Фармацевтические соединения для применения в терапии инфекции clostridium difficile
Номер патента: 24069
Опубликовано: 31.08.2016
Авторы: Шнайдер Гисберт, Пратсинис Анна, Леру Жан-Кристоф, Кастагнер Бастьен, Иварссон Маттиас








Формула / Реферат
1. Соединение, характеризующееся общей формулой (4), (5) или (6)

где R1 выбран из группы, включающей
полиэтиленгликоль,
полиглицерин,
каждый X независимо выбран из ОРО32-, OPSO22- или OSO3- и Y является CH или N.
2. Мио-инозитолпентакисфосфат-2-PEG(400) (7).
3. Соединение, описываемое общей формулой (15)

где каждый X независимо выбран из OPO32-, OPSO22- или OSO3-, при условии, что не все X являются OPO32- и не все X являются OSO3-.
4. Соединение по п.3, где соединение характеризуется общей формулой (15а) или (15b)

5. Соединение по любому из предыдущих пп.3 или 4, характеризующееся формулой (16а) или (16b)

где
a) X2 является OSO3-, а каждый из X1, X3, X4, X5 и X6 независимо выбран из OPO32-, OPSO22- или OSO3-,
b) X1, X3 и X5 являются OPO32-, а X2, X4 и X6 являются OSO3-,
c) X1, X3 и X5 являются OSO3-, а X2, X4 и X6 являются ОРО32-,
d) X4, X5 и X6 являются OSO3-, а X1, X2 и X3 являются ОРО32-,
e) X4, X5 и X6 являются ОРО32-, а X1, X2 и X3 являются OSO3-, или
f) X2 и X5 являются OPO32-, а X1, X3, X4 и X6 являются OSO3-,
g) X2 и X5 являются OSO3-, a X1, X3, X4 и X6 являются ОРО32-,
h) X2 и X3 являются ОРО32-, а X1, X4, X5 и X6 являются OSO3-, или
i) X2 и X3 являются OSO3-, а X1, X4, X5 и X6 являются ОРО32-.
6. Применение соединения, описываемого общей формулой (4), (5) или (6)

где R1 выбран из группы, включающей
полиэтиленгликоль,
полиглицерин,
каждый X независимо выбран из ОРО32-, OPSO22- или OSO3- и Y является CH или N;
или соединения по любому из предыдущих пп.3-5 в качестве лекарственного препарата для профилактики или терапии инфекции С. difficile.
7. Лекарственная форма, содержащая соединение, описываемое общей формулой (4), (5) или (6)

где R1 выбран из группы, включающей
полиэтиленгликоль,
полиглицерин,
каждый X независимо выбран из OPO32-, OPSO22- или OSO3- и Y является CH или N,
или содержащая соединение по любому из предыдущих пп.3-5.
8. Лекарственная форма по п.7, дополнительно содержащая антибиотик.
9. Лекарственная форма по п.8, отличающаяся тем, что антибиотиком является метронидазол, ванкомицин или фидаксомицин.
10. Лекарственная форма по любому из пп.7-9 в виде таблетки, капсулы, раствора, порошка или сиропа.
Текст
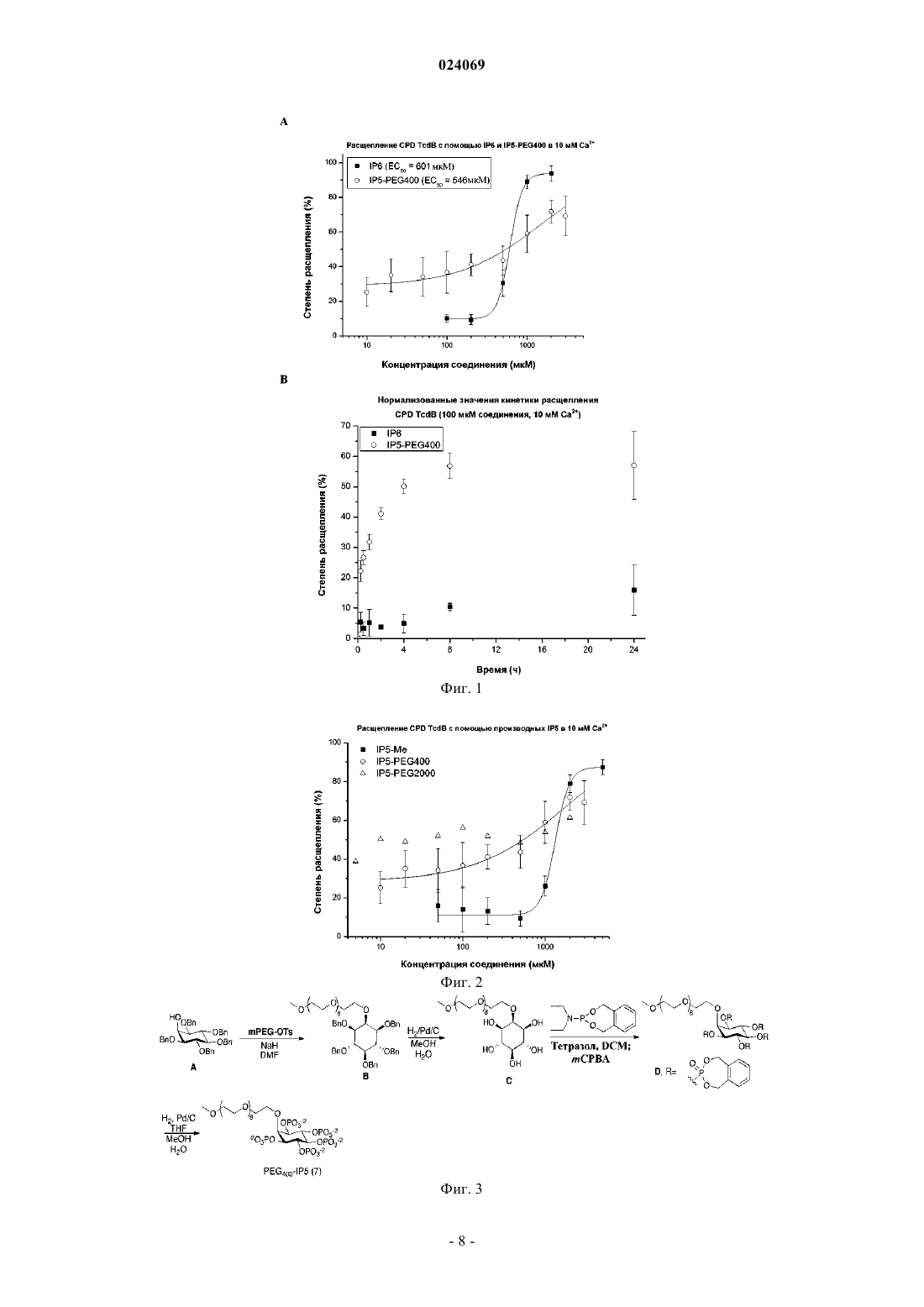
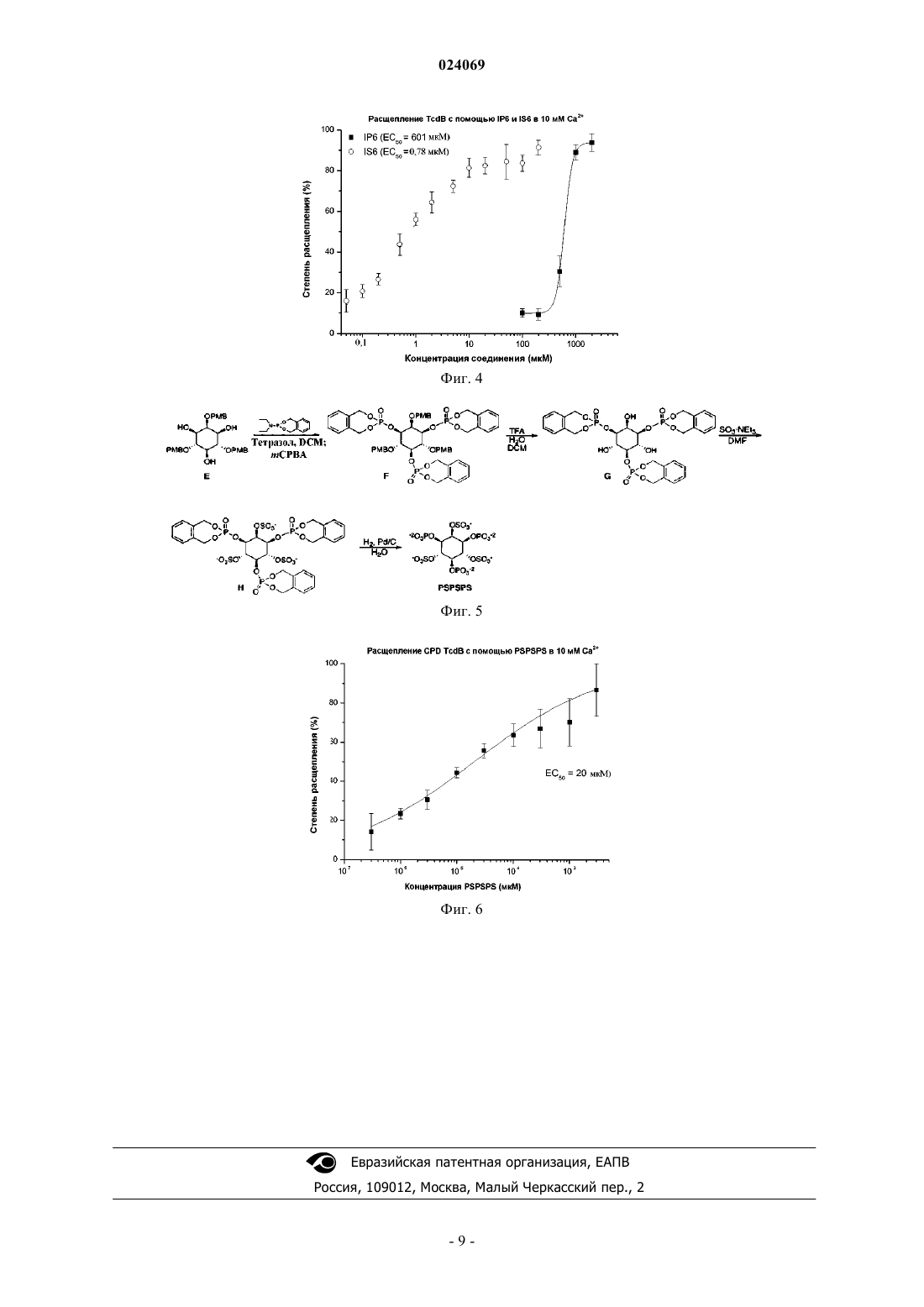
ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ В ТЕРАПИИ ИНФЕКЦИИ CLOSTRIDIUM DIFFICILE где каждый X независимо может быть OPO32-, OPSO22- или OSO3-; R1 включает обеспечивающую растворимость функциональную группу, такую как полиэтиленгликолевый фрагмент и Y являетсяCH или N. Настоящее изобретение также относится к применению указанных соединений в терапии инфекции, вызванной Clostridium difficile. Изобретение относится к кишечным активаторам токсина Clostridium difficile, в частности к полифосфатным производным, полисульфатным производным или смешанным полифосфатным/сульфатным производным шестичленных циклических полиолов.Clostridium difficile является одним из видов грамположительных бактерий, который вызывает сильную диарею у пациентов-людей. Инфекция С. difficile (CDI), как правило, поражает пациентов при лечении антибиотиками, так как бактерия способна колонизировать толстую кишку только у пациентов с истощенной бактериальной флорой. Появление устойчивых к антибиотикам штаммов С. difficile вызывает все более высокий уровень заболеваемости и смертности в связи с распространением новых, более вирулентных штаммов, с недавними вспышками в Северной Америке и Европе. С. difficile бессимптомно колонизирует 2-5% взрослого населения. Бактерии формируют споры, которые трудно нейтрализовать с помощью обычных способов дезинфекции. Как результат, инфекции С.difficile являются общей причиной длительной госпитализации; патоген считается ведущей причиной госпитальной диареи в США. Современной предпочтительной терапией является пероральное применение метронидазола или в случае неудачи первого варианта терапии - ванкомицина. Так как клинические симптомы CDI вызываются двумя токсичными белками, секретируемыми С. difficile в толстой кишке, а не присутствием непосредственно самой бактерии, в последнее время были предприняты усилия, нацеленные на эти токсины(например, с использованием полимерных связующих), но они также потерпели неудачу в клинических испытаниях. Энтеротоксин (токсин A, TcdA) и цитотоксин (токсин В, TcdB) С. difficile являются основными факторами, обуславливающими появление симптомов заболевания (для обзора биологии токсинов см.Voth and Ballard, Clinical Microbiology Reviews 2005, 18, 247-263). Вкратце, оба токсина состоят из четырех доменов: первый домен, являющийся посредником в процессе прикрепления токсина к клеткам; второй, способствующий перемещению в цитозоль; третий домен, вызывающий расщепление токсичного домена в результате автопротеолиза, и, наконец, токсичный домен или собственно "боеголовка", который обуславливает физиологические эффекты токсина в пораженной клетке.Reineke et al. (Nature 2007, 446, 415) определили мио-инозитолгексакисфосфат (IP6) как естественный триггер автопроцессинга TcdA/TcdB в цитозоле клетки. Egerer et al. (PLoS Pathog. 2010, 6, e1000942) и Shen et al. (Nat. Struct. Mol. Biol. 2011, 18, 364) предложили нацелить IP6-индуцированный механизм автопроцессинга в качестве средства терапевтического вмешательства на опосредованную токсином патогенность.Kreimeyer et al. предложили фармацевтическое применение IP6 для вмешательства в CDI (NaunynSchmiedeberg's Arch. Pharmacol. 2011, 383, 253). Однако этот подход не возможен, так как наличие высоких концентраций кальция в толстом кишечнике осаждает IP6 и предотвращает его активность. Таким образом, целью настоящего изобретения является создание усовершенствованных вариантов лечения пациентов, страдающих CDI. Эту цель достигают за счет отдельных объектов независимых пунктов формулы изобретения. Определения Полимер из данной группы мономеров представляет собой гомополимер (составленный из нескольких одинаковых мономеров); сополимер из данного набора мономеров представляет собой гетерополимер, образованный по меньшей мере двумя из группы. Настоящее изобретение основано на новой конструкции низкомолекулярных аналогов IP6, которые представлены в виде перорального терапевтического средства для индукции расщепления токсина в просвете толстой кишки, тем самым отщепляя боеголовку до того, как она достигает своего пункта назначения, и делая его безвредным. Так как сам по себе IP6 не может быть использован для этих целей, поскольку он не растворяется при высоких концентрациях кальция, обнаруживаемых в просвете толстой кишки, настоящее изобретение относится к новым аналогам IP6 с повышенной растворимостью. В соответствии с первым аспектом настоящего изобретения предложено фармацевтическое соединение, характеризующееся общей формулой (4), (5) или (6) где R1 выбран из группы, включающей полиэтиленгликоль,полиглицерин,каждый X независимо выбран из OPO32-, OPSO22- или OSC3- и Y является CH или N. Обеспечивающая растворимость функциональная группа R1 обеспечивает растворимость молекулы-1 024069 в водном растворе в присутствии 10 ммоль/л Са 2+. Молекула согласно настоящему изобретению имеет более высокую растворимость, чем IP6 в концентрациях кальция выше 1 ммоль/л; в соответствии с предпочтительным вариантом осуществления растворимость молекулы по настоящему изобретению превышает 10 ммоль/л. В некоторых вариантах осуществления указанный полиэтиленгликоль описан формулой (R3-(OCH2-CH2)n-), при этом R3 является водородом, метилом или этилом и n имеет значение от 3 до 200. В некоторых вариантах осуществления n имеет значение от 3 до 20. В некоторых вариантах осуществления n имеет значение от 10 до 30. В некоторых вариантах осуществления n имеет значение от 9 до 45. В некоторых вариантах осуществления указанный полиэтиленгликоль является разветвленным полиэтиленгликолем. В некоторых вариантах осуществления R1 является полиглицерином, описанным формулой R3-O(CH2-CHOH-CH2O)n-), при этом R3 является водородом, метилом или этилом, и n имеет значение от 3 до 200. В некоторых альтернативах этих вариантов осуществления n имеет значение от 3 до 20. В некоторых альтернативах этих вариантов осуществления n имеет значение от 10 до 30. В некоторых альтернативах этих вариантов осуществления n имеет значение от 9 до 45. В некоторых вариантах осуществления R1 является разветвленным полиглицерином, описанным формулой (R3-O-(CH2-CHOR5-CH2-O)n-), при этом R5 является водородом или цепью линейного глицерина, описанной формулой (R3-O-(CH2-CHOH-CH2-O)n-), a R3 является водородом, метилом или этилом. В некоторых вариантах осуществления R1 является гиперразветвленным полиглицерином, описанным формулой (R3-O-(CH2-CHOR5-CH2-O)n-), при этом R5 является водородом или глицериновой цепью,описанной формулой (R3-O-(CH2-CHOR6-CH2-O)n-), при этом R6 является водородом или глицериновой цепью, описанной формулой (R3-O-(CH2-CHOR7-CH2-O)n-), при этом R7 является водородом или цепью линейного глицерина, описанной формулой (R-О-(CH2-CHOH-CH2-О)n-), и при этом R3 является водородом, метилом или этилом. Гиперразветвленный глицерин и способы его синтеза описаны в Oudshorn et al., Biomaterials (2006),27, 5471-5479; Wilms et al., Acc. Chem. Res. (2010) 43, 129-41, и источниках, указанных там же. В одном варианте осуществления (6) включает поли(этиленгликоль) PEG400 в качестве обеспечивающей растворимость функциональной группы, при этом R1 является CH3(OCH2-CH2)9-О-, мио-инозитолпентакисфосфат-2-PEG(400) (7) является PEG400-аналогом мио-инозитолгексакисфосфата. Было доказано, что (7) характеризуется улучшенной способностью расщеплять TcdB CPD в присутствии кальция. Другой вариант осуществления относится к PEG2000-аналогу для (7), т.е. PEG с примерно 45 этиленгликолевыми мономерами. Другой вариант осуществления относится к аналогу сциллоинозитолгексакисфосфата, описанного формулой (8) где R1 имеет то же самое значение, которое описано выше. В некоторых вариантах осуществления соединение по настоящему изобретению может быть описано формулой (8), a R1 является полиэтиленгликолевым фрагментом. Обеспечивающая растворимость функциональная группа, полиэтиленгликолевая (PEG) цепь, показанная здесь в качестве неограничивающего примера, прикреплена к молекуле, чтобы сделать ее растворимой в просвете толстой кишки, при концентрациях кальциях, указанных в данном контексте. В соответствии с вторым аспектом настоящего изобретения предложено фармацевтическое соединение, которое характеризуется представленной общей формулой (15)-2 024069 где каждый X независимо выбран из OPO32-, OPSO22-, or OSO3-, при условии, что не все X являютсяOPO32- и не все X являются OSO3-. В соответствии с третьим аспектом настоящего изобретения предложено соединение, характеризующееся в предыдущем абзаце формулой (15), для применения в качестве лекарственного препарата, в частности для применения в профилактике или терапии инфекций, вызванных Clostridium dificile. В некоторых вариантах осуществления соединение в соответствии с этим вторым аспектом настоящего изобретения характеризуется общей формулой (15 а) или (15b), где X имеет описанные выше значения, В некоторых вариантах осуществления соединение в соответствии с этим вторым аспектом настоящего изобретения характеризуется общей формулой (16 а) или (16b)a) X2 является OSO3-, и X1, X3, X4, X5 и X6 независимо выбраны из OPO32-, OPSO22- или OSO3-,b) X1, X3 и X5 являются OPO32-, а X2, X4 и X6 являются OSO3- (соединение 16 а-b или 16b-b),c) X1, X3 и X5 являются OSO3-, а X2, X4 и X6 являются ОРО 32- (соединение 16 а-с или 16b-с),d) X4, X5 и X6 являются OSO3-, а X1, X2 и X3 являются ОРО 32- (соединение 16 а-d или 16b-d),e) X4, X5 и X6 являются OPO32-, а X1, X2 и X3 являются OSO3- (соединение 16 а-е или 16b-е),f) X2 и X5 являются OPO32-, а X1, X3, X4 и X6 являются OSO3- (соединение 16a-f или 16b-f),g) X2 и X5 являются OSO3-, а X1, X3, X4 и X6 являются OPO32- (соединение 16a-g или 16b-g),h) X2 и X3 являются OPO32-, а X1, X4, X5 и X6 являются OSO3- (соединение 16a-h или 16b-h), илиi) X2 и X3 являются OSO3-, а X1, X4, X5 и X6 являются OPO32- (соединение 16a-i или 16b-i). Соединения, определенные выше, могут быть синтезированы в соответствии со стандартными способами. Синтез соединения 16 а-b описан в примерах настоящего изобретения. В соответствии с другим аспектом настоящего изобретения предложено соединение по любому из вышеуказанных аспектов настоящего изобретения в самом широком данном определении, или как указано в любом из вариантов осуществления, для применения в качестве лекарственного препарата. Согласно еще одному аспекту настоящего изобретения предложено соединение по любому из вышеуказанных аспектов настоящего изобретения в самом широком данном определении, или как определено в любом из вариантов осуществления, для применения при лечении и профилактике инфекции С.difficile. Соединение согласно настоящему изобретению может быть дано пациенту с уже поставленным диагнозом CDI или пациенту, который предположительно страдает от CDI. Альтернативно, соединение можно применять в качестве профилактического средства для пациентов, которые подвержены риску заражения инфекцией, таких как пациенты, проходящие лечение антибиотиками в стационарных условиях. Соединения согласно настоящему изобретению просто синтезировать, они устойчивы к распаду в желудочно-кишечном тракте и вряд ли будут всасываться в кровяной поток, таким образом, избегая возможных побочных эффектов. Соединения согласно настоящему изобретению не нуждаются в проникновении в мембраны млекопитающих или бактерий для того, чтобы быть активными, что делает их более эффективными in vivo. Кроме того, маловероятно, что соединения согласно настоящему изобретению оказывают селективное давление на бактерии и, следовательно, избегают проблем, связанных с устойчивостью. Согласно еще одному аспекту настоящего изобретения предложена фармацевтическая композиция для применения в способе для профилактики или лечения инфекции С. difficile, содержащая соединение в соответствии с любым из вышеописанных аспектов настоящего изобретения. Предпочтительные фармацевтические композиции содержат от примерно 1 до примерно 95% ак-3 024069 тивного ингредиента, предпочтительно от примерно 20 до примерно 90% активного ингредиента. Фармацевтическая композиция по указанным выше аспектам настоящего изобретения может быть введена отдельно или в комбинации с одним или несколькими другими терапевтическими средствами. Комбинированная терапия может принимать форму комбинированных препаратов из соединения настоящего изобретения и одного или нескольких других антибиотиков. Введение может быть поочередным; альтернативно, лекарственные средства могут быть даны независимо друг от друга или в виде комбинированного препарата. Согласно предпочтительному варианту осуществления фармацевтическая композиция содержит соединение по настоящему изобретению согласно любому из вышеупомянутых аспектов настоящего изобретения и дополнительно метронидазол, ванкомицин и/или фидаксомицин. Согласно еще одному аспекту настоящего изобретения предложена лекарственная форма, включающая в себя соединение согласно любому из вышеупомянутых аспектов настоящего изобретения. Предпочтителен пероральный состав, в частности, таблетка, сироп, раствор, капсула или порошок. В соответствии с предпочтительным вариантом осуществления такая лекарственная форма дополнительно содержит обладающие антибиотической активностью соединение, такое как (в качестве неограничивающего примера) метронидазол, ванкомицин или фидаксомицин. Согласно еще с одним аспектом настоящего изобретения предложена схема лечения для профилактики и лечения CDI, включающая введение соединения согласно настоящему изобретению. Введение может быть осуществлено любым из описанных в данном документе способов. Также, в объем настоящего изобретения подпадает способ профилактики или лечения CDI, включающий введение соединения согласно настоящему изобретению субъекту, нуждающемуся в этом. Аналогично, предложено соединение согласно настоящему изобретению для получения лекарственного препарата для профилактики и лечения CDI. Лекарственные препараты согласно настоящему изобретению получают методами, известными в данной области, в частности с помощью традиционных способов смешивания, покрытия, гранулирования, растворения или лиофилизации. Везде, где существуют альтернативы для отдельных элементов, таких как R1, X, Y и т.д. изложенных в настоящем документе в качестве "вариантов осуществления", следует понимать, что такие альтернативы могут быть легко скомбинированы для формирования отдельных вариантов осуществления всей представленной молекулы как таковой или для применения в способе или согласно медицинским показаниям, указанным в данном документе. Краткое описание фигур На фиг. 1 показана концентрационная зависимость расщепления домена цистеиновой протеазыTcdB в присутствии соединения-активатора (7) (незаштрихованные круги) или IP6 in vitro (1A), и соответствующие кинетики (1 В) в 10 мМ Са +. На фиг. 2 показана концентрационная зависимость расщепления домена цистеиновой протеазыTcdB в присутствии соединения-активатора (7) (незаштрихованные круги), его PEG2000-аналога (незаштрихованные треугольники), и его метильного аналога (черные квадраты). На фиг. 3 показан синтез соединения 7. На фиг. 4 показана концентрационная зависимость расщепления домена цистеиновой протеазыTcdB в присутствии 10 мМ Ca2+ для IS6 (незаштрихованные круги) и IP6 (черные квадраты). На фиг. 5 показан синтез соединения (16 а-b). На фиг. 6 показана концентрационная зависимость расщепления домена цистеиновой протеазыTcdB в присутствии 10 мМ Са 2+ для соединения-активатора (16 а-b). Примеры 1. Синтез соединения (7). Синтез протекал согласно последовательности, изображенной на фиг. 3. Соединение В. К суспензии гидрида натрия (4,3 ммоль, 103,7 мг) в 10 мл диметилформамида (DMF) по каплям добавляли раствор соединения A [Martin, S. F. et al., J. Org. Chem. 1994, 59, 4805] (2,16 ммоль,1363 мг) в DMF (10 мл). По завершении добавления смесь перемешивали 30 мин при комнатной температуре с последующим добавлением MeO-PEG-OTs (при этом OTs представляет собой толуолсульфонат)(3,2 ммоль, 1,64 г в 10 мл DMF). Реакционную смесь оставляли перемешиваться в течение ночи, затем ее гасили водой (5 мл). Смесь экстрагировали дихлорметаном (DCM). Растворитель выпаривали и остаток хроматографировали на силикагеле шестью порциями по 100 мл этилацетататексана в соотношениях 20:80; 30:70; 40:60; 60:40; 80:20; 100:0. В результате хроматография происходило разделение на фракции продукта с различными размерами PEG, включая соединение В со средним значением в 9 PEG-звеньев. 1H ЯМР (400 МГц; CDCl3):7,29-7,13 (m, 25H), 4,83 (d, J=10,8 Гц, 2H), 4,79 (s, 2 Н), 4,74 (d, J=10,8 Гц,2H), 4,63 (d, J=11,7 Гц, 2 Н), 4,59 (d, J=11,6 Гц, 2 Н), 3,97-3,88 (m, 5H), 3,64-3,42 (m, 31H), 3,38 (t, J=9,2 Гц,1H), 3,29-3,25 (m, 5H). Соединение С. Соединение В растворяли в смеси тетрагидрофурана (THF, 4 мл), метанола (7 мл) и воды (3 мл) с последующим добавлением избытка 10%-го палладия на древесном угле. Смесь помещали в водородную атмосферу и перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь продували азотом, фильтровали и выпаривали растворитель. Сырую смесь очищали на кар-4 024069H NMR (400 МГц; D2O):3,96-3,94 (m, 2H), 3,89 (t, J=2,8 Гц, 1H), 3,78-3,69 (m, 28H), 3,69-3,60 (m, 5 Н),3,56 (dd, J=10,0, 2,8 Гц, 2H), 3,40 (s, 3H), 3,25 (t, J=9,2 Гц, 1 Н). Соединение D. Соединение С (0,2 ммоль, 119 мг) суспендировали в тетразоле (3,630 ммоль, 8,1 мл,0,45 М в CH3CN) и DCM (10 мл), затем добавляли N,N-диэтил-1,5-дигидро-2,4,3-бензодиоксафосфепин 3-амин (1,8 ммоль, 434 мг) и смесь перемешивали при комнатной температуре в течение ночи. Затем смесь охлаждали до -10C и добавляли раствор мета-хлорпероксибезнзойной кислоты (mCPBA, предварительной просушенной над Na2SO4, 4,8 ммоль, 1189 мг) в DCM (2 мл). Смесь оставляли перемешиваться при -10C в течение дополнительных 10 мин, а затем доводили до комнатной температуры и перемешивали в течение 1-го ч. Смесь промывали разбавленным сульфитом натрия и экстрагировали с помощью DCM. Органические слои сушили с помощью Na2SO4, фильтровали и концентрировали. Остаток хроматографировали на силикагеле с градиентом 1-5% метанола в DCM. 1H ЯМР (400 МГц; CDCl3):7,41-7,27 (m, 18H), 7,21 (dd, J =6,9, 1,7 Гц, 2 Н), 5,59-5,51 (m, 6H), 5,44 (t, J=14,2 Гц, 2 Н), 5,37-5,29 (m, 6H),5,25-4,95 (m, 10H), 4,74 (ddd, J=9,9, 7,6, 2,1 Гц, 2 Н), 4,61 (t, J=2,2 Гц, 1H), 4,03 (t, J=4,9 Гц, 2 Н), 3,68-3,50(m, 22H), 3,48 (dd, J=6,1, 4,0 Гц, 2 Н), 3,40-3,34 (m, 7H). 13C ЯМР (101 МГц; CDCl3):135,78, 135,72,135,4, 135,14, 134,99, 129,38, 129,27, 129,26, 129,22, 129,14, 129,11, 128,96, 128,95, 77,6, 76,05, 76,01, 73,8,71,9, 70,67, 70,60, 70,57, 70,54, 70,53, 70,49, 70,38, 70,34, 70,29, 69,46, 69,39, 69,33, 69,24, 69,16, 69,01,68,95, 59,0. Соединение (7). Соединение D растворяли в смеси THF (1 мл), метанола (1,5 мл) и воды (2 мл), с последующим добавлением избытка 10%-го палладия на древесном угле. Смесь помещали под водородную атмосферу и перемешивали при комнатной температуре в течение ночи. Затем смесь продували азотом, фильтровали и концентрировали. Соединение доводили до pH 7 добавлением разбавленного водного NaOH (170 мкл, 0,1 М). Остаток очищали на колонке с сефадексом (PD-10, GE Healthcare, Sephadex G25 М, кат.17-0851-01) путем элюирования 10 мл воды. Все фракции (1,5 мл) лиофилизировали и анализировали с помощью 1H ЯМР. Фракции, содержащие продукт, дополнительно очищали на картридже для обратно-фазовой хроматографии (Sep-Pak, Waters, 1 г, C18, кат.WAT 036905) путем элюирования 10 мл воды. Все фракции (1,5 мл) лиофилизировали и анализировали с помощью 1H ЯМР-анализа. 1H ЯМР (400 МГц; D2O):4,44 (q, J =9,4 Гц, 2 Н), 4,20 (s, 1H), 4,16-4,10 (m, 3H), 4,06 (t, J =4,7 Гц, 2 Н), 3,833,63 (m, 26H), 3,40 (s, 3H). 31 Р ЯМР (162 МГц; D2O):1,5, 1,2, 0,8. 2. Определение EC50 в присутствии 10 мМ Са 2+. Подлежащее тестированию соединение добавляли к рекомбинантному His-меченному домену цистеиновой протеазы токсина В С. difficile с SEQ ID 1 в присутствии 10 мМ Са 2+ в 100 мМ Tris, pH 7,4 и инкубировали в течение 2 ч при 37C. Расщепленные фрагменты белка разделяли с помощью SDS-PAGE и визуализировали с помощью окрашивания Кумасси. Степень расщепления количественно оценивали по интенсивностям бэндов белка с помощью программного обеспечения ImageJ. Сигналы нормализировали по результатам расщепления положительных и отрицательных контролей. Результаты этого анализа для IP6 и соединения (7) показаны на фиг. 1 А. Исходя из результатов,видно, что 50% расщепления фрагмента токсина достигали при схожих концентрация IP6 и (7). Активность IP6 почти полностью исчезала при 100 мкМ, тогда как (7) сохраняло остаточную активность. PEGцепь (7) вероятно обеспечивала молекуле более широкий интервал растворимости. 3. Сравнение кинетик расщепления. Подлежащее тестированию соединение добавляли к His-меченому домену цистеиновой протеазы токсина В С. difficile (с той же последовательностью, которая приведена выше) в присутствии 10 мМ Са 2+ в 100 мМ Tris pH 7,4 и инкубировали 24 ч при 37C, при этом забирали аликвоты через равные промежутки времени. Расщепленные фрагменты белка разделяли, визуализировали и анализировали как указано выше. Результаты этого анализа для IP6 и соединения (7) показаны на фиг. 1 В. Исходя из результатов, видно, что степень расщепления через 4 ч была в 5 раз выше, чем с IP6. 4. Синтез соединения (16 а-b). Соединение F. Раствор 2,4,6-три-O-(4-метоксибензил)-мио-инозитола (Е) [D. Lampe, С. Liu, В. V. L.Potter, J. Med Chem. 1994, 37, 907] (0,541 г, 1 ммоль, 1 экв.) в сухом CH2Cl2 (20 мл, 0,05 m) в атмосфере азота обрабатывали тетразолом в ацетонитриле 0,45 m (20,0 мл, 9,0 ммоль, 9 экв.) и о-ксилилен-N,Nдиэтилфосфорамидитом (6 ммоль, 1,44 г, 6 экв.). Реакционную смесь перемешивали при комнатной температуре 2 дня. Раствор mCPBA (12 ммоль, 2,07 г, 12 экв.), высушенный над Na2SO4, добавляли при 10C и реакционную смесь перемешивали при комнатной температуре в течение дополнительных 45 мин. Затем смесь разбавляли в EtOAc, промывали насыщенным раствором водного NaHCO3 и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали in vacuo. Очистка флеш-хроматографией (SiO2, CH2Cl2/MeOH постепенно от 0 до 4%, трижды) давала 2,4,6-три-O-(4 метоксибензил)-1,3,5-три-O-(о-ксилиленфосфо)мио-инозитол (F) в виде белого твердого вещества (98%). 1(ppm) 159,3, 159,1, 135,4, 135,3, 135,2, 130,8, 130,3, 129,7, 129,6, 129,2, 129,13, 129,12, 129,0, 128,9, 128,6,113,74, 113,57, 80,6 (d, Jcp 6,0 Гц), 78,1 (dd, Jcp 6,9, 3,2 Гц), 77,6, 77,1 (m), 76,0, 74,9, 68,8, 68,70, 68,68,68,62, 68,34, 68,28, 55,4, 55,3; 31 Р ЯМР (160 МГц, 1H-не связано, CDCl3)(ppm) 1,10,-1,32. Соединение G. Соединение F (97 мг, 0,089 ммоль) растворяли в 1 мл дихлорметана. Добавляли 6 мл смеси 5:1 трифторуксусной кислоты и воды. Перемешивали 25 мин и затем разбавляли 10 мл толуола и концентрировали под вакуумом. Полученный остаток растирали с гексаном и дихлорметаном и затем сушили под сильным вакуумом. Получали 68 мг неочищенного соединения G, которое непосредственно использовали на следующем этапе. Соединение Н. Соединение G (39 мг, 0,054 ммоль) растворяли в 3 мл DMF и добавляли SO3Et3N(195 мг, 1,07 ммоль). Раствор перемешивали в течение ночи при 50C и концентрировали на роторном испарителе. Остаток растворяли в 6 мл воды, фильтровали и загружали на три картриджа Vac 6cc, 1 гtC18 Sep-Pak (Waters). Колонки элюировали градиентом 0-40% МеОН/Н 2 О. Получали 32 мг Н. 1H ЯМРMeOD/CDCl3):131,6, 131,2, 125,26, 125,21, 125,09, 124,93, 124,87, 124,78, 70,25, 70,22, 70,19, 69,95,69,90, 65,18, 65,11, 65,07, 65,00, 64,85, 64,78, 42,4, 4,2; 31 Р ЯМР (162 МГц; MeOD/CDCl3):-7,8, -8,9. Соединение PSPSPS 16 а)b): соединение Н (32 мг) растворяли в 3 мл Н 2 О. Добавляли небольшую порцию Pd на активированном угле (10%), смесь помещали под Н 2-атмосферу и перемешивали 4 ч. Затем смесь продували с помощью N2 и добавляли каплю NH4OH. Смесь фильтровали через целит и испаряли на роторном испарителе. Остаток растворяли в 1 мл воды, загружали на картридж Vac 6cc 1 г tC18 SepPak (Waters) и элюировали водой. Элюированные фракции лиофилизировали и анализировали с помощью 1H ЯМР. Получали 16 мг PSPSPS2Et3NH+xNH4+. 1H-ЯМР (400 МГц; D2O):4,93-4,78 (m, 3H), 4,554,39 (m, 3H), 3,13 (q, J=7,3 Гц, 14 Н), 1,21 (t, J=7,3 Гц, 21 Н). 31P ЯМР (162 МГц; D2O):-0,3, -0,7. Расщепление, индуцированное соединением PSPSPS 16 а-b), определяли в присутствии кальция как описано в примере 2, и результаты показаны на фиг. 7. Расщепление, индуцированное этим производным, составляло 50% при концентрации 20 мкМ, что намного эффективнее, чем для IP6 (601 мкМ). Из этого результата видно, что присутствие нескольких сульфатных групп усиливало активность соединения в присутствии кальция. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, характеризующееся общей формулой (4), (5) или (6) где R1 выбран из группы, включающей полиэтиленгликоль,полиглицерин,каждый X независимо выбран из ОРО 32-, OPSO22- или OSO3- и Y является CH или N. 2. Мио-инозитолпентакисфосфат-2-PEG(400) (7). 3. Соединение, описываемое общей формулой (15) где каждый X независимо выбран из OPO32-, OPSO22- или OSO3-, при условии, что не все X являются OPO32- и не все X являются OSO3-. 4. Соединение по п.3, где соединение характеризуется общей формулой (15 а) или (15b) 5. Соединение по любому из предыдущих пп.3 или 4, характеризующееся формулой (16 а) или (16b)a) X2 является OSO3-, а каждый из X1, X3, X4, X5 и X6 независимо выбран из OPO32-, OPSO22- илиi) X2 и X3 являются OSO3-, а X1, X4, X5 и X6 являются ОРО 32-. 6. Применение соединения, описываемого общей формулой (4), (5) или (6)R1 выбран из группы, включающей полиэтиленгликоль,полиглицерин,каждый X независимо выбран из ОРО 32-, OPSO22- или OSO3- и Y является CH или N; или соединения по любому из предыдущих пп.3-5 в качестве лекарственного препарата для профилактики или терапии инфекции С. difficile. 7. Лекарственная форма, содержащая соединение, описываемое общей формулой (4), (5) или (6) где R1 выбран из группы, включающей полиэтиленгликоль,полиглицерин,каждый X независимо выбран из OPO32-, OPSO22- или OSO3- и Y является CH или N,или содержащая соединение по любому из предыдущих пп.3-5. 8. Лекарственная форма по п.7, дополнительно содержащая антибиотик. 9. Лекарственная форма по п.8, отличающаяся тем, что антибиотиком является метронидазол, ванкомицин или фидаксомицин. 10. Лекарственная форма по любому из пп.7-9 в виде таблетки, капсулы, раствора, порошка или сиропа.
МПК / Метки
МПК: C07F 9/117
Метки: диффициле, инфекции, соединения, терапии, применения, фармацевтические, clostridium
Код ссылки
<a href="https://eas.patents.su/10-24069-farmacevticheskie-soedineniya-dlya-primeneniya-v-terapii-infekcii-clostridium-difficile.html" rel="bookmark" title="База патентов Евразийского Союза">Фармацевтические соединения для применения в терапии инфекции clostridium difficile</a>
Соединения актагардина и их применение для лечения или профилактики инфекции clostridium difficile

Номер патента: 21628
Опубликовано: 30.07.2015
Авторы: Кортес Баргалло Хесус, Доусон Майкл Джон, Вадман Сьёрд Николаас
МПК: A61P 1/12, A61K 38/16, A61P 31/04...
Метки: clostridium, соединения, актагардина, лечения, диффициле, применение, инфекции, профилактики
Формула / Реферат:
1. Способ лечения или профилактики инфекции Clostridium difficile у субъекта, включающий введение лантибиотика типа В, выбранного из группы, состоящей из актагардина и его вариантов, содержащих 1, 2, 3 или 4 аминокислоты, замененные другой аминокислотой по положению, выбранному из положений 2, 3, 4, 5, 8, 10, 11, 13, 15, 16 или 18; их производных и солей.2. Способ по п.1, отличающийся тем, что лантабиотик типа В вводят для лечения инфекции...
Новые соединения, содержащие их фармацевтические композиции и способы их применения

Номер патента: 13371
Опубликовано: 30.04.2010
Авторы: Макфэдден Джилл М., Медгалчи Сьюзен М., Тхупари Джаган, Таунсенд Крэйг А., Кухаджа Фрэнсис П.
МПК: A61K 31/38, C07D 333/32
Метки: фармацевтические, содержащие, применения, соединения, способы, композиции, новые
Формула / Реферат:
1. Соединение, выбранное из группы, включающей2. Соединение формулы3. Фармацевтическая композиция, содержащая фармацевтический разбавитель и соединение по п.2.4. Способ лечения рака, включающий введение пациенту, нуждающемуся в этом, композиции по п.3.5. Соединение формулы6. Фармацевтическая композиция, содержащая фармацевтический разбавитель и соединение по п.5.7. Способ лечения рака, включающий введение пациенту, нуждающемуся в этом,...
Замещенные ароматические соединения, композиции на их основе и их фармацевтические применения

Номер патента: 22445
Опубликовано: 29.01.2016
Авторы: Ганьон Лин, Бьенвеню Жан-Франсуа, Зашари Було, Груи Брижитт, Пенни Кристофер, Перрон Валери
МПК: A61K 31/192, A61K 31/41, A61K 31/44...
Метки: композиции, замещенные, применения, фармацевтические, основе, соединения, ароматические
Формула / Реферат:
1. Соединение, представленное формулой IIили его фармацевтически приемлемая соль, гдеn равно 1;Q представляет собой С1-С4алкил, необязательно замещенный одним заместителем Ra;R1 представляет собой С5-С6алкил, С5-С6алкенил или С5-С6алкинил,R2 представляет собой Н, атом галогена, галогенС1-С4алкил, ORb, SRb или NRcRd;R3 представляет собой Н, атом галогена, галогенС1-С4алкил, С1-С4алкил, ORb, SRb или NRcRd;R4 представляет собой Н, атом галогена,...
Соединения индазола, фармацевтические препараты для ингибирования протеинкиназ и способы их применения

Номер патента: 4460
Опубликовано: 29.04.2004
Авторы: Джонсон Майкл Дэвид, Райх Зигфрид Хайнц, Палмер Синтия Луиза, Тенг Мин, Борчардт Аллен Дж., Бендер Стивен Ли, Уоллас Майкл Бреннан, Хуа Йе, Варни Майкл Дэвид, Кания Роберт Стивен, Луу Хип Те, Криппс Стефан Джеймс, Томас Кристин, Джонсон Теодор Отто Младший, Браганза Джон Ф., Коллинз Майкл Рэймонд, Темпчик-Расселл Анна Мария
МПК: C07D 231/56, A61K 31/416, A61P 35/00...
Метки: применения, протеинкиназ, препараты, фармацевтические, соединения, ингибирования, способы, индазола
Формула / Реферат:
1. Соединение формулы I в которой R1 представляет собой 6-12-членный арил или 5-12-членный гетероарил или группу формулы CH=CH-R3 или CH=N-R3, где R3 является незамещенным C1-C8алкилом, C1-C8алкилом, C2-C8алкенилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом или 5-12-членным гетероарилом, и R1 является необязательно замещенным алкилом C1-C8; и R2 представляет собой 6-12-членный арил или 5-12-членный гетероарил,...
Соединения, обладающие антибактериальной активностью в отношении clostridium

Номер патента: 21567
Опубликовано: 30.07.2015
Авторы: Гийемон Жером Эмиль Жорж, Люни Насер, Рабуассон Пьер Жан-Мари Бернар
МПК: A61P 31/04, A61K 31/53, C07D 487/04...
Метки: обладающие, clostridium, отношении, антибактериальной, активностью, соединения
Формула / Реферат:
1. Соединение формулы (I)включая любые его стереохимические изомерные формы и таутомеры, гдеR1 и R2, каждый независимо, выбраны из водорода, галогена, гидрокси, C1-6алкила, полигалоген-C1-6алкила, C1-6алкилокси или полигалоген-C1-6алкилокси;R3 представляет собой гидрокси, амино, моно- или ди(С1-4алкил)амино;R4 представляет собой водород или C1-4алкил;X представляет собой азот или CR5, где R5 представляет собой водород, галоген или C1-4алкил;при...
Предыдущий патент: Производные бициклических гетероциклов для лечения пульмонарной артериальной гипертонии
Следующий патент: Способ и композиция для улучшенного извлечения углеводородов
Случайный патент: Игровая система