Способ синтеза n-[3-(-3-цианопиразоло[1,5a] пиримидин-7-ил)-фенил]-n-этилацетамида
Номер патента: 10830
Опубликовано: 30.12.2008
Авторы: Тарканьи Габор, Добаи Ласло, Цоке Каталин, Надьне Багди Юдит, Цибула Ласло, Веркне Папп Эва







Формула / Реферат
1. Способ синтеза терапевтически применимого N-[3-(3-цианопиразоло[1,5-а]пиримидин-7-ил)фенил]-N-этилацетамида формулы (IV), содержащего менее 0,2% примесей, включающий взаимодействие 3-амино-4-пиразолкарбонитрила формулы (III) с эквимолярным количеством гидрохлорида
N-{3-[3-(диметиламино)-1-оксо-2-пропенил]фенил}-N-этилацетамида формулы (II) в органических растворителях или в смеси из воды и органических растворителей при pH 3,0-3,5, с последующим выделением и перекристаллизацией продукта.
2. Способ по п.1, в котором в качестве органического растворителя используют метанол, этанол, изопропанол или н-пропанол.
3. Способ по любому из пп.1, 2, в котором небольшими порциями в реакционную смесь добавляют соединения формулы (II).
4. Способ по любому из пп.1-3, в котором реакцию проводят при 15-50шC.
5. Способ по любому из пп.1-3, в котором реакцию проводят при 45-50шC.
6. Способ по любому из пп.1-5, в котором реакцию проводят в течение 0,5-1,0 ч.
7. Гидрохлорид N-{3-[3-(диметиламино)-1-оксо-2-пропенил]фенил}-N-этилацетамида формулы (II), имеющий чистоту более 99,5%.
8. Способ синтеза гидрохлорида N-{3-[3-(диметиламино)-1-оксо-2-пропенил]фенил}-N-этилацетамида формулы (II), имеющего чистоту более 99,5%, включающий суспендирование
N-{3-[3-(диметиламино)-1-оксо-2-пропенил]фенил}-N-этилацетамида формулы (I) в ацетоне в атмосфере азота при 20-25шC и добавление эквимолярного количества концентрированной соляной кислоты в суспензию.
Текст
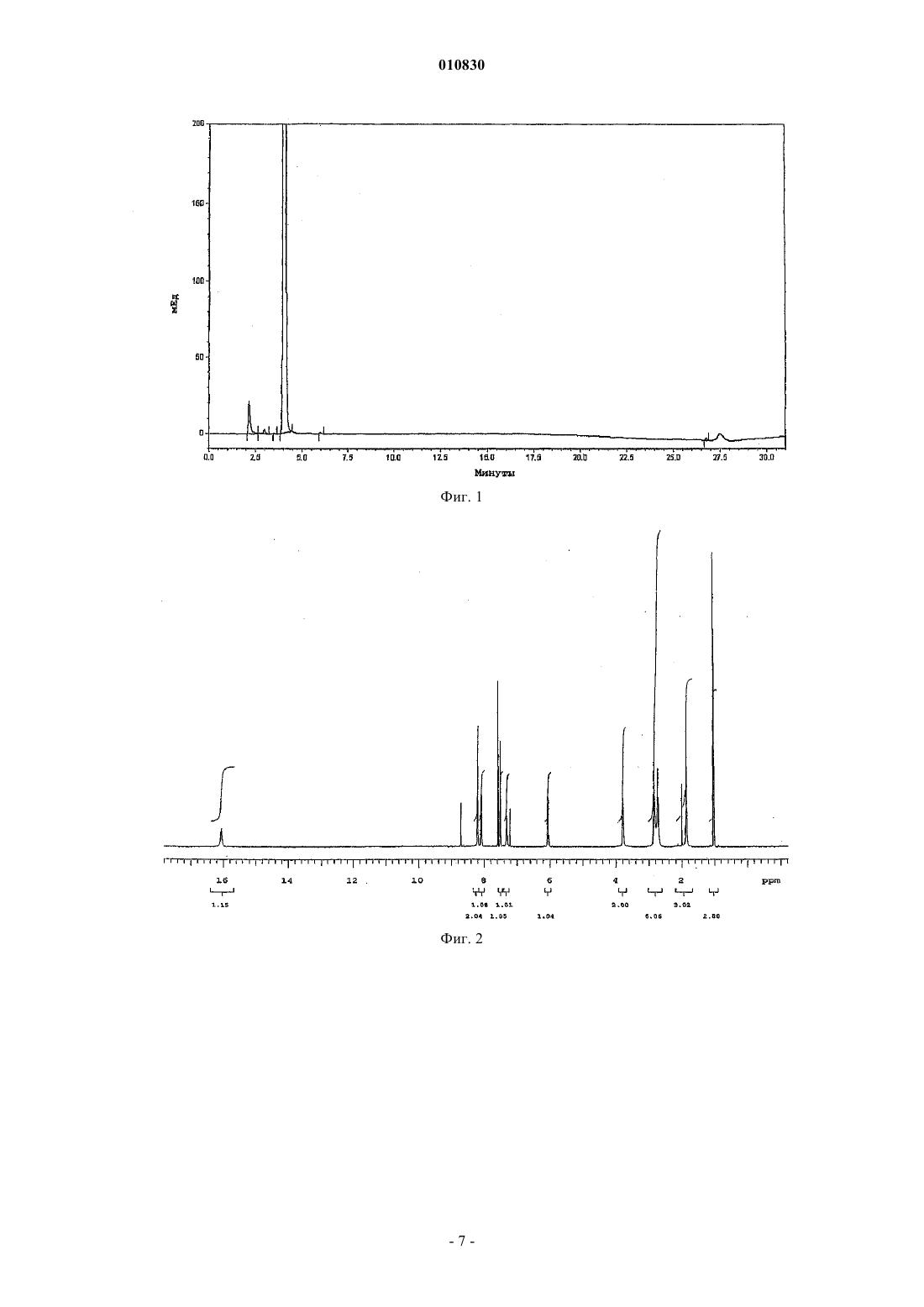
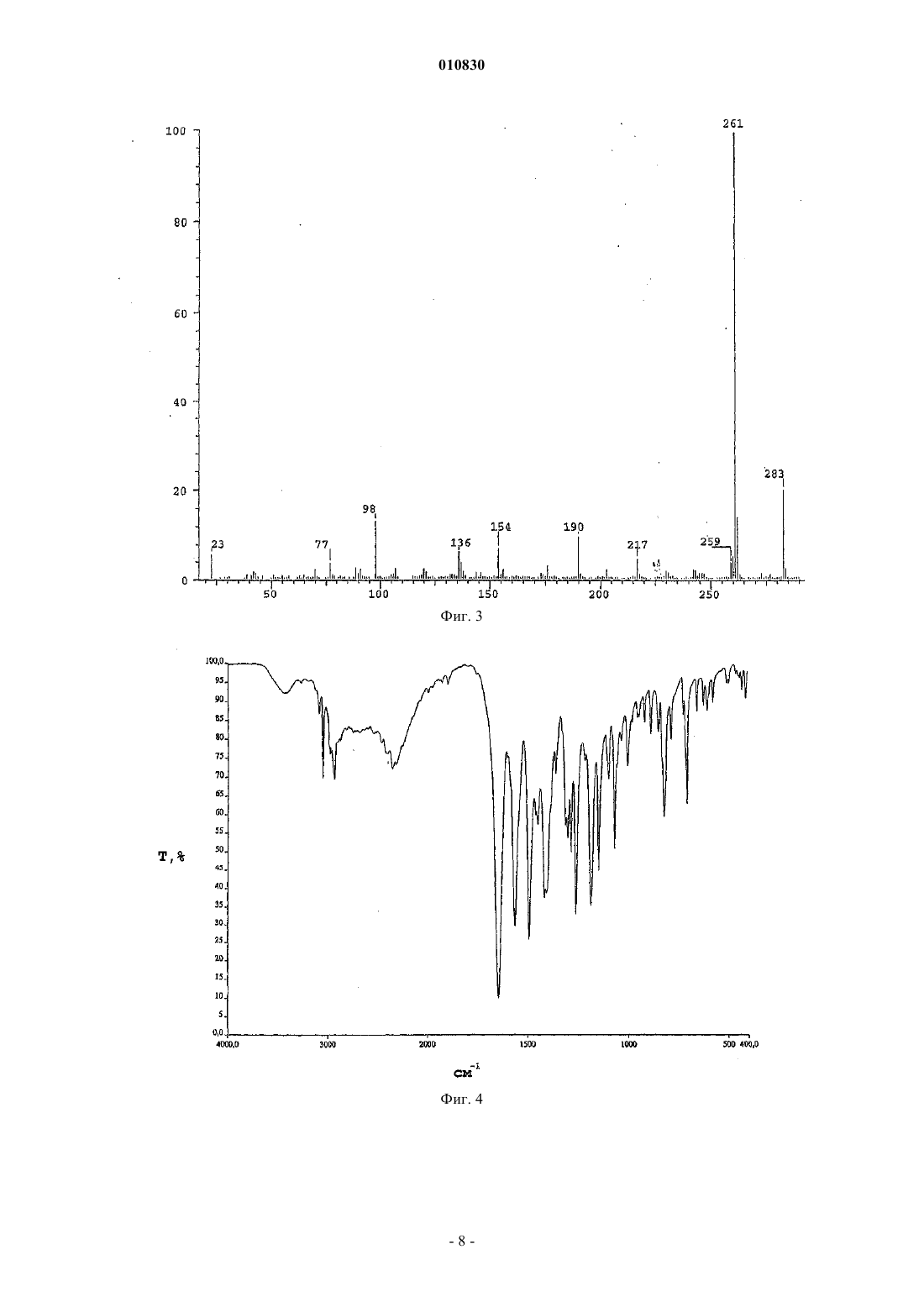
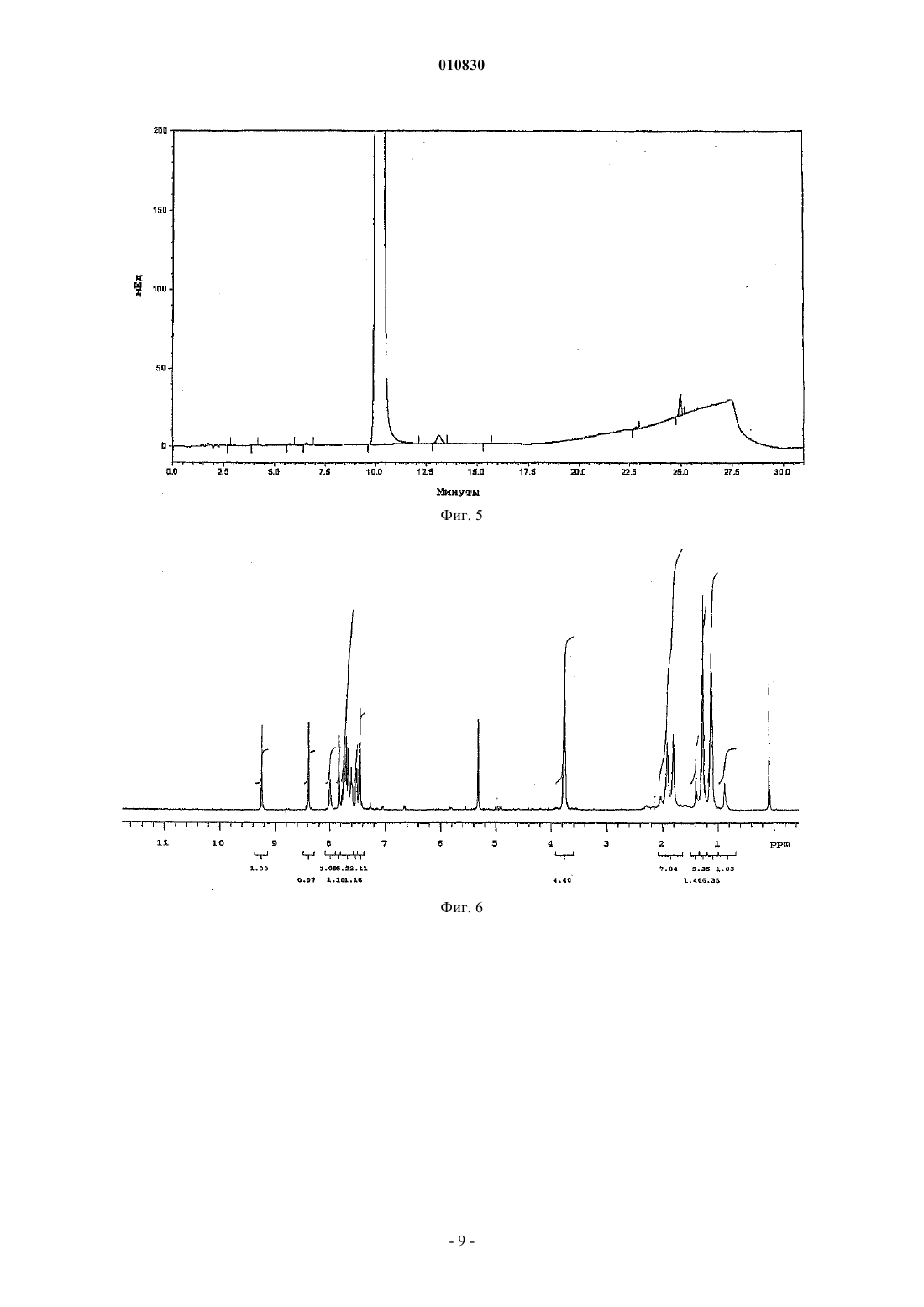
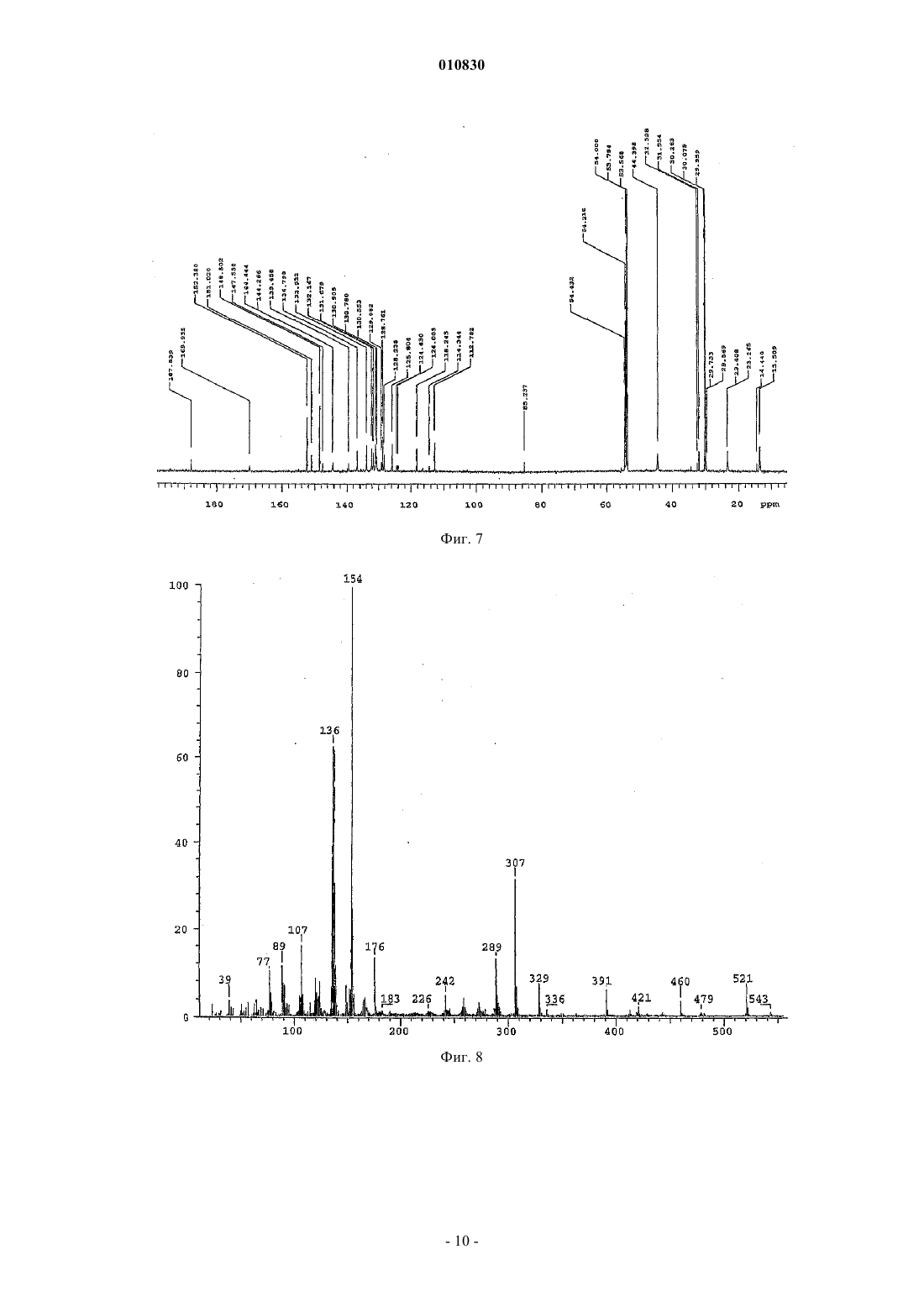
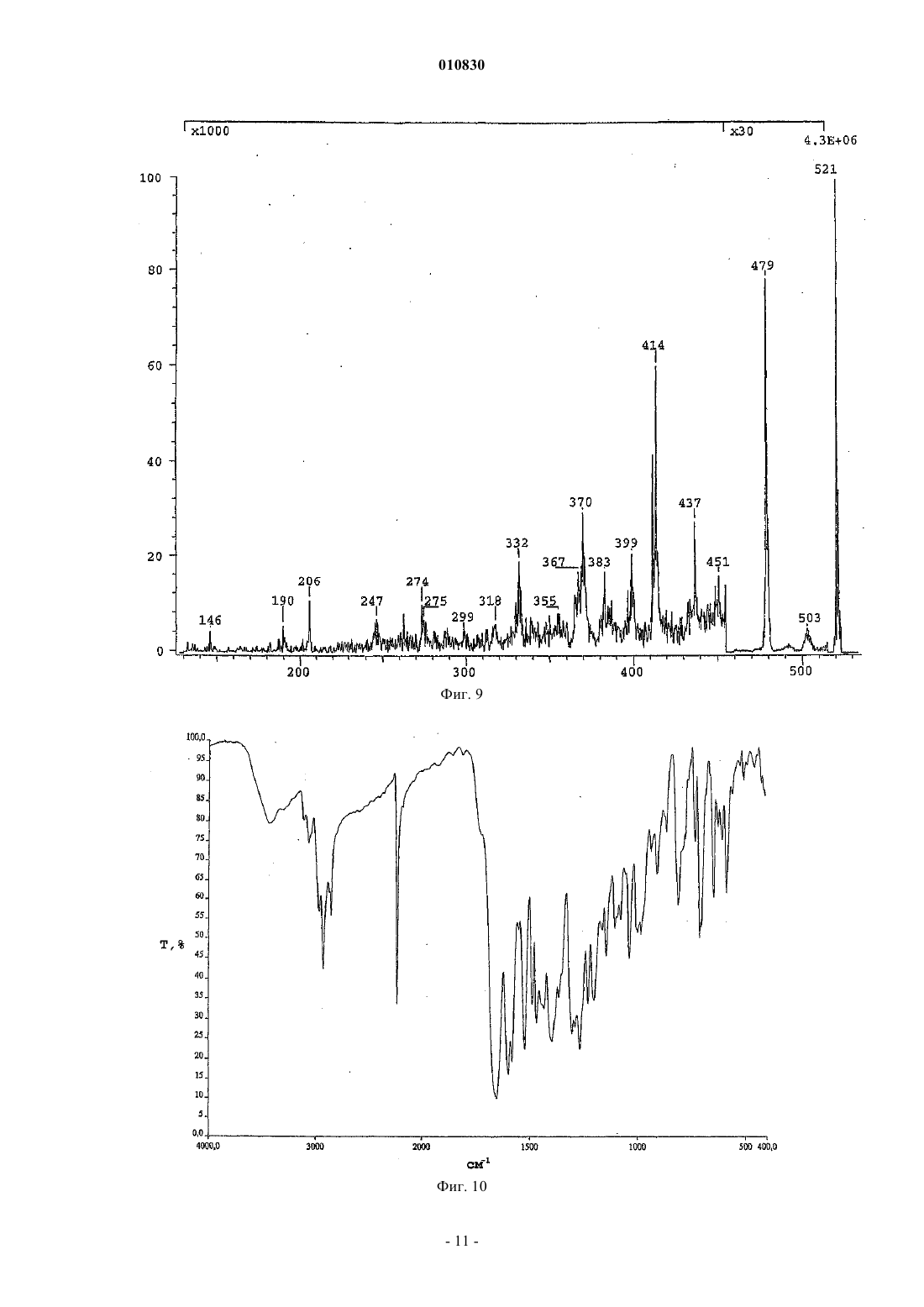
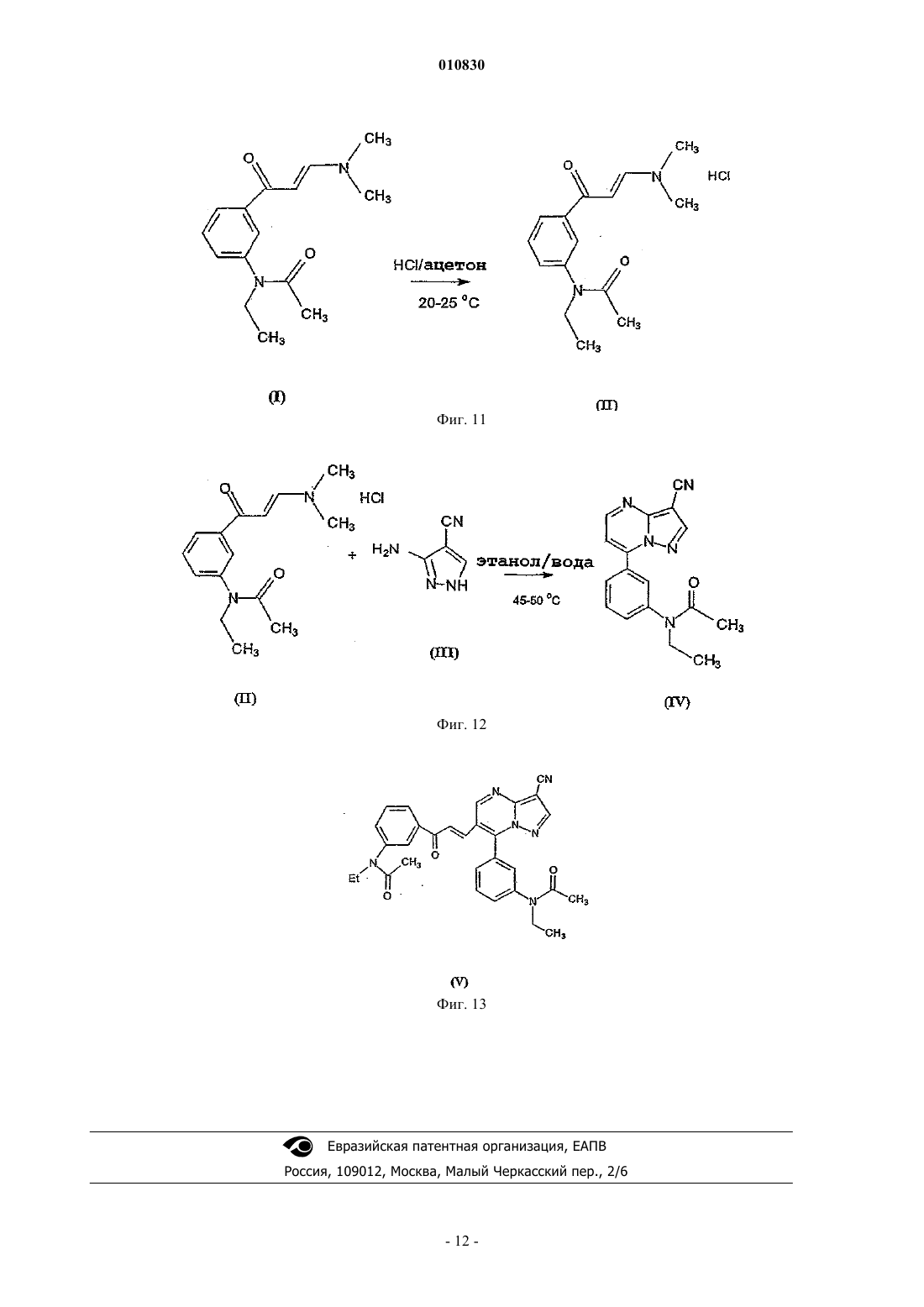
010830 Область техники, к которой относится изобретение Изобретение касается N-[3-(3-цианопиразоло[1,5-a]пиримидин-7-ил)фенил]-N-этилацетамида формулы (IV), содержащего менее 0,2% примесей, который пригоден для терапевтического применения. Кроме того, изобретение касается способа синтеза терапевтически применимого соединения формулы (IV), содержащего менее 0,2% примесей, реакцией нового промежуточного соединения гидрохлорида N-3-[3-(диметиламино)-1-оксо-2-пропенил]фенил-N-этилацетамида формулы (II) с основанием 3-амино-4-пиразолкарбонитрила формулы (III) в свободной от кислоты среде. Изобретение также касается нового гидрохлоридаN-3-[3-(диметиламино)-1-оксо-2 пропенил]фенил-N-этилацетамида формулы (II). Изобретение также касается способа синтеза нового гидрохлорида N-3-[3-(диметиламино)-1-оксо 2-пропенил]фенил-N-этилацетамида формулы (II) с чистотой более 99,5%. Кроме того, изобретение касается N-3-(3-цианопиразоло[1,5-а]пиримидин)-6-ил-3-[(3-Nэтилацетамидофенил)-3-оксопропен-1-ил]-(пиримидин-7-ил)фенил-N-этилацетамида формулы (V),представляющего собой новую выделенную примесь, которая образуется с выходом 0,07% при синтезе соединения формулы (IV). Уровень техники Соединение формулы (IV) (залеплон) является анксиолитическим, антиэпилептическим, седативным, снотворным средством, действующим как агонист ГАМКА-рецепторов. Соединение формулы (IV) описано в патенте US 4626538, пример 14, табл. 8. Конечный продукт получают при кипячении основания N-3-[3-(диметиламино)оксо-2-пропенил]фенил-N-этилацетамида формулы (I) с основанием 3-амино-4-пиразолкарбонитрила формулы (III) в ледяной уксусной кислоте в течение 8 ч. Точные физико-химические свойства, чистота и выход соединения не описаны в этом патенте, приведена только температура кипения, которая равна 186-187 С. Способ из патента US 4626538 усовершенствован в патенте EP 776898. Усовершенствованный способ синтеза соединения формулы (IV) состоит в следующем. Основание N-3-[3-(диметиламино)оксо-2-пропенил]фенил-N-этилацетамида формулы (I) подвергают реакции с основанием 3-амино-4-пиразолкарбонитрила формулы (III) в смеси из уксусной кислоты и воды (от 10 до 85 об.%), предпочтительно в их смеси 1:2 по объему. Чистота полученного при этом продукта составляет 98,86-99,40% согласно HPLC. Выход наиболее чистого (99,4%) соединения составляет 83,5%, как описано в примере 1. Исходными материалами являются основания во всех примерах. Способ синтеза соединения формулы (IV) заявлен как исходящий не только из промежуточных оснований, но также и их солей. В вышеуказанном патенте EP не описан какой-либо способ, в котором используется соль в качестве исходного материала, и в нем отсутствует описание применимых промежуточных солей или их синтез. В патентной заявке WO 2002/100828 также описан способ синтеза соединения формулы (IV), включающий реакцию основания N-3-[3-(диметиламино)оксо-2-пропенил]фенил-N-этилацетамида формулы (I) с основанием 3-амино-4-пиразолкарбонитрила формулы (III) в смеси из воды и водорастворимого органического растворителя в кислых условиях. Кислые условия создаются добавлением органической или неорганической кислоты - среди прочего соляной кислоты, в реакционную смесь. Количество кислоты может быть эквимолярно одному из исходных оснований или может применяться в большом избытке. В пп.12-15 заявлено применение кислотной соли основания формулы (I) в качестве исходного материала и отдельно добавление кислоты, однако с одной стороны, в приведенных пп.1 и 6 только основание используется в качестве исходного материала, с другой стороны - описание не подтверждает применения соли в качестве исходного материала. В патентной заявке WO 2002/100828, как и в патенте EP 776898, не приведено примера или описания получения, выделения и предпочтительного воплощения солей оснований формулы (I) или (III). B примере 12 используется 1 экв. соляной кислоты, что является нижним пределом количества вносимой кислоты, как заявлено в п.21. Согласно этому примеру продукт получают после 24-часовой реакции с выходом 82%, а чистота составляет лишь 98,95%. В наших экспериментах при проведении синтеза в соответствии с вышеуказанным примером 12 и с использованием 1 экв. соляной кислоты значение pH находилось в районе 1,5 на всем протяжении реакции. В тех реакциях, в которых количество вносимой кислоты превышало 1 экв., как описано в примерах 1-21, значение pH было меньше 1,0, т.е. используются сильно кислые условия реакции. В патентной заявке WO 2003/011228 описан новый региоизомер соединения формулы (IV)N-[3-(3-цианопиразоло[1,5-a]пиримидин-5-ил)фенил]-N-этилацетамид, который образуется при синтезе соединения формулы (IV). Значительное количество этого региоизомера 0,2-0,5% образуется, к примеру,в способе синтеза соединения формулы (IV), описанном в патентной заявке US 2003/0040522. Поскольку вышеприведенные значения химической чистоты меньше, чем те, что требуются в фармацевтической промышленности в настоящее время, целью изобретателей было найти новый способ синтеза соединения формулы (IV), который был бы пригоден для получения продукта высокой чистоты(удовлетворяющего самым строгим требованиям по качеству) с хорошим выходом.-1 010830 Сущность изобретения Основой предлагаемого изобретения является следующее открытие. pH реакционной среды нужно поддерживать между 3,0 и 3,5 во время реакции, для того чтобы достичь наибольшей избирательности химической реакции. В результате исследования оптимальное значение pH смеси, которое необходимо для поддержания высокой чистоты соединения формулы (IV), поддерживается самопроизвольным образом при применении высокочистой соляно-кислой соли формулы (II). Во время исследования также было обнаружено, что высокая чистота промежуточных соединений,особенно самых последних, является предпосылкой высокой чистоты конечного продукта. Высококачественное основание 3-амино-4-пиразол-карбонитрила формулы (III) доступно коммерчески. Основание формулы (I) может быть получено путем многостадийного синтеза, а основная форма соединения надлежащей чистоты может быть выделена только с потерей большого количества материала. Неожиданно оказалось, что из солей основания формулы (I) соляно-кислая соль, которая до этого не была охарактеризована, может быть легко получена из основания формулы (I) с высокой чистотой и очень хорошим выходом. В примерах из патентов и патентных заявок предшествующего уровня техники, касающихся синтеза соединения формулы (IV), реакция проводится в сильнокислых условиях, а в некоторых случаях используется более 1 экв. кислоты. Для начала авторы повторили эти известные методики. Оказалось, что при таких сильнокислых условиях в каждом случае полученное соединение формулы (IV) содержит более 0,2% региоизомера N-[3-(3-цианопиразоло[1,5-а]пиримидин-5-ил)фенил]-N-этилацетамида. Тогда был разработан новый способ синтеза высокочистого соединения формулы (IV). Неожиданно оказалось, что соединение формулы (IV) может быть синтезировано при реакции соляно-кислой соли формулы (II) высокочистого основания формулы (I), которая была получена с хорошим выходом предлагаемым способом, с основанием формулы (III) без добавления кислоты в реакционную смесь, более того, выход и чистота продукта намного выше, чем это описано на предшествующем уровне техники. Синтез высокочистого соединения формулы (IV), содержащего менее 0,2% примесей, не был описан в литературе, как не был описан и способ настоящего изобретения для синтеза соединения формулы(IV) без добавления кислоты в реакционную смесь. Изобретение касается способа синтеза терапевтически применимого соединения формулы (IV), содержащего менее 0,2% примесей, в реакции новой соли формулы (II) с основанием формулы (III) в свободной от кислоты среде. Изобретение также касается нового гидрохлоридаN-3-[3-(диметиламино)-1-оксо-2 пропенил]фенил-N-этилацетамида формулы (II). Кроме того, изобретение касается способа синтеза нового гидрохлорида N-3-[3-(диметиламино)-1 оксо-2-пропенил]фенил-N-этилацетамида формулы (II) с чистотой более 99,5%, включающего суспендирование соединения формулы (I) в ацетоне при 20-25 С в атмосфере азота и добавление эквимолярного количества концентрированной соляной кислоты. Раскрытие сущности изобретения"Неочищенный конечный продукт" означает конечный продукт, полученный при воспроизведении методик синтеза соединения формулы (IV) из предшествующего уровня техники."Сильнокислые условия" означают проведение реакции при значении pH ниже 1,0. В соответствии с настоящим изобретением новую соль формулы (II) получают путем суспендирования основания формулы (I) в ацетоне при комнатной температуре в атмосфере азота и добавления эквимолярного количества концентрированной соляной кислоты. После перемешивания выпавшие в осадок кристаллы отфильтровывают, промывают и сушат. Чистота полученного при этом продукта составляет более 99,5%. В соответствии с настоящим изобретением синтез терапевтически применимого соединения формулы (IV), содержащего менее 0,2% примесей, заключается в следующем. Основание формулы (III) растворяют в смеси воды и спирта и добавляют твердую соль формулы(II) небольшими порциями в подогретый раствор. Во время добавления продолжают сильное перемешивание. В соответствии с другим воплощением соль формулы (II) растворяют в растворителе и вносят раствор в реакционную смесь. Сначала раствор, а затем полученную суспензию непрерывно перемешивают,после чего выделяют продукт. Обычно суспензию охлаждают и продолжают перемешивание. Выпавшие в осадок кристаллы отфильтровывают, промывают и сушат. Полученный при этом кристаллический материал растворяют в теплом метаноле в атмосфере азота, раствор фильтруют, охлаждают, выпавший в осадок продукт отфильтровывают, промывают и сушат. Способ по настоящему изобретению предпочтительно выполняется в органическом растворителе или в смеси воды и водорастворимого органического растворителя. В качестве органического растворителя можно использовать короткоцепочечные алифатические спирты, предпочтительно метанол, этанол,изопропанол, н-пропанол. Способ по изобретению предпочтительно выполняется при 15-50C в течение 0,5-1 ч. Основание 3-амино-4-цианопиразола формулы (III) предпочтительно растворяют в соответст-2 010830 вующей смеси растворителей и в раствор добавляют соляно-кислую соль формулы (II) небольшими порциями. После охлаждения выпавший в осадок продукт отфильтровывают и сушат. Чистота полученного при этом продукта формулы (IV) составляет более 99,8% методом HPLC. Применение способа настоящего изобретения - реакции между новой высокочистой соляно-кислой солью формулы (II) и соединением формулы (III) приводит к значительно более высокому качеству (чистота более 99,4%) соединения формулы (IV), чем может быть достигнуто согласно предшествующему уровню техники, т.е. при проведении реакции в водном растворе уксусной кислоты. Высокая чистота, которая особенно выгодна для фармацевтической промышленности, конечного продукта формулы (IV), полученного способом предложенного изобретения, частично обусловлена высокой чистотой новой соляно-кислой соли формулы (II), частично свободной от кислоты реакционной смесью, частично добавлением новой соляно-кислой соли формулы (II) небольшими порциями и частично коротким временем реакции. Большое преимущество применения новой соляно-кислой соли формулы (II), которая была впервые получена и использована нами при синтезе соединения формулы (IV), состоит в том, что применяется высокочистый исходный материал при синтезе соединения формулы (IV) с высокой чистотой, требуемой в фармацевтической промышленности. Согласно HPLC чистота основания I обычно составляет 90-95%,тогда как чистота соляно-кислой соли формулы (II), полученной из этого основания, составляет более 98,5%. Следует подчеркнуть, что в способе настоящего изобретения при синтезе соединения формулы (IV) значение pH реакции равно 3,0-3,5. Большое преимущество настоящего способа заключается в том, что не нужно добавлять дополнительное количество кислоты в реакцию, поскольку используемая в качестве исходного материала новая соляно-кислая соль формулы (II) содержит эквимолярную кислоту, более того, значение pH реакционной смеси остается между 3,0-3,5 на протяжении всей реакции вследствие добавления в нее соляно-кислой соли формулы (II) небольшими порциями, в результате чего условия реакции в этом способе синтеза соединения формулы (IV) являются намного более мягкими по сравнению с методиками предшествующего уровня техники, в которых реакция проводится при pH ниже 1,0 или самое большее при pH 1,5. Большим преимуществом применения новой соляно-кислой соли формулы (II), впервые полученной и использованной авторами в синтезе соединения формулы (IV), является то, что региоизомер соединения формулы (IV) образуется только с выходом менее 0,1%. Конечный продукт формулы (IV), полученный по методикам предшествующего уровня техники, содержит, как заявлено в описании, в некоторых случаях 0,5% региоизомера в виде примеси. Дальнейшим преимуществом применения новой соляно-кислой соли формулы (II), впервые полученной и использованной в синтезе соединения формулы (IV), помимо образования небольшого количества региоизомера формулы (IV), является то, что примесь формулы (V), которая не упоминается в литературе или в раскрытых патентах и была выделена авторами изобретения, образуется только в следовых количествах (0,07%). Аналитические характеристики примеси формулы (V) приведены в примере 2. Наши аналитические измерения подтвердили структуру соединения формулы (V), химическое название которогоN-3-(3-цианопиразоло[1,5-а]пиримидин)-6-ил-3-[(3-N-этилацетамидофенил)-3-оксопропен-1-ил](пиримидин-7-ил)фенил-N-этилацетамид. Вышеуказанную примесь формулы (V) можно было бы выделить из неочищенных конечных продуктов, полученных методами, описанными в литературе в синтезе соединения формулы (IV), поскольку эти неочищенные продукты содержат большее количество примеси формулы (V). Примесь формулы (V) выделяли из маточного раствора от перекристаллизации методом препаративной HPLC. Неожиданно оказалось, что примесь формулы (V) образуется с выходом всего лишь 0,07% в способе данного изобретения при синтезе соединения формулы (IV). Авторы изобретения поняли, что примесь формулы (V) может быть уменьшена добавлением в реакционную смесь соляно-кислой соли формулы (II) небольшими порциями в способе этого изобретения для синтеза соединения формулы (IV), а также соблюдением надлежащего значения pH, температуры,соотношения растворителя и продолжительности реакции. Это особенно выгодно для фармацевтической промышленности. Итак, преимущества способа данного изобретения для синтеза соединения формулы (IV), в сравнении с известными способами, заключаются в следующем: конечный продукт формулы (IV) получают с хорошим выходом, он содержит менее 0,2% примесей в соответствии с требованиями GMP к производству фармацевтических препаратов; очень высокая чистота соляно-кислой соли формулы (II); конечный продукт формулы (IV) получают без добавления кислоты; конечный продукт формулы (IV) содержит намного меньше (макс. 0,1%) региоизомера в качестве примеси по сравнению с продуктами, полученными по методикам предшествующего уровня техники; конечный продукт формулы (IV) содержит намного меньше (макс. 0,07%) примеси формулы (V),выделенной нами, по сравнению с продуктами, полученными по методикам предшествующего уровня-3 010830 техники; высокую чистоту препарата можно надежно контролировать при производстве конечного продукта формулы (IV), поскольку можно проверять не только чистоту конечного продукта формулы (IV), но и чистоту конечного промежуточного соединения формулы П. Примеры Следующие примеры являются иллюстративными и не предназначаются для ограничения области притязаний изобретения. В примерах новое соединение формулы (II), конечный продукт формулы (IV) и его примесь формулы (V), полученные способом данного изобретения, исследовали следующими аналитическими методами. Чистота продукта, полученного способом по изобретению, определялась методом HPLC. Установка для HPLC - Thermo Separation Products, насос - Spectra System P2000, детектор - Spectra System UV2000Control, сбор и обработка данных - программа ChromQuest, вер. 2.51. Условия хроматографии: колонка YMC-Pack Pro C18, 1504,6 мм внутр. диам., 5 мкм. Элюент A: смесь из 140 мл ацетонитрила + 30 мл метанола + 30 мл тетрагидрофурана + 800 мл 0,1 M ацетата аммония; элюент B: смесь из 400 мл ацетонитрила + 100 мл метанола + 100 мл тетрагидрофурана + 400 мл 0,1 M ацетата аммония; из них состояла система растворителей для линейного градиента, приведенная в таблице. Скорость протока - 1 мл/мин, температура колонки - комнатная, длина волны детектора - 235 нм. Растворение анализируемого образца - в смеси из 77% элюента A + 23% элюента В. Вводимый объем 10 мкл. Система растворителей для градиента HPLC Спектры 1 Н-ЯМР и 13 С-ЯМР регистрировали на спектрометре Varian INOVA (500 МГц для Н-ЯМР). Измерения проводились в дейтерий-пиридине или дейтерий-дихлорметане при 30C. Химические сдвигиприводятся в ppm с использованием тетраметилсилана (TMS) в качестве внутреннего стандарта (TMS - 0,00 ppm). B спектрах ЯМР применяются следующие сокращения: s - синглет, d - дублет,t - триплет, q - квартет, mмультиплет, dd - двойной дублет, br - широкий мультиплет, dm - дублетный мультиплет. Спектры MS высокого разрешения для соединений измеряли методом FAB-MS. Установка - Finnigan MAT 955Q. Метод ионизации FAB - ионная пушка Cs+ на 20 кВ, матричный растворитель 3-нитробензиловый спирт. Оценка спектров - m/z. Спектры FAB дочерних ионов получали методом тандемной MS. ИК-спектры регистрировали на спектрофотометре Perkin-Elmer 1000, используя таблетки бромида калия при спектральном разрешении в 4 см-1. Краткое описание чертежей Для того чтобы облегчить понимание изобретения, приводятся следующие фигуры. Фиг. 1. Профиль элюции конечного продукта формулы (II), полученный методом HPLC. Абсцисса: время элюции в мин, ордината: поглощение в мЕд. Фиг. 2. Спектр 1 Н-ЯМР соединения формулы (II) в дейтерий-пиридине, химические сдвиги приведены в ppm. Фиг. 3. Спектр FAB-MS соединения формулы (II). Абсцисса: m/z, ордината: интенсивность иона в %. Фиг. 4. ИК-спектр соединения формулы (II). Абсцисса: длина волны (см-1), ордината: пропускание в %. Фиг. 5. Профиль элюции конечного продукта формулы (IV), полученный методом HPLC. Абсцисса: время элюции в мин, ордината: поглощение в мЕд. Фиг. 6. Спектр 1 Н-ЯМР соединения формулы (V) в дейтерий-дихлорметане, химические сдвиги приведены в ppm. Фиг. 7. Спектр 13 С-ЯМР соединения формулы (V) в дейтерий-дихлорметане, химические сдвиги приведены в ppm. Фиг. 8. Спектр FAB-MS соединения формулы (V). Абсцисса: m/z, ордината: интенсивность иона в %. Фиг. 9. Спектр FAB дочерних ионов соединения формулы (V). Абсцисса: m/z, ордината: интенсивность иона в %. Фиг. 10. ИК-спектр соединения формулы (V). Абсцисса: длина волны (см-1), ордината: пропускание в %. Фиг. 11. Схема синтеза соединения (II). Фиг. 12. Схема синтеза соединения (IV). Фиг. 13. Структурная формула соединения (V). 1-4 010830 Пример 1. N-3-[3-(Диметиламино)-1-оксо-2-пропенил]фенил-N-этилацетамида гидрохлорид формулы (II). В атмосфере азота суспендировали 31,5 г N-3-[3-(диметиламино)-1-оксо-2-пропенил]фенил-Nэтилацетамида формулы (I) в 300 мл ацетона при комнатной температуре и по каплям добавляли 10,4 мл концентрированной соляной кислоты. После добавления 0,5 мл концентрированной соляной кислоты получали однородный раствор, а выпадение кристаллов начиналось после добавления 3-4 мл концентрированной соляной кислоты. После добавления всего количества концентрированной соляной кислоты реакционную смесь перемешивали при комнатной температуре в течение 30 мин и еще 1 ч при 0-5C. Выпавшие в осадок кристаллы отфильтровывали, промывали дважды 10 мл холодного (3-5 С) ацетона и высушивали при менее 50C, получая 31,15 г (87%) указанного в заголовке соединения. Температура плавления: 136-137 С. Аналитические характеристики полученного при этом продукта. Профиль элюции гидрохлоридаN-3-[3-(диметиламино)-1-оксо-2-пропенил]фенил-Nэтилацетамида формулы (II), полученный методом HPLC, представлен на фиг. 1. Абсцисса: время элюции в мин, от 0 до 31 мин; ордината: поглощение в мЕд. Концентрация образца составляла 0,5 мг/мл. Время удержания гидрохлоридаN-3-[3-(диметиламино)-1-оксо-2-пропенил]фенил-N-этилацетамида формулы (II) составило 4,0 мин. Определение осуществлялось по нормализованной площади. Согласно HPLC чистота продукта составила 98,5%. Спектр 1 Н-ЯМР соединения формулы (II) в дейтерий-пиридине представлен на фиг. 2, на которой химические сдвигиприведены в ppm с использованием тетраметилсилана (TMS) в качестве внутреннего стандарта (TMS=0,00 ppm). Характерны следующие химические сдвиги: 1 Н-ЯМР: 1,03 (t, 3H), 1,87 (s, 3H), 2,74 (s, br, 3H), 2,84 (s, br, 3H), 3,79 (q, br, 2H), 6,07 (d,3JH,H=12,5 Гц, 1H), 7,29-7,33 (m, br, 1H), 7,50-7,54 (m, 1H), 8,10 (d, 3JH,H=12,5 Гц, 1H), 8,19-8,23 (m, 2H),16,07 (s, br, 1H). Спектр FAB-MS соединения формулы (II) представлен на фиг. 3. Характерен ион протонированной молекулы при m/z = 261, тогда как ион молекулы с катионом натрия при m/z = 283. Пики других фрагментов: образование иона 3-(диметиламино)-1-оксо-2-пропенила (m/z = 98), расщепление боковой цепи 2-диметиламиноэтилена (m/z = 190), расщепление ацетильной группы (m/z = 217), элиминация этана из иона протонированной молекулы (m/z = 231). Пики при m/z = 77, 136 и 154 могут исходить от матричного растворителя. ИК-спектр соединения формулы (II) представлен на фиг. 4. Характерны следующие ИК-полосы поглощения (см-1): Другие характерные полосы поглощения (см-1): 3050, 2932, 1562, 1492, 1259, 1185, 1146,1067. Пример 2. N-[3-(3-Цианопиразоло[1,5-а]пиримидин-7-ил)фенил]-N-этилацетамид формулы (IV). Пропускали азот через смесь из 150 мл очищенной воды и 50 мл этанола и в атмосфере азота вносили 10,8 г 3-амино-4-пиразолкарбонитрила формулы (III) в эту смесь. После растворения реакционную смесь нагревали до 45-50C и добавляли 29,6 г гидрохлорида N-3-[3-(диметиламино)-1-оксо-2 пропенил]фенил-N-этилацетамида формулы (II) тремя равными порциями через каждые 15 мин. Реакционную смесь, которая вначале представляла собой раствор, а затем превращалась в суспензию, перемешивали при этой температуре в течение 1 ч в атмосфере азота. Затем полученную суспензию охлаждали до 0-5C и перемешивали при этой температуре в течение 2 ч. Выпавшие в осадок кристаллы отфильтровывали, промывали 25 мл ледяного этанола и 225 мл ледяной воды. Продукт высушивали под вакуумом при температуре менее 50C. Полученный при этом продукт растворяли в 280 мл метанола в атмосфере азота при 65-67C, для того чтобы удалить механические загрязнения. Раствор фильтровали, а фильтрат охлаждали до 0-5C. Смесь выдерживали при этой температуре в течение 2 ч, после чего выпавшие в осадок кристаллы отфильтровывали, промывали 210 мл ледяного метанола и высушивали под вакуумом при менее 50C, получая 28,2 г (92,5%) указанного в заголовке соединения. Аналитические характеристики полученного при этом продукта. Профиль элюции соединения формулы (IV), полученный методом HPLC, представлен на фиг. 5. Абсцисса: время элюции в мин, от 0 до 31 мин; ордината: поглощение в мЕд. Концентрация образца составляла 1,0 мг/мл. Время удержания соединения формулы (IV) составило 10,2 мин. Определение проводилось методом внешнего стандарта, по разведенному в тысячу раз образцу стандартного раствора. Согласно HPLC чистота продукта формулы (IV) составила 99,8%. Время удержания N-3-(3-цианопиразоло[1,5-а]пиримидин)-6-ил-3-[(3-N-этилацетамидофенил)-3 оксопропен-1-ил]-(пиримидин-7-ил)фенил-N-этилацетамида формулы (V), представляющего собой примесь, которая образуется с выходом 0,07% и была охарактеризована нами, составляет 24,9 мин, как-5 010830 видно из фиг. 5. Эта примесь также была выделена из неочищенного конечного продукта, полученного в соответствии с известными методиками синтеза соединения формулы (IV), причем конечные продукты по этим методикам содержали большее количество (0,1%) примеси формулы (V). Спектры 1 Н-ЯМР и 13 С-ЯМР соединения формулы (V) в дейтерий-дихлорметане представлены на фиг. 6 и 7, на которых химические сдвигиприведены в ppm с использованием тетраметилсилана(TMS) в качестве внутреннего стандарта (TMS=0,00 ppm). Характерны следующие химические сдвиги соединения формулы (V): 1 Н-ЯМР: 1,12 (t, br, 3H), 1,27 (t, br, 3H), 1,79 (s, br, 3H), 3,76 (q, br, 4H), 7,43-7,86 (m, 9H), 7,98-8,02(С), 169,9 (С), 187,8 (С). Спектр FAB-MS соединения формулы (V) представлен на фиг. 8. Характерен ион протонированной молекулы при m/z = 521. Пики при m/z = 107, 136, 137, 154, 289 и 307 могут исходить от матричного растворителя. Спектр FAB дочерних ионов соединения формулы (V) представлен на фиг. 9. Пики фрагментов(m/z = 479 и 437) могут исходить от иона протонированной молекулы при элиминации одной или двух кетеновых групп соответственно. ИК-спектр соединения формулы (V) представлен на фиг. 10. Характерны следующие ИК полосы поглощения (см-1): Другие характерные полосы поглощения (см -1): 2926, 2854, 1520, 1394, 1265, 1199, 1035, 641, 583. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ синтеза терапевтически применимого N-[3-(3-цианопиразоло[1,5-а]пиримидин-7 ил)фенил]-N-этилацетамида формулы (IV), содержащего менее 0,2% примесей, включающий взаимодействие 3-амино-4-пиразолкарбонитрила формулы (III) с эквимолярным количеством гидрохлоридаN-3-[3-(диметиламино)-1-оксо-2-пропенил]фенил-N-этилацетамида формулы (II) в органических растворителях или в смеси из воды и органических растворителей при pH 3,0-3,5, с последующим выделением и перекристаллизацией продукта. 2. Способ по п.1, в котором в качестве органического растворителя используют метанол, этанол,изопропанол или н-пропанол. 3. Способ по любому из пп.1, 2, в котором небольшими порциями в реакционную смесь добавляют соединения формулы (II). 4. Способ по любому из пп.1-3, в котором реакцию проводят при 15-50C. 5. Способ по любому из пп.1-3, в котором реакцию проводят при 45-50C. 6. Способ по любому из пп.1-5, в котором реакцию проводят в течение 0,5-1,0 ч. 7. Гидрохлорид N-3-[3-(диметиламино)-1-оксо-2-пропенил]фенил-N-этилацетамида формулы (II),имеющий чистоту более 99,5%. 8. Способ синтеза гидрохлоридаN-3-[3-(диметиламино)-1-оксо-2-пропенил]фенил-Nэтилацетамида формулы (II), имеющего чистоту более 99,5%, включающий суспендированиеN-3-[3-(диметиламино)-1-оксо-2-пропенил]фенил-N-этилацетамида формулы (I) в ацетоне в атмосфере азота при 20-25C и добавление эквимолярного количества концентрированной соляной кислоты в суспензию.
МПК / Метки
МПК: C07D 487/04
Метки: n-[3-(-3-цианопиразоло[1,5a, способ, синтеза, пиримидин-7-ил)-фенил]-n-этилацетамида
Код ссылки
<a href="https://eas.patents.su/13-10830-sposob-sinteza-n-3-3-cianopirazolo15a-pirimidin-7-il-fenil-n-etilacetamida.html" rel="bookmark" title="База патентов Евразийского Союза">Способ синтеза n-[3-(-3-цианопиразоло[1,5a] пиримидин-7-ил)-фенил]-n-этилацетамида</a>
Способ синтеза (2r,2-альфа-r, 3а)-2-[1-[3,5-бис (трифторметил)фенил]этокси]-3-(4-фторфенил)-1,4-оксазина

Номер патента: 4743
Опубликовано: 26.08.2004
Авторы: Конрад Карен М., Цай Фух-Ронг, Брэндс Карел М.Ес, Зао Мэттью М.
МПК: C07D 265/32
Метки: синтеза, 3а)-2-[1-[3,5-бис, трифторметил)фенил]этокси]-3-(4-фторфенил)-1,4-оксазина, 2r,2-альфа-r, способ
Формула / Реферат:
1. Способ получения соединения формулы который включает контактирование соединения формулы с соединением формулы где Y выбирают из MgCl, MgBr, MgI и Li, в первом растворителе с последующим гидрированием во втором растворителе. 2. Способ по п.1, где Y означает MgBr. 3. Способ по п.1, где первый растворитель выбирают из группы, включающей толуол, тетрагидрофуран, 1,2-диметоксиэтан (ДМЭ), диэтиловый эфир, диизопропиловый эфир, МТБЭ,...
Способ получения 5-[2-этокси-5-(4-метилпиперазин-1-илсульфонил)-фенил]-1-метил-3-н-пропил,- 1,6-дигидро-7н-пиразоло[4,3-d]пиримидин-7-она

Номер патента: 102
Опубликовано: 27.08.1998
Авторы: Вуд Элберт Шо, Данн Питер Джеймз
МПК: C07D 487/04
Метки: 5-[2-этокси-5-(4-метилпиперазин-1-илсульфонил)-фенил]-1-метил-3-н-пропил, способ, получения, 1,6-дигидро-7н-пиразоло[4,3-d]пиримидин-7-она
Формула / Реферат:
1. Способ получения 5-[2-этокси-5-(4-метилпиперазин-1-ил-сульфонил)-фенил]-1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло-[4,3-d] пиримидин-7-она формулы (I) отличающийся тем, что соединение формулы (II) подвергают реакции циклизации в щелочной, нейтральной или кислой среде. 2. Способ по п.1, отличающийся тем, что циклизацию осуществляют в присутствии основания, предпочтительно в растворителе, возможно в присутствии перекиси водорода или...
Новый способ синтеза сложных эфиров n-[(s)-1-карбоксибутил]-(s)-аланина и их применение для синтеза периндоприла

Номер патента: 9980
Опубликовано: 28.04.2008
Авторы: Бреар Фабьенн, Фуже Клод
МПК: C07C 229/16, C07C 229/12, C07C 227/32...
Метки: синтеза, n-[(s)-1-карбоксибутил]-(s)-аланина, сложных, применение, эфиров, способ, новый, периндоприла
Формула / Реферат:
1. Способ синтеза соединений формулы (I) в которой R представляет собой линейную или разветвленную С1-С6-алкильную группу, который характеризуется тем, что морфолинон формулы (III) в которой P представляет собой защитную группу для функциональной аминогруппы, подвергают реакции с аллилбромидом или аллилтрифлатом в присутствии основания, получая соединение формулы (IV), имеющее (3S,5S)-конфигурацию в которой P имеет значения, указанные выше,...
Полиморфы n-метил-n-(3-{3-[2-тиенилкарбонил]пиразол [1,5-а]пиримидин-7-ил}фенил)ацетамида и относящиеся к ним композиции и способы

Номер патента: 4706
Опубликовано: 24.06.2004
Авторы: О'доннелл Патрик Б., Тил Вилльям Дж.
МПК: C07D 487/04, A61P 25/20, A61K 31/519...
Метки: n-метил-n-(3-{3-[2-тиенилкарбонил]пиразол, композиции, относящиеся, способы, ним, полиморфы, 1,5-а]пиримидин-7-ил}фенил)ацетамида
Формула / Реферат:
1. По существу чистый полиморф формы I N-метил-N-(3-{3-[2-тиенилкарбонил]пиразол[1,5-a]пиримидин-7-ил}фенил)ацетамида, показывающий преобладающую эндотерму при около 192-198шC по данным измерения с помощью дифференциального сканирующего калориметра. 2. По существу чистый полиморф формы I по п.1, который включает менее чем около 6% полиморфа формы II по массе. 3. По существу чистый полиморф формы I по п.2, который включает менее чем около 2%...
Новый способ синтеза (2s, 3аs, 7аs )-пергидроиндол-2-карбоновой кислоты и её сложных эфиров, а также их применение для синтеза периндоприла

Номер патента: 7948
Опубликовано: 27.02.2007
Авторы: Дюбюффе Тьерри, Ланглуа Паскаль
МПК: C07D 209/42
Метки: 3аs, сложных, периндоприла, кислоты, синтеза, также, пергидроиндол-2-карбоновой, эфиров, способ, применение, новый
Формула / Реферат:
1. Способ синтеза соединений формулы (I) в которой R представляет собой атом водорода или защитную группу для кислотной группы, характеризующийся тем, что 1-(1-циклогексен-1-ил)пирролидин формулы (III) подвергают реакции с соединением формулы (IV) в которой R является таким, как определено для формулы (I), a R' представляет собой защитную группу для аминовой группы, которая является отличной от R, с получением соединения формулы (V) в...
Предыдущий патент: Клопидогрель и его полиморфные формы
Следующий патент: Полиморфная форма d гидробромида метил-(+)-(s)-альфа-(2-хлорфенил)-6,7-дигидротиено [3,2-c] пиридин-5(4н)- ацетата
Случайный патент: Самогерметизирующийся выдачной затвор для гибких упаковок