Энантио- и стереоспецифические синтезы β-амино-α-гидроксиамидов
Номер патента: 22038
Опубликовано: 30.10.2015
Авторы: Альбанесе-Уолкер Дженнифер, Божен Стефан Л., Смит Рэнди М., Пуарье Марк, Чэнь Юнган, Феррейра Амадео, Ляо Хунбяо, Беннет Фрэнк, Траверз Джон, Ко Шэнь-Чунь, Демоншо Патрис, Арасаппан Ашок, Нджороге Ф.Джордж, Ван Лицзюнь, Миллер Стивен П., Мэттьюз Кеннет С., Лекхал Аззеддине, Хантер Томас Дж., Леонг Уилльям У., Трзаска Скотт Т., Кун Цзяньшэ





Формула / Реферат
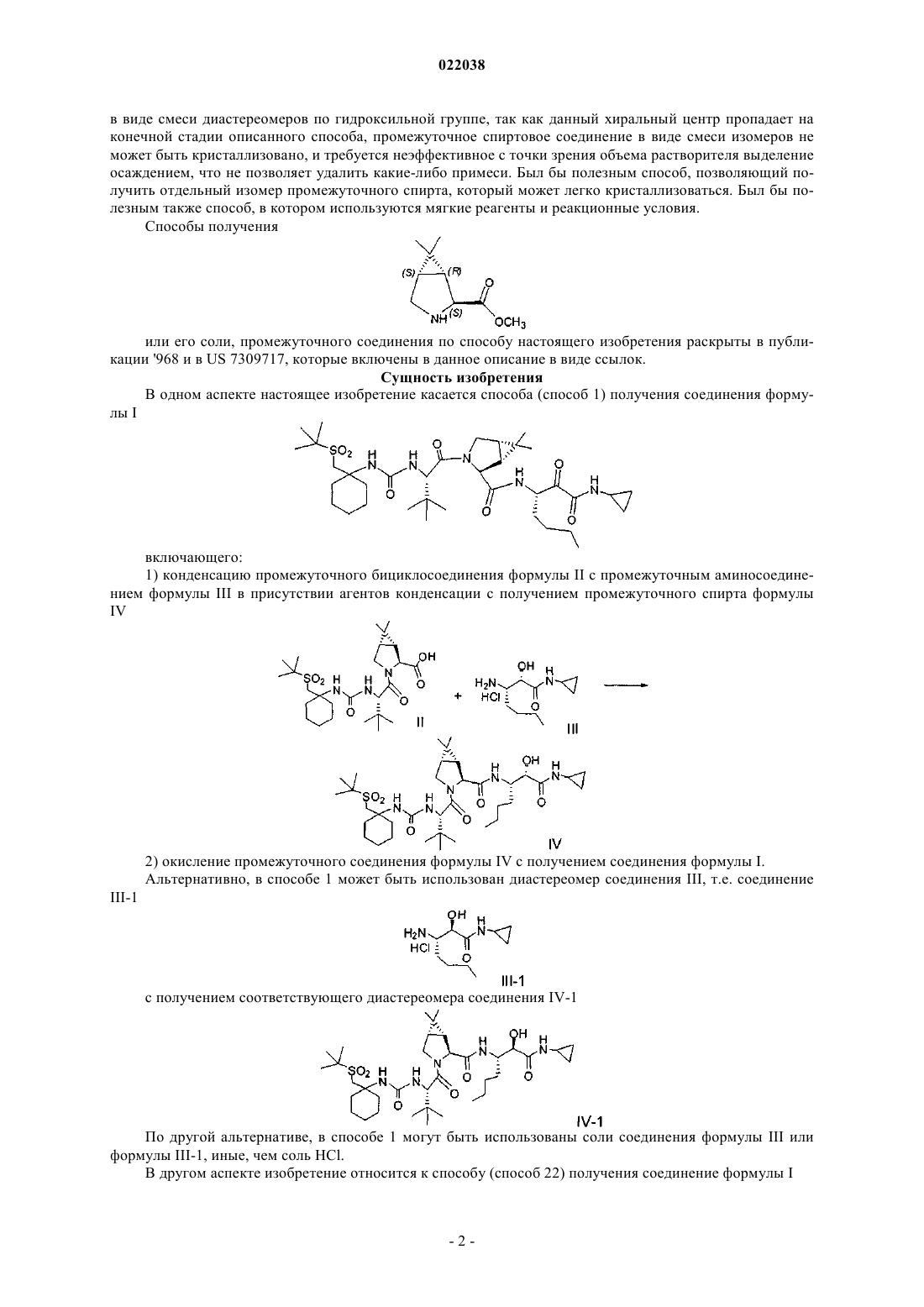
1. Способ получения соединения формулы I

включающий:
(а) конденсацию промежуточного бициклосоединения формулы II с промежуточным аминосоединением формулы III в присутствии агентов конденсации с получением промежуточного спирта формулы IV

(b) окисление промежуточного соединения формулы IV с получением соединения формулы I.
2. Способ по п.1, где соединения формул II и III подвергают конденсации, используя 1-этил-3-(3-диметиламинопропил)карбодиимид и 1-гидроксибензотриазол в присутствии N,N-диизопропилэтиламина.
3. Способ по п.1, где соединение формулы IV окисляют гипохлоритом натрия в присутствии каталитического или стехиометрического количества 2,2,6,6-тетраметил-1-пиперидинилоксила (TEMPO).
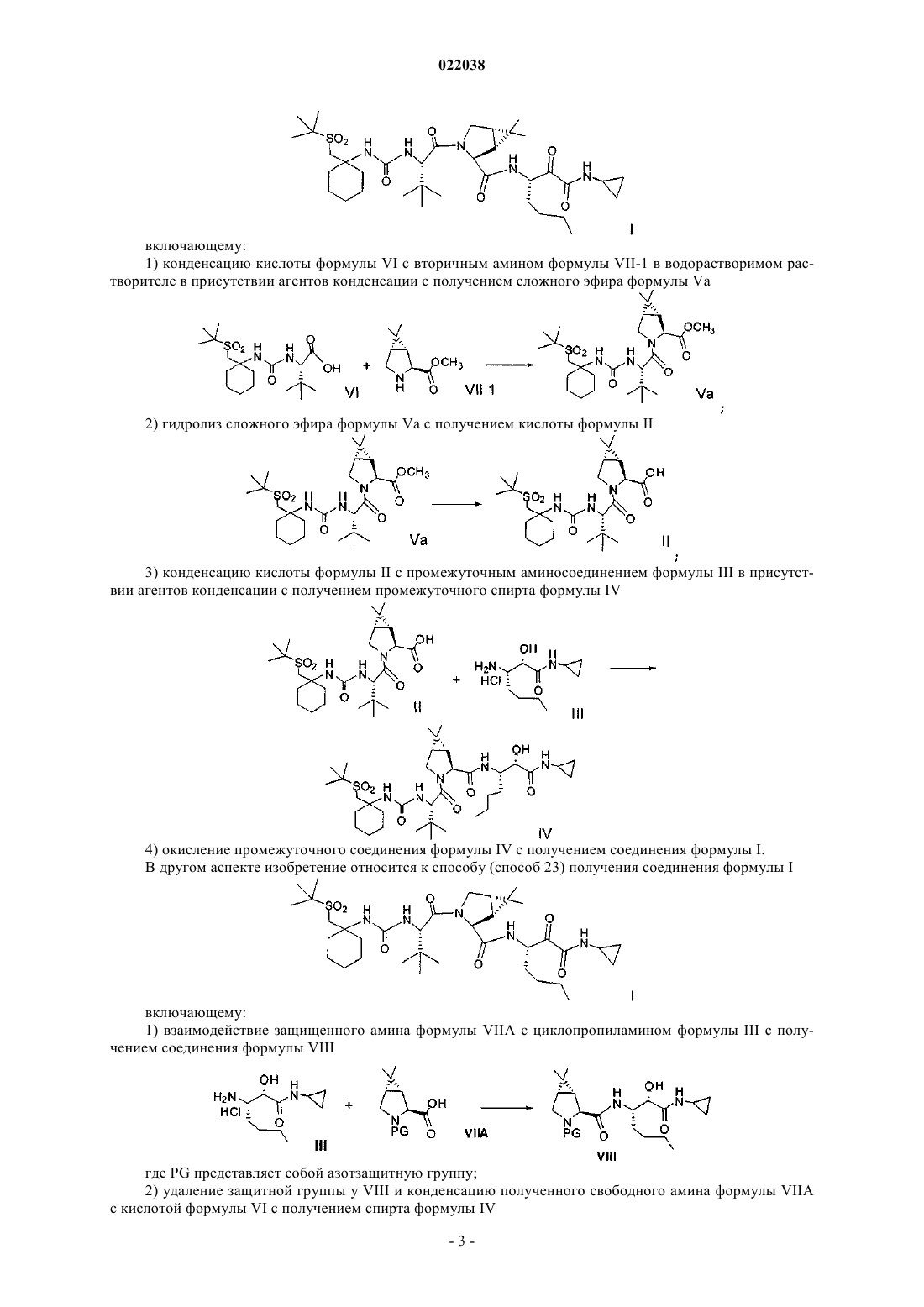
4. Способ получения соединения формулы I

включающий:
(а) конденсацию кислоты формулы VI с вторичным амином формулы VII-1 в водорастворимом растворителе в присутствии агентов конденсации с получением соединения формулы Va

(b) гидролиз сложного метилового эфира формулы Va с получением кислоты формулы II

(с) конденсацию кислоты формулы II с промежуточным аминосоединением формулы III в присутствии агентов конденсации с получением промежуточного спирта формулы IV

(d) окисление промежуточного соединения формулы IV с получением соединения формулы I.
5. Способ по п.4, где соединения формул VI и VII подвергают конденсации, используя 1-этил-3-(3-диметиламинопропил)карбодиимид и 1-гидроксибензотриазол в присутствии N-метилморфолина в ацетонитриле.
6. Способ по п.4, где соединения формул II и III подвергают конденсации, используя 1-этил-3-(3-диметиламинопропил)карбодиимид и 1-гидроксибензотриазол в присутствии N,N-диизопропилэтиламина.
7. Способ по п.4, где соединение формулы IV подвергают окислению гипохлоритом натрия в присутствии каталитического или стехиометрического количества 2,2,6,6-тетраметил-1-пиперидинилоксила (TEMPO).
Текст
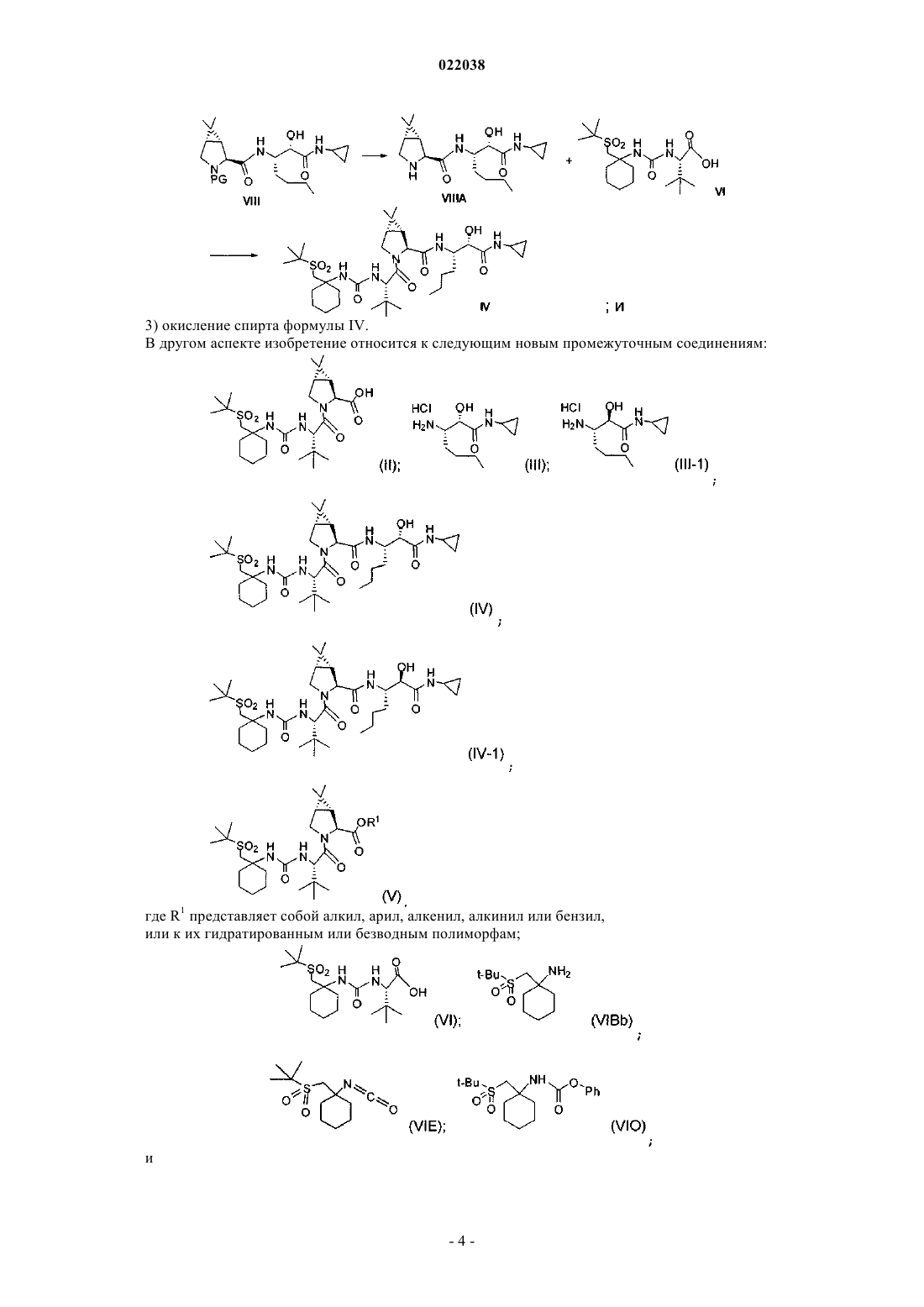
Описан способ получения соединения формулы I(71)(73) Заявитель и патентовладелец: МЕРК ШАРП ЭНД ДОМЭ КОРП. Перекрестные ссылки к родственным заявкам Заявка на данный патент основана и по ней испрашивается приоритет в соответствии с предварительными заявками США 61/229613, 61/229636, 61/229648, 61/229652 и 61/229618, каждая из которых была подана 29 июля 2009 г. и каждая из указанных заявок включена в данное описание в качестве ссылки в полном своем объеме. Область изобретения Настоящее изобретение относится к способу получения соединения формулы I, которое показало наличие активности в качестве ингибитора протеазы HCV. Настоящее изобретение относится также к способу получения промежуточных соединений, используемых при получении соединения формулы I,указываемого здесь также как (1R,5S)-N-[1(S)-[2-(циклопропиламино)-1,2-диоксоэтил]пентил]-3-[2(S)1-1,1-(диметилэтил)сульфонил]метил]циклогексил]амино]карбонил]амино]-3,3-диметил-1 оксобутил]-6,6-диметил-3-азабицикло[3.1.0]гексан-2(S)-карбоксамид. Предпосылки изобретения Указание какой-либо публикации в данном разделе или в любом разделе данного изобретения не является признанием того, что такая публикация является прототипом настоящего патента. Соединение формулы I в общем и конкретно раскрыто в опубликованном патенте США 2007/0042968, опубликованном 22 февраля 2007 г. (публикация '968), включенном в данное описание в качестве ссылки. Способы, подходящие для получения соединения формулы I, в целом описаны в публикации '968. В частности, в публикации '968 обсуждено получение сульфонкарбаматного соединения, например, соединения формулы 837, содержащего в качестве заместителя циклический сульфон (абзацы [0395]-[0403]). Способ представлен на следующей реакционной схеме: Способ, раскрытый в публикации '968, приводит к получению промежуточного спирта на стадии S7 в виде смеси диастереомеров по гидроксильной группе, так как данный хиральный центр пропадает на конечной стадии описанного способа, промежуточное спиртовое соединение в виде смеси изомеров не может быть кристаллизовано, и требуется неэффективное с точки зрения объема растворителя выделение осаждением, что не позволяет удалить какие-либо примеси. Был бы полезным способ, позволяющий получить отдельный изомер промежуточного спирта, который может легко кристаллизоваться. Был бы полезным также способ, в котором используются мягкие реагенты и реакционные условия. Способы получения или его соли, промежуточного соединения по способу настоящего изобретения раскрыты в публикации '968 и в US 7309717, которые включены в данное описание в виде ссылок. Сущность изобретения В одном аспекте настоящее изобретение касается способа (способ 1) получения соединения формулы I включающего: 1) конденсацию промежуточного бициклосоединения формулы II с промежуточным аминосоединением формулы III в присутствии агентов конденсации с получением промежуточного спирта формулы 2) окисление промежуточного соединения формулы IV с получением соединения формулы I. Альтернативно, в способе 1 может быть использован диастереомер соединения III, т.е. соединение с получением соответствующего диастереомера соединения IV-1 По другой альтернативе, в способе 1 могут быть использованы соли соединения формулы III или формулы III-1, иные, чем соль HCl. В другом аспекте изобретение относится к способу (способ 22) получения соединение формулы I включающему: 1) конденсацию кислоты формулы VI с вторичным амином формулы VII-1 в водорастворимом растворителе в присутствии агентов конденсации с получением сложного эфира формулы Va 2) гидролиз сложного эфира формулы Va с получением кислоты формулы II 3) конденсацию кислоты формулы II с промежуточным аминосоединением формулы III в присутствии агентов конденсации с получением промежуточного спирта формулы IV 4) окисление промежуточного соединения формулы IV с получением соединения формулы I. В другом аспекте изобретение относится к способу (способ 23) получения соединения формулы I включающему: 1) взаимодействие защищенного амина формулы VIIA с циклопропиламином формулы III с получением соединения формулы VIII где PG представляет собой азотзащитную группу; 2) удаление защитной группы у VIII и конденсацию полученного свободного амина формулы VIIA с кислотой формулы VI с получением спирта формулы IV 3) окисление спирта формулы IV. В другом аспекте изобретение относится к следующим новым промежуточным соединениям: где R1 представляет собой алкил, арил, алкенил, алкинил или бензил,или к их гидратированным или безводным полиморфам; Подробное описание изобретения В одном варианте осуществления изобретения настоящее изобретение относится к способу получения соединения формулы I, как описано в способе 1. В другом варианте осуществления изобретения настоящее изобретение относится к способу 1, где соединение формулы II получают путем преобразования соединения формулы Va в соответствующую кислоту. По способу 1 получения соединения формулы I конденсацию промежуточной кислоты формулы II и амина формулы III осуществляют, используя стандартные способы конденсации пептидов, известные в данной области, например, используя 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCi) и 1 гидроксибензотриазол (HOBt) в присутствии основания, такого как N,N-диизопропилэтиламин (DIPEA). Использование отдельного диастереомера амина III дает отдельный диастереомер соединения формулыIV, который кристаллизуют с высокой степенью чистоты из смеси этилацетат/вода. По способу, описанному в публикации '968, образовывалась смесь диастереомеров на стадии 7; смесь можно было не кристаллизовать, и было с точки зрения требуемого объема нецелесообразно выделять осадок, поскольку это не позволяло удалить какие-либо примеси. Способ в соответствии с настоящей формулой изобретения позволяет осуществить с точки зрения требуемого объема более эффективное выделение, уменьшает использование растворителя, повышает объемную способность реактора и возможность эффективного удаления примесей на этой стадии. Отдельный диастереомер амина III может быть заменен в способе на диастереомер III-1. Более того,хотя соль HCl и является предпочтительной для соединения III или III-1, может быть использована любая подходящая кислотно-аддитивная соль. Свободную кислоту формулы II получают, исходя из сложного эфира формулы V, способами, хорошо известными в данной области, например, путем обработки NaOH. Предпочтительными сложными эфирами формулы V являются такие сложные эфиры, где R1 представляет собой алкил или бензил; более предпочтительно R1 представляет собой метил. На стадии 2 способа 1, спирт формулы IV подвергают окислению до кетона формулы I одним из многих возможных способов окисления, включая:i) основанные на TEMPO (2,2,6,6-тетраметил-1-пиперидинилокси, свободный радикал) способы окисления, где TEMPO представляет собой стехиометрический реагент, где TEMPO не соответствует его полностью окисленной форме; стехиометрический или каталитический реагент, где TEMPO используют с дополнительным агентом окисления; дополнительные агенты окисления включают хлорную известь (гипохлорит натрия); перманганат (любая соль); диоксид марганца; оксон, перкислоту, такую как метаперхлорбензойную кислоту; пероксид, такой как пероксид водорода или алкил-пероксид водорода, такой как третбутилпероксид водорода; кислород, либо чистый, либо в виде компонента газовой смеси (например, воздух); щелочно-земельные металлы степени окисления +4, такие как соли церия; соли железа степени окисления +3; соли палладия степени окисления +2 либо отдельно, либо в виде кооксиданта с дополнительным стехиометрическим окислительным агентом; галогены степени окисления от 0 до +7; соединения N-йод, N-бром или N-хлор сукцинидного типа; хинон, такой как бензохинон;iii) основанное на сульфоксиде или селеноксиде окисление, такое как окисление по Шверну или по Моффатту;iv) окисление гипервалентными галогенами, такими как перйодинат Десса-Мартина или IBX (йодоксибензойная кислота), либо стехиометрически, либо каталитически со стехиометрическим кооксидантом;v) окисление в присутствии переходных металлов, таких как Ag, Ru, V, Mo, Cu, Co, Cr, Pb, Fe, Pd или Mn, либо стехиометрически, либо каталитически со стехиометрическим кооксидантом;vi) окисление по Оппенауэру, где стехиометрическим окислителем является кетон и катализатор,такой как Al(OR)3;viii) окисление в присутствии диоксиранов (например, DMDO). По предпочтительному способу окисления в качестве окислителя используют хлорную известь и в качестве катализатора - TEMPO. Соединение формулы IV обладает плохой растворимостью во многих растворителях: в качестве одного примера подходящего растворителя для окисления можно указать влажный метилацетат. Способ 22 получения соединения формулы I предпочтительно включает гидролиз сложного мети-5 022038 лового эфира V в свободную кислоту II способами, известными в данной области, например, путем обработки основанием, таким как NaOH, затем конденсацией кислоты II с амином III и окисление полученного спирта, как описано в способе 1. В способе 23 получения соединения формулы I используются промежуточные соединения, подобные тем, которые описаны в способе 22, но объединенные в другом порядке. Защищенный по аминогруппе амин формулы VIIA, где PG представляет собой группу, такую как трет-бутилкарбонил (ВОС) или карбобензилокси (CBZ), вводят во взаимодействие с аминоспиртом формулы III с получением соединения формулы VIII, которое затем подвергают удалению защитных групп и взаимодействию с кислотой формулы VI с получением спирта формулы IV. Спирт формулы IV затем окисляют с получением соединения формулы I, как описано в способе 1. В описании были использованы следующие аббревиатуры, помимо указанных выше: Me = метил,Et = этил, Bu = бутил, Ms = мезил = метансульфонил, Ts = тозил = толуолсульфонил, Ph = фенил; ТГФ = тетрагидрофуран; LDA = диизопропиламид лития; TMSCl = хлортриметил силан; МТВЕ = метиловый трет-бутиловый эфир; RT = комнатная температура; ТФУ = трифторуксусная кислота; 2-МеТГФ = 2 метилтетрагидрофуран. Как здесь используется, если не указано иного, следующие термины определены далее."Алкил" обозначает алифатическую углеводородную группу, которая может быть прямой или разветвленной и содержащей от приблизительно 1 до приблизительно 20 атомов углерода в цепи. Предпочтительные алкильные группы содержат от приблизительно 1 до приблизительно 12 атомов углерода в цепи. Более предпочтительные алкильные группы содержат от приблизительно 1 до приблизительно 6 атомов углерода в цепи. Разветвление обозначает, что одна или несколько из низших алкильных групп,такая как метил, этил или пропил, присоединены к линейной алкильной цепи."Алкенил" обозначает прямую или разветвленную алифатическую углеводородную группу, имеющую по меньшей мере одну углерод-углеродную двойную связь и содержащую от приблизительно 2 до приблизительно 15 атомов углерода в цепи. Предпочтительные алкенильные группы содержат от приблизительно 2 до приблизительно 12 атомов углерода в цепи и более предпочтительно от приблизительно 2 до приблизительно 6 атомов углерода в цепи. "Низший алкенил" обозначает от приблизительно 2 до приблизительно 6 атомов углерода в цепи, которая может быть прямой или разветвленной. Неограничивающие примеры подходящих алкенильных групп включают этенил, пропенил, н-бутенил, 3-метилбут-2 енил, н-пентенил, октенил и деценил. Предпочтительной алкенильной группой является аллил."Алкинил" обозначает алифатическую углеводородную группу, имеющую по меньшей мере одну углерод-углеродную тройную связь, и которая может быть прямой или разветвленной и содержащей от приблизительно 2 до приблизительно 15 атомов углерода в цепи. Предпочтительные алкинильные группы содержат от приблизительно 2 до приблизительно 12 атомов углерода в цепи и более предпочтительно от приблизительно 2 до приблизительно 4 атомов углерода в цепи. Разветвление обозначает, что одна или несколько из низших алкильных групп, такая как метил, этил или пропил, присоединены к линейной алкинильной цепи. "Низший алкинил" обозначает от приблизительно 2 до приблизительно 6 атомов углерода в цепи, которая может быть прямой или разветвленной. Неограничивающие примеры подходящих алкинильных группы включают этинил, пропинил, 2-бутинил и 3-метилбутинил."Арил" обозначает ароматическую моноциклическую или полициклическую кольцевую систему,содержащую от приблизительно 6 до приблизительно 14 атомов углерода, предпочтительно от приблизительно 6 до приблизительно 10 атомов углерода. Арильная группа необязательно может быть замещена одним или несколькими "заместителями кольцевой системы", которые могут быть одинаковыми или различными и имеют значения, указанные выше. Неограничивающие примеры подходящих арильных групп включают фенил и нафтил. Примеры солей добавления кислот включают ацетаты, аскорбаты, бензоаты, бензолсульфонаты,бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, фумараты, гидрохлориды, гидробромиды, гидройодиды, лактаты, малеаты, метансульфонаты, нафталинсульфонаты, нитраты, оксалаты, фосфаты, пропионаты, салицилаты, сукцинаты, сульфаты, тартраты, тиоцианаты, толуолсульфонаты(также известные как тозилаты) и т.п. Кроме того, кислоты, которые обычно рассматриваются как подходящие для образования фармацевтически приемлемых солей из основных фармацевтических соединений, обсуждены, например, P. Stahl et al., Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties,Selection and Use. (2002), Zurich: Wiley-VCH; S. Berge et al., Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceutics (1986), 33, 201-217; Anderson et al., The Practice of MedicinalChemistry (1996), Academic Press, New York; и в публикации The Orange Book (FoodDrug Administration, Washington, D.С. на их веб-сайте). Указанные описания включены в настоящее описание в виде ссылок. В реактор помещали соединение VI (87,6 кг), соединение VII-1 (48,2 кг), HOBt (6 кг) и CH3CN (615 кг). Реакционную смесь охлаждали до температуры приблизительно 5C и добавляли NMM (35 кг) иEDCi (53,4 кг). Реакционную смесь нагревали до 20-25C в течение приблизительно 1 ч и затем до 3540C, при этом добавляли воду для кристаллизации соединения Va. Реакционную смесь охлаждали до 5C и выдерживали при этой температуре в течение приблизительно 4 ч. Соединение Va фильтровали и промывали водой. Данные XRD для гидратированного полиморфа Va являются следующими: Влажную лепешку соединения Va помещали в чистый сосуд и растворяли в этилацетате при температуре 25-30C. Раствор промывали водным раствором HCl, водным раствором K2CO3 и насыщенным раствором соли. Раствор затем упаривали в вакууме, поддерживая температуру в интервале между 35 и 50C. Добавляли дополнительное количество этилацетата и раствор нагревали при 65-70C. Поддерживая температуру при 65-70C, добавляли н-гептан, затем охлаждали полученный раствор до 0-5C. Соединение Va фильтровали и промывали смесью этилацетат/н-гептан. Влажную лепешку сушили в вакууме при температуре 55-60C с получением 96,6 кг кристаллического соединения Va (мольный выход = 79,2%). МС вычислено: 541,32; МС найдено (ES+, М+Н): 542,35. Данные XRD для безводного полиморфа Va являются следующими: В реактор для работы под давлением помещали уксусную кислоту (1,1 кг), метанол (55 кг) и соединение IIIE (10,9 кг). В отдельном сосуде суспендировали Pd/C (50%-ная влажность, 0,5 кг) в метаноле (5 кг). Суспензию Pd/C помещали в раствор, содержащий соединение IIIE. Полученную смесь помещали в атмосферу водорода под давлением 80 фунт на кв. дюйм (55 кПа) и перемешивали при температуре 60C в течение 7 ч. Реакционную смесь затем продували азотом и катализатор Pd/C отфильтровывали. Полученный раствор концентрировали в вакууме и нагревали до приблизительно 20C. Добавляли МТВЕ и полученный раствор кипятили с обратным холодильником. Добавляли концентрированную HCl (3 л) и продукт кристаллизовали путем охлаждения реакционной смеси до приблизительно 3C. Желаемое соединение фильтровали, промывали МТВЕ и сушили в вакууме, поддерживая температуру ниже 40C, с получением 5,5 кг соединения III (мольный выход = 83,0%). МС вычислено (свободное основание): 200,15; МС найдено (ES+, М+Н): 201,12. Получение соединения II Соединение Va (119,3 кг) растворяли в 2-МеТГФ (720 кг) и воде (180 кг). К этому раствору добавляли 50% NaOH (21,4 кг), поддерживая температуру между 20 и 30C. Реакционную смесь затем перемешивали в течение приблизительно 7 ч при температуре между 50 и 60C. Реакционную смесь охлаждали до температуры между 20 и 30C. рН реакционной смеси устанавливали равным 1,5-3,0 с помощью разбавленной фосфорной кислоты, поддерживая температуру между 20 и 30C. Полученную смесь перемешивали в течение 10 мин, отстаивали в течение 30 мин, и нижний, водный, слой отделяли и удаляли. Верхний, органический, слой промывали водой, затем концентрировали дистилляцией при атмосферном давлении. Концентрированный раствор подвергали замене растворителя на CH3CN путем продолжительной дистилляции при атмосферном давлении и кристаллизовали охлаждением до 0C. Кристаллический про-8 022038 дукт фильтровали, промывали CH3CN и сушили в вакууме при температуре между 45 и 55C с получением 97,9 кг соединения II (мольный выход = 83,7%). МС вычислено: 527,30; МС найдено (ES+, М+Н): 528,29. Получение соединения IV Соединение II (21,1 кг), соединение III (9,9 кг), HOBt (3,2 кг) и EDCi (11,2 кг) помещали в сосуд, затем добавляли CH3CN (63 кг), этилацетат (20 кг) и воду (1,5 кг). Реакционную смесь перемешивали и гетерогенную смесь охлаждали до температуры в интервале от -5 до +5C. В реакционную смесь добавляли DIPEA (11,2 кг), поддерживая температуру между -5 и +5C, и смесь перемешивали при температуре от -5 до +5C в течение 1 ч. Полученную реакционную смесь нагревали до 20-30C и перемешивали в течение 2-3 ч. Полученный продукт экстрагировали водным раствором HCl, водным раствором K2CO3 и водой. Желаемый продукт кристаллизовали из этилацетата путем охлаждения кипящего раствора (78C) до приблизительно 0C. Кристаллический продукт фильтровали и сушили при температуре 30C в вакууме с получением 23,1 кг соединения IV (мольный выход = 81,3%). МС вычислено: 709,44; МС найдено (ES+, М+Н): 710,47. Получение соединения I Соединение IV (22,5 кг), TEMPO (5 кг), NaOAc (45 кг), метилацетат (68 л), МТВЕ (158 л), воду (23 л) и уксусную кислоту (22,5 л) помещали в реактор. Реакционную смесь перемешивали при температуре 20-30C, давая твердым продуктам раствориться, и затем охлаждали до 5-15C. В реакционную смесь помещали раствор NaOCl (1,4 мол.экв.), поддерживая температуру приблизительно 10C. После завершения добавления NaOCl реакционную смесь перемешивали при температуре 10C в течение 2 ч. Реакцию гасили промыванием буферированным водным раствором смеси аскорбат натрия/HCl, затем промывали водой. Реакционную смесь подвергали замене растворителя на ацетон в вакууме, поддерживая температуру ниже 20C; желаемый продукт кристаллизовали путем добавления воды и сушили в вакууме, поддерживая температуру ниже 40C с получением 18,6 кг соединения I (мольный выход = 82,7%). МС вычислено: 707,43: МС найдено (ES+, М+Н): 708,44. Получение соединения VIR В воду (45 л) добавляли Na2WO4 (2,2 кг, 0,013 экв.), затем трет-бутилметилсульфид (54,8 кг, 1 экв.). Добавляли при перемешивании Н 2 О 2 (115 кг, 2,2 экв.), в атмосфере N2, при этом температуру реакции поддерживали ниже 30C. После завершения реакции (отслеживалась с помощью ВЭЖХ) добавлялиNaCl (50 кг), затем МТВЕ (214 л) и реакционную смесь выдерживали при температуре 223C в течение 15 мин. Органический слой отделяли и водный слой вновь экстрагировали МТВЕ (110 л). Объединенные органические слои упаривали для уменьшения объема и дистиллят медленно охлаждали до 10-15C для кристаллизации желаемого продукта. Полученную смесь охлаждали до 73C и добавляли гептан (110 л); через 1 ч смесь охлаждали до 03C. Через 1 ч твердый продукт выделяли центрифугированием, дважды промывали гептаном (233 л) и сушили при 30C. Продукт VIR получали с выходом 85%. МС вычислено: 136; МС найдено: 137 (М+Н). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы I(а) конденсацию промежуточного бициклосоединения формулы II с промежуточным аминосоединением формулы III в присутствии агентов конденсации с получением промежуточного спирта формулы(b) окисление промежуточного соединения формулы IV с получением соединения формулы I. 2. Способ по п.1, где соединения формул II и III подвергают конденсации, используя 1-этил-3-(3 диметиламинопропил)карбодиимид и 1-гидроксибензотриазол в присутствииN,Nдиизопропилэтиламина. 3. Способ по п.1, где соединение формулы IV окисляют гипохлоритом натрия в присутствии каталитического или стехиометрического количества 2,2,6,6-тетраметил-1-пиперидинилоксила (TEMPO). 4. Способ получения соединения формулы I(а) конденсацию кислоты формулы VI с вторичным амином формулы VII-1 в водорастворимом растворителе в присутствии агентов конденсации с получением соединения формулы Va(b) гидролиз сложного метилового эфира формулы Va с получением кислоты формулы II(с) конденсацию кислоты формулы II с промежуточным аминосоединением формулы III в присутствии агентов конденсации с получением промежуточного спирта формулы IV(d) окисление промежуточного соединения формулы IV с получением соединения формулы I. 5. Способ по п.4, где соединения формул VI и VII подвергают конденсации, используя 1-этил-3-(3 диметиламинопропил)карбодиимид и 1-гидроксибензотриазол в присутствии N-метилморфолина в ацетонитриле. 6. Способ по п.4, где соединения формул II и III подвергают конденсации, используя 1-этил-3-(3 диметиламинопропил)карбодиимид и 1-гидроксибензотриазол в присутствииN,Nдиизопропилэтиламина. 7. Способ по п.4, где соединение формулы IV подвергают окислению гипохлоритом натрия в присутствии каталитического или стехиометрического количества 2,2,6,6-тетраметил-1-пиперидинилоксила
МПК / Метки
МПК: C07D 209/52, C07C 315/00, C07C 237/02, C07K 5/06
Метки: стереоспецифические, beta;-амино-α-гидроксиамидов, синтезы, энантио
Код ссылки
<a href="https://eas.patents.su/12-22038-enantio-i-stereospecificheskie-sintezy-beta-amino-alpha-gidroksiamidov.html" rel="bookmark" title="База патентов Евразийского Союза">Энантио- и стереоспецифические синтезы β-амино-α-гидроксиамидов</a>
Способы получения 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила

Номер патента: 7953
Опубликовано: 27.02.2007
Авторы: Виллемс Йоаннес Йозефус Мария, Лендерс Рубен Герардус Жорж, Медар Барт Петрус Анна Мария Йозеф, Гийемон Жером Эмиль Жорж, Хэрес Ян, Паскье Элизабет Тереза Жанна, Жанссен Поль Адриан Ян, Схилс Дидье Филипп Робер
МПК: A61K 31/505, C07C 253/20, C07C 253/30...
Метки: получения, 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, способы
Формула / Реферат:
1. Способ получения 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила формулы (I) его N-оксида, фармацевтически приемлемой кислотно-аддитивной соли, четвертичного амина или стереохимически изомерной формы, который включает осуществление взаимодействия промежуточного соединения формулы (II) его подходящей кислотно-аддитивной соли или стереохимически изомерной формы с промежуточным соединением формулы (III) его...
Производные [[ 2-(амино-3,4-диоксо-1-циклобутен-1-ил)амино ]алкил]кислоты, применяемые для лечения боли

Номер патента: 9993
Опубликовано: 30.06.2008
Авторы: Брандт Майкл Ричард, Залеска Маргарет Мария, Мойер Джон Аллен
МПК: A61K 31/198, A61K 31/4245, A61K 31/197...
Метки: алкил]кислоты, 2-(амино-3,4-диоксо-1-циклобутен-1-ил)амино, производные, лечения, боли, применяемые
Формула / Реферат:
1. Способ лечения боли у млекопитающего, включающий введение млекопитающему, нуждающемуся в лечении боли, эффективного для лечения боли количества по меньшей мере одного соединения, имеющего формулу где R1 представляет собой водород, алкил, содержащий 1-6 атомов углерода, или фенилалкил, содержащий 7-12 атомов углерода; R2 представляет собой водород, алкил, содержащий 1-6 атомов углерода, алкенил, содержащий 2-6 атомов углерода, или фенилалкил,...
Фумарат 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила

Номер патента: 11036
Опубликовано: 30.12.2008
Авторы: Стапперс Альфред Элизабет, Копманс Алекс Херман, Петерс Йозеф, Вандекрюйс Рогер Петрус Гереберн, Стевенс Пол Теодор Агнес
МПК: A61P 31/18, A61K 31/505
Метки: 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, фумарат
Формула / Реферат:
1. Соединение формулы (I) его N-оксид или стереохимически изомерная форма. 2. Соединение по п.1, где указанное соединение представляет собой 3. Соединение по п.1 или 2 для использования в качестве лекарственного средства. 4. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента - терапевтически эффективное количество соединения по п.1 или 2. 5. Фармацевтическая композиция по п.4, где...
Гидрохлорид 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила

Номер патента: 13686
Опубликовано: 30.06.2010
Авторы: Стапперс Альфред Элизабет, Вандекрюйс Рогер Петрус Гереберн, Копманс Алекс Херман, Петерс Йозеф, Стевенс Пол Теодор Агнес
МПК: C07D 239/02, A61K 31/505, A61P 31/18...
Метки: гидрохлорид, 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила
Формула / Реферат:
1. Твердая фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения формулы (I-а)или его N-оксида.2. Фармацевтическая композиция по п.1, в которой соединение формулы (I-а) представляет собой полиморфную форму А, характеризующуюся пиками порошковой рентгеновской дифракции в положениях два тета 9,7±0,2°, 13,5±0,2° и 15,0±0,2°.3. Фармацевтическая...
2-[(5-хлор-2-{[3-метил-1-(1-метилэтил)-1н-пиразол-5-ил]амино}-4-пиридинил)амино]-n-(метилокси)бензамид в качестве ингибиторов fak и содержащая его фармацевтическая композиция

Номер патента: 21927
Опубликовано: 30.09.2015
Авторы: Пэн Синь, Адамс Джерри Лерой, Каспарек Иржи, Фейт Томас Х., Се Жэнь, Линь Хун, Джонсон Нейл В., Меллинджер Марк
МПК: A61K 31/535
Метки: фармацевтическая, 2-[(5-хлор-2-{[3-метил-1-(1-метилэтил)-1н-пиразол-5-ил]амино}-4-пиридинил)амино]-n-(метилокси)бензамид, качестве, содержащая, композиция, ингибиторов
Формула / Реферат:
1. Соединение 2-[(5-хлор-2-{[3-метил-1-(1-метилэтил)-1H-пиразол-5-ил]амино}-4-пиридинил)амино]-N-(метилокси)бензамид или его фармацевтически приемлемая соль.2. Соединение по п.1, представляющее собой 2-[(5-хлор-2-{[3-метил-1-(1-метилэтил)-1H-пиразол-5-ил]амино}-4-пиридинил)амино]-N-(метилокси)бензамид.3. Фармацевтическая композиция для лечения злокачественной опухоли, содержащая терапевтически эффективное количество соединения по п.1 или 2 и...
Предыдущий патент: Копреципитат, включающий ингибитор фосфодиэстеразы-5 (pde-5-ингибитор) и фармацевтически совместимый носитель, его получение и применение
Следующий патент: Полимерные красители
Случайный патент: Способ контактирования расплавленной мочевины с потоком газа