацию и т.п. В способе получения настоящего изобретения остаток А предпочтительно представлен галогенидом(таким как хлорид, бромид или йодид), фосфатом (таким как пирофосфат, монофосфат, дифосфат или трифосфат), сульфатом, нитратом или органическим карбоксилатом (таким как ацетат, бензоат, лактат и т.п.). Более предпочтительно остаток А представлен галогенидом, в частности хлоридом. В способе получения настоящего изобретения остаток X предпочтительно представлен галогенидом, более предпочтительно хлоридом, бромидом или йодидом. Предпочтительный остаток X представляет собой бромид. В способе получения настоящего изобретения группа, представленная посредством R1, предпочтительно является линейным или разветвленным алкилом, содержащим 1-6 атомов углерода, арилом, арилалкилом или алкиларилом, содержащим 7-8 атомов углерода. Группа R1 также может содержать обычные органические заместители, такие как галогенидная группа, нитрогруппа, гидроксигруппа и т.п. Группу R1 предпочтительно выбирают из метила, этила, изопропила, фенила и бензила. Реакцию (1) способа получения настоящего изобретения предпочтительно проводят в органическом растворителе. Типичные примеры органических растворителей, которые являются полезными в способе получения настоящего изобретения, представляют собой предпочтительно полярные апротонные растворители, такие как кетоны (например, ацетон или метилэтилкетон), тетрагидрофуран, диметилформамид,диметилсульфоксид, диоксан, ацетонитрил и т.п. Реакцию (1) способа получения согласно настоящему изобретению проводят в условиях горячего или прогретого состояния, предпочтительно при температуре кипения реакционного раствора. Реакцию (1) способа получения по настоящему изобретению предпочтительно проводят в присутствии основного органического или неорганического соединения. Полезными примерами основных органических веществ являются алифатические или ароматические амины, такие как моно-, ди- или триалкиламины, моно-, ди- или триалканоламины, бензиламин, N-метилбензиламин и т.п. Полезными примерами неорганических оснований являются сильные основания, такие как NaOH или KOH, или слабые основания, такие как NH4OP, Na2CO3 и т.п. Реакцию (1) способа получения по настоящему изобретению предпочтительно проводят в присутствии активирующего вещества, например йодида калия, йодида цезия, йодида тетрабутиламмония или йодида триметилфениламмония. Гидролиз (2) сложного эфира, полученного посредством реакции (1) способа получения по настоящему изобретению, проводят известными в данной области способами. Предпочтительно гидролиз проводят в полярных протонных органических растворителях, например в метаноле, этаноле, пропаноле,изопропаноле, бутаноле и т.п., или в апротонных органических растворителях, таких как метилэтилкетон, тетрагидрофуран, диметилформамид, диметилсульфоксид, диоксан, ацетонитрил и т.п., и в присутствии основания, такого как NaOH или KOH. Получение соединений формулы (А) может быть выполнено в соответствии со способом, описанным в патенте ЕР 0975623 В 1. Получение соединений формулы (В) может быть выполнено так, как описано в журнале Journal ofthe American Chemical Society (1940), 62: 1435-8. Аналгетическая активность соединения (I) была продемонстрирована с помощью экспериментальной модели на крысах, представленной аллодинией, вызванной лигированием (перевязыванием) седалищного нерва. Как известно специалистам в данной области, вышеупомянутая экспериментальная модель может рассматриваться как модель, прогнозирующая активность в человеке. Экспериментальная модель лигирования седалищного нерва у крыс имитирует невропатию, которая воспроизводит ряд реакций, аналогичных реакциям, наблюдаемым у человека при многочисленных травматических и патологических состояниях, связанных с невропатической болью. Причиной тому является то, что лигирование седалищного нерва может инициировать синдром, связанный с активированием особых контуров, предназначенных для регулирования восприятия боли, и охарактеризованный возникновением аллодинии, гипералгии и спонтанной боли. Такая модель общеизвестна для создания эффективного инструмента для изучения лекарственных средств, которые должны быть использованы в лечении невропатической боли у человека, и, в частности, в регулировании состояний, таких как аллодиния и гипералгия. Примерами патологий у человека, охарактеризованных дисфункциями, которые описаны в вышеупомянутой экспериментальной модели и охарактеризованы присутствием невропатической боли, являются диабеты, рак, иммунодефицит, травма, ишемия, множественный склероз, ишиалгия, тригеминальная невралгия и постгерпетический синдром. Подтверждение связывания серотонинергических рецепторов было проведено посредством биохимического теста на очищенных мембранах из рекомбинантных клеток человека, которые стабильно экспрессируют специфические рецепторы, или из тканей животных, выбранных так, как описано конкретно для каждого типа серотонинергического рецептора в публикациях Martin G.R. and Humphrey P.P.A. Neuropharmacol., 1994, 33: 261; Hoyer D. Eur. J. Pharmacol., 1985, 118: 1; Bonhaus D.W. et al. Br. J. Pharmacol.,1995, 115: 622; Wolf W.A. et al. J. Neurochem., 1997, 69: 1449; Boess F.G. et al. Neuropharmacol., 1997, 36: 637; Mialet J. et al. Br. J. Pharmacol., 2000, 129: 771; Rees S. et al. FEBS Lett., 1994, 355: 242; Monsma F.J. etet al. Am. J. Physiol., 1998, 275: C1621. Как известно специалистам в данной области, такой тест создает прогностическую модель молекулярного взаимодействия и селективности в отношении к выбранным рецепторам. Подтверждение взаимодействия с калиевыми каналами было сделано посредством теста функционального состояния клеток (метод фиксации потенциала (patch clamp, в котором используют рекомбинантные клетки человека HEK-293, которые стабильно экспрессируют ионный канал hERG, как описано в публикации Zhou Z. et al. Biophys. J., 1998, 74: 230. Как известно специалистам в данной области, этот тест создает прогностическую модель вызывания изменений функциональности сердца и, в частности, воздействий на ритм сердца вследствие изменений Qt-времени. Тесты 1. Аллодиния, вызванная лигированием седалищного нерва у крыс. Используют самцов крыс CD, имеющих "по прибытии" массу 200-250 г. Аллодинию вызывают лигированием под анестезией седалищного нерва левой задней ноги [Seltzerdisorders of pain sensation like those seen in man, Pain, 1988, 33: 87-107]. По меньшей мере через две недели после лигирования седалищного нерва выбирают крыс, которые показали снижение по меньшей мере на 50% порога реакции, зарегистрированного до вмешательства. Болевой порог измеряют посредством машины фон Фрея, которая позволяет посредством применения постепенного увеличения давления на лапу левой задней ноги крысы регистрировать ноцицептивную реакцию, выраженную в граммах, соответствующую моменту, при котором животное отводит ногу. Через 30 мин, 1, 2 и 4 ч после лечения болевой порог, измеренный у контрольного животного, сравнивают с болевым порогом, измеренным у животных, подвергнутых лечению тестовым продуктом (соединение 1 из табл. 1). Контрольных животных подвергают обработке средой для лекарственного средства (метилцеллюлоза), использованной для введения тестового продукта. Результаты проиллюстрированы на фиг. 1. 2. Связывание серотонинергических рецепторов. Подтверждение связывания серотонинергических рецепторов делают с использованием очищенных мембран из тканей выбранных животных или из рекомбинантных клеток человека, которые стабильно экспрессируют специфические рецепторы, как описано в публикациях Martin G.R. and Humphrey P.P.A.man L.P. et al. Am. J. Physiol., 1998, 275: C1621. При использованных условиях проведения эксперимента соединение 1 из табл. 1 показывает селективное ингибирование связывания 5-НТ 4-рецептора. Полученные результаты представлены в табл. 2. Таблица 2 3. Взаимодействие с калиевыми каналами (hERG). Подтверждение взаимодействия с калиевыми каналами выполняют посредством теста с фиксацией потенциала, описанного в публикации Zhou Z. et al. Biophys. J., 1998, 74:230, с использованием рекомбинантной клеточной линии человека HEK-293, которая стабильно экспрессирует ионный канал hERG. При использованных условиях проведения эксперимента соединение 1 из табл. 1 показывает значение IC5050 мкмоль. Полученные результаты представлены в табл. 3. Таблица 3 Примеры Пример 1. Получение соединения 1 из табл. 1. 4-2-[4-([(1-Изопропил-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]этилбензойная кислота. 1 а. 1-[4-(2-Бромэтил)фенил]этанон. Раствор, содержащий ацетилхлорид (85 мл; 0,85 моль) и бромэтилбензол (82 мл; 0,60 моль), добавляют при 0 С к смеси, содержащей безводный трихлорид алюминия (72 г; 0,54 моль), дихлорметан (300 мл) и ацетилхлорид (42,5 мл; 0,6 моль). Смесь перемешивают при холодных условиях в течение 3 ч и затем добавляют к холодному раствору 6 N HCl (600 мл). Получающуюся в результате смесь перемешивают при комнатной температуре в течение ночи. Две фазы, образованные после оставления смеси стоять при комнатной температуре в течение нескольких минут, разделяют. Кислотную фазу экстрагируют дважды посредством 300 мл дихлорметана. Объединенные органические фазы промывают три раза посредством 100 мл воды и один раз посредством 100 мл насыщенного раствора NaCl и затем сушат над безводным Na2SO4. Растворитель затем выпаривают при пониженном давлении, что дает 138 г сырого продукта, который впоследствии очищают фракционной перегонкой при пониженном давлении. Таким образом получают 79,0 г (0,35 моль) 1-[4-(2 бромэтил)фенил]этанона (Т=120 С; Р=7 мм рт. ст.). 1H ЯМР ( м.д. (ppm), CDCl3, 300 МГц): 2,59 (с, 3 Н), 3,23 (т, J=7,31 Гц, 2 Н), 3,59 (т, J=7,31 Гц, 2 Н),7,31 (д, J=7,89 Гц, 2 Н), 7,92 (д, J=8,18 Гц, 2 Н). 2 а. 4-(2-Бромэтил)бензойная кислота. Раствор, содержащий 1-[4-(2-бромэтил)фенил]этанон (38,2 г; 0,17 моль) в диоксане (100 мл), медленно добавляют к раствору, содержащему бром (80,6 г; 0,50 моль), гидроксид натрия (55,4 г; 1,39 моль),воду (470 мл) и диоксан (340 мл), перемешиваемому при 0 С. Смесь перемешивают при той же температуре в течение 3 ч с последующим добавлением, пока смесь по-прежнему холодная, 120 мл концентрированной HCl. Получающуюся в результате смесь перемешивают при комнатной температуре в течение ночи. Твердое вещество, полученное из смеси, отфильтровывают и промывают на фильтре посредством 1N-го раствора HCl (200 мл). Так получают 20,3 г 4-(2-бромэтил)бензойной кислоты (0,09 моль) и используют для последующей реакции без дополнительной очистки.H ЯМР ( м.д., 300 МГц, ДМСО-d6): 3,21 (т, J=6,95 Гц, 2 Н), 3,77 (т, J=7,14 Гц, 2 Н), 7,40 (д, J=8,42 Гц, 2 Н), 7,89 (д, J=8,05 Гц, 2 Н), 12,83 (с, 1 Н). 3 а. Этил 4-(2-бромэтил)бензоат. Смесь, содержащую 4-(2-бромэтил)бензойную кислоту (10 г; 43,7 ммоль), 9,5 N-й хлористоводородный этанол (50 мл; 475 ммоль) и абсолютный этанол (50 мл), перемешивают при кипячении с обратным холодильником в течение 4 ч. Затем смесь охлаждают до комнатной температуры и растворитель выпаривают при пониженном давлении. Остаток смывают насыщенным раствором Na2CO3 (100 мл) и затем экстрагируют этилацетатом (3100 мл). Объединенные органические фазы промывают сначала водой (50 мл) и затем насыщенным раствором NaCl (30 мл). Затем раствор сушат над безводным Na2SO4. В конце концов, растворитель выпаривают при пониженном давлении, что дает 10,8 г этил 4-(2-бромэтил)бензоата (42 ммоль), который используют в последующей реакции без дополнительной очистки. 1H ЯМР ( м.д., 300 МГц, CDCl3): 1,39 (т, J=6,87 Гц, 3 Н), 3,22 (т, J=7,45 Гц, 2 Н), 3,58 (т, J=7,45 Гц,2H), 4,37 (кв., J=6,90 Гц, 2 Н), 7,28 (д, J=8,15 Гц, 2 Н), 8,00 (д, J=8,18 Гц, 2 Н). 4 а. Этил 4-2-[4-([(1-изопропил-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1 ил]этилбензоат. Смесь,содержащую 1-изопропил-N-(пиперидин-4-илметил)-1 Н-индазол-3-карбоксамидгидрохлорид (45,9 г, 136 ммоль), полученный так, как описано в патенте ЕР 0975623, этил 4-(2 бромэтил)бензоат (37 г; 144 ммоль), 2-бутанон (1600 мл) и триэтиламин (18,9 мл; 136 ммоль), перемешивают при кипячении с обратным холодильником. После проведения реакции в течение 1,5 ч добавляют вторую аликвоту триэтиламина (9,5 мл; 68 ммоль) при поддерживании кипячения с обратным холодильником. После проведения реакции в течение дополнительных 1,5 ч добавляют третью аликвоту триэтиламина (9,5 мл; 68 ммоль) при поддерживании кипячения с обратным холодильником. По истечении дополнительных 1,5 ч к реакционной смеси добавляют йодид калия (6,2 г; 37,3 ммоль), по-прежнему поддерживая кипячение с обратным холодильником. В конце концов, затем смесь перемешивают при кипячении с обратным холодильником в течение ночи. Получающуюся в результате смесь затем охлаждают до комнатной температуры и выливают в воду(2 л). Разделяют две фазы и водную фазу экстрагируют 2-бутаноном (2400 мл). Объединенные органические фазы затем промывают водой (50 мл) и насыщенным раствором NaCl (50 мл). Раствор сушат над безводным Na2SO4 и затем растворитель выпаривают при пониженном давлении. Получают 58,9 г сырого продукта, который очищают хроматографией с гравитационным градиентометром на нейтральном оксиде алюминия (1400 г), с использованием смеси из хлороформа и гексана, взятых в соотношении 8/2 в качестве элюента. Таким образом получают 14,2 г этил 4-2-[4-([(1-изопропил-1 Н-индазол-3 ил)карбонил]аминометил)пиперидин-1-ил]этилбензоата (29,8 ммоль). 1H ЯМР ( м.д., 300 МГц, CDCl3): 1,32-1,51 (м, 2H), 1,38 (т, J=7,16 Гц, 3 Н), 1,61 (д, J=6,72 Гц, 6 Н),1,64-1,91 (м, 3 Н), 2,05 (тд, J=11,55, 2,34 Гц, 2 Н), 2,54-2,66 (м, 2 Н), 2,81-2,91 (м, 2 Н), 3,02 (д, J=11,69 Гц,2 Н), 3,42 (т, J=6,43 Гц, 2 Н), 4,36 (кв., 2 Н), 4,87 (септет, 1 Н), 7,15 (т, J=6,28 Гц, 1 Н), 7,22-7,31 (м, 3 Н), 7,357,49 (м, 2 Н), 7,96 (д, 2 Н), 8,39 (дт, J=8,18, 1,02 Гц, 1 Н). 5 а. 4-2-[4-([(1-Изопропил-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1 ил]этилбензойная кислота. Раствор, содержащий этил 4-2-[4-([(1-изопропил-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]этилбензоат (6,2 г; 13 ммоль), тетрагидрофуран (30 мл), абсолютный этанол (30 мл) и 1 N-й гидроксид натрия (28,5 мл; 28,5 ммоль), перемешивают при комнатной температуре в течение 3 дней. Получающуюся в результате смесь нейтрализуют до рН 7 путем добавления ледяной уксусной кислоты(приблизительно 1,7 мл) и затем перемешивают при комнатной температуре в течение ночи. Твердое вещество, таким образом полученное, отфильтровывают, пока оно холодное, и затем промывают на фильтре смесью тетрагидрофурана и абсолютного этанола, взятых в соотношении 1/1. Твердое вещество перекристаллизовывают несколько раз из смеси этилацетата и этанола в соотношении 4/3. Таким образом получают 3,7 г 4-2-[4-([(1-изопропил-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]этилбензойной кислоты (8,3 ммоль). 1(т, J=6,22 Гц, 2 Н), 5,06 (септет, J=6,62 Гц, 1 Н), 7,21-7,28 (м, 1 Н), 7,31 (д, J=8,05 Гц, 2 Н), 7,42 (ддд, J=8,51,7,04, 1,28 Гц, 1 Н), 7,76 (д, J=8,42 Гц, 1 Н), 7,84 (д, J=8,05 Гц, 2 Н), 8,12-8,24 (м, 2 Н), 10,78 (ушир.с., 1 Н). Температура плавления (m.p.)=179-181 С; масс-спектрометрия (MS) показывает основной пик 449(МН+); элементный анализ для C26H32N4O3. Пример 2. Таблетка, содержащая в качестве действующего начала соединение 1 настоящего изобретения, имеет следующую композицию: Пример 3. Ампула, содержащая в качестве действующего начала соединение 1 по настоящему изобретению,имеет следующий состав: Пример 4. Фармацевтическая композиция в форме гранул, содержащая в качестве действующего начала соединение 1 по настоящему изобретению, имеет следующий состав: в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;Y представляет собой СН или N и р представляет собой целое число от 0 до 3,и его соли либо соли присоединения фармацевтически приемлемых органических или неорганических кислот или соли присоединения фармацевтически приемлемых органических или неорганических оснований. 2. Соединение по п.1, где R представляет собой линейную или разветвленную алкильную группу,содержащую от 2 до 3 атомов углерода. 3. Соединение по п.1, где Y представляет собой СН. 4. Соединение по п.1, где р представляет собой целое число от 0 до 1. 5. Соединение по п.1, где R, Y, р и положение группы 6. Соединение по любому одному из предшествующих пунктов, где упомянутую фармацевтически приемлемую органическую или неорганическую кислоту выбирают из группы, включающей щавелевую кислоту, малеиновую кислоту, метансульфокислоту, паратолуолсульфокислоту, янтарную кислоту, лимонную кислоту, винную кислоту, молочную кислоту, хлористо-водородную кислоту, фосфорную кислоту, серную кислоту, аспарагиновую кислоту и глутаминовую кислоту. 7. Соединение по любому одному из пп.1-5, где упомянутое фармацевтически приемлемое органическое или неорганическое основание выбирают из группы, включающей метиламин, диметиламин,триметиламин, этиламин, диэтиламин, триэтиламин, пропиламин, дипропиламин, трипропиламин, этилендиамин, моноэтаноламин, диэтаноламин, триэтаноламин, гуанидин, морфолин, пиперидин, пирролидин,пиперазин,1-бутилпиперидин,1-этил-2-метилпиперидин,N-метилпиперазин,1,4 диметилпиперазин, N-бензилфенилэтиламин, N-метилглюкозамин, трис-(гидроксиметил)аминометан,аргинин, лизин, аммиак, гидроксид натрия, гидроксид кальция, гидроксид калия, гидроксид алюминия,гидроксид железа, гидроксид магния и гидроксид цинка. 8. Фармацевтическая композиция, содержащая эффективное количество соединения формулы (I) в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;Y представляет собой СН или N и р представляет собой целое число от 0 до 3,и его солей либо солей присоединения фармацевтически приемлемых органических или неорганических кислот или солей присоединения фармацевтически приемлемых органических или неорганических оснований и по меньшей мере один фармацевтически приемлемый эксципиент. 9. Фармацевтическая композиция по п.8, в которой упомянутый по меньшей мере один фармацевтически приемлемый эксципиент выбирают из группы, включающей разделительные средства, связывающие вещества, разрыхлители, наполнители, разбавители, красители, флюидизирующие вещества,регуляторы сыпучести, скользящие вещества, консервирующие вещества, стабилизаторы, увлажнители,абсорбенты, поверхностно-активные вещества, буферные растворы, соли для регулирования осмотического давления, эмульгаторы, ароматизирующие вещества и подслащивающие вещества. 10. Фармацевтическая композиция по п.8, в которой соединение формулы (I) определено в соответствии с любым одним из пп.1-7. 11. Фармацевтическая композиция по любому одному из пп.8-10, где фармацевтическую композицию выбирают из группы, включающей таблетки, капсулы, таблетки, покрытые оболочкой, гранулы,растворы и сиропы для перорального введения, антисептические пластыри, растворы, пасты, кремы и мази для трансдермального введения, суппозитории для ректального введения, стерильные растворы для инъекционного или аэрозольного введения, либо в лекарственной форме с мгновенным высвобождением, либо в лекарственной форме с замедленным высвобождением. 12. Фармацевтическая композиция по любому одному из пп.8-11, где упомянутая фармацевтическая композиция содержит количество соединения формулы (I) или его соли присоединения кислоты либо соли присоединения основания, которое надежно обеспечивает уровень введения от 0,001 до 100 мг/кг/день соединения формулы (I), представленного в свободной форме. 13. Фармацевтическая композиция по п.12, в которой упомянутое количество будет надежно обеспечивать уровень введения от 0,05 до 50 мг/кг/день. 14. Фармацевтическая композиция по п.12, в которой упомянутое количество будет надежно обеспечивать уровень введения от 0,1 до 10 мг/кг/день. 15. Способ получения соединения формулы (I) в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;Y представляет собой СН или N и р представляет собой целое число от 0 до 3,и его солей или солей присоединения фармацевтически приемлемых органических или неорганических кислот либо солей присоединения фармацевтически приемлемых органических или неорганических оснований,отличающийся тем, что он включает:(1) реакцию между соединением формулы (А) в которой R имеет значение, описанное выше, и А представляет собой остаток в виде аниона неорганической или органической кислоты,и соединением формулы (В)R1 представляет собой линейную или разветвленную алкильную, арильную, арилалкильную или алкиларильную группу, содержащую от 1 до 10 атомов углерода; иX представляет собой атом галогена, и(2) последующий гидролиз сложного эфира, полученного реакцией (1). 16. Способ получения по п.15, в котором упомянутый остаток А выбирают из группы, включающей галогенид, фосфат, сульфат, нитрат и органический карбоксилат. 17. Способ получения по п.16, в котором упомянутый остаток А выбирают из группы, включающей хлорид, бромид и йодид. 18. Способ получения по п.15, в котором упомянутый остаток X выбирают из группы, включающей хлорид, бромид и йодид. 19. Способ получения по п.15, в котором упомянутую группу R1 выбирают из группы, включающей линейный или разветвленный алкил, содержащий 1-6 атомов углерода, арил, арилалкил и алкиларил,содержащий 7-8 атомов углерода. 20. Способ получения по любому одному из пп.15-19, в котором упомянутую реакцию (1) проводят в органическом растворителе, выбранном из группы, включающей полярные апротонные растворители. 21. Способ получения по п.20, в котором упомянутый органический растворитель выбирают из группы, включающей ацетон, метилэтилкетон, тетрагидрофуран, диметилформамид, диметилсульфоксид, диоксан и ацетонитрил. 22. Способ получения по любому одному из пп.15-21, в котором упомянутую реакцию (1) проводят в присутствии основного органического или неорганического соединения. 23. Способ получения по любому одному из пп.15-22, в котором упомянутую реакцию (1) проводят в присутствии активирующего вещества, выбранного из группы, включающей йодид калия, йодид цезия,йодид тетрабутиламмония и йодид триметилфениламмония. 24. Применение соединения формулы (I) в которой R представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода;Y представляет собой СН или N и р представляет собой целое число от 0 до 3,и его солей или солей присоединения фармацевтически приемлемых органических или неорганических кислот либо солей присоединения фармацевтически приемлемых органических или неорганических оснований для приготовления фармацевтической композиции для лечения невропатической боли. 25. Применение соединения по п.24 для приготовления фармацевтической композиции для лечения невропатической боли, вызванной диабетами, раком, иммунодефицитом, травмой, ишемией, рассеянным склерозом, ишиалгией, тригеминальной невралгией и постгерпетическим синдромом.
МПК / Метки
МПК: C07D 401/14, C07D 401/12, A61P 29/00, A61K 31/4545
Метки: средство, боли, лекарственное, активное, невропатической, отношении
Код ссылки
<a href="https://eas.patents.su/13-19405-lekarstvennoe-sredstvo-aktivnoe-v-otnoshenii-nevropaticheskojj-boli.html" rel="bookmark" title="База патентов Евразийского Союза">Лекарственное средство, активное в отношении невропатической боли</a>
Карбаматные соединения для использования при предотвращении или лечении невропатической боли, гистаминовой головной боли и головной боли, связанной с мигренью

Номер патента: 5648
Опубликовано: 28.04.2005
Авторы: Роджерс Кэтрин Э., Плата-Саламан Карлос Р., Зао Бою, Шэнк Ричард П., Кодд Эллен Э.
МПК: A61P 25/06, A61K 31/27
Метки: головной, карбаматные, гистаминовой, соединения, боли, предотвращении, связанной, невропатической, использования, лечении, мигренью
Формула / Реферат:
1. Способ предотвращения или лечения невропатической боли, гистаминовой головной боли и головной боли, связанной с мигренью, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества энантиомера формулы (I), по существу свободного от других энантиомеров, или энантиомерной смеси, где количество энантиомера формулы (I) составляет по меньшей мере около 90%: Формула (I) где фенил замещен от одного до пяти...
Фармацевтическая композиция для лечения острой, хронической боли и/или невропатической боли и мигреней

Номер патента: 4930
Опубликовано: 28.10.2004
Авторы: Сандз Стивен Брэдли, Уэтски Эрик Джейкоб, Коу Джоутам Уодзуэрт, О'нилл Брайан Томас, Харриган Эдмунд Патрик
МПК: A61K 45/06, A61P 25/06
Метки: фармацевтическая, мигреней, композиция, боли, лечения, хронической, невропатической, острой
Формула / Реферат:
1. Фармацевтическая композиция для лечения острой, хронической и/или невропатической боли и мигрени, содержащая (а) частичный агонист никотиновых рецепторов или его фармацевтически приемлемую соль, где частичный агонист никотиновых рецепторов представляет собой арильное конденсированное азаполициклическое соединение; (б) анальгетический агент или его фармацевтически приемлемую соль и (в) фармацевтически приемлемый носитель; где вышеуказанные...
Производные изоксазола, их фармацевтические композиции и способ лечения невропатической боли

Номер патента: 13905
Опубликовано: 30.08.2010
Авторы: Моригги Ерманно, Наполетано Мауро
МПК: A61P 25/02, A61K 31/42
Метки: способ, изоксазола, лечения, композиции, производные, невропатической, боли, фармацевтические
Формула / Реферат:
1. Применение производного изоксазола формулы (I)или его фармацевтически приемлемых солей при получении лекарственного средства для лечения невропатической боли.2. Применение фармацевтической композиции, включающей в качестве активного соединения соединение формулы (I) по п.1 либо его фармацевтически приемлемую соль и, по меньшей мере, фармацевтически приемлемый наполнитель при лечении невропатической боли.3. Применение фармацевтической...
Применение производных индазола для лечения невропатической боли

Номер патента: 8144
Опубликовано: 27.04.2007
Авторы: Поленцани Лоренцо, Каццолла Никола, Гильельмотти Анджело, Ализи Алессандра
МПК: A61K 31/454, A61P 25/02, A61K 31/4545...
Метки: невропатической, применение, лечения, производных, индазола, боли
Формула / Реферат:
1. Применение соединения формулы I где X представляет собой СН или N, и когда X представляет собой СН, R представляет собой Н, ОН, прямую или разветвленную алкильную цепь, имеющую от 1 до 3 атомов углерода, прямую или разветвленную алкоксицепь, имеющую от 1 до 3 атомов углерода, или атом галогена, и когда X представляет собой N, R представляет собой Н, или его кислотно-аддитивной соли с фармацевтически приемлемой органической или неорганической...
Применение производных 2н-[1,3]-оксазино [3,2-a] индола для лечения невропатической боли

Номер патента: 8927
Опубликовано: 31.08.2007
Авторы: Поленцани Лоренцо, Каццолла Никола, Гульельмотти Анджело, Ализи Алессандра
МПК: A61P 25/02, A61K 31/5365
Метки: боли, невропатической, производных, 3,2-a, 2н-[1,3]-оксазино, индола, применение, лечения
Формула / Реферат:
1. Применение соединения формулы I где R представляет Н, линейную или разветвленную алкильную цепь, имеющую от 1 до 12 атомов углерода, или арилалкильную группу, где алкильный фрагмент имеет от 1 до 4 атомов углерода, или циклогексилметил, и его кислотно-аддитивных солей с фармацевтически приемлемыми органическими или неорганическими кислотами для приготовления фармацевтической композиции, активной при лечении невропатической боли. 2....
Предыдущий патент: Способ покрытия замороженных изделий
Следующий патент: Способ и устройство для контроля доступа к сосудам, а также устройство для экстракорпоральной обработки крови с устройством для контроля доступа к сосудам
Случайный патент: Ротавирусная вакцина, индуцирующая гетеротипический перекрестный иммунитет