Способ получения амидов пиразолкарбоновой кислоты
Номер патента: 19475
Опубликовано: 31.03.2014
Авторы: Джордано Фанни, Лаггер Мартин, Мюллер Адриан, Грибков Денис










Формула / Реферат
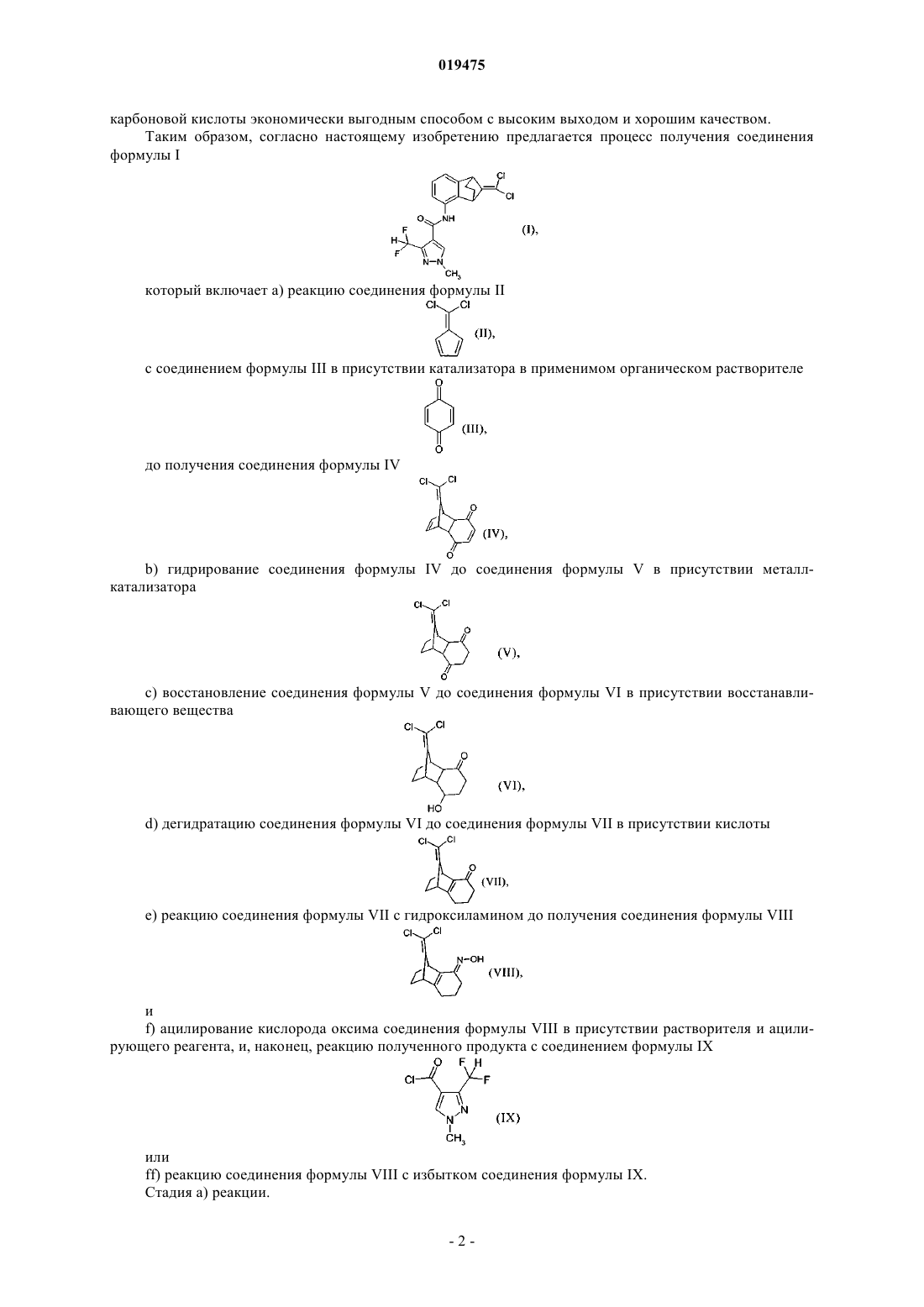
1. Способ получения соединения формулы I
который включает:
а) реакцию соединения формулы II
с соединением формулы III в присутствии катализатора в подходящем органическом растворителе
с получением соединения формулы IV
b) гидрирование соединения формулы IV до соединения формулы V в присутствии металл-катализатора
с) восстановление соединения формулы V до соединения формулы VI в присутствии восстанавливающего вещества
d) дегидратацию соединения формулы VI до соединения формулы VII в присутствии кислоты
е) реакцию соединения формулы VII с гидроксиламином с получением соединения формулы VIII
и
f) ацилирование кислорода оксима соединения формулы VIII в присутствии растворителя и ацилирующего реагента, и затем реакцию полученного продукта с соединением формулы IX
или
ff) реакцию соединения формулы VIII с избытком соединения формулы IX.
2. Способ по п.1, который включает f) ацилирование кислорода оксима соединения формулы VIII в присутствии растворителя и ацилирующего реагента, и затем реакцию полученного продукта с соединением формулы IX.
3. Способ по п.1, который включает ff) реакцию соединения формулы VIII с избытком в 2-3 эквивалента соединения формулы IX.
4. Соединение формулы IV
и его изомеры.
5. Соединение формулы V
и его изомеры.
6. Соединение формулы VI
и его изомеры.
7. Соединение формулы VII
и его изомеры.
8. Соединение формулы VIII
и его изомеры.
Текст
СПОСОБ ПОЛУЧЕНИЯ АМИДОВ ПИРАЗОЛКАРБОНОВОЙ КИСЛОТЫ Предметом настоящего изобретения является способ получения (9-дихлорметилен-1,2,3,4 тетрагидро-1,4-метанонафталин-5-ил)амида 3-дифторметил-1-метил-1H-пиразол-4-карбоновой кислоты. Соединение (9-дихлорметилен-1,2,3,4-тетрагидро-1,4-метанонафталин-5-ил)амида 3-дифторметил 1-метил-1H-пиразол-4-карбоновой кислоты и его микробицидные свойства описаны, например, в публикации WO 2007/048556. Получение (9-дихлорметилен-1,2,3,4-тетрагидро-1,4-метанонафталин-5-ил)амида 3-дифторметил-1 метил-1 Н-пиразол-4-карбоновой кислоты известно из публикации WO 2007/048556. Упомянутое соединение может быть получено согласно схемам 1 и 4 путем а) реакции соединения формулы А(В), где R' и R" обозначают, например, (С 1-С 4)алкил, до получения соединения формулы С соединения формулы Сb) гидрирования соединения формулы С до соединения формулы D в присутствии применимого металл-катализатора с) озонирования соединения формулы D и его последующей обработки восстанавливающим веществом до получения соединения формулы Еd) реакции соединения формулы Е в присутствии трифенилфосфина/тетрахлорида углерода до получения 2,9-дихлорметилиден-5-нитробензонорборнена формулы F е) гидрирования соединения формулы F до 2,9-дихлорметилиден-5-амино-бензонорборнена формулы G в присутствии металл-катализатораf) и путем реакции соединения формулы G с соединением формулы Н до получения (9-дихлорметилен-1,2,3,4-тетрагидро-1,4-метанонафталин-5-ил)амида 3-дифторметил 1-метил-1 Н-пиразол-4-карбоновой кислоты. Существенными недостатками этого предыдущего процесса получения является реакция озонолиза,которую сложно проводить, и дорогостоящая стадия d), для которой требуется применение трифенилфосфина. Упомянутые недостатки делают данный процесс неэкономичным и определенно не предназначенным для крупносерийного производства. Поэтому цель настоящего изобретения - предложить новый процесс производства (9-дихлорметилен-1,2,3,4-тетрагидро-1,4-метанонафталин-5-ил)амида 3-дифторметил-1-метил-1 Н-пиразол-4-карбоновой кислоты, который устраняет недостатки известного процесса и дает возможность получать (9 дихлорметилен-1,2,3,4-тетрагидро-1,4-метанонафталин-5-ил)амид 3-дифторметил-1-метил-1 Н-пиразол-4-1 019475 карбоновой кислоты экономически выгодным способом с высоким выходом и хорошим качеством. Таким образом, согласно настоящему изобретению предлагается процесс получения соединения формулы I который включает а) реакцию соединения формулы II до получения соединения формулы IVb) гидрирование соединения формулы IV до соединения формулы V в присутствии металлкатализатора с) восстановление соединения формулы V до соединения формулы VI в присутствии восстанавливающего веществаd) дегидратацию соединения формулы VI до соединения формулы VII в присутствии кислоты е) реакцию соединения формулы VII с гидроксиламином до получения соединения формулы VIIIf) ацилирование кислорода оксима соединения формулы VIII в присутствии растворителя и ацилирующего реагента, и, наконец, реакцию полученного продукта с соединением формулы IXff) реакцию соединения формулы VIII с избытком соединения формулы IX. Стадия а) реакции. Соединение формулы II известно и раскрыто, например, в журнале Chemical Communications, 20,1293 (1971). Соединение формулы II может, например, быть получено путем реакции циклопентадиена сCCl4 в присутствии металлкатализатора, выбранного из комплексов рутения, меди, железа, палладия и родия, до получения соединения формулы X и реакции соединения формулы X с основанием в применимом растворителе до получения соединения формулы II. Соединение формулы III известно и доступно в продаже. Соединение формулы IV (и его эндо- и экзоизомеры) является новым и специально разработано для процесса по настоящему изобретению, поэтому оно представляет собой дополнительную цель настоящего изобретения. Применимыми инертными органическими растворителями для стадии а) реакции являются, например, толуол, ксилол, бензол, метилциклогексан, дихлорметан или хлорбензол, предпочтительно толуол. Реакцию можно успешно проводить в присутствии кислот Льюиса в качестве катализаторов. В качестве катализатора также могут использоваться некоторые сильные кислоты Бренстеда, например метансульфоновая кислота, а также закрепленные на твердой подложке кислоты Бренстеда, например AMBERLYST. Кислоты Льюиса были более эффективными, чем кислоты Бренстеда. Применимыми кислотами Льюиса являются, например, SnCl4, AlCl3 или FeCl3. Для увеличения выхода можно добавить лиганд-донор, особенно если в качестве катализатора используется AlCl3 или FeCl3. Предпочтительными лигандами-донорами являются диэтиловый эфир, тетрагидрофуран, нитрометан или нитробензол. Предпочтительным катализатором для стадии а) реакции является AlCl3 (успешно используемый в количестве 1-100 мол.%, предпочтительным количеством является 10-20 мол.%) в присутствии тетрагидрофурана. Тетрагидрофуран можно добавлять в количестве от 1 до 3 эквивалентов, в частности 1,1 эквивалента по отношению к используемому AlCl3. В предпочтительном варианте изобретения упомянутый предпочтительный катализатор можно успешно приготовить путем добавления тетрагидрофурана к суспензии AlCl3 в растворителе (например, в толуоле) при температуре от -10 до 60C, предпочтительно при температуре 25C. Раствор AlCl3/тетрагидрофурана можно добавить к смеси соединений формул II и III в растворителе (например, в толуоле) при температуре от -20 до 30C, предпочтительно 10C. Раствор AlCl3/диэтилового эфира можно добавить к смеси соединений формул II и III в хлорбензоле при температуре от -50 до -30C, предпочтительно -35C. В другом предпочтительном варианте изобретения твердый AlCl3 можно добавить к реакционной смеси, содержащей соединения формул II и III, а также тетрагидрофуран или диэтиловый эфир, при упомянутых выше значениях температуры. Стадия b) реакции. Соединение формулы V и его изомеры являются новыми и специально разработаны для процесса по настоящему изобретению, поэтому они представляют собой дополнительную цель настоящего изобретения. Применимые гетерогенные металл-катализаторы для стадии b) реакции представляют собой мелкодисперсные металлы групп 8, 9 и 10 периодической системы элементов, как вариант на твердой подложке, например активированном угле, оксиде алюминия или оксиде кальция, предпочтительно Pd/C, Pt/C,Rh/C или катализаторе в виде губчатого никеля (никелевого сплава) (например, скелетный никелевый катализатор). Используя Pd/C, Pt/C и Rh/C, гидрирование может быть успешно проведено при давлении водорода 1000-15000 гПа и температуре от 0 до 60C, предпочтительно при температуре 30-35C или комнатной температуре, в то время как скелетный никелевый катализатор требует более высокого давления водорода, например 1000-30000 гПа. Предпочтительным катализатором является Rh/C, в частности с 0,03-0,5 мол.% нагрузки и при давлении водорода 1000-15000 гПа, предпочтительно при давлении водорода 2000-5000 гПа, в частности при давлении водорода 3000 гПа. Стадию b) реакции проводят в присутствии растворителя. Применимыми органическими растворителями на стадии b) реакции для спиртов,простых и сложных эфиров являются - как вариант - хлорированные ароматические и алифатические углеводороды, например пропан-2-ол, пентанол, тетрагидрофуран, толуол, ксилол, этиловый эфир уксусной кислоты или трет-бутил метиловый эфир, в частности тетрагидрофуран. Реакцию гидрирования можно проводить при значениях температуры от низких до повышенных, предпочтительно при значениях температуры от 0 до 80C, более предпочтительно от 20 до 60C, в частности при температуре 3035C. Гидрирования также можно достичь благодаря использованию гомогенных гидрирующих катализаторов (комплексы иридия, родия или рутения, например (Ph3P)3RhCl), а также путем реакции гидрирования с переносом при помощи, например, пропан-2-ола, циклогексадиена или диимида (HN=NH), созданного in situ. Стадия с) реакции. Соединение формулы VI и его изомеры являются новыми и специально разработаны для процесса по настоящему изобретению, поэтому они представляют собой дополнительную цель настоящего изо-3 019475 бретения. Применимыми восстанавливающими веществами являются, например, водород с металлкатализатором, NaBH4, моноацетоксиборгидрид (NaBH3OAc), LiAlH4, бис(2-метоксиэтокси)алюмогидрид натрия (Red-Al), диизобутилалюминий гидрид (DIBAL-H) или боргидрид (BH3SMe2,BH3 тетрагидрофуран) или гидрирование с переносом от формиата или спирта. Особо предпочтительным является NaBH4. Стадию с) восстановления можно в некоторых случаях также проводить в присутствии гидрирующего катализатора, используемого для стадии b) реакции. Восстановление при помощиNaBH4 успешно проводят в растворителе или смесях растворителей, в частности в спирте, например в метаноле, этаноле, изопропаноле, смеси тетрагидрофурана/метанола, смеси тетрагидрофурана/этанола,предпочтительно в метаноле/тетрагидрофуране. Предпочтительными значениями температуры являются значения от -20 до +40C, в частности 030C. Возможно также использование водорода в присутствии катализатора в качестве восстанавливающего вещества. Стадия d) реакции: Применимыми кислотами для стадии d) реакции являются сильные кислоты, такие как фосфорная кислота, полифосфорные кислоты, концентрированная H2SO4, метансульфоновая кислота, n-толуолсульфоновая кислота, иммобилизованные кислоты (закрепленные на полимерных носителях), напримерAmberlyst, предпочтительно концентрированная H2SO4. В зависимости от применяемой кислоты, реакцию можно проводить при значениях температуры от 10 до 150C. Предпочтительный диапазон температур для применения концентрированной H2SO4 в качестве растворителя составляет от 10 до 25C. Для концентрированной H2SO4 весовое отношение исходного материала к концентрированной H2SO4 составляет от 1 : 0,2 до 1 : 10, предпочтительно 1 : 1 или менее, в этом случае необходимо применение растворителя, а предпочтительный диапазон температур составляет 70-90C. Соединение формулы VI добавляют к кислоте в твердой форме, или кислоту добавляют к раствору соединения формулы VI в органическом растворителе. Реакцию можно поддерживать с помощью азеотропной дистилляции воды, как вариант при пониженном давлении, особенно если используется каталитическое количество кислоты. Применимыми органическими растворителями для стадии d) реакции являются, например, толуол,ксилол, метилциклогексан, хлорбензол или дихлорбензол, предпочтительно толуол. Как и любое элиминирование, настоящую реакцию можно провести путем обращения гидроксила в соответствующую замещаемую группу, такую как галоген (Br, Cl, путем реакции, например, с PCl5, PBr3, SOCl2) или сульфонат (путем реакции, например, с метансульфонилхлоридом в присутствии основания), после чего следует обработка основанием, кислотой или кислотой Льюиса (например, KOH, NaOH NaOtBu, KOtBu или третичные амины, в том числе ароматические, такие как пиридин). Соединение формулы VII может встречаться в виде своих следующих изомеров или смесей: Необходимости в выделении или очистке конкретного изомера или смеси изомера формулы VII нет. Соединение формулы VII и его изомеры являются новыми и специально разработаны для процесса по настоящему изобретению, поэтому они представляют собой дополнительную цель настоящего изобретения. Стадия е) реакции. Гидроксиламин может быть использован в качестве нейтрального основания в воде (50 % раствор доступен в продаже) или создан in situ из своих солей, таких как гидрохлорид или сульфат, путем обработки основанием (например, триэтиламин, пиридин, NaOH или KOH, ацетат натрия, карбонат калия или натрия). Гидроксиламин предпочтительно используется в виде своего сульфата или гидрохлорида и в количестве от 1 до 2 эквивалентов, в частности от 1,1 до 1,3 эквивалента по отношению к соединению формулы VII. Применимыми основаниями для этой стадии реакции являются, например, пиридин, третичные амины, такие как триэтиламин, NaOH или KOH, ацетат натрия, карбонат калия или натрия. Особо предпочтительными является ацетат натрия или NaOH. Основание используется в количестве от 1 до 2 эквивалентов, предпочтительно 1-1,5 эквивалента по отношению к соединению формулы VII. Применимыми растворителями являются спирты (предпочтительно безводные), диметилформамид, N-метил-2 пирролидон или CH3CN, в частности безводный этанол или безводный метанол. Особо предпочтительным растворителем является безводный этанол. Стадию е) реакции можно успешно проводить при значениях температуры от 10 до 40C, предпочтительно при температуре 25C или комнатной температуре. Реакцию можно также проводить в двухфазной системе (органический растворитель/вода, органическими растворителями являются, например, толуол, ксилол, метилциклогексан) при значениях температуры 50-100C, используя вышеупомянутые основания и источники гидроксиламина в присутствии катализаторов межфазного переноса, выбранных из карбоновых кислот (например, уксусной, пропионовой, изомасляной, триметилуксусной, валериановой, изовалериановой, бензойной, 2-этилгексановой),которые применяются в количестве 2-50 мол.%. Предпочтительное количество катализатора составляет 5-10 мол.%, предпочтительная температура составляет 80-90C, предпочтительными катализаторами являются бензойная и 2-этилгексановая кислота. В случае применения ацетата натрия в качестве основания нет необходимости в катализаторе межфазного переноса. Это предпочтительный вариант процесса. Соединение формулы VIII может встречаться в виде своих следующих изомеров или смесей: Необходимости в выделении или очистке конкретного изомера или смеси изомера формулы VIII нет. Соединение формулы VII и его изомеры являются новыми и специально разработаны для процесса по настоящему изобретению, поэтому они представляют собой дополнительную цель настоящего изобретения. Стадия f) реакции. Соединение формулы IX известно и доступно в продаже. Соединение раскрыто, например, в публикации US-5,093,347. Эта стадия состоит из двух химических трансформаций: реакции кислорода оксима с хлорангидридом (например, ацетил хлоридом, пивалоил хлоридом, бензоил хлоридом или хлорацетил хлоридом) или ацил ангидридом, например ангидридом уксусной кислоты, предпочтительно пивалоил хлоридом, после чего следует трансформация ацилированного производного соединения in situ до получения соединения формулы I путем реакции с 1 эквивалентом соединения формулы IX, предпочтительно в присутствии кислоты (обычно HCl или MeSO3H, наиболее предпочтительно HCl). Для первой стадии ацилирования(ацилирование оксима) может использоваться дополнительный эквивалент соединения формулы IX. Предпочтительным для процедуры рециркуляции является использование только одного типа ацилирующего реагента. Ацилирование успешно проводят в присутствии основания. Основание используется в количестве от 1 до 1,5 эквивалента по отношению к соединению формулы IX, в частности в эквимолярных количествах. Применимыми основаниями для стадии f) реакции являются пиридин или третичные амины, такие как триэтиламин. Триэтиламин является особо предпочтительным в качестве основания. Предпочтительные значения температуры реакции для стадии f) реакции находятся в диапазоне от 60 до 120C, в частности 80-100C, наиболее предпочтительно 85-95C. Применимыми растворителями являются толуол, диоксан, тетрагидрофуран, ксилол, хлорбензол или ацетонитрил. Наиболее предпочтительным растворителем является диоксан. Стадия ff) реакции является особо предпочтительным вариантом процесса согласно настоящему изобретению: соединение формулы VIII реагирует непосредственно с избытком соединения формулы IX. Соединение формулы IX используется в избытке, предпочтительно в количестве от 2 до 3 эквивалентов,предпочтительно 2,1 эквивалента по отношению к соединению формулы VIII. Применение дополнительного ацилирующего реагента не является необходимым для этого варианта. 3-дифторметил-1-метил-1 Нпиразол-4-карбоновая кислота, образованная в этом варианте процесса в качестве побочного продукта,может быть восстановлена и трансформирована в соединение формулы IX. Поскольку для проведения реакции не требуется использование дополнительных ацилирующих реагентов, настоящий вариант процесса является экономически очень выгодным. Для этого варианта процесса не требуется присутствие кислоты. Кроме того, реакцию также можно проводить без основания с незначительным снижением выхода (3-5%). Предпочтительный вариант процесса включает:f) ацилирование кислорода оксима соединения формулы VIII в присутствии растворителя и ацилирующего реагента, и наконец реакцию полученного продукта с соединением формулы IX. Другой предпочтительный вариант процесса включает ff) реакцию соединения формулы VIII с избытком в 2-3 эквивалента соединения формулы IX. В особо предпочтительном варианте процесса по настоящему изобретению стадию а) реакции проводят в присутствии SnCl4, AlCl3 или FeCl3 в качестве катализатора; в качестве металл-катализатора для стадии b) реакции используются Pd/C, Pt/C, Rh/C или скелетный никелевый катализатор; в качестве восстанавливающего вещества на стадии с) реакции используются NaBH4, моноацетоксиборгидрид (NaBH3OAc), LiAlH4, бис(2-метоксиэтокси)алюмогидрид натрия (Red-Al), диизобутилалюминий гидрид (DIBAL-H) или боргидрид (BH3SMe2, BH3 тетрагидрофуран); в качестве кислот на стадии d) реакции используются фосфорная кислота, полифосфорные кислоты,концентрированная H2SO4, метансульфоновая кислота или n-толуолсульфоновая кислота; на стадии е) реакции гидроксиламин используется в виде своего гидрохлорида; а на стадии ff) соединение формулы VIII реагирует непосредственно с избытком в 2 -3 эквивалента соединения формулы IX. Стадию а) реакции в настоящем предпочтительном варианте процесса предпочтительно проводят сAlCl3 или FeCl3 в качестве катализатора и в присутствии лиганда-донора, выбранного из диэтилового эфира, тетрагидрофурана, бис(2-метокси)-этилового эфира, нитрометана, нитробензола и пиридина. В особо предпочтительном варианте процесса по настоящему изобретению стадию а) реакции проводят в присутствии AlCl3 в качестве катализатора; в качестве металл-катализатора на стадии b) реакции используется Rh/C; в качестве восстанавливающего вещества на стадии с) реакции используется NaBH4; в качестве кислоты на стадии d) реакции используется концентрированная H2SO4; на стадии е) реакции гидроксиламин используется в виде своего гидрохлорида; а на стадии ff) соединение формулы VIII реагирует непосредственно с избытком в 2-3 эквивалента соединения формулы IX. Соединение формулы VI является новым и представляет собой дополнительную цель настоящего изобретения. Соединение формулы VI может быть обращено в соединение формулы X, которое может вступать в реакцию до получения соединения формулы XI. Это показано на следующей схеме реакции 1. Схема реакции 1X обозначает водород, может также быть получено, начиная с соединения формулы VIII, как показано на схеме 2. Схема 2 На схеме 2 R обозначает метил, трет-бутил, CH2Cl или фенил. Соединение формулы XI является ценным промежуточным продуктом для получения соединения формулы I. Соединение формулы I может быть получено путем реакции соединения формулы XI с соединением формулы IX. Соединение формулы XI также может быть получено непосредственно (одна стадия) из соединения формулы VIII нагреванием соединения формулы VIII в присутствии кислоты (НС 1) в соответствующем растворителе(например, диоксане). Также настоящая трансформация может быть осуществлена нагреванием соединения формулы VIII в присутствии каталитического количества Pd/C (0,5-5 мол. %) в высококипящем растворителе, таком как триглим, при температуре 180C. Примеры получения Пример Р 1. Получение соединения формулы IV. Раствор катализатора. К перемешанной суспензии AlCl3 (60,0 г, 0,45 моль) в толуоле (200 г) по капле при температуре 2025C в инертной атмосфере (азота) добавляли тетрагидрофуран (46,0 г, 0,64 моль). Чистый раствор катализатора хранили при комнатной температуре. Циклоприсоединение Дильса-Альдера. В стеклянный реактор загружали холодный раствор 6,6-дихлорфульвена в толуоле (858 г, 0,479 моль, 8,2%) и 1,4-бензохинон (56,9 г, 0,526 моль). Содержимое реактора охлаждали до -9C, одновременно перемешивая его в инертной атмосфере(азота). Раствор катализатора (40 г, содержит 7,8 г AlCl3) добавляли в реактор в течение 30 мин при температуре -9C, затем в течение 60 мин добавляли дополнительное количество раствора катализатора (10 г, содержит 2,0 г AlCl3). После перемешивания в течение 3,5 ч при температуре -9C, реакционную смесь гасили, добавляя по капле этанол (70 мл) при температуре -9C. Реакционную массу перемешивали при температуре -9C в течение 30 мин и отфильтровывали. Продукт промывали холодной смесью этанола/толуола (2:1, 360 мл) и сушили под вакуумом. Выход 102 г (83%). 1(CDCl3, 75 МГц)47,5; 49,6; 103,4; 134,8; 142,6; 147,6; 196,6. Пример Р 2. Получение соединения формулы V. Колбу с двумя горлами емкостью 1 л наполняли соединением формулы IV (36,6 г, 0,143 моль) и 5%-Rh/C (3,0 г, 0,42 мол.% Rh, содержание воды 58,0%). Колбу опорожняли и дважды повторно наполняли азотом, а затем добавляли тетрагидрофуран (600 мл). Затем удаляли реакционную смесь до закипания тетрагидрофурана и повторно дважды наполняли водородом из баллона. Потребление водорода контролировали с помощью пузырькового счетчика. Для быстрого гидрирования крайне важным является интенсивное перемешивание реакционной смеси. Конверсию контролировали при помощи 1H ЯМР, она завершалась через 7 ч. В это время потребление водорода становилось очень медленным. Реакционную смесь отфильтровывали через пористый стеклянный фильтр. Фильтрационный осадок, содержащий нерастворенный продукт, несколько раз промывали тетрагидрофураном для его растворения. Объединенный фильтрат упаривали, а оставшийся кристаллизованный остаток перемешивали с метанолом (150 мл) в течение примерно 15 мин при комнатной температуре, затем охлаждали в ванне со льдом, перемешивали в течение дополнительных 15 мин, отфильтровывали, промывали метанолом и сушили на воздухе. Выход 32,7 г (88%). 1H ЯМР (CDCl3, 400 МГц)1,47-1,53 (m, 2 Н), 1,72-1,79 (m, 2 Н), 2,51-2,60 (m, 2 Н), 2,82-2,92 (m, 2 Н),3,20 (m, 2 Н), 3,37 (m, 2 Н). 13 С ЯМР (CDCl3, 100 МГц)23,7; 38,8; 43,9; 50,5; 106,9; 144,0; 207,8. Пример Р 3. Получение соединения формулы VI. Смесь соединения формулы V (47,3 г, 0,183 моль), метанола (300 мл) и тетрагидрофурана (300 мл) охлаждали до 0-5C в ванне со льдом. В течение 1,5 ч порциями добавляли боргидрид натрия (2,17 г,0,0573 моль). Реакционную смесь оставляли нагреваться до комнатной температуры, а растворитель удаляли путем ротационного выпаривания. Остаток разделяли между метил-трет-бутиловым эфиром (1000 мл) и 0,5 N HCl (300 мл). Органическую фазу отделяли, отфильтровывали и упаривали. Остаток сушили под вакуумом. Выход 46,9 г (98 %, смесь изомеров 9:1 при гидроксиле). 1 Н ЯМР (CDCl3, 400 МГц)(основной изомер) 1,58-1,72 (m, 3H), 1,84 (bs, 1H), 2,04 (m, 2 Н), 2,202,35 (m, 2 Н), 2,48-2,55 (m, 1 Н), 2,74 (m, 2 Н), 3,12 (m, 1H), 3,28 (m, 1 Н), 4,41 (m, 1 Н). Пример Р 4. Получение соединения формулы VII. Тонкоизмельченное соединение формулы VI (26,25 г, 0,1005 моль) добавляли в течение 10 мин к интенсивно перемешиваемой 96 %-ной серной кислоте (80 мл) при комнатной температуре (охлаждение с помощью водяной бани). Реакционную смесь перемешивали при аналогичной температуре в течение 30 мин и затем медленно, энергично перемешивая, выливали в смесь льда (200 г), охлажденной до температуры замерзания воды (200 мл) и метил-трет-бутилового эфира (250 мл). Органическую фазу отделяли, а водную фазу экстрагировали метил-трет-бутиловым эфиром (70 мл). Объединенный экстракт промывали 3 %-ным раствором бикарбоната натрия (150 мл), а затем рассолом (100 мл). Органическую фазу отделяли, а растворитель удаляли путем ротационного выпаривания. Остаток экстрагировали в кипящий гексан(100 + 10 + 10 мл). Горячий раствор отфильтровывали через пористый стеклянный фильтр (легкое вакуумирование) и оставляли кристаллизоваться при комнатной температуре. Через 1 ч кристаллизационную смесь далее охлаждали до 0C (ванна со льдом) и удерживали при этой температуре в течение 30 мин. Крупные образовавшиеся кристаллы отфильтровывали, промывали гексаном (30 мл) и сушили на воздухе. Остаточный раствор упаривали до объема 15 мл и собирали дополнительную порцию. ВыходH ЯМР (CDCl3, 400 МГц)(основной изомер) 1,23-1,32 (m, 2H), 1,88-2,14 (m, 4 Н), 2,23-2,30 (m,1 Н), 2,35-2,57 (m, 3H), 3,49 (m, 1H), 3,87 (m, 1 Н). 13 С ЯМР (CDCl3, 100 МГц)23,3; 24,2; 25,0; 25,7; 37,4; 42,2; 49,6; 102,3; 140,7; 149,2; 167,1; 193,7. Пример Р 5-а. Получение соединения формулы VIII. Смесь соединения формулы VII (24,6 г, 0,101 моль), гидрохлорида гидроксиламина (8,43 г, 0,121 моль), пиридина (12,0 г, 0,152 моль) и абсолютного этанола перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь разделяли между этилацетатом (500 мл) и водой (500 мл). Органическую фазу отделяли, дважды промывали водой (500 мл) и упаривали. Кристаллизованный остаток сушили под вакуумом. Выход 25,6 г (99 %). 1(m, 2 Н), 2,14-2,31 (m, 3H), 2,50 (m, 1 Н), 3,36 (d, J= 3,4 Гц, 1H), 3,64 (d, J= 3,3 Гц, 1 Н), 10,70 (s, 1H). Пример Р 5-b. Получение соединения формулы VIII (в двухфазной системе). Смесь соединения VII(30,0 г, 0,123 моль), сульфата гидроксиламина (12,15 г, 0,074 моль), ацетата натрия (12,15 г, 0,148 моль),толуола (100 мл) и воды (15 мл) перемешивали в стеклянном реакторе при температуре 85C в течение 3 ч. К реакционной смеси добавляли воду (30 мл), а затем по капле добавляли водный раствор гидроксида натрия (18,1 г, 0,136 моль, 30%), при этом удерживая температуру в диапазоне 80-85C. Водную фазу отделяли (горячую), а органическую фазу частично упаривали (65 мл толуола удаляли). Полученную суспензию охлаждали до -10C, перемешивали при указанной температуре в течение часа и отфильтровывали. Продукт промывали холодным толуолом (20 мл) и сушили под вакуумом при температуре 60C. Выход 29,6 г (92%, чистота продукта 99%). Пример Р 6. Получение соединения формулы I (стадия реакции f). Смесь соединения формулы VIII (5,16 г, 0,02 моль), пивалоил хлорида (2,41 г, 0,02 моль), триэтиламина (2,04 г, 0,02 моль) и диоксана (80 мл) перемешивали при температуре 40C в течение 30 мин. Затем добавляли раствор HCl в диоксане (2 мл, 0,01 моль, 2,0 М) и DFPA-Cl (соединение формулы IX) (3,89 г,0,02 моль). Реакционную смесь нагревали при температуре 85C в течение 1,5 ч. После охлаждения до комнатной температуры основную часть растворителя удаляли путем ротационного выпаривания, а остаток разделяли между этилацетатом (100 мл) и водой (100 мл). Органическую фазу отделяли, промывали 1 N NaOH (100 мл), затем дважды промывали водой (100 мл) и упаривали. Остаток сушили под вакуумом. Выход 6,60 г (70%, чистота продукта 85% на основании количественного 1H ЯМР-анализа). Неочищенное вещество (5,00 г) растворяли в смеси ксилола (10 мл) и метилциклогексана (5 мл) при температуре 80C. Раствор медленно с помешиванием охлаждали до температуры 5C (ванна со льдом). Осадок отфильтровывали, промывали холодным ксилолом (1 г) и сушили под вакуумом. Выход 3,0 г (50%, чистота продукта 99+ % на основании количественного 1 Н ЯМР-анализа). 1(m, 1H), 6,91 (t, JH.F = 54,2 Гц, 1H), 7,02 (d, J= 7,3 Hz, 1H), 7,16 (t, J= 7,8 Гц, 1H), 7,79 (d, J= 8,2 Гц, 1H),8,01 (s, 1H), 8,15 (m, 1H). Пример Р 7. Получение соединения формулы I (стадия реакции ff): К перемешанному раствору соединения формулы VIII (5,00 г, 0,0193 моль) и триэтиламина (1,76 г, 0,0174 моль) в диоксане (25 мл) в течение 15 мин добавляли соединение формулы IX (7,91 г, 0,0406 ммоль), удерживая температуру в диапазоне 25-35C. Реакционную смесь медленно нагревали до температуры 82C и удерживали при этой температуре в течение 3 ч. После охлаждения до комнатной температуры большую часть растворителя удаляли путем ротационного выпаривания, а остаток перемешивали в течение 25 мин с метил-третбутиловым эфиром (150 мл) и водой (35 мл). Добавляли раствор NaOH (2,4 г, 0,06 моль) в воде (10 мл), и смесь перемешивали в течение дополнительных 30 мин. Водную фазу отделяли, а органическую фазу экстрагировали 1 N NaOH (5 мл). Объединенные водные экстракты окисляли 32 %-ой HCl (5 мл, 0,05 моль), охлаждали до 0C, и полученную суспензию перемешивали при этой температуре в течение 30 мин. Белый осадок отфильтровывали, промывали водой (20 мл) и сушили для получения чистой на 99% кислоты DFPA. Выход 3,60 г (96%). Органическую фазу промывали 1 N HCl (50 мл, 0,05 моль), затем водой (50 мл). Растворитель медленно удаляли путем ротационного выпаривания, а кристаллизованный остаток сушили под вакуумом при температуре 50C. Выход 7,66 г (96 %, чистота 95 %). Это вещество перемешивали с метилциклогексаном (50 мл) при температуре кипения в течение 30 мин. Суспензию медленно охлаждали до 45C. Кристаллизованное вещество отфильтровывали, промывали метилциклогексаном и сушили на воздухе до получения чистого соединения формулы I. Выход 7,07 г (92%, чистота 99% согласно количественному 1 Н ЯМР-анализу). В зависимости от чистоты соединения формулы IX, конечный продукт может содержать небольшие количества побочных продуктов формулы B1 и В 2: ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы I который включает: а) реакцию соединения формулы II с получением соединения формулы IVb) гидрирование соединения формулы IV до соединения формулы V в присутствии металлкатализатора с) восстановление соединения формулы V до соединения формулы VI в присутствии восстанавливающего веществаd) дегидратацию соединения формулы VI до соединения формулы VII в присутствии кислоты е) реакцию соединения формулы VII с гидроксиламином с получением соединения формулы VIIIf) ацилирование кислорода оксима соединения формулы VIII в присутствии растворителя и ацилирующего реагента, и затем реакцию полученного продукта с соединением формулы IXff) реакцию соединения формулы VIII с избытком соединения формулы IX. 2. Способ по п.1, который включает f) ацилирование кислорода оксима соединения формулы VIII в присутствии растворителя и ацилирующего реагента, и затем реакцию полученного продукта с соединением формулы IX. 3. Способ по п.1, который включает ff) реакцию соединения формулы VIII с избытком в 2-3 эквивалента соединения формулы IX. 4. Соединение формулы IV
МПК / Метки
МПК: C07C 45/62, C07C 45/64, C07C 231/14, C07C 45/69, C07C 49/743, C07C 45/66, C07C 251/44, C07C 49/693
Метки: амидов, пиразолкарбоновой, способ, получения, кислоты
Код ссылки
<a href="https://eas.patents.su/11-19475-sposob-polucheniya-amidov-pirazolkarbonovojj-kisloty.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения амидов пиразолкарбоновой кислоты</a>
Способ получения амидов и сложных эфиров пиридинкарбоновой кислоты

Номер патента: 13797
Опубликовано: 30.06.2010
Авторы: Веверс Ян-Хендрик, Кнелль Маркус, Бринк Моника
МПК: C07D 213/81, C07D 213/79
Метки: эфиров, пиридинкарбоновой, способ, сложных, кислоты, получения, амидов
Формула / Реферат:
1. Способ получения амидов и сложных эфиров пиридинкарбоновой кислоты Iгде Hal означает атом галогена;X означает О или NR2;R1 означает C1-С6-алкильную или арильную группу, где обе группы могут быть замещены одним или несколькими атомами галогена, нитро, циано, C1-С6-алкильными, C1-С6-галоалкильными или C1-С6-алкоксильными группами;R2 означает атом водорода или C1-С6-алкильную группу;который включает следующие стадии:(а) нагревание смеси,...
Анилиды пиразолкарбоновой кислоты и содержащее их средство для борьбы с патогенными грибами

Номер патента: 12667
Опубликовано: 30.12.2009
Авторы: Шивек Франк, Штратманн Зигфрид, Шэфер Петер, Блеттнер Карстен, Ломанн Ян Клаус, Швёглер Аня, Грамменос Вассилиос, Мюллер Бернд, Гроте Томас, Дитц Йохен, Райнхаймер Йоахим, Гевер Маркус, Хюнгер Удо, Штирль Райнхард
МПК: C07D 231/14, A01N 43/56
Метки: кислоты, пиразолкарбоновой, грибами, патогенными, содержащее, борьбы, средство, анилиды
Формула / Реферат:
1. Анилиды пиразолкарбоновой кислоты формулы (I) в которой переменные имеют следующие значения: n - 0 или 2; m - 2 или 3; X1 - фтор или хлор, причем в случае n=2 оба остатка X1 могут иметь различные значения; X2 - галоген, причем остатки X2 могут иметь различные значения; Y - CN, NO2, C1-C4-алкил, C1-C4-галогеналкил, метокси или метилтио; р - 0 или 1; R1 - фтор, хлор, бром, C1-C4-алкил или C1-C4-галогеналкил; R2 - водород или галоген; R3 -...
Способ получения амидов 2-галогенпиридинкарбоновых кислот

Номер патента: 7106
Опубликовано: 30.06.2006
Авторы: Шрёдер Йохен, Исак Хайнц, Майер Хорст, Гольш Дитер
МПК: C07D 213/82, C07D 213/81
Метки: 2-галогенпиридинкарбоновых, способ, амидов, кислот, получения
Формула / Реферат:
1. Способ получения амидов 2-галогенпиридинкарбоновых кислот первичных ароматических моноаминов I, которые в орто-положении, по отношению к аминогруппе, имеют заместители, отличающиеся от водорода, путем взаимодействия хлоридов 2-галогенпиридинкарбоновых кислот II с ароматическими моноаминами I, отличающийся тем, что реакцию проводят в смеси растворителей, состоящей из воды и минимум одного не смешивающегося с водой органического растворителя,...
Способ получения амидов

Номер патента: 13638
Опубликовано: 30.06.2010
Авторы: Эренфройнд Йозеф, Тоблер Ханс, Зайферт Готтфрид, Целлер Мартин, Корси Камилла, Жиордано Фанни, Боннет Пол, Шах Шаилеш С., Вальтер Харальд, Джоунз Ян, Джордж Нэйл
МПК: C07C 211/45, C07C 205/06, A01N 43/26...
Метки: способ, амидов, получения
Формула / Реферат:
1. Способ получения соединения формулы (I)где R1и R2, каждый независимо друг от друга, означают водород или C1-C5-алкил и R3 означает CF3или CF2H,который включает в себя:(a) взаимодействие соединения формулы (II)где R1и R2 имеют значения, которые определены для формулы (I),по меньшей мере с одним восстановителем с образованием соединения формулы (III)где R1и R2 имеют значения, которые определены для формулы (I); и(b) взаимодействие полученного...
Производные амидов 6,7-дигидро-5н-имидазо[1,2-а]имидазол-3-карбоновой кислоты

Номер патента: 17688
Опубликовано: 28.02.2013
Авторы: Хайм-Ритер Александер, Ю Ян, Коган Дерек, Чэнь Чжидон, Ю Сюй, Лю Вэйминь, Маккиббен Брайан, Чантц Матт Аарон, Бентцин Йёрг Мартин, Хоран Джошуа Кортни, Брюнетте Стивен Ричард, Лемьё Рене М., Мосс Нил, Сюйн Чжаомин, Миллер Крейг Эндрью, Гао Донхон А., Лолор Майкл Дейвид, Ковальски Дженнифер А., Барбоса Антонио Хосе Дель Мораль
МПК: A61P 29/00, A61K 31/519, C07D 471/04...
Метки: производные, 6,7-дигидро-5н-имидазо[1,2-а]имидазол-3-карбоновой, кислоты, амидов
Формула / Реферат:
1. Соединение формулы Iв которойR1 выбран из группы, включающей -CN, -OCF3, галоген, гетероарил, необязательно замещенный галогеном, или C1-С3-алкил, необязательно замещенный галогеном, и фенил, необязательно замещенный галогеном;R2 выбран из группы, включающей:(А) Н,(B) С1-С3-алкил, необязательно замещенный одной или двумя группами, выбранными из группы, включающей:a) С3-С6-циклоалкил,b) -OR9,c) -NR9R10,d) -SOR9,e) -SO2R9,f) -C(O)NH2,g)...
Предыдущий патент: Безопочная формовочная машина
Следующий патент: Композиции и способы ингибирования экспрессии генов eg5 и vegf
Случайный патент: Производные индола в качестве антагонистов возбуждающих аминокислот